

Abstract
Alzheimer’s disease (AD) is the most common neurodegenerative disorder with heterogeneous etiology. Intracellular neurofibrillary tangles caused by tau (τ) protein phosphorylation and extracellular senile plaques caused by aggregation of amyloid-beta (Aβ) peptide are characteristic histopathological hallmarks of AD. Oxidative stress (OS) is also suggested to play a role in the pathophysiology of AD. The antioxidant glutathione (GSH) is able to mitigate OS through the detoxification of free radicals. The clearance of these free radicals is reported to be affected when there is a decline in GSH levels in AD. These radicals further react with the methionine-35 (M-35) residue of Aβ and facilitate its subsequent oligomerization. This review presents a plausible model indicating the role of master antioxidant GSH to protect M35 of Aβ1–40/Aβ1–42 from oxidation in pathological conditions as compared to healthy controls.
Introduction
Alzheimer’s disease is the most common neurodegenerative disease, affecting millions of people worldwide and is manifested with behavioral changes as well as cognitive deficits.1 The dominant pathological hallmarks of AD are the following: the presence of senile plaques caused by aggregation of Aβ peptide; neurofibrillary tangles (NFTs) resulting from the hyperphosphorylation of τ protein; and anatomical features like brain atrophy caused by neuronal loss.2 Clinical symptoms of AD include progressive cognitive decline involving memory loss and loss of higher executive functioning.3 Both amyloid plaques and NFTs have been implicated in the cognitive impairment associated with AD.4 There are various hypotheses explaining the pathophysiology of AD, such as amyloid hypothesis,5 tau propagation hypothesis,6 neuroinflammation hypothesis,7 mitochondrial dysfunction,8 and oxidative stress hypothesis.9 Accruing evidence has readily suggested oxidative stress (OS) as one of the major axes contributing to the pathophysiology of AD progression.10−12
The brain consumes 20% of the body’s energy to maintain its biochemical integrity, or metabolic homeostasis, contributing to the normal functioning of the central nervous system.13 Consumption of oxygen leads to the generation of free radicals which creates an ionic imbalance14 further resulting in aberrant intracellular calcium signaling and the initiation of an apoptotic cascade.15 The deleterious effects of these free radicals are usually nullified by the body’s endogenous antioxidant defense system, leading to maintenance of redox balance.2 However, dysregulation of neuroenergetics can lead to OS, an imbalance caused by failure of the biological antioxidant system to remove reactive oxygen species (ROS) from the system.16 Accumulation of ROS leads to neuronal death and loss of synapse, thus having a profound effect on brain function.17 The byproduct of metabolic reactions is free radicals such as ROS, reactive nitrogen species (RNS), and some radicals having a carbon or sulfur center.2 The ROS are mostly generated as a byproduct of aerobic respiration.18 The electron structure of oxygen makes it susceptible to radical formation by addition of free electrons, further leading to different ROS formation. Though ROS, such as superoxide (O2•–), hydrogen peroxide (H2O2), and the most reactive hydroxyl radical (•OH), and RNS play a crucial role in cell signaling and neuronal development, hyperoxia can lead to their overproduction and exceed the scavenging capacity of antioxidant molecules which are involved in their clearance.2,19 A high level of OS is neurotoxic and can lead to impairments in long-term potentiation, alter synaptic transmission and mechanisms of brain plasticity, and cause changes in the brain microarchitecture.2
Oxidative stress can be proposed to temporally precede the onset of pathogenicity of AD progression. The imbalance in pro-oxidant/antioxidant status leads to the activation of various cell signaling pathways as compensatory mechanisms against lesion formation in AD pathogenesis. The mitochondria are particularly susceptible to OS due to their role in producing energy and is hence considered the hub of ROS production.20,21 Metabolic pathways involved in mitochondrial dysfunction include the tricarboxylic acid cycle and also the terminal part of the oxidative phosphorylation involving the reduction of molecular oxygen.20 The mitogen-activated protein kinase (MAPK) pathway is an important signal transduction responsible for eliciting physiological responses like cell proliferation, differentiation, and also apoptosis and is conserved from yeast to humans.22 OS has been seen to interfere with one or more components of the MAPK pathway.23 The extracellular-signal regulated kinase (ERK) cassette of the MAPK pathway has been observed to be abnormally activated in AD and contribute to its pathogenicity.24 Another signaling pathway, the canonical Wnt signaling pathway (Wnt/β-catenin pathway) also participates in diverse physiological processes.25 Overproduction of ROS has been observed to affect the functioning of this pathway to promote cellular aging and senescence.26 Nuclear factor-erythroid factor 2-related factor (Nrf2) is a transcription regulator and forms a line of defense against oxidative insults.27 Under conditions of OS, it can translocate to the nucleus from the cytoplasm and transactivate cytoprotective genes.28 Nrf2 signaling has been seen to be impaired in many neurodegenerative diseases, including AD.29 OS can impair the translocation of Nrf2 from the cytosol to the nucleus, thus inhibiting Nrf2 from exerting its antioxidative effect.30 Thus, OS directly, and also indirectly impede functioning of various cellular and metabolic pathways that in turn affects the physiological state of the cell under stress.
Protective mechanisms against OS in the brain circumscribe the antioxidant enzyme system and low molecular-weight antioxidants.2 GSH is a thiol-containing tripeptide and the most important and abundant low molecular weight antioxidant in the brain.2 The ratio of GSH to oxidized glutathione (glutathione disulfide; GSSG) is maintained over 100 to provide adequate protection against oxidative insults.31 An important factor of AD-related increase in OS can be attributed to the depleted levels of GSH.1 A putative underlying mechanism for AD pathology can be attributed for the regulation of OS by GSH.
Due to the heterogeneous etiology of AD and multiple factors playing a role in disease progression, its treatment strategy is still under extensive research.32,33 The existing therapeutic approach for AD is based on symptomatic treatments that try to counterbalance neurotransmitter disturbance which eventually relieves the symptoms.34,35 The Food and Drug Administration (FDA) approved drugs such as achetylcholinesterase inhibitors (AChEIs) and N-methyl d-aspartate (NMDA) antagonists were in use for the treatment against AD.36 Recently, a new drug based on passive immunotherapy [Aβ-targeting monoclonal antibodies (mAbs)] has also been approved.37 It is based on the amyloid hypothesis and targets the aggregated forms of Aβ.35 These drugs provide only symptomatic treatment and cannot block the progression of AD. Therefore, therapeutic strategies which are capable to stop or modify the course of AD are also under research.32,34,38 A list of drugs approved for treatment of AD is provided in Table 1.
Table 1. Summary of Drugs Approved for AD.
| drug | action | working | used for | year of approval | ref |
|---|---|---|---|---|---|
| acetylcholinesterase inhibitors (AChE) | |||||
| 1. galantamine | AChE inhibitor | halt the breakdown of acetylcholine and stimulates nicotinic receptors which releases more acetylcholine | mild-moderate | 1996 | (45) |
| 2. rivastigmine | AChE and Butyrylcholinesterase inhibitor | prevents the breakdown of butyrylcholine and acetylcholine | mild-moderate | 1997 | (46) |
| 3. donepezil | AChE inhibitor | averts the breakdown of acetylcholine | mild-moderate | 2001 | (47) |
| antagonist of NMDA receptor | |||||
| 4. memantine | NMDA antagonist | obstructs the toxic effects linked with excessive glutamate; controls glutamate activation | moderate-severe | 2003 | (48) |
| Aβ-targeting monoclonal antibodies | |||||
| 5. aducanumab | binding of aggregated Aβ plaques | eliminates abnormal beta-amyloid to help in reducing the no. of plaques | mild | 2021 | (37) |
In this review, we briefly discuss the pathogenesis of AD focusing on the OS theory. The primary aim of this review is to establish the role of GSH rendering a protective environment against M35 residue oxidation and subsequent oligomer formation of the Aβ peptide. Outcomes from several studies are analyzed to corroborate M35 oxidation and Aβ aggregation.39−44 Literature from PubMed, Google Scholar, and Web of Science database was used to extract and analyze relevant articles from the year 1987 to 2021 for manuscript preparation.
