

Abstract
The activation of P2Y1 receptors in platelets contributes to platelet aggregation, and selective antagonists are sought as potential antithrombotic agents. We reported (Kim et al. J. Med. Chem. 2000, 43, 746–755) that acyclic analogues of adenine nucleotides, containing two phosphate groups on a symmetrically branched aliphatic chain, attached at the 9-position of adenine, are moderately potent P2Y1 receptor antagonists. In this study we have varied the chain structure, to include asymmetric substitution, olefinic, and cyclopropyl groups. These antagonists inhibited the stimulation of phospholipase C in turkey erythrocyte membranes induced by 30 nM 2-MeS-ADP in the micromolar range. In the series of symmetrically branched aliphatic groups substituted with two phosphate groups, the optimal antagonist potency occurred with the 2-methylpropyl group. A 2-chloro-N6-methyladenine derivative, 2-[2-(2-chloro-6-methylaminopurin-9-yl)methyl]propane-1,3-bisoxy(diammoniumphosphate) (7), was a full antagonist at the P2Y1 receptor with an IC50 value of 0.48 μM. Esterification of one of the phosphate groups or substitution with O-acetyl greatly reduced the antagonist potency at the P2Y1 receptor. Removal of a methylene group of 7 or inclusion of an olefinic or cyclopropyl group also reduced potency. A pair of enantiomeric glycerol derivatives demonstrated a 5-fold stereoselectivity for the S-isomer. Stereoisomerically defined analogues of 7 containing a cyclopropyl group in place of the branched carbon were less potent than 7 as antagonists, with IC50 values of 2–3 μM. No agonist activity was observed for these analogues. A new rhodopsin-based molecular model of the P2Y1 receptor indicated that the optimal docked orientation of the two monophosphate moieties relative to the adenine N6 (compared to a rigid, bicyclic analogue) was consistent with the dependence of antagonist potency on chain length. The 3′-phosphate was predicted to occupy a restricted space, deeper in the binding cleft than the 5′-phosphate location. In summary, modification of the flexible spacer chain linking bisphosphate groups to the adenine moiety provided many moderately potent antagonists.
Introduction
P2 receptors, which are activated by purine and/or pyrimidine nucleotides, consist of two families: G protein-coupled receptors, termed P2Y, and ligand-gated cation channels, termed P2X. Numerous subtypes have been cloned within each family regulating a diverse range of functions in the central and peripheral nervous systems, the cardiovascular system, the endocrine system, lungs, intestines, muscle, and the immune system.1–3
The P2Y1 receptor has been cloned from chick, turkey, human, rat, and mouse.4–6 ADP induces aggregation of human platelets by acting at both P2Y1 and P2Y12 receptors.7–10 The P2Y1 receptor antagonist N6-methyl-2′-deoxyadenosine 3′,5′-bisphosphate has been shown to inhibit ADP-induced platelet aggregation.11,12 A mouse line lacking expression of the P2Y1 receptor has been reported,13,14 and the absence of the P2Y1 receptors in platelets of these mice interfered with ADP-promoted aggregation.
We have developed selective P2Y1 receptor agonists and antagonists of high affinity for use as pharmacological probes.15–20 With simplified pharmacophores we are exploring the steric and electronic constraints of the receptor binding site and the structural basis of receptor activation. Various naturally occurring bisphosphates of adenosine (e.g., adenosine 3′,5′-bisphosphate) act as competitive antagonists at the P2Y1 receptor,15 and N6-methyl and 2-chloro groups on the adenine moiety have been found to increase the potency and selectivity of the bisphosphate derivatives acting as antagonists.16 Among ribose modifications, both rigidifying (e.g., bicyclic carbocycles)19 and adding flexibility (e.g., acyclic modifications)20 have led to effective P2Y1 receptor ligands. Rigid rings in the bicyclic “methanocarba” series (Figure 1) have defined a preference at the receptor binding site for the northern (N) conformation. Compound 1 ((1R,2S,4S,5S)-1-[(phosphato)methyl]-4-(6-aminopurin-9-yl)bicyclo [3.1.0]-hexane-2-phosphate), an (N)-methanocarba analogue, is a potent agonist (EC50 = 155 nM), 120-fold more potent than the corresponding southern (S) isomer. Compound 2, the corresponding 2-Cl-N6-methyl analogue, is a potent P2Y1 receptor antagonist (IC50 = 52 nM).
