

Abstract
An unusual form of lung disease has begun to affect some children with systemic juvenile idiopathic arthritis (JIA), coincident with increasing utilization of interleukin‐1 (IL‐1) and IL‐6 antagonists. Many children with systemic JIA–associated lung disease (SJIA‐LD) have a history of clinical and laboratory features resembling drug reaction with eosinophilia and systemic symptoms (DRESS), a presentation now convincingly associated with HLA–DRB1*15. Treatment of DRESS typically requires drug discontinuation, a daunting prospect for clinicians and families who rely upon these agents. Here we review SJIA‐LD and its associated DRESS‐like phenotype. We suggest an alternative explanation, the cytokine plasticity hypothesis, proposing that IL‐1 and IL‐6 blockers modulate the milieu in which T cells develop, leading to a pathologic immune response triggered through exposure to common microbes, or to other exogenous or endogenous antigens, rather than to the drugs themselves. This hypothesis differs from DRESS in mechanism but also in clinical implications, predicting that control of pathogenic T cells could permit continued use of IL‐1 and IL‐6 antagonists in some individuals. The spectrum posed by these two hypotheses provides a conceptual framework that will guide investigation into the pathogenesis of SJIA‐LD and may open up new therapeutic avenues for patients with systemic JIA.
Introduction
The pediatric rheumatology community is worried. Some children with systemic juvenile idiopathic arthritis (JIA) are developing an unusual, sometimes life‐threatening, condition termed systemic JIA–associated lung disease (SJIA‐LD) (1, 2, 3, 4). The emergence of SJIA‐LD has coincided with increasing use of blockers of interleukin‐1 (IL‐1; anakinra, canakinumab, and rarely rilonacept) or IL‐6 (tocilizumab), and most children with SJIA‐LD have a history of exposure to one or more of these agents (2, 3). Recent work by Saper et al linked these reactions to the HLA class II allele DRB1*15, prompting the suggestion that SJIA‐LD arises through an IL‐1/IL‐6 blocker–induced drug‐induced hypersensitivity syndrome (DIHS) or even drug reaction with eosinophilia and systemic symptoms (DRESS) (5). These observations place clinicians, patients, and families in an extremely difficult conundrum. IL‐1 and IL‐6 antagonists have transformed systemic JIA from the most destructive arthritis of childhood into a disease with an excellent prognosis for most patients (6, 7, 8, 9, 10, 11). But if these biologics are potentially harmful, even life‐threatening, what are we to do?
Here we review the available data supporting the suggestion that allergic‐type responses and SJIA‐LD reflect hypersensitivity to IL‐1 and IL‐6 antagonists—a proposal we term the “DRESS hypothesis”—and present an alternative, the “cytokine plasticity hypothesis,” that differs in mechanism and in clinical implications. We present this alternative not because we believe that the DRESS hypothesis is necessarily incorrect but because articulating alternate explanations is sound scientific practice. Considering these hypotheses, potentially together with others, will shape the scientific agenda and thereby help us to understand the relationship between IL‐1 and IL‐6 blockade, DIHS‐type reactions, and SJIA‐LD, while guiding clinical decision‐making for patients with systemic JIA and its adult counterpart, adult‐onset Still's disease.
Systemic JIA–associated lung disease
Following isolated case reports of pulmonary involvement in systemic JIA in the pre‐biologic era (12, 13), Kimura and colleagues reported in 2013 a series of 25 patients with systemic JIA and lung disease, including pulmonary arterial hypertension, interstitial lung disease, and alveolar proteinosis. Mortality was high (68%). Most patients had been treated with IL‐1 and/or IL‐6 antagonists, raising the concern that these agents might have contributed to lung complications (1). In 2019, two groups reported an additional 70 patients with SJIA‐LD (2, 3). Where lung histology was available, in many cases it resembled pulmonary alveolar proteinosis (PAP), a condition in which alveolar macrophages fail to clear surfactant and other material from the airspace. Risk factors for SJIA‐LD included onset of systemic JIA before age 2 years, a history of macrophage activation syndrome (MAS), trisomy 21, and use of IL‐1 and/or IL‐6 blockers. Intriguingly, almost 40% of children had experienced anaphylactic reactions to tocilizumab, which is very unusual in other contexts. These studies noted increasing incidence of SJIA‐LD over the past two decades, roughly paralleling the increased use of IL‐1 and IL‐6 blockers (2, 3). In one center in the US, SJIA‐LD affected 5 of 74 new patients with systemic JIA (7%), though the European Pharmachild Registry of 306 systemic JIA patients treated with anakinra identified only one with SJIA‐LD (0.3%) (3, 14).
These reports raised several questions: Does exposure to IL‐1 or IL‐6 blockade cause SJIA‐LD? What underlies the apparent geographic variability in SJIA‐LD incidence? Might IL‐1 or IL‐6 blockade permit an as‐yet undefined pathogen to drive SJIA‐LD? Could there be confounding by indication—in other words, if IL‐1 and IL‐6 blockade are used preferentially in the patients who are also the most likely to develop SJIA‐LD, could the association be true but not causal? Could reduced reliance on corticosteroids, methotrexate, and other agents play a role? One of us entertained these possibilities two years ago (4). Do we have better insight now?
Importantly, one of the 18 patients (6%) with SJIA‐LD reported by Schulert and colleagues had not received IL‐1 or IL‐6 antagonists (3). Among the 61 patients reported by Saper and colleagues, 15 (25%) had no exposure before disease onset (2), a result echoed in a European cohort in which 7 of 26 subjects with SJIA‐LD (27%) had never received IL‐1 or IL‐6 blockade (ref. 15 and Bracaglia C: personal communication).
What do we know about the pathogenesis of SJIA‐LD? In all series, patients with a history of MAS are at greater risk for SJIA‐LD (1, 2, 3). Schulert and colleagues reported that patients with SJIA‐LD exhibit higher levels of serum IL‐18; increased levels of interferon‐γ (IFNγ), IL‐18, CXCL9, and CXCL10 in bronchoalveolar lavage fluid; and lung transcriptional profiles demonstrating up‐regulation of IFNγ and T cell activation networks, features that echo aspects of the pathogenesis of MAS (3). Using a mouse model of MAS induced by repeated administration of the Toll‐like receptor 9 agonist CpG, Schulert's group identified CD4+ T cell–predominant IFNγ‐dependent pulmonary inflammation, although not PAP (16). By contrast, T cell overexpression of T‐bet, the Th1 master transcription factor, impairs macrophage development and results in PAP‐like lung disease in mice (17). Clinical experience indicating that SJIA‐LD and MAS may respond favorably to blocking IFNγ with emapalumab supports the notion of a pathogenic role of Th1‐type inflammation in both conditions (18, 19).
Chen and colleagues recently reported the results of serum proteomic analysis of patients with SJIA‐LD and related conditions, identifying 20 proteins with elevated levels and 6 with decreased levels in SJIA‐LD independent of MAS (20). Unexpectedly, up‐regulated proteins included chemokines associated with Th2 responses: CCL11/eotaxin 1 and CCL17/thymus activation‐related chemokine, the latter sometimes elevated in DRESS (21, 22, 23, 24, 25). Also up‐regulated were the lung adhesion molecule intercellular adhesion molecule 5 (ICAM‐5) and the ICAM‐5–cleaving matrix metalloproteinase 7. ICAM‐5 was elevated in other forms of interstitial lung disease, but since it is not otherwise increased in systemic JIA the authors propose ICAM‐5 as a valuable biomarker of SJIA‐LD. Intriguingly, in some subjects the SJIA‐LD proteome profile occurred in the absence of high MAS activity, suggesting the possibility of an “independent origin” of MAS and SJIA‐LD, meaning that MAS and SJIA‐LD are driven by distinct pathogenic mechanisms, rather than a “sequential” model in which MAS directly predisposes to SJIA‐LD (20).
