

Abstract
Herein we describe Hyperion, a new program for computing relativistic picture-change-corrected magnetic resonance parameters from scalar relativistic active space wave functions, with or without spin–orbit coupling (SOC) included a posteriori. Hyperion also includes a new orbital decomposition method for assisting active space selection for calculations of hyperfine coupling. For benchmarking purposes, we determine hyperfine coupling constants of selected alkali metal, transition metal, and lanthanide atoms, based on complete active space self-consistent field spin–orbit calculations in OpenMolcas. Our results are in excellent agreement with experimental data from atomic spectroscopy as well as theoretical predictions from four-component relativistic calculations.
1. Introduction
Magnetic resonance spectroscopy techniques, such as electron paramagnetic resonance (EPR) and nuclear magnetic resonance (NMR), are capable of providing very accurate information on the interactions between electron spins, nuclear spins, and external magnetic fields. The information available from EPR and NMR spectra is often encoded via iterative fitting of the experimental data to a model spin Hamiltonian in a set of effective parameters, each related to a specific type of coupling between magnetic entities. Quantities probing interactions between electron and nuclear spins, namely, EPR hyperfine coupling constants (HFCCs) and paramagnetic NMR shifts, depend strongly on unpaired electron (spin) density1,2 and are central to the study of various phenomena encountered in molecular systems of interest. Examples include using HFCCs as proxies for covalency in actinide complexes,3 quantifying spin decoherence in molecular qubits via theoretically determined hyperfine coupling (HFC) tensors,4 and employing contact NMR shifts—which have an intrinsic dependence on HFC parameters5—to understand the solution structure and magnetic properties of paramagnetic MRI contrast agents.6
The steady evolution of electronic structure algorithms has made it computationally feasible to study heavy-element complexes fully ab initio. As the atomic number increases, relativistic effects become more important, to the extent where they cannot be regarded as mere perturbations of the Schrödinger picture; instead, a four-component Dirac formalism becomes necessary. The onset of the relativistic regime is especially important for magnetic interactions, which couple the electronic and positronic degrees of freedom of a Dirac spinor, therefore requiring an explicit description of the latter. Hence, theoretical frameworks developed for magnetic properties such as HFC must account for relativistic effects—both spin-independent (scalar relativistic, SR) and spin-dependent (e.g., spin–orbit coupling, SOC)—to ensure a wide range of applicability and to keep up with the latest experimental advances.
HFC between an unpaired electron spin S and a nuclear spin IN is most frequently modeled non-relativistically as the sum of anisotropic dipolar coupling, known as the spin-dipole (SD) mechanism, and isotropic Fermi coupling (FC)7
| 1 |
where we use μ0, ge, μB, gN, μN, and IN to denote the vacuum permeability, electron g-factor, Bohr magneton, nuclear g-factor, nuclear magneton, and nuclear spin vector, respectively; rN = r – RN is the position vector of the unpaired electron with respect to the magnetic nucleus. The strength of FC is proportional to the spin population at the magnetic nucleus, ρNα–β, thus providing a convenient probe for spin delocalization in a molecule. Although the single-configurational view of electronic structure suggests that this term arises solely through s-type atomic orbital (AO) contributions to the singly occupied molecular orbital (SOMO), several factors complicate this interpretation.
Differences between the interaction of core spin-up and spin-down electrons with the unpaired spin give rise to spin polarization (SP),2 which gives additional contributions to the isotropic HFC and cannot be described by a single electronic configuration. Thus, the quantitative interpretation of HFCCs requires quantum chemical techniques to model the electronic structure accurately and capture SP effects. For this purpose, there are two possible solutions: spin-unrestricted single-configurational methods, such as unrestricted Hartree–Fock (UHF) and unrestricted Kohn–Sham density functional theory (UKS-DFT), which offer low computational cost at the expense of broken spin symmetry, and more expensive spin-adapted multiconfigurational methods, which preserve spin symmetry and are designed to handle electron correlation.
Unrestricted DFT is currently the most widely used approach for determining HFCCs, due to the lower computational cost relative to wave-function-based (ab initio) algorithms. Hybrid functionals give good predictions for HFCCs of organic radicals and transition metal complexes;8,9 however, the accuracy of these results is believed to be caused by fortuitous error cancellation.10 Moreover, a recent study11 shows that the best choice of functional for HFC is system-dependent. Aside from HFC-related shortcomings, the single-configurational framework underlying Kohn–Sham DFT is inappropriate for describing static correlation, an important feature of f-element complexes. Such systems require a multiconfigurational approach, usually in the form of active space wave function optimization techniques such as complete active space self-consistent field (CASSCF), in order to obtain meaningful predictions of energies and molecular properties.
Although far from black-box, active space algorithms are not only cheaper than fully correlated alternatives such as full configuration interaction or coupled cluster methods but also more flexible, as the active space is user-defined. This framework can be leveraged to obtain accurate theoretical HFCCs by including, in addition to static correlation, a selection of dynamical correlation effects (e.g., SP) that significantly influence HFC. The challenge, then, is developing a strategy for choosing computationally feasible active spaces that result in accurate theoretical HFCCs. Previous studies employing multireference configuration interaction (MR-CI) algorithms have already analyzed the convergence of HFCCs with respect to CI excitation level, orbital selection threshold, and basis set completeness.2,12−19 However, most of the established trends are only applicable to the non-relativistic regime; in particular, the observations about AO contributions to HFCCs result directly from the delta distribution form of the FC term (eq 1, right), which is specific to the non-relativistic Schrödinger–Pauli framework.20 Relativistic treatments of HFC hence require new work to establish updated guidelines for theoretical investigations.
Fully relativistic four-component approaches are unfeasible for all but the simplest systems,21,22 unless combined with a low-cost electronic structure algorithm such as Dirac–Hartree–Fock (DHF)23 or DFT.24 As such, multiple strategies that decouple the upper (electronic) and lower (positronic) components of the Dirac Hamiltonian have been developed to lower the cost of relativistic calculations for application to real molecular systems. Use of a decoupling transformation is, in effect, a change in the reference frame of the wave function, and therefore, it must also be applied to the property operators; ignoring this second step leads to the so-called picture-change error (PCE).25 Due to the picture-change correction, relativistic property operators are different from their non-relativistic counterparts and, in the case of relativistic HFC, the isotropic contribution is no longer proportional to the spin density at the nucleus.
Approximate (quasi-relativistic) decoupling techniques, such as the regular approximation (RA) and Douglas–Kroll–Hess (DKH), are now fairly widespread and provide excellent predictions for the energies of most relativistic systems.26,27 These two-component approaches can be simplified further by disregarding spin-dependent effects, yielding a one-component scalar relativistic formalism. For magnetic resonance applications, the zeroth-order regular approximation (ZORA) is widely used in combination with DFT.28−31 Despite providing a good description of valence properties,32,33 ZORA affords large errors for core-dependent properties of heavy-element systems (e.g., core-hole X-ray excitations or absolute nuclear shielding tensors);32,34 DKH is significantly more accurate in this respect. Indeed, HFCCs obtained using SR-DKH2 operators are in good agreement with experiment,35,36 although Nguyen Lan et al.37 show that a higher-order decoupling transformation (DKH3) is needed for a converged relativistic HFC picture. Quasi-relativistic HFCCs can also be derived via the infinite-order regular approximation (IORA), and when combined with a multiconfigurational wave function method, this approach produces highly accurate atomic hyperfine structure constants for alkali and coinage metals.38
Exact two-component decoupling schemes are also available, together with one-component SR variants. For the purpose of theoretical HFC, these offer two main improvements over quasi-relativistic theories: there is no uncertainty regarding the appropriate order of decoupling, and picture-change corrections are straightforward to implement via matrix multiplication. The exact-2-component (X2C)39−41 and normalized elimination of the small component (NESC)42 approaches are two popular, fully numerical, choices for theoretical studies of relativistic HFC. In such cases, it is worth keeping in mind that the form of the relativistic HFC operator changes on a case-by-case basis, since the picture-change transformation is only defined in matrix form.
Our goal was to devise a general methodology for determining relativistic, picture-change-corrected HFCCs for chemical systems of arbitrary size and complexity. With this in mind, we developed Hyperion, a Python-based program that computes SR-X2C-decoupled magnetic resonance parameters from complete active space (CAS) or restricted active space (RAS) wave functions, with or without spin–orbit coupling (SOC) added a posteriori (CASSCF-SO/RASSCF-SO); a method based on the same theoretical formalism has been developed in parallel by Autschbach and co-workers and implemented as part of OpenMolcas.41 Although our code has to date been tested only with OpenMolcas, Hyperion is a stand-alone package that can be straightforwardly extended to allow inputs from other quantum chemical software. Herein, we demonstrate the use of Hyperion to obtain HFCCs of selected atoms and benchmark our results against experimental data from atomic spectroscopy and predictions from four-component calculations. As well as showing the performance of Hyperion, we demonstrate various strategies for tackling specific combinations of electron correlation, SR, and SOC effects, highlighting both merits and limitations of CASSCF-SO/RASSCF-SO methods. The comparatively small number of electrons and high symmetry of atoms mean that high levels of theory are achievable with relatively little computational cost, but as our goal is to extend our approach to molecules in the near future, we work with usual electronic structure approximations, such as contracted basis sets and RAS subspaces of limited size, which are the only feasible strategies for molecular calculations.
2. Theory
2.1. SR-X2C Magnetic Properties
Here,
operators are denoted using the hat symbol (T̂) and vector quantities
are shown in bold (A). One-component and two-component
operators are represented using normal font (ĤSF-X2C,UU(1)), while bold-italic notation (HSF-X2C,UU) is used for their matrix representations in a scalar AO basis.
We denote four-component operators as matrix operators (ĤmDE) to emphasize their 2 × 2 block structure. Finally,
we employ double-struck notation (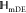 ) for matrix representations of four-component
operators.
) for matrix representations of four-component
operators.
