

Abstract
Under mild conditions (room temperature, 80 psi of H2) Cp*Rh(2-(2-pyridyl)phenyl)H catalyzes the selective hydrogenation of the C═C bond in α,β-unsaturated carbonyl compounds, including natural product precursors with bulky substituents in the β position and substrates possessing an array of additional functional groups. It also catalyzes the hydrogenation of many isolated double bonds. Mechanistic studies reveal that no radical intermediates are involved, and the catalyst appears to be homogeneous, thereby affording important complementarity to existing protocols for similar hydrogenation processes.
Graphical Abstract

INTRODUCTION
The catalytic hydrogenation of unsaturated organic compounds is a transformation of widespread importance in both academia and industry, particularly as applied for the preparation of pharmaceuticals, fragrances, and other fine chemicals.1 The 1,4-reduction of α,β-unsaturated carbonyl compounds has attracted particular attention, and much effort has been devoted to its development.2 Hydrogen sources other than molecular hydrogen,3 including silicon hydrides,4 formates,5 and alcohols,6 are often efficient and have become widely used.7,8 To date, the majority of methods possessing both high efficiency and selectivity have used transition metal promoters (Ir,6 Pd,9 Co,10,11 Ni,12 and others13) with a variety of supporting ligands,14,5 though there have also been recent disclosures of transition-metal-free hydrogenation reagents involving Se powder,15 borane,16 and electrons.17–19
Nevertheless, most 1,4-reductions still suffer from drawbacks, such as poor tolerance of sensitive functional groups and a lack of effectiveness with highly substituted, sterically encumbered substrates. Indeed, such steric constraints can sometimes lead to an undesired stereochemical outcome and/or prevent hydrogenation entirely. Such issues arose in the Snyder group’s recent total syntheses of the coccinellid alkaloids, including targets such as exochomine,20 arborisidine,21 and chilocorine C.22,23 For example, one of the final steps in the exochomine work required the selective 1,4-reduction of the hindered enone 1 to 2 (Scheme 1A). However, the dithiolane group, benzylic ketone, and acyl pyrrole all proved prone to reduction and/or side-product formation; the desired product was best obtained by reducing 1 with silanes in the presence of stoichiometric Mn(dpm)3, a procedure adapted from those reported by Magnus24 and Shenvi.25 Given this, and other related, examples, a robust catalytic procedure for the 1,4-reduction of α,β-unsaturated carbonyl compounds using H2 was viewed as highly desirable.
Scheme 1.
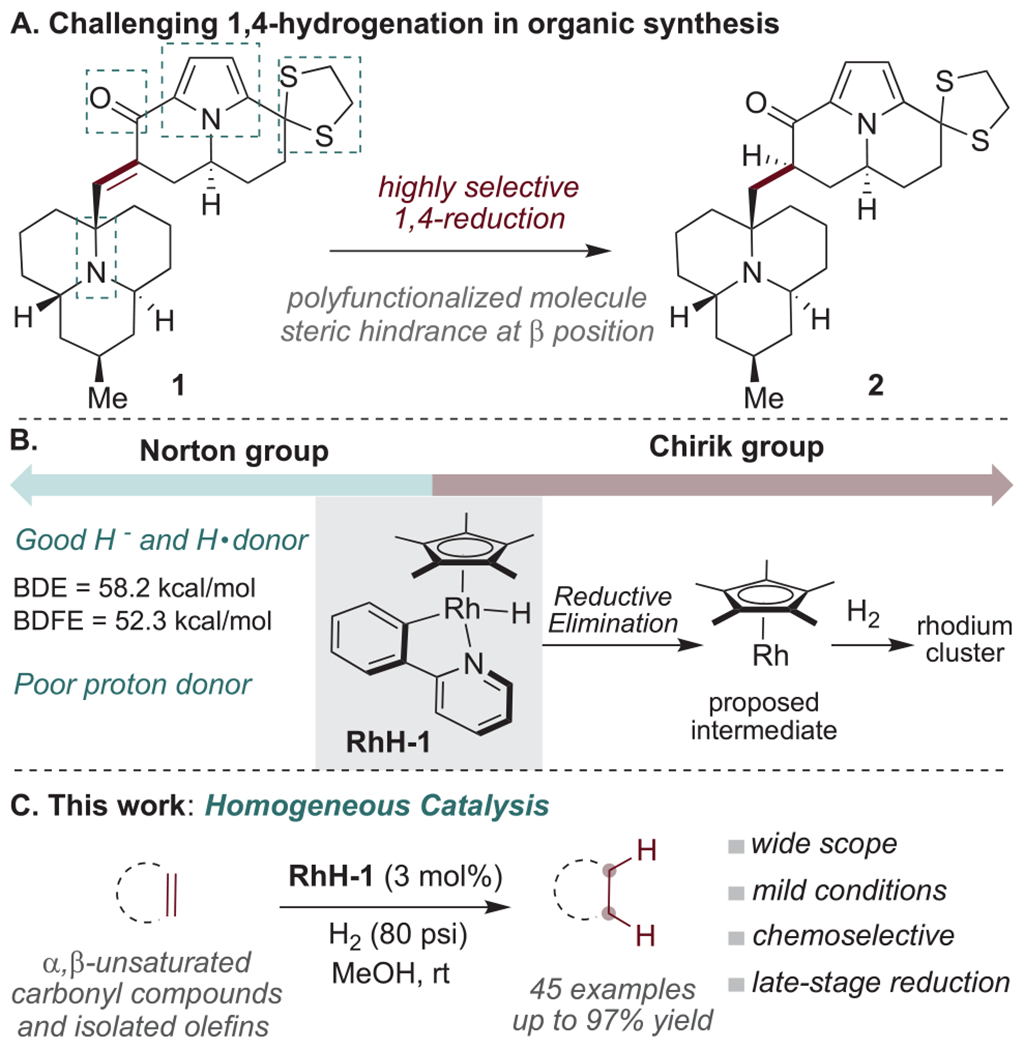
(A) A Challenging Substrate for 1,4-Hydrogenation, with Alternative Sites of Reaction; (B) Chirik Proposal for Mechanism of Action of (η5-C5Me5)Rh(ppy)H; (C) Homogeneous Hydrogenation of α,β-Unsaturated Carbonyl Compounds and Isolated Olefins Catalyzed by RhH-1
The Norton group initially attempted to employ the Cr [CpCr(CO)3H] and Co [Co(dmgBF2)2L2] (L = H2O, THF, etc.) catalysts that they had used26–30 for H· transfer from H2 to solve this problem, but neither gave any 2 from 1. We then considered (η5-C5Me5)Rh(ppy)H (ppy = 2-(2-pyridyl)-phenyl), RhH-1, developed in the Norton laboratory and shown to be a fast hydride and hydrogen atom donor, but a relatively poor proton donor (Scheme 1B).31,32 Related Cp*Rh systems have been shown to effectively catalyze arene and olefin hydrogenation.33–35 Indeed, in 2019 the