Role of Oxidative Stress in Pathogenesis of Alzheimer’s Disease
Clinical diagnosis of AD is characterized by an aggregation of fibrillary Aβ peptide (senile plaques) and NFT of hyperphosphorylated τ protein in the brain. This leads to dysfunction and loss of synapses and subsequent neuronal death.49 Much evidence has accrued over the years to lend cognizance to the fact that oxidative damages precede plaque formation.12,17,50
Magnetic resonance spectroscopy (MRS) studies have also yielded linkage between the biological alterations and pathophysiology of AD.51 In a study assessing brain GSH levels using 1H MRS in healthy old subjects, a negative correlation was found between the GSH levels and brain amyloidosis in the temporal and parietal regions, indicating the potential role of OS in amyloid plaque formation.52 GSH has also been implicated as a potential biomarker to diagnose mild cognitive impairment (MCI), the prodromal stage of AD.53 These findings can be suggestive of a GSH-based therapy, which can alleviate the symptoms of AD if diagnosed during its MCI stage.
OS causes aberrant signaling pathways which can hyperphosphorylate the microtubule protein τ, leading to the abnormal formation of NFTs in the AD brain.54 Hyperphosphorylated τ is found in an insoluble form with neuropil threads and filamentous tangles. Such τ proteins lose the ability to bind and stabilize microtubules in the axon, leading to loss of neuronal function. NFTs have been observed to aggregate much more in the CA1 and CA4 regions of the hippocampus, which are particularly vulnerable to OS.55
Mitochondrial dysfunction8 and impaired glucose metabolism are mutually related to OS.49 Both phenomena lead to diminished adenosine triphosphate (ATP) production, which has a direct effect on the neuron’s ability to maintain ionic gradients, thus affecting action potentials and in turn neurotransmission.49 Trace elements like iron, aluminum, and mercury have received considerable attention in AD due to their ability to generate free radicals and cause redox imbalance. The •OH radical produced by Fenton’s reaction and a disrupted iron metabolism is also indicated in the brain of AD patients.9 Excessive free iron levels can generate OS and lead to the formation of free radicals in the brain.56 Furthermore, iron levels are also reported to affect GSH concentrations, causing first an increase and then a decrease in the antioxidant’s levels.57 The increased OS and presence of free radicals can then lead to amyloid aggregation by reacting with the M35 residue and leading to production of ROS like methionine sulfoxide (MetSO).
Metal Ions and Their Interaction with Αβ Peptide
The αβ peptide, like other proteins, aggregates under different solvent conditions and metal ions have been implicated to play a role in the aggregation and fibrillation of αβ, even stabilizing the oligomeric state of αβ.58 Deterioration of metal homeostasis has also been observed to be a crucial factor in AD pathology.59 The metal ion effect has hence come to be an important parameter for studying metal-dependent amyloid aggregation.58
Neurotoxic lesions caused by αβ precipitation have been posited to arise from interactions with neocortical pro-oxidative metal ions like Zn, Cu, and Fe.60 Αβ has the ability to reduce Cu2+ and Fe3+, and these reduced metal ions can react further with molecular oxygen to produce superoxide radicals, and on further reduction H2O2.61 H2O2 can react with reduced metals to produce the extremely toxic OH radical through Fenton’s reaction. The reduction of metal in this reaction is mediated by the M35 residue whose sulfide group acts as an electron donor.61 In the absence of proper antioxidative mechanisms, the generation of these ROS contributes further to OS, leading to modified αβ accumulation which are resistant to clearance.62 This leads to lipid and protein peroxidation and DNA damage, ultimately causing neuronal death.
Iron is another pro-oxidant but is also involved in maintaining the normal physiology of neurons.63 However, dysregulation of iron metabolism can adversely affect brain homeostasis. In a quantitative susceptibility mapping (QSM) study, the amount of iron load in the left hippocampus in the clinical population was significantly increased in MCI patients as compared to AD patients. However, the same was not observed between healthy controls and MCI patients. This suggests a plausible role of iron in the progression of MCI to AD (paper in press from our laboratory). Thus, iron accumulation may contribute to the etiology of AD progression.
Amyloid Beta Protein
The Aβ peptide is an ∼4 kDa protein containing 39–43 amino acids derived from the cleavage of the larger amyloid-β precursor protein (APP). It was first isolated as the principal component of amyloid deposits in the brain and cerebrovasculature of patients with AD and Down syndrome.64 Proteolytic cleavage of APP by β and γ secretase enzymes generates Aβ fragments of different lengths. The two main forms are Aβ1–40 and Aβ1–42, the latter being the primary component of extracellular senile plaques and forms the basis of the amyloid cascade hypothesis.65 Residues 1–28 of Aβ peptide, corresponding to the N-terminal domain, are largely hydrophilic whereas the C-terminal domain (residues 29–39/42) is hydrophobic and derived from the APP transmembrane domain. The C-terminal domain predominately exists as an oligomeric cross β-sheet in solution and is probably responsible for the tendency of Aβ1–42 to fold into β-pleated sheet and form aggregates found in amyloid plaques.66 OS has been found to be high in regions like the hippocampus and cortex which have greater deposits of Aβ42, thus implicating the role of OS in amyloid aggregation. Protein oxidation and lipid peroxidation was also found to induce in synaptosomes and neurons in C. elegans expressing human Aβ42.67
Role of Methionine-35 in Amyloid Aggregation
Chemistry of Methionine
Methionine is one of the two sulfur-containing amino acids and serves as the initiating amino acid in the synthesis of most eukaryotic proteins. The presence of sulfur in methionine makes it hydrophobic, but some exposed groups are also susceptible to oxidative damage. The low oxidation potential of the methionine sulfur makes the amino acid readily susceptible to oxidation.68 Oxidation of methionine is a two-step process and involves the transfer of two electrons (Figure 1). Methionine is a reducing agent and donates its electrons to first form MetSO through a reversible reaction.68 The asymmetric center around the sulfur atom leads to the formation of two diastereomers: the R and S form of MetSO. Methionine sulfoxide reductases (Msr) are enzymes that reduce methionine sulfoxides back to methionine based on the susceptibility of these diastereomers for the enzyme. Impaired activity of Msr can lead to accumulation of ROS and are associated with age-related neurodegeneration.69 The activity of Msr has been found to decrease in the brain of patients with AD.70 Failure of Msr activity leads to elevated ROS levels, and this in turn can cause further oxidation of MetSO to form methionine sulfone (MES).71 This latter reaction is biologically unfavorable and irreversible.67 MES is also a ROS and enhances the oxidative insults on the brain. Prolonged oxidation of M35 may lead to Aβ oligomerization and induce pathogenesis of AD.
