

Abstract
The pseudooligosaccharide acarbose is a potent inhibitor of amylases, glucosidases, and cyclodextrin glycosyltransferase and is clinically used for the treatment of so-called type II or insulin-independent diabetes. The compound consists of an unsaturated aminocyclitol, a deoxyhexose, and a maltose. The unsaturated aminocyclitol moiety (also called valienamine) is primarily responsible for the inhibition of glucosidases. Due to its structural similarity to maltotetraose, we have investigated whether acarbose is recognized as a substrate by the maltose/maltodextrin system of Escherichia coli. Acarbose at millimolar concentrations specifically affected the growth of E. coli K-12 on maltose as the sole source of carbon and energy. Uptake of radiolabeled maltose was competitively inhibited by acarbose, with a Ki of 1.1 μM. Maltose-grown cells transported radiolabeled acarbose, indicating that the compound is recognized as a substrate. Studying the interaction of acarbose with purified maltoporin in black lipid membranes revealed that the kinetics of acarbose binding to LamB is asymmetric. The on-rate of acarbose is approximately 30 times lower when the molecule enters the pore from the extracellular side than when it enters from the periplasmic side. Acarbose could not be utilized as a carbon source since the compound alone was not a substrate of amylomaltase (MalQ) and was only poorly attacked by maltodextrin glucosidase (MalZ).
The maltose regulon of Escherichia coli encompasses genes that are controlled by the positive regulator MalT and by cyclic AMP/CAP, the global regulator for carbon metabolism. Some of these genes are organized in clusters. The malA region at 76.5 min contains the malPQ operon encoding essential metabolic enzymes, maltodextrin phosphorylase and amylomaltase, respectively, and the divergently transcribed malT gene. Likewise, the malB region at 91.4 min contains the genes encoding the components of the transport system, organized in two divergently oriented operons: malEFG and malK lamB malM (22; for a recent review, see reference 6).
With the exception of malT, the expression of these genes is induced when the MalT protein resides in the active conformation that is acquired by the simultaneous binding of maltotriose and ATP. Then, the protein binds to specific sites upstream of the respective promoters (MalT boxes), but transcription is not initiated unless cyclic AMP/CAP also binds upstream of the MalT boxes. This results in repositioning of MalT binding, thereby inducing the bending of the DNA, which eventually turns on transcription (24).
The uptake of maltose and maltodextrins is accomplished by the combined action of five proteins: a specific channel protein in the outer membrane (maltoporin or LamB), a substrate-specific binding protein in the periplasm (MalE or maltose-binding protein), and a transport complex localized to the cytoplasmic membrane (MalFGK2) (reviewed in reference 6). The latter is a member of the superfamily of ATP-binding cassette transporter proteins (7, 15) and is composed of one copy each of the transmembrane proteins MalF and MalG and two copies of the ATP-hydrolyzing subunit MalK (9). While the crystal structures of maltoporin and MalE have been solved (25, 32), structural information on the membrane-bound complex is not yet available. However, crystals of the isolated MalK subunit from Salmonella typhimurium that diffract to a resolution of 3 Å were recently obtained (26).
Maltodextrins and maltose (≤10 μM) cross the outer membrane through maltoporin molecules which are organized as homotrimers. Each subunit contains a channel that is formed by an 18-stranded, antiparallel β-barrel. Within the channels, the substrates are in contact with a “greasy slide” of aromatic residues that provide a path for translocation (12).
In the periplasm, the ligands are readily complexed with maltose-binding protein that can exist in an open or a closed conformation, respectively. Binding of the substrate stabilizes the closed conformation (31). Only then can MalE productively interact with cytoplasmically exposed peptide loops of the membrane-integral components MalF and MalG. Through subsequent conformational changes of the latter, the presence of substrate is signaled, resulting in hydrolysis of ATP at the MalK subunits at the cytoplasmic side of the membrane. In turn, MalF and MalG are likely to be set into motion, and this leads to the substrate eventually being translocated across the membrane (10).
In the cytoplasm, maltose and maltodextrins are attacked by the products of three genes, malQ, encoding an amylomaltase, malP, encoding a maltodextrin phosphorylase, and malZ, encoding a maltodextrin glucosidase. MalQ is essential for growth on maltose, while MalP is required for the utilization of maltodextrins only (6).
Acarbose is a pseudooligosaccharide that is produced by strains of the genus Actinoplanes and is used to treat patients with diabetes. It is an effective inhibitor of α-amylases, glucosidases, and sucrases (36) and consists of an unsaturated aminocyclitol moiety (Fig. 1, ring A), a deoxyhexose (ring B) (together also called acarviosine), and a normal maltose (rings C and D). Prompted by its structural similarity to maltotetraose, we have studied the effects of acarbose on the metabolism of maltose and maltodextrins in whole cells of E. coli and on individual components of the maltose/maltodextrin system. Our results demonstrate that acarbose is efficiently transported but not metabolized by E. coli due to its poor performance as a substrate of maltodextrin-degrading enzymes. Also, the effects of acarbose on the channel properties of maltoporin were investigated in detail.
FIG. 1.

Structure of acarbose. The individual sugar residues are designated A to D (see the text for details).
MATERIALS AND METHODS
Chemicals.
Acarbose and [14C]acarbose (2.89 MBq/mg; purity, ≥96%) were generous gifts of Bayer AG (Wuppertal, Germany). Acarbose was of 98% purity and contained trace amounts of glucose (9 μg/g) and maltose (42 μg/g) but was devoid of maltotriose. Radiolabeled acarbose contained the 14C isotope in place of all carbon atoms in the acarviosine moiety (rings A and B in Fig. 1). [14C]maltose (13.3 GBq/mmol) was purchased from ICN (Eschwege, Germany).
Bacterial strains, media, and growth conditions.
