

Abstract
Chemotherapy is often ineffective in advanced stage and aggressive histologic subtypes of endometrial cancer. Overexpression of the receptor tyrosine kinase AXL has been found to be associated with therapeutic resistance, metastasis, and poor prognosis. However, the mechanism of how inhibition of AXL improves response to chemotherapy is still largely unknown. Thus, we aimed to determine whether treatment with AVB-500, a selective inhibitor of GAS6-AXL, improves endometrial cancer cell sensitivity to chemotherapy particularly through metabolic changes. We found that both GAS6 and AXL expression were higher by immunohistochemistry in patient tumors with a poor response to chemotherapy compared to tumors with a good response to chemotherapy. We showed that chemotherapy resistant endometrial cancer cells (ARK1, uterine serous carcinoma and PUC198, grade 3 endometrioid adenocarcinoma) had improved sensitivity and synergy with paclitaxel and carboplatin when treated in combination with AVB-500. We also found that in vivo intraperitoneal models with ARK1 and PUC198 cells had decreased tumor burden when treated with AVB-500 + paclitaxel compared to paclitaxel alone. Treatment with AVB-500 + paclitaxel decreased AKT signaling which resulted in a decrease in basal glycolysis. Finally, multiple glycolytic metabolites were lower in the tumors treated with AVB-500 + paclitaxel than in tumors treated with paclitaxel alone. Our study provides strong pre-clinical rationale for combining AVB-500 with paclitaxel in aggressive endometrial cancer models.
Keywords: AXL, AVB-500, endometrial cancer, chemo-resistance, glycolysis
Introduction
In the United States, endometrial cancer is rising in both incidence and mortality with an expected 66,570 new cases and 12,940 deaths in 2021 [1]. Seventy-five percent of patients are diagnosed with disease confined to the uterine corpus which confers a greater than 90% 5-year survival [1]. However, prognosis for advanced endometrial cancer is poor with a 5-year overall survival of 40–65% for stage III and 15–17% for stage IV disease [2].
Though surgery is the mainstay of initial management, some patients require platinum and taxane chemotherapy and/or radiation based on their histology, grade, stage, and other patient specific factors. Patients with more aggressive subtypes, such as high grade endometrioid histology and uterine serous carcinoma (USC), are more likely to be diagnosed at an advanced stage and often do not respond well to chemotherapy [3–5]. Alternative or adjunctive treatments are desperately needed for these patients.
One target under active investigation is AXL. AXL is a member of the TAM family of receptor tyrosine kinases and is activated by its only ligand, growth arrest specific 6 (GAS6). In multiple tumor types, overexpression of AXL is related to therapeutic resistance, metastasis, and overall poor prognosis [6]. Previous work from our lab demonstrated that patients with chemotherapy resistant USC had higher AXL expression than patients with chemotherapy sensitive USC [7, 8]. High-grade endometrial carcinomas, which tend to be more chemotherapy resistant, are also more likely to demonstrate high AXL expression compared to low-grade endometrial carcinomas [7]. In addition, AXL knockdown improved sensitivity to paclitaxel, a chemotherapeutic agent frequently used to treat endometrial cancer [8]. This data supports AXL as a potential therapeutic target in endometrial cancer.
AXL has been shown to play a role in cellular metabolism. Augustine et al found that transgenic mice with AXL overexpression were obese, diabetic, and insulin resistant [9, 10]. A subsequent study by Lijnen et al showed that AXL inhibition with R428 resulted in reduced weight gain in mice fed a high fat diet. This led to the hypothesis that GAS6-AXL signaling may be important for lipid and glucose metabolism in peripheral organs [10, 11]. More recently, Li et al found that high glycolytic activity in tumor cells was associated with poor survival in patients with non-small cell lung cancer [12]. Furthermore, they found that AXL inhibition led to increased cell death in tumor cells with a glycolytic phenotype. Though there have been previous reports on AXL affecting glucose metabolism, there have been no reports on whether AXL inhibition improves response to chemotherapy in any cancer through modulation of glycolysis or related pathways.
The most promising anti-AXL therapeutic is AVB-500, a novel Fc-sAXL fusion protein, which has a 200 times greater affinity for GAS6 than the native AXL [6, 13]. Recently, a phase Ib clinical trial investigated AVB-500 + paclitaxel in platinum-resistant high grade serous ovarian cancer and showed tolerability of this combination [13]. However, whether the combination of AVB-500 + paclitaxel would also be effective in aggressive, endometrial cancer has not been determined.
Through in vitro and in vivo models, we demonstrate that AVB-500 improves endometrial cancer sensitivity to paclitaxel. Furthermore, we show that treatment with AVB-500 + paclitaxel, via decreased signaling through phosphoinositide 3-kinase/AKT (PI3K/AKT), compromises the tumor cell’s ability to undergo glycolysis. This decreased ability to support adenosine triphosphate (ATP) generation and production of necessary metabolic intermediates compromises tumor cell resilience to cytotoxic stress, thereby introducing a potential mechanism for the observed increase in chemo-sensitivity seen with this treatment combination.
Materials and Methods
Clinical Samples
A tissue microarray containing specimens from patients with primary and metastatic USC treated at Washington University in St. Louis, MO, was developed with Institutional Review Board approval (#201409005) in accordance with ethical guidelines per the U.S. Common Rule. All specimens were collected at the time of diagnosis before the initiation of treatment. Written informed consent was obtained for tissue banking. Patients were considered to have had a good response to chemotherapy if their disease recurred greater than 6 months after the last chemotherapy regimen. Patients were considered to have a poor response to chemotherapy if their disease recurred within 6 months of the last chemotherapy regimen.
Overall survival (OS) was defined as time in months from date of surgery to date of death or last contact. Progression free survival (PFS) was defined as time in months from date of surgery to date of disease progression or death. Kaplan-Meier methods were used to generate time-to-event curves. A Cox-proportional hazard model was used to estimate the hazard of high GAS6 expression.