Figure 1.
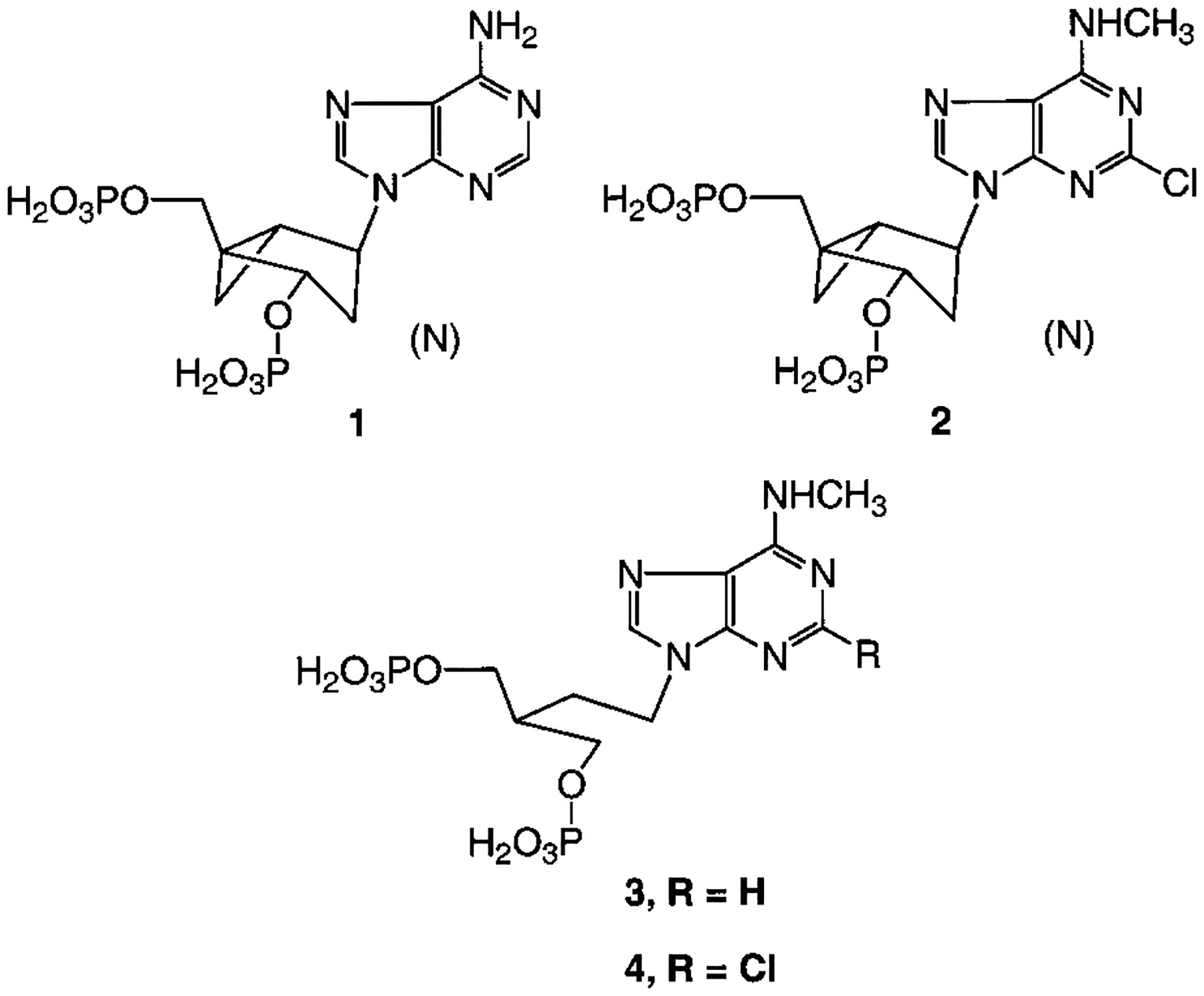
Structures of rigid and acyclic nucleotide analogues found to be receptor agonist (1) or antagonists (2-4).