DRB1*15:XX and DIHS/DRESS‐like reactions
A recent report by Saper and colleagues sought to better understand adverse outcomes in children receiving IL‐1/IL‐6 antagonists by comparing 66 subjects with systemic JIA and apparent drug reactions to 65 “drug‐tolerant” systemic JIA controls (5). Features of DRESS among the first group included eosinophilia, elevation of hepatic transaminases, and non‐evanescent rash, including facial edema characteristic of DRESS but unusual in systemic JIA. Critically, DRB1*15 was vastly overrepresented among patients with systemic JIA and DIHS‐like reactions (75–93%) compared to drug‐tolerant patients, of whom 0% carried DRB1*15:01 and 18% carried any allele of DRB1*15. (DRB1*15:01 predominates in White subjects whereas 15:03 and 15:06 are more common in non‐White subjects [26]; we follow the convention of Saper and colleagues and use the term DRB1*15:XX to encompass all of these [5].) The fact that none of the 65 “drug‐tolerant” patients carried DRB1*15:01, an allele carried by 6–28% of individuals in the US (26), suggests that the large majority of systemic JIA patients with this risk allele react adversely to the introduction of drug. Among patients with DIHS‐like reactions, 82% with SJIA‐LD and 72% without SJIA‐LD expressed DRB1*15:XX; this allele is therefore a risk factor for DIHS‐like reactions, not for SJIA‐LD independent of such reactions. However, because SJIA‐LD occurred predominantly among patients with DIHS‐like reactions, these phenotypes are closely linked. MAS was also far more common in patients with DIHS‐like reactions (64% versus 3% in drug‐tolerant controls), prompting the authors to speculate that DRESS might directly provoke MAS, although potential mechanisms were not explored (5). Echoing these findings, in the recent study by Chen and colleagues, DRB1*15:XX was present in 87% of the subjects with SJIA‐LD in whom HLA typing was available, and all had prior exposure to IL‐1 or IL‐6 blockade (20).
To classify patients as having DRESS, Saper and colleagues (5) used the well‐accepted Registry of severe cutaneous adverse reactions (RegiSCAR) criteria incorporating 8 clinical features: fever, lymphadenopathy, eosinophilia, atypical lymphocytosis, rash, skin histology, organ involvement (including hepatic transaminase elevation), and persistence of symptoms for 15 days or more after drug withdrawal (27, 28). Since many of these features overlap with those of active systemic JIA, a typical systemic JIA patient could score in the range of “possible/probable” DRESS, potentially complicating the use of the RegiSCAR criteria in this context. The authors were careful to point out, however, that most patients met RegiSCAR criteria floridly and many exhibited facial edema and eosinophilia not typical of systemic JIA (29). Saper et al (5) observed that patients who discontinued biologics did better than those in whom treatment continued or was reintroduced, although many SJIA‐LD patients remain stable or even improve despite ongoing exposure to IL‐1/IL‐6 blockade (ref. 3 and personal experience of the authors), unusual for DRESS, which typically progresses until the offending agent is discontinued. How can we understand what is going on in these patients, using the critical insight provided by the newly‐recognized DRB1*15:XX association as the stepping‐off point?
HLA class II and CD4+ T cells in systemic JIA
DRB1*15:XX is part of the class II major histocompatibility complex (MHC) system. MHC II presents peptide antigens to CD4+ T cells, allowing them to recognize antigen, become activated, and undergo clonal expansion. As they do so, they differentiate from naïve (Th0) cells into more specialized cell subsets, including Th1 (making cytokines such as IFNγ), Th2 (making cytokines such as IL‐4), Th17 (making cytokines such as IL‐17), and Treg cells, depending on the milieu in which the T cell develops.
DRB1*15:XX is not the first MHC II allele linked to systemic JIA or its complications. In 2015, Ombrello and colleagues observed that DRB1*11 and its associated haplotype (DRB1*11; DQA1*05‐DQB1*03) predisposes to development of systemic JIA, drawing renewed attention to the potential involvement of CD4+ T cells in this disorder, despite its autoinflammatory‐like phenotype (30, 31). However, DRB1*11 was not associated with development of DIHS‐like responses, and its odds ratio for incident systemic JIA (~2.3) was much lower than that of DRB1*15:XX with apparent DIHS (~40.8) (5, 30). Thus, these two MHC II alleles play very different roles: DRB1*11 predisposes to systemic JIA but not otherwise to SJA‐LD, whereas DRB1*15:XX predisposes for DIHS‐type responses in patients who already have systemic JIA.
A key concept in T cell biology is phenotypic plasticity (for review, see refs. 32, 33, 34, 35, 36). CD4+ T cells exposed to cytokines and other cues can convert from Treg cells to Th17 cells and vice versa (37, 38, 39), from Th17 cells to Th1 cells (40, 41, 42), and from Th1 and Th17 cells to Th2 cells (43, 44). This propensity grounds the so‐called “biphasic hypothesis” of systemic JIA, which proposes that the IL‐1/IL‐6‐rich environment of early systemic JIA skews T cells away from beneficial Treg cells and toward pathogenic Th17 cells, thereby mediating chronic arthritis (31, 45). While the biphasic hypothesis remains unconfirmed, some supportive evidence has emerged. Omoyinmi and colleagues observed enrichment of circulating Th1 and Th17 cells in patients with systemic JIA (46). Henderson and colleagues found a Th17 gene expression signature among T cells in systemic JIA synovial fluid and blood, predominantly among unusual IL‐17–producing Treg cells in acute disease and among effector Th17 cells in chronic disease, although the latter expressed little IL‐17 protein (47). These findings plausibly reflect the known role of IL‐1 and IL‐6 in Th17 polarization and maintenance, since early IL‐1 blockade largely abrogated Treg cell IL‐17 expression (38, 47, 48, 49). In chronic systemic JIA, effector Th cells also up‐regulated TNF, IFNα, and IFNγ pathways, underscoring the complexity of the adaptive immune response (47). Not explored in that study was whether IL‐1 blockade led to an alternative cytokine polarization pattern, in either the Treg or Th cells, except that the Treg cells appeared not to express IFNγ protein. Analogously, Kessel and colleagues found IL‐17–expressing γδ T cells in systemic JIA, potentially reflecting cytokine‐driven lymphocyte polarization beyond conventional CD4+ T cells (50). To date, no study has provided comparable analysis of T cell cytokine/gene expression in systemic JIA with DIHS‐like features.
The DRESS hypothesis revisited
With this background, we can better consider how DRB1*15:01 might lead to DIHS‐like reactions to IL‐1 and IL‐6 blockers. Saper and colleagues propose that anakinra, canakinumab, and tocilizumab, or their excipients (the vehicle and its associated components) drive pathogenic T cell responses as antigens or by otherwise changing antigen presentation (5). Strengths of this hypothesis are the DRESS‐like phenotype of many drug‐exposed patients with systemic JIA; the improvement reported in some patients after drug discontinuation; the clinical deterioration observed in some patients who continue to receive the drug; and the extremely striking MHC II association, strongly implicating an antigen‐directed response.
