

Abstract
The interleukin (IL)-36 cytokine family plays an essential role in inflammatory processes in the skin and is implicated in the pathogenesis of psoriasis. This study explored the role of IL-36 in psoriasis and investigated the molecular mechanism involved in tumour necrosis factor-α (TNFα)/IL-17A-mediated IL-36 induction. In human keratinocytes IL-36 expression was strongly upregulated by combined TNFα and IL-17A stimulation. Moreover, IκBζ, encoded by NFKBIZ, was identified as a key regulator required for TNFα/IL-17A-induced IL-36γ expression. TNFα/IL-17A-induced IL-36γ expression also involved the nuclear factor κB (NF-κB), p38 mitogen-activated protein kinase and ERK1/2 signalling pathways. Furthermore, a specific NF-κB DNA-binding site in the promoter region of IL36G responsible for the TNFα/IL-17A-induced IL36G gene expression was identified. Finally, in a cohort of patients with psoriasis receiving anti-IL-17A treatment, a positive correlation was found between the expression of NFKBIZ and IL36G. In conclusion, these data reveal a novel crucial regulatory mechanism by which TNFα and IL-17A regulate IL-36γ expression.
Key words: psoriasis, IL-36, IκBζ, keratinocytes, IL-17A
The interleukin (IL)-36 cytokine family consists of 4 members: IL-36α, IL-36β, IL-36γ and IL-36Ra which are encoded by IL36A, IL36B, IL36G and IL36RN, respectively (1, 2). The IL-36 cytokines bind to the IL-36 receptor. Binding of IL-36α, IL-36β or IL-36γ to the IL-36 receptor leads to recruitment and binding of the co-receptor IL-1RAcP (3). Activation of this receptor complex leads to activation of the nuclear factor κB (NF-κB) and mitogen-activated protein kinase (MAPK) signalling pathways, resulting in induced expression of inflammatory cytokines, such as IL-1β, IL-6 and IL-8 (4). In contrast, binding of IL-36Ra to the IL-36 receptor does not result in recruitment of IL-1RAcP and therefore exerts antagonistic effects (5). The role of IL-36Ra as a receptor antagonist is supported by the discovery of loss-of-function mutations in IL36RN in patients with generalized pustular psoriasis (6). All IL-36 cytokines possess multiple roles in host immunity and inflammatory processes, making them potential treatment targets in inflammatory diseases (7, 8).
SIGNIFICANCE
Psoriasis is a common chronic skin disease. The skin cells play a crucial role in the psoriasis disease mechanism by producing key disease promoting factors such as interleukin-36γ and IκBζ in response to interleukin-17A and tumour necrosis factor-α. In this study, we identify IκBζ as an essential regulatory factor for interleukin-17A- and tumour necrosis factor α-mediated induction of interleukin-36γ in human skin cells. Moreover, we identified a specific DNA sequence in the promoter region of IL36G (the gene encoding interleukin-36γ) which was responsible for the interleukin-36γ expression. In addition, in a cohort of psoriasis patients receiving anti-interleukin-17A treatment, we demonstrated a strong correlation between the expression of IκBζ and interleukin-36γ.
Psoriasis is a chronic inflammatory skin disease characterized by a strong Th17 response and an increased expression of a number of proinflammatory cytokines, including IL-17A, TNFα, and IL-23 (9). Stimulation of keratinocytes with IL-17A and TNFα increases the expression of IL-36α, IL-36β and IL-36γ. Furthermore, these IL-36 cytokines increase the expression of TNFα, IL-8, IL-6 as well as IL-36 cytokines themselves (10). Accumulating evidence suggests that IL-36 cytokines are important in psoriasis. The IL-36 cytokines IL-36α, IL-36γ and IL-36Ra have been found to be highly overexpressed in lesional skin from psoriatic patients and seem to correlate with IL-17A expression (8, 10). Interestingly, the level of IL-36γ correlates with the extent of skin inflammation and the IL-36γ serum level has been found to correlate with disease activity in psoriasis (10, 11). In addition, a recent study identified polymorphisms in the IL36G gene, which were associated with plaque psoriasis (12).
Interestingly, transgenic expression of IL-36α promotes psoriasis-like skin abnormalities in mice (13). Moreover, Th17 cell-derived cytokines induce the expression of IL-36 cytokines both in skin of mice and in human keratinocytes (10). In the imiquimod-induced psoriasis model, blocking of the IL-36 receptor resulted in less severe skin lesions, decreased neutrophil infiltration and reduced chemokine (CXCL-1, CXCL-2, CXCL-5) and cytokine (IL-36γ, IL-17A, TNFα, IL-22) expression (14). Thus, IL-36 appears to have a central role in psoriatic skin disease and, together with IL-17, drives potent feedback loops reinforcing proinflammatory gene expression. Despite its importance for skin inflammation, however, the regulatory mechanisms involved in IL-36 expression remain unknown. This study explores and characterizes these underlying molecular mechanisms.