Immunohistochemistry
Tissue microarray slides were deparaffinized with xylene, rehydrated, and stained with anti-GAS6 primary antibody (R&D Systems) or anti-AXL primary antibody (R&D Systems). Two tumor cores per patient were evaluated for GAS6 and AXL expression [14]. Similarly, slides derived from PUC198 in vivo tumor were deparaffinized with xylene, rehydrated, and stained with anti-pAKT primary antibody (Cell Signaling Technologies). Multiple tumors for each treatment condition were evaluated for pAKT expression. For all immunohistochemistry assays, intensity of staining and percentage of positive cells were blindly scored by two reviewers and averaged for a final score.
Chemoresistant Endometrial Cancer Cell Lines
Established and previously characterized USC cell line, ARK1, was provided by Shi-Wen Jiang (Mercer University School of Medicine, Savannah, GA) [15]. ARK1 cells were cultured in RPMI (Sigma Aldrich) supplemented with 10% heat inactivated fetal bovine serum (FBS) (Sigma Aldrich) and 1% penicillin and streptomycin (Thermo Fisher Scientific). PUC198 are primary cells derived from ascites of a patient with platinum refractory high-grade endometrioid endometrial adenocarcinoma. Cells were obtained with informed consent. PUC198 were cultured in RPMI supplemented with 20% FBS and 1% penicillin and streptomycin. AN3CA and KLE were obtained from the American Type Culture Collection (Rockville, MD, USA), and Ishikawa cells were provided by Stuart Adler (Washington University School of Medicine, St. Louis, MO, USA). AN3CA, Ishikawa, and KLE were maintained in DMEM (Sigma Aldrich) supplemented with 10% FBS and 1% penicillin and streptomycin. ARK1shAXL (contains AXL-targeting shRNA) and ARK1shSCRM (contains scrambled sequence used as non-targeting shRNA control) were generated as previously described and maintained in RPMI with 10% FBS and 1% penicillin and streptomycin [8]. All cells were maintained at 37 ᵒC in a 5% CO2 incubator. IDEXX BioAnalytics authenticated ARK1 cell line by short tandem repeat profiling and inter-species contamination testing.
Cell viability (XTT) and synergism assays
On the day before treatment, 5×103 cells were plated in each well of a 96-well plate. After 24 hours, cells were treated with 0.03 nM to 640 nM paclitaxel (Sigma-Aldrich) or 0.24 µM to 1000 µM Carboplatin (Sigma-Aldrich) with or without 1 µM AVB-500 (Aravive Biologics). After 72 hours, a 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide inner salt (XTT) based assay (Alpha Aesar) was performed to measure cell viability. The 96-well plates were placed in the 37 ᵒC incubator for three hours. XTT metabolism was quantified by measuring absorbance at 450 nm (Tecan infinite M200 Pro). Experimental values were normalized to values obtained for cells treated with vehicle only.
Synergism analysis was calculated by the Loewe model with Combenefit software [16]. When the synergy score is less than −10, the drug interaction is likely to be antagonistic and when larger than 10, the interaction is likely to be synergistic. Cell viability and synergism experiments were performed in triplicate.
Tumor Xenograft Models
In vivo experiments were conducted according to Institutional Animal Care and Use Committee Policy. All animal studies were also approved and supervised by the Washington University Institutional Animal Care and Use Committee in accordance with the Animal Welfare Act, the Guide for the Care and Use of Laboratory Animals and NIH guidelines (protocol 20–0378). ARK1 cells (1×107) were injected intraperitoneally (IP) into female (NOD) SCID mice (Jackson Laboratory) aged 6 to 8 weeks. After tumor engraftment, the mice were treated with either vehicle, 30 mg/kg AVB-500, 20 mg/kg paclitaxel, or a combination of AVB-500 and paclitaxel. Chemotherapy was delivered weekly and AVB-500 was given every 3 days, both by IP injection. After 21 days of treatment, the mice were sacrificed, and aggregate tumor weight, volume, and number of tumor nodules were recorded for each.
PUC198 cells (5×106) were injected IP into female NSG mice (Jackson Laboratory) aged 6 to 8 weeks. After tumor engraftment, the mice were treated with either vehicle, 30 mg/kg AVB-500, 10 mg/kg paclitaxel, or a combination of 30 mg/kg AVB-500 and 10 mg/kg paclitaxel. Chemotherapy was delivered twice weekly and AVB-500 was given every 3 days, both by IP injection. After 25 days of treatment, all mice in the vehicle and AVB-500 treatment groups were sacrificed, and three mice in each of the paclitaxel and paclitaxel + AVB-500 groups were also sacrificed. Aggregate tumor weight, volume, and number of tumor nodules were recorded for each. The remaining mice in the paclitaxel + AVB-500 group received maintenance therapy every 3 days with 30 mg/kg AVB-500. The remaining paclitaxel only mice received vehicle maintenance. After 42 days of maintenance therapy, the mice were sacrificed, the pattern of tumor progression was assessed, and tumor nodules ≥ 1 mm were quantified.
Western Blot and Antibodies
Protein lysates were harvested in protein lysis buffer with dithiothreitol and were quantified by using the Bradford assay (BioRad). Western blot analysis was performed by either reducing SDS-PAGE according to standard methods or with the Jess Automated Western Blotting System (Protein Simple), which uses automated capillary technology to measure protein expression. For standard Western Blotting, equal protein loading was confirmed by probing blots for β-actin. For the Jess Automated Western blotting system, protein levels were visualized and quantified using Compass for SimpleWestern Software (Protein Simple). Protein samples were analyzed in triplicate and expression is normalized to β-actin. Western blots were probed with the indicated antibodies (Supplementary Table 1).