The DRESS hypothesis also faces some conceptual hurdles. Anakinra (IL‐1 receptor antagonist [IL‐1Ra]) shares no antigen‐length peptides with canakinumab or tocilizumab. Indeed, aside from an N‐terminal methionine, anakinra is identical to endogenous IL‐1Ra and should therefore have limited antigenic potential. The only excipient shared by these agents is polysorbate‐80, an emulsifier that prevents protein aggregation. Polysorbate‐80 is found in most monoclonal antibodies and in many vaccines. Since polysorbate‐80 is a heterogeneous chemical product, it is conceivable that something might be different about the preparation employed in IL‐1/IL‐6 antagonists; however, its ubiquity renders this excipient a poor candidate trigger for DRESS.
Importantly, drugs can cause DRESS even if they are not themselves the antigen (51). First, they can bind covalently to endogenous proteins; these haptenated antigens are then recognized by T cells as foreign. Second, drugs can bind non‐covalently to the MHC and/or the T cell receptor (TCR) outside of the antigen binding pocket, leading to T cell activation in a manner reminiscent of bacterial superantigens, agents that can even display HLA class II allele specificity (52). Third, drugs can bind within or otherwise modify the MHC groove, altering peptide specificity such that certain self‐peptides now appear foreign. IL‐1 and IL‐6 blockers could each be capable of one of these interactions with DRB1*15:XX, despite their structural differences, or they could induce DIHS via a new type of interaction yet to be described. The possibility that any—actually, all—of these drugs interact with DRB1*15:XX to stimulate CD4+ T cells can (and should!) be tested directly in vitro, for example by using activation‐induced marker or similar sensitive assays to identify antigen‐specific CD4+ T cells in patients (53).
Anakinra, canakinumab, and tocilizumab are used in diseases other than systemic JIA, such as rheumatoid arthritis, polyarticular JIA, giant cell arteritis, and autoinflammatory diseases. In the Canakinumab Antiinflammatory Thrombosis Outcome Study (CANTOS) trial, almost 7,000 individuals with atherosclerotic cardiovascular disease and persistent C‐reactive protein elevation were treated with canakinumab for 48 months (54). Given a carrier frequency of DRB1*15:XX of 15–29% in the US (26), one might have predicted DIHS/DRESS to develop regularly in these patient groups—something that simply hasn't happened, apart from isolated case reports cited by Saper and colleagues (5). They did observe 4 patients with Kawasaki disease (KD) with suspected anakinra reactions (defined as an increase in eosinophil count of >50% compared to baseline), of whom 3 of 4 carried DRB1*15:XX, in contrast to 2 of 15 “drug‐tolerant” patients with KD (5). This small series is intriguing but might well reflect something shared in the hyperinflammatory milieu of systemic JIA and KD—that is, a “drug–disease” interaction. We present below one proposal for such an interaction.
An additional concern is that most HLA associations with DIHS/DRESS are with MHC class I alleles, although well‐described exceptions exist, including the association of DRB1*15:01 with Stevens‐Johnson syndrome/toxic epidermal necrolysis in Han Chinese subjects (55, 56). Why MHC I predominates over MHC II is not entirely clear but may have to do with another interesting observation: DRESS is often accompanied by reactivation of human herpesvirus 6 (HHV‐6) and other herpesviruses such as Epstein‐Barr virus (EBV). One theory is that the expanded MHC I–restricted CD8+ T cell clones that recognize reactivating herpesviruses coincidently cross‐recognize drug, inducing DRESS via molecular mimicry (57). Whether reactivation or de novo infection with HHV‐6, EBV, or other herpesviruses occurs in patients with systemic JIA and DIHS needs to be explored, particularly since CD8+ T cell responses could underlie the increase in IFNγ production and T cell activation profile observed in SJIA‐LD (3). It is worth noting that the DRESS hypothesis requires that 3 structurally distinct biologics each represent an outlier among known drug–MHC–DRESS associations, and then each (coincidentally) with the same MHC II allele.
A final issue, noted above, is that ~25% of patients with SJIA‐LD have never been exposed to IL‐1 or IL‐6 blockers, seemingly excluding any pathogenic role for biologic therapy in this subset of patients. It will be interesting to investigate whether eosinophilia or other DRESS‐like manifestations occur in patients with systemic JIA not exposed to biologics, and if so whether such patients express DRB1*15:XX. Such patients would not have been detected by the recent study by Saper et al (5) since its case–control design required all patients to be drug‐exposed.
The cytokine plasticity hypothesis
Boiled down, the key feature of the DRESS hypothesis is that IL‐1 and IL‐6 blockers drive DIHS via antigen presentation—either as the source of peptide or by altering the peptide–MHC II–TCR complex. We propose instead an alternative we term the cytokine plasticity hypothesis, that it is the biologic activity of IL‐1 and IL‐6 blockers that sets the stage for DIHS‐like reactions and even SJIA‐LD in predisposed patients.
The association of DRB1*15:XX implies a pathogenic role for CD4+ T cells. The antigens recognized by these T cells could originate from self‐proteins, commensal organisms, pathogens, or other exposures. For the sake of the cytokine plasticity hypothesis, the identity of the antigen is not crucial. Rather, as with the biphasic hypothesis, the key is the inherent susceptibility of CD4+ T cells to change phenotype in response to environmental cues.
Prior to treatment, patients with systemic JIA will have a diverse population of CD4+ T cells, potentially including expanded Th and Treg cell clones. Through genetic, environmental, age‐dependent, and stochastic factors, patients will differ from one another in the balance of Th effector profiles among these clones. Some clones exhibit Th17 features, as identified by Henderson and colleagues (47), whereas others exhibit Th1 or Th2 differentiation. The cytokine plasticity hypothesis proposes that biologics shift the balance of some of these T cells away from the IL‐1/IL‐6‐driven Th17 phenotype. In patients predisposed to develop SJIA‐LD, we speculate that the phenotypic switch is predominantly from Th17 to Th1, with associated production of IFNγ. Such a Th switch could be facilitated by other risk factors epidemiologically associated with SJIA‐LD, including (again speculatively) young age, high levels of circulating IL‐18 and other mediators (58, 59, 60), and trisomy 21. For example, trisomy 21 is associated with up‐regulation of IFNγ and its receptor (61), expansion of Th1 and Th1/Th17 T cells (62), and higher levels of other proinflammatory cytokines including TNF and IL‐6 (63). By contrast, for clones exhibiting a Th2 bias, blocking IL‐1 or IL‐6 might promote Th17‐to‐Th2 conversion leading to a DIHS‐type response. Under the right conditions, such clonal skew could happen even in the absence of exogenous manipulation, enabling the cytokine plasticity hypothesis to account for patients who develop SJIA‐LD without exposure to IL‐1 or IL‐6 antagonists.