The following E. coli strains were used in this study: K-12 (wild type, DSM 498), HS3018 [MC4100 malT(Con)-1 ΔmalE444] (13), CB39 (MC4100 malQ::Tn10) (11), and TK38 (MC4100 malZ Specr) (from W. Boos via M. Ehrmann). The cells were usually grown in minimal medium (M63) (18) supplemented with the indicated carbon sources (0.5%). Fresh media were routinely inoculated from a fully grown culture at a 1/100 dilution, and growth was monitored spectrophotometrically at 650 nm until the late exponential phase was reached.
Purification of LamB.
LamB of E. coli was purified essentially by a published procedure (2, 30).
Preparation of osmotic shock fluid.
Proteins were released from the periplasm of maltose-grown cells as described previously (19).
Transport assay.
The uptake of radiolabeled sugars was measured as described previously (27).
Binding assay.
Shock fluid from maltose-grown cells was concentrated by ultrafiltration, extensively dialyzed against 10 mM Tris-HCl (pH 7.2), and assayed for binding of [14C]maltose by an established procedure (23).
Enzyme assay.
Crude extracts were prepared from cells grown in rich medium supplemented with 0.2% maltose, as described previously (13). The activities of MalQ (amylomaltase) and MalZ (maltodextrin glucosidase) were assayed by monitoring the release of glucose by using the GOD-POD method (5).
Identification of sugars released by cell extracts.
Aliquots from the enzyme assays were applied to thin-layer chromatography plates and developed as above. Saccharide spots were visualized by dipping the plate into methanol containing 2% H2SO4, dyeing it, and charring it for 10 min at 180°C as described previously (13).
Experiments with black lipid membranes.
Black lipid bilayer membranes were formed from a 1% solution of diphytanoyl phosphatidylcholine (Avanti Polar Lipids, Alabaster, Ala.) in n-decane, as described previously (4). The instrumentation consisted of a Teflon chamber with two aqueous compartments connected by a small circular hole with a surface area of 0.3 mm2, across which the membranes were formed. The aqueous salt solutions (Merck, Darmstadt, Germany) were used unbuffered and had a pH around 6. The LamB protein was added from the concentrated stock solution to the aqueous phase bathing a membrane in the black state. The temperature was kept at 25°C throughout. The reconstitution of pores in the black lipid membrane was observed on a strip chart recorder. The membrane current was measured with a pair of Ag/AgCl electrodes with salt bridges switched in series with a voltage source and a current amplifier (Keithley 427). The feedback resistors of the current amplifier were between 0.01 and 10 GΩ. The amplified signal was monitored with a strip chart recorder to measure the absolute magnitude of the membrane current and to calculate the stability constant for carbohydrate binding (2).
The binding of acarbose and maltotetraose to LamB was measured in titration experiments similar to those described previously (3). In former experiments, the carbohydrate was added to the aqueous phase on both sides of the membrane. In the present study, we expanded the protocol in such a way that carbohydrate was also added to one side of the membrane only: either to the cis side (side ′; the side of the addition of LamB, carbohydrate concentration c′) or to the trans side (side "; the opposite side of the membrane, carbohydrate concentration c"). In the case where carbohydrate is added to both sides of the membrane (c′ = c" = c), the relative conductance inhibition is given by
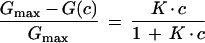 |
1 |
where Gmax is the conductance of a LamB-containing membrane prior to the addition of carbohydrate, and G(c) is the conductance in the presence of carbohydrate. This means that the titration curves can be analyzed by using Lineweaver-Burk plots, as shown in previous publications for carbohydrate-specific porins (2, 29).
The stability constant, K, for the carbohydrate binding to the channel is given by
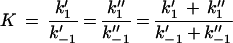 |
2 |
where k′1 and k"1 represent the on-rate constants for carbohydrate binding from the cis side and the trans side, respectively, to the binding site. The off-rate constants are given by k′−1 and k"−1. In the case of symmetric kinetics of carbohydrate binding to maltoporin (k1′ = k1" and k−1′ = k−1") equation 2 reduces to
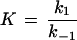 |
3 |
In contrast, when the carbohydrate is added to only one side of the membrane (c′ = c; c" = 0), the relative conductance inhibition is given by
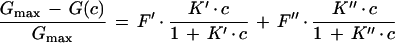 |
4 |
Here, attention must be paid to the possibly nonrandom orientation of channels when LamB is added only to the cis side of the membrane. F′ and F" are the percentages of LamB channels oriented in one or the other way in the lipid bilayer membrane (F′ + F" = 1; for random orientation, F′ = F" = 0.5). K′ and K" are then given by
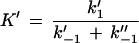 |
5 |
and
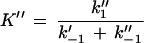 |
6 |
Note that K′ and K" cannot be compared to the stability constant K given in equation 2, since they do not represent the stability constants for carbohydrate binding to the channel. Rather, K′/K" reflects the ratio of the on-rates of the binding processes from the two different sides.
In the case of symmetric carbohydrate binding kinetics to maltoporin (k1′ = k1" and k−1′ = k−1") equation 4 reduces to
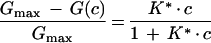 |
7 |
and K* is given by
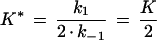 |
8 |
RESULTS
Acarbose specifically inhibits the growth of E. coli K-12 on maltose.
Cells of E. coli K-12 were grown in M63 minimal/maltose medium (0.5%) to the mid-exponential phase, and acarbose was added at 0.5, 1, and 2 mM, respectively. As shown in Fig. 2, growth with 0.5 mM acarbose ceased within 1 h whereas initial lysis of cells was observed at higher acarbose concentrations. In contrast, control cells with no addition of acarbose and cells grown in glucose in the presence of the same concentrations of acarbose displayed undisturbed growth. These results demonstrate that the inhibitory effect of acarbose is specifically imposed under conditions that induce the maltose system. Similar results were obtained when the experiments were repeated with an E. coli strain carrying a mutation (malF500) that renders the transport of maltose independent of the binding protein (reference 35 and data not shown).