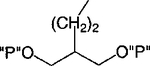
The present study explores the SAR of nucleotide analogues containing acyclic aliphatic chains, such as 3 and 4, which act as P2Y1 receptor antagonists.20 Compound 3 (2-[2-(6-methylaminopurin-9-yl)ethyl]-propane-1,3-bisphosphate) and its 2-chloro- analogue were found to be selective antagonists of the P2Y1 receptor with EC50 values of 1.60 and 0.84 μM, respectively. Compound 4 was shown to be inactive at the rat P2X1 receptor.21 We have introduced shorter chains than in the previous study20 and also elements of rigidity, such as olefinic and cyclopropyl groups. Molecular modeling of the receptor and its docked ligands, by homology to the recently reported high-resolution structure of rhodopsin,22 was carried out. In general, modifying the flexible spacer chain linking bisphosphate groups to the adenine moiety, within spatial requirements as defined by the docked positions of the phosphates relative to the adenine N6, provided many moderately potent antagonists.
Results
Chemical Synthesis.
Acyclic and cyclopropyl analogues 5–20 of adenosine bisphosphates were prepared in the ammonium salt form and tested biologically as antagonists at the P2Y1 receptor (Table 1). The nucleotide analogues were characterized by high-resolution mass spectrometry and by HPLC in two different solvent systems (Table 2).
Table 1.
In Vitro Pharmacological Data for Antagonism of Turkey Erythrocyte P2Y1 Receptors, Indicated by the Inhibition of PLC Stimulation Elicited by 30 nM 2-MeSADP, for at Least Three Separate Determinations. “P” = Phosphate