How might DRB1*15:XX predispose to DIHS/DRESS‐like reactions and/or SJIA‐LD under the cytokine plasticity hypothesis? One pathway is suggested by the association of HLA–DRB1*15:01 with allergic bronchopulmonary aspergillosis (ABPA) (64, 65). Experimental data show that this risk allele skews T cells to differentiate toward Th2 rather than Th1 cells when encountering Aspergillus fumigatus, resulting in allergy‐like hypersensitivity instead of pathogen clearance (66). In patients with systemic JIA carrying DRB1*15:XX, IL‐1 or IL‐6 blockade might favor expansion of related Th2 clones in response to Aspergillus, leading to a DIHS‐type response. Importantly, this possibility generates testable predictions: these systemic JIA patients might have elevated Aspergillus‐specific IgE titers, CD4+ T cells activatable in vitro in response to Aspergillus‐derived peptides, or tetramer‐binding CD4+ T cells specific for Aspergillus‐derived peptides presented by DRB1*15:XX. Of course, the universe of allergens/antigens is vast. Cross‐reactivity to foreign antigens has been proposed to drive the risk of multiple sclerosis associated with DRB1*15:XX, implicating peptides from EBV and from the abundant gut gram negative anaerobe Akkermansia muciniphila, including some peptides that (as with ABPA) favor Th2‐predominant responses, although the mechanisms underlying this MHC class II‐influenced Th2 skewing remain unknown (66, 67). Endogenous antigens, potentially modulated with the inflammatory milieu of systemic JIA, might also be presented. The central point is that a wide range of DRB1*15:XX‐presented antigens are plausible drivers of SJIA‐LD under the cytokine plasticity hypothesis.
Like the DRESS hypothesis, the cytokine plasticity hypothesis confronts certain challenges. The clonal shifts proposed remain to be confirmed experimentally. Acute anaphylactic reactions to tocilizumab are difficult to explain under the cytokine plasticity hypothesis, although IgE‐mediated reactions are also uncharacteristic of DRESS (68). Why are patients with systemic JIA (and possibly KD) susceptible to this complication while those with other inflammatory diseases remain protected? How does the Th1 and/or Th2 shift cause alveolar macrophage dysfunction manifesting as PAP—also an unusual pulmonary manifestation of DRESS (69)? Most of these mechanistic unknowns are shared with the DRESS hypothesis.
The cytokine plasticity hypothesis also has strengths (Table 1). Most importantly, it explains how biologic‐naïve patients might develop the same phenotype as biologic‐exposed children. By invoking non‐drug antigens as the disease trigger, it explains how reactions to 3 structurally unrelated biologics could share the same HLA association, and potentially why the incidence of DRESS‐like reactions and of SJIA‐LD varies across regions of the world, with age, and with duration from first exposure to drug (7, 14, 70, 71). To the extent that corticosteroids and other nonbiologic immunomodulators keep pathogenic T cells in check, the cytokine plasticity hypothesis offers a plausible mechanism by which reduced reliance on these agents might contribute to the increasing incidence of SJIA‐LD. Finally, whereas DRESS progresses with continued drug exposure, the cytokine plasticity hypothesis implies that some patients could remain stable or even improve despite ongoing IL‐1/IL‐6 blockade if the pathogenic cytokine/cellular milieu is otherwise remediated—a difference of major clinical importance.
Table 1.
Comparison of two hypotheses to explain how IL‐1 or IL‐6 blocking agents might contribute to systemic JIA–associated DIHS and/or systemic JIA–associated lung disease*
| Explained by DRESS hypothesis | Explained by cytokine plasticity hypothesis | |
|---|---|---|
| Eosinophilia | Yes | Yes |
| Rash | Yes | Yes |
| HLA class II association | Usually class I | Yes |
| Drug exposure in many | Yes | Yes |
| No drug exposure in some | No | Yes |
| Structurally unrelated but biologically similar drugs | No | Yes |
| Low frequency of DIHS in other diseases treated with these drugs | No | Yes |
| Anakinra is a self protein | No | Yes |
| Many patients can continue drug | No | Yes |
| Possibility of foreign antigen | No | Yes |
| Pulmonary alveolar proteinosis | Unclear | Unclear |
IL‐1 = interleukin‐1; JIA = juvenile idiopathic arthritis; DIHS = drug‐induced hypersensitivity syndrome; DRESS = drug reaction with eosinophilia and systemic symptoms.
Two hypotheses and next steps
We present two hypotheses to explain the association of adverse reactions to IL‐1 or IL‐6 blockade with DRB1*15:XX (Figure 1). Other hypotheses could also be entertained. For example, the HLA association might reflect not DRB1*15 itself but instead another linked gene (C2, C4, HSP, and TNF are encoded nearby). SJIA‐LD might arise through an infection yet to be identified, potentially one to which systemic JIA patients with particular HLA alleles and receiving cytokine antagonists are especially susceptible. Even if not exhaustive of all possible explanations, the contrast between the DRESS hypothesis and the cytokine plasticity hypothesis provides a useful foundation to develop a scientific agenda for future research (Table 2).
Figure 1.
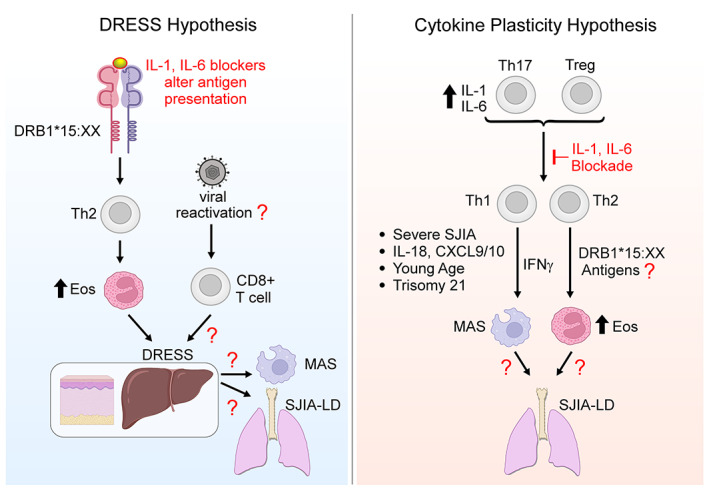
Two hypotheses regarding the relationship between systemic juvenile idiopathic arthritis (SJIA), major histocompatibility complex (MHC) class II, and adverse outcomes after interleukin‐1 (IL‐1) and IL‐6 blockade. In the drug reaction with eosinophilia and systemic symptoms (DRESS) hypothesis, IL‐1 and IL‐6 antagonists alter antigen presentation by MHC class II to CD4+ T cells, leading to Th2‐dominated DRESS. Many DRESS reactions involve herpesvirus reactivation and thus CD8+ T cell activation, factors yet to be studied in systemic JIA. DRESS may then predispose to macrophage activation syndrome (MAS) and/or systemic JIA–associated lung disease (SJIA‐LD) through pathways still to be defined. Under the cytokine plasticity hypothesis, elevated IL‐1 and IL‐6 levels in systemic JIA lead to Th17 skewing in CD4+ Th and Treg cells. Blocking IL‐1 or IL‐6 converts these cells to interferon‐γ (IFNγ)–producing Th1 cells and/or IL‐4–producing Th2 cells, in particular CD4+ T cells recognizing HLA–DRB1*15:XX–presented antigens (exogenous or endogenous). Some patients may undergo analogous transitions without exposure to therapeutics. The resulting clones lead to DRESS‐like reactions and/or SJIA‐LD through pathways still to be defined. Risk factors include severe systemic JIA; increased levels of IL‐18, CXCL9, and CXCL10; young age; and trisomy 21. The critical difference between these hypotheses is the contribution of the IL‐1 and IL‐6 blocking agents. Eos = eosinophils. Created in part using BioRender.