FIG. 2.

Effect of acarbose on the growth of E. coli K-12 on maltose. Cells were grown at 37°C in minimal medium (M63) in the presence of maltose (0.5%) or glucose (0.5%). At the time indicated by the arrow, acarbose was added at increasing concentrations and growth was continued for 3 h. Maltose-grown cells: ○, no addition; ●, 0.5 mM acarbose; □, 1 mM acarbose; ■, 2 mM acarbose. Glucose-grown cells: ▵, no addition; ▴ 2 mM acarbose. OD650, optical density at 650 nm.
Acarbose is a competitive inhibitor of [14C]maltose uptake.
To identify the components of the maltose system that are affected by acarbose, we first studied the uptake of [14C]maltose in vivo. The initial rate of transport was examined in experiments where the substrate was varied in the presence of fixed concentrations of acarbose. The Lineweaver-Burk transformation from this analysis (Fig. 3) revealed a pattern of intersecting lines, indicating competitive inhibition by the compound. From these data, an inhibition constant (Ki) of 1.1 μM was calculated.
FIG. 3.

Inhibition of [14C]maltose uptake by acarbose. Cells were grown in M63-maltose medium to the late exponential phase, harvested, washed twice in M63 salts, and resuspended to an optical density at 650 nm of 7.8. Aliquots (10 μl) were diluted in M63 salts (1 ml), and the reaction was started by adding radiolabeled maltose. At 15-s intervals, aliquots (180 μl) were withdrawn, the cells were collected by rapid filtration through OE67 membrane filters (pore size, 0.45 μm; Schleicher & Schuell), washed once with ice-cold M63 salts, and counted. Shown is a Lineweaver-Burk plot of maltose affinity recorded in the presence of different acarbose concentrations (○, 0 μM; ●, 0.5 μM; □, 2 μM; ■, 10 μM). Initial rates of transport were calculated per 109 cells.
[14C]acarbose is a substrate of the maltose transport system.
Next, we examined the capability of maltose-grown cells to transport [14C]acarbose that has replaced all carbon atoms of the acarviosine moiety (Fig. 1, rings A and B) by the 14C isotope. The results are shown in Fig. 4. Acarbose was transported at a similar rate to radiolabeled maltose when supplied at the same specific radioactivity. Moreover, uptake of acarbose was abolished by the addition of 100 μM maltose, strongly indicating that the compound is taken up via the maltose transport system. This notion was further substantiated by the finding that cells precultured in the presence of glycerol failed to exhibit acarbose transport activity (not shown).
FIG. 4.

Uptake of [14C]acarbose. Cells were grown and prepared for transport assays as described in legend to Fig. 3, except that the final cell suspension was adjusted to an optical density at 650 nm of 7.5. Aliquots (10 μl) were diluted in 1 ml of M63 salts, and the reactions were initiated by the addition of radiolabeled acarbose (□, ■) or radiolabeled maltose (○, ●) (final concentrations, 5.7 μM; 22 kBq). The solid symbols represent uptake of the sugars in the presence of 0.1 mM maltose and 0.1 mM acarbose, respectively. The open symbols represent transport of maltose and acarbose, respectively, in the absence of competing (unlabeled) sugars.
Interaction of acarbose with MalE and maltoporin.
The above results clearly implied that the individual components of the maltose transport system must recognize acarbose as a substrate. For MalE, this was verified by demonstrating the inhibitory action of acarbose on the binding of radiolabeled maltose. By using an osmotic shock fluid that was prepared from cells grown in M63-maltose medium, inhibition of 5μM [14C]maltose-binding activity by acarbose was half-maximal at 17 μM acarbose (data not shown).
To gain more detailed insight into the mode of interaction of acarbose with a known maltodextrin-binding site, we have chosen maltoporin as a model for two reasons: (i) structural data on the architecture of the substrate binding sites are available, and (ii) interaction with sugar molecules can conveniently be studied by monitoring changes in the channel properties of the protein, embedded in planar lipid membranes. First, we carried out titration experiments by addition of acarbose to both sides of the membrane. The channels were blocked by acarbose in a dose-dependent manner, as shown in Fig. 5 for a similar experiment where acarbose was added to one side of the membrane (see below). The stability constants were evaluated by using equation 1 and are summarized in Table 1. The data reveal that the stability constants for acarbose lie in the same range as those obtained for the corresponding maltooligosaccharide, maltotetraose (2).
FIG. 5.

Titration of membrane conductance induced by maltoporin with acarbose. The membrane was formed from diphytanoyl phosphatidylcholine/n-decane. Acarbose was added to the trans side of the membrane at the concentrations shown at the top of the figure. The temperature was 25°C, and the applied voltage was 20 mV.
TABLE 1.
Kinetic constants of acarbose and maltotetraose binding to maltoporin
| Addition to: | Maltotetraose
|
Acarbose
|
|||||
|---|---|---|---|---|---|---|---|
| K (M−1)a | K* (M−1)c | K (M−1)a | F′ (%)d | K′ (M−1)d | F" (%)d | K" (M−1)d | |
| Both sides | 9,000b | 15,100 ± 4,800 | |||||
| cis side | 3,900 ± 440 | 55.2 ± 9.8 | 10,800 ± 2,200 | 44.8 ± 9.8 | 553 ± 245 | ||
| trans side | 4,500 ± 450 | 51.3 ± 8.4 | 12,000 ± 1,500 | 48.7 ± 8.4 | 315 ± 99 | ||
Stability constants correspond to equation 1.