The starting point for any relativistic treatment of magnetic properties is the four-component Dirac equation under a scalar potential V and a vector potential A
| 2 |
where σ is the 3-vector of Pauli spin matrices, p̂ is the electron linear momentum operator, me is the electron mass, and c is the speed of light in a vacuum; SI units are used throughout this work. We represent all four-component operators in block form, with each matrix element denoting a two-component operator. Note that, in two-component equations, σ = 2s, where s is the electron spin vector.
The Dirac wave function, also known as a four-component spinor,
has an upper component ψU, sometimes referred to
as the large component, as well as a lower component (or small component)
ψL. In order for eq 2 to approach the correct non-relativistic limit, the
basis sets chosen for ψU and ψL must
obey the restricted kinetic balance (RKB) condition,43−46 namely, that, for an upper component basis set {ϕμ}, the lower component basis set is 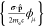 . Although using RKB alone is formally justified
only in the absence of magnetic fields, this approach is reasonable,
as the magnetic-field-dependent terms are treated as a perturbation
herein (vide infra). By substituting the RKB condition
into the four-component Hamiltonian, we arrive at the modified Dirac
equation47,48
. Although using RKB alone is formally justified
only in the absence of magnetic fields, this approach is reasonable,
as the magnetic-field-dependent terms are treated as a perturbation
herein (vide infra). By substituting the RKB condition
into the four-component Hamiltonian, we arrive at the modified Dirac
equation47,48
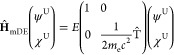 |
3 |
with
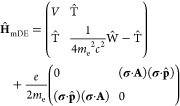 |
4 |
| 5 |
| 6 |
Notice that the lower component of eq 2 has been replaced by the pseudo-upper component χU, which can be represented in the same basis as that used for ψU. Additionally, the modified Dirac Hamiltonian (eq 4) has been separated into A-independent and A-dependent contributions; we henceforth treat the A-dependent term as a first-order perturbation, Ĥ(1). Using the Dirac relation, we rewrite Ŵ as
| 7 |
and discard the σ-dependent term to obtain a spin-free four-component operator. By ignoring the spin-dependent contribution, this relativistic formalism becomes compatible with the one-component framework employed by most electronic structure packages, including OpenMolcas. The resulting spin-free (SF) modified Dirac operator is
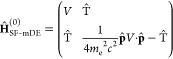 |
8 |
which shall serve as the zeroth-order (unperturbed) Hamiltonian within our theoretical framework.
Scalar relativistic
exact-2-component (SR-X2C) theory49−52 can be employed to determine
a unitary transformation matrix 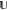 (eq 9) that decouples the upper
and pseudo-upper components by
block-diagonalizing the matrix representation of ĤSF-mDE(0), herein denoted as
(eq 9) that decouples the upper
and pseudo-upper components by
block-diagonalizing the matrix representation of ĤSF-mDE(0), herein denoted as 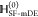 . The electronic problem
can then be solved
by diagonalization of the upper–upper matrix, HSF-X2C,UU, without any reference to the other two-component
blocks.
. The electronic problem
can then be solved
by diagonalization of the upper–upper matrix, HSF-X2C,UU, without any reference to the other two-component
blocks.
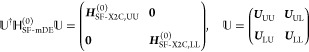 |
9 |
The SF-X2C-decoupled
perturbation is obtained
in an analogous manner, by applying the same unitary transformation
to 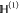 . For simplicity, we employ
operator notation
throughout the remainder of this section; however, it should be noted
that, within the Hyperion implementation, the decoupling
transformation of Ĥ(1) and indeed all
subsequent steps in the calculation of expectation values are carried
out in matrix form. As this work pertains to electronic properties,
only the upper–upper block of the transformed operator
. For simplicity, we employ
operator notation
throughout the remainder of this section; however, it should be noted
that, within the Hyperion implementation, the decoupling
transformation of Ĥ(1) and indeed all
subsequent steps in the calculation of expectation values are carried
out in matrix form. As this work pertains to electronic properties,
only the upper–upper block of the transformed operator
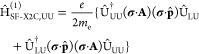 |
10 |
is of interest, and we henceforth drop the UU subscript from Hamiltonian operator notation.
The interaction between an unpaired electron and a nuclear spin, i.e., HFC, can be derived from eq 10 by substituting in the vector potential AN induced by a point-like magnetic dipole N
| 11 |
Alternatively, the nuclear magnetic dipole can be modeled as a Gaussian distribution (eqs 12 and 13)—this is also implemented in Hyperion; however, results herein employ the point nucleus expression for simplicity.
| 12 |
| 13 |
The electronic HFC perturbation operator within the SF-X2C framework is therefore
| 14 |
| 15 |
| 16 |
where we distinguish between a spin-dependent contribution, ĤN,SF-X2CFC+SD, and an imaginary, spin-independent contribution, ĤN,SF-X2C. The former reduces to the sum of the FC and SD operators in the non-relativistic limit, while the latter represents the interaction between the electronic orbital angular momentum and the nuclear spin and is known as the paramagnetic spin–orbit coupling (PSO) term.
It is worth highlighting that eq 15 does not include a delta-function, that would imply only contributions at the nucleus and characteristic of traditional interpretations of FC; as shown by Kutzelnigg,20 this hallmark only arises in the non-relativistic limit of a two-component framework. As a result, relativistic spin-dependent HFC cannot be interpreted as a combination of classical dipolar coupling (SD term) and an FC term sampling the spin density at nucleus N.7
An important aspect that bears discussion is the spin–orbit contribution to HFC. Herein, SOC effects are included in the zeroth-order Hamiltonian, and as such, the spin–orbit HFC contribution is a first-order property,34 computed as the expectation of the PSO operator, eq 16. An alternative approach involves modeling SOC as a perturbation and mapping the HFC operator onto a true spin effective Hamiltonian,;53 in this case, the spin–orbit HFC term only appears in the second-order response, and the second-order HFC energy includes both the PSO interaction and SOC effects on the wave function.54 Therefore, even though both approaches yield spin–orbit contributions to the HFC energy, the two are not directly comparable. The perturbative SOC formalism is the de facto choice in non-relativistic HFC studies;9,31 meanwhile, relativistic four- and two-component methods already account for SOC in the wave function; hence, the HFC energy includes a first-order PSO term.55 Within a one-component formalism such as that employed by OpenMolcas and Hyperion, SOC can be modeled using either approach. Nevertheless, as we are targeting a variety of chemical systems, including atoms and molecules where strong SOC significantly alters the manifold of electronic states, we do not implement the perturbative SOC method within Hyperion. Instead, effects of SOC are included a posteriori in the zeroth order wave function via the restricted active space state interaction (RASSI) formalism56 before calculation of the hyperfine coupling.
2.2. Spin Hamiltonian Parametrizations
The cornerstone of EPR theory is the spin Hamiltonian, an effective operator describing all of the relevant magnetic interactions as couplings in a (2S + 1)-dimensional space, where S is the spin quantum number of the system. The HFC spin Hamiltonian is
| 17 |
where the HFC tensor aN quantifies the strength and anisotropy of the interaction. Quantum mechanical determinations of EPR parameters are based on mapping ab initio operators onto the spin Hamiltonian and differentiating to obtain the tensor components of aN:
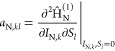 |
18 |
where 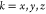 . In a spin-free/scalar
relativistic framework,
provided the electronic system is spin-only (no SOC), the tensor components
are computed as
. In a spin-free/scalar
relativistic framework,
provided the electronic system is spin-only (no SOC), the tensor components
are computed as
| 19 |
where we have defined the vector operator ĥN as
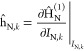 |
20 |
Note that in the case of spin-free electronic structure approaches, such as spin-free CASSCF, the spin projection expectation value ⟨Ŝz⟩ is exactly equal to S.
This spin-only parametrization
is not appropriate, however, for systems exhibiting non-negligible
SOC, as S is no longer a good quantum number. Instead,
the size of the model space is chosen as the number of low-lying electronic
states in the SO-coupled energy spectrum, resulting in an effective 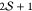 multiplet, where
multiplet, where 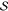 is the
pseudospin quantum number. Replacing S with
is the
pseudospin quantum number. Replacing S with 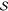 in eq 17 yields the pseudospin
Hamiltonian, from which the
elements of symmetrized tensor aNaNT are computed via the method of Chibotaru57
in eq 17 yields the pseudospin
Hamiltonian, from which the
elements of symmetrized tensor aNaNT are computed via the method of Chibotaru57
| 21 |
where |I⟩ and |J⟩ denote eigenstates in the pseudospin manifold. A similar approach is widely used to derive g-tensors for strongly SO-coupled systems.58 The eigenvectors and eigenvalues of the symmetrized tensors correspond to the principal axes and the squared principal values of the original tensors; as a result, the pseudospin parametrization yields unsigned HFCCs. Although the present work is focused on HFCC magnitude, we note in passing that sign information becomes important if the parameters determined theoretically are employed in further computational work, such as simulations of EPR spectra.
Finally, Hyperion is also capable of determining SF-X2C g-values, using a similar methodology to that described above for HFCCs. The key expressions employed in g-value calculations can be easily obtained by replacing eqs 11 and 17 with the vector potential induced by an external magnetic field and with the electron-Zeeman spin Hamiltonian, respectively. This additional capability will be assessed as part of a future study on molecules.
2.3. HFC Orbital Decomposition
One aspect of theoretical determination of HFCCs using multiconfigurational methods is choosing the most appropriate active space. This is largely a task of trial and error, for which it is hard to define general rules. Hence, we have developed an orbital decomposition method to assess the involvement of particular MOs in the spin-dependent HFCC of a spin-free state ΨSS. To do so, we use the second quantization formalism to represent the expectation value of each vector operator component ĥN,kFC+SD as a sum over pairs of MOs {μν}
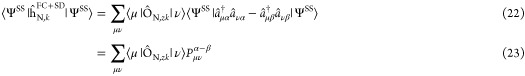 |
22 |
where ↠and â denote spin–orbital creation and annihilation operators, respectively, and Pμνα–β is a spin density matrix element. We have used ÔN,zk to denote the l = z components of a rank-2 tensor operator defined as
| 24 |
Note that ÔN represents the spatial part of hN,SF-X2CFC+SD, as derived via eqs 15 and 20.