MATERIALS AND METHODS
Psoriatic patients
Biopsies from 14 psoriatic patients treated with secukinumab were taken as described previously (15). Briefly, 4-mm skin punch biopsies were taken on days 0, 4, 14, 42 and 84 during treatment. A target lesion was chosen, and biopsies collected from lesional and non-lesional psoriatic skin at baseline. These biopsies were used for correlation analysis of NFKBIZ and IL36G expression. In another cohort of 12 psoriatic patients and 7 healthy controls 4-mm skin punch biopsies were collected. The biopsies were taken from the centre of a chronic plaque and non-lesional psoriatic skin from either the upper or the lower extremities. Biopsies from lesional and non-lesional psoriatic skin were taken as paired samples from the same body region. The study was approved by the Regional Ethical Committee of Region Midtjylland, Denmark, and was conducted in compliance with the Declaration of Helsinki. Signed informed consent was obtained from each patient.
Cell cultures
Primary human keratinocytes were obtained by trypsinization of skin from patients undergoing plastic surgery, as described (16). Second-passage keratinocytes were grown in keratinocyte serum-free medium (KSFM) (Life Technologies, Austin, TX, USA) at 37°C and 5% CO2 in an incubator 24 h before stimulation with TNFα (10 ng/ml) and IL-17A (100 ng/ml), the medium was replaced by medium without growth factors. In some experiments, cells were preincubated with a p38 MAPK inhibitor SB202190 (10 µM), an inhibitor of nuclear factor κB kinase 2 (IKK2) SC-514 (50 µM), a JNK inhibitor SP600125 (20 µM), or an ERK1/2 inhibitor PD98059 (50 µM) (all from Calbiochem, La Jolla, CA, USA) for 45 min before stimulation.
IL36G reporter plasmid construct
The human IL36G promoter was cloned into the pGL4.10[luc2] vector by Genscript (Piscataway, NJ, USA), generating an IL36G_1632-Luc2 reporter plasmid as described previously (17). Truncation of this promoter fragment was made by Genscript creating 3 new plasmids: IL36G_1427-Luc2, IL36G_1151-Luc2 and IL36G_887-Luc2. Five transcription factor binding sites were mutated in IL36G_1427-Luc2: IL36G_ΔOCT1/3 (containing a mutated OCT1/3 binding site, –573 to –559 bp), IL36G_ΔCEBP/β (containing a mutated CEPB/β binding site, –467 to –453 bp), IL36G_ΔNF-κB/a (containing a mutated NF-κB binding site, –337 to –323 bp), IL36G_ΔNF-κB/b (containing a mutated NF-κB binding site, –337 to –323 bp) and IL36G_ΔOCT1 (containing a mutated OCT1 binding site, –160 to –142 bp). Details of the constructs are shown in Table SI1.
Electrophoretic mobility shift assay
The oligonucleotides used were: NF-κB: 5´-ACTCTGGGAAATT-CCCTTAGTT-´3 and NF-κB mutant: 5´-ACTCTATACAGG-GAACGTAGTT-´3 (the mutation is underlined). Isolation of the nuclear fraction of human keratinocytes, labelling of oligonucleotides and the gel shift assay were performed as described previously (18). Briefly, the oligonucleotides were labelled by T4 polynucleotide kinase (Promega, Madison, WI, USA) in T4 kinase buffer (Promega) and then purified on a nick column (Illustra™ NICK columns sephadex™ G-50 DNA grade, GE Healthcare, Amersham, UK). Nuclear protein (1–2 µg) was incubated with the 32P-labelled oligonucleotides, separated on a 6% DNA Retardation Gel (Invitrogen, Carlsbad, CA, USA) and visualized on X-ray film. Supershifts were performed with antibody against p50 and p65 (cat. number sc-7178 and sc-7151, respectively; Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Statistical analysis
Statistical analysis was performed using Student’s t-test. If the combined TNFα and IL-17A stimulation was significantly higher than the additive effect of stimulation with TNFα and IL-17A alone, the effect was considered synergistic. A probability of p < 0.05 was regarded as statistically significant.
Supplementary Materials and Methods can be found in Appendix S11.
RESULTS
Interleukin-36 gene expression is increased in psoriatic skin
The gene expression pattern of IL36 in psoriatic skin has been investigated previously, but conflicting data exist (8, 19). The current study examined the mRNA expression of IL36A, IL36B, IL36G and IL36RN in skin biopsies from 12 psoriatic patients and 7 healthy controls. The mRNA expression of all 4 IL36 members was significantly increased in lesional psoriatic skin compared with non-lesional skin from the same patient (Fig. S11). Unlike mRNA expression of IL36A, IL36G and IL36RN, transcription of IL36B was much weaker and did not significantly differ between samples from lesional psoriatic skin compared with healthy skin controls. Furthermore, in agreement with previous data (11), IL36G and IL36RN mRNA expression was significantly increased in non-lesional psoriatic skin compared with skin of healthy controls (Fig. S1c and d1). These studies strongly indicate a role of IL-36 cytokines in the pathogenesis of psoriasis.