Taken from reference 2.
K* corresponds to equation 8.
Calculated from equation 4. K′ and K" correspond to equations 5 and 6, respectively.
The binding kinetics of acarbose to maltoporin is asymmetric.
Recently, the three-dimensional structure of maltoporin was solved and the amino acid residues involved in the binding of carbohydrates were identified (12, 25). The data indicated that maltooligosaccharides are attached to the binding site in a fixed orientation: the nonreducing end is directed toward the periplasmic opening of the channel. Thus, to elucidate the kinetics of acarbose binding to maltoporin, we repeated the above experiments except that the compound was added to only one side of the membrane. From the result of such an experiment, it can be deduced whether the kinetics of acarbose binding to the periplasmic side of the channel differs from that of binding to the extracellular side (see equations 5 and 6). Figure 6 shows the relative conductance inhibition dependent on the acarbose concentration under these conditions. The data could not be fitted by using equation 7, assuming symmetric binding of acarbose (Fig. 6, line 1). Equation 4 led to a much better fit of the experimental data (line 2), strongly indicating that the binding kinetics of acarbose to the binding site is asymmetric. According to equation 4, we obtained two constants, K′ and K", for acarbose binding to maltoporin. One (K′) had a high value of about 11,000 M−1, whereas the other (K") was much lower (400 M−1). The definitions of K′ and K" (see equations 5 and 6 for details) mean that the ratio of the two reflects the ratios of the two on-rates of the binding process. Thus, the on-rates of acarbose binding to the binding site inside LamB differ by about a factor of 30. In contrast, the experimental data obtained with maltotetraose could be fitted with equation 7, which is consistent with symmetric binding kinetics to the protein. Furthermore, the calculated constants K* (equation 8) did not differ between cis and trans experiments (Table 1).
FIG. 6.

The relative conductance inhibition dependent on the acarbose concentration at one side of the membrane (trans). The data were derived from the experiment in Fig. 5. Line 1 corresponds to the fit with equation 1, assuming symmetrical binding of acarbose to maltoporin. Line 2 is the fit with equation 4. It is composed of the sum of two independent binding processes reflecting the binding from the periplasmic side (line 3; F′ = 61.3%, K′ = 12,600 M−1) and from the extracellular side (line 4; F" = 38.7%, K" = 392 M−1). Fits were done by least-squares analysis.
We also investigated the influence of potential on acarbose binding. Acarbose is a secondary amine with a Ka of 5.1 (36). The acarbose stock solution we used had a pH around 6.7, meaning that the majority of the acarbose molecules were uncharged under our experimental conditions. The influence of the membrane potential on acarbose binding is shown in Fig. 7. Open LamB channels are voltage independent up to 100 mV, as shown previously (2). The decrease in current caused by acarbose-mediated channel blockage was only slightly dependent on the applied membrane potential. Only at very high acarbose concentrations was the current-voltage curve slightly asymmetric. The current through the channels was higher when the side of the membrane where acarbose was added was negative (Fig. 7). The maximum difference between the two polarities was observed at ±100 mV and at high acarbose concentrations. At an acarbose concentration of 9.9 mM, the current at +100 mV was about 20% smaller than at −100 mV. It is noteworthy that a small asymmetry was also observed at +20 mV compared to −20 mV. Here the difference was only 10% at 9.9 mM acarbose. From these results, we concluded that the low-affinity binding of acarbose was not caused by the membrane potential or by charged acarbose molecules.
FIG. 7.

Current-voltage curves of a membrane containing 890 LamB channels. The different curves were measured at acarbose concentrations ranging from 0 to 9.9 mM. The voltage is given relative to the cis side of the membrane, the side to which LamB and acarbose were added. The membrane was formed from diphytanoyl phosphatidylcholine/n-decane. The temperature was 25°C.
Finally, it should also be noted that these experiments revealed a more or less random orientation of reconstituted channels in artificial membranes, indicating that acarbose molecules could enter them from either the periplasmic or extracellular side.
Acarbose is not used as a carbon source but acts as a weak inducer of the maltose regulon.
The observed inhibitory effect of acarbose on the growth of cells on maltose (Fig. 2) already suggested that E. coli might be incapable of using acarbose as a carbon source. Consistent with this notion was the failure to grow cells on M63 medium supplemented with 1% acarbose. Under these conditions, acarbose would also have to act as an inducer of the maltose regulon. To test for a potential inducing activity, we measured the transport rates of cells that were grown in minimal/glycerol medium supplemented with 0.5% acarbose. In contrast to control cells that were precultured in the presence of 0.5% maltose, no uptake of radiolabeled maltose or of acarbose could be observed (not shown).
However, immunoblot analysis of total protein of cells that were grown in the presence of acarbose (1%) revealed elevated levels of maltose-binding protein (∼30% relative to those in a maltose-induced culture). By analyzing cultures that were grown in the presence of 1 μM maltose, we excluded the possibility that trace amounts of maltose contaminating the acarbose preparation (see Materials and Methods) could raise MalE above the basal level, thereby accounting for the observed effect (results not shown).
Acarbose alone is not attacked by amylomaltase but serves as a weak substrate of maltodextrin glucosidase.