Chirik group found that RhH-1 can catalyze the hydrogenation to ammonia of amides,36 nitrides,37 and related ligands.38 Very recently, the use of the same precatalyst for the hydrogenation of N-heteroarenes has been reported by the same group.39 They have proposed that, upon heating or irradiation, the reductive elimination of 2-phenylpyridine from RhH-1 can lead to the formation of catalytically active multimetallic clusters (and eventually nanoparticles), under varied H2 pressures (4–36 atm) at elevated temperatures (80–100 °C).
To our delight, we found that (η5-C5Me5)Rh(ppy)H (RhH-1) does indeed show activity for the hydrogenation of the C═C bonds of enones. Herein we describe a highly selective and mild procedure for catalyzing the C═C hydrogenation of α,β-unsaturated carbonyl compounds and isolated olefins (Scheme 1C) which works on an array of substrates with high chemoselectivity and functional group tolerance. We follow-up these studies of scope with mechanistic investigations which reveal that our catalyst appears to be behaving in a homogeneous, rather than heterogeneous, manner.
RESULTS AND DISCUSSION
As shown in Table 1, we selected chalcone 3 as a test enone to develop and optimize our rhodium-catalyzed 1,4-hydrogenation method. With 3 mol % RhH-1 and 80 psi of H2 gas in MeOH (0.05 M) at 23 °C, the reaction took 24 h to reach completion, affording reduction product 4 in 94% isolated yield (Table 1, entry 1). Lowering the catalyst loading or the pressure of H2 gas eroded the yield during the same time period (entries 2 and 3). Further, changing the catalyst to the benzo[h]quinoline derivative RhH-2 gave a slightly lower yield (entry 4), while the use of solvents other than MeOH also proved deleterious (entries 5–7). As shown by control experiments, both the H2 gas and the rhodium promoter are essential (entries 8 and 9).
Table 1.
Optimization of the Reaction Conditionsa

|
3 (0.20 mmol), RhH-1 catalyst (3 mol %), H2 (80 psi), MeOH (4 mL) at room temperature, 24 h.
NMR yields using CH2Br2 as internal standard.
Isolated yield.
As shown by the reaction scope in Table 2, the method displays excellent chemoselectivity with various α,β-unsaturated carbonyl compounds, which in all cases underwent 1,4-reduction exclusively to form the indicated products in high yields (with the colored bond marking the site of hydrogenation). As can be discerned, chalcones containing both electron-rich and electron-poor arenes are reduced appropriately, to 4–9, and the reduction of the precursor to 5 can be scaled up without compromising the overall yield. Related substrates containing aromatic heterocycles such as imidazole or thiophene also react smoothly, giving good yields of 10 and 11. Vinyl phenyl ketones with substituents at the α or β position also undergo 1,4-hydrogenation, making 12 and 13 in good yield. In addition, the vinyl methyl ketones in the substrates leading to 14–16 are selectively reduced in excellent yields, while the trisubstituted double bonds in 15 and 16 remain untouched. No 1,6-reduction product was detected along with 16. Pleasingly, the α,β-unsaturated esters, vinyl amide, and vinyl sulfones within products 17–22 all posed no problems even with steric hindrance at the α position (as in the precursor of 17). The substituted cyclic, α,β-unsaturated ester in 22 was not reduced.
Table 2.

|
Under the conditions of entry 1 in Table 1.
Isolated yields, average of two independent runs.
Using iPrOH as solvent instead of MeOH.
Gram scale (10 mmol reaction).
