

Abstract
The last several years have seen unprecedented advances in deciphering the genetic etiology of autism spectrum disorders (ASDs). Heritability studies have repeatedly affirmed a contribution of genetic factors to the overall disease risk. Technical breakthroughs have enabled the search for these genetic factors via genome-wide surveys of a spectrum of potential sequence variations, from common single-nucleotide polymorphisms to essentially private chromosomal abnormalities. Studies of copy-number variation have identified significant roles for both recurrent and nonrecurrent large dosage imbalances, although they have rarely revealed the individual genes responsible. More recently, discoveries of rare point mutations and characterization of balanced chromosomal abnormalities have pinpointed individual ASD genes of relatively strong effect, including both loci with strong a priori biological relevance and those that would have otherwise been unsuspected as high-priority biological targets. Evidence has also emerged for association with many common variants, each adding a small individual contribution to ASD risk. These findings collectively provide compelling empirical data that the genetic basis of ASD is highly heterogeneous, with hundreds of genes capable of conferring varying degrees of risk, depending on their nature and the predisposing genetic alteration. Moreover, many genes that have been implicated in ASD also appear to be risk factors for related neurodevelopmental disorders, as well as for a spectrum of psychiatric phenotypes. While some ASD genes have evident functional significance, like synaptic proteins such as the SHANKs, neuroligins, and neurexins, as well as fragile × mental retardation–associated proteins, ASD genes have also been discovered that do not present a clear mechanism of specific neurodevelopmental dysfunction, such as regulators of chromatin modification and global gene expression. In their sum, the progress from genetic studies to date has been remarkable and increasingly rapid, but the interactive impact of strong-effect genetic lesions coupled with weak effect common polymorphisms has not yet led to a unified understanding of ASD pathogenesis or explained its highly variable clinical expression. With an increasingly firm genetic foundation, the coming years will hopefully see equally rapid advances in elucidating the functional consequences of ASD genes and their interactions with environmental/experiential factors, supporting the development of rational interventions.
Pervasive developmental disorders are a cluster of common, complex conditions characterized by impairments in social interactions and communication, as well as repetitive or stereotypic behaviors. This cluster of neurodevelopmental abnormalities is more commonly referred to as autism spectrum disorders (ASDs), which by DSM-IV criteria include narrowly defined autism, social disintegration disorder, Asperger’s disorder, and pervasive developmental disorder not otherwise specified. Few empirical data suggest distinct etiologies among subgroups, however, and the ongoing revisions to the Diagnostic and Statistical Manual of Mental Disorders reduce these phenotypic subgroups to a single ASD diagnostic classification.
The prevalence of ASD has been steadily rising, accompanied by widespread speculation concerning the factors that may be responsible.1–6 In the 1970s and 1980s, prevalence was estimated at ~0.5 per 1000.7,8 The most recent population-based study from 2008 suggests at least a tenfold increase, with the prevalence of ASD among eight-year-old children across 11 sites now estimated at 11.3 per 1,000 (1 in 88) but varying widely among sites (range = 4.8–21.2 per 1000).9 A portion of the increase in prevalence over the past decades is likely attributable to changes in diagnostic classifications.6 Even among similar study designs and populations, however, a detectable and consistent rise in estimated prevalence is evident over a relatively short period of time, from 6.7 per 1000 based on 2000 data10 to 9.0 per 1000 based on 2006 data11 and 11.0 per 1000 based on 2008 data.9 This increase has been speculated to arise in part from increasing awareness of the disorder,9 and quantifying the changes in prevalence over time in the absence of an accepted, quantitative diagnostic biomarker is a significant challenge. Nevertheless, these data leave little doubt that ASD is a common, devastating developmental disorder with a severe public health impact both in the United States and globally.
Given the increasing prevalence, the relative importance of genetic versus environmental factors in the etiology of ASD is a matter of ongoing debate. It is likely that these factors act in interaction, given the overwhelming evidence indicating indicate a substantial genetic component. The high heritability of autism has been well established since the late 1980s. A meta-analysis of twin studies and case reports found an average monozygous twin concordance of 64% and an average dizygous twin concordance of 9%, compared to a population prevalence estimated to be 0.05% at the time of the study.12 A large family-based study estimated recurrence risk—the probability that any given child born after the first autistic child in a family will show signs of autism—at 8.6%8, although more recent studies have found higher concordance among dizygous twins.13,14 Heritability estimates for ASD have thus ranged from as high as 90% under a multifactorial threshold model15 to a recent study with a much lower estimate of ~37%, albeit with a wide confidence interval.14 A more detailed discussion of heritability estimates is outside the scope of this review. Despite the wide range in point estimates of heritability, it is clear that ASD has a large heritable component. It is also clear, however, that expressivity of the disorder is significantly influenced by nongenetic factors. Given that the human genome, unlike the environment that we experience, represents a defined, finite space in which to search for risk factors, there has been ample motivation for investigating the genetic underpinnings of ASD. This search has been facilitated by the dramatic increase in the power of modern genetic technologies, and in the following review, we offer one vantage on what has been a remarkable half-decade of progress in ASD genetics. We also temper this enthusiasm with the knowledge that the pathogenic mechanisms by which the contributing genes operate remain unknown in most individuals, and that novel, broadly applicable therapeutic interventions have not yet been developed based upon this progress.
We focus here on five seminal findings in the last several years in defining the genetic architecture of ASD. First, it has become clear that the genetic contribution to ASD comprises a diversity of mutational mechanisms, including rare single-nucleotide variants,16–23 copy-number variations (CNVs),24–31 chromosomal abnormalities,26,32 and common polymorphic variation (see Figure 1).33–36 Second, highly penetrant de novo variations that disrupt normal gene dosage can have a major impact on ASD risk.19,22,23,26–31,37 Third, regions of recurrent rearrangements occur in a measurable proportion of ASD subjects,38 and their phenotypic impact can, at times, be attributed to a single strong-effect locus within the region.26,39–41 Fourth, genes that contribute to ASD represent diverse biological pathways and include not only participants in synaptic function21,42 but also genes involved in global regulation of transcript expression and epigenetic modification.18,19,26,43 Finally, many genes associated with ASD confer shared risk to a wide range of neurodevelopmental abnormalities and psychopathology.26,44 Collectively, these data have provided an emerging portrait of a genetic architecture of ASD in which a highly heterogeneous collection of genes can contribute to a similar clinical presentation; at the same time, specific genetic lesions can be associated with widely variable clinical outcomes. In this review, we describe recent breakthroughs in the genetic etiology of ASD and their implications for our understanding of the models of ASD pathogenesis.
Figure 1. Genes and loci reported to significantly affect autism risk.