We consider each term from the summation
in eq 23 separately,
combine the k = x, y, z components of ÔN,z via the vector norm, and divide the result by the
spin projection to obtain a two-dimensional symmetric matrix 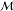 with elements
with elements
| 25 |
The 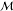 matrix
contains the same information as
the spin-dependent HFCC, albeit in a modified form wherein MO degrees
of freedom are not integrated out. We note that the factor of ⟨Ŝz⟩–1, which reduces
to S–1 for spin-free states, mimics
the expression for HFC operators in the spin-only formalism. Within
this representation, the diagonal elements
matrix
contains the same information as
the spin-dependent HFCC, albeit in a modified form wherein MO degrees
of freedom are not integrated out. We note that the factor of ⟨Ŝz⟩–1, which reduces
to S–1 for spin-free states, mimics
the expression for HFC operators in the spin-only formalism. Within
this representation, the diagonal elements 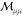 can be interpreted as individual orbital
contributions, while the coupling between two different orbitals is
quantified by
can be interpreted as individual orbital
contributions, while the coupling between two different orbitals is
quantified by 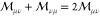 .
.
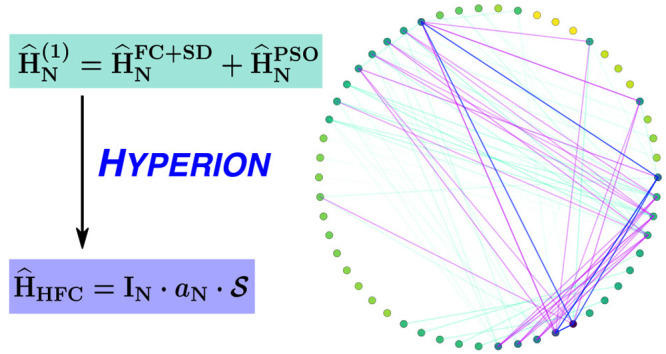
Inspired by density matrix renormalization group (DMRG) entanglement
diagrams,59 we designed a similar pictorial
representation of orbital involvement in HFC (Figure 1), using the matrix elements of 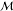 . Individual
orbital contributions are shown
as markers on the circumference of a circle, while pairwise contributions
are shown as chords, color-coded by order of magnitude.
. Individual
orbital contributions are shown
as markers on the circumference of a circle, while pairwise contributions
are shown as chords, color-coded by order of magnitude.
Figure 1.

HFC orbital
decomposition diagrams obtained from relativistic determinations
of HFCCs using Hyperion. (a) 133Cs atom, RASCI(45,52)
using CASSCF(11,15)-optimized orbitals; (b) 197Au atom,
RASCI(45,54) using RASSCF(19,26)-optimized orbitals. Lines across
the diagram correspond to off-diagonal matrix elements of 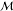 . Individual
orbital contributions, as indicated
by circular markers, correspond to diagonal matrix elements
. Individual
orbital contributions, as indicated
by circular markers, correspond to diagonal matrix elements 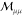 .
.
It is worth emphasizing that this model is based
on a CI-type wave
function that is not relaxed after applying the first-order HFC perturbation
and that our orbital decomposition diagrams only show a static picture
of HFC. To further understand this, we adopt the direct/indirect terminology used by Engels14 to describe
the influence of specific configurations in a CI wave function. The
HFC orbital decomposition matrix 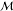 for
a given electronic state is computed
via its spin density matrix, which is completely determined by the
CI expansion. The direct influence of an (active)
orbital or pair of orbitals, as indicated by
for
a given electronic state is computed
via its spin density matrix, which is completely determined by the
CI expansion. The direct influence of an (active)
orbital or pair of orbitals, as indicated by 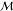 , is
then proportional to the total CI contribution
from all configurations where the orbitals of interest are singly
occupied. However, the spin density associated with one orbital can
vary between different active space selections, due to the indirect effect of all of the other correlated orbitals.
Therefore, the active space selection should not be based solely on
the orbital decomposition diagram. HFC calculations should be carried
out both with and without a specific orbital in the CAS/RAS to determine
the magnitude of its indirect effect.
, is
then proportional to the total CI contribution
from all configurations where the orbitals of interest are singly
occupied. However, the spin density associated with one orbital can
vary between different active space selections, due to the indirect effect of all of the other correlated orbitals.
Therefore, the active space selection should not be based solely on
the orbital decomposition diagram. HFC calculations should be carried
out both with and without a specific orbital in the CAS/RAS to determine
the magnitude of its indirect effect.
3. Computational Details
All electronic structure calculations use a local version of OpenMolcas60 v19.11 adapted to print the X2C decoupling matrices, while the spatial HFC integrals are evaluated analytically with Libcint.61 Full ANO-RCC basis sets62−66 are used throughout to ensure sufficient flexibility and to maintain consistency with previous relativistic HFC studies.39,41,67 The electronic wave function is optimized using either the complete active space self-consistent field (CASSCF) approach68 or a restricted active space (RAS) approach69 with (RASSCF) or without orbital optimization (RASCI). For RASSCF/RASCI calculations, the maximum allowed number of RAS1 holes and the maximum allowed number of RAS3 electrons are both set to 2. Spherical symmetry in the optimized MOs is enforced via the ATOM keyword, which prevents rotations among orbitals belonging to different representations of the full rotation group. Where necessary, SOC is added a posteriori using the RASSI approach,56 which employs a Pauli spin–orbit Hamiltonian,70 represented using atomic mean field integrals (AMFIs).71 To keep RAS calculations tractable, the RAS2 subspace is restricted to the partially occupied shells. As such, for 2S1/2 systems, only the singly occupied s orbital is included in RAS2, while, for transition metals or lanthanides with partially filled d or f shells, respectively, RAS2 is made up of the valence nd or nf and (n + 1)s orbitals as required.
4. Results and Discussion
4.1. Spin Polarization vs Electron Correlation
We take this opportunity to discuss a conceptual aspect that is often encountered in computational HFC literature: the overlap between spin polarization and electron correlation. Both effects have physical interpretations related to the interaction between electrons in a many-electron system; however, a distinction arises in electronic structure theory due to the use of single-configurational (also known as mean-field) references with respect to which perturbations such as SP, static correlation, and dynamic correlation are described.
Unrestricted Hartree–Fock (UHF) theory is the simplest wave-function-based electronic structure framework that accounts for SP, albeit not accurately, due to spin contamination. The UHF wave function can be expressed as a perturbation expansion from a restricted open-shell Hartree–Fock (ROHF) reference, with the first-order term comprising only single excitations.72 It can be deduced that SP arises mainly from singly excited configurations; a spin-adapted CI ansatz with singles (S–CI) should therefore be able to provide a more accurate, non-spin-contaminated description of SP.2
Electron correlation—the instantaneous interaction of electrons, which is not captured by a mean-field ansatz—is also routinely described by a CI wave function, which is most accurate when all possible configurations obtained by distributing all electrons among the available orbitals are included (full CI, FCI). The correlation energy is defined as the difference between the FCI and the reference Hartree–Fock energies. For open-shell systems represented in a spin-adapted framework (i.e., with a ROHF reference), it follows that electron correlation includes an SP contribution,72 whose magnitude directly relates to the overall weighting of single excitations in the CI expansion. Note, however, that, if a UHF reference is used instead, the boundary between SP and “other” correlation becomes unclear. This is fortunately not a concern herein, as we employ spin-adapted wave function methods exclusively.
In a comprehensive review of the spin-polarization model for HFC, Chipman2 refers to second- and higher-order correlation effects/true correlation, thus dividing spin-polarizing single excitations from other excited configurations sampled by FCI. While this split view helps balance computational efficiency with accuracy in multiconfigurational calculations, it has also led to a prevalence in the HFC literature of single-excitation-only results, with no discussion of higher-order effects.41,67 This is likely a safe approximation for systems such as simple organic radicals;73 however, there is no reason to assume a priori that the SP contribution to HFC is more important than contributions from higher-order excitations. In fact, early work shows that high-level CI approximations are required even for second period atoms to achieve quantitative agreement with experiment.16 For the atomic systems studied here, we attempt to include most correlation afforded by a chosen active space using either CASSCF or, where this is unfeasible, RASSCF/RASCI with single and double excitations.
4.2. Alkali Metals
We first apply our methodology to study the hyperfine structure of atoms in the alkali series. This is a popular test set for theoretical HFC computations, requiring a good description of electron correlation, as well as an appropriate treatment of relativistic effects for the heavier elements.22,74,75Table 1 shows predicted (unsigned) HFCCs for the 2S1/2 ground state of each alkali atom, determined via active space electronic structure methods. We note that our CASSCF(1,1) results are very close to the four-component Hartree–Fock data reported by Talukdar,22 suggesting that, at least for the direct contribution to HFC, relativistic effects are correctly accounted for with the X2C decoupling in Hyperion. In order to include correlation contributions to the spin density, the active space must be expanded beyond the SOMO; the CAS size is limited to a maximum of 18 orbitals, and we turn to RAS methods to explore electron correlation effects in a larger orbital space. Note that, due to the computational scaling of RAS algorithms, we restrict the RAS2 subspace to the SOMO. HFCCs converge the fastest when the CASSCF algorithm is used, with CASSCF(11,15) results within approximately 10% of experimental HFC data for alkali Na–Fr. This contrasts with RASSCF and RASCI results, which display slower convergence with active space size and only reach CASSCF accuracy when the majority of MOs are correlated.