Critically, the scope of the Rh-catalyzed hydrogenation extends to dienes and to cyclic enones. Both of the conjugated double bonds leading to 23 and 24 were hydrogenated with high efficiency, giving these materials in almost quantitative yields. The C═C bonds of cyclic enones are also hydrogenated in 1,4-fashion (leading to 25–28). The cyclic enones found in products 29 and 30, possessing β ethoxy substituents, are not hydrogenated, although the isolated C═C bonds are. The chemoselectivity of the Rh-catalyzed hydrogenation is further illustrated by the fact that acetals (24 and 26), esters (22 and 25), aryl halides (21 and 23), and even unprotected alcohol (6 and 28) are well tolerated, leaving ample room for further derivatization, as desired.
Although we did not observe byproducts with 1,2-reduction for any of the substrates used in Table 2, we did find that the 1,4-reduction products (34–36) from α,β-unsaturated aldehydes (31–33) undergo slow, further 1,2-reduction to afford 37–39 (Table 3). We note that both aliphatic and aromatic substituents seem to be tolerated at the β position. Of particular interest, the hydrogenation of the intermediate aldehyde is considerably slower than the 1,4-hydrogenation of the initial enone, as judged by the reaction times required.
Table 3.

|
Under the conditions of entry 1 in Table 1.
Isolated yields, average of two independent runs.
Triethylamine (1.0 equiv) was added.
With these initial results in hand, we then returned to the highly substituted substrates that had caused difficulty for the Snyder group in their exochomine synthesis (cf. Scheme 1A).20 For example, Stryker’s reagent (H6Cu6L6) had given a sluggish reaction, with the principal product being the result of 1,6-reduction across the pyrrole ring. A similar 1,6-reduction result was obtained after one-electron reduction by SmI2; by contrast, catecholborane and DIBAL-H gave the 1,2-reduction product, while DIBAL-H with Cu(I), RedAl, Pd°/n-Bu3SnH, and sulfonylhydrazides (NBSH) gave no reaction. Specifically, we tried to hydrogenate somewhat simpler predecessors of 40 and 41 with our rhodium catalyst RhH-1 under our optimal conditions, and found that selective 1,4-hydrogenation of the C═C bonds of these two enones could be achieved in the presence of a dithiolane and a pyrrole, providing both 40 and 41 in high yields and diastereoselectivities (Table 3). The presence of the tert-butyl substituents caused no issues in these transformations, and the addition of Et3N did not suppress the formation of 40 (suggesting that the tertiary amine found in exochomine itself would not be problematic if executed on even more advanced intermediates).
In view of the hydrogenation of the isolated double bonds leading to 29 and 30, we have further explored the utility of RhH-1 as a catalyst for the hydrogenation of other such olefins (cf. Table 4). As shown, carbon–carbon double bonds with a variety of electronic and steric properties gave high yields of the hydrogenated products 42–49. Functional groups that are not affected under these conditions include a boronic ester, a brominated arene, an unprotected indole, and free alcohols (both primary and tertiary). The trisubstituted C═C bond that belongs to the allylic alcohol precursor of 49 was reduced, but the remote, trisubstituted double bond remained untouched; in this case, the hydroxyl group might be serving as a directing group. Complete reduction of diphenylacetylene to 50 was observed in 6 h, while the terminal alkyne in mestranol is hydrogenated to afford 51 in high yield (94%).
Table 4.

|
Under the conditions of entry 1 in Table 1.
Isolated yields, average of two independent runs.
Reaction time is 6 h.
The observed chemoselectivity of our Rh-catalyzed hydrogenation reactions suggested to us that they may also be useful in late-stage reductions during the synthesis of fine chemicals and/or pharmaceutical agents. Indeed, we can carry out such reactions (Scheme 2): for example, cyproheptadine is exclusively hydrogenated at the less substituted double bond, producing a good yield of the reduced pharmaceutical 52; reduction of the trisubstituted olefin in brucine delivers 53 in 94% yield. The late-stage hydrogenation of levofloxacin was similarly achieved with ease to give 54 in high yield, along with decarboxylation of the β-keto acid that occurs with standard hydrogenation protocols.40
Scheme 2.

Late-stage Hydrogenation of Pharmaceuticals
In view of the effectiveness of RhH-1 in catalyzing the hydrogenation of unactivated alkenes, we have attempted its use on the exomethylene of 55, a substrate whose stereochemical outcomes in catalytic hydrogenation have been investigated in detail by Shenvi.25 As shown in Scheme 3, we obtained in THF mostly the cis-disposed reduction product 56 (which is also the predominant product with traditional catalysts).41–43 We obtained a near-equimolar mixture of the epimers 56 and 57 when the reaction was conducted in MeOH. Under no conditions did our catalyst prefer to form the more stable product 57, one which Shenvi has found to be the kinetic product with Mn(dpm)3 or Co(dpm)2 as the catalyst.44
Scheme 3.