Tumour necrosis factor-α and interleukin-17A synergistically induce interleukin-36 expression in human keratinocytes
TNFα and IL-17A are key cytokines in the pathogenesis of psoriasis, and combined stimulation of human keratinocytes with the 2 cytokines results in a synergistic induction of a number of psoriasis-associated genes (20–26). Thus, in order to investigate the effect of TNFα, IL-17A and their combination on IL-36 expression, human keratinocytes were stimulated for various timepoints. Combined TNFα and IL-17A (TNFα/IL-17A) stimulation for 6 and 12 h significantly increased IL36A mRNA expression (Fig. 1a). The same tendency was observed after TNFα/IL-17A stimulation for 24 h. A significant induction of IL36B expression was seen after stimulation with IL-17A and TNFα/IL-17A for 12 h and after stimulation with TNFα and TNFα/IL-17A for 24 h (Fig. 1b). Interestingly, combined stimulation with TNFα and IL-17A for 12 and 24 h resulted in a synergistic and significant induction of IL36B mRNA. IL36G expression was significantly increased by IL-17A stimulation alone for all time-points investigated, and a synergistic induction of IL36G expression was observed for all time-points after TNFα/IL-17A stimulation (Fig. 1c). A significant synergistic induction of IL36RN mRNA upon TNFα/IL-17A stimulation was demonstrated after 24 h. Combined TNFα and IL-17A stimulation for 6 and 12 h, significantly, but not synergistically, increased the IL36RN expression compared with vehicle treatment (Fig. 1d). In addition, stimulation with TNFα for 24 h resulted in significant induction of IL36RN mRNA.
Fig. 1.

Tumour necrosis factor-á (TNFá) and interleukin (IL)-17A induce IL-36 in keratinocytes in a synergistic manner. (a–d) Cultured human keratinocytes were stimulated with TNFá and/or IL-17A for different time-points. The mRNA expression of (a) IL36A, (b) IL36B, (c) IL36G and (d) IL36RN was analysed by qPCR (n = 6). RPLP0 was used as a reference gene for normalization. (e) Protein levels of IL-36á, IL-36â, IL-36ã and IL-36Ra were analysed by western blotting. â-actin was used as a loading control (n = 3). Results from qPCR are expressed as mean ± standard deviation. *p < 0.05 compared with vehicle (Veh), **p < 0.05 compared with the additive values of TNFá and IL-17A stimulation separately.
This study also analysed whether the increased mRNA expression was paralleled by increased protein production of IL-36 cytokines. The protein level was measured over a 48-h period and, in agreement with the mRNA data, a highly increased induction of IL-36α, IL-36γ and IL-36Ra after TNFα/IL-17A stimulation was demonstrated (Fig. 1e). In contrast, only a minor increase in the protein level of IL-36β was observed (Fig. 1e). As TNFα/IL-17A stimulation resulted in the highest fold induction of IL36G expression, the molecular mechanism underlying this induction was further investigated.
Interleukin-36γ is induced by a p38 mitogen-activated protein kinase-, ERK1/2- and nuclear factor κB (NF-dependent mechanism)
To further investigate the molecular mechanism mediating the synergistic effect of TNFα and IL-17A on IL36G gene expression, human keratinocytes were preincubated with inhibitors of the MAPK or the NF-κB signalling pathway. Preincubation with the p38 MAPK inhibitor SB202190, the ERK1/2 inhibitor PD98059, or the NF-κB inhibitor SC-514 significantly reduced TNFα/IL-17A-induced IL36G expression (Fig. 2a). In contrast, preincubation with the JNK1/2 inhibitor SP600125 did not significantly affect IL36G expression. IL-36γ protein level was also examined after preincubation with the indicated inhibitors. Similar alterations in IL-36γ protein levels were found as observed for IL36G mRNA expression (Fig. 2b). JNK1/2 inhibition resulted in a significant increase in TNFα-mediated induction of IL36G mRNA expression, but did not seem to influence IL-17A- and TNFα/IL-17A-mediated induction of IL36G mRNA. The same effect of the JNK1/2 inhibitor on IL-36γ expression was observed at the protein level (Fig. 2b).
Fig. 2.

Characterization of tumour necrosis factor-α (TNFα)/interleukin (IL)-17A-induced IL-36γ expression. Human keratinocytes were incubated with the p38 mitogen-activated protein kinase (MAPK) inhibitor SB202190, the ERK1/2 inhibitor PD98059, the IKK2 inhibitor SC-514, the JNK inhibitor SP600125 or with vehicle (Veh) for 45 min before stimulation with TNFα and/or IL-17A for 24 h. (a) IL36G mRNA expression was measured by qPCR (n = 3). Results are expressed as mean ± standard deviation. RPLP0 mRNA expression was used for normalization. (b) The protein level of IL-36γ was measured by western blotting with β-actin as a loading control (n = 3). *p < 0.05 compared with TNFα- and/or IL-17A-stimulated cells without inhibitors.