The above results clearly demonstrated that acarbose is recognized as a substrate of the maltose transport system, thereby providing at least in part an explanation for its inhibitory action on cells growing with maltose as sole source of energy and carbon. To elucidate the fate of acarbose in the cytoplasm, we measured the release of glucose from acarbose in a crude cell extract of strain HS3018, expressing the mal genes constitutively (13). As shown in Fig. 8A (lane 2), the amount of glucose liberated from 10 mM acarbose was <0.05 μmol/mg of protein, while in the presence of maltotetraose, 1.5 μmol/mg was released (lane 1). Moreover, when the degradation of maltotetraose (10 mM) was analyzed in the presence of 1 mM acarbose, only a slight reduction in the glucose-liberating activity was observed (lane 3). These results indicate that acarbose is neither a good substrate nor a strong inhibitor of the maltodextrin-metabolizing enzymes amylomaltase (MalQ) and maltodextrin glucosidase (MalZ). MalQ liberates glucose from the reducing end of maltotriose and longer maltodextrins and transfers the remaining maltosyl or dextrinyl moiety onto the nonreducing end of glucose, maltose, or longer molecules (20). MalZ removes glucose from the reducing end of maltodextrin chains but cannot cleave maltose (34). Thus, when the experiment is performed in the presence of excess maltose, the activity of MalQ can be more specifically analyzed. Again, acarbose had only a small inhibitory effect on the release of glucose from a mixture of maltose (10 mM) and maltotetraose (1 mM) (Fig. 8A, compare lanes 4 and 5). However, when the formation of glucose was measured with mixtures of maltose and acarbose, substantial activity was observed only in the presence of excess maltose (compare lanes 6 and 7), indicating that MalQ can use acarbose as a glucosyl or maltosyl donor but not as an acceptor (the residual activity in lane 7 is due to MalZ action [Fig. 8B, lane 7]). To confirm these results, the same set of experiments was performed with extracts from strains that lack either MalQ or MalZ activity due to mutations in the encoding genes. As shown in Fig. 8B and C, the results confirmed the above conclusions. In extracts of strain CB39, expressing only the malZ gene, acarbose could serve as a weak substrate (about 30% compared to maltotetraose [Fig. 8B, lanes 1 and 2]) and also inhibited the degradation of maltotetraose by about 40% (lane 3). However, the absolute activity measured with maltotetraose was only 27% compared to that of strain HS3018, which expresses both genes. This finding suggests that in the wild-type strain the glucose-liberating activity from maltotetraose was due mainly to MalQ action. This conclusion was supported by the data obtained with extracts from strain TK38 that carries an intact malQ gene but lacks malZ (Fig. 8C). The results from this analysis were also confirmed by visualizing the sugars present in each assay after separation on a thin-layer chromatography plate (data not shown). Together, these findings indicate that acarbose remains largely untouched by the maltose- and maltodextrin-degrading enzymes of E. coli and thus accumulates in the cytoplasm.
FIG. 8.

Glucose-releasing activities of amylomaltase (MalQ) and maltodextrin glucosidase (MalZ) in the presence of maltotetraose, acarbose, or maltose. Cell extracts (80 μl) of strains HS3018 (malQ+ malZ+) (A), CB39 (malQ malZ+) (B), and TK38 (malQ+ malZ) (C), were incubated with the indicated sugars for 30 min and assayed in duplicate for the release of glucose by the GOD-POD method (5). Values represent the mean of two independent experiments. Lanes: 1, maltotetraose (10 mM); 2, acarbose (10 mM); 3, maltotetraose (10 mM) and acarbose (1 mM); 4, maltose (10 mM) and maltotetraose (1 mM); 5, maltose (10 mM), and maltotetraose (1 mM), and acarbose (1 mM); 6, maltose (10 mM) and acarbose (1 mM); 7, acarbose (10 mM) and maltose (1 mM). Standard deviations are indicated by error bars.
DISCUSSION
We have shown that acarbose, a pseudooligosaccharide, is a substrate of the binding-protein-dependent transport system for maltose and maltodextrins of E. coli. This conclusion was drawn from several lines of experimental evidence: (i) acarbose specifically inhibits the growth of E. coli cells on maltose, (ii) acarbose is a competitive inhibitor of maltose uptake, (iii) [14C]acarbose is transported by E. coli cells, and, most notably, (iv) the uptake of acarbose is blocked by maltose. Since only the carbon atoms of the acarviosine moiety were replaced by the 14C isotope, either acarbose itself or a degradation product encompassing the A and B sugars is transported by the system. In fact, E. coli produces a periplasmic α-amylase, the product of the malS gene, that cleaves maltodextrins except maltose from the nonreducing end (28), which might be a candidate for extracellular breakdown of acarbose. However, up to 0.1 mM acarbose did not affect the activity of purified MalS (31a) (in Fig. 4, transport was assayed at 5.7 μM). Thus, it appears safe to conclude that acarbose itself is the transported sugar.
The above results imply that acarbose structurally mimics a natural substrate of the transporter, most probably maltotetraose, and thus should interact with the components involved in substrate recognition, such as maltoporin, maltose-binding protein, and MalF/MalG. Experimentally, this view was confirmed by demonstrating that acarbose (i) competes with maltose for the binding site on MalE, (ii) inhibits the growth of a binding-protein-independent mutant on maltose, and (iii) interferes with the channel properties of maltoporin. The last results led us to conclude that maltoporin functions as a channel for acarbose uptake in the periplasmic space. In comparison to maltotetraose, there is a difference in the interaction with the binding site inside the channel. Structurally, acarbose and maltotetraose differ at their nonreducing ends (Fig. 1). The three-dimensional structure of the substrate-loaded maltoporin shows that maltooligosaccharides are bound to the binding site in only one orientation (12): the nonreducing end is directed to the periplasmic space, which means that this part of the molecule enters the pore from the extracellular side. When maltotetraose was added to only one side of the black lipid membrane, the carbohydrate could enter the pore either from the periplasmic or extracellular side, assuming random orientation of pores. In this case, we could fit the titration data with a simple formula (equation 7), which means that the binding kinetics are the same from either side. In contrast, when acarbose was added to one side of the membrane only, a two-phase binding curve was observed, which could be fitted by equation 4. The K′/K" ratio reflects the on-rates of the binding processes from the different sides. Because of the structural comparison of acarbose and maltotetraose, we conclude that the higher on-rate was associated with the binding process from the periplasmic side of the channel and that the low on-rate belonged to the binding process of acarbose that entered the LamB channel from the extracellular side. The difference might be due to improper contact of the acarviosine moiety to amino acid residues within the sugar-binding site. The proportions of the two binding curves were almost 50%, indicating that the channels are probably randomly oriented under our experimental conditions.