Table 1. Unsigned Isotropic HFCCs in MHz Computed for the Ground State of Alkali Atomsa.
| atom | wave function | RAS1 | RAS2 | RAS3 | |A| |
|---|---|---|---|---|---|
| 7Li | 4c-HF | 288.2 | |||
| CASSCF(1,1) | 1 × s | 282.3 | |||
| CASSCF(3,10) | 4 × s, 2 × p | 367.9 | |||
| CASSCF(3,15) | 4 × s, 2 × p, 1 × d | 367.9 | |||
| CASSCF(3,14) | 5 × s, 3 × p | 395.7 | |||
| RASSCF(3,29) | 1 × s | 1 × s | 6 × s, 7 × p | 392.8 | |
| Experimental | 401.7 | ||||
| 23Na | 4c-HF | 633.4 | |||
| CASSCF(1,1) | 1 × s | 639.2 | |||
| CASSCF(9,13) | 2 × s, 2 × p, 1 × d | 724.6 | |||
| CASSCF(9,14) | 3 × s, 2 × p, 1 × d | 794.2 | |||
| CASSCF(11,15) | 4 × s, 2 × p, 1 × d | 801.9 | |||
| RASSCF(11,35) | 2 × s, 1 × p | 1 × s | 6 × s, 6 × p, 1 × d | 820.9 | |
| RASSCF(11,40) | 2 × s, 1 × p | 1 × s | 6 × s, 6 × p, 2 × d | 839.6 | |
| Experimental | 885.8 | ||||
| 39K | 4c-HF | 151.0 | |||
| CASSCF(1,1) | 1 × s | 152.9 | |||
| CASSCF(9,13) | 2 × s, 2 × p, 1 × d | 184.0 | |||
| CASSCF(9,14) | 3 × s, 2 × p, 1 × d | 197.1 | |||
| CASSCF(11,15) | 4 × s, 2 × p, 1 × d | 203.3 | |||
| CASSCF(11,16) | 5 × s, 2 × p, 1 × d | 210.7 | |||
| RASSCF(19,39) | 3 × s, 2 × p | 1 × s | 6 × s, 6 × p, 1 × d | 203.6 | |
| Experimental | 230.8 | ||||
| 85Rb | 4c-HF | 666.9 | |||
| CASSCF(1,1) | 1 × s | 691.9 | |||
| CASSCF(9,13) | 2 × s, 2 × p, 1 × d | 831.2 | |||
| CASSCF(11,15) | 4 × s, 2 × p, 1 × d | 906.5 | |||
| RASSCF(19,26) | 1 × s, 1 × p, 1 × d | 1 × s | 1 × s, 1 × p, 1 × d, 1 × f | 859.1 | |
| RASCI(37,54) | 4 × s, 3 × p, 1 × d | 1 × s | 5 × s, 6 × p, 1 × d, 1 × f | 908.8 | |
| Experimental | 1011.9 | ||||
| 133Cs | 4c-HF | 1495.5 | |||
| CASSCF(1,1) | 1 × s | 1539.1 | |||
| CASSCF(9,13) | 2 × s, 2 × p, 1 × d | 1863.9 | |||
| CASSCF(9,14) | 3 × s, 2 × p, 1 × d | 2024.6 | |||
| CASSCF(11,15) | 4 × s, 2 × p, 1 × d | 2041.3 | |||
| RASCI(35,40) | 5 × s, 4 × p | 1 × s | 5 × s, 4 × p, 1 × d | 2083.2 | |
| RASCI(35,47) | 5 × s, 4 × p | 1 × s | 5 × s, 4 × p, 1 × d, 1 × f | 2095.7 | |
| Experimental | 2298.1 | ||||
| 223Fr | 4c-HF | 5518.0 | |||
| CASSCF(1,1) | 1 × s | 5240.1 | |||
| CASSCF(9,13) | 2 × s, 2 × p, 1 × d | 6277.0 | |||
| CASSCF(11,15) | 4 × s, 2 × p, 1 × d | 6685.7 | |||
| RASCI(43,57) | 6 × s, 5 × p | 1 × s | 5 × s, 6 × p, 1 × d, 1 × f | 6940.0 | |
| Experimental | 7654.0 | ||||
The RAS1, RAS2, and RAS3 columns indicate the number of atomic shells—separated by angular momentum—included in each subspace. Experimental HFCCs and four-component Hartree-Fock (4c-HF) HFCCs are reproduced from ref (22).
The balance of doubly occupied and virtual (unoccupied) orbitals in the CAS/RAS has a crucial influence on HFCC accuracy; it is not sufficient to augment a minimal active space with core orbitals, as appropriate virtual orbitals are needed to correlate them. A similar observation was made in a recent coupled-cluster study22 that emphasizes the need for high-energy unoccupied orbitals to correlate inner-core electrons. From our results, we deduce that radial correlation, introduced via virtual shells with the same angular momentum as the core–shells,76 has the most significant effect on HFCCs. Compare, for example, the HFCCs obtained from CASSCF(9,13) and CASSCF(9,14); the virtual s shell included in the latter leads to a 6–8% improvement in accuracy. For calculations that include orbital optimization (i.e., CASSCF or RASSCF), a good basic principle for active space selection is to include one radially correlating virtual shell for each doubly occupied shell in the active space. However, the exponential scaling of CASSCF severely restricts this strategy, and as such, the largest CAS selections reported herein include only one virtual shell of each angular momentum.
It is also interesting to analyze the influence of polarization basis functions. The inclusion of one virtual d shell in CASSCF affects the HFCCs of all alkali by 4–5% (except Li), suggesting an angular correlation76 effect related to the presence of core p electrons. Extrapolating, we postulate that, for core–shells with the highest angular momentum l, virtual shells with an angular momentum of at least l + 1 are needed to capture angular correlation. Note that, although previous theoretical work highlights the slow convergence of correlation energy with maximum angular momentum77—indicating that an accurate description of correlation likely requires much higher angular momenta—similar effects on HFCCs have only been explored for light atoms.13,16 Nevertheless, testing this hypothesis here is unfeasible given the computational limitations of CASSCF and RASSCF algorithms; we therefore limit our approach to include one virtual d shell for atoms Na–Fr and additionally one virtual f shell for Rb–Fr. Nonetheless, we come within 8–12% total relative error for these atoms.
Spin-dependent HFCCs are known to be particularly sensitive to correlation effects from inner-core electrons; to investigate this, we performed RASSCF and RASCI calculations with RAS1 subspaces spanning most of the core region. All RASCI calculations are performed with CASSCF(11,15)-optimized orbitals for consistency. Orbital decomposition analysis of RASCI results (Figure 1a) reveals a trend of decreasing HFC contribution with increasing angular momentum. Most s and p orbitals are strongly coupled by the HFC operator and make significant (>10 MHz) direct contributions to the HFCC. The influence of (virtual) polarization functions (4–5% increase in HFCC accuracy) is not reflected by the relatively insignificant contributions (<10–3 MHz) shown in the orbital decomposition diagram. Therefore, such functions have a predominantly indirect effect, displacing spin density from orbitals with significant HFC contributions. Occupied d and f shells show similarly small orbital decomposition contributions, and additionally, our calculations suggest their inclusion leads to insignificant variations in the computed HFCC. As such, we conclude that core d and f orbitals of alkali atoms can be safely left out of the RAS1 subspace for the purpose of HFCC determinations. We note that the error with respect to experimental HFCCs plateaus around 10% for RASCI (Figure 2), which could be a consequence of the restricted excitation level and/or the basis set size. These results are nevertheless very encouraging, considering the highly correlated nature of atomic systems, as well as the fact that our calculations explore a relatively limited parameter space (contracted basis sets, RAS1/RAS3 subspaces limited to no more than 35 orbitals).
Figure 2.

Evolution of HFCC absolute error with active space size for alkali atoms. Absolute errors are computed with respect to experimental data from ref (22).
Lastly, the issue of orbital optimization in RAS calculations bears discussion. The super-CI algorithm employed by OpenMolcas “folds” single excitations involving inactive and secondary orbitals into the active MO coefficients,68 thus capturing more correlation than a CI-only calculation with the same parameters. Theoretically, orbital optimization should allow for accurate HFCCs to be obtained with relatively little computational expense, since the active space needs only be large enough to capture the significant correlation contributions to HFC; indeed, this proves true for CASSCF; however, for RASSCF, the results are unpredictable. In the case of light alkali (Li–K), it was possible to run RASSCF calculations involving the entire core s and p manifold, together with polarization functions, which produced HFCCs in excellent agreement with experiment. On the other hand, this approach is prohibitive for heavier atoms—even if memory is not a limitation, orbital optimization is slow and prone to convergence issues. We therefore only employ RASCI for large active space calculations on Rb–Fr. RASSCF calculations involving fewer active orbitals were also attempted; however, the resulting HFCCs were on average less accurate than those from RASCI. It appears that the excitation level restriction severely hinders the efficiency of orbital optimization; stochastic CASSCF78 and DMRG-CASSCF methods79 are likely to provide more accurate HFCCs; however, due to additional uncertainty introduced by approximate CI solvers, such algorithms are not explored herein.
4.3. Coinage Metals
The treatment of HFC in coinage metals is very similar to that of the alkali metals, given they both have single-reference 2S1/2 ground states. A notable point of difference is the valence d shell which, unlike the core-like d shells in alkali atoms, imparts a significant contribution to the HFCC. Correlating the entire valence region is therefore not feasible with CASSCF, and in this case, RASSCF is the only option. We find that the benefits of a larger active space outweigh the limitations due to excitation level restrictions, leading to a net improvement in HFCC accuracy from CASSCF to RASSCF (Table 2). This contrasts with the behavior observed for the alkali metals, indicating that it is perhaps the nature of the correlated orbitals (valence vs core), rather than the CI approximation, that makes the biggest difference to theoretical HFCCs.