Seahorse Metabolic Analysis
Metabolic analysis of live cells was performed with the Seahorse XF96 extracellular flux analyzer (Agilent Technologies). Seeding density was optimized for each cell type (10,000–40,000 cells per well). Cells were incubated overnight at 37ᵒC in 5% CO2 then exposed to treatment for 24 hours unless otherwise indicated. Cell culture medium was replaced 1 hour prior to the assay with RPMI or DMEM without phenol red supplemented with 1 mM sodium pyruvate, 10 mM glucose, and 2 mM glutamine. Basal glycolytic rate was measured with the Seahorse XF Glycolytic Rate Test Kit (Agilent Technologies) according to the manufacturer’s instructions. All experiments were performed in triplicate and data were normalized to cell number. Seahorse Wave Desktop Software was used for data analysis (Agilent Technologies).
Quantitative PCR
RNA from human cell lines was extracted with a Qiagen RNeasy Mini Kit (Qiagen). Reverse-transcriptase PCR was performed with Superscript IV First-Strand Synthesis System (Invitrogen). Quantitative PCR (qPCR) reactions were performed with Fast SYBR Green Master Mix (Applied Biosystems) with human primer pairs (Supplementary Table 2) using the 7500 Fast Real-Time PCR System (Applied Biosystems). Relative expression of mRNA was calculated by the Applied Biosystems comparative delta-delta CT method. All mRNA expression levels were normalized to GAPDH. All qPCR experiments were performed in triplicate.
Hexokinase Activity Assay
ARK1 and PUC198 cells were plated in triplicate at 5×105 cells per 10 cm plate. Forty-eight hours later, cells were treated with vehicle, 2 uM AVB-500, 12.5 nM paclitaxel, or 12.5 nM paclitaxel + 2 uM AVB-500. After 24 hours of treatment, cells were trypsinized and washed with PBS. Protein concentration of cell lysates were measured using Bradford Protein Assay (Bio-Rad). Hexokinase (HK) activity was measured fluorometrically using PicoProbe Hexokinase Activity Assay Kit (BioVision, Catalog #K769–100) per manufacturer’s instructions. The fluorescence intensity at Ex/Em =535/587 nm was detected using an Infinite M200Pro plate reader (Tecan) in kinetic mode every 1 minute for 40 minutes at 25 °C. Data were analyzed by linear regression of fluorescence curve in linear range using Prism software and presented as HK specific activity (mU/mg). One unit of HK is the amount of enzyme that generates 1.0 µmol of NADPH per minute at pH 8.0 at 25 °C.
In vivo Metabolomics Analysis
[U‐13C] D-glucose (1 g/kg, Cambridge Isotope Laboratories) was injected into each mouse IP one hour before sacrifice. Tumor was collected, snap frozen in liquid nitrogen, and prepared for liquid chromatography/mass spectrometry (LC/MS). Ultra-high-performance LC (UHPLC)/MS was performed with an Agilent 1290 Infinity II LC system coupled with an Agilent 6530 QTOF or a Thermo Scientific Vanquish Horizon UHPLC system interfaced with a Thermo Scientific Orbitrap ID-X Tribrid Mass Spectrometer. Hydrophilic interaction liquid chromatography (HILIC) was performed by using a HILICON iHILIC-(P) classic column with the following specifications: 100 mm x 2.1 mm, 5 µm. Mobile-phase solvents were composed of A = 20 mM ammonium bicarbonate, 0.1% ammonium hydroxide and 2.5 μM medronic acid in water:acetonitrile (95:5) and B = 2.5 μM medronic acid in acetonitrile:water (95:5). The following linear gradient was applied at a flow rate of 250 μL min−1: 0–1 min: 90% B, 1–12 min: 90–35% B, 12–12.5 min: 35–25% B, 12.5–14.5 min: 25% B. The column was re-equilibrated with 20 column volumes of 90% B. The injection volume was 2 μL for all experiments. Data were collected with the same settings as previously reported [17]. LC/MS data were processed and analyzed with the open-source Skyline program [18]. Metabolites were identified by matching the retention times and fragmentation patterns from research samples to authentic standards from an in-house database. Samples were normalized on the basis of tissue mass. To extract metabolites, 40 μL of 2:2:1 methanol:acetonitrile:water was added per 1 mg of tissue.
Statistical Analysis
Prism 8.0 (GraphPad Software) was used to analyze data. Student’s t-tests and Mann-Whitney tests were used where appropriate. Results are reported as mean ± standard deviation unless otherwise noted. Data was considered statistically significant when p < 0.05. Asterisks represent P values: * < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001.
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files. Additional data are available upon request from the corresponding author.
Results
Increased GAS6 and AXL expression is associated with a poor response to chemotherapy in USC
To determine whether GAS6 or AXL expression correlated with response to chemotherapy, we examined both GAS6 and AXL expression by immunohistochemistry (IHC) in a panel of USC tumors from patients at the time of diagnosis, who subsequently underwent treatment with chemotherapy. The majority of patients in the USC tissue microarray (TMA) had International Federation of Gynecology and Obstetrics (FIGO) stage III/IV disease (39/61, 63.9%) and 92% of patients received carboplatin and paclitaxel adjuvant chemotherapy (Supplementary Table 3A). Of the 61 patients in the TMA, 43 had a good response to chemotherapy and 14 had a poor response to chemotherapy (defined progression of disease greater than or less than six months after final chemotherapy treatment, respectively). Response to chemotherapy was unknown for 4 patients. We found that mean GAS6 and AXL expression were significantly higher among patients with a poor response to chemotherapy than among those with a good response to chemotherapy (GAS6: 40.9% vs. 30.4%, p=0.012; AXL: 60.7% vs. 42.7%, p=0.013) (Figure 1A, B; Supplementary Table 3B).
Figure 1:

High GAS6 and AXL expression in uterine serous tumors is associated with poor response to chemotherapy. (A) Uterine serous tumors with poor response to chemotherapy are associated with higher GAS6 (40.9% vs. 30.4%, p=0.012) and (B) AXL expression (60.7% vs. 42.7%, p=0.013) than uterine serous tumors with a good response to chemotherapy by immunohistochemistry. (C, D) Low GAS6 expression (<42%) is associated with significantly improved PFS and OS compared to high GAS6 expression (≥ 42%). (E) AXL expression is significantly higher in P53 mutated endometrial cancer tumor samples in the TCGA Firehose Legacy dataset.