A relatively low on-rate belonging to the binding process of acarbose entering the pore from the extracellular side seems to contradict the high rate of acarbose transport as shown in Fig. 4. The transport assays were performed with a concentration of acarbose (5.7 μM) that is below the half-saturation constants of acarbose (k1/2 = 1/K = 62 μM [this study]) and maltose (k1/2 = 1/K = 10 mM [2]). This means that the permeation of substrate through the pores is determined by the on-rate of the binding process (1a). However, kinetic studies of maltooligosaccharide binding to LamB of Salmonella typhimurium, which is very similar to LamB of E. coli, demonstrated that the on-rate of maltose is more than 1 order of magnitude lower than the on-rate of maltotetraose (16a). Thus, this finding might explain why the transport rate of acarbose is comparable to that of maltose in spite of a decreased on-rate constant for extracellular binding.
Since the conditions needed to grow crystals of both maltose-binding protein and maltoporin are well established, it should be feasible to elucidate the mode of interaction by which acarbose binds to these proteins. For MalE, such studies are in progress.
Structural information for several enzymes that are inhibited by acarbose suggests that various modes of interaction exist. When complexed with α-amylase from barley, acarbose was found to bind to the active site by the A, B, and C rings, while interaction with a starch granule-binding site located at the surface occurred via two sugar residues only (17). At the latter residues, binding involved stacking of acarbose rings on tryptophan residues, a typical feature of protein-carbohydrate interactions that is also found in the substrate-binding sites of MalE and maltoporin (25, 32). In glycogen phosphorylase, acarbose binds in an orientation such that the A ring makes no contact with the protein (14). In contrast, all four rings are hydrogen bonded in the active site of cyclodextrin glycosyltransferase (33) and pancreatic α-amylase (1), although the binding mode differs between the two enzymes.
Although acarbose is efficiently transported, E. coli is incapable of utilizing it as the sole source of carbon and energy. Moreover, growth on maltose was severely inhibited by acarbose. Our results (Fig. 8) strongly suggest that this is mainly because acarbose is only poorly attacked by the enzymes involved in maltose and maltodextrin degradation. Acarbose alone was not degraded by amylomaltase, but in the presence of excess maltose the enzyme could use acarbose to some extent as a glucosyl or maltosyl donor molecule. This indicates that the acarviosine moiety at the nonreducing end of the compound cannot serve as an acceptor for dextrinyl residues. Maltodextrin glucosidase, which, unlike other glucosidases, primarily removes glucose (and to some extent maltose) from the reducing end of a maltodextrin chain with a minimum length of maltotriose (34), could utilize acarbose as a weak substrate. Moreover, the compound inhibited enzyme action on maltotetraose, which is consistent with its sequence homology to cyclodextrinyl transferases (6, 33, 34).
Since no release of glucose was monitored with an extract of a malZ mutant, it is safe to assume that the compound cannot serve as a substrate of maltodextrin phosphorylase (MalP). MalP produces glucose-1-phosphate by sequential phosphorolysis from the nonreducing end of maltopentaose and larger maltodextrins (6). Maltotetraose and maltotriose are not attacked. Rather, as with glycogen phosphorylase (14), the compound might be a potent inhibitor of MalP.
From the above results, it can be concluded that the vast majority of acarbose molecules that enter the cell are not attacked by the metabolic enzymes involved in maltodextrin degradation and thus accumulate in the cytoplasm. Such a scenario is reminiscent of the phenotype of E. coli malQ mutants that cannot grow on maltose or maltotriose despite the presence of MalZ (6, 16). This finding was interpreted to mean that accumulation of maltose is toxic to the cell. In fact, when spread on indicator plates in the presence of maltose, malQ mutants display an unusual colony morphology and readily give rise to regularly shaped offspring (papillae) that are transport deficient and thus relieved of the toxic cause (16). Interestingly, irregularly shaped colonies were also found with cells of a malQ mutant that were plated on indicator plates supplemented with 1% acarbose (not shown). Thus, the observed interference of acarbose with the growth of E. coli on maltose might not only be caused by inhibition of maltose transport activity but also be due to a toxic effect of acarbose when accumulated in the cell. The observation that higher concentrations of acarbose caused cell lysis (Fig. 2) would be consistent with this view.
Induction of the maltose regulon requires maltotriose as an effector of the MalT protein. Thus, other carbon sources that have been demonstrated to induce the system, including free glucose and maltose, are thought to be converted into maltotriose by a hypothetical cytoplasmic enzyme (8, 11, 13; discussed in detail in reference 6). The finding that acarbose raised the level of maltose-binding protein is in line with the observation that small amounts of glucose were released by the action of maltodextrin glucosidase (Fig. 8B). Nevertheless, the level of induction proved to be insufficient to measure any transport activity.
In summary, we have shown that acarbose, a pseudooligosaccharide similar in structure to maltotetraose, is efficiently transported by the maltose-maltodextrin transport system of E. coli. Moreover, acarbose is not a substrate of amylomaltase and is only poorly attacked by maltodextrin glucosidase, thereby providing an explanation for the failure of the cells to utilize it as a source of carbon and energy. Thus, in contrast to the components of the transport system that are involved in substrate binding, the degrading enzymes exhibit an elaborate substrate specificity. In this respect, acarbose might be of potential use in further elucidating the molecular mechanism by which MalZ exerts its function.