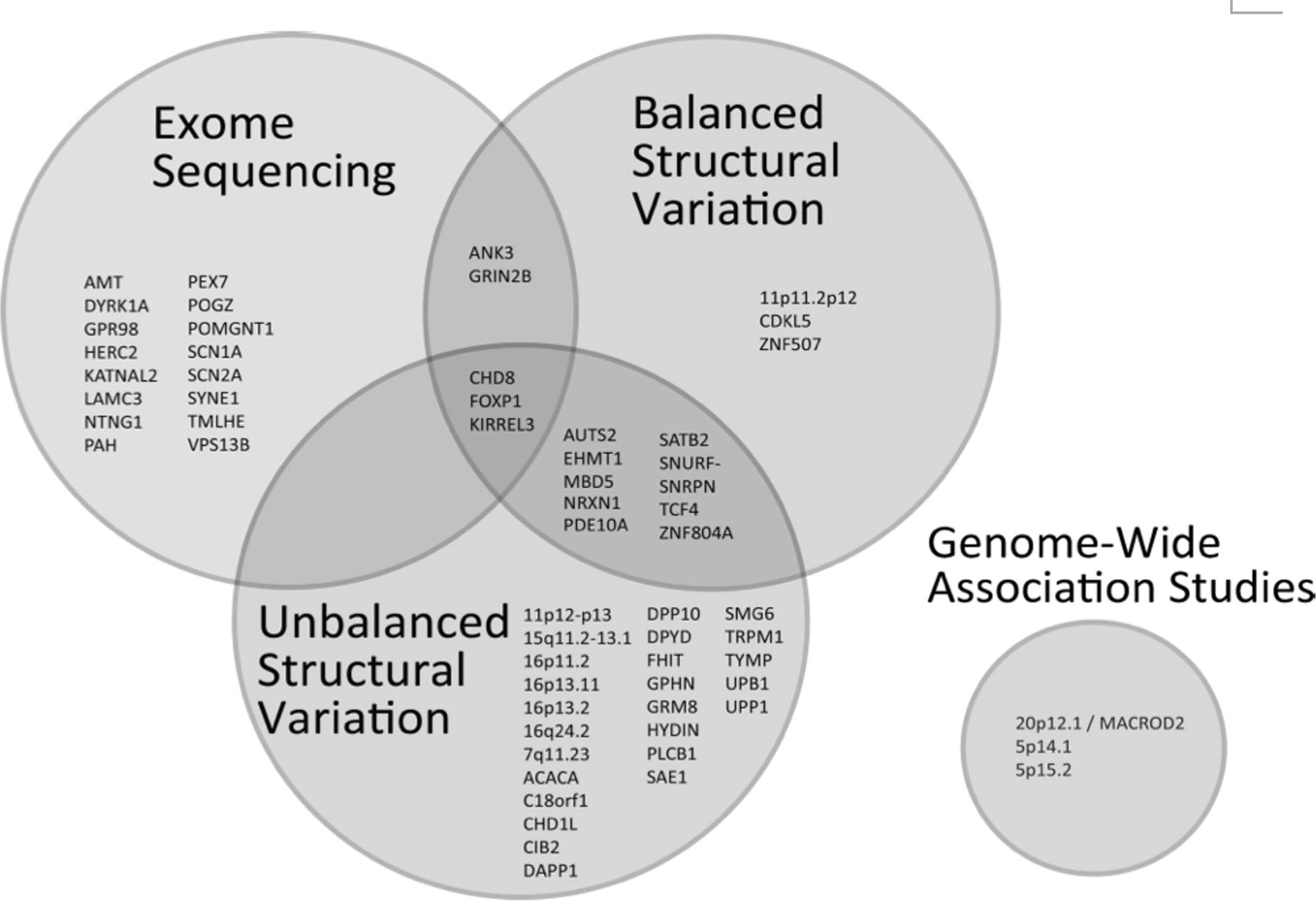
Protein-coding single nucleotide variants and small insertions/deletions discovered through exome sequencing, unbalanced structural variation (whole-gene or chromosomal region deletions and duplications), and balanced structural variation (translocations, inversions, and chromothripsis) affect risk through overlapping sets of genes and loci. Signals reported in genome-wide association studies are also shown.
GENE DISCOVERY
The number of definitive genetic risk factors for ASD that have been discovered in recent years compared to the preceding decade of research has been remarkable. Thanks to revolutionary technical leaps such as oligonucleotide microarrays and massively parallel sequencing, coupled with clever methodological innovations, genetic studies have elucidated a large number of unambiguous risk loci in ASD, ranging from large genomic regions to individual nucleotides. Moreover, there are many additional loci that, given the modest sample sizes studied to date in ASD, have not met the stringent criteria of reproducible statistical significance but that represent promising new leads warranting further investigation.
Prior to emergence of the capacity to perform unbiased genome-wide surveys of individual nucleotides and genome structure, precious little was known about the genetic architecture of ASD other than its comorbidity with several other Mendelian disorders, including Fragile X syndrome, Rett’s syndrome, tuberous sclerosis, neurofibromatosis, Angelman syndrome, and macrocephaly associated with PTEN mutations.45–52 In addition, candidate gene studies, while largely unreliable due to many false-positive findings, did identify associations of a handful of genes that continue to be associated with ASD in some studies: those expressed as neuroligins,53 SHANK,54 and CNTNAP.55
The development of array-based technologies to detect relative DNA dosage changes, as well as technology capable of simultaneously genotyping hundreds of thousands to millions of single-nucleotide polymorphisms (SNPs), first opened the door to unbiased genome-wide assessment of loci conferring risk to ASD in an association-based design. The next technical advance came with the advent of massively parallel sequencing and, with it, methods to reduce the complexity of the genome by enriching for targeted regions, such as whole-exome capture, which surveys the approximately 1% of the genome comprising known protein-coding sequences. We highlight the most significant findings of the last several years from each major approach taken in ASD genetics. This is not meant to be a comprehensive review of the existing studies or of each gene implicated, but rather a review of the seminal findings from each approach, with a synthesis of the most significant loci elucidated from those studies.