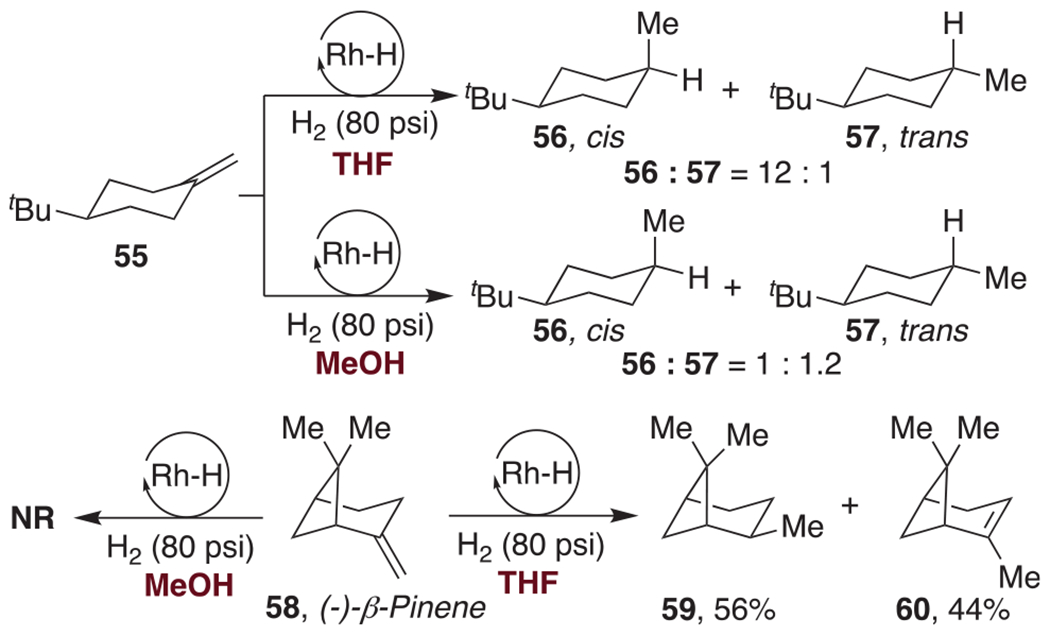
Effect of Solvent on Exomethylene Reduction
The outcome of such hydrogenation reactions is determined by both substituent effects and by solvent. With β-pinene (58), for example, there is no hydrogenation in MeOH, but in THF the equatorial methyl product 59 is obtained in 56% yield along with 44% of the isomerization product 60. The isomerization surely arises from the reversibility of the olefin insertion.
MECHANISM
In order to probe the difference between the mechanism of enones and that of isolated olefins, we have compared the results of deuterium labeling experiments with the α,β-unsaturated ketone in 61 with the results of such experiments with the isolated terminal olefin in 63. The extent of label incorporation is shown in Scheme 4. When CD3OD was used as solvent, 0.75 D was transferred to the α position of 62, while no deuterium was detected in 64. The protonation of a Rhenolate intermediate with methanol is faster than reductive elimination of the enolate ligand with the H on rhodium.45 The H2 gas supplies the H atom added to the β carbon of 61, and both of the H atoms added to 63; the solvent supplies only the H atom found at the other α carbon of enone substrate 61! Reaction of the same substrate 61 with D2 instead of H2 gas resulted in 90% deuteration of 65 with only 0.25 hydrogen at the β position. Given that both the H2 gas and the MeOH are present in large excess relative to the substrate and the catalyst, there is little scrambling between the H2/D2 and the solvent during these experiments.46,47
Scheme 4.

Deuterium Labelling Experiments
Mechanisms that explain the results in Scheme 4 are shown in Scheme 5, with substrates bearing enones on the left and those with isolated olefins on the right. The generation of the active catalyst probably begins with the reductive elimination of phenylpyridine from RhH-1 (as suggested by Chirik),39 followed by the addition of H2 to the Cp*Rh(I). The resulting Cp*RhH2 has been drawn by the Maitlis,33 Finke,34 and Chirik39 groups in catalytic cycles, but to our knowledge has never been isolated or identified.
Scheme 5.

Mechanistic Proposal
To determine if the generation of carbon-centered radicals by MHAT (metal hydrogen atom transfer) was involved in this RhH-1 catalyzed hydrogenation, we treated 71 and 74 with H2 in the presence of RhH-1. Although RhH-1 has a low bond dissociation free energy (BDFE = 52.3 kcal/mol)32 no trace of the radical cyclization products 73 or 76 was observed (the predominant products are shown in Scheme 6, i.e. 72 and 75).
Scheme 6.

Evidence against MHAT and Radical Formation
Heterogeneous and homogeneous catalysis have been established by Finke and Chirik in related Cp*Rh systems,33–35,39,48–51 but it appears that our catalyst is homogeneous. All materials dissolved with our reactions being clear and red within 20 min. We observed no precipitates even after 24 h of reaction time, and the kinetic plot in Scheme 7 suggests a clean first-order transformation of the tested substrate (S15, it is the precursor to compound 15) to product with a rate constant of about 2.3 × 10−5 M−1 s−1. A sigmoidal curve is typical of metal-particle formation. Furthermore, we found the addition of excess mercury after 200 min did not change the observed rate constant, a result typical of a homogeneous catalyst. Finally, TEM analysis (see the Supporting Information section for full details) of the residue when solvent was removed after the reaction showed neither heterogeneous metal particles nor rhodium clusters.52
Scheme 7.