Table 2.
Key research foci and questions for systemic JIA investigators*
| Clinical research |
| Develop classification criteria for DIHS/DRESS‐like reactions in systemic JIA and for SJIA‐LD |
| Collect HLA class II typing on large, diverse, well‐characterized populations of systemic JIA patients |
| Translational research |
| Determine if HHV‐6 (or other viral reactivation) occurs in DIHS/DRESS‐like reactions in systemic JIA |
| Determine if DIHS/DRESS‐like reactions or SJIA‐LD are associated with prior Aspergillus or EBV exposure or the presence of Akkermansia muciniphila in gut microbiome |
| Basic research |
| Determine if IL‐1/IL‐6 blockers activate CD4 T cells in a DRB1*15:XX‐dependent manner |
| Characterize the Th cell gene/cytokine expression profile of “high‐risk” systemic JIA versus typical systemic JIA |
| Understand mechanisms driving pulmonary alveolar proteinosis in systemic JIA |
JIA = juvenile idiopathic arthritis; DIHS = drug‐induced hypersensitivity syndrome; DRESS = drug reaction with eosinophilia and systemic symptoms; SJIA‐LD = systemic JIA–associated lung disease; HHV‐6 = human herpesvirus 6; EBV = Epstein‐Barr virus; IL‐1 = interleukin‐1.
The main differences between these ideas are the roles of the IL‐1 and IL‐6 blocking drugs and the identities of the DRB1*15:XX‐presented antigens. Under the first hypothesis, the drugs cause disease via antigen presentation. Under the second, the drugs alter the cytokine milieu in which Th and other cells develop and differentiate. A key experimental question is whether patients with systemic JIA and DIHS/DRESS‐like syndromes and/or SJIA‐LD have T cells that react to these drugs, or not. If the second hypothesis is correct, defining the critical antigens could begin with investigation of DRB1*15‐presented pathogens, including Aspergillus, EBV, and Akkermansia muciniphila, and by testing whether onset of DIHS‐like responses and/or SJIA‐LD coincides with EBV seroconversion, as observed recently for multiple sclerosis (72).
Notably, the cytokine and chemokine milieu of systemic JIA, MAS, DIHS/DRESS‐like syndromes, and SJIA‐LD may not differentiate the two hypotheses. Under either, patients with DIHS‐like reactions will likely exhibit a Th2 profile, patients with MAS/SJIA‐LD will exhibit a Th1 profile, and some patients may have both. It will be essential to evaluate patients who have not been previously exposed to IL‐1 or IL‐6 blockers and yet go on to develop DIHS‐like reactions and/or SJIA‐LD, as well as to study patients with SJIA‐LD risk factors, such as young age, a history of MAS, and trisomy 21. Such patients might for example exhibit a Th1‐predominant profile even before development of SJIA‐LD, in contrast to the Th17 profile described in other patients with systemic JIA (46, 47). Importantly, basic investigations of the mechanisms of PAP in systemic JIA are needed, as neither hypothesis clearly delineates how this pathology arises, although the observation that PAP arises in mice whose T cells overexpress the Th1 transcription factor T‐bet represents an enticing clue (17).
Fundamentally, what the field needs most is detailed information about more patients. This will require coordinated efforts of clinicians and basic and translational investigators. Thankfully, the Childhood Arthritis and Rheumatology Research Alliance, the Paediatric Rheumatology International Trials Organisation, the Paediatric Rheumatology European Society, and other collaborative networks are well‐positioned to collect clinical data on a broad range of patients with systemic JIA. For investigative discovery, it will be informative to collect HLA typing on all subjects with systemic JIA. Organized efforts to harvest T cells from both DRB1*15:XX‐positive and ‐negative subjects will be critical to determine if subjects with DIHS‐like reactions have drug‐reactive T cells and to explore other candidate antigens. Analyzing the apparently rare patients with systemic JIA who express DRB1*15:XX but do not develop DIHS‐like reactions in response to IL‐1 or IL‐6 blockade could reveal protective factors, for instance more liberal use of corticosteroids and other immunomodulators. Basic investigations in DRB1*15:01 (DR2)–transgenic mice in conjunction with existing models of systemic JIA might also be informative, as it has been in ABPA (66).
While the field works to understand disease pathogenesis in greater depth, the immediate challenge is how to care for our patients. Should clinical HLA typing be performed on all patients with systemic JIA, and should detection of DRB1*15:XX be considered a contraindication to IL‐1 and IL‐6 blockade? Should patients who develop eosinophilia, transaminase elevation, or rash while receiving IL‐1 or IL‐6 blockade stop the drug immediately and thereafter avoid these biologics altogether? If so, what other therapies could be substituted? Avoiding the offending agents makes sense under the DRESS hypothesis, based on the observation from Saper and colleagues that the 26% of their patients with DRESS‐like reactions who stopped IL‐1 or IL‐6 blockade demonstrated resolution of eosinophilia, rash, and transaminase elevation, whereas some of those who continued treatment had worse outcomes, including death (5). The difficulty is that IL‐1 and IL‐6 blocking agents are exquisitely effective for systemic JIA, with few available alternatives. Many rheumatologists (and families) are therefore understandably hesitant to discontinue them, particularly in the sickest children—a key confounder in the comparison between patients who tolerated stopping biologics and patients who had to continue or even restart these agents. Saper and colleagues reported resolution of SJIA‐LD in 3 patients in whom IL‐1 or IL‐6 blockade was stopped within 6 weeks of detection of SJIA‐LD; they further suggested that longer duration of biologic exposure might increase the risk of SJIA‐LD, although this is again confounded by indication, as patients with more severe systemic JIA are more likely to be receiving biologics longer (73). In contrast, Schulert et al elected to maintain IL‐1 or IL‐6 blockade in all 18 patients in their SJIA‐LD series, with clinical stability in 14 patients and even improved lung disease in 3 (3). The cytokine plasticity hypothesis suggests that it is premature to assume that drug discontinuation is the only correct answer to DIHS‐like reactions and/or SJIA‐LD, although in some patients it may be useful to interrupt therapy.
The framework of the cytokine plasticity hypothesis provides a new way to approach clinical questions. Highly targeted biologics could be too specific for some patients with systemic JIA, shifting the cytokine milieu without providing sufficient global immune suppression. Perhaps patients at particular risk of DIHS‐like reactions and SJIA‐LD (those with DRB1*15:XX, young age, severe disease with MAS, or trisomy 21) are not good candidates for monotherapy with IL‐1 or IL‐6 antagonists. Instead, these patients might benefit from therapeutic strategies that combine these biologics with corticosteroids and/or agents that target T cells (e.g., methotrexate, mycophenolate mofetil, tacrolimus, abatacept) to address both innate and adaptive immune contributors. Where possible, we think it is reasonable to perform HLA typing on all patients with systemic JIA at disease onset and to monitor all patients with care when using IL‐1 or IL‐6 blockers since adverse reactions can occur even in patients lacking the risk alleles. However, we would not avoid starting IL‐1 and IL‐6 antagonists while awaiting these results and believe that IL‐1 or IL‐6 blockade should remain an important component of initial therapy for most patients with new‐onset systemic JIA, even if risk alleles are present.