Table 2. Unsigned Isotropic HFCCs in MHz Computed for the Ground States of Cu, Ag, and Aua.
| atom | wave function | RAS1 | RAS2 | RAS3 | |A| |
|---|---|---|---|---|---|
| 63Cu | CASSCF(11,6) | 1 × s, 1 × d | 4137.3 | ||
| CASSCF(19,14) | 3 × s, 2 × p, 1 × d | 4604.9 | |||
| RASSCF(19,19) | 1 × s, 1 × p, 1 × d | 1 × s | 1 × s, 1 × p, 1 × d | 4611.3 | |
| RASSCF(19,26) | 1 × s, 1 × p, 1 × d | 1 × s | 1 × s, 1 × p, 1 × d, 1 × f | 4831.4 | |
| RASSCF(19,35) | 1 × s, 1 × p, 1 × d | 1 × s | 1 × s, 1 × p, 1 × d, 1 × f, 1 × g | 4813.7 | |
| RASSCF(29,45) | 3 × s, 2 × p, 1 × d | 1 × s | 3 × s, 2 × p, 1 × d, 1 × f, 1 × g | 5004.8 | |
| RASCI(29,50) | 3 × s, 2 × p, 1 × d | 1 × s | 5 × s, 6 × p, 1 × d, 1 × f | 5036.5 | |
| Experimental | 5866.9 | ||||
| 107Ag | CASSCF(11,6) | 1 × s, 1 × d | 1325.1 | ||
| CASSCF(19,14) | 3 × s, 2 × p, 1 × d | 1455.1 | |||
| RASSCF(19,19) | 1 × s, 1 × p, 1 × d | 1 × s | 1 × s, 1 × p, 1 × d | 1481.1 | |
| RASSCF(19,26) | 1 × s, 1 × p, 1 × d | 1 × s | 1 × s, 1 × p, 1 × d, 1 × f | 1594.6 | |
| RASCI(37,54) | 4 × s, 3 × p, 1 × d | 1 × s | 5 × s, 6 × p, 1 × d, 1 × f | 1620.6 | |
| RASCI(47,59) | 4 × s, 3 × p, 2 × d | 1 × s | 5 × s, 6 × p, 1 × d, 1 × f | 1619.4 | |
| Experimental | 1712.5 | ||||
| 197Au | CASSCF(11,6) | 1 × s, 1 × d | 2362.1 | ||
| CASSCF(19,14) | 3 × s, 2 × p, 1 × d | 2536.0 | |||
| RASSCF(19,19) | 1 × s, 1 × p, 1 × d | 1 × s | 1 × s, 1 × p, 1 × d | 2608.3 | |
| RASSCF(19,26) | 1 × s, 1 × p, 1 × d | 1 × s | 1 × s, 1 × p, 1 × d, 1 × f | 2733.6 | |
| RASCI(45,54) | 5 × s, 4 × p, 1 × d | 1 × s | 4 × s, 5 × p, 1 × d, 1 × f | 2704.6 | |
| Experimental | 3049.7 |
The RAS1, RAS2, and RAS3 columns indicate the number of atomic shells—separated by angular momentum—included in each subspace. Experimental HFCCs are reproduced from ref (75).
All RASCI calculations reported herein were performed using a RASSCF(19,26)-optimized orbital space for all coinage metals. Surprisingly, correlating most of the s and p manifold changes the initial RASSCF HFCCs very little; the largest variations are observed in calculations that correlate additional polarization shells. We observe improvements of 2–6% upon including one f shell in the orbital optimization step (cf. the RASCI results in Tables 2 and S2). Despite the significant challenges associated with these elements, Hyperion is still able to achieve relative errors of less than 15%.
Some notable differences between the coinage metals and the alkali are highlighted by the orbital decomposition analysis. Figure 1 shows orbital decomposition diagrams obtained from RASCI calculations on 133Cs and 197Au atoms; while both systems have a 6s1 ground configuration, their core configurations set them apart, which is reflected by the observed HFC. Compared to 133Cs, 197Au exhibits stronger coupling between d orbitals, as well as more significant contributions from the f polarization shell (however, the latter could be a consequence of including this shell in the orbital optimization step). Nevertheless, in both cases, the largest contributions to HFC are concentrated around orbitals 5s–7p, with additional non-negligible couplings involving the most diffuse s and p functions.
4.4. Groups VI-B (Cr) and VIII-B (Fe)
The hyperfine structure of transition metal (TM) atoms with partially filled d shells is by far the most challenging to model due to a number of competing factors. The orbital angular momentum couples to the nuclear spin through the PSO mechanism, and the resulting HFCC contribution is similar in magnitude to the spin-dependent FC+SD contribution (Figure S3). Both SOC and spin density must therefore be modeled accurately. On the one hand, the RASSI approach requires a sufficient number of spin-adapted states in order to represent the SOC states accurately, where the number of optimized roots corresponds to the lowest-energy Russell–Saunders (LS) terms (Table 3). Additional LS terms were included for 101Ru, 183W, and 189Os to obtain a converged ordering of SO energies at the minimal CASSCF-SO level.
Table 3. Number of Spin-Adapted Roots Optimized for Each Spin S and Corresponding LS Terms.
| atom | number of roots | LS terms |
|---|---|---|
| 53Cr | 1 (S = 3), 6 (S = 2) | 7S, 5D, 5S |
| 95 Mo | 1 (S = 3), 6 (S = 2) | 7S, 5D, 5S |
| 183W | 1 (S = 3), 5 (S = 2), 3 (S = 1) | 7S, 5D, 3P |
| 57Fe | 12 (S = 2) | 5D, 5F |
| 101Ru | 7 (S = 2), 7 (S = 1) | 5F, 3F |
| 189Os | 12 (S = 2), 10 (S = 1) | 5D, 5F, 3P, 3F |
On the other hand, the electronic states of TM atoms
exhibit significant
mixing between ndN(n + 1)s2 and ndN+1(n + 1)s1 configurations;
these have competing influences on the form of the valence s orbital,
as optimized singly occupied s functions are usually more radially
expanded than doubly occupied s functions.76 The 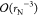 dependence of the HFC operator amplifies
such differences, leading to computed HFCCs that are very sensitive
to variations in the CI expansion. This proves particularly problematic
when CASSCF/RASSCF orbitals are averaged over states dominated by
different configurations, such as the low-lying 5D (3d64s2) and 5F (3d74s1) terms of 57Fe.82,83 Note that CASSCF(8,6)-SO
HFCCs for 101Ru are in much better agreement with experiment
than 57Fe HFCCs (Figure 3); this is because the 101Ru 5F term is sufficiently energetically separated and can be modeled
without requiring additional quintet roots in the spin-adapted CASSCF
step. Similar state-averaging effects are observed in the quintet
levels of 53Cr and 95Mo.
dependence of the HFC operator amplifies
such differences, leading to computed HFCCs that are very sensitive
to variations in the CI expansion. This proves particularly problematic
when CASSCF/RASSCF orbitals are averaged over states dominated by
different configurations, such as the low-lying 5D (3d64s2) and 5F (3d74s1) terms of 57Fe.82,83 Note that CASSCF(8,6)-SO
HFCCs for 101Ru are in much better agreement with experiment
than 57Fe HFCCs (Figure 3); this is because the 101Ru 5F term is sufficiently energetically separated and can be modeled
without requiring additional quintet roots in the spin-adapted CASSCF
step. Similar state-averaging effects are observed in the quintet
levels of 53Cr and 95Mo.
Figure 3.

Unsigned HFCCs computed by Hyperion for selected energy levels of Cr group and Fe group atoms. Experimental HFCCs are reproduced from ref (80) (53Cr), (81) (95Mo, 183W), (82) and (83) (57Fe), (84) (101Ru), and (85) (189Os).
Although relatively small in magnitude, the spin-dependent part of TM HFCCs depends on SP and higher-order correlation, similar to the spin-only HFCCs of alkali and coinage metals. Unlike the 2S1/2 systems, however, the expanded RAS2 subspace precludes large RAS optimizations. Therefore, calculations that correlate the entire core region were only feasible with RASCI for 3d TMs; however, this still gave very good results (Figure 3). RASCI-SO HFCCs computed for 57Fe are overall improved compared to CASSCF(8,6)-SO; meanwhile, RASCI-SO HFCCs for all 53Cr energy levels except 5S2 are slightly worse than those obtained from CASSCF-SO. We note that the RASCI-SO energies determined for 53Cr do not match the ordering observed experimentally and that the wrong ground state is predicted, indicating inaccuracies in the electronic wave function that are reflected in the calculated HFCCs. RASSCF-SO calculations correlating the valence shells were performed for all six atoms, yielding HFCCs that are overall less accurate compared to CASSCF-SO for the 3d and 5d TMs. The improvement with RASSCF observed for 95Mo and 101Ru can be justified by their well separated ground terms, which prevents state contamination during the RASSCF optimization.
Overall, it appears that minimal CASSCF-SO provides the most balanced model for the hyperfine structure of TMs, with errors around 25%. Approaches that include more correlation effects can theoretically improve the accuracy of the FC+SD term; in practice, however, the state-averaging formalism employed in CASSCF-SO, combined with the number of optimized spin-free states—which are likely characterized by different dynamical correlation effects—worsen the quality of the SO-coupled wave functions,86,87 which propagates to the computed HFCCs.
4.5. Lanthanides
The hyperfine structure of lanthanide (Ln) atoms is dominated by the PSO term, with previous work suggesting minor contributions from core polarization.88 As the 6s orbital and the 4f manifold are energetically well separated, CI effects between the two are negligible and the HFC response is expected to arise predominantly from the 4f shell. Hence, we compute the HFCCs of multiple levels in 4fN6s2 Ln atoms using a minimal CASSCF(N,7)-SO, with the number of optimized roots (Table 4) selected using a similar approach to section 4.4.
Table 4. CASSCF(N,7)-SO-Optimized LS Terms for Each Ln Atom.
| atom | LS terms |
|---|---|
| 141Pr | 4I |
| 143Nd | 5I |
| 147Pm | 6H |
| 147Sm | 7F, 5D |
| 151Eu | 8S |
| 159Tb | 6H, 4I |
| 161Dy | 5I |
| 165Ho | 4I |
| 167Er | 3H, 3F, 1G |
| 169Tm | 2F |
Predicted HFCCs (Table 5) are in remarkable agreement with experiment overall.89 The poorest agreement is observed for 151Eu and 165Ho; the former has a spin-only septet ground state, which only exhibits spin-dependent HFC (no PSO contribution), and hence requires a more sophisticated treatment of correlation. Inaccuracies in the latter could also be due to missing CI effects, as indicated in previous work,89 but increasing the active space with CASSCF or RASSCF methods had no appreciable impact on the calculated HFCCs here.