Mercury Poisoning Study
CONCLUSION
In summary, (η5-C5Me5)Rh(2-(2-pyridyl)phenyl)H (RhH-1) catalyzes the selective 1,4-reduction of α,β-unsaturated carbonyl compounds—even ones with bulky substituents—under H2. It also catalyzes the hydrogenation of many isolated alkenes under mild reaction conditions. The system shows excellent selectivity and functional group compatibility, and appears to operate mechanistically under conditions that are homogeneous. The rhodium catalyst is a significant complement to the existing toolbox for metal-catalyzed selective hydrogenations and offers an opportunity to overcome longstanding challenges in the hydrogenation of polyfunctionalized molecules.
Supplementary Material
ACKNOWLEDGMENTS
We thank Sara Triana Hamilton and Prof. Ah-Hyung (Alissa) Park for helping with the TEM analysis. We are grateful to BASF for providing RhCl3. Dr. Jiawei Chen is acknowledged for helpful discussions, and members of the Snyder group are thanked for their contributions of additional α,β-unsaturated substrates which were explored in the course of these studies. We also thank Dr. Hunter B. Vibbert for the calculation of the equilibrium constant of 56/57. This research has been supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award R01-GM124295.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c04683.
Experimental procedures, spectral data (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c04683
The authors declare no competing financial interest.
Contributor Information
Yiting Gu, Department of Chemistry, Columbia University, New York City, New York 10027, United States.
Jack R. Norton, Department of Chemistry, Columbia University, New York City, New York 10027, United States.
Farbod Salahi, Department of Chemistry, University of Chicago, Chicago, Illinois 60637, United States.
Vladislav G. Lisnyak, Department of Chemistry, University of Chicago, Chicago, Illinois 60637, United States.
Zhiyao Zhou, Department of Chemistry, University of Chicago, Chicago, Illinois 60637, United States.
Scott A. Snyder, Department of Chemistry, University of Chicago, Chicago, Illinois 60637, United States.
REFERENCES
- (1).(a) Andersson PG, Munslow IJ, Eds. Modern Reduction Methods; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2008. [Google Scholar]; (b) Lovering F; Bikker J; Humblet C Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]; (c) Busacca CA; Fandrick DR; Song JJ; Senanayake CH The Growing Impact of Catalysis in the Pharmaceutical Industry. Adv. Synth. Catal 2011, 353, 1825–1864. [Google Scholar]; (d) Etayo P; Vidal-Ferran A Rhodium-catalysed Asymmetric Hydrogenation as a Valuable Synthetic Tool for the Preparation of Chiral Drugs. Chem. Soc. Rev 2013, 42, 728–754. [DOI] [PubMed] [Google Scholar]; (e) Parker PD; Hou X; Dong VM Reducing Challenges in Organic Synthesis with Stereoselective Hydrogenation and Tandem Catalysis. J. Am. Chem. Soc 2021, 143, 6724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Carey FA; Sundberg RJ Reduction of Carbonyl and Other Functional Groups. Advanced Organic Chemistry: Part B: Reactions and Synthesis; Springer US: Boston, MA, 1977; pp 129–161. [Google Scholar]; (b) Patai S; Rappoport Z The Chemistry of Enones; John Wiley & Sons: Chichester, 1989. [Google Scholar]; (c) Nashimura S Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis; Wiley-Interscience: New York, 2001. [Google Scholar]; (d) de Vries JG; Elsevier CJ The Handbook of Homogeneous Hydrogenation; Wiley-VCH: Weinheim, 2007. [Google Scholar]
- (3).Friedfeld MR; Zhong H; Ruck RT; Shevlin M; Chirik PJ Cobalt-Catalyzed Asymmetric Hydrogenation of Enamides Enabled by Single-Electron Reduction. Science 2018, 360, 888–893. [DOI] [PubMed] [Google Scholar]