SINGLE-NUCLEOTIDE VARIANTS
Genome Wide Association Studies
Both common-variant and rare-variant hypotheses have been suggested to explain the genetics of ASD, but the advent of genome-wide, SNP-based microarrays enabled a much earlier test of the former in an unbiased genome-wide association study (GWAS), with carefully determined thresholds for statistical significance. The method has been extremely productive in identifying disease-associated SNPs across a broad array of human diseases. As of 23 January 2014, 1,789 GWAS studies have been published, identifying 7,196 trait-associated SNPs at genome-wide statistical significance (www.genome.gov/gwastudies ).56 The ultimate success of GWAS in psychiatric genetics was initially uncertain, but a seminal publication by Purcell and colleagues57 established a reproducibly significant effect of polygenic variation in schizophrenia and bipolar disorder. Published meta-analyses have begun to uncover genome-wide significant loci in schizophrenia,58 and as power is increased from the accumulation of sufficiently large sample sizes, ongoing large-scale studies across psychiatric disorders suggest the comparable impact of common variation in neuropsychiatric disorders on many other traits.44 These studies also suggest a significant degree of shared genetic risk among multiple neurodevelopmental and psychiatric disorders. Discoveries of subtle effects from common variation in ASD alone have been far more incremental; it is unknown whether these results reflect the genetic architecture of the disorder or a limitation due to sample size and the power of studies to date. GWAS and family-linkage analyses from SNP microarrays in three studies performed thus far33–35 identified independent genome-wide significant associations, but a meta-analysis of these studies did not support consistent effects of any of the loci.59 More recently, Devlin and colleagues36 have showed that common variants exerting weak additive effects collectively represent a major component of ASD risk. It remains noteworthy that none of the existing ASD GWAS studies approached the power that has been achieved from the amalgamation of samples in other disorders, and that the overall contribution of common variation in ASD likely remains an underestimate of its true effect.
Exome Sequencing
Since 2010, the continued decline in the price of next-generation sequencing, combined with the banking of genetic material and phenotypic data from ASD probands and their families by the Simons Simplex Collection,60 the Autism Genetic Resource Exchange,61 and the Autism Consortium of Boston (www.autismconsortium.org), has allowed the emergence of whole-exome sequencing (WES) as a powerful new tool for exploring the genetic etiology of ASD. Studies of the contribution of rare and de novo coding variation in exomes have uncovered several previously unreported gene associations, confirmed advanced parental age as a significant risk factor for increased de novo mutation rates, and implicated particular biological pathways. Different hypotheses as to the nature of causal coding variations have motivated exome sequencing under a variety of study designs.
Rare complete knockouts in outbred populations
Healthy individuals are estimated to carry ~100 total loss-of-function (LoF) mutations per genome.62 A recessive model of ASD inheritance would suggest that some ASD individuals have chance combinations of deleterious LoF alleles for critical genes, resulting in a “complete knockout” homozygous, compound heterozygous, or hemizygous state. WES has been employed in a case-control study design to demonstrate that across all genes in the genome, the occurrence of such rare complete knockouts is indeed enriched approximately twofold relative to controls in subjects with ASD, with a trend toward greater enrichment for brain-expressed genes.17 For genes on the X chromosome, male cases also showed enrichment relative to male controls for hemizygous inactivating mutations. Complete knockouts were nonetheless rare in cases and in controls, and a recessive LoF mode of inheritance was estimated to explain just ~5% of ASD cases (3% by autosomes, and 2% by the X chromosome). The excess of affected males in ASD motivated an effort to sequence only the X chromosome exome in concordant ASD male sibling pairs, which identified several possibly causal LoF mutations, including three in genes otherwise associated with intellectual disability or with autism via CNVs63—that is, genetic variations involving the gain or loss of DNA segments.
Complete knockouts in chromosomes identical by descent
The combination of recessive LoF alleles into a homozygous complete-knockout state is more likely to occur in genomes rich in runs of homozygosity, as these genomes may represent the convergence in an individual of two copies of the same ancestral chromosome being inherited from both parental lineages. Studies illuminating the contribution of such recessive alleles to ASD have taken advantage of the elevated homozygosity found in consanguineous families, isolated populations, and a small fraction of individuals whose parents are not known to be related. One study, which specifically targeted multiplex consanguineous families with multiple ASD individuals in order to enrich for such homozygous-inactivating mutations, performed WES and then filtered for rare alleles that were homozygous in all affected individuals and heterozygous or absent in all unaffected individuals.16 Functional experiments in that study suggested that partial loss of function mutations in two genes (AMT and PEX7) might be causal for ASD whereas total loss of function in these genes is associated with more severe developmental phenotypes.
Due to a limited ancestral gene pool, isolated and endogamous populations have frequent runs of homozygosity even when parents are not nominally related. Mapping and WES were used in multiplex Amish and Mennonite families to identify candidate genomic regions, and variants in those regions were filtered for novel variants (absent from dbSNP) and heterozygotes in all healthy population-matched controls.64 The fraction of the genome covered by runs of homozygosity is highly variable since even in families not known to be consanguineous, some common ancestry may exist. One study selected the 16 patients with the highest degree of homozygosity from 1000 Autism Genetic Resource Exchange families and performed WES.65 After functional filtering, candidate mutations were validated by targeted sequencing in parents and in unaffected siblings to verify segregation with ASD. This approach identified candidate missense mutations in four genes that, in a separate cohort, also proved to have a higher mutation burden in cases than controls. Together, these studies support a role for mutations in identical-by-descent alleles both in consanguineous families and, to some extent, in the general population.
Dominant de novo coding variations
Sporadic cases of autism with unrelated parents are likely to be enriched for de novo mutations. Although de novo mutations may not be inherited or may arise from germ-line mosaics, they do contribute to estimates of ASD heritability because they are usually shared between monozygous twins. Several studies have performed whole exome sequencing of trios, sometimes including unaffected siblings, in order to identify de novo coding mutations present in heterozygous states in affected children,19–23 with some genes later targeted for resequencing.18 One study performed whole-genome sequencing of concordant monozygous twins and their parents.24 Advanced parental age has been associated with a higher total number of de novo coding variations in children, both affected and unaffected.19,21–23 Maternal and paternal age were found to be strongly correlated with one another,22,23 but the limited number of de novo variations that were successfully phased proved to reside overwhelmingly on the paternal chromosome,19,22 implicating paternal age as a major contributor to the higher rate of point mutations. All studies that compared the rate of de novo mutations in affected versus unaffected siblings failed to find evidence of a significantly elevated total number of de novo mutations in cases. Rather, those de novo mutations that were detected in the affected sibs showed an enrichment for variants of functional importance: nonsense mutations occurred at a much higher rate in cases than controls,19,21–23 with variable evidence among studies for a significant enrichment of missense mutations.23 Similarly, the de novo mutations in cases showed a relative enrichment for occurrence in genes whose products showed protein-protein interactions with one another,22 were regulated by FMRP,21 or were involved in chromatin regulation.19,43
Dominant, incompletely penetrant inherited mutations
In contrast to the family-based studies discussed above, one study sought to identify ASD genes through a case-control design coupled with filtering for genes annotated to belong to neurodevelopment pathways.66 This study identified one candidate gene with mutations present in multiple cases, but validation sequencing revealed that the mutation was inherited from unaffected parents in some cases, implying either false-positive association or a dominant allele with incomplete penetrance.