Median PFS was 33.6 months in patients with low GAS6 expressing tumors and 10.6 months among patients with high GAS6 expressing tumors (P=0.003) (Figure 1C). Similarly, median OS was significantly longer in patients with low GAS6 expressing tumors compared to those with high GAS6 expressing tumors (39.5 months vs. 27.7 months, P=0.003) (Figure 1D).
We also performed Cox proportional hazards model to estimate the hazard of death due to high GAS6 expression. In a univariate model, high GAS6 expression had a hazard ratio of 2.84 (CI: 1.39 – 5.81; P=0.004). When we controlled for age and stage, the hazard ratio was 2.21 (CI: 0.97 – 5.02; P=0.06). Based on previous publications on the regulation of P53 by AXL [19, 20], we used the TCGA Firehose Legacy dataset and found a significantly higher association between TP53 mutations and AXL expression by RPPA in endometrial cancer specimens (Figure 1E).
AVB-500 increases uterine cancer cell sensitivity to, and synergizes with, paclitaxel and carboplatin in vitro
To determine whether treatment with GAS6-AXL inhibitor AVB-500 increases endometrial cancer cell sensitivity to chemotherapy, we focused on two endometrial cancer histologies that are often chemotherapy resistant particularly in advanced stages: USC and high-grade endometrioid adenocarcinoma of the endometrium. ARK1 cells were derived from chemotherapy-resistant USC. PUC198 cells are primary cells derived from the ascites of a patient with progressive platinum-refractory grade 3 endometrioid adenocarcinoma of the endometrium. These were both found to have high AXL expression (Supplementary Figure 1). We performed viability assays by treating the cells with increasing concentrations of paclitaxel alone or in combination with AVB-500. AVB-500 increased sensitivity of both ARK1 and PUC198 to paclitaxel and to carboplatin (Figure 3A-D). These results indicate that GAS6-AXL pathway inhibition with AVB-500 increases uterine cancer cell sensitivity to chemotherapy.
Figure 3:

GAS6/AXL inhibition improves chemotherapy response of ARK1 and PUC198 intra-peritoneal xenograft tumors in vivo. (A) ARK1 total tumor weight, (B) ARK1 tumor nodules, (C) ARK1 tumor volume. (D) PUC198 total tumor weight, (E) PUC 198 tumor nodules ≥ 1mm
Given that AVB-500 increased sensitivity of two cell lines to both paclitaxel and carboplatin, we asked whether AVB-500 synergized with these two chemotherapy drugs. To answer this question, we treated cells with a range of drug concentrations and measured viability. We found that AVB-500 synergized with both paclitaxel and carboplatin in ARK1 and PUC198 cells at several doses (Figure 3E and 3F).
GAS6-AXL inhibition improves response to chemotherapy in vivo
We next sought to determine whether AVB-500 would improve response to chemotherapy in vivo. To recapitulate a platinum-resistant endometrial cancer clinical scenario, we chose to treat the mice with single agent paclitaxel rather than carboplatin + paclitaxel. Thus, we injected mice IP with ARK1 cells, allowed tumors to establish, then treated mice with either vehicle, AVB-500, paclitaxel, or AVB-500 + paclitaxel. We observed significantly lower tumor weight (0.025 g vs. 0.079 g, P<0.001), fewer tumor nodules (6.3 vs. 13.0, P=0.007), and smaller tumor volume (16.8 mm3 vs. 72.3 mm3, P=0.002) in mice treated with AVB-500+paclitaxel than in mice treated with paclitaxel alone (Figure 3A-C).
We then repeated the in vivo IP injection with PUC198 cells, allowed tumors to establish, then treated mice with either vehicle, AVB-500, paclitaxel, or AVB-500 + paclitaxel. We observed significantly lower tumor weight and fewer tumor nodules ≥ 1mm in mice treated with AVB-500 + paclitaxel compared to those treated with paclitaxel alone (Figure 3D-E). Subjectively, we also observed fewer peritoneal and mesenteric nodules < 1mm in the combination treated mice compared to paclitaxel alone, however these nodules were too numerous to quantify.
After 25 days of treatment, a portion of the mice in both the paclitaxel and AVB-500 + paclitaxel treatment groups went on to receive vehicle and AVB-500 maintenance, respectively, to see if AVB-500 maintenance therapy affected patterns of disease progression (Supplementary Figure 2A). We found that after 42 days of maintenance, the AVB-500 maintenance treatment mice had significantly fewer tumor nodules ≥ 1mm suggesting that GAS6-AXL signaling is important for metastasis (Supplementary Figure 2B). We also subjectively observed fewer peritoneal and mesenteric nodules < 1mm in the AVB-500 maintenance mice but these were too numerous to quantify (Supplementary Figure 2C).