Tumour necrosis factor-α/interleukin-17A-induced IL36G gene expression is mediated by IκBζ
IκBζ is a transcriptional coactivator, which is rapidly induced by IL-17A. It is known to be critical for IL-17A-driven effects by exerting its transcription-enhancing activity on inflammatory secondary response genes (27). In order to characterize the role of IκBζ in TNFα- and/or IL-17A-mediated IL36G gene expression, siRNA was used to knockdown NFKBIZ, the gene encoding IκBζ. A strong knockdown of IκBζ expression at both the protein and mRNA level was shown in keratinocytes transfected with IκBζ siRNA compared with keratinocytes transfected with control siRNA (siCon) (Fig. 3a and b). IκBζ siRNA transfection before IL-17A and TNFα/IL-17A stimulation significantly diminished IL36G expression compared with control siRNA-transfected cells, suggesting that IκBζ plays a central role in expression of IL36G (Fig. 3c). Interestingly, IκBζ was not involved in TNFα/IL-17A-induced mRNA expression of IL36A, IL36B and IL36RN (Fig. S21), demonstrating that, in the regulation of the IL-36 cytokines, IκBζ seems to be specific for IL-17A- and TNFα/IL-17A-induced IL36G expression.
Fig. 3.

IκBζ is involved in tumour necrosis factor-α (TNFα)/interleukin (IL)-17A-induced IL36G mRNA expression. Human keratinocytes were transfected with IκBζ or control siRNA (siCon) 24 h before stimulation with TNFα or/and IL-17A for (a and b) 1 h or (c) 24 h. IκBζ knockdown was measured by (a) western blotting (n = 6) using β-actin as a loading control or by (b) qPCR (n = 4) using RPLP0 as a reference gene. (c) qPCR was used to analyse IL36G mRNA expression (n = 6). RPLP0 was used as a reference gene for normalization. Results from qPCR are expressed as mean ± SD. *p < 0.05 compared with the corresponding siCon samples.
Identification of the specific nuclear factor κB DNA-binding site essential for tumour necrosis factor-α/interleukin-17A-induced IL36G promoter activity
IκBζ, which is devoid of a DNA-binding region, associates with DNA-binding NF-κB proteins p50 or p52, and thereby gains access to DNA-regulatory regions of distinct NF-κB target genes (28). Because IL-36γ was regulated by a mechanism involving IκBζ, the current study next examined the IL36G promoter region. Thus, 1632 bp of the IL36G promotor were ligated in front of the firefly luciferase gene, and this plasmid construct was used to transiently transfect human keratinocytes. TNFα and/or IL-17A stimulation of transfected cells significantly induced IL36G promoter activity (Fig. 4a), indicating that IL36G is regulated at the transcriptional level after TNFα and/or IL-17A stimulation. To uncover the region(s) of the IL36G promoter involved in this induction, the full-length IL36G-1632-Luc2 promoter was further analysed using truncated constructs: IL36G_1427-Luc2, IL36G_1151-Luc2 and IL36G_887-Luc2. Using these deletion constructs, the current study demonstrated that truncation to IL36G_887-Luc2 resulted in a significant decrease in IL36G promoter activity (Fig. 4b). Because a small, but insignificant, reduction in IL36G promoter activity was observed upon truncation of the plasmid from IL36G_1427 to IL36G_1151 (Fig. 4b), the IL36G_1427-Luc2 plasmid was chosen for further investigation, to ensure that no potentially important transcription factor binding sites were overlooked.
Fig. 4.

Mutations in a specific nuclear factor κB (NF-κB) DNA binding site abolish IL36G promoter activity. (a) Human keratinocytes were transfected with a luciferase plasmid containing 1632 bp of the IL36G promoter together with an internal control (Renilla). Then keratinocytes were stimulated with TNFα and/or IL-17A for 24 h (n = 5). *p < 0.05 compared with vehicle (Veh). (b) Cells were transfected with different IL36G-promoter plasmids before stimulated with TNFα/IL-17A for 24 h (n = 7). *p<0.05 compared with cells transfected with the IL36G_1632 plasmid. (c) The IL36G promoter constructs mutated in different combinations are shown. (d) Keratinocytes were transfected with IL36G_1427 (WT) or mutated (Δ) forms of the IL36G_1427-promoter plasmid together with Renilla, before stimulated with TNFα/IL-17A for 24 h (n = 9). *p < 0.05 compared with TNFα/IL-17A-stimulated WT cells. All results are expressed as mean ± SD. (e) Keratinocytes were stimulated with TNFα or/and IL-17A for 1 h before NF-κB DNA-binding activity was analysed by electrophoretic mobility shift assay. Antibodies against p65 and p50 caused a supershift of the nuclear complex. In separate experiments, both wildtype and mutated NF-κB oligo were analysed. Representative gels from 3 (left) and 2 (right) different experiments are shown.