Table 5. Unsigned Isotropic HFCCs in MHz Determined for Ln Atomsa.
| atom | level | |AFC+SD| | |APSO| | |Atot| | |Aexpt| |
|---|---|---|---|---|---|
| 141Pr | 4I9/2 | 28.6 | 933.9 | 962.5 | 926.2 |
| 4I11/2 | 13.9 | 759.4 | 773.4 | 730.4 | |
| 4I13/2 | 0.4 | 654.8 | 655.1 | 613.2 | |
| 4I15/2 | 13.3 | 587.0 | 573.8 | 541.6 | |
| 143Nd | 5I4 | 6.2 | 207.5 | 201.3 | 195.7 |
| 5I5 | 3.8 | 163.1 | 159.3 | 153.7 | |
| 5I6 | 1.7 | 137.7 | 135.9 | 130.6 | |
| 5I7 | 0.3 | 121.8 | 122.1 | 117.6 | |
| 5I8 | 2.4 | 111.2 | 113.6 | 110.5 | |
| 147Pm | 6H7/2 | 42.0 | 472.3 | 430.4 | 447.1 |
| 147Sm | 7F1 | 34.8 | 70.4 | 35.6 | 33.5 |
| 7F2 | 28.6 | 70.4 | 41.8 | 41.2 | |
| 7F3 | 20.4 | 70.4 | 50.0 | 50.2 | |
| 7F4 | 11.2 | 70.4 | 59.2 | 59.7 | |
| 7F5 | 1.7 | 70.4 | 68.7 | 69.1 | |
| 7F6 | 7.7 | 70.4 | 78.1 | 78.4 | |
| 151Eu | 8S7/2 | 1.8 | 0.0 | 1.8 | 20.1 |
| 159Tb | 6H15/2 | 25.2 | 720.0 | 745.2 | 673.8 |
| 6H13/2 | 2.8 | 773.4 | 770.7 | 682.9 | |
| 6H11/2 | 34.0 | 856.8 | 822.9 | 729.0 | |
| 161Dy | 5I8 | 2.7 | 124.9 | 127.6 | 116.2 |
| 5I7 | 0.3 | 136.8 | 137.1 | 126.8 | |
| 5I6 | 1.9 | 154.6 | 152.7 | 139.6 | |
| 5I5 | 4.3 | 183.2 | 178.9 | 162.0 | |
| 5I4 | 6.9 | 233.2 | 226.2 | 205.2 | |
| 165Ho | 4I15/2 | 28.6 | 1253.7 | 1225.1 | 800.6 |
| 4I13/2 | 0.7 | 1398.4 | 1399.0 | 937.2 | |
| 4I11/2 | 29.8 | 1621.9 | 1651.7 | 1035.1 | |
| 4I9/2 | 61.5 | 1994.5 | 2056.0 | 1137.7 | |
| 167Er | 3H6 | 9.2 | 136.2 | 127.0 | 120.5 |
| 3F4 | 16.8 | 147.1 | 130.4 | 121.8 | |
| 3H5 | 4.7 | 158.0 | 162.7 | 159.5 | |
| 3H4 | 5.2 | 168.4 | 173.6 | 173.4 | |
| 3F3 | 4.6 | 149.8 | 145.2 | 143.5 | |
| 3F2 | 14.4 | 217.9 | 203.5 | 167.1 | |
| 169Tm | 2F7/2 | 48.4 | 433.0 | 384.7 | 374.1 |
| 2F5/2 | 115.8 | 577.4 | 693.2 | 704.6 |
Experimental HFCCs are reproduced from ref (89).
5. Conclusions
We have presented a new computational package, Hyperion, that enables the evaluation of relativistic picture-change-corrected magnetic resonance parameters from CASSCF-SO and RASSCF-SO wave functions, along with a new orbital decomposition method to assist in choosing appropriate active spaces for HFCC calculations. We used this code to study the hyperfine structure of alkali metal, transition metal, and lanthanide atoms in order to understand the range of applicability afforded by this approach. Our best predicted HFCCs are within 10% accuracy for alkali, 15% for coinage metals, and 20% for lanthanides (although the vast majority of Ln HFCCs deviate less than 10% from the experimental value). The hyperfine structure of group VI-B (Cr) and group VIII-B (Fe) transition metals proved to be the most challenging to model; however, despite the larger percentage errors, we obtain theoretical HFCCs that closely follow experimentally observed trends. Based on these results, we devised a number of guidelines for modeling HFC in systems exhibiting important correlation effects, strong SOC, or a combination of both. In future work, these guidelines will be refined by using Hyperion to study HFC in molecular systems.
Acknowledgments
The authors thank The University of Manchester Computational Shared Facility for computational resources, The Ministry of Defence (PhD studentship to L.B.), and The Royal Society (University Research Fellowship URF191320 to N.F.C.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jctc.2c00257.
Discussion of active space selection criteria using orbital decomposition analysis and additional Hyperion results for alkali and transition metal atoms (PDF)
The authors declare no competing financial interest.
Notes
The Hyperion code is available as free open-source software at https://gitlab.com/chilton-group/hyperion.
Supplementary Material
References
- Mar G. N. L.NMR of Paramagnetic Molecules; Academic Press: 1973; pp 85–126. [Google Scholar]
- Chipman D. M. The spin polarization model for hyperfine coupling constants. Theoretica Chimica Acta 1992, 82, 93–115. 10.1007/BF01113132. [DOI] [Google Scholar]
- Formanuik A.; Ariciu A.-M. M.; Ortu F.; Beekmeyer R.; Kerridge A.; Tuna F.; McInnes E. J. L.; Mills D. P. Actinide covalency measured by pulsed electron paramagnetic resonance spectroscopy. Nat. Chem. 2017, 9, 578–583. 10.1038/nchem.2692. [DOI] [PubMed] [Google Scholar]
- Chen J.; Hu C.; Stanton J. F.; Hill S.; Cheng H. P.; Zhang X. G. Decoherence in Molecular Electron Spin Qubits: Insights from Quantum Many-Body Simulations. J. Phys. Chem. Lett. 2020, 11, 2074–2078. 10.1021/acs.jpclett.0c00193. [DOI] [PubMed] [Google Scholar]
- Martin B.; Autschbach J. Temperature dependence of contact and dipolar NMR chemical shifts in paramagnetic molecules. J. Chem. Phys. 2015, 142, 054108. 10.1063/1.4906318. [DOI] [PubMed] [Google Scholar]
- Parker D.; Suturina E. A.; Kuprov I.; Chilton N. F. How the Ligand Field in Lanthanide Coordination Complexes Determines Magnetic Susceptibility Anisotropy, Paramagnetic NMR Shift, and Relaxation Behavior. Acc. Chem. Res. 2020, 53, 1520–1534. 10.1021/acs.accounts.0c00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennati M. EPR Interactions - Hyperfine Couplings. eMagRes. 2017, 6, 271–282. 10.1002/9780470034590.emrstm1503. [DOI] [Google Scholar]
- Hermosilla L.; Calle P.; García De La Vega J. M.; Sieiro C. Density functional theory predictions of isotropic hyperfine coupling constants. J. Chem. Phys. 2005, 109, 1114–1124. 10.1021/jp0466901. [DOI] [PubMed] [Google Scholar]
- Kossmann S.; Kirchner B.; Neese F. Performance of modern density functional theory for the prediction of hyperfine structure: Meta-GGA and double hybrid functionals. Mol. Phys. 2007, 105, 2049–2071. 10.1080/00268970701604655. [DOI] [Google Scholar]
- Engels B.Calculation of NMR and EPR Parameters: Theory and Applications; Wiley-VCH: 2004; Chapter 30, pp 483–492. [Google Scholar]
- Witwicki M.; Walencik P. K.; Jezierska J. How accurate is density functional theory in predicting spin density? An insight from the prediction of hyperfine coupling constants. J. Mol. Model. 2020, 26, 10. 10.1007/s00894-019-4268-0. [DOI] [PubMed] [Google Scholar]
- Engels B.; Peyerimhoff S. D.; Davidson E. R. Calculation of hyperfine coupling constants an ab initio MRD-CI study for nitrogen to analyse the effects of basis sets and CI parameters. Mol. Phys. 1987, 62, 109–127. 10.1080/00268978700102091. [DOI] [Google Scholar]
- Engels B.; Peyerimhoff S. D. The hyperfine coupling constants of the X3Σ– states of NH influence of polarization functions and configuration space on the description of spin polarization. Mol. Phys. 1989, 67, 583–600. 10.1080/00268978900101301. [DOI] [Google Scholar]
- Engels B. Estimation of the influence of the configurations neglected within truncated multi-reference CI wavefunctions on molecular properties. Chem. Phys. Lett. 1991, 179, 398–404. 10.1016/0009-2614(91)85173-T. [DOI] [Google Scholar]
- Feller D.; Davidson E. R. A multireference CI determination of the isotropic hyperfine constants for first row atoms B-F. J. Chem. Phys. 1988, 88, 7580–7587. 10.1063/1.454322. [DOI] [Google Scholar]
- Bauschlicher C. W.; Langhoff S. R.; Partridge H.; Chong D. P. Theoretical study of the nitrogen atom hyperfine coupling constant. J. Chem. Phys. 1988, 89, 2985–2992. 10.1063/1.455004. [DOI] [Google Scholar]
- Bauschlicher C. W. Theoretical study of the nitrogen-atom hyperfine coupling constant. II. J. Chem. Phys. 1990, 92, 518–521. 10.1063/1.458600. [DOI] [Google Scholar]
- Carmichael I. Ab initio calculation of the hyperfine coupling constants in B2. J. Chem. Phys. 1989, 91, 1072–1078. 10.1063/1.457179. [DOI] [Google Scholar]
- Munzarova M. L.; Kubacek P.; Kaupp M. Mechanisms of EPR hyperfine coupling in transition metal complexes. J. Am. Chem. Soc. 2000, 122, 11900–11913. 10.1021/ja002062v. [DOI] [Google Scholar]
- Kutzelnigg W. Origin and meaning of the Fermi contact interaction. Theoretica Chimica Acta 1988, 73, 173–200. 10.1007/BF00528203. [DOI] [Google Scholar]