AVB-500 + paclitaxel decreases basal glycolysis via reduced AKT signaling
To determine the mechanism by which AVB-500 increases uterine cancer cell sensitivity to chemotherapy, we first examined activation of the serine/threonine kinase AKT pathway, which is one of the most frequently activated protein kinases in human cancer. AKT is well known to play a role in cancer cell survival, treatment resistance, and glucose metabolism [21]. ARK1 cells with AXL inactivation had less p-AKT expression than AXL expressing cells by western blot (Figure 4A). Given that AKT signaling plays an important role in the regulation of glucose metabolism, we hypothesized that AXL deficient cells and cells with low AXL expression would impair uterine cancer cell ability to perform glycolysis. We performed a glycolytic rate assay using a Seahorse extracellular flux analyzer. We found that basal glycolysis in ARK1shAXL was decreased compared to ARK1shSCRM (Figure 4B) and that basal glycolysis was decreased in cells with low AXL expression (KLE, Ishikawa, and AN3CA) (previously published by Divine et al. [7]) compared to cells with high AXL expression (ARK1 and PUC198) (Figure 4C). Based on our findings that AXL promotes the glycolytic phenotype via AKT signaling in endometrial cancer cells, we hypothesized that treatment of ARK1 and PUC198 cells with AVB-500 + paclitaxel would decrease p-AKT expression to a larger degree than treatment with paclitaxel alone (Figure 4D). Furthermore, immunohistochemistry revealed that p-AKT expression in PUC198 tumor from mice treated with AVB-500+paclitaxel was significantly decreased compared to mice treated with paclitaxel alone (Supplementary Figure 3). We then performed a glycolytic rate assay and found that after 24 hours of treatment with AVB-500+paclitaxel, basal glycolytic rates in ARK1 and PUC198 cells were lower than those in cells treated with paclitaxel alone (Figure 4E). AVB-500 alone had no effect on basal glycolytic rate. These results suggest that AVB-500 increases uterine cancer cell sensitivity to chemotherapy by reducing glycolysis.
Figure 4:

AVB-500 + paclitaxel leads to reduced glycolysis via decreased AKT activation. (A) WB detection of AXL, AKT, and p-AKT expression in (Left) ARK1shSCRM & shAXL and (Middle & Right) PUC198shSCRM & shAXL. Left and middle WB performed with Jess automated capillary blotting system. Right WB by traditional methods. (B) Basal glycolysis of ARK1shSCRM and shAXL. (C) Basal glycolysis of multiple endometrial cancer cell lines. (D) Protein expression of p-AKT in ARK1 and PUC198, treated for 24 hours with vehicle, AVB-500 2 µM, paclitaxel 12 nM, or AVB 2 µM + paclitaxel 12.5 nM. Cell lysates were analyzed using the Jess automated capillary blotting system and normalized to β-Actin. Representative band images are shown. (E) Basal glycolysis of ARK1 and PUC198 after 24 hour treatment with vehicle, 2 uM AVB-500, 12.5 nM paclitaxel, or 12.5 nM paclitaxel + AVB-500 2 µM.
AVB-500 + paclitaxel decreases hexokinase and phosphofructokinase mRNA expression and hexokinase enzyme activity in vitro
Activation of the PI3K/AKT pathway has been shown to alter glucose metabolism though direct phosphorylation of key metabolic enzymes as well as through control of transcription factors that regulate expression of components of the metabolic pathway [21]. We asked whether treatment with AVB-500 + paclitaxel would result in changes in the transcription of key glycolytic enzymes, hexokinase (HK) and phosphofructokinase (PFK). Figure 5 shows qPCR results for HK and PFK mRNA levels in ARK1 (Figure 5A-B) and PUC198 cells (Figure 5C-D) treated for 24 hours with vehicle, AVB-500, paclitaxel, or AVB-500 + paclitaxel. Both ARK1 and PUC198 cells show a significant decrease in HK mRNA expression in cells treated with AVB-500 + paclitaxel compared to treatment with paclitaxel alone. Similarly, ARK1 cells show a significant decrease in PFK mRNA expression in cells treated with AVB-500 + paclitaxel compared to treatment with paclitaxel alone. PUC198 cells demonstrate a trend towards decreased PFK expression in combination treated cells but this was not significant.
Figure 5:
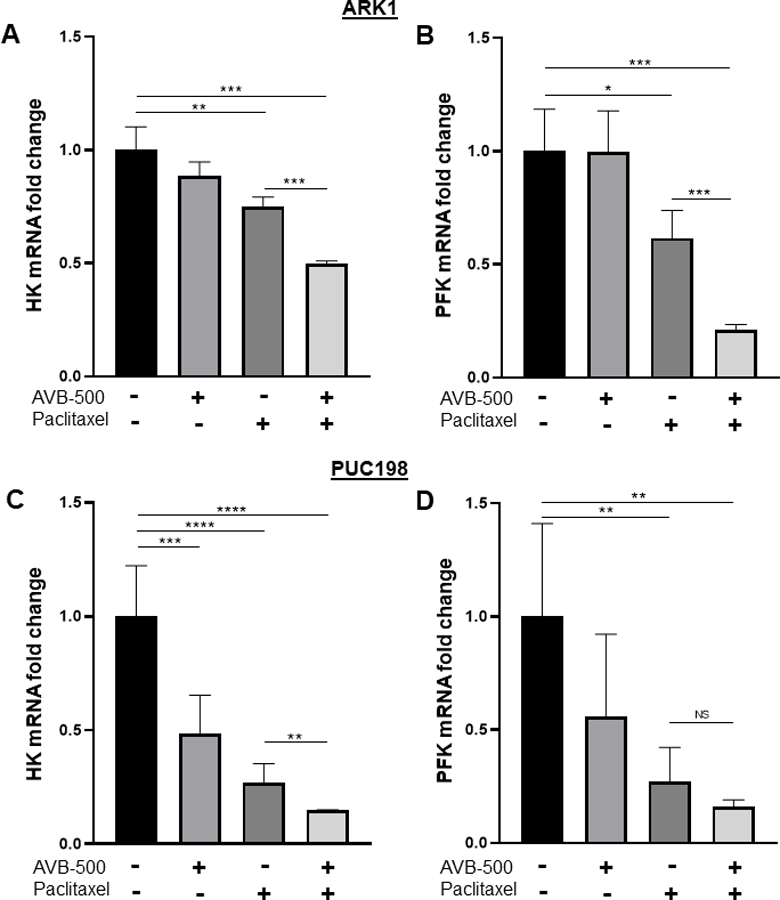
Quantitative PCR (qPCR) reveals that treatment with AVB-500 + paclitaxel decreases mRNA transcript for hexokinase (HK) and phosphofructokinase (PFK) to a larger degree then treatment with paclitaxel alone in ARK1 and PUC198 cells. A) HK mRNA in ARK1, B) PFK mRNA in ARK1, C) HK mRNA in PUC198 and D) PFK mRNA in PUC198.