Figure 1.

Structure of methionine. (A) Structure of methionine with the thiol-ether group highlighted. (B) Methionine oxidation. Prolonged oxidation leads to the generation of the reactive oxygen species methionine sulfone.72 The figure is prepared based on the earlier work.72
In Vitro Studies
Many studies have focused on the role of M35 in AD since it is one of the most readily oxidized constituents of proteins in vivo, especially under OS conditions.73 One of the earliest studies explaining the mechanism behind β-amyloid aggregation and toxicity utilized electron paramagnetic resonance (EPR) to study the properties of Aβ in solution.40 Synthetic Aβ1–40 and Aβ25–35 could independently induce the conversion of the unstable N-tert-butyl-α-phenylnitrone (PBN) to its stable paramagnetic conformation in phosphate-buffered saline, suggesting the presence of a free radical in the Aβ peptides.41,74 Both Aβ1–40 and Aβ25–35 yielded characteristic EPR signals, but in the presence of nitrogen-saturated solutions all EPR signals disappeared, suggesting the role of molecular oxygen for free radical formation. M35 being the only redox active amino acid of Aβ peptide was posited to play a critical role in the neurotoxicity of Aβ.40 In another in vitro study, the substitution of M35 with other amino acids like Ile, Lys, Leu, and Tyr was found to inhibit the aggregation of synthetic Aβ25–35.75
In a spin trapping EPR study, deletion of M35 in structural variants Aβ25–35, Aβ25–34 arrested free radical production and hence protein oxidation and neurotoxicity due to Aβ25–35.39 The carbonyl oxygen of isoleucine (Ile)-31 of Aβ can react with the sulfur atom of M35 and change the electronic environment around it, leading to the production of a sulfuranyl free radical on M35.44 Replacement of Ile31 with the helix-breaker proline resulted in a diminished helical environment as assessed by circular dichroism (CD).44 This altered the aggregation and neurotoxic properties of Aβ1–42, suggesting an important role of helix-associated interactions for the aggregation of Aβ.
Mutation of glycine 37 to the negatively charged aspartic acid (Aβ1–42 G37D) causes removal of M35 from the lipid bilayer. The addition of this modified peptide to the membranes removes allylic H-atoms from M35 for abstraction, hence eliminating lipid peroxidation.43 The G37D peptide also did not demonstrate any aggregation properties as assessed by the thioflavin T (ThT) assay and fibril morphology. Thus, Aβ1–42 G37D successfully negated the oxidative and neurotoxic attributes of Aβ1–42, concluding that the interaction of M35 with the lipid bilayer is a requirement for Aβ1–42-mediated lipid peroxidation.43 Lipid peroxidation and the subsequent production of 4-hydroxynonenal (4-HNE) is assumed to be the first harmful event to occur with Aβ1–42 in the AD brain.17
The form of Aβ also plays a key role in the induction of Aβ in the aggregation pathway and eventual plaque deposition. Aβ42 has been found to be more neurotoxic and form fibrils fasters than its counterpart Aβ40.76 Using the technique of photoinduced cross-linking of unmodified proteins (PICUP), oxidation of M35 was observed to regulate the oligomer size distribution of Aβ42 into one that is distinct from Aβ40. This posits that M35 oxidation plays a key role in deciding the oligomerization pathway that Aβ40/42 participates in.76
Even though several studies have implicated M35 oxidation in Aβ aggregation, some NMR studies have also shown conflicting results.77,78 A solution NMR study on oxidized and reduced M35 residues from different Aβ peptides reported that M35 oxidation significantly hindered the rate of Aβ aggregation by influencing hydrophobic and electrostatic associations.77 M35 oxidation has also been found to reduce the total amount of fibers generated for the two dominant forms of Aβ.78 Yet, an NMR analysis of the interaction between the Cα and Cδ M35 and the carboxyl carbon of Ala42 observed that the interaction between the two can be stabilized, leading to aggregation and enhancement of OS.79
In Vivo Studies
Protein carbonyls, a measure of protein oxidation, have been found to be elevated in the brain of human AD subjects, localized to the regions replete in Aβ plaques.17 In addition, lipid peroxidation in the brain produces 4-HNE which binds to the glutamate transporter, GLT-1, and changes its conformation and activity.80 Accumulation of glutamate in the synaptic cleft leads to excitotoxicity.81
Studies conducted on C. elegans shows that when the codon for M35 in Aβ1–42 was replaced with the codon for cysteine, the transgenic animals showed no increase in the levels of protein carbonyls.42
Genetic factors associated with autosomal dominant familial AD include mutations in the APP, PS-1/2 genes and APOEε4 allele. In a transgenic mouse study with the Swedish/Indiana mutations of the APP gene, the mouse exhibited significantly elevated levels of OS markers at nine months of age (since Aβ deposition has been found to begin significantly during this age bracket).82,83 When Met-631 of APP, which corresponds to the M35 position of Aβ1–42, was substituted by leucine, OS markers were found to be mitigated in the 9 months old transgenic mice, corroborating with the findings from the C. elegans study and demonstrating that M35 residue of Aβ1–42 is critical for the neurotoxic aggregation associated with the protein.82 Additionally, Aβ1–42 M35L substitution in mice has also been observed to deposit similar amounts of Aβ1–42 as well as Aβ1–40 in punctuate forms, supporting the theses of this review that oxidation of M35 residue of Aβ1–42 is implicated for plaque formation.82
Post-Mortem Studies
In a study involving analysis of Aβ structural variants in sporadic and familial AD cases, the senile-plaque resident Aβ1–40 showed a high concentration of MetSO.84 Measurement of antioxidants like GSH, catalase, superoxide dismutase, use of oxidative markers from the frontal cortex of post-mortem AD patient brains have shown significant reduction in these antioxidants and OS to be more localized to the synaptosomes (Table 2).85 Earlier research has demonstrated depleted GSH/GSSG and complex I level along with increased amyloidosis in the frontal cortex of AD patients, suggesting the impact of oxidative damage in the prognosis of AD.86 GSH can be thus considered to provide a protective mechanism against the oxidation of M35 residue of Aβ (Figure 2). Significant reduction in GSH levels increases the oxidative load on the hippocampus which may be involved for the initiation of MCI and consequently AD. This is further corroborated by the fact that analyses of frozen hippocampal samples from MCI patients’ brains have shown notable GSH depletion.87 The enzyme GSH-transferase can sequester harmful aldehydes like 4-HNE. The levels of this enzyme were found to be depleted in the brain and ventricular cerebrospinal fluid of post-mortem AD patients.88 Such autopsy studies further implicate the role of upstream processes like oxidative stress and GSH depletion to influence downstream processes like M35 oxidation leading to Aβ oligomer formation and aggregation.