- Arratia-Perez R.; Case D. A.. Relativistic effects on molecular hyperfine interactions: Application to XeF and CsO. J. Chem. Phys. 1983, 79.4939. 10.1063/1.445587 [DOI] [Google Scholar]
- Talukdar K.; Sasmal S.; Nayak M. K.; Vaval N.; Pal S. Correlation trends in the magnetic hyperfine structure of atoms: A relativistic coupled-cluster case study. Phys. Rev. A 2018, 98, 022507. 10.1103/PhysRevA.98.022507. [DOI] [Google Scholar]
- Quiney H. M.; Belanzoni P. Relativistic calculation of hyperfine and electron spin resonance parameters in diatomic molecules. Chem. Phys. Lett. 2002, 353, 253–258. 10.1016/S0009-2614(01)01483-X. [DOI] [Google Scholar]
- Gohr S.; Hrobárik P.; Repiský M.; Komorovský S.; Ruud K.; Kaupp M. Four-Component Relativistic Density Functional Theory Calculations of EPR g - And Hyperfine-Coupling Tensors Using Hybrid Functionals: Validation on Transition-Metal Complexes with Large Tensor Anisotropies and Higher-Order Spin-Orbit Effects. J. Phys. Chem. A 2015, 119, 12892–12905. 10.1021/acs.jpca.5b10996. [DOI] [PubMed] [Google Scholar]
- Barysz M.; Sadlej A. J. Expectation values of operators in approximate two-component relativistic theories. Theor. Chem. Acc. 1997, 97, 260–270. 10.1007/s002140050260. [DOI] [Google Scholar]
- Saue T. Relativistic Hamiltonians for chemistry: A primer. ChemPhysChem 2011, 12, 3077–3094. 10.1002/cphc.201100682. [DOI] [PubMed] [Google Scholar]
- Liu W.Handbook of Relativistic Quantum Chemistry; Springer Nature: 2016. [Google Scholar]
- Van Lenthe E.; Van Der Avoird A.; Wormer P. E. Density functional calculations of molecular hyperfine interactions in the zero order regular approximation for relativistic effects. J. Chem. Phys. 1998, 108, 4783–4796. 10.1063/1.475889. [DOI] [Google Scholar]
- Belanzoni P.; van Lenthe E.; Baerends E. J. An evaluation of the density functional approach in the zero order regular approximation for relativistic effects: Magnetic interactions in small metal compounds. J. Chem. Phys. 2001, 114, 4421–4433. 10.1063/1.1345509. [DOI] [Google Scholar]
- Aquino F.; Pritchard B.; Autschbach J. Scalar relativistic computations and localized orbital analyses of nuclear hyperfine coupling and paramagnetic NMR chemical shifts. J. Chem. Theory Comput. 2012, 8, 598–609. 10.1021/ct2008507. [DOI] [PubMed] [Google Scholar]
- Verma P.; Autschbach J. Relativistic density functional calculations of hyperfine coupling with variational versus perturbational treatment of spin-orbit coupling. J. Chem. Theory Comput. 2013, 9, 1932–1948. 10.1021/ct301114z. [DOI] [PubMed] [Google Scholar]
- Autschbach J. The accuracy of hyperfine integrals in relativistic NMR computations based on the zeroth-order regular approximation. Theor. Chem. Acc. 2004, 112, 52–57. 10.1007/s00214-003-0561-0. [DOI] [Google Scholar]
- Autschbach J. The role of the exchange-correlation response kernel and scaling corrections in relativistic density functional nuclear magnetic shielding calculations with the zeroth-order regular approximation. Mol. Phys. 2013, 111, 2544–2554. 10.1080/00268976.2013.796415. [DOI] [Google Scholar]
- Autschbach J.Relativistic Methods for Chemists; Springer: Dordrecht, The Netherlands, 2010; pp 521–598. [Google Scholar]
- Malkin I.; Malkina O. L.; Malkin V. G.; Kaupp M. Scalar relativistic calculations of hyperfine coupling tensors using the Douglas-Kroll-Hess method. Chem. Phys. Lett. 2004, 396, 268–276. 10.1016/j.cplett.2004.08.037. [DOI] [PubMed] [Google Scholar]
- Malkin E.; Malkin I.; Malkina O. L.; Malkin V. G.; Kaupp M. Scalar relativistic calculations of hyperfine coupling tensors using the Douglas-Kroll-Hess method with a finite-size nucleus model. Phys. Chem. Chem. Phys. 2006, 8, 4079–4085. 10.1039/B607044B. [DOI] [PubMed] [Google Scholar]
- Nguyen Lan T.; Kurashige Y.; Yanai T. Scalar Relativistic Calculations of Hyperfine Coupling Constants Using Ab Initio Density Matrix Renormalization Group Method in Combination with Third-Order Douglas-Kroll-Hess Transformation: Case Studies on 4d Transition Metals. J. Chem. Theory Comput. 2015, 11, 73–81. 10.1021/ct5007778. [DOI] [PubMed] [Google Scholar]
- Filatov M.; Cremer D. Relativistically corrected hyperfine structure constants calculated with the regular approximation applied to correlation corrected ab initio theory. J. Chem. Phys. 2004, 121, 5618–5622. 10.1063/1.1785772. [DOI] [PubMed] [Google Scholar]
- Autschbach J. Relativistic Effects on Electron-Nucleus Hyperfine Coupling Studied with an Exact 2-Component (X2C) Hamiltonian. J. Chem. Theory Comput. 2017, 13, 710–718. 10.1021/acs.jctc.6b01014. [DOI] [PubMed] [Google Scholar]
- Wodyński A.; Kaupp M. Density Functional Calculations of Electron Paramagnetic Resonance g - And Hyperfine-Coupling Tensors Using the Exact Two-Component (X2C) Transformation and Efficient Approximations to the Two-Electron Spin-Orbit Terms. J. Phys. Chem. A 2019, 123, 5660–5672. 10.1021/acs.jpca.9b03979. [DOI] [PubMed] [Google Scholar]
- Feng R.; Duignan T. J.; Autschbach J. Electron-Nucleus Hyperfine Coupling Calculated from Restricted Active Space Wavefunctions and an Exact Two-Component Hamiltonian. J. Chem. Theory Comput. 2021, 17, 255–268. 10.1021/acs.jctc.0c01005. [DOI] [PubMed] [Google Scholar]
- Filatov M.; Zou W.; Cremer D. Analytic calculation of isotropic hyperfine structure constants using the normalized elimination of the small component formalism. J. Phys. Chem. A 2012, 116, 3481–3486. 10.1021/jp301224u. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y.; Binning R. C.; Sando K. M. Dirac-Fock discrete-basis calculations on the beryllium atom. Chem. Phys. Lett. 1983, 101, 111–114. 10.1016/0009-2614(83)80314-5. [DOI] [Google Scholar]
- Stanton R. E.; Havriliak S. Kinetic balance: A partial solution to the problem of variational safety in Dirac calculations. J. Chem. Phys. 1984, 81, 1910. 10.1063/1.447865. [DOI] [Google Scholar]
- Dyall K. G.; Fægri K. Kinetic balance and variational bounds failure in the solution of the Dirac equation in a finite Gaussian basis set. Chem. Phys. Lett. 1990, 174, 25–32. 10.1016/0009-2614(90)85321-3. [DOI] [Google Scholar]
- Sun Q.; Liu W.; Kutzelnigg W. Comparison of restricted, unrestricted, inverse, and dual kinetic balances for four-component relativistic calculations. Theor. Chem. Acc. 2011, 129, 423–436. 10.1007/s00214-010-0876-6. [DOI] [Google Scholar]
- Dyall K. G. An exact separation of the spin-free and spin-dependent terms of the Dirac-Coulomb-Breit Hamiltonian. J. Chem. Phys. 1994, 100, 2118. 10.1063/1.466508. [DOI] [Google Scholar]
- Dyall K. G.Introduction to relativistic quantum chemistry; Oxford University Press: New York, 2007. [Google Scholar]
- Kutzelnigg W.; Liu W. Quasirelativistic theory equivalent to fully relativistic theory. J. Chem. Phys. 2005, 123, 241102. 10.1063/1.2137315. [DOI] [PubMed] [Google Scholar]
- Kutzelnigg W.; Liu W. Quasirelativistic theory I. Theory in terms of a quasi-relativistic operator. Mol. Phys. 2006, 104, 2225–2240. 10.1080/00268970600662481. [DOI] [Google Scholar]
- Liu W.; Kutzelnigg W. Quasirelativistic theory. II. Theory at matrix level. J. Chem. Phys. 2007, 126, 114107. 10.1063/1.2710258. [DOI] [PubMed] [Google Scholar]
- Liu W.; Peng D. Exact two-component Hamiltonians revisited. J. Chem. Phys. 2009, 131, 031104. 10.1063/1.3159445. [DOI] [PubMed] [Google Scholar]
- Neese F. Quantum chemistry and EPR parameters. eMagRes. 2017, 6, 1–22. 10.1002/9780470034590.emrstm1505. [DOI] [Google Scholar]
- Arbuznikov A. V.; Vaara J.; Kaupp M. Relativistic spin-orbit effects on hyperfine coupling tensors by density-functional theory. J. Chem. Phys. 2004, 120, 2127–2139. 10.1063/1.1636720. [DOI] [PubMed] [Google Scholar]
- Hennum A. C.; Klopper W.; Helgaker T. Direct perturbation theory of magnetic properties and relativistic corrections for the point nuclear and Gaussian nuclear models. J. Chem. Phys. 2001, 115, 7356–7363. 10.1063/1.1405009. [DOI] [Google Scholar]