Using the Genomatix software we identified 5 transcription factor binding sites previously reported to be associated with IL-17A-mediated effects. These binding sites were exchanged by point mutations in IL36G_1427-Luc2 to identify their respective roles in the induction of IL36G: IL36G_ΔOCT1/3, IL36G_ΔCEBP/β, IL36G_ΔNF-κB/a, IL36G_ΔNF-κB/b and IL36G_ΔOCT1 (Fig. 4c). Interestingly, using these mutated constructs the current study found that a specific point mutation in the proximal NF-κB-binding site ΔNF-κB/b significantly decreased TNFα/IL-17A-induced IL36G promoter activity (Fig. 4d). This demonstrates that the proximal, but not the more distal, NF-κB-binding site plays a crucial role in TNFα/IL-17A-mediated activation of the IL36G promoter.
Knowing that TNFα/IL-17A-induced activation of the IL36G promoter is dependent on one specific NF-κB DNA binding site, the current study next investigated whether NF-κB binds to this specific DNA binding site. An oligonucleotide matching the putative NF-κB DNA binding site was designed. Stimulation with TNFα and/or IL-17A clearly increased the DNA binding of NF-κB to its specific binding site (bp –286 to –272) with the strongest binding activity resulting from TNFα stimulation alone and from combined TNFα and IL-17A stimulation (Fig. 4e). The specificity of the bound complex was determined by supershift assay, as incubation of the DNA-protein complex with anti-p65 and anti-p50 antibodies resulted in a shift of the bands indicating the specificity (Fig. 4e). Furthermore, no DNA binding was seen when the nuclear extracts were incubated with an oligonucleotide mutated in the NF-κB binding site. Hence, the specificity of the binding sequence is essential for NF-κB DNA binding (Fig. 4e).
Expression of NFKBIZ and IL36G is positively correlated in psoriatic skin during anti-IL-17A treatment
Because IκBζ was shown to be essential for TNFα/IL-17A-induced expression of IL-36γ and because IL36G expression was highly increased in lesional psoriatic skin, the study next analysed for a potential correlation between NFKBIZ and IL36G mRNA expression during anti-IL-17A treatment in patients with psoriasis. In a cohort of 14 patients with psoriasis all treated with secukinumab, an anti-IL-17A drug, as described previously (15), a positive correlation was demonstrated between NFKBIZ and IL36G mRNA expression in lesional psoriatic skin during secukinumab treatment (Fig. 5). These data support the in vitro data showing that IκBζ is a transcriptional regulator of IL36G expression and suggest a role for IκBζ-mediated regulation of IL36G in psoriatic skin.
Fig. 5.
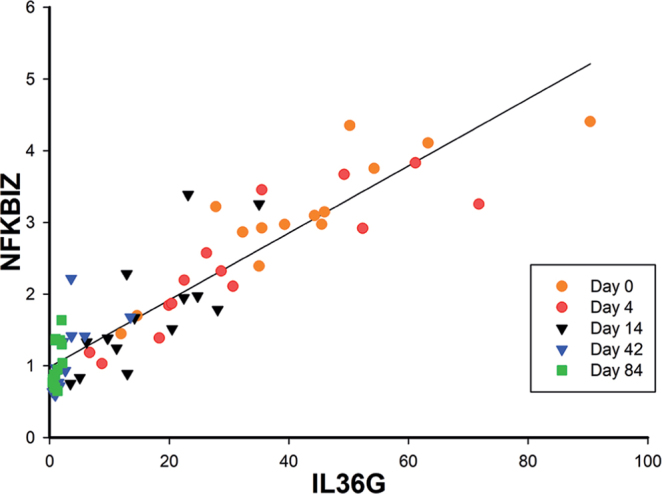
NFKBIZ and IL36G expression are positively correlated in psoriatic skin during anti-interleukin (IL)-17A treatment. Quantitative PCR was used to analyse the mRNA expression of NFKBIZ and IL36G in punch biopsies taken from lesional psoriatic skin during secukinumab treatment. RPLP0 was used as a reference gene for normalization. The correlation between the mRNA expression levels of NFKBIZ and IL36G was analysed as indicated in the figure.
DISCUSSION
Accumulating evidence suggests that IL-36 signalling plays a pivotal role in the pathogenesis of psoriasis (8, 10, 14, 29). IL-36 cytokines are expressed by a variety of cell types, but are abundantly produced by keratinocytes upon IL-17A and TNFα stimulation (10, 19, 30–32). IL-17A and TNFα are key effector cytokines in the pathogenesis of psoriasis, and numerous proinflammatory cytokines are synergistically induced in keratinocytes after combined IL-17A and TNFα stimulation (20, 31). Although different studies have investigated the expression of IL-36 cytokine family members after IL-17A and TNFα stimulation (19, 30, 32) a comprehensive study investigating the precise mechanism by which IL-17A and TNFα regulate the expression of IL-36 in human keratinocytes has not been conducted. In this study, we characterize IL-36 in psoriasis and delineate the underlying molecular mechanism through which IL-17A cooperates with TNFα to induce a synergistic response on the IL-36γ expression in human keratinocytes.