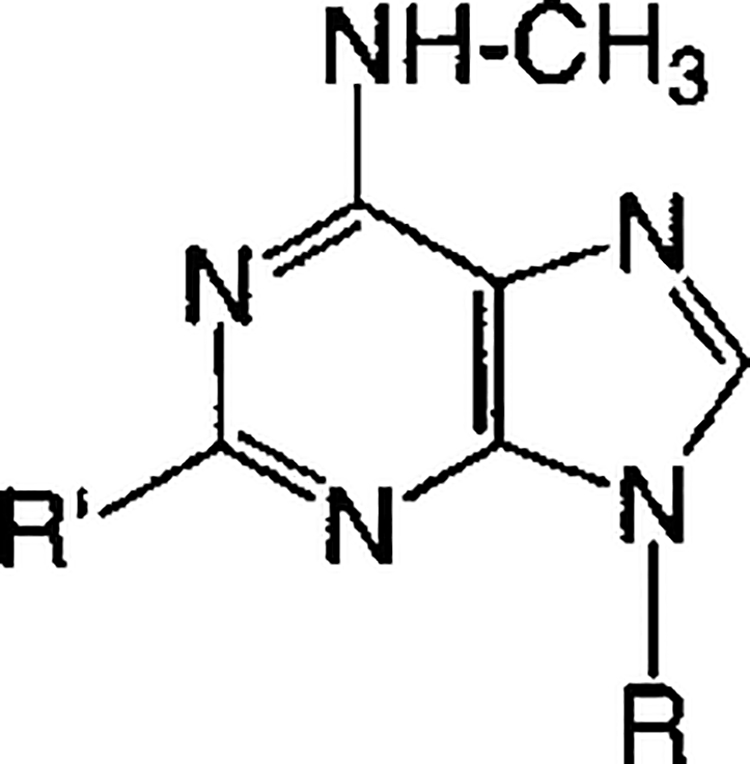
| ||||
|---|---|---|---|---|
| Compound | R = | R′ = | Antagonist Effect,a % of max. | IC50, μM (n = 3, unless noted) |
| 5 |
|
Cl | 100 | 4.56 ± 1.38 |
| 6 |
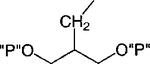
|
H | 100 | 1.09 ± 0.51 |
| 7 |
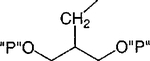
|
Cl | 100 | 0.48 ±0.13 |
| 8 |
|
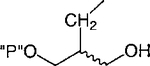
Cl | 68 ±7 | 22.6+ 14.6 |
| 9 |
|
Cl | 78 ±7 | 31 + 11 |
| 10 |
|
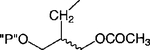
Cl | 65 ±8 | 37.6 + 12.6 |
| 11 |
|
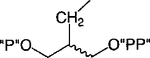
Cl | 100 | 0.23 ±0.10 |
| 3 |
|
H | 100 | 1.60 ± 0.47 |
| 4 |
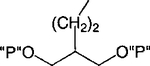
|
Cl | 99 ± 1 | 0.84 ±0.13 |
| 12 |
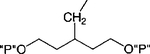
|
Cl | 100 | 2.04 ± 0.94 (4) |
| 13 |
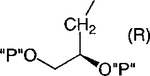
|
Cl | 100 | 7.86 ±3.13 |
| 14 |
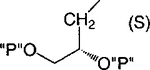
|
Cl | 100 | 1.47 ± 0.43 |
| 15 |
|
Cl | 100 | 2.10 ±0.97 |
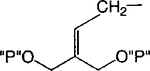
| 16 |
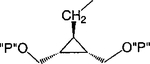
|
Cl | 100 | 1.74 ± 0.80 |
| 17 |
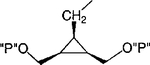
|
Cl | 100 | 2.17 ±0.85 |
| 18 |
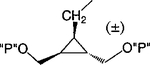
|
Cl | 100 | 2.70 ± 0.66 |
| 19 |
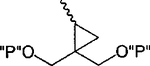
|
Cl | 100 | 2.36 ± 0.63 |
| 20 |
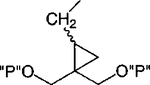
|
Cl | 100 | 1.90 ±0.24 |
At 100 μM, none of the acyclic adenosine bisphosphate nucleotide analogues displayed agonist activity at the P2Y1 receptor in turkey erythrocyte membranes.