Given that AKT activation also drives glycolysis through direct phosphorylation of key enzymes, we asked whether HK enzyme activity would be impacted by AVB-500 + paclitaxel. We found that PUC198 cells had significantly deceased HK activity when treated with AVB-500 + paclitaxel compared to when treated with paclitaxel alone (Supplementary Figure 4). These results, taken together, support the multi-faceted nature of PI3K/AKT mediated regulation of glucose metabolism.
In vivo ARK1 treatment with AVB-500 + paclitaxel decreases glycolytic metabolite abundance more than treatment with paclitaxel alone
We next investigated whether AVB-500 + paclitaxel had similar effects on tumor cell glycolysis in vivo. This approach allowed for exploration of alterations in tumor cell metabolism which is more comparable than cell-based assays to those that occur in human endometrial cancer. Thus, we injected ARK1 cells IP into mice, treated with vehicle, paclitaxel, AVB-500, or AVB-500 + paclitaxel. Tumors were isolated and LC/MS was used to measure pool sizes of metabolites. Treatment with AVB-500 + paclitaxel decreased multiple glycolytic metabolites, including fructose-1,6-bisphosphate by 4.20 fold (P=0.002), 3-phosphoglyceric acid by 7.76 fold (P=0.014), phosphoenolpyruvate by 8.85 fold (P=0.013), and lactate by 2.78 fold (P=0.024) when compared to treatment with paclitaxel alone (Figure 6). These results support the hypothesis that AVB-500 increases uterine cancer cell sensitivity to chemotherapy, in part, by impairing ability of the tumor cells to perform glycolysis.
Figure 6:

In vivo ARK1 treatment with AVB-500+paclitaxel decreases glycolytic metabolite abundance compared to treatment with paclitaxel alone. A) Summary of changes in glycolytic metabolite abundance with AVB-500+paclitaxel treatment compared to paclitaxel alone. B) fructose-1,6-bisphosphate, C) 3-phosphoglyceric acid, D) phosphoenolpyruvate, and E) lactate
Discussion
High grade endometrioid and non-endometrioid endometrial tumors are often resistant to chemotherapy [3–5]. To improve outcomes for these patients, we need to identify strategies to overcome chemo-resistance. Here, we present several lines of evidence that GAS6-AXL pathway inhibition with AVB-500 improves endometrial cancer chemo-sensitivity. First, we show that high GAS6 and high AXL expression by IHC correlate with worse clinical response to chemotherapy. Second, we demonstrate that treatment with AVB-500 restores sensitivity to carboplatin and paclitaxel in vitro, and that these drug combinations act in a synergistic manner. Third, we show that treatment with AVB-500 and paclitaxel decreases tumor burden more than treatment with paclitaxel alone in vivo. Finally, we demonstrate that AVB-500 + paclitaxel compromises the tumor cell’s ability to undergo glycolysis through decreased activation of PI3K/AKT. Reduced PI3K/AKT pathway activation results in decreased HK and PFK transcription and HK enzyme activity (Supplementary Figure 5). These transcriptional and post-translational changes contribute to the observed decrease in tumor cell glycolysis in vitro and in vivo. The decreased ability of endometrial tumor cells to produce ATP and other necessary metabolic intermediates is a potential mechanism for the observed restoration of chemo-sensitivity seen with this treatment combination. These findings provide strong pre-clinical evidence for combining AVB-500 with paclitaxel in endometrial cancer clinical trials.
AXL expression has been associated with treatment resistance in multiple cancer types, including endometrial cancer [8, 22–25]. Our study demonstrated that there was a trend for high GAS6 as an independent factor for overall survival. Future directions include further analysis with a greater number of specimens. Previous work from our lab demonstrated three mechanisms by which this occurs. The first mechanism is through AXL mediated limitation of intracellular accumulation of chemotherapy via upregulation of drug efflux pumps [8] [22–24, 26, 27]. Additionally, AXL has been shown to reduce paclitaxel mediated cell cycle arrest and apoptosis [8, 27–32]. Finally, AXL signaling supports the epithelial-mesenchymal transition (EMT) thereby promoting tumor metastasis. Previous work from our lab showed that genetic inactivation of AXL in ARK1 cells led to decreased cellular invasion, migration, tumor burden, metastatic colonization in vivo, and increased sensitivity to paclitaxel compared to AXL expressing cells [7]. AXL’s critical role in endometrial cancer metastasis is further supported by our ARK1 and PUC198 in vivo models which collectively showed decreased tumor burden in mice who received AVB-500 + paclitaxel vs. paclitaxel alone, as well as decreased metastatic disease in mice who received AVB-500 maintenance treatment vs. vehicle.
Our study introduces glucose metabolism as a novel mechanism by which AXL promotes chemo-resistance. Glycolysis, in addition to producing ATP, provides metabolic intermediates as substrates for pathways that support production of proteins, lipids, and nucleotides that are required for tumor cell growth and proliferation. The PI3K/AKT cascade is one of the most commonly activated pathways in human cancer and has important effects on glucose metabolism. It has been shown that AKT promotes glycolysis though direct post-translational regulation of nutrient transporters and metabolic enzymes as well as through control of transcription factors [21].
GAS6-AXL signaling is known to activate the PI3K/AKT signaling network which in turn promotes glycolysis [33, 34]. Li et al was the first to show that AXL inhibition in non-small cell lung tumor samples with a glycolytic phenotype led to increased cell death compared to tumor samples with an oxidative phenotype [12]. These findings are supported by our results that cell lines with high AXL expression exhibit higher basal glycolysis than cell lines with low AXL expression. Furthermore, our study shows that GAS6-AXL pathway inhibition with AVB-500 plus paclitaxel resulted in decreased endometrial tumor cell basal glycolysis in vitro as well as glycolytic intermediates in vivo. Tian et al also demonstrated that AXL’s role in the resistance of ovarian cancer cells to cisplatin is through the promotion of glycolysis and that inhibition of AXL can improve the therapeutic effect of cisplatin [35]. Our study contributes to the growing body of literature supporting AXL’s role in the promotion of glycolysis as a mechanism of chemo-resistance. We theorize that GAS6-AXL pathway inhibition with AVB-500 leads to decreased production of ATP and other key metabolic intermediates, thereby increasing susceptibility to cytotoxic stress imparted by chemotherapy.