We found that all 4 IL-36 family members were inducible in primary human keratinocytes upon TNFα/IL-17A stimulation; however, only induction of IL-36γ was mediated by an IκBζ-dependent mechanism. Because IκBζ is described to function as an activator of NF-κB target genes and because the promoter regions of IL36A, IL36B and IL36RN in contrast to IL36G do not contain putative NF-κB DNA binding sites, this might explain why only IL36G is regulated through an IκBζ-dependent mechanism. In addition to what was observed in the keratinocytes after TNFα/IL-17A stimulation, we also found an increased level of IL-36Ra in lesional psoriatic skin compared with non-lesional psoriatic skin and healthy skin. This observation might be explained by the fact that IL-36Ra, being an antagonist, is produced in high levels to dampen the inflammatory response. Previous studies by our group and others demonstrated IκBζ to play an important role in psoriasis development by mediating IL-17A signalling resulting in induction of proinflammatory mediators in keratinocytes (27, 33). Moreover, recent data identified IκBζ as a crucial regulator of IL-36α- and IL-36γ-driven psoriasis-associated gene expression in human keratinocytes (29). Thus, IκBζ seems to be a key regulatory protein, not only for IL-17A-driven effects, but also for the signalling of specific IL-17A-induced signature genes, such as IL-36α and IL-36γ in human keratinocytes (29). In a previous study conducted in human colonic myofibroblasts, IL-1β was shown to induce the expression of IL-36γ by a mechanism involving the intracellular signalling pathways p38 MAPK, ERK1/2 and NF-κB (34). In agreement with this, we found that TNFα/IL-17A-induced IL-36γ expression not only was mediated by an IκBζ-dependent mechanism, but also involved the p38 MAPK, ERK1/2 and NF-κB signalling pathways, because inhibition of each of these signalling pathways significantly attenuated IL-36γ expression. These intracellular pathways all play a role in psoriasis, and have been shown to have increased activity in psoriatic skin (35–37).
Investigating IL-36 expression in skin samples from psoriatic patients, we found that all 4 IL-36 family members were increased in lesional compared with non-lesional psoriatic skin. However, IL36B had a relatively modest expression level in psoriatic skin compared with the other IL-36 family members. Among the proinflammatory IL-36 members we observed a 2-fold increase in IL36B expression between lesional and non-lesional psoriatic skin, whereas we observed a ~1,000-fold and a ~50-fold induction of IL36A and IL36G, respectively, suggesting that these IL-36 family members might be the dominating members in the pathogenesis of plaque psoriasis. This is also supported by previous data by Mahil et al. (14), which suggests a prominence of IL-36γ in the early pathogenesis of psoriasis. Likewise, D’Erme et al. (38) highlight IL-36γ as a valuable biomarker in psoriasis and, just recently, polymorphisms in the IL36G gene have been associated with plaque psoriasis (12). Thus, several studies, including this one, indicate that, among the IL-36 family members, IL-36γ especially might be an essential cytokine in the pathogenesis of plaque psoriasis. In this context, the development of new drugs targeting IL-36γ as a therapeutic strategy for plaque psoriasis might represent an alternative to anti-IL-17A or anti-TNFα therapy. Another approach could be to target IκBζ, which constitutes a regulatory key point not only for IL-17- and IL-36-driven effects (27, 29), but also for TNFα/IL-17A-mediated induction of IL-36γ in human keratinocytes, as shown in this study. In further support of IκBζ being involved in the regulation of IL-36γ, we demonstrated a clear correlation between the expression of NFKBIZ and IL36G in psoriatic skin during secukinumab treatment.
In conclusion, this study delineates the molecular mechanism by which TNFα and IL-17A mediate the induction of IL-36γ in human keratinocytes, and identify IκBζ as a master regulator of IL-36γ expression, whereas IκBζ seems to be dispensable for the regulation of the other IL-36 family members.
The authors have no conflicts of interest to declare.