- (4).Sugiura M; Sato N; Kotani S; Nakajima M Lewis Base-Catalyzed Conjugate Reduction and Reductive Aldol Reaction of d,,-Unsaturated Ketones Using Trichlorosilane. Chem. Commun 2008, 36, 4309–4311. [DOI] [PubMed] [Google Scholar]
- (5).Broggi J; Jurčík V; Songis O; Poater A; Cavallo L; Slawin AMZ; Cazin CSJ The Isolation of [Pd{OC(O)H}(H)(NHC)-(PR3)] (NHC = N-Heterocyclic Carbene) and Its Role in Alkene and Alkyne Reductions Using Formic Acid. J. Am. Chem. Soc 2013, 135, 4588–4591. [DOI] [PubMed] [Google Scholar]
- (6).(a) Wang Y; Huang Z; Leng X; Zhu H; Liu G; Huang Z Transfer Hydrogenation of Alkenes Using Ethanol Catalyzed by a NCP Pincer Iridium Complex: Scope and Mechanism. J. Am. Chem. Soc 2018, 140, 4417–4429. [DOI] [PubMed] [Google Scholar]; (b) Huang Z; Wang Y; Leng X; Huang Z An Amine-Assisted Ionic Monohydride Mechanism Enables Selective Alkyne cis-Semihydrogenation with Ethanol: From Elementary Steps to Catalysis. J. Am. Chem. Soc 2021, 143, 4824–4836. [DOI] [PubMed] [Google Scholar]
- (7).Zheng C; You S-L Transfer Hydrogenation with Hantzsch Esters and Related Organic Hydride Donors. Chem. Soc. Rev 2012, 41, 2498–2518. [DOI] [PubMed] [Google Scholar]
- (8).(a) Wang D; Astruc D The Golden Age of Transfer Hydrogenation. Chem. Rev 2015, 115, 6621–6686. [DOI] [PubMed] [Google Scholar]; (b) Garg N; Sarkar A; Sundararaju B Recent Developments on Methanol as Liquid Organic Hydrogen Carrier in Transfer Hydrogenation Reactions. Coord. Chem. Rev 2021, 433, 213728. [Google Scholar]
- (9).Ding B; Zhang Z; Liu Y; Sugiya M; Imamoto T; Zhang W Chemoselective Transfer Hydrogenation of D,,-Unsaturated Ketones Catalyzed by Pincer-Pd Complexes Using Alcohol as a Hydrogen Source. Org. Lett 2013, 15, 3690–3693. [DOI] [PubMed] [Google Scholar]
- (10).Mendelsohn LN; MacNeil CS; Tian L; Park Y; Scholes GD; Chirik PJ Visible-Light-Enhanced Cobalt-Catalyzed Hydrogenation: Switchable Catalysis Enabled by Divergence between Thermal and Photochemical Pathways. ACS Catal. 2021, 11, 1351–1360. [Google Scholar]
- (11).Beltran F; Bergamaschi E; Funes-Ardoiz I; Teskey CJ Photocontrolled Cobalt Catalysis for Selective Hydroboration of α,,-Unsaturated Ketones. Angew. Chem., Int. Ed 2020, 59, 21176–21182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Alonso F; Osante I; Yus M Conjugate Reduction of α-Unsaturated Carbonyl Compounds Promoted by Nickel Nanoparticles. Synlett 2006, 2006, 3017–3020. [Google Scholar]; (b) Shevlin M; Friedfeld MR; Sheng H; Pierson NA; Hoyt JM; Campeau L-C; Chirik PJ Nickel-Catalyzed Asymmetric Alkene Hydrogenation of g,,-Unsaturated Esters: High-Throughput Experimentation-Enabled Reaction Discovery, Optimization, and Mechanistic Elucidation. J. Am. Chem. Soc 2016, 138, 3562–3569. [DOI] [PubMed] [Google Scholar]
- (13).(a) Wei D; Darcel C Iron Catalysis in Reduction and Hydrometalation Reactions. Chem. Rev 2019, 119, 2550–2610. [DOI] [PubMed] [Google Scholar]; (b) Li M; Xia H-F; Yang L-Y; Hong T; Xie L-J; Li S; Wu J Synthesis of N-Aryl β-Amino Acid Derivatives via Cu(II)-Catalyzed Asymmetric 1,4-Reduction in Air. RSC Adv. 2019, 9, 9187–9192. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mendes-Burak J; Ghaffari B; Copéret C Selective Hydrogenation of d-Unsaturated Carbonyl Compounds on Silica-Supported Copper Nanoparticles. Chem. Commun 2019, 55, 179–181. [DOI] [PubMed] [Google Scholar]
- (14).(a) Alig L; Fritz M; Schneider S First-Row Transition Metal (De)Hydrogenation Catalysis Based on Functional Pincer Ligands. Chem. Rev 2019, 119, 2681–2751. [DOI] [PubMed] [Google Scholar]; (b) Chirik PJ Iron- and Cobalt-Catalyzed Alkene Hydrogenation: Catalysis with Both Redox-Active and Strong Field Ligands. Acc. Chem. Res 2015, 48, 1687–1695. [DOI] [PubMed] [Google Scholar]; (c) Sklyaruk J; Zubar V; Borghs JC; Rueping M Methanol as the Hydrogen Source in the Selective Transfer Hydrogenation of Alkynes Enabled by a Manganese Pincer Complex. Org. Lett 2020, 22, 6067–6071. [DOI] [PubMed] [Google Scholar]; (d) Wang H; Zhang Y; Yang T; Guo X; Gong Q; Wen J; Zhang X Chiral Electron-Rich PNP Ligand with a Phospholane Motif: Structural Features and Application in Asymmetric Hydrogenation. Org. Lett 2020, 22, 8796–8801. [DOI] [PubMed] [Google Scholar]