How best to treat patients who do progress to SJIA‐LD remains an enormous and important challenge for the field. In addition to the T cell targeting agents mentioned above, clinicians are currently using JAK inhibitors, the anti‐IFNγ monoclonal antibody emapalumab, and other approaches. Prospective studies to evaluate these therapeutic approaches are necessary and should be supported by our field's collaborative research groups. Most importantly, patients and families need reassurance that their care team is fully informed about these emerging concerns, paying close attention to the known risk factors, and working alongside them to develop thoughtful, tailored treatment plans.
Conclusions
Pediatric rheumatology faces a conundrum posed by the twin complications of DIHS‐like reactions and SJIA‐LD. We present two reasonable explanations for these phenomena—the DRESS hypothesis and the cytokine plasticity hypothesis. Our hope is that consideration of these alternatives, potentially together with others, will stimulate targeted studies across clinical, translational, and basic domains. Our patients are counting on us.
AUTHOR CONTRIBUTIONS
Drs. Binstadt and Nigrovic drafted the article, revised it critically for important intellectual content, and approved the final version to be published.
Supporting information
Disclosures Form
ACKNOWLEDGMENTS
The authors thank Lauren Henderson, David Hoytema van Konijnenburg, Marc Jenkins, Ronald Laxer, Pui Lee, Shawn Mahmud, Elizabeth Mellins, Michael Ombrello, Elizabeth Phillips, Vivian Saper, and the reviewers for their insightful ideas and constructive comments.
Dr. Binstadt's work was supported by the National Heart, Lung, and Blood Institute, NIH (award R01‐HL‐121093), the National Institute of Allergy and Infectious Diseases, NIH (award R21‐AI‐149334), and an Innovative Research Award from the Rheumatology Research Foundation. Dr. Nigrovic's work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIH (awards 2R01‐AR‐065538, R01‐AR‐075906, R01‐AR‐073201, and 2P30‐AR‐070253), the Fundación Bechara, and the Arbuckle Family Fund for Arthritis Research.
Author disclosures are available at https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fart.42137&file=art42137‐sup‐0001‐Disclosureform.pdf.
Contributor Information
Bryce A. Binstadt, Email: binstadt@umn.edu.
Peter A. Nigrovic, Email: peter.nigrovic@childrens.harvard.edu.
REFERENCES
- 1. Kimura Y, Weiss JE, Haroldson KL, Lee T, Punaro M, Oliveira S, et al. Pulmonary hypertension and other potentially fatal pulmonary complications in systemic juvenile idiopathic arthritis. Arthritis Care Res (Hoboken) 2013;65:745–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Saper VE, Chen G, Deutsch GH, Guillerman RP, Birgmeier J, Jagadeesh K, et al. Emergent high fatality lung disease in systemic juvenile arthritis. Ann Rheum Dis 2019;78:1722–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schulert GS, Yasin S, Carey B, Chalk C, Do T, Schapiro AH, et al. Systemic juvenile idiopathic arthritis‐associated lung disease: characterization and risk factors. Arthritis Rheumatol 2019;71:1943–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nigrovic PA. Storm warning: lung disease in systemic juvenile idiopathic arthritis [editorial]. Arthritis Rheumatol 2019;71:1773–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saper VE, Ombrello MJ, Tremoulet AH, Montero‐Martin G, Prahalad S, Canna S, et al. Severe delayed hypersensitivity reactions to IL‐1 and IL‐6 inhibitors link to common HLA‐DRB1*15 alleles. Ann Rheum Dis 2022;81:406–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nigrovic PA, Mannion M, Prince FH, Zeft A, Rabinovich CE, van Rossum MA, et al. Anakinra as first‐line disease‐modifying therapy in systemic juvenile idiopathic arthritis: report of forty‐six patients from an international multicenter series. Arthritis Rheum 2011;63:545–55. [DOI] [PubMed] [Google Scholar]
- 7. Ter Haar NM, van Dijkhuizen EH, Swart JF, van Royen‐Kerkhof A, El Idrissi A, Leek AP, et al. Treatment to target using recombinant interleukin‐1 receptor antagonist as first‐line monotherapy in new‐onset systemic juvenile idiopathic arthritis: results from a five‐year follow‐up study. Arthritis Rheumatol 2019;71:1163–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Quartier P, Allantaz F, Cimaz R, Pillet P, Messiaen C, Bardin C, et al. A multicentre, randomised, double‐blind, placebo‐controlled trial with the interleukin‐1 receptor antagonist anakinra in patients with systemic‐onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis 2011;70:747–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Benedetti F, Brunner HI, Ruperto N, Kenwright A, Wright S, Calvo I, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med 2012;367:2385–95. [DOI] [PubMed] [Google Scholar]
- 10. Yokota S, Imagawa T, Mori M, Miyamae T, Aihara Y, Takei S, et al. Efficacy and safety of tocilizumab in patients with systemic‐onset juvenile idiopathic arthritis: a randomised, double‐blind, placebo‐controlled, withdrawal phase III trial. Lancet 2008;371:998–1006. [DOI] [PubMed] [Google Scholar]
- 11. Ruperto N, Brunner HI, Quartier P, Constantin T, Wulffraat N, Horneff G, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med 2012;367:2396–406. [DOI] [PubMed] [Google Scholar]
- 12. Padeh S, Laxer RM, Silver MM, Silverman ED. Primary pulmonary hypertension in a patient with systemic‐onset juvenile arthritis. Arthritis Rheum 1991;34:1575–9. [DOI] [PubMed] [Google Scholar]
- 13. Schultz R, Mattila J, Gappa M, Verronen P. Development of progressive pulmonary interstitial and intra‐alveolar cholesterol granulomas (PICG) associated with therapy‐resistant chronic systemic juvenile arthritis (CJA). Pediatr Pulmonol 2001;32:397–402. [DOI] [PubMed] [Google Scholar]
- 14. Giancane G, Papa R, Vastert S, Bagnasco F, Swart JF, Quartier P, et al. Anakinra in patients with systemic juvenile idiopathic arthritis: long‐term safety from the Pharmachild registry. J Rheumatol 2022;49:398–407. [DOI] [PubMed] [Google Scholar]
- 15. Bracaglia C, Minoia F, Kessel C, Vastert S, Pardeo M, Arduini A, et al. Systemic juvenile idiopathic arthritis associated lung disease in Europe [abstract]. Pediatr Rheumatol Online J 2021;19 Suppl:P53. [Google Scholar]