Table 2. Comparison of Different Anti-Oxidative Markers between Healthy Subjects and AD/MCI Patients.
| antioxidative enzyme(s)/ marker(s) | comparison group | % change vs controls | total N (disease/ control) | brain region/ body tissue | ref |
|---|---|---|---|---|---|
| superoxide dismutase | AD | ↓6.6% | 36/36 | red blood cells | (90) |
| AD | ↓88%, ↓71% | 10/9 | frontal cortex, temporal cortex | (91) | |
| AD | ↓27%, ↓27%, ↓35% | 12/12, 14/11, 8/5 | cerebellum, frontal cortex, hippocampus | (92) | |
| MCI | ↓20% | 6/6 | hippocampus | (87) | |
| MCI, AD | ↓∼18%, ↓∼23% | 15/15, 15/15 | serum | (93) | |
| AD | significant decrease | n = 18 | hippocampus | (94) | |
| catalase | AD | ↓34% | 10/9 | temporal cortex | (91) |
| AD | significant decrease | n = 18, n = 22 | hippocampus, frontal cortex | (94) | |
| glutathione-S-transferase | MCI | ↓30% | 6/6 | hippocampus | (87) |
| AD | significant decrease | n = 18 | hippocampus | (94) | |
| glutathione peroxidase | MCI, AD | ↓∼64%, ↓∼71% | 15/15, 15/15 | serum | (93) |
| AD | significant decrease | n = 19 | cerebellum | (94) | |
| glutathione reductase | AD | significant decrease | n = 18, n = 19 | hippocampus, cerebellum | (94) |
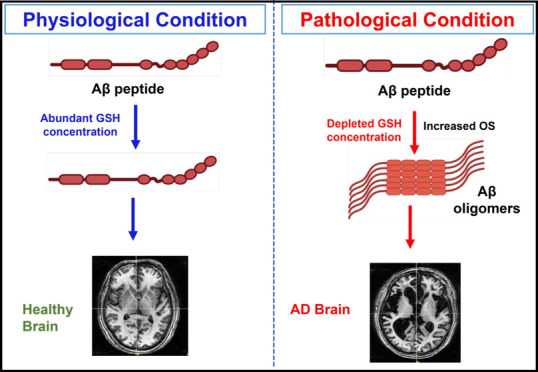
Figure 2.

Schematic representation of the impact of GSH in (A) healthy brain and (B) pathological condition in AD brain. (A) In a normal brain under physiological conditions, glutathione levels are maintained. GSH participates in the GSH cycle.89 The GSH:GSSG ratio is maintained at 100:1 via the cycle by recycling GSH from oxidized glutathione (GSSG) intracellularly. ROS is actively detoxified by GSH, which acts as a modulator for the radicals, and ROS cannot cause further oxidation of the M35 residue of Aβ. The redox homeostasis is thus maintained in the healthy brain. (B) A model of Alzheimer’s disease pathology due to oxidative stress. Depleted GSH levels can lead to increased levels of free radicals. These free radicals can then oxidize the M35 residue leading to several biochemical modifications12 like lipid peroxidation and protein modifications as well as formation of amyloid plaques, which can consequently form neurofibrillary tangles. Red star indicates ROS and oxidative damage. Representative image of structure of residues 35–42 of Aβ1–42 has been adapted with permission from ref (79). Copyright 2008 Elsevier B.V. Representative MRI images of healthy and AD brain are taken from NINS laboratory data.
Therapeutic Avenue: Glutathione Antioxidant System
Glutathione is a tripeptide and the body’s most common antioxidant molecule. Originating from glutamic acid, cysteine, and glycine, all mammalian cells have a global presence of GSH. GSH serves as a scavenger against both ROS and RNS. The sulfyhydryl (−SH) group of GSH enables it to chelate metal ions as well as participate in redox cycling reactions.95 It can thus sequester free radicals and metal ions, reducing the oxidative load on the brain and in turn render protection to M35 of Aβ from increased oxidation. Consequently, depletion in brain GSH concentration might serve as an indicator of increased OS.96 This translates to the incidence of neuroinflammatory processes, mitochondrial dysfunction, and is also associated with several neuropathological conditions, including AD.53 In a study using immature cortical HT22 neurons, it was shown that the depletion of GSH triggers the activation of the lipid-peroxidising enzyme 12-lipooxygenase (12-LOX), inducing an apoptotic cascade and ultimately neuronal death.97 12-LOX is directly inhibited by GSH, providing a crucial role of GSH in mitigating the effects of OS. Maintaining GSH concentrations in the brain (1–2 mM)98 through dietary supplementations and targeting pathways impeded in AD may provide molecular targets for pharmacologically relevant clinical interventions. This may aid in halting the conversion from healthy conditions to MCI.
The basic tenet of the OS hypothesis underlying AD pathology is age-related loss of cognitive functioning due to the increasing and irreversible accumulation of oxidative damage to neurons and glia. Aging is thus considered the most significant risk factor for AD.99 In a study with AD patients, the levels of antioxidant enzymes’ gene expression were measured in the hippocampus, inferior parietal lobule, and cerebellum.100 The levels of GSH-peroxidase and GSSG reductase were found to be elevated in the hippocampus and inferior parietal lobule but not in the cerebellum. The region-specific differences in expression of these enzymes are indicative of the variation of magnitude of ROS-mediated injury in different areas of AD brain. 4-HNE is found elevated in AD brain and CSF,101 indicating lipid peroxidation due to oxidative damage. GSH has also been observed to protect cultured neurons from oxidative damage resulting due to β-amyloidosis, iron, and/or HNE accumulation.102
In transgenic mouse models, a decrease in the levels of GSH has been found to precede β-amyloidosis as well as plaque formation.103,104 In a proton MRS (1H-MRS) study correlating the levels of cortical GSH and brain amyloidosis (measured using Pittsburgh Compound-B), GSH levels were found to be negatively correlated with amyloidosis in the temporal and parietal brain regions.52 This inverse association implicates the role of OS in Aβ plaque formation. Another study involving 1H-MRS showed that the reduction in GSH was correlated to a decline in the global cognitive status of patients as assessed by different neuropsychological tests like mini mental status examination (MMSE) and clinical dementia ratio (CDR).53 A negative correlation was also observed between the trail making tests (TMT B-A) and GSH levels in the hippocampus and right frontal cortex. GSH levels in these brain regions can serve as a predictive biological marker for the intensity of cognitive impairment. Depleted GSH levels have also been associated with mitochondrial dysfunction and neurodegeneration.94,8,105 Corroborating the data, it can be implied that OS plays a critical role in the pathogenesis of AD, and the GSH antioxidant system can be used therapeutically for different molecular targets.
Oxidative stress and the oxidation of M35 residue play a collaborative role in accelerating the onset of AD symptoms. Necrotic death of neurons due to OS contributes significantly to the biochemical and pathogenic changes observed in AD brains. Treatments that facilitate increased biosynthesis of GSH or suppress its degradation can be developed.106 Sodium-dependent GSH transporters in the endothelial cells of the blood brain barrier (BBB) are involved in the bidirectional transport of GSH into and out of the brain.107 Pharmacologically targeting these transporters may provide means for the direct uptake of GSH via the BBB and into the brain. It has already been indicated that an iron chelator in combination with a GSH enhancing drug can used as a mode of delivery to restore cognitive reserve in patients with AD and MCI.38 Further clinical trials utilizing GSH supplements can therefore be conducted to assess its therapeutic potential against AD patients.
Conclusion and Future Directions
Alzheimer’s disease is a highly devastating condition that impedes one’s normal day-to-day life. The integral role of OS in the pathophysiology and progression of AD is now a well-accepted notion. Aβ1–42 associated OS is heavily implicated as its Met35 residue is particularly vulnerable to oxidation due to pathological conditions.
The GSH antioxidant system is an important scavenger of ROS and RNS and any imbalance in GSH homeostasis is ensnared in the prognosis of AD and several other neurodegenerative disorders. GSH is easily quantified using 1H-MRS methods, and evaluating its brain region-specific concentration can provide a benchmark in the comparison of normal brain versus specific areas of brain that are implicated in different neurodegenerative diseases.