Table 2.
Synthetic Data for Nucleotide Derivatives, Including Structural Verification Using High Resolution Mass Spectroscopy and Purity Verification Using HPLC
| no. | formula | FAB (M − H+) | HPLC (rt; min)a | yield (%)b | ||
|---|---|---|---|---|---|---|
| calcd | found | system A (purity %) | system B (purity %) | |||
| 5 | C9H13O8N5ClP2 | 415.9928 | 415.9940 | 4.981 (97) | 8.900 (98) | 63 |
| 6 | C10H16O8N5P2 | 396.0474 | 396.0482 | 7.779 (99) | 6.639 (98) | 70 |
| 7 | C10H15O8N5ClP2 | 430.0084 | 430.0094 | 6.655 (95) | 6.453 (97) | 41 |
| 8 | C10H14O5N5ClP | 350.0421 | 350.0423 | 8.963 (97) | 2.942 (99) | 79 |
| 9 | C12H19O8N5ClP2 | 458.0397 | 458.0416 | 13.579 (95) | 5.534 (99) | 76 |
| 10 | C12H16O6N5ClP | 392.0527 | 392.0517 | 7.452 (99) | 4.206 (99) | 59 |
| 11 | C10H16O11N5ClP3 | 509.9748 | 509.9751 | 6.062 (99) | 7.970 (96) | 72 |
| 12 | C12H19O8N5ClP2 | 458.0397 | 458.0385 | 7.865 (99) | 6.205 (100) | 69 |
| 13 | C9H13O8N5ClP2 | 415.9928 | 415.9945 | 5.960 (100) | 8.466 (99) | 44 |
| 14 | C9H13O8N5ClP2 | 415.9928 | 415.9938 | 5.314 (99) | 7.610 (100) | 55 |
| 15 | C11H15O8N5ClP2 | 442.0084 | 442.0086 | 5.279 (99) | 8.565 (99) | 63 |
| 16 | C12H19O8N5ClP2 (FAB positive) | 458.0397 | 458.0392 | 10.648 (96) | 8.564 (98) | 77 |
| 17 | C12H19O8N5ClP2 (FAB positive) | 458.0397 | 458.0415 | 10.391 (99) | 8.457 (99) | 57 |
| 18 | C12H17O8N5ClP2 | 456.0241 | 456.0256 | 7.882 (100) | 6.176 (100) | 71 |