REFERENCES
- 1.Gresnigt MS, van de Veerdonk FL. Biology of IL-36 cytokines and their role in disease. Semin Immunol 2013; 25: 458–465. [DOI] [PubMed] [Google Scholar]
- 2.Dunn E, Sims JE, Nicklin MJ, O’Neill LA. Annotating genes with potential roles in the immune system: six new members of the IL-1 family. Trends Immunol 2001; 22: 533–536. [DOI] [PubMed] [Google Scholar]
- 3.Towne JE, Garka KE, Renshaw BR, Virca GD, Sims JE. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J Biol Chem 2004; 279: 13677–13688. [DOI] [PubMed] [Google Scholar]
- 4.Gabay C, Towne JE. Regulation and function of interleukin-36 cytokines in homeostasis and pathological conditions. J Leukoc Biol 2015; 97: 645–652. [DOI] [PubMed] [Google Scholar]
- 5.Towne JE, Renshaw BR, Douangpanya J, Lipsky BP, Shen M, Gabel CA, et al. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36alpha, IL-36beta, and IL-36gamma) or antagonist (IL-36Ra) activity. J Biol Chem 2011; 286: 42594–42602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Onoufriadis A, Simpson MA, Pink AE, Di Meglio P, Smith CH, Pullabhatla V, et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am J Hum Genet 2011; 89: 432–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chu M, Wong CK, Cai Z, Dong J, Jiao D, Kam NW, et al. Elevated expression and pro-inflammatory activity of IL-36 in patients with systemic lupus erythematosus. Molecules 2015; 20: 19588–19604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boutet MA, Bart G, Penhoat M, Amiaud J, Brulin B, Charrier C, et al. Distinct expression of interleukin (IL)-36alpha, beta and gamma, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn’s disease. Clin Exp Immunol 2016; 184: 159–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Meglio P, Villanova F, Nestle FO. Psoriasis. Cold Spring Harb Perspect Med 2014; 4: a015354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carrier Y, Ma HL, Ramon HE, Napierata L, Small C, O’Toole M, et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J Invest Dermatol 2011; 131: 2428–2437. [DOI] [PubMed] [Google Scholar]
- 11.Keermann M, Koks S, Reimann E, Abram K, Erm T, Silm H, et al. Expression of IL-36 family cytokines and IL-37 but not IL-38 is altered in psoriatic skin. J Dermatol Sci 2015; 80: 150–152. [DOI] [PubMed] [Google Scholar]
- 12.Traks T, Keermann M, Prans E, Karelson M, Loite U, Koks G, et al. Polymorphisms in IL36G gene are associated with plaque psoriasis. BMC Med Genet 2019; 20: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blumberg H, Dinh H, Trueblood ES, Pretorius J, Kugler D, Weng N, et al. Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J Exp Med 2007; 204: 2603–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mahil SK, Catapano M, Di Meglio P, Dand N, Ahlfors H, Carr IM, et al. An analysis of IL-36 signature genes and individuals with IL1RL2 knockout mutations validates IL-36 as a psoriasis therapeutic target. Sci Transl Med 2017; 9: eaan2514. [DOI] [PubMed] [Google Scholar]
- 15.Bertelsen T, Ljungberg C, Litman T, Huppertz C, Hennze R, Rønholt K, et al. IκBζ is a key player in the antipsoriatic effects of secukinumab. J Allergy Clin Immunol 2020; 145: 379–390. [DOI] [PubMed] [Google Scholar]
- 16.Johansen C. Generation and culturing of primary human keratinocytes from adult skin. J Vis Exp 2017; 130: 56863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bachmann M, Scheiermann P, Hardle L, Pfeilschifter J, Muhl H. IL-36gamma/IL-1F9, an innate T-bet target in myeloid cells. J Biol Chem 2012; 287: 41684–41696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johansen C, Iversen L, Ryborg A, Kragballe K. 1alpha,25-dihydroxyvitamin D3 induced differentiation of cultured human keratinocytes is accompanied by a PKC-independent regulation of AP-1 DNA binding activity. J Invest Dermatol 2000; 114: 1174–1179. [DOI] [PubMed] [Google Scholar]
- 19.Johnston A, Xing X, Guzman AM, Riblett M, Loyd CM, Ward NL, et al. IL-1F5, -F6, -F8, and -F9: a novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J Immunol 2011; 186: 2613–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johansen C, Bertelsen T, Ljungberg C, Mose M, Iversen L. Characterization of TNF-alpha- and IL-17A-mediated synergistic induction of DEFB4 gene expression in human keratinocytes through IκBζ. J Invest Dermatol 2016; 136: 1608–1616. [DOI] [PubMed] [Google Scholar]