STRUCTURAL VARIATIONS
Studies of variation in genome structure have advanced our understanding of the genetic architecture of ASD perhaps more than any other approach. Structural variations (SVs) are broadly divided into two categories: unbalanced SVs, which result in regional genomic dosage change (that is, in CNVs), and balanced chromosomal rearrangements (BCRs), which involve changes in the chromosome structure without significant gain or loss of genetic material. Because array comparative genomic hybridization techniques and SNP microarrays have allowed for numerous studies of readily detectable CNVs, almost all SV studies to date have focused on this class of variants. The elucidation of genes disrupted by BCRs has traditionally been more problematic, as these were detectable only by cytogenetic techniques such as karyotyping, and characterization of the precise breakpoints required laborious serial positional-cloning techniques. Innovations in massively parallel sequencing, however, have now enabled rapid delineation of these rearrangement events to base-pair resolution.67,68 All initial studies of SVs were based on karyotyping, which is limited to microscopically visible changes in the chromosomes, typically on the order of 3–10 Mb, depending on banding resolution.69 Previous studies by Scherer and colleagues70 established that roughly 7% of ASD cases harbored cytogenetically visible chromosome abnormalities, and subsequent efforts reported a consistent result (5.8%) in independent ASD cases.32 Several early studies used array-based technologies to establish CNVs as a major source of genomic variation,71–73 and such structural changes in the chromosomes have been robustly established as highly penetrant contributors to the etiology of ASD, as reviewed below.
Copy number variations
Reports of the impact of large, de novo CNVs in ASD have been remarkably consistent74 and have been replicated in virtually every study conducted.25,32,75 Genomic imbalances associated with ASD generally fit into two classifications: recurrent and nonrecurrent events. Recurrent events are predominantly mediated by non-allelic homologous recombination (NAHR), have similar rearrangement breakpoints localized to flanking segmental duplications, result in reciprocal dosage imbalances (deletion and duplication) in different individuals, and occur at measurable frequency in ASD as well as in other disorders and controls. A now classic example is the 16p11.2 microdeletion/microduplication syndrome.76 Other regions of recurrent CNVs that have met significant statistical thresholds for association with ASD include 15q11–13,77 22q11.2,78 and 1q21.1,31 with other recurrent reciprocal dosage regions, such as 17p12, being observed in ASD but not yet meeting thresholds of statistical significance. Other loci are frequently disrupted by CNVs in ASD (and in related neurodevelopmental disorders that do not fit strict diagnostic criteria for ASD) and less commonly in the general population, but since the source of this disruption is not NAHR, the CNVs themselves are of different sizes and have independent breakpoints. Examples of microdeletion/duplication regions with frequent disruption by nonrecurrent CNVs include 9q34.3,79 2q23.1,40 1q21,80 2q33.1,26,81–83 and 16q24.2.29
The initial CNV studies, and most that followed, revealed association with genomic segments that were often large and included many genes and regulatory elements. Much like linkage-based approaches, these studies rarely pointed directly to individual genes. More recently, the establishment of large patient cohorts has enabled the identification of the minimum segments of overlap of nonrecurrent CNVs that define a critical region disrupted across many cases. In several recent examples, this strategy has revealed a single gene to be a major contributor for a particular microdeletion or microduplication syndrome: MBD5 in 2q23.1,39,40,84 SATB2 in 2q33.1,82 EHMT1 in 9q34.3,85 CHD1L in 1q21.1,31 and ACACA in 17p21,31 to name just a few. These discoveries represent a significant advance in ASD genetics as well as in the general annotation of the morbid human genome, as they propose the hypothesis that many of the recurrent changes to genomic segments associated with human disease are attributable to one or a few strong-effect genes in these regions and that those genes may eventually serve as targets for rational intervention. While this same approach is not applicable to recurrent NAHR-mediated imbalances, given that they are a consequence of recurrent breakpoints localized to repeated regions, one study performed a series of individual gene knockdowns and overexpression in zebrafish to implicate a single contributory locus, KCTD13, in neuroanatomical phenotypes of the recurrent 16p11.2 microdeletion/duplication syndrome.86 Similar approaches in other model organisms could provide a feasible approach to dissecting NAHR-mediated recurrent rearrangements, although direct association to the ASD phenotype is more difficult in such organisms; the studies rely, instead, on a measurable morphological trait or biomarker.
Studies of recurrent CNV have also provided significant insight into the impact of dysregulation of gene dosage for a number of ASD-associated loci. In many such regions, risk for similar phenotypes is conferred by reciprocal copy-number changes, suggesting the presence of genes whose normal function requires tight dosage control. For example, although associated with a broad spectrum of psychiatric and developmental phenotypes as well as with anthropometric traits,87–89 reciprocal gains and losses of 16p11.2 are both associated with increased risk of ASD.76,90 Similar results have been reported for other loci such as 22q11.2, 15q11–13, 1q21.1, and 2q23.1.25,26,38 The mechanisms by which these reciprocal alterations confer similar phenotypes is at present unknown; future transcriptome and network studies of ASD, however, may help to elucidate such pathogenic processes.
Balanced chromosomal rearrangements
In contrast to the many CNV studies of ASD, there are precious few studies evaluating the impact of BCRs. This type of structural variation can involve balanced translocations, inversions, or excision/insertion events, and in each instance one or more genes can be disrupted at the breakpoints in a relatively balanced manner. BCRs have typically been identified clinically at microscopic resolution by karyotyping, which has significantly limited the capacity to define individual loci disrupted at the breakpoints without labor and cost-intensive positional cloning studies. Even so, positional cloning studies have uncovered a number of highly penetrant gene defects contributing to ASD, including those in AUTS2,91, NRXN1,92, and EHMT1.85 Recently, methodological manipulation of next-generation sequencing data has enabled detection of BCRs at sequence-based resolution.26,67,68,93–95 Snyder and colleagues67 initially demonstrated the feasibility of the detection of all classes of SV by large-insert, mate-pair sequencing using long-read 454 technology. Talkowski and colleagues95 recently performed the first large-scale sequencing study of constitutional BCRs, which revealed a highly complex chromosomal architecture associated with some BCRs in ASD, including rearrangements as extreme as those recently dubbed in cancer as chromothripsis, which have previously been considered as catastrophic events that were exclusively somatic in origin.96 Subsequent sequencing of de novo BCRs associated with ASD and related neurodevelopmental phenotypes has revealed a large number of genes whose disruption potentially contributes to these disorders. In one study of 38 subjects, 33 independent genes were found to be disrupted by BCR breakpoints, many of which were novel loci not previously associated with ASD, including as CHD8, KIRREL3, METTL2B, and many others.26 The primary hurdle in these studies, much like exome-sequencing studies, is to distinguish, among rare or private loss-of-function mutations, those that represent highly penetrant pathogenic variation or moderately penetrant oligogenic/polygenic risk factors from those that are benign and well tolerated. This study used a convergent genomics approach, nominating loci from BCR sequencing and then interpreting the significance of the locus by analyzing gene expression, CNV, and GWAS. For many of the 33 genes, this approach identified a significantly increased CNV burden among independent cases compared to controls. Importantly, the GWAS portion of the study also found, when considering those 33 genes, a significant association of ASD with common SNP alleles, suggesting that in some individuals the outright inactivation of a gene, and in many more persons the polymorphic variation of the same gene, can contribute to the risk of neurodevelopmental abnormality. These data firmly support the hypothesis that many ASD-associated SVs disrupt genes that are under tight dosage regulation, and that subtle or profound perturbations of their normal dosage for expression can confer risk regarding a spectrum of neurodevelopmental phenotypes, including ASD.