- 16. Gao DK, Salomonis N, Henderlight M, Woods C, Thakkar K, Grom AA, et al. IFN‐γ is essential for alveolar macrophage‐driven pulmonary inflammation in macrophage activation syndrome. JCI Insight 2021;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iriguchi S, Kikuchi N, Kaneko S, Noguchi E, Morishima Y, Matsuyama M, et al. T‐cell‐restricted T‐bet overexpression induces aberrant hematopoiesis of myeloid cells and impairs function of macrophages in the lung. Blood 2015;125:370–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Benedetti F, Brogan P, Bracaglia C, Pardeo M, Marucci G, Sacco E, et al. Emapalumab (anti‐interferon‐γ monoclonal antibody) in patients with macrophage activation syndrome (MAS) complicating systemic juvenile idiopathic arthritis (sJIA) [abstract]. Arthritis Rheumatol 2020;72 Suppl 4. URL: https://acrabstracts.org/abstract/emapalumab‐anti‐interferon‐gamma‐monoclonal‐antibody‐in‐patients‐with‐macrophage‐activation‐syndrome‐mas‐complicating‐systemic‐juvenile‐idiopathic‐arthritis‐sjia/. [Google Scholar]
- 19. Arduini A, Pardeo M, De Matteis A, Marucci G, Caiello I, Prencipe G, et al. Efficacy of emapalumab in chronic/relapsing macrophage activation syndrome in systemic juvenile idiopathic arthritis with lung and liver involvement [abstract]. Pediatr Rheumatol Online J 2021;19 Suppl:P146. [Google Scholar]
- 20. Chen G, Deutsch GH, Schulert G, Zheng H, Jang S, Trapnell B, et al. Identification of distinct inflammatory programs and biomarkers in systemic juvenile idiopathic arthritis and related lung disease by serum proteome analysis. Arthritis Rheumatol 2022;74:1273–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ogawa K, Morito H, Hasegawa A, Daikoku N, Miyagawa F, Okazaki A, et al. Identification of thymus and activation‐regulated chemokine (TARC/CCL17) as a potential marker for early indication of disease and prediction of disease activity in drug‐induced hypersensitivity syndrome (DIHS)/drug rash with eosinophilia and systemic symptoms (DRESS). J Dermatol Sci 2013;69:38–43. [DOI] [PubMed] [Google Scholar]
- 22. Ogawa K, Morito H, Hasegawa A, Miyagawa F, Kobayashi N, Watanabe H, et al. Elevated serum thymus and activation‐regulated chemokine (TARC/CCL17) relates to reactivation of human herpesvirus 6 in drug reaction with eosinophilia and systemic symptoms (DRESS)/drug‐induced hypersensitivity syndrome (DIHS). Br J Dermatol 2014;171:425–7. [DOI] [PubMed] [Google Scholar]
- 23. Komatsu‐Fujii T, Chinuki Y, Niihara H, Hayashida K, Ohta M, Okazaki R, et al. The thymus and activation‐regulated chemokine (TARC) level in serum at an early stage of a drug eruption is a prognostic biomarker of severity of systemic inflammation. Allergol Int 2018;67:90–95. [DOI] [PubMed] [Google Scholar]
- 24. Choudhary R, Vinay K, Srivastava N, Bishnoi A, Kamat D, Parsad D, et al. Clinical, biochemical, and serologic predictors of drug reaction with eosinophilia and systemic symptoms syndrome: a prospective case‐control study. J Am Acad Dermatol 2021;85:901–09. [DOI] [PubMed] [Google Scholar]
- 25. Wang F, He D, Tang X, Zhang X. Chemokine expression in diverse nonimmediate drug hypersensitivity reactions: focus on thymus activation‐regulated chemokine, cutaneous T‐cell‐attracting chemokine, and interleukin‐10. Ann Allergy Asthma Immunol 2014;113:204–8. [DOI] [PubMed] [Google Scholar]
- 26. Maiers M, Gragert L, Klitz W. High‐resolution HLA alleles and haplotypes in the United States population. Hum Immunol 2007;68:779–88. [DOI] [PubMed] [Google Scholar]
- 27. Kardaun SH, Sidoroff A, Valeyrie‐Allanore L, Halevy S, Davidovici BB, Mockenhaupt M, et al. Variability in the clinical pattern of cutaneous side‐effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol 2007;156:609–11. [DOI] [PubMed] [Google Scholar]
- 28. Kardaun SH, Sekula P, Valeyrie‐Allanore L, Liss Y, Chu CY, Creamer D, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Br J Dermatol 2013;169:1071–80. [DOI] [PubMed] [Google Scholar]
- 29. Musette P, Janela B. New insights into drug reaction with eosinophilia and systemic symptoms pathophysiology. Front Med (Lausanne) 2017;4:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ombrello MJ, Remmers EF, Tachmazidou I, Grom A, Foell D, Haas JP, et al. HLA‐DRB1*11 and variants of the MHC class II locus are strong risk factors for systemic juvenile idiopathic arthritis. Proc Natl Acad Sci U S A 2015;112:15970–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nigrovic PA. Autoinflammation and autoimmunity in systemic juvenile idiopathic arthritis. Proc Natl Acad Sci U S A 2015;112:15785–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stadhouders R, Lubberts E, Hendriks RW. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J Autoimmun 2018;87:1–15. [DOI] [PubMed] [Google Scholar]
- 33. Mazzoni A, Maggi L, Liotta F, Cosmi L, Annunziato F. Biological and clinical significance of T helper 17 cell plasticity. Immunology 2019;158:287–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bluestone JA, Mackay CR, O'Shea JJ, Stockinger B. The functional plasticity of T cell subsets. Nat Rev Immunol 2009;9:811–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. DuPage M, Bluestone JA. Harnessing the plasticity of CD4(+) T cells to treat immune‐mediated disease. Nat Rev Immunol 2016;16:149–63. [DOI] [PubMed] [Google Scholar]
- 36. Cerboni S, Gehrmann U, Preite S, Mitra S. Cytokine‐regulated Th17 plasticity in human health and diseases. Immunology 2021;163:3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Koenen HJ, Smeets RL, Vink PM, van Rijssen E, Boots AM, Joosten I. Human CD25highFoxp3pos regulatory T cells differentiate into IL‐17‐producing cells. Blood 2008;112:2340–52. [DOI] [PubMed] [Google Scholar]
- 38. Deknuydt F, Bioley G, Valmori D, Ayyoub M. IL‐1β and IL‐2 convert human Treg into T(H)17 cells. Clin Immunol 2009;131:298–307. [DOI] [PubMed] [Google Scholar]
- 39. Gagliani N, Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 2015;523:221–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, et al. Fate mapping of IL‐17‐producing T cells in inflammatory responses. Nat Immunol 2011;12:255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C, et al. Single‐cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell 2015;163:1400–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schnell A, Huang L, Singer M, Singaraju A, Barilla RM, Regan BM, et al. Stem‐like intestinal Th17 cells give rise to pathogenic effector T cells during autoimmunity. Cell 2021;184:6281–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moon HG, Tae YM, Kim YS, Jeon SG, Oh SY, Gho YS, et al. Conversion of Th17‐type into Th2‐type inflammation by acetyl salicylic acid via the adenosine and uric acid pathway in the lung. Allergy 2010;65:1093–103. [DOI] [PubMed] [Google Scholar]
- 44. Panzer M, Sitte S, Wirth S, Drexler I, Sparwasser T, Voehringer D. Rapid in vivo conversion of effector T cells into Th2 cells during helminth infection. J Immunol 2012;188:615–23. [DOI] [PubMed] [Google Scholar]
- 45. Nigrovic PA. Is there a window of opportunity for treatment of systemic juvenile idiopathic arthritis? [review]. Arthritis Rheumatol 2014;66:1405–13. [DOI] [PubMed] [Google Scholar]