Recent research studies with significant representation of clinical population show that GSH levels are depleted with a concurrent increase in iron load in AD progression. Hence, a combination of GSH and iron chelator, namely “Sanjeevani”, could be utilized as a fixed dual composition drug in upcoming clinical trials for restoring the cognitive reserve in AD and MCI patients. Importantly, a diet rich in antioxidants along with a healthy lifestyle might reduce the risk and prevent the onset and progression of this highly debilitating disease.
Acknowledgments
Prof. Pravat K. Mandal (Principal Investigator) thanks the partial financial support from various agencies: Tata Innovation Fellowship (Department of Biotechnology, Ministry of Science and Technology, Government of India) (Award BT/HRD/01/05/2015), Indo Australian grant strategic funding to PKM (Grant BT/Indo-Aus/10/31/2016), and the Ministry of Information Technology (Grant 4(5)/201-ITEA).
Author Contributions
P.K.M. (Principal Investigator) conceptualized the idea and was involved in writing the manuscript and figure preparation. R.G.R. and A.S. were involved in writing the manuscript, preparation of figures, and participated in the discussion for manuscript preparation.
The authors declare no competing financial interest.
References
- Saharan S.; Mandal P. K. The Emerging Role of Glutathione in Alzheimer’s Disease. Journal of Alzheimer’s Disease 2014, 40, 519–529. 10.3233/JAD-132483. [DOI] [PubMed] [Google Scholar]
- Salim S. Oxidative Stress and the Central Nervous System. J. Pharmacol Exp Ther 2017, 360 (1), 201–205. 10.1124/jpet.116.237503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarawneh R.; Holtzman D. M. The clinical problem of symptomatic Alzheimer disease and mild cognitive impairment. Cold Spring Harb Perspect Med. 2012, 2 (5), a006148. 10.1101/cshperspect.a006148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson P. T.; Braak H.; Markesbery W. R. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. Journal of neuropathology and experimental neurology 2009, 68 (1), 1–14. 10.1097/NEN.0b013e3181919a48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J.; Selkoe D. J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science (New York, N.Y.) 2002, 297 (5580), 353–356. 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Lewis J.; Dickson D. W. Propagation of tau pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta neuropathologica 2016, 131 (1), 27–48. 10.1007/s00401-015-1507-z. [DOI] [PubMed] [Google Scholar]
- Akiyama H.; Barger S.; Barnum S.; Bradt B.; Bauer J.; Cole G. M.; Cooper N. R.; Eikelenboom P.; Emmerling M.; Fiebich B. L.; et al. Inflammation and Alzheimer’s disease. Neurobiology of aging 2000, 21 (3), 383–421. 10.1016/S0197-4580(00)00124-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M. T.; Beal M. F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443 (7113), 787–795. 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Markesbery W. R. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol. Med. 1997, 23 (1), 134–147. 10.1016/S0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- Mandal P. K.; Tripathi M.; Sugunan S. Brain oxidative stress: detection and mapping of anti-oxidant marker ’Glutathione’ in different brain regions of healthy male/female, MCI and Alzheimer patients using non-invasive magnetic resonance spectroscopy. Biochem. Biophys. Res. Commun. 2012, 417 (1), 43–48. 10.1016/j.bbrc.2011.11.047. [DOI] [PubMed] [Google Scholar]
- Huang W. J.; Zhang X.; Chen W. W. Role of oxidative stress in Alzheimer’s disease. Biomed Rep 2016, 4 (5), 519–522. 10.3892/br.2016.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheignon C.; Tomas M.; Bonnefont-Rousselot D.; Faller P.; Hureau C.; Collin F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. 10.1016/j.redox.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raichle M. E.; Gusnard D. A. Appraising the brain’s energy budget. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (16), 10237–10239. 10.1073/pnas.172399499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uttara B.; Singh A. V.; Zamboni P.; Mahajan R. T. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Current neuropharmacology 2009, 7 (1), 65–74. 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görlach A.; Bertram K.; Hudecova S.; Krizanova O. Calcium and ROS: A mutual interplay. Redox Biology 2015, 6, 260–271. 10.1016/j.redox.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betteridge D. J. What is oxidative stress?. Metabolism 2000, 49 (2), 3–8. 10.1016/S0026-0495(00)80077-3. [DOI] [PubMed] [Google Scholar]
- Butterfield D. A.; Boyd-Kimball D. Oxidative Stress, Amyloid-beta Peptide, and Altered Key Molecular Pathways in the Pathogenesis and Progression of Alzheimer’s Disease. J. Alzheimers Dis 2018, 62 (3), 1345–1367. 10.3233/JAD-170543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P.; Jha A. B.; Dubey R. S.; Pessarakli M. Reactive Oxygen Species, Oxidative Damage, and Antioxidative Defense Mechanism in Plants under Stressful Conditions. Journal of Botany 2012, 2012, 217037. 10.1155/2012/217037. [DOI] [Google Scholar]
- Gandhi S.; Abramov A. Y. Mechanism of oxidative stress in neurodegeneration. Oxid Med. Cell Longev 2012, 2012, 428010. 10.1155/2012/428010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su B.; Wang X.; Nunomura A.; Moreira P. I.; Lee H. G.; Perry G.; Smith M. A.; Zhu X. Oxidative stress signaling in Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5 (6), 525–532. 10.2174/156720508786898451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieber M.; Chandel N. S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24 (10), R453–R462. 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison D. K. MAP kinase pathways. Cold Spring Harb Perspect Biol. 2012, 4 (11), ao11254. 10.1101/cshperspect.a011254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son Y.; Cheong Y. K.; Kim N. H.; Chung H. T.; Kang D. G.; Pae H. O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can. ROS Activate MAPK Pathways?. J. Signal Transduct 2011, 2011, 792639. 10.1155/2011/792639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; Lee H. G.; Raina A. K.; Perry G.; Smith M. A. The role of mitogen-activated protein kinase pathways in Alzheimer’s disease. Neurosignals 2002, 11 (5), 270–281. 10.1159/000067426. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Wang X. Targeting the Wnt/β-catenin signaling pathway in cancer. Journal of Hematology & Oncology 2020, 13 (1), 165. 10.1186/s13045-020-00990-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimaian A.; Majidinia M.; Bannazadeh Baghi H.; Yousefi B. The crosstalk between Wnt/β-catenin signaling pathway with DNA damage response and oxidative stress: Implications in cancer therapy. DNA Repair 2017, 51, 14–19. 10.1016/j.dnarep.2017.01.003. [DOI] [PubMed] [Google Scholar]
- Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol Toxicol 2013, 53, 401–426. 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg M.; Patil J.; D’Angelo B.; Weber S. G.; Mallard C. NRF2-regulation in brain health and disease: implication of cerebral inflammation. Neuropharmacology 2014, 79, 298–306. 10.1016/j.neuropharm.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies D. A.; Adlimoghaddam A.; Albensi B. C. Role of Nrf2 in Synaptic Plasticity and Memory in Alzheimer’s Disease. Cells 2021, 10 (8), 1884. 10.3390/cells10081884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey C. P.; Glass C. A.; Montgomery M. B.; Lindl K. A.; Ritson G. P.; Chia L. A.; Hamilton R. L.; Chu C. T.; Jordan-Sciutto K. L. Expression of Nrf2 in neurodegenerative diseases. Journal of neuropathology and experimental neurology 2007, 66 (1), 75–85. 10.1097/nen.0b013e31802d6da9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lushchak V. I. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. Journal of Amino Acids 2012, 2012, 736837. 10.1155/2012/736837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiannopoulou K. G.; Papageorgiou S. G. Current and Future Treatments in Alzheimer Disease: An Update. J. Cent Nerv Syst. Dis 2020, 12, 1. 10.1177/1179573520907397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines J. L. Alzheimer Disease: Perspectives from Epidemiology and Genetics. Journal of Law, Medicine & Ethics 2018, 46 (3), 694–698. 10.1177/1073110518804230. [DOI] [PubMed] [Google Scholar]
- Yiannopoulou K. G.; Papageorgiou S. G. Current and future treatments for Alzheimer’s disease. Therapeutic advances in neurological disorders 2013, 6 (1), 19–33. 10.1177/1756285612461679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P.-P.; Xie Y.; Meng X.-Y.; Kang J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduction and Targeted Therapy 2019, 4 (1), 29. 10.1038/s41392-019-0063-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atri A. Current and Future Treatments in Alzheimer’s Disease. Semin Neurol 2019, 39 (02), 227–240. 10.1055/s-0039-1678581. [DOI] [PubMed] [Google Scholar]
- Sevigny J.; Chiao P.; Bussière T.; Weinreb P. H.; Williams L.; Maier M.; Dunstan R.; Salloway S.; Chen T.; Ling Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537 (7618), 50–56. 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- Mandal P. K.; Samkaria A.; Maroon J. C. AD Hypotheses and Suggested Clinical Trials. ACS Chem. Neurosci. 2021, 12 (21), 3968–3971. 10.1021/acschemneuro.1c00627. [DOI] [PubMed] [Google Scholar]
- Varadarajan S.; Yatin S.; Kanski J.; Jahanshahi F.; Butterfield D. A. Methionine residue 35 is important in amyloid beta-peptide-associated free radical oxidative stress. Brain Res. Bull. 1999, 50 (2), 133–141. 10.1016/S0361-9230(99)00093-3. [DOI] [PubMed] [Google Scholar]
- Hensley K.; Carney J. M.; Mattson M. P.; Aksenova M.; Harris M.; Wu J. F.; Floyd R. A.; Butterfield D. A. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc. Natl. Acad. Sci. U. S. A. 1994, 91 (8), 3270–3274. 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley K.; Aksenova M.; Carney J. M.; Harris M.; Butterfield D. A. Amyloid beta-peptide spin trapping. II: Evidence for decomposition of the PBN spin adduct. Neuroreport 1995, 6 (3), 493–496. 10.1097/00001756-199502000-00022. [DOI] [PubMed] [Google Scholar]
- Yatin S. M.; Varadarajan S.; Link C. D.; Butterfield D. A. In vitro and in vivo oxidative stress associated with Alzheimer’s amyloid beta-peptide (1–42). Neurobiology of aging 1999, 20 (3), 325–330. 10.1016/S0197-4580(99)00056-1. [DOI] [PubMed] [Google Scholar]
- Kanski J.; Aksenova M.; Butterfield D. A. The hydrophobic environment of Met35 of Alzheimer’s Abeta(1–42) is important for the neurotoxic and oxidative properties of the peptide. Neurotox Res. 2002, 4 (3), 219–223. 10.1080/10298420290023945. [DOI] [PubMed] [Google Scholar]
- Kanski J.; Aksenova M.; Schöneich C.; Butterfield D. A. Substitution of isoleucine-31 by helical-breaking proline abolishes oxidative stress and neurotoxic properties of Alzheimer’s amyloid beta-peptide. Free Radic Biol. Med. 2002, 32 (11), 1205–1211. 10.1016/S0891-5849(02)00821-3. [DOI] [PubMed] [Google Scholar]
- Scott L. J.; Goa K. L. Galantamine. Drugs 2000, 60 (5), 1095–1122. 10.2165/00003495-200060050-00008. [DOI] [PubMed] [Google Scholar]
- Farlow M. R. Update on Rivastigmine. Neurologist 2003, 9, 230–234. 10.1097/01.nrl.0000087724.73783.5f. [DOI] [PubMed] [Google Scholar]
- Birks J. S.; Harvey R. J. Donepezil for dementia due to Alzheimer’s disease. Cochrane database of systematic reviews 2018, 6 (6), CD001190. 10.1002/14651858.CD001190.pub3. [DOI] [PubMed] [Google Scholar]
- van Marum R. J. Update on the use of memantine in Alzheimer’s disease. Neuropsychiatric disease and treatment 2009, 5, 237–247. 10.2147/NDT.S4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield D. A.; Halliwell B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci 2019, 20 (3), 148–160. 10.1038/s41583-019-0132-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guglielmotto M.; Giliberto L.; Tamagno E.; Tabaton M. Oxidative stress mediates the pathogenic effect of different Alzheimer’s disease risk factors. Frontiers in aging neuroscience 2010, 2, Review. 10.3389/neuro.24.003.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P. K. Magnetic resonance spectroscopy (MRS) and its application in Alzheimer’s disease. Concepts in Magnetic Resonance Part A 2007, 30A (1), 40–64. 10.1002/cmr.a.20072. [DOI] [Google Scholar]
- Chiang G. C.; Mao X.; Kang G.; Chang E.; Pandya S.; Vallabhajosula S.; Isaacson R.; Ravdin L. D.; Shungu D. C. Relationships among Cortical Glutathione Levels, Brain Amyloidosis, and Memory in Healthy Older Adults Investigated In Vivo with (1)H-MRS and Pittsburgh Compound-B PET. AJNR Am. J. Neuroradiol 2017, 38 (6), 1130–1137. 10.3174/ajnr.A5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P. K.; Saharan S.; Tripathi M.; Murari G. Brain Glutathione Levels – A Novel Biomarker for Mild Cognitive Impairment and Alzheimer’s Disease. Biol. Psychiatry 2015, 78 (10), 702–710. 10.1016/j.biopsych.2015.04.005. [DOI] [PubMed] [Google Scholar]
- DeTure M. A.; Dickson D. W. The neuropathological diagnosis of Alzheimer’s disease. Molecular neurodegeneration 2019, 14 (1), 32. 10.1186/s13024-019-0333-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Michaelis E. K. Selective neuronal vulnerability to oxidative stress in the brain. Frontiers in aging neuroscience 2010, 2, 1. 10.3389/fnagi.2010.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carocci A.; Catalano A.; Sinicropi M. S.; Genchi G. Oxidative stress and neurodegeneration: the involvement of iron. Biometals 2018, 31 (5), 715–735. 10.1007/s10534-018-0126-2. [DOI] [PubMed] [Google Scholar]