FUNCTIONAL AND CLINICAL IMPLICATIONS
Much of the neurobiological work in ASD has suggested a pathogenic process that disrupts the development and function of neuronal synapses, yet one that leads specifically to ASD—whose clinical definition is distinct from other neurodevelopmental syndromes and genomic disorders. In many ways, genetic discoveries to date have supported this hypothesis, but they have also offered a much broader and more complicated perspective on ASD etiology and its resultant clinical implications. In some ways they have come to challenge established dogma.
Mutations in genes encoding members of protein families involved in cell signaling, cell adhesion, and synaptic function or plasticity—including SHANK,25,54,97–101 neurexins,92,102,103 neuroligin proteins,53,104 glutamate receptors,20,26 BDNF,105 and KIRREL3,26 along with members of mTOR and FMRP signaling pathways106—have been strongly aligned with the standard hypothesis. All of these proteins play a role in the interconnected network of proteins related to synaptic function, and many have been specifically implicated in ASD.107
A significant number of genetic discoveries in ASD suggest, however, that diverse biological pathways contribute to this disorder and that many ASD genes have a wider etiological role in a spectrum of human developmental abnormalities and psychopathology. Gene-network analyses and gene-set enrichment analyses—performed on genes associated with ASD via mutations or transcriptomics—have implicated a variety of networks and pathways as associated with autism.25,42,43,108 Among the most statistically significant risk factors discovered to date is CHD8, a chromodomain helicase involved in chromatin remodeling and transcriptional repression that was discovered as direct disruption by translocation in an ASD subject26 and as a de novo loss-of-function mutation in multiple ASD exome-sequencing studies.18,19,22,23 CHD8 was also associated with an increased CNV burden (from both deletion and duplication) in ASD subjects and with macrocephaly and various developmental outcomes.18,26 Other genes, such as TCF4, also present a complex picture from both neurobiological and clinical perspectives. Point mutations in TCF4, which encodes a transcriptional regulator, have been shown to result in Pitt-Hopkins syndrome;109,110 structural variations have been associated with ASD and other neurodevelopmental phenotypes;26,109 and common variants have been among the most significant genome-wide significant risk factors for schizophrenia and other psychiatric disorders.44,111 A similar mutational spectrum (translocation, deletion, duplication, LoF mutation, non-synonymous mutation) has been shown to disrupt MBD5 and result in ASD, intellectual disability, epilepsy, and even later-onset neurobehavioral regression.26,39,40,84,112,113 MBD5 encodes a member of the methyl CpG-binding domain family of proteins that includes MeCP2, a known causal locus in Rett’s syndrome. Numerous additional genes encoding transcriptional regulators and chromatin modifiers have emerged from SV and exome-sequencing studies as implicated in ASD and also in diverse other developmental and psychiatric phenotypes.
How these proteins might comport with a final common pathway model of ASD associated with synaptic dysfunction is unclear, given that their role in gene regulation is not specific to synapse-related proteins; this area of research is active and ongoing. Similarly, the links between ASD and other developmental phenotypes (and also psychiatric traits) are now well recognized from numerous CNV, BCR, and GWAS studies, as reviewed above. One open area of study is to determine whether there are definable endophenotypes with distinct genetic etiologies embedded among these broader disease phenotypes. Another is to define the genetic modifiers or environmental effects that may predispose an individual toward specific phenotypic outcomes. Given the complex biological pathways implicated in ASD etiology to date, integration of the complete genetic background of potentially at-risk individuals, leading to defined risks for specific phenotypic outcomes, is a much needed avenue of research in the coming years.
CONCLUSIONS
As reviewed above, the discovery of novel genetic factors contributing to ASD etiology has yielded remarkable insight in the last several years. Much debate remains as to the precise genetic architecture of ASD. The models being investigated range from ones in which common variation explains the largest proportion of disease risk to ones that implicate rare, but relatively penetrant, risk factors that individually explain little of the overall disease risk but that collectively are quite important. However, the advancement of genomic technologies and the capacity to perform unbiased genome-wide surveys for genomic variants that contribute risk to ASD—unfettered by prior neurobiological hypotheses—has largely put the question of whether ASD has a genetic basis to rest, which in itself is perhaps the most significant breakthrough of the last half-decade in ASD genetics.
In all of the large studies reviewed above, each of the individual genetic alterations identified explains a meaningful, but still small, proportion of the overall disease variance, with the most common risk factors, such as 16p11.2 alterations, accounting for about 1% of all ASD cases. However, it is important to recognize several considerations. All approaches discussed (GWAS, exome, and SV) have been applied in underpowered studies that could be expected to detect only the strongest, most frequent contributors. The evidence from many disorders, however—including, most recently, from psychiatric disorders such as schizophrenia—indicates that polygenic effects are relatively weak but are increasingly detectable with adequate statistical power. Also, published studies of rare exome variants have investigated the risk associated with de novo loss-of-function mutations and have not performed tests adequately powered for the full mutational spectrum, including synonymous, non-synonymous, and inherited variants, though such studies are ongoing. Similarly, studies of SVs have generally not accounted for small or multi-allelic CNVs, or for recurrent BCRs. Finally, submicroscopic balanced events and small CNVs are not accessible to current methodologies and have not been evaluated in any capacity to date, although such studies are also ongoing. The discoveries to date in the genetics of ASD thus show great promise, with equally high anticipation of further contributions from future studies, especially those that address the interaction of genetic background and environmental factors that drive phenotypic outcomes. The discovery of a strong association between ASD and genes involved in epigenetic regulation opens at least one route to exploring potential environmental influences on such regulation. Nonetheless, despite all of these advances in identifying the genetic predispositions to ASD, the ultimate target of rational therapeutics remains a distant goal.