- Malmqvist P. Å.; Roos B. O.; Schimmelpfennig B. The restricted active space (RAS) state interaction approach with spin-orbit coupling. Chem. Phys. Lett. 2002, 357, 230–240. 10.1016/S0009-2614(02)00498-0. [DOI] [Google Scholar]
- Chibotaru L. F.Advances in Chemical Physics; John Wiley & Sons, Ltd: 2013; pp 397–519. [Google Scholar]
- Bolvin H. An alternative approach to the g-matrix: Theory and applications. ChemPhysChem 2006, 7, 1575–1589. 10.1002/cphc.200600051. [DOI] [PubMed] [Google Scholar]
- Boguslawski K.; Tecmer P.; Legeza Ö.; Reiher M. Entanglement Measures for Single- and Multireference Correlation. J. Phys. Chem. Lett. 2012, 3, 3129–3135. 10.1021/jz301319v. [DOI] [PubMed] [Google Scholar]
- Aquilante F.; Ferré N.; Autschbach J.; Conti I.; Baiardi A.; Vico L. D.; Battaglia S.; Veniamin A.; Ernst D.; Norell J.; Lindh R.; Delcey M.; Galván I. F.; Freitag L.; Garavelli M.; Gong X.; Knecht S.; Nenov A.; Lundberg M.; Schapiro I.; Odelius M.; Phung Q. M.; Ungur L.; Segatta F.; Olivucci M.; Seijo L.; Pedersen T. B.; Pedrazagonzález L.; Segarra-martí J.; Pierloot K.; Vacher M.; Reiher M.; Valentini A.; Veryazov V.; Conti I.; Galván I. F.; Freitag L. Modern quantum chemistry with [Open ] Molcas Modern quantum chemistry with [Open ] Molcas. J. Chem. Phys. 2020, 152, 214117. 10.1063/5.0004835. [DOI] [PubMed] [Google Scholar]
- Sun Q. Libcint: An efficient general integral library for Gaussian basis functions. J. Comput. Chem. 2015, 36, 1664–1671. 10.1002/jcc.23981. [DOI] [PubMed] [Google Scholar]
- Roos B. O.; Lindh R.; Malmqvist P. Å.; Veryazov V.; Widmark P. O. Main Group Atoms and Dimers Studied with a New Relativistic ANO Basis Set. J. Phys. Chem. A 2004, 108, 2851–2858. 10.1021/jp031064+. [DOI] [Google Scholar]
- Roos B. O.; Veryazov V.; Widmark P. O. Relativistic atomic natural orbital type basis sets for the alkaline and alkaline-earth atoms applied to the ground-state potentials for the corresponding dimers. Theor. Chem. Acc. 2004, 111, 345–351. 10.1007/s00214-003-0537-0. [DOI] [Google Scholar]
- Roos B. O.; Lindh R.; Malmqvist P. Å.; Veryazov V.; Widmark P. O. New relativistic ANO basis sets for actinide atoms. Chem. Phys. Lett. 2005, 409, 295–299. 10.1016/j.cplett.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Roos B. O.; Lindh R.; Malmqvist P. Å.; Veryazov V.; Widmark P. O. New relativistic ANO basis sets for transition metal atoms. J. Phys. Chem. A 2005, 109, 6575–6579. 10.1021/jp0581126. [DOI] [PubMed] [Google Scholar]
- Roos B. O.; Lindh R.; Malmqvist P. Å.; Veryazov V.; Widmark P. O.; Borin A. C. New relativistic atomic natural orbital basis sets for lanthanide atoms with applications to the Ce diatom and LuF3. J. Phys. Chem. A 2008, 112, 11431–11435. 10.1021/jp803213j. [DOI] [PubMed] [Google Scholar]
- Sharkas K.; Pritchard B.; Autschbach J. Effects from spin-orbit coupling on electron-nucleus hyperfine coupling calculated at the restricted active space level for Kramers doublets. J. Chem. Theory Comput. 2015, 11, 538–549. 10.1021/ct500988h. [DOI] [PubMed] [Google Scholar]
- Roos B. O. The complete active space SCF method in a Fock-matrix-based super-CI formulation. Int. J. Quantum Chem. 1980, 18, 175–189. 10.1002/qua.560180822. [DOI] [Google Scholar]
- Malmqvist P. Å.; Rendell A.; Roos B. O. The restricted active space self-consistent-field method, implemented with a split graph unitary group approach. J. Phys. Chem. 1990, 94, 5477–5482. 10.1021/j100377a011. [DOI] [Google Scholar]
- Li Z.; Xiao Y.; Liu W. On the spin separation of algebraic two-component relativistic Hamiltonians. J. Chem. Phys. 2012, 137, 154114. 10.1063/1.4758987. [DOI] [PubMed] [Google Scholar]
- Heß B. A.; Marian C. M.; Wahlgren U.; Gropen O. A mean-field spin-orbit method applicable to correlated wavefunctions. Chem. Phys. Lett. 1996, 251, 365–371. 10.1016/0009-2614(96)00119-4. [DOI] [Google Scholar]
- Kollmar H.; Staemmler V. Violation of Hundas rule by spin polarization in molecules. Theoretica Chimica Acta 1978, 48, 223–239. 10.1007/BF00549021. [DOI] [Google Scholar]
- Giner E.; Tenti L.; Angeli C.; Ferré N. Computation of the Isotropic Hyperfine Coupling Constant: Efficiency and Insights from a New Approach Based on Wave Function Theory. J. Chem. Theory Comput. 2017, 13, 475–487. 10.1021/acs.jctc.6b00827. [DOI] [PubMed] [Google Scholar]
- Tterlikkis L.; Mahanti S.; Das T. Theoretical Analysis of the Hyperfine Structure of Alkali Atoms. Phys. Rev. 1968, 176, 10–19. 10.1103/PhysRev.176.10. [DOI] [Google Scholar]
- Lindgren I.; Rosén A.. Case Studies in Atomic Physics; Elsevier: 1975; pp 197–298. [Google Scholar]
- Helgaker T.; Taylor P. R.. Modern Electronic Structure Theory; World Scientific: 1995; pp 725–856. [Google Scholar]
- Kutzelnigg W. r12-Dependent terms in the wave function as closed sums of partial wave amplitudes for large l. Theoretica Chimica Acta 1985, 68, 445–469. 10.1007/BF00527669. [DOI] [Google Scholar]
- Li Manni G.; Smart S. D.; Alavi A. Combining the Complete Active Space Self-Consistent Field Method and the Full Configuration Interaction Quantum Monte Carlo within a Super-CI Framework, with Application to Challenging Metal-Porphyrins. J. Chem. Theory Comput. 2016, 12, 1245–1258. 10.1021/acs.jctc.5b01190. [DOI] [PubMed] [Google Scholar]
- Battaglia S.; Keller S.; Knecht S. Efficient Relativistic Density-Matrix Renormalization Group Implementation in a Matrix-Product Formulation. J. Chem. Theory Comput. 2018, 14, 2353–2369. 10.1021/acs.jctc.7b01065. [DOI] [PubMed] [Google Scholar]
- Jarosz A.; Stefańska D.; Elantkowska M.; Ruczkowski J.; Buczek A.; Furmann B.; Głowacki P.; Krzykowski A.; Pitkowski; Stachowska E.; Dembczyński J. High precision investigations of the hyperfine structure of metastable levels in a chromium atom. Journal of Physics B: Atomic, Molecular and Optical Physics 2007, 40, 2785–2797. 10.1088/0953-4075/40/13/019. [DOI] [Google Scholar]
- Büttgenbach S.Hyperfine Structure in 4d- and 5d-Shell Atoms; Springer-Verlag: Berlin, Heidelberg, 1982. [Google Scholar]
- Childs W. J.; Goodman L. S. Hyperfine Interactions and the Magnetic Fields Due to Core Polarization in Fe57. Phys. Rev. 1966, 148, 74–78. 10.1103/PhysRev.148.74. [DOI] [Google Scholar]
- Dembczyński J.; Ertmer W.; Johann U.; Stinner P. High precision measurements of the hyperfine structure of seven metastable atomic states of 57Fe by laser-Rf double-resonance. Zeitschrift für Physik A Atoms and Nuclei 1980, 294, 313–317. 10.1007/BF01434138. [DOI] [Google Scholar]
- Forest D. H.; Powis R. A.; Cochrane E. C.; Griffith J. A.; Tungate G. High resolution laser spectroscopy of naturally occurring ruthenium isotopes. Journal of Physics G: Nuclear and Particle Physics 2014, 41, 025106. 10.1088/0954-3899/41/2/025106. [DOI] [Google Scholar]
- Kröger S.; Başar G.; Baier A.; Guthöhrlein G. H. Hyperfine Structure and Isotope Shift of Osmium I. Phys. Scr. 2002, 65, 56–68. 10.1238/Physica.Regular.065a00056. [DOI] [Google Scholar]
- Ganyushin D.; Neese F. A fully variational spin-orbit coupled complete active space self-consistent field approach: Application to electron paramagnetic resonance g-tensors. J. Chem. Phys. 2013, 138, 104113. 10.1063/1.4793736. [DOI] [PubMed] [Google Scholar]
- Seed J. A.; Birnoschi L.; Lu E.; Tuna F.; Wooles A. J.; Chilton N. F.; Liddle S. T. Anomalous magnetism of uranium(IV)-oxo and -imido complexes reveals unusual doubly degenerate electronic ground states. Chem. 2021, 7, 1666–1680. 10.1016/j.chempr.2021.05.001. [DOI] [Google Scholar]
- Childs W. J.; Crowsswhite H.; Goodman L. S.; Pfeufer V. Hyperfine structure of 4fN6s2 configurations in 159Tb, 161,163Dy, and 169Tm. Journal of the Optical Society of America B 1984, 1, 22–29. 10.1364/JOSAB.1.000022. [DOI] [Google Scholar]
- Cheng K. T.; Childs W. J. Ab initio calculation of 4fN6s2 hyperfine structure in neutral rare-earth atoms. Phys. Rev. A 1985, 31, 2775–2784. 10.1103/PhysRevA.31.2775. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.