- 21.Gabr MA, Jing L, Helbling AR, Sinclair SM, Allen KD, Shamji MF, et al. Interleukin-17 synergizes with IFNgamma or TNFalpha to promote inflammatory mediator release and intercellular adhesion molecule-1 (ICAM-1) expression in human intervertebral disc cells. J Orthop Res 2011; 29: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goldberg M, Nadiv O, Luknar-Gabor N, Agar G, Beer Y, Katz Y. Synergism between tumor necrosis factor alpha and interleukin-17 to induce IL-23 p19 expression in fibroblast-like synoviocytes. Mol Immunol 2009; 46: 1854–1859. [DOI] [PubMed] [Google Scholar]
- 23.Honda K, Wada H, Nakamura M, Nakamoto K, Inui T, Sada M, et al. IL-17A synergistically stimulates TNF-alpha-induced IL-8 production in human airway epithelial cells: a potential role in amplifying airway inflammation. Exp Lung Res 2016; 42: 205–216. [DOI] [PubMed] [Google Scholar]
- 24.Liu Y, Mei J, Gonzales L, Yang G, Dai N, Wang P, et al. IL-17A and TNF-alpha exert synergistic effects on expression of CXCL5 by alveolar type II cells in vivo and in vitro. J Immunol 2011; 186: 3197–3205. [DOI] [PubMed] [Google Scholar]
- 25.Nonaka M, Ogihara N, Fukumoto A, Sakanushi A, Kusama K, Pawankar R, et al. Synergistic induction of macrophage inflammatory protein-3alpha;/CCL20 production by interleukin-17A and tumor necrosis factor-alpha; in nasal polyp fibroblasts. World Allergy Organ J 2009; 2: 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shinjo T, Iwashita M, Yamashita A, Sano T, Tsuruta M, Matsunaga H, et al. IL-17A synergistically enhances TNFalpha-induced IL-6 and CCL20 production in 3T3-L1 adipocytes. Biochem Biophys Res Commun 2016; 477: 241–246. [DOI] [PubMed] [Google Scholar]
- 27.Johansen C, Mose M, Ommen P, Bertelsen T, Vinter H, Hailfinger S, et al. IκBζ is a key driver in the development of psoriasis. Proc Natl Acad Sci U S A 2015; 112: E5825–E5833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamamoto M, Yamazaki S, Uematsu S, Sato S, Hemmi H, Hoshino K, et al. Regulation of Toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein IκBζ. Nature 2004; 430: 218–222. [DOI] [PubMed] [Google Scholar]
- 29.Muller A, Hennig A, Lorscheid S, Grondona P, Schulze-Osthoff K, Hailfinger S, et al. IkappaBzeta is a key transcriptional regulator of IL-36-driven psoriasis-related gene expression in keratinocytes. Proc Natl Acad Sci U S A 2018; 115: 10088–10093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfaff CM, Marquardt Y, Fietkau K, Baron JM, Luscher B. The psoriasis-associated IL-17A induces and cooperates with IL-36 cytokines to control keratinocyte differentiation and function. Sci Rep 2017; 7: 15631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiricozzi A, Guttman-Yassky E, Suarez-Farinas M, Nograles KE, Tian S, Cardinale I, et al. Integrative responses to IL-17 and TNF-alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol 2011; 131: 677–687. [DOI] [PubMed] [Google Scholar]
- 32.Mercurio L, Morelli M, Scarponi C, Eisenmesser EZ, Doti N, Pagnanelli G, et al. IL-38 has an anti-inflammatory action in psoriasis and its expression correlates with disease severity and therapeutic response to anti-IL-17A treatment. Cell Death Dis 2018; 9: 1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muromoto R, Hirao T, Tawa K, Hirashima K, Kon S, Kitai Y, et al. IL-17A plays a central role in the expression of psoriasis signature genes through the induction of IkappaB-zeta in keratinocytes. Int Immunol 2016; 28: 443–452. [DOI] [PubMed] [Google Scholar]
- 34.Takahashi K, Nishida A, Shioya M, Imaeda H, Bamba S, Inatomi O, et al. Interleukin (IL)-1β Is a Strong Inducer of IL-36γ expression in human colonic myofibroblasts. PloS One 2015; 10: e0138423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johansen, C.E., Flindt, K., Kragballe, J., Henningsen, M., Westergaard, K., Kristiansen, L. Iversen. Inverse regulation of the nuclear factor-kB binding to the p53 and interleukin-8 kB response elements in lesional psoriatic skin. J Invest Dermatol 2005; 124: 1284–1292. [DOI] [PubMed] [Google Scholar]
- 36.Johansen C, Kragballe K, Westergaard M, Henningsen J, Kristiansen K, Iversen L. The mitogen-activated protein kinases p38 and ERK1/2 are increased in lesional psoriatic skin. Br J Dermatol 2005; 152: 37–42. [DOI] [PubMed] [Google Scholar]
- 37.Lizzul PF, Aphale A, Malaviya R, Sun Y, Masud S, Dombrovskiy V, et al. Differential expression of phosphorylated NF-kappaB/RelA in normal and psoriatic epidermis and downregulation of NF-kappaB in response to treatment with etanercept. J Invest Dermatol 2005; 124: 1275–1283. [DOI] [PubMed] [Google Scholar]
- 38.D’Erme AM, Wilsmann-Theis D, Wagenpfeil J, Holzel M, Ferring-Schmitt S, Sternberg S, et al. IL-36gamma (IL-1F9) is a biomarker for psoriasis skin lesions. J Invest Dermatol 2015; 135: 1025–1032. [DOI] [PubMed] [Google Scholar]