Text Box 1. Technical terms.
array comparative genomic hybridization: a technology used to identify copy number variations in a DNA sample
Autism Genetic Resource Exchange: a biobank and data repository for autism research
balanced chromosomal rearrangement (BCR): the exchange of DNA between two or more chromosomes without cytogenetically detectable gain or loss of DNA
copy-number variation (CNV): genetic variation involving the gain or loss of segments of DNA.
genome-wide association study (GWAS): the use of genome-wide SNP chip genotyping technologies to discover genetic variations associated with a phenotype
loss of function (LoF): the loss of a gene’s ability to perform its native function, due to deletion or mutation
non-allelic homologous recombination (NAHR): a process by which genomic segments located between segments of homologous sequence can be gained or lost due to the improper pairing, breakage and rejoining of those homologous flanking regions
next-generation sequencing: highly parallel short-read DNA sequencing technology
runs of homozygosity: regions of an individual’s genome where all maternal and paternal alleles are identical, often due to consanguinity
single-nucleotide polymorphism (SNP): a change in a single DNA base pair, found in the population
single-nucleotide variant: a change in a single DNA base pair change, including private and de novo mutations not normally seen in the population
structural variation (SV): any rearrangement in the structure of genomic DNA, including CNVs and BCRs
whole-exome sequencing (WES): the use of next-generation sequencing on only the exonic, or protein-coding, portion of a genome
whole-genome sequencing: the use of next-generation sequencing on a whole genome
Acknowledgments:
Funded, in part, by the Simons Foundation for Autism Research and Nancy Lurie Marks Family Foundation (Drs. Talkowski and Gusella); NARSAD, March of Dimes, and Charles Hood Foundation (Dr. Talkowski); Autism Speaks (Dr. Gusella); and National Institutes of Health (MH095867 to Dr. Talkowski and GM061354 to Dr. Gusella).
Footnotes
Declaration of interest: Other than for the research support as described in the funding acknowledgments, the authors report no conflicts of interest. The authors alone are responsible for the content and writing of the article.
Contributor Information
Michael E. Talkowski, Harvard Medical School, Massachusetts General Hospital, Boston, MA.; Department of Neurology, Massachusetts General Hospital, Boston, MA. Psychiatric and Neurodevelopmental Genetics Unit, Massachusetts General Hospital, Boston, MA.
Eric Vallabh Minikel, Molecular Neurogenetics Unit, Center for Human Genetic Research, Massachusetts General Hospital, Boston, MA..
James F. Gusella, Harvard Medical School, Massachusetts General Hospital, Boston, MA.; Department of Neurology, Massachusetts General Hospital, Boston, MA. Molecular Neurogenetics Unit, Center for Human Genetic Research, Massachusetts General Hospital, Boston, MA.
REFERENCES
- 1.Hertz-Picciotto I, Delwiche L. The rise in autism and the role of age at diagnosis. Epidemiol 2009;20:84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atladóttir HO, Parner ET, Schendel D, Dalsgaard S, Thomsen PH, Thorsen P. Time trends in reported diagnoses of childhood neuropsychiatric disorders: a Danish cohort study. Arch Pediatr Adolesc Med 2007;161:193–8. [DOI] [PubMed] [Google Scholar]
- 3.Taylor B Vaccines and the changing epidemiology of autism. Child Care Health Dev 2006;32:511–9. [DOI] [PubMed] [Google Scholar]
- 4.Shattuck PT. Diagnostic substitution and changing autism prevalence. Pediatrics 2006;117:1438–9. [DOI] [PubMed] [Google Scholar]
- 5.Fombonne E, Zakarian R, Bennett A, Meng L, McLean-Heywood D. Pervasive developmental disorders in Montreal, Quebec, Canada: prevalence and links with immunizations. Pediatrics 2006;118:e139–150. [DOI] [PubMed] [Google Scholar]
- 6.Elsabbagh M, Divan G, Koh Y-J, et al. Global prevalence of autism and other pervasive developmental disorders. Autism Res 2012;5:160–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wing L, Yeates SR, Brierley LM, Gould J. The prevalence of early childhood autism: comparison of administrative and epidemiological studies. Psychol Med 1976;6:89–100. [DOI] [PubMed] [Google Scholar]
- 8.Ritvo ER, Freeman BJ, Pingree C, et al. The UCLA-University of Utah epidemiologic survey of autism: prevalence. Am J Psychiatry 1989;146:194–9. [DOI] [PubMed] [Google Scholar]
- 9.Autism and Developmental Disabilities Monitoring Network Surveillance Year 2008 Principal Investigators; Centers for Disease Control and Prevention. Prevalence of autism spectrum disorders—Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008. MMWR Surveill Summ 2012;61:1–19. [PubMed] [Google Scholar]
- 10.Autism and Developmental Disabilities Monitoring Network Surveillance Year 2000 Principal Investigators; Centers for Disease Control and Prevention. Prevalence of autism spectrum disorders—Autism and Developmental Disabilities Monitoring Network, six sites, United States, 2000. MMWR Surveill Summ 2007;56:1–11. [PubMed] [Google Scholar]
- 11.Autism and Developmental Disabilities Monitoring Network Surveillance Year 2006 Principal Investigators; Centers for Disease Control and Prevention. Prevalence of autism spectrum disorders--Autism and Developmental Disabilities Monitoring Network, United States, 2006. MMWR Surveill Summ 2009;58:1–20. [PubMed] [Google Scholar]
- 12.Smalley SL, Asarnow RF, Spence MA. Autism and genetics. A decade of research. Arch Gen Psychiatry 1988;45:953–61. [DOI] [PubMed] [Google Scholar]
- 13.Hoekstra RA, Bartels M, Verweij CJH, Boomsma DI. Heritability of autistic traits in the general population. Arch Pediatr Adolesc Med 2007;161:372–7. [DOI] [PubMed] [Google Scholar]
- 14.Hallmayer J, Cleveland S, Torres A, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry 2011;68:1095–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bailey A, Le Couteur A, Gottesman I, et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med 1995;25:63–77. [DOI] [PubMed] [Google Scholar]
- 16.Yu TW, Chahrour MH, Coulter ME, et al. Using whole-exome sequencing to identify inherited causes of autism. Neuron 2013;77:259–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lim ET, Raychaudhuri S, Sanders SJ, et al. Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron 2013;77:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Roak BJ, Vives L, Fu W, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012;338:1619–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Roak BJ, Vives L, Girirajan S, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012;485:246–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Roak BJ, Deriziotis P, Lee C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 2011;43:585–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iossifov I, Ronemus M, Levy D, et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012;74:285–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neale BM, Kou Y, Liu L, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012;485:242–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanders SJ, Murtha MT, Gupta AR, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012;485:237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michaelson JJ, Shi Y, Gujral M, et al. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 2012;151:1431–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pinto D, Pagnamenta AT, Klei L, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010;466:368–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Talkowski ME, Rosenfeld JA, Blumenthal I, et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 2012;149:525–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sebat J, Lakshmi B, Malhotra D, et al. Strong association of de novo copy number mutations with autism. Science 2007;316:445–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lionel AC, Vaags AK, Sato D, et al. Rare exonic deletions implicate the synaptic organizer Gephyrin (GPHN) in risk for autism, schizophrenia and seizures. Hum Mol Genet 2013;22:2055–66. [DOI] [PubMed] [Google Scholar]