- (15).Li H-C; An C; Wu G; Li G-X; Huang X-B; Gao W-X; Ding J-C; Zhou Y-B; Liu M-C; Wu H-Y Transition-Metal-Free Highly Chemoselective and Stereoselective Reduction with Se/DMF/H2O System. Org. Lett 2018, 20, 5573–5577. [DOI] [PubMed] [Google Scholar]
- (16).Huang X; Hu J; Wu M; Wang J; Peng Y; Song G Catalyst-Free Chemoselective Conjugate Addition and Reduction of α,β-Unsaturated Carbonyl Compounds via a Controllable Boration/Protodeboronation Cascade Pathway. Green Chem. 2018, 20, 255–260. [Google Scholar]
- (17).Shi Z; Li N; Lu H-K; Chen X; Zheng H; Yuan Y; Ye K-Y Recent Advances in the Electrochemical Hydrogenation of Unsaturated Hydrocarbons. Curr. Opin. Electrochem 2021, 28, 100713. [Google Scholar]
- (18).Qin Y; Lu J; Zou Z; Hong H; Li Y; Li Y; Chen L; Hu J; Huang Y Metal-Free Chemoselective Hydrogenation of Unsaturated Carbon-Carbon Bonds via Cathodic Reduction. Org. Chem. Front 2020, 7, 1817–1822. [Google Scholar]
- (19).Huang B; Li Y; Yang C; Xia W Electrochemical 1,4-Reduction of α,β-Unsaturated Ketones with Methanol and Ammonium Chloride as Hydrogen Sources. Chem. Commun 2019, 55, 6731–6734. [DOI] [PubMed] [Google Scholar]
- (20).Gao AX; Hamada T; Snyder SA The Enantioselective Total Synthesis of Exochomine. Angew. Chem., Int. Ed 2016, 55, 10301–10306. [DOI] [PubMed] [Google Scholar]
- (21).Zhou Z; Gao AX; Snyder SA Total Synthesis of (+)-Arborisidine. J. Am. Chem. Soc 2019, 141, 7715–7720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lisnyak VG; Snyder SA A Concise, Enantiospecific Total Synthesis of Chilocorine C Fueled by a Reductive Cyclization/Mannich Reaction Cascade. J. Am. Chem. Soc 2020, 142, 12027–12033. [DOI] [PubMed] [Google Scholar]
- (23).Lisnyak VG; Sherwood TC; Snyder SA The Development of Reaction Cascades to Synthesize Dimeric Coccinellid Alkaloids. Acc. Chem. Res 2021, 54, 1610–1622. [DOI] [PubMed] [Google Scholar]
- (24).Magnus P; Waring MJ; Scott DA Conjugate Reduction of t-Unsaturated Ketones Using an MnIII Catalyst, Phenylsilane and Isopropyl Alcohol. Tetrahedron Lett. 2000, 41, 9731. [Google Scholar]
- (25).Iwasaki K; Wan KK; Oppedisano A; Crossley SWM; Shenvi RA Simple, Chemoselective Hydrogenation with Thermodynamic Stereocontrol. J. Am. Chem. Soc 2014, 136, 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Smith DM; Pulling ME; Norton JR Tin-Free and Catalytic Radical Cyclizations. J. Am. Chem. Soc 2007, 129, 770–771. [DOI] [PubMed] [Google Scholar]
- (27).Estes DP; Norton JR; Jockusch S; Sattler W Mechanisms by Which Alkynes React with CpCr(CO)3H. Application to Radical Cyclization. J. Am. Chem. Soc 2012, 134, 15512–15518. [DOI] [PubMed] [Google Scholar]
- (28).Kuo JK; Hartung J; Han A; Norton JR Direct Generation of Oxygen-Stabilized Radicals by H· Transfer from Transition Metal Hydrides. J. Am. Chem. Soc 2015, 137, 1036–1039. [DOI] [PubMed] [Google Scholar]
- (29).Li G; Kuo JL; Han A; Abuyuan JM; Young LC; Norton JR; Palmer JH Radical Isomerization and Cycloisomerization Initiated by H· Transfer. J. Am. Chem. Soc 2016, 138, 7698–7704. [DOI] [PubMed] [Google Scholar]
- (30).Lorenc C; Vibbert HB; Yao C; Norton JR; Rauch M H· Transfer-Initiated Synthesis of γ-Lactams: Interpretation of Cycloisomerization and Hydrogenation Ratios. ACS Catal. 2019, 9, 10294–10298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).RhH-1 can be easily synthesized in large quantities by NaOAc promoted C–H activation of 2-phenylpyridine with [Cp*RhCl2]2, followed by treatment with either NaBH4 in THF or NaOMe in MeOH. For the detailed procedure, please see ref 32.