| 19 | C11H15O8N5ClP2 | 442.0084 | 442.0089 | 5.036 (96) | 8.592 (98) | 61 |
| 20 | C12H17O8N5ClP2 | 456.0241 | 456.0251 | 7.211 (98) | 6.522 (100) | 55 |
Purity of each derivative was ≥95%, as determined using HPLC with two different mobile phases. System A: gradient of 0.1 M TEAA/CH3CN from 95/5 to 40/60. System B: gradient of 5 mM TBAP/CH3CN from 80/20 to 40/60.
The percent yields refer to yield for deprotection sequence.
The synthesis of these nucleotide analogues and the corresponding nucleoside precursors are shown in Schemes 1–10. Symmetrical (Schemes 1, 2, and 4) and chiral acyclic aliphatic (Scheme 5), olefinic (Scheme 6), and cyclopropyl moieties (Schemes 7–10) were included. In general, introduction of the 2-chloro-N6-methyl- (or 2-H) adenine moiety was accomplished either by direct alkylation of a mesylate with 2-chloro-N6-methylaminopurine or by a Mitsunobu reaction utilizing 2,6-dichloropurine, followed by displacement of 6-chloro with methylamine. During the displacement, the diols were protected by silylation (e.g., TBS, tert-butyldimethylsilyl) or masked as a precursor olefin (Scheme 4).
Scheme 1.
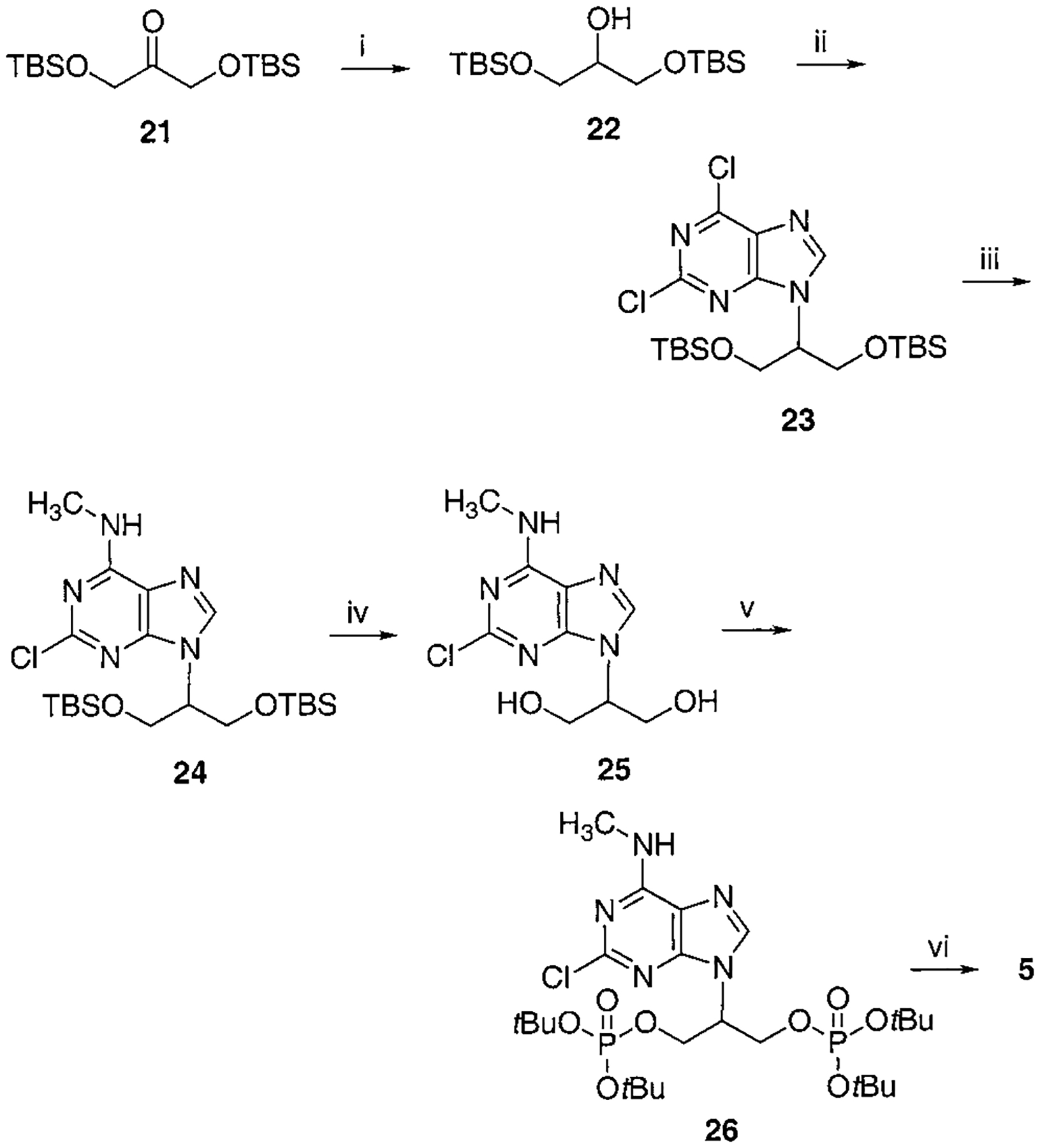
Synthesis of Isopropyl Bisphosphate, Compound 5a
a Reaction conditions: (i) NaBH4, EtOH, 0 °C, 30 min, 91%; (ii) 2,6-dichloropurine, DEAD, PPh3, THF, rt, overnight, 68%; (iii) CH3NH2, THF, rt, 4 h, 88%; (iv) Dowex 50x-8 200, MeOH, reflux, overnight, 75%; (v) Et2NP(OtBu)2, tetrazole, THF, rt, 20 min, then MCPBA, 73%; (vi) Dowex 50x-8 200, MeOH:H2O = 1:1, 80 °C, 16 h, 63%.
Scheme 10.