Our data provide pre-clinical in vitro and in vivo evidence that AVB-500 in combination with paclitaxel is a promising drug combination for the treatment of chemotherapy resistant endometrial cancer. AVB-500 is currently being investigated in multiple phase I/II clinical trials in clear cell renal cancer, pancreatic cancer, and advanced urothelial cancer. Within the realm of gynecologic malignancies, AVB-500 + paclitaxel demonstrated safety and efficacy in the treatment of recurrent platinum-resistant ovarian cancer which has led to an ongoing phase III trial [36]. Our data indicate that AVB-500 + paclitaxel should also be studied in clinical trials for chemotherapy resistant endometrial cancer. Additionally, continued identification of viable biomarkers is critical to the success of targeted therapies such as AVB-500. Given AXL’s role in glucose homeostasis, one or more glycolytic intermediates may prove to be a useful biomarker to help predict sensitivity in patients who are eligible to receive this treatment combination.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 2:

AVB-500 increases uterine cancer cell sensitivity to Paclitaxel and Carboplatin. (A) Results of XTT cell viability assays of ARK1 cells in increasing concentrations of Paclitaxel and (B) Carboplatin with or without 1µM AVB-500. (C) Results of XTT cell viability assays of PUC198 cells in increasing concentrations of Paclitaxel and (D) Carboplatin with or without 1µM AVB-500. (E) Loewe synergism analysis of ARK1 cells and (F) PUC198 cells treated with increasing doses of Paclitaxel and AVB-500 as well as Carboplatin and AVB-500. Synergy scores with standard deviation are shown for each dose combination.
Acknowledgements
The authors thank Deborah Frank for assistance in manuscript editing and Peinan Zhao for assistance with statistical analysis.
Funding
Supported by Research Scholar Grant RSG-19-080-01 -TBG from the American Cancer Society (KCF). NIH grant R35 ES028365 (GP)
Footnotes
Disclosure: Dr. Fuh receives personal fees from Aravive Biologics
References
- 1.Society, A.C. Key Statistics for Endometrial Cancer 2021. [cited 2021 February 3]; Available from: https://www.cancer.org/cancer/endometrial-cancer/about/key-statistics.html#:~:text=The%20American%20Cancer%20Society%20estimates,or%20corpus)%20will%20be%20diagnosed.
- 2.Wang Q, Peng H, Qi X, Wu M, and Zhao X, Targeted therapies in gynecological cancers: a comprehensive review of clinical evidence. Signal Transduct Target Ther, 2020. 5(1): p. 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davidson BA, Foote J, Clark LH, Broadwater G, Ehrisman J, Gehrig P, et al. , Tumor grade and chemotherapy response in endometrioid endometrial cancer. Gynecol Oncol Rep, 2016. 17: p. 3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fader AN, Drake RD, O’Malley DM, Gibbons HE, Huh WK, Havrilesky LJ, et al. , Platinum/taxane-based chemotherapy with or without radiation therapy favorably impacts survival outcomes in stage I uterine papillary serous carcinoma. Cancer, 2009. 115(10): p. 2119–27. [DOI] [PubMed] [Google Scholar]
- 5.Hoskins PJ, Swenerton KD, Pike JA, Wong F, Lim P, Acquino-Parsons C, et al. , Paclitaxel and carboplatin, alone or with irradiation, in advanced or recurrent endometrial cancer: a phase II study. J Clin Oncol, 2001. 19(20): p. 4048–53. [DOI] [PubMed] [Google Scholar]
- 6.Kariolis MS, Miao YR, Diep A, Nash SE, Olcina MM, Jiang D, et al. , Inhibition of the GAS6/AXL pathway augments the efficacy of chemotherapies. J Clin Invest, 2017. 127(1): p. 183–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Divine LM, Nguyen MR, Meller E, Desai RA, Arif B, Rankin EB, et al. , AXL modulates extracellular matrix protein expression and is essential for invasion and metastasis in endometrial cancer. Oncotarget, 2016. 7(47): p. 77291–77305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palisoul ML, Quinn JM, Schepers E, Hagemann IS, Guo L, Reger K, et al. , Inhibition of the Receptor Tyrosine Kinase AXL Restores Paclitaxel Chemosensitivity in Uterine Serous Cancer. Mol Cancer Ther, 2017. 16(12): p. 2881–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Augustine KA, Rossi RM, Van G, Housman J, Stark K, Danilenko D, et al. , Noninsulin-dependent diabetes mellitus occurs in mice ectopically expressing the human Axl tyrosine kinase receptor. J Cell Physiol, 1999. 181(3): p. 433–47. [DOI] [PubMed] [Google Scholar]
- 10.Zhao M, Jung Y, Jiang Z, and Svensson KJ, Regulation of Energy Metabolism by Receptor Tyrosine Kinase Ligands. Front Physiol, 2020. 11: p. 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lijnen HR, Christiaens V, and Scroyen L, Growth arrest-specific protein 6 receptor antagonism impairs adipocyte differentiation and adipose tissue development in mice. J Pharmacol Exp Ther, 2011. 337(2): p. 457–64. [DOI] [PubMed] [Google Scholar]
- 12.Li Z, Wang Z, Tang Y, Lu X, Chen J, Dong Y, et al. , Liquid biopsy-based single-cell metabolic phenotyping of lung cancer patients for informative diagnostics. Nat Commun, 2019. 10(1): p. 3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuh KC, Bookman MA, Liu JF, Coleman RL, Herzog TJ, Thaker PH, et al. , Phase 1b study of AVB-500 in combination with paclitaxel or pegylated liposomal doxorubicin platinum-resistant recurrent ovarian cancer. Gynecol Oncol, 2021. [DOI] [PubMed]