- Aracena P.; Aguirre P.; Munoz P.; Nunez M. T. Iron and glutathione at the crossroad of redox metabolism in neurons. Biol. Res. 2006, 39 (1), 157–165. 10.4067/S0716-97602006000100017. [DOI] [PubMed] [Google Scholar]
- Lee M.; Kim J. I.; Na S.; Eom K. Metal ions affect the formation and stability of amyloid β aggregates at multiple length scales. Phys. Chem. Chem. Phys. 2018, 20 (13), 8951–8961. 10.1039/C7CP05072K. [DOI] [PubMed] [Google Scholar]
- Rajasekhar K.; Mehta K.; Govindaraju T. Hybrid Multifunctional Modulators Inhibit Multifaceted Aβ Toxicity and Prevent Mitochondrial Damage. ACS Chem. Neurosci. 2018, 9 (6), 1432–1440. 10.1021/acschemneuro.8b00033. [DOI] [PubMed] [Google Scholar]
- Bush A. I. The metallobiology of Alzheimer’s disease. Trends Neurosci 2003, 26 (4), 207–214. 10.1016/S0166-2236(03)00067-5. [DOI] [PubMed] [Google Scholar]
- Rajasekhar K.; Chakrabarti M.; Govindaraju T. Function and toxicity of amyloid beta and recent therapeutic interventions targeting amyloid beta in Alzheimer’s disease. Chem. Commun. 2015, 51 (70), 13434–13450. 10.1039/C5CC05264E. [DOI] [PubMed] [Google Scholar]
- Maynard C. J.; Bush A. I.; Masters C. L.; Cappai R.; Li Q.-X. Metals and amyloid-β in Alzheimer’s disease. International Journal of Experimental Pathology 2005, 86 (3), 147–159. 10.1111/j.0959-9673.2005.00434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. L.; Fan Y. G.; Yang Z. S.; Wang Z. Y.; Guo C. Iron and Alzheimer’s Disease: From Pathogenesis to Therapeutic Implications. Front Neurosci 2018, 12, 1. 10.3389/fnins.2018.00632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M. P.; LeVine H. 3rd Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis 2010, 19 (1), 311–323. 10.3233/JAD-2010-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.-f.; Xu T.-h.; Yan Y.; Zhou Y.-r.; Jiang Y.; Melcher K.; Xu H. E. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacologica Sinica 2017, 38 (9), 1205–1235. 10.1038/aps.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrow C. J.; Zagorski M. G. Solution structures of beta peptide and its constituent fragments: relation to amyloid deposition. Science (New York, N.Y.) 1991, 253 (5016), 179–182. 10.1126/science.1853202. [DOI] [PubMed] [Google Scholar]
- Butterfield D. A.; Boyd-Kimball D. The critical role of methionine 35 in Alzheimer’s amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity. Biochim. Biophys. Acta 2005, 1703 (2), 149–156. 10.1016/j.bbapap.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Drazic A.; Winter J. The physiological role of reversible methionine oxidation. Biochimica et Biophysica Acta (BBA) -. Proteins and Proteomics 2014, 1844 (8), 1367–1382. 10.1016/j.bbapap.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Moskovitz J. Methionine sulfoxide reductases: ubiquitous enzymes involved in antioxidant defense, protein regulation, and prevention of aging-associated diseases. Biochim. Biophys. Acta 2005, 1703 (2), 213–219. 10.1016/j.bbapap.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Gabbita S. P.; Aksenov M. Y.; Lovell M. A.; Markesbery W. R. Decrease in peptide methionine sulfoxide reductase in Alzheimer’s disease brain. J. Neurochem 1999, 73 (4), 1660–1666. 10.1046/j.1471-4159.1999.0731660.x. [DOI] [PubMed] [Google Scholar]
- Schöneich C. Methionine oxidation by reactive oxygen species: reaction mechanisms and relevance to Alzheimer’s disease. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2005, 1703 (2), 111–119. 10.1016/j.bbapap.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Shumilina E.; Dobrovolska O.; Dikiy A.. Evolution of Structural and Coordination Features Within the Methionine Sulfoxide Reductase B Family. The Structural Basis of Biological Energy Generation; Springer, 2014; pp 199–215, 10.1007/978-94-017-8742-0_11. [DOI] [Google Scholar]
- Vogt W. Oxidation of methionyl residues in proteins: tools, targets, and reversal. Free Radic Biol. Med. 1995, 18 (1), 93–105. 10.1016/0891-5849(94)00158-G. [DOI] [PubMed] [Google Scholar]
- Hensley K.; Aksenova M.; Carney J. M.; Harris M.; Butterfield D. A. Amyloid beta-peptide spin trapping. I: Peptide enzyme toxicity is related to free radical spin trap reactivity. Neuroreport 1995, 6 (3), 489–492. 10.1097/00001756-199502000-00021. [DOI] [PubMed] [Google Scholar]
- Pike C. J.; Walencewicz-Wasserman A. J.; Kosmoski J.; Cribbs D. H.; Glabe C. G.; Cotman C. W. Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25–35 region to aggregation and neurotoxicity. J. Neurochem 1995, 64 (1), 253–265. 10.1046/j.1471-4159.1995.64010253.x. [DOI] [PubMed] [Google Scholar]
- Bitan G.; Tarus B.; Vollers S. S.; Lashuel H. A.; Condron M. M.; Straub J. E.; Teplow D. B. A molecular switch in amyloid assembly: Met35 and amyloid beta-protein oligomerization. J. Am. Chem. Soc. 2003, 125 (50), 15359–15365. 10.1021/ja0349296. [DOI] [PubMed] [Google Scholar]
- Hou L.; Shao H.; Zhang Y.; Li H.; Menon N. K.; Neuhaus E. B.; Brewer J. M.; Byeon I. J.; Ray D. G.; Vitek M. P.; et al. Solution NMR studies of the A beta(1–40) and A beta(1–42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J. Am. Chem. Soc. 2004, 126 (7), 1992–2005. 10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]
- Gu M.; Viles J. H. Methionine oxidation reduces lag-times for amyloid-β(1–40) fiber formation but generates highly fragmented fibers. Biochim. Biophys. Acta 2016, 1864 (9), 1260–1269. 10.1016/j.bbapap.2016.04.009. [DOI] [PubMed] [Google Scholar]
- Masuda Y.; Uemura S.; Nakanishi A.; Ohashi R.; Takegoshi K.; Shimizu T.; Shirasawa T.; Irie K. Verification of the C-terminal intramolecular β-sheet in Aβ42 aggregates using solid-state NMR: Implications for potent neurotoxicity through the formation of radicals. Bioorg. Med. Chem. Lett. 2008, 18 (11), 3206–3210. 10.1016/j.bmcl.2008.04.060. [DOI] [PubMed] [Google Scholar]
- Masliah E.; Alford M.; DeTeresa R.; Mallory M.; Hansen L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1996, 40 (5), 759–766. 10.1002/ana.410400512. [DOI] [PubMed] [Google Scholar]
- Butterfield D. A.; Pocernich C. B. The glutamatergic system and Alzheimer’s disease: therapeutic implications. CNS Drugs 2003, 17 (9), 641–652. 10.2165/00023210-200317090-00004. [DOI] [PubMed] [Google Scholar]