- 29.Handrigan GR, Chitayat D, Lionel AC, et al. Deletions in 16q24.2 are associated with autism spectrum disorder, intellectual disability and congenital renal malformation. J Med Genet 2013;50:163–73. [DOI] [PubMed] [Google Scholar]
- 30.Beunders G, Voorhoeve E, Golzio C, et al. Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am J Hum Genet 2013;92:210–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Girirajan S, Dennis MY, Baker C, et al. Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am J Hum Genet 2013;92:221–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marshall CR, Noor A, Vincent JB, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet 2008;82:477–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weiss LA, Arking DE, Daly MJ, Chakravarti A. A genome-wide linkage and association scan reveals novel loci for autism. Nature 2009;461:802–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang K, Zhang H, Ma D, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 2009;459:528–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anney R, Klei L, Pinto D, et al. A genome-wide scan for common alleles affecting risk for autism. Hum Mol Genet 2010;19:4072–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klei L, Sanders SJ, Murtha MT, et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol Autism 2012;3:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Girirajan S, Johnson RL, Tassone F, et al. Global increases in both common and rare copy number load associated with autism. Hum Mol Genet 2013;22:2870–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cooper GM, Coe BP, Girirajan S, et al. A copy number variation morbidity map of developmental delay. Nat Genet 2011;43:838–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams SR, Mullegama SV, Rosenfeld JA, et al. Haploinsufficiency of MBD5 associated with a syndrome involving microcephaly, intellectual disabilities, severe speech impairment, and seizures. Eur J Hum Genet 2010;18:436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Talkowski ME, Mullegama SV, Rosenfeld JA, et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet 2011;89:551–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosenfeld JA, Ballif BC, Torchia BS, et al. Copy number variations associated with autism spectrum disorders contribute to a spectrum of neurodevelopmental disorders. Genet Med 2010;12:694–702. [DOI] [PubMed] [Google Scholar]
- 42.Ben-David E, Shifman S. Networks of neuronal genes affected by common and rare variants in autism spectrum disorders. PLoS Genet 2012;8:e1002556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ben-David E, Shifman S. Combined analysis of exome sequencing points toward a major role for transcription regulation during brain development in autism. Mol Psychiatry 2013;18:1054–6. [DOI] [PubMed] [Google Scholar]
- 44.Cross-Disorder Group of the Psychiatric Genomics Consortium; Genetic Risk Outcome of Psychosis (GROUP) Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 2013;381:1371–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miles JH. Autism spectrum disorders—a genetics review. Genet Med 2011;13:278–94. [DOI] [PubMed] [Google Scholar]
- 46.Brown WT, Jenkins EC, Friedman E, et al. Autism is associated with the fragile-X syndrome. J Autism Dev Disord 1982;12:303–8. [DOI] [PubMed] [Google Scholar]
- 47.Lam CW, Yeung WL, Ko CH, et al. Spectrum of mutations in the MECP2 gene in patients with infantile autism and Rett syndrome. J Med Genet 2000;37:E41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Betancur C Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res 2011;1380:42–77. [DOI] [PubMed] [Google Scholar]
- 49.Butler MG, Dasouki MJ, Zhou X-P, et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet 2005;42:318–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steffenburg S, Gillberg CL, Steffenburg U, Kyllerman M. Autism in Angelman syndrome: a population-based study. Pediatr Neurol 1996;14:131–6. [DOI] [PubMed] [Google Scholar]
- 51.Gillberg C, Forsell C. Childhood psychosis and neurofibromatosis—more than a coincidence? J Autism Dev Disord 1984;14:1–8. [DOI] [PubMed] [Google Scholar]
- 52.Bolton PF, Griffiths PD. Association of tuberous sclerosis of temporal lobes with autism and atypical autism. Lancet 1997;349:392–5. [DOI] [PubMed] [Google Scholar]
- 53.Jamain S, Quach H, Betancur C, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet 2003;34:27–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Durand CM, Betancur C, Boeckers TM, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet 2007;39:25–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Strauss KA, Puffenberger EG, Huentelman MJ, et al. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med 2006;354:1370–7. [DOI] [PubMed] [Google Scholar]
- 56.Hindorff LA, Sethupathy P, Junkins HA, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 2009;106:9362–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Purcell SM, Wray NR, Stone JL, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009;460:748–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Genome-wide association study identifies five new schizophrenia loci. Nat Genet 2011;43:969–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Devlin B, Melhem N, Roeder K. Do common variants play a role in risk for autism? Evidence and theoretical musings. Brain Res 2011;1380:78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fischbach GD, Lord C. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron 2010;68:192–5. [DOI] [PubMed] [Google Scholar]
- 61.Lajonchere CM. Changing the landscape of autism research: the autism genetic resource exchange. Neuron 2010;68:187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.MacArthur DG, Balasubramanian S, Frankish A, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 2012;335:823–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nava C, Lamari F, Héron D, et al. Analysis of the chromosome X exome in patients with autism spectrum disorders identified novel candidate genes, including TMLHE. Transl Psychiatry 2012;2:e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Puffenberger EG, Jinks RN, Wang H, et al. A homozygous missense mutation in HERC2 associated with global developmental delay and autism spectrum disorder. Hum Mutat 2012;33:1639–46. [DOI] [PubMed] [Google Scholar]
- 65.Chahrour MH, Yu TW, Lim ET, et al. Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet 2012;8:e1002635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bi C, Wu J, Jiang T, et al. Mutations of ANK3 identified by exome sequencing are associated with autism susceptibility. Hum Mutat 2012;33:1635–8. [DOI] [PubMed] [Google Scholar]
- 67.Korbel JO, Urban AE, Affourtit JP, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science 2007;318:420–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Talkowski ME, Ernst C, Heilbut A, et al. Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research. Am J Hum Genet 2011;88:469–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Manning M, Hudgins L. Use of array-based technology in the practice of medical genetics. Genet Med 2007;9:650–3. [DOI] [PubMed] [Google Scholar]
- 70.Xu J, Zwaigenbaum L, Szatmari P, Scherer SW. Molecular cytogenetics of autism. Curr Genomics 2004;5:347–64. [Google Scholar]