ACKNOWLEDGMENTS
We thank A. Crueger (Bayer AG, Wuppertal, Germany) for generous gifts of acarbose and [14C]acarbose, W. Boos and M. Ehrmann (Konstanz, Germany) for providing strains, M. Ehrmann for analyzing the effect of acarbose on purified MalS, E. Bakker (Osnabrück, Germany) for his help in the initial phase of this study, and R. Benz (Würzburg, Germany) for general support and helpful discussions.
This work was supported by the Deutsche Forschungsgemeinschaft (Project B9 of the Sonderforschungsbereich 176; SCHN274/6-1/6-2), and by the Fonds der Chemischen Industrie.
REFERENCES
- 1.Al Kazaz M, Desseaux V, Marchis-Mouren G, Prodanov E, Santimone M. The mechanism of porcine pancreatic α-amylase. Inhibition of maltopentaose hydrolysis by acarbose, maltose and maltotriose. Eur J Biochem. 1998;252:100–107. doi: 10.1046/j.1432-1327.1998.2520100.x. [DOI] [PubMed] [Google Scholar]
- 1a.Andersen C, Jordy M, Benz R. Evaluation of the rate constant of sugar transport through maltoporin (LamB) of Escherichia coli from the sugar-induced current noise. J Gen Physiol. 1995;105:385–401. doi: 10.1085/jgp.105.3.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benz R, Schmid A, Vos-Scheperkeuter G H. Mechanism of sugar transport through the sugar-specific LamB channel of Escherichia coli outer membrane. J Membr Biol. 1987;100:12–29. doi: 10.1007/BF02209137. [DOI] [PubMed] [Google Scholar]
- 3.Benz R, Schmid A, Nakae T, Vos-Scheperkeuter G H. Pore formation by LamB of Escherichia coli in lipid bilayer membranes. J Bacteriol. 1986;165:978–986. doi: 10.1128/jb.165.3.978-986.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benz R, Janko K, Boos W, Läuger P. Formation of large, ion-permeable membrane channels by the matrix protein (porin) of Escherichia coli. Biochim Biophys Acta. 1978;511:305–319. doi: 10.1016/0005-2736(78)90269-9. [DOI] [PubMed] [Google Scholar]
- 5.Bergmeyer H U. Methods of enzymatic analysis. 3rd ed. Weinheim, Germany: Wiley-VCH; 1974. [Google Scholar]
- 6.Boos W, Shuman H. Maltose/maltodextrin system of Escherichia coli: transport, metabolism, and regulation. Microbiol Mol Biol Rev. 1998;62:204–229. doi: 10.1128/mmbr.62.1.204-229.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boos W, Lucht J M. Periplasmic binding protein-dependent ABC transporters. In: Neidhardt F C, Curtiss III R, Ingraham J L, Lin E C C, Low K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Escherichia coli and Salmonella: cellular and molecular biology. 2nd ed. Washington, D.C: American Society for Microbiology; 1996. pp. 1175–1209. [Google Scholar]
- 8.Bukau B, Ehrmann M, Boos W. Osmoregulation of the maltose regulon in Escherichia coli. J Bacteriol. 1986;166:884–891. doi: 10.1128/jb.166.3.884-891.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davidson A L, Nikaido H. Purification and characterization of the membrane-associated components of the maltose transport system from Escherichia coli. J Biol Chem. 1991;266:8946–8951. [PubMed] [Google Scholar]
- 10.Davidson A L, Shuman H A, Nikaido H. Mechanism of maltose transport in Escherichia coli: transmembrane signaling by periplasmic binding proteins. Proc Natl Acad Sci USA. 1992;89:2360–2364. doi: 10.1073/pnas.89.6.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Decker K, Peist R, Reidl J, Kossmann M, Brand B, Boos W. Maltose and maltotriose can be formed endogenously in Escherichia coli from glucose and glucose-1-phosphate independently of enzymes of the maltose system. J Bacteriol. 1993;175:5655–5665. doi: 10.1128/jb.175.17.5655-5665.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dutzler R, Wang Y-F, Rizkallah P J, Rosenbusch J P, Schirmer T. Crystal structure of various maltooligosaccharides bound to maltoporin reveal a specific sugar translocation pathway. Structure. 1996;4:127–134. doi: 10.1016/s0969-2126(96)00016-0. [DOI] [PubMed] [Google Scholar]
- 13.Ehrmann M, Boos W. Identification of endogenous inducers of the mal regulon of Escherichia coli. J Bacteriol. 1987;169:3539–3545. doi: 10.1128/jb.169.8.3539-3545.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldsmith E J, Fletterick R J, Withers S G. The three-dimensional structure of acarbose bound to glycogen phosphorylase. J Biol Chem. 1987;262:1449–1455. [PubMed] [Google Scholar]
- 15.Higgins C F. ABC transporter: from microorganisms to man. Annu Rev Cell Biol. 1992;8:67–113. doi: 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- 16.Hofnung M, Hatfield D, Schwartz M. malB region in Escherichia coli K-12: characterization of new mutations. J Bacteriol. 1974;117:40–47. doi: 10.1128/jb.117.1.40-47.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16a.Jordy M, Andersen C, Schülein K, Ferenci T, Benz R. Rate constants of sugar transport through two LamB mutants of Escherichia coli: comparison to wild-type maltoporin and to LamB of Salmonella typhimurium. J Mol Biol. 1996;259:666–678. doi: 10.1006/jmbi.1996.0348. [DOI] [PubMed] [Google Scholar]