- 14.Rankin EB, Fuh KC, Taylor TE, Krieg AJ, Musser M, Yuan J, et al. , AXL is an essential factor and therapeutic target for metastatic ovarian cancer. Cancer Res, 2010. 70(19): p. 7570–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El-Sahwi K, Bellone S, Cocco E, Cargnelutti M, Casagrande F, Bellone M, et al. , In vitro activity of pertuzumab in combination with trastuzumab in uterine serous papillary adenocarcinoma. Br J Cancer, 2010. 102(1): p. 134–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Veroli GY, Fornari C, Wang D, Mollard S, Bramhall JL, Richards FM, et al. , Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics, 2016. 32(18): p. 2866–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho K, Schwaiger-Haber M, Naser FJ, Stancliffe E, Sindelar M, and Patti GJ, Targeting unique biological signals on the fly to improve MS/MS coverage and identification efficiency in metabolomics. Analytica Chimica Acta, 2021. 1149: p. 338210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adams KJ, Pratt B, Bose N, Dubois LG, St John-Williams L, Perrott KM, et al. , Skyline for Small Molecules: A Unifying Software Package for Quantitative Metabolomics. J Proteome Res, 2020. 19(4): p. 1447–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vaughan CA, Singh S, Windle B, Yeudall WA, Frum R, Grossman SR, et al. , Gain-of-Function Activity of Mutant p53 in Lung Cancer through Up-Regulation of Receptor Protein Tyrosine Kinase Axl. Genes Cancer, 2012. 3(7–8): p. 491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Polo A, Luo Z, Gerarduzzi C, Chen X, Little JB, and Yuan ZM, AXL receptor signalling suppresses p53 in melanoma through stabilization of the MDMX-MDM2 complex. J Mol Cell Biol, 2017. 9(2): p. 154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoxhaj G and Manning BD, The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer, 2020. 20(2): p. 74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilson C, Ye X, Pham T, Lin E, Chan S, McNamara E, et al. , AXL inhibition sensitizes mesenchymal cancer cells to antimitotic drugs. Cancer Res, 2014. 74(20): p. 5878–90. [DOI] [PubMed] [Google Scholar]
- 23.Giles KM, Kalinowski FC, Candy PA, Epis MR, Zhang PM, Redfern AD, et al. , Axl mediates acquired resistance of head and neck cancer cells to the epidermal growth factor receptor inhibitor erlotinib. Mol Cancer Ther, 2013. 12(11): p. 2541–58. [DOI] [PubMed] [Google Scholar]
- 24.Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, et al. , An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res, 2013. 19(1): p. 279–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quinn JM, Greenwade MM, Palisoul ML, Opara G, Massad K, Guo L, et al. , Therapeutic Inhibition of the Receptor Tyrosine Kinase AXL Improves Sensitivity to Platinum and Taxane in Ovarian Cancer. Mol Cancer Ther, 2019. 18(2): p. 389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. , Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet, 2012. 44(8): p. 852–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin JZ, Wang ZJ, De W, Zheng M, Xu WZ, Wu HF, et al. , Targeting AXL overcomes resistance to docetaxel therapy in advanced prostate cancer. Oncotarget, 2017. 8(25): p. 41064–41077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tuck C, Zhang T, Potapova T, Malumbres M, and Novak B, Robust mitotic entry is ensured by a latching switch. Biol Open, 2013. 2(9): p. 924–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Potapova TA, Daum JR, Byrd KS, and Gorbsky GJ, Fine tuning the cell cycle: activation of the Cdk1 inhibitory phosphorylation pathway during mitotic exit. Mol Biol Cell, 2009. 20(6): p. 1737–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakurikar N, Eichhorn JM, and Chambers TC, Cyclin-dependent kinase-1 (Cdk1)/cyclin B1 dictates cell fate after mitotic arrest via phosphoregulation of antiapoptotic Bcl-2 proteins. J Biol Chem, 2012. 287(46): p. 39193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Wang X, Bi S, Zhao K, and Yu C, Inhibition of Mer and Axl receptor tyrosine kinases leads to increased apoptosis and improved chemosensitivity in human neuroblastoma. Biochem Biophys Res Commun, 2015. 457(3): p. 461–6. [DOI] [PubMed] [Google Scholar]
- 32.Keating AK, Kim GK, Jones AE, Donson AM, Ware K, Mulcahy JM, et al. , Inhibition of Mer and Axl receptor tyrosine kinases in astrocytoma cells leads to increased apoptosis and improved chemosensitivity. Mol Cancer Ther, 2010. 9(5): p. 1298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu C, Wei Y, and Wei X, AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol Cancer, 2019. 18(1): p. 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paccez JD, Vogelsang M, Parker MI, and Zerbini LF, The receptor tyrosine kinase Axl in cancer: biological functions and therapeutic implications. Int J Cancer, 2014. 134(5): p. 1024–33. [DOI] [PubMed] [Google Scholar]
- 35.Tian M, Chen XS, Li LY, Wu HZ, Zeng D, Wang XL, et al. , Inhibition of AXL enhances chemosensitivity of human ovarian cancer cells to cisplatin via decreasing glycolysis. Acta Pharmacol Sin, 2020. [DOI] [PMC free article] [PubMed]
- 36.Fuh KC, Bookman MA, Coleman RL, Herzog TJ, Thaker PH, Liu JF, Lane MW, Rangwala RA, McIntyre GF, Monk BJ, and Moore KN, Phase I study of GAS6/AXL inhibitor (AVB-500) in recurrent, platinum resistant ovarian carcinoma Abstract presented at: Society of Gynecological Oncology 2021 Virtual Annual Meeting on Women’s Cancer; March 19–21, 2021. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files. Additional data are available upon request from the corresponding author.