- Butterfield D. A.; Galvan V.; Lange M. B.; Tang H.; Sowell R. A.; Spilman P.; Fombonne J.; Gorostiza O.; Zhang J.; Sultana R.; et al. In vivo oxidative stress in brain of Alzheimer disease transgenic mice: Requirement for methionine 35 in amyloid beta-peptide of APP. Free Radic Biol. Med. 2010, 48 (1), 136–144. 10.1016/j.freeradbiomed.2009.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson R. A. S.; Lange M. B.; Sultana R.; Galvan V.; Fombonne J.; Gorostiza O.; Zhang J.; Warrier G.; Cai J.; Pierce W. M.; et al. Differential expression and redox proteomics analyses of an Alzheimer disease transgenic mouse model: effects of the amyloid-β peptide of amyloid precursor protein. Neuroscience 2011, 177, 207–222. 10.1016/j.neuroscience.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Näslund J.; Schierhorn A.; Hellman U.; Lannfelt L.; Roses A. D.; Tjernberg L. O.; Silberring J.; Gandy S. E.; Winblad B.; Greengard P.; et al. Relative abundance of Alzheimer A beta amyloid peptide variants in Alzheimer disease and normal aging. Proc. Natl. Acad. Sci. U. S. A. 1994, 91 (18), 8378–8382. 10.1073/pnas.91.18.8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari M. A.; Scheff S. W. Oxidative Stress in the Progression of Alzheimer Disease in the Frontal Cortex. Journal of Neuropathology & Experimental Neurology 2010, 69 (2), 155–167. 10.1097/NEN.0b013e3181cb5af4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M.; Owen A. D.; Toffa S. E.; Cooper J. M.; Dexter D. T.; Jenner P.; Marsden C. D.; Schapira A. H. Mitochondrial function, GSH and iron in neurodegeneration and Lewy body diseases. J. Neurol Sci. 1998, 158 (1), 24–29. 10.1016/S0022-510X(98)00095-1. [DOI] [PubMed] [Google Scholar]
- Sultana R.; Piroddi M.; Galli F.; Butterfield D. A. Protein levels and activity of some antioxidant enzymes in hippocampus of subjects with amnestic mild cognitive impairment. Neurochem. Res. 2008, 33 (12), 2540–2546. 10.1007/s11064-008-9593-0. [DOI] [PubMed] [Google Scholar]
- Lovell M. A.; Xie C.; Markesbery W. R. Decreased glutathione transferase activity in brain and ventricular fluid in Alzheimer’s disease. Neurology 1998, 51 (6), 1562–1566. 10.1212/WNL.51.6.1562. [DOI] [PubMed] [Google Scholar]
- Johnson W. M.; Wilson-Delfosse A. L.; Mieyal J. J. Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients 2012, 4 (10), 1399–1440. 10.3390/nu4101399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snaedal J.; Kristinsson J.; Gunnarsdóttir S.; Ólafsdóttir Á.; Baldvinsson M.; Jóhannesson T. Copper, Ceruloplasmin and Superoxide Dismutase in Patients with Alzheimer’s Disease. Dementia and Geriatric Cognitive Disorders 1998, 9 (5), 239–242. 10.1159/000017067. [DOI] [PubMed] [Google Scholar]
- Marcus D. L.; Thomas C.; Rodriguez C.; Simberkoff K.; Tsai J. S.; Strafaci J. A.; Freedman M. L. Increased Peroxidation and Reduced Antioxidant Enzyme Activity in Alzheimer’s Disease. Exp. Neurol. 1998, 150 (1), 40–44. 10.1006/exnr.1997.6750. [DOI] [PubMed] [Google Scholar]
- Chen L.; Richardson J. S.; Caldwell J. E.; Ang L. C. Regional Brain Activity of Free Radical Defense Enzymes in Autopsy Samples from Patients with Alzheimer’s Disease and from Nondemented Controls. International Journal of Neuroscience 1994, 75 (1–2), 83–90. 10.3109/00207459408986291. [DOI] [PubMed] [Google Scholar]
- Padurariu M.; Ciobica A.; Hritcu L.; Stoica B.; Bild W.; Stefanescu C. Changes of some oxidative stress markers in the serum of patients with mild cognitive impairment and Alzheimer’s disease. Neurosci. Lett. 2010, 469 (1), 6–10. 10.1016/j.neulet.2009.11.033. [DOI] [PubMed] [Google Scholar]
- Venkateshappa C.; Harish G.; Mahadevan A.; Srinivas Bharath M. M.; Shankar S. K. Elevated Oxidative Stress and Decreased Antioxidant Function in the Human Hippocampus and Frontal Cortex with Increasing Age: Implications for Neurodegeneration in Alzheimer’s Disease. Neurochem. Res. 2012, 37 (8), 1601–1614. 10.1007/s11064-012-0755-8. [DOI] [PubMed] [Google Scholar]
- Jozefczak M.; Remans T.; Vangronsveld J.; Cuypers A. Glutathione is a key player in metal-induced oxidative stress defenses. International journal of molecular sciences 2012, 13 (3), 3145–3175. 10.3390/ijms13033145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottino F.; Lucignani M.; Napolitano A.; Dellepiane F.; Visconti E.; Rossi Espagnet M. C.; Pasquini L. In Vivo Brain GSH: MRS Methods and Clinical Applications. Antioxidants (Basel) 2021, 10 (9), 1407. 10.3390/antiox10091407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Maher P.; Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron 1997, 19 (2), 453–463. 10.1016/S0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- Mandal P. K. In vivo proton magnetic resonance spectroscopic signal processing for the absolute quantitation of brain metabolites. Eur. J. Radiol 2012, 81 (4), e653 10.1016/j.ejrad.2011.03.076. [DOI] [PubMed] [Google Scholar]
- Guerreiro R.; Bras J. The age factor in Alzheimer’s disease. Genome Med. 2015, 7, 106. 10.1186/s13073-015-0232-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksenov M. Y.; Tucker H. M.; Nair P.; Aksenova M. V.; Butterfield D. A.; Estus S.; Markesbery W. R. The expression of key oxidative stress-handling genes in different brain regions in Alzheimer’s disease. J. Mol. Neurosci 1998, 11 (2), 151–164. 10.1385/JMN:11:2:151. [DOI] [PubMed] [Google Scholar]
- Zarkovic K. 4-hydroxynonenal and neurodegenerative diseases. Mol. Aspects Med. 2003, 24 (4–5), 293–303. 10.1016/S0098-2997(03)00024-4. [DOI] [PubMed] [Google Scholar]
- Mark R. J.; Lovell M. A.; Markesbery W. R.; Uchida K.; Mattson M. P. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J. Neurochem 1997, 68 (1), 255–264. 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- Resende R.; Moreira P. I.; Proença T.; Deshpande A.; Busciglio J.; Pereira C.; Oliveira C. R. Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radic Biol. Med. 2008, 44 (12), 2051–2057. 10.1016/j.freeradbiomed.2008.03.012. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Rodriguez C.; Spaulding J.; Aw T. Y.; Feng J. Age-dependent and tissue-related glutathione redox status in a mouse model of Alzheimer’s disease. J. Alzheimers Dis 2012, 28 (3), 655–666. 10.3233/JAD-2011-111244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A.; Mårtensson J.; Stole E.; Auld P. A.; Meister A. Glutathione deficiency leads to mitochondrial damage in brain. Proc. Natl. Acad. Sci. U. S. A. 1991, 88 (5), 1913–1917. 10.1073/pnas.88.5.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocernich C. B.; Butterfield D. A. Elevation of glutathione as a therapeutic strategy in Alzheimer disease. Biochim. Biophys. Acta 2012, 1822 (5), 625–630. 10.1016/j.bbadis.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi D.; Megha K.; Mishra R.; Mandal P. K. Glutathione in Brain: Overview of Its Conformations, Functions, Biochemical Characteristics, Quantitation and Potential Therapeutic Role in Brain Disorders. Neurochem. Res. 2020, 45 (7), 1461–1480. 10.1007/s11064-020-03030-1. [DOI] [PubMed] [Google Scholar]