- 17.Kadziola A, Søgaard M, Svensson B, Haser R. Molecular structure of a barley α-amylase-inhibitor complex: implications for starch binding and catalysis. J Mol Biol. 1998;278:205–217. doi: 10.1006/jmbi.1998.1683. [DOI] [PubMed] [Google Scholar]
- 18.Miller J H. Experiments in molecular genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1972. [Google Scholar]
- 19.Nossal N G, Heppel L A. The release of enzymes by osmotic shock from Escherichia coli in exponential phase. J Biol Chem. 1966;241:3055–3062. [PubMed] [Google Scholar]
- 20.Palmer N T, Ryman B E, Whelan W J. The action pattern of amylomaltase from Escherichia coli. Eur J Biochem. 1976;69:105–115. doi: 10.1111/j.1432-1033.1976.tb10863.x. [DOI] [PubMed] [Google Scholar]
- 21.Quian M, Haser R, Buisson G, Duée E, Payan F. The active center of a mammalian α-amylase with a carbohydrate inhibitor refined to 2.2-Å resolution. Biochemistry. 1994;33:6284–6294. doi: 10.1021/bi00186a031. [DOI] [PubMed] [Google Scholar]
- 22.Raibaud O, Roa M, Braun-Breton C, Schwartz M. Structure of the malB region in Escherichia coli K12. 1. Genetic map of the malK-lamB operon. Mol Gen Genet. 1979;174:241–248. doi: 10.1007/BF00267796. [DOI] [PubMed] [Google Scholar]
- 23.Richarme G, Kepes A. Study of binding protein-ligand interaction by ammonium sulfate-assisted adsorption on cellulose esters filters. Biochim Biophys Acta. 1983;742:16–24. doi: 10.1016/0167-4838(83)90353-9. [DOI] [PubMed] [Google Scholar]
- 24.Richet E, Vidal-Ingigliardi D, Raibaud O. A new mechanism for coactivation of transcription initiation: repositioning of an activator triggered by binding of a second activator. Cell. 1991;66:1185–1195. doi: 10.1016/0092-8674(91)90041-v. [DOI] [PubMed] [Google Scholar]
- 25.Schirmer T, Keller T A, Wang Y-F, Rosenbusch J P. Structural basis for sugar translocation through maltoporin channels at 3.1 Å resolution. Science. 1995;267:512–514. doi: 10.1126/science.7824948. [DOI] [PubMed] [Google Scholar]
- 26.Schmees G, Hoener z. Bentrup K, Schneider E, Vinzenz C, Ermler U. Crystallization and preliminary X-ray analysis of the bacterial ATP-binding-cassette (ABC)-protein MalK. Acta Crystallogr. 1999;D55:285–286. doi: 10.1107/S0907444998008518. [DOI] [PubMed] [Google Scholar]
- 27.Schneider E, Walter C. A chimeric nucleotide-binding protein, encoded by a hisP-malK hybrid gene, is functional in maltose transport in Salmonella typhimurium. Mol Microbiol. 1991;5:1375–1383. doi: 10.1111/j.1365-2958.1991.tb00784.x. [DOI] [PubMed] [Google Scholar]
- 28.Schneider E, Freundlieb S, Tapio S, Boos W. Molecular characterization of the MalT-dependent periplasmic α-amylase of Escherichia coli encoded by malS. J Biol Chem. 1992;267:5148–5154. [PubMed] [Google Scholar]
- 29.Schülein K, Benz R. LamB (maltoporin) of Salmonella typhimurium: isolation, purification and comparison of sugar binding with LamB of Escherichia coli. Mol Microbiol. 1990;4:625–632. doi: 10.1111/j.1365-2958.1990.tb00631.x. [DOI] [PubMed] [Google Scholar]
- 30.Schülein K, Andersen C, Benz R. The deletion of 70 amino acids near the N-terminal end of the sucrose-specific porin ScrY causes its functional similarity to LamB in vivo and in vitro. Mol Microbiol. 1995;17:757–767. doi: 10.1111/j.1365-2958.1995.mmi_17040757.x. [DOI] [PubMed] [Google Scholar]
- 31.Sharff A J, Rodseth L E, Spurlino J C, Quiocho F A. Crystallographic evidence of a large ligand-induced hinge-twist motion between the two domains of the maltodextrin binding protein involved in active transport and chemotaxis. Biochemistry. 1992;31:10657–10663. doi: 10.1021/bi00159a003. [DOI] [PubMed] [Google Scholar]
- 31a.C. Spiess and M. Ehrmann. Personal communication.
- 32.Spurlino J C, Lu G-Y, Quiocho F A. The 2.3-Å resolution structure of the maltose- or maltodextrin-binding protein, a primary receptor of bacterial active transport and chemotaxis. J Biol Chem. 1991;266:5202–5219. doi: 10.2210/pdb1mbp/pdb. [DOI] [PubMed] [Google Scholar]
- 33.Strokopytov B, Penninga D, Rozeboom H J, Kalk K H, Dijkhuizen L, Dijkstra B W. X-ray structure of cyclodextrin glycosyltransferase complexed with acarbose. Implications for the catalytic mechanism of glycosidases. Biochemistry. 1995;34:2234–2240. doi: 10.1021/bi00007a018. [DOI] [PubMed] [Google Scholar]
- 34.Tapio S, Yeh F, Shuman H A, Boos W. The malZ gene of Escherichia coli, a member of the maltose regulon, encodes a maltodextrin glucosidase. J Biol Chem. 1991;266:19450–19458. [PubMed] [Google Scholar]
- 35.Treptow N A, Shuman H A. Genetic evidence for substrate and periplasmic-binding-protein-recognition by the MalF and MalG proteins, cytoplasmic membrane components of the Escherichia coli maltose transport system. J Bacteriol. 1985;163:654–660. doi: 10.1128/jb.163.2.654-660.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Truscheit E, Frommer W, Junge B, Müller L, Schmidt D, Wingender W. Chemistry and biochemistry of α-glucosidase inhibitors. Angew Chem Int Ed. 1981;20:744–761. [Google Scholar]