- 71.Iafrate AJ, Feuk L, Rivera MN, et al. Detection of large-scale variation in the human genome. Nat Genet 2004;36:949–51. [DOI] [PubMed] [Google Scholar]
- 72.Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature 2006;444:444–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sebat J, Lakshmi B, Troge J, et al. Large-scale copy number polymorphism in the human genome. Science 2004;305:525–8. [DOI] [PubMed] [Google Scholar]
- 74.Cook EH Jr, Scherer SW. Copy-number variations associated with neuropsychiatric conditions. Nature 2008;455:919–23. [DOI] [PubMed] [Google Scholar]
- 75.Sanders SJ, Ercan-Sencicek AG, Hus V, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 2011;70:863–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Weiss LA, Shen Y, Korn JM, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med 2008;358:667–75. [DOI] [PubMed] [Google Scholar]
- 77.Van Bon BWM, Mefford HC, Menten B, et al. Further delineation of the 15q13 microdeletion and duplication syndromes: a clinical spectrum varying from non-pathogenic to a severe outcome. J Med Genet 2009;46:511–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vorstman JAS, Morcus MEJ, Duijff SN, et al. The 22q11.2 deletion in children: high rate of autistic disorders and early onset of psychotic symptoms. J Am Acad Child Adolesc Psychiatry 2006;45:1104–13. [DOI] [PubMed] [Google Scholar]
- 79.Smalley SL. Autism and tuberous sclerosis. J Autism Dev Disord 1998;28:407–14. [DOI] [PubMed] [Google Scholar]
- 80.Mefford HC, Sharp AJ, Baker C, et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med 2008;359:1685–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.De Ravel TJ, Balikova I, Thiry P, Vermeesch JR, Frijns J-P. Another patient with a de novo deletion further delineates the 2q33.1 microdeletion syndrome. Eur J Med Genet 2009;52:120–2. [DOI] [PubMed] [Google Scholar]
- 82.Rosenfeld JA, Ballif BC, Lucas A, et al. Small deletions of SATB2 cause some of the clinical features of the 2q33.1 microdeletion syndrome. PloS One 2009;4:e6568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Balasubramanian M, Smith K, Basel-Vanagaite L, et al. Case series: 2q33.1 microdeletion syndrome—further delineation of the phenotype. J Med Genet 2011;48:290–8. [DOI] [PubMed] [Google Scholar]
- 84.Hodge JC, Mitchell E, Pillalamarri V, et al. Disruption of MBD5 contributes to a spectrum of psychopathology and neurodevelopmental abnormalities. Mol Psychiatry 2013. Apr 16 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kleefstra T, Kramer JM, Neveling K, et al. Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am J Hum Genet 2012;91:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Golzio C, Willer J, Talkowski ME, et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 2012;485:363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McCarthy SE, Makarov V, Kirov G, et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet 2009;41:1223–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jacquemont S, Reymond A, Zufferey F, et al. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature 2011;478:97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Walters RG, Jacquemont S, Valsesia A, et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature 2010;463:671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kumar RA, KaraMohamed S, Sudi J, et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet 2008;17:628–38. [DOI] [PubMed] [Google Scholar]
- 91.Sultana R, Yu C-E, Yu J, et al. Identification of a novel gene on chromosome 7q11.2 interrupted by a translocation breakpoint in a pair of autistic twins. Genomics 2002;80:129–34. [DOI] [PubMed] [Google Scholar]
- 92.Kim H-G, Kishikawa S, Higgins AW, et al. Disruption of neurexin 1 associated with autism spectrum disorder. Am J Hum Genet 2008;82:199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen W, Kalscheuer V, Tzschach A, et al. Mapping translocation breakpoints by next-generation sequencing. Genome Res 2008;18:1143–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen W, Ullmann R, Langnick C, et al. Breakpoint analysis of balanced chromosome rearrangements by next-generation paired-end sequencing. Eur J Hum Genet 2010;18:539–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chiang C, Jacobsen JC, Ernst C, et al. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nat Genet 2012;44:390–397, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stephens PJ, Greenman CD, Fu B, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011;144:27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sato D, Lionel AC, Leblond CS, et al. SHANK1 deletions in males with autism spectrum disorder. Am J Hum Genet 2012;90:879–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Leblond CS, Heinrich J, Delorme R, et al. Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders. PLoS Genet 2012;8:e1002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Berkel S, Tang W, Treviño M, et al. Inherited and de novo SHANK2 variants associated with autism spectrum disorder impair neuronal morphogenesis and physiology. Hum Mol Genet 2012;21:344–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Moessner R, Marshall CR, Sutcliffe JS, et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet 2007;81:1289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gauthier J, Spiegelman D, Piton A, et al. Novel de novo SHANK3 mutation in autistic patients. Am J Med Genet B Neuropsychiatr Genet 2009;150B:421–4. [DOI] [PubMed] [Google Scholar]
- 102.Gauthier J, Siddiqui TJ, Huashan P, et al. Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Hum Genet 2011;130:563–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vaags AK, Lionel AC, Sato D, et al. Rare deletions at the neurexin 3 locus in autism spectrum disorder. Am J Hum Genet 2012;90:133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Glessner JT, Wang K, Cai G, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009;459:569–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ernst C, Marshall CR, Shen Y, et al. Highly penetrant alterations of a critical region including BDNF in human psychopathology and obesity. Arch Gen Psychiatry 2012;69:1238–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Auerbach BD, Osterweil EK, Bear MF. Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 2011;480:63–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ebert DH, Greenberg ME. Activity-dependent neuronal signalling and autism spectrum disorder. Nature 2013;493:327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Voineagu I, Wang X, Johnston P, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011;474:380–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rosenfeld JA, Leppig K, Ballif BC, et al. Genotype-phenotype analysis of TCF4 mutations causing Pitt-Hopkins syndrome shows increased seizure activity with missense mutations. Genet Med 2009;11:797–805. [DOI] [PubMed] [Google Scholar]
- 110.Pitt D, Hopkins I. A syndrome of mental retardation, wide mouth and intermittent overbreathing. Aust Paediatr J 1978;14:182–4. [DOI] [PubMed] [Google Scholar]
- 111.Ripke S, Wray NR, Lewis CM, et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry 2013;18:497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chung BHY, Stavropoulos J, Marshall CR, Weksberg R, Scherer SW, Yoon G. 2q23 de novo microdeletion involving the MBD5 gene in a patient with developmental delay, postnatal microcephaly and distinct facial features. Am J Med Genet A 2011;155A:424–9. [DOI] [PubMed] [Google Scholar]
- 113.Mullegama SV, Rosenfeld JA, Orellana C, et al. Reciprocal deletion and duplication at 2q23.1 indicates a role for MBD5 in autism spectrum disorder. Eur J Hum Genet 2013. May 1 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]