- (32).(a) Hu Y; Li L; Shaw AP; Norton JR; Sattler W; Rong Y Synthesis, Electrochemistry, and Reactivity of New Iridium(III) and Rhodium(III) Hydrides. Organometallics 2012, 31, 5058–5064. [Google Scholar]; (b) Hu Y; Norton JR Kinetics and Thermodynamics of H−/H·/H+ Transfer from a Rhodium(III) Hydride. J. Am. Chem. Soc 2014, 136, 5938–5948. [DOI] [PubMed] [Google Scholar]
- (33).Maitlis PM (Pentamethylcyclopentadienyl) rhodium and -iridium Complexes: Approaches to New Types of Homogeneous Catalysts. Acc. Chem. Res 1978, 11, 301–307. [Google Scholar]
- (34).Bayram E; Linehan JC; Fulton JL; Szymczak NK; Finke RG Determination of the Dominant Catalyst Derived from the Classic [RhCp*Cl2]2 Precatalyst System: Is it Single-Metal Rh1Cp*-Based, Subnanometer Rh4 Cluster-Based, or Rh(0)n Nanoparticle-Based Cyclohexene Hydrogenation Catalysis at Room Temperature and Mild Pressures? ACS Catal. 2015, 5, 3876–3886. [Google Scholar]
- (35).Bayram E; Linehan JC; Fulton JL; Roberts JAS; Szymczak NK; Smurthwaite TD; Özkar S; Balasubramanian M; Finke RG Is It Homogeneous or Heterogeneous Catalysis Derived from [RhCp*Cl2]2? In Operando XAFS, Kinetic, and Crucial Kinetic Poisoning Evidence for Subnanometer Rh4 Cluster-Based Benzene Hydrogenation Catalysis. J. Am. Chem. Soc 2011, 133, 18889–18902. [DOI] [PubMed] [Google Scholar]
- (36).Pappas I; Chirik PJ Catalytic Proton Coupled Electron Transfer from Metal Hydrides to Titanocene Amides, Hydrazides and Imides: Determination of Thermodynamic Parameters Relevant to Nitrogen Fixation. J. Am. Chem. Soc 2016, 138, 13379–13389. [DOI] [PubMed] [Google Scholar]
- (37).Kim S; Zhong H; Park Y; Loose F; Chirik PJ Catalytic Hydrogenation of a Manganese(V) Nitride to Ammonia. J. Am. Chem. Soc 2020, 142, 9518–9524. [DOI] [PubMed] [Google Scholar]
- (38).Bezdek MJ; Chirik PJ Pyridine(Diimine) Chelate Hydrogenation in a Molybdenum Nitrido Ethylene Complex. Organometallics 2019, 38, 1682–1687. [Google Scholar]
- (39).Kim S; Loose F; Bezdek MJ; Wang X; Chirik PJ Hydrogenation of N -Heteroarenes Using Rhodium Precatalysts: Reductive Elimination Leads to Formation of Multimetallic Clusters. J. Am. Chem. Soc 2019, 141, 17900–17908. [DOI] [PubMed] [Google Scholar]
- (40).(a) Hanson RW Decarboxylation of a Keto Acids. J. Chem. Educ 1987, 64, 591–595. [Google Scholar]; (b) Logue MW; Pollack RM; Vitullo VP Nature of the Transition State for the Decarboxylation of e-keto Acids. J. Am. Chem. Soc 1975, 97, 6868–6869. [Google Scholar]
- (41).Mitchell TRB Stereochemistry of Hydrogenation of 4-t-Butylmethylenecyclohexane. J. Chem. Soc. B 1970, 823. [Google Scholar]
- (42).Siegel S; Dmuchovsky B Stereochemistry and the Mechanism of Hydrogenation of Cyclo-Alkenes. IV. 4-tert-Butyl-1-Methylcyclohexene and 4-tert-Butyl-1-Methylenecyclohexane on Platinum Oxide and a Palladium Catalyst. J. Am. Chem. Soc 1962, 84, 3132–3136. [Google Scholar]
- (43).van Tamelen EE; Timmons RJ The Effect of Substrate Steric Properties on The Stereochemical Course of Diimide Reductions. J. Am. Chem. Soc 1962, 84, 1067–1068. [Google Scholar]
- (44).DFT calculations show that the trans-57 is 1.3 kcal/mol more stable than the cis-56. See Supporting Information for full details.
- (45).(a) Arisawa M; Yamaguchi M Rhodium Enolate Complexes as Synthons and Catalysts in Organic Chemistry. In PATAI’S Chemistry of Functional Groups; John Wiley & Sons, Ltd.: 2016. [Google Scholar]; (b) Sibi MP; Tatamidani T; Patil K Enantioselective rhodium Enolate Protonation. A New Methodology for the synthesis of P-Amino Acids. Org. Lett 2005, 7, 2571–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).The small amount of deuterium (0.1 D) incorporated under D2 into the α position of 65 probably arises from slow H/D exchange between Cp*RhD2 and methanol.
- (47).The incorporation of more than one deuterium into the terminal methyl group of 66 under D2 implies that the insertion of the C═C into the Rh–D bond is reversible.
- (48).Jaska CA; Manners I Catalytic Dehydrocoupling of Amine-Borane and Phosphine-Borane Adducts: The Mechanism Is Heterogeneous in One Case and Homogeneous in the Other. J. Am. Chem. Soc 2004, 126, 1334–1335. [DOI] [PubMed] [Google Scholar]
- (49).Jaska CA; Manners I Heterogeneous or Homogeneous Catalysis? Mechanistic Studies of the Rhodium-Catalyzed Dehydrocoupling of Amine-Borane and Phosphine-Borane Adducts. J. Am. Chem. Soc 2004, 126, 9776–9785. [DOI] [PubMed] [Google Scholar]
- (50).Gill DS; White C; Maitlis PM Pentamethylcyclopentadienyl-Rhodium and -Iridium Complexes. Part 16. Homogeneous Hydrogenation Catalysts. J. Chem. Soc., Dalton Trans 1978, 6, 617–626. [Google Scholar]
- (51).Hagen CM; Widegren JA; Maitlis PM; Finke RG Is It Homogeneous or Heterogeneous Catalysis? Compelling Evidence for Both Types of Catalysts Derived from [Rh(η5-C5Me5)Cl2]2 as a Function of Temperature and Hydrogen Pressure. J. Am. Chem. Soc 2005, 127, 4423–4432. [DOI] [PubMed] [Google Scholar]
- (52).Neither the Hg nor the TEM experiment is definitive, but we believe that the absence of both Hg poisoning and Rh clusters/particles collectively suggests that our catalyst is homogeneous. For further discussion of this point, please see refs 34 and 50.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.