- 46. Omoyinmi E, Hamaoui R, Pesenacker A, Nistala K, Moncrieffe H, Ursu S, et al. Th1 and Th17 cell subpopulations are enriched in the peripheral blood of patients with systemic juvenile idiopathic arthritis. Rheumatology (Oxford) 2012;51:1881–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Henderson LA, Hoyt KJ, Lee PY, Rao DA, Jonsson AH, Nguyen JP, et al. Th17 reprogramming of T cells in systemic juvenile idiopathic arthritis. JCI Insight 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL‐6 programs T(H)‐17 cell differentiation by promoting sequential engagement of the IL‐21 and IL‐23 pathways. Nat Immunol 2007;8:967–74. [DOI] [PubMed] [Google Scholar]
- 49. Harbour SN, DiToro DF, Witte SJ, Zindl CL, Gao M, Schoeb TR, et al. TH17 cells require ongoing classic IL‐6 receptor signaling to retain transcriptional and functional identity. Sci Immunol 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kessel C, Lippitz K, Weinhage T, Hinze C, Wittkowski H, Holzinger D, et al. Proinflammatory cytokine environments can drive interleukin‐17 overexpression by γ/δ T cells in systemic juvenile idiopathic arthritis. Arthritis Rheumatol 2017;69:1480–94. [DOI] [PubMed] [Google Scholar]
- 51. White KD, Chung WH, Hung SI, Mallal S, Phillips EJ. Evolving models of the immunopathogenesis of T cell‐mediated drug allergy: the role of host, pathogens, and drug response. J Allergy Clin Immunol 2015;136:219–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Scholl PR, Diez A, Karr R, Sekaly RP, Trowsdale J, Geha RS. Effect of isotypes and allelic polymorphism on the binding of staphylococcal exotoxins to MHC class II molecules. J Immunol 1990;144:226–30. [PubMed] [Google Scholar]
- 53. Reiss S, Baxter AE, Cirelli KM, Dan JM, Morou A, Daigneault A, et al. Comparative analysis of activation induced marker (AIM) assays for sensitive identification of antigen‐specific CD4 T cells. PLoS One 2017;12:e0186998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017;377:1119–31. [DOI] [PubMed] [Google Scholar]
- 55. Yang SC, Chen CB, Lin MY, Zhang ZY, Jia XY, Huang M, et al. Genetics of severe cutaneous adverse reactions. Front Med (Lausanne 2021;8:652091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang W, Hu FY, Wu XT, An DM, Yan B, Zhou D. Genetic predictors of Stevens‐Johnson syndrome and toxic epidermal necrolysis induced by aromatic antiepileptic drugs among the Chinese Han population. Epilepsy Behav 2014;37:16–9. [DOI] [PubMed] [Google Scholar]
- 57. Niu J, Jia Q, Ni Q, Yang Y, Chen G, Yang X, et al. Association of CD8(+) T lymphocyte repertoire spreading with the severity of DRESS syndrome. Sci Rep 2015;5:9913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schulert GS. The IL‐18/IFNγ axis in systemic JIA and MAS‐new answers, more questions. Rheumatology (Oxford) 2021;60:3045–47. [DOI] [PubMed] [Google Scholar]
- 59. Hinze T, Kessel C, Hinze CH, Seibert J, Gram H, Foell D. A dysregulated interleukin‐18‐interferon‐γ‐CXCL9 axis impacts treatment response to canakinumab in systemic juvenile idiopathic arthritis. Rheumatology (Oxford) 2021;60:5165–74. [DOI] [PubMed] [Google Scholar]
- 60. Bracaglia C, de Graaf K, Marafon DP, Guilhot F, Ferlin W, Prencipe G, et al. Elevated circulating levels of interferon‐γ and interferon‐γ‐induced chemokines characterise patients with macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Ann Rheum Dis 2017;76:166–72. [DOI] [PubMed] [Google Scholar]
- 61. Sullivan KD, Lewis HC, Hill AA, Pandey A, Jackson LP, Cabral JM, et al. Trisomy 21 consistently activates the interferon response. Elife 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Araya P, Waugh KA, Sullivan KD, Nunez NG, Roselli E, Smith KP, et al. Trisomy 21 dysregulates T cell lineages toward an autoimmunity‐prone state associated with interferon hyperactivity. Proc Natl Acad Sci U S A 2019;116:24231–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sullivan KD, Evans D, Pandey A, Hraha TH, Smith KP, Markham N, et al. Trisomy 21 causes changes in the circulating proteome indicative of chronic autoinflammation. Sci Rep 2017;7:14818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Patel G, Greenberger PA. Allergic bronchopulmonary aspergillosis. Allergy Asthma Proc 2019;40:421–24. [DOI] [PubMed] [Google Scholar]
- 65. Chauhan B, Santiago L, Hutcheson PS, Schwartz HJ, Spitznagel E, Castro M, et al. Evidence for the involvement of two different MHC class II regions in susceptibility or protection in allergic bronchopulmonary aspergillosis. J Allergy Clin Immunol 2000;106:723–9. [DOI] [PubMed] [Google Scholar]
- 66. Koehm S, Slavin RG, Hutcheson PS, Trejo T, David CS, Bellone CJ. HLA‐DRB1 alleles control allergic bronchopulmonary aspergillosis‐like pulmonary responses in humanized transgenic mice. J Allergy Clin Immunol 2007;120:570–7. [DOI] [PubMed] [Google Scholar]
- 67. Wang J, Jelcic I, Muhlenbruch L, Haunerdinger V, Toussaint NC, Zhao Y, et al. HLA‐DR15 molecules jointly shape an autoreactive T cell repertoire in multiple sclerosis. Cell 2020;183:1264–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Park EH, Lee EY, Shin K, Kim HA. Tocilizumab‐induced anaphylaxis in patients with adult‐onset Still's disease and systemic juvenile idiopathic arthritis: a case‐based review. Rheumatol Int 2020;40:791–98. [DOI] [PubMed] [Google Scholar]
- 69. Taweesedt PT, Nordstrom CW, Stoeckel J, Dumic I. Pulmonary manifestations of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome: a systematic review. Biomed Res Int 2019;2019:7863815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Brunner HI, Quartier P, Alexeeva E, Constantin T, Kone‐Paut I, Marzan K, et al. Efficacy and safety of canakinumab in patients with systemic juvenile idiopathic arthritis with and without fever at baseline: results from an open‐label, active‐treatment extension study. Arthritis Rheumatol 2020;72:2147–58. [DOI] [PubMed] [Google Scholar]
- 71. Pardeo M, Rossi MN, Marafon DP, Sacco E, Bracaglia C, Passarelli C, et al. Early treatment and IL1RN single‐nucleotide polymorphisms affect response to anakinra in systemic juvenile idiopathic arthritis. Arthritis Rheumatol 2021;73:1053–61. [DOI] [PubMed] [Google Scholar]
- 72. Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, et al. Longitudinal analysis reveals high prevalence of Epstein‐Barr virus associated with multiple sclerosis. Science 2022;375:296–301. [DOI] [PubMed] [Google Scholar]
- 73. Saper V, Prahalad S, Canna S, Abul‐Aziz R, Alvarez M, Bingham C, et al. Effect of drug withdrawl on interleukin‐1 or interleukin‐6 inhibitor associated diffuse lung disease [abstract]. Arthritis Rheumatol 2021;73 Suppl 10. URL: https://acrabstracts.org/abstract/effect‐of‐drug‐withdrawal‐on‐interleukin‐1‐or‐interleukin‐6‐inhibitor‐associated‐diffuse‐lung‐disease/. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosures Form