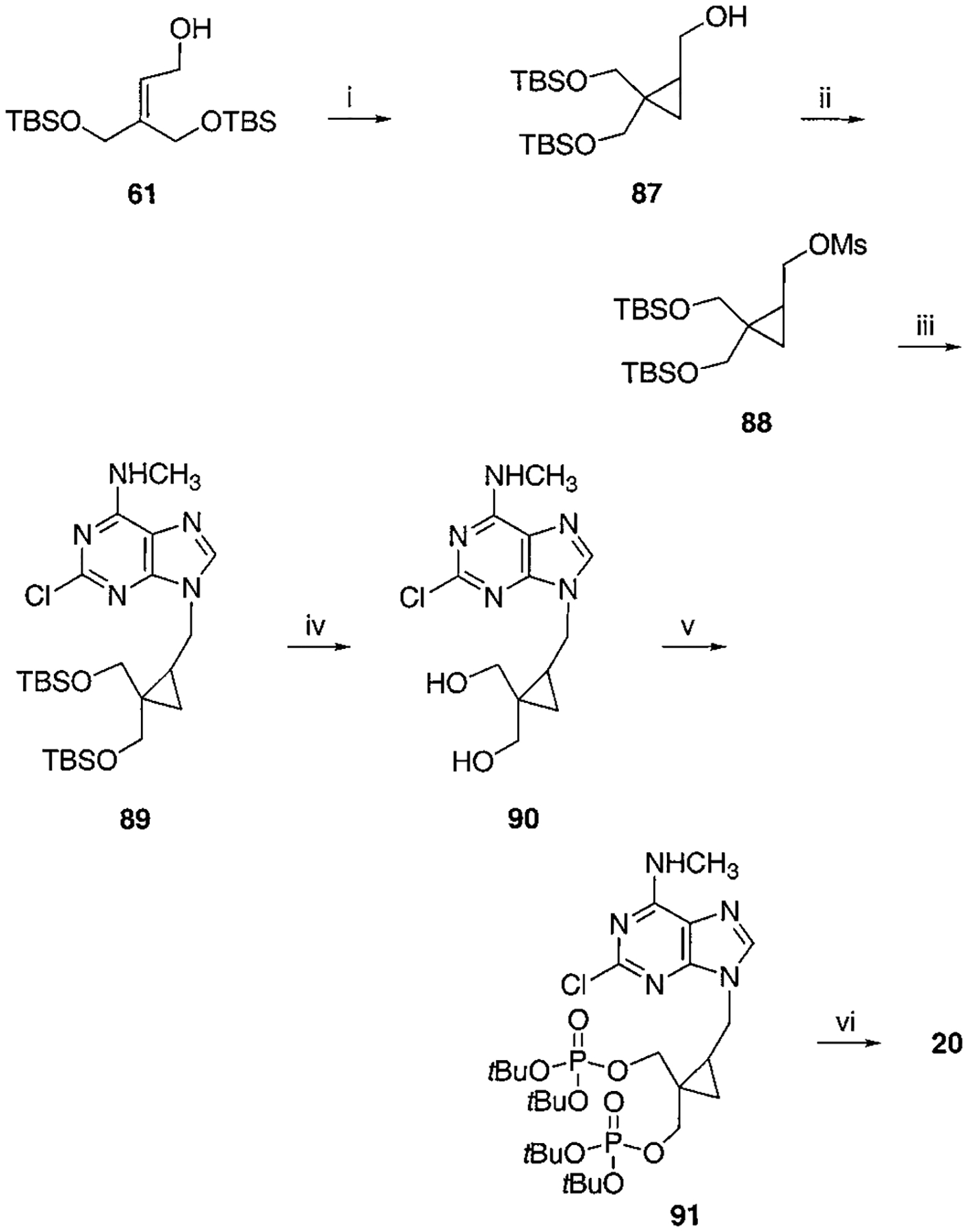
Synthesis of Cyclopropyl Bisphosphate Derivative, Compound 20a
a Reaction conditions: (i) Et2Zn, CH2I2, CH2Cl2, rt, overnight, 96%; (ii) MsCl, TEA, CH2Cl2, rt, 1 h, 93%; (iii) 2-chloro-N6-methylaminopurine, K2CO3, 18-crown-6, DMF, 60 °C, overnight, 62%; (iv) AcOH:THF:H2O = 1:2:2, 40 °C, overnight, 55%; (v) Et2NP(OtBu)2, tetrazole, THF, rt, 30 min then MCPBA, 40%; (v) 5% TFA:CH2Cl2:THF = 10:20:1, rt, 30 min, 55%.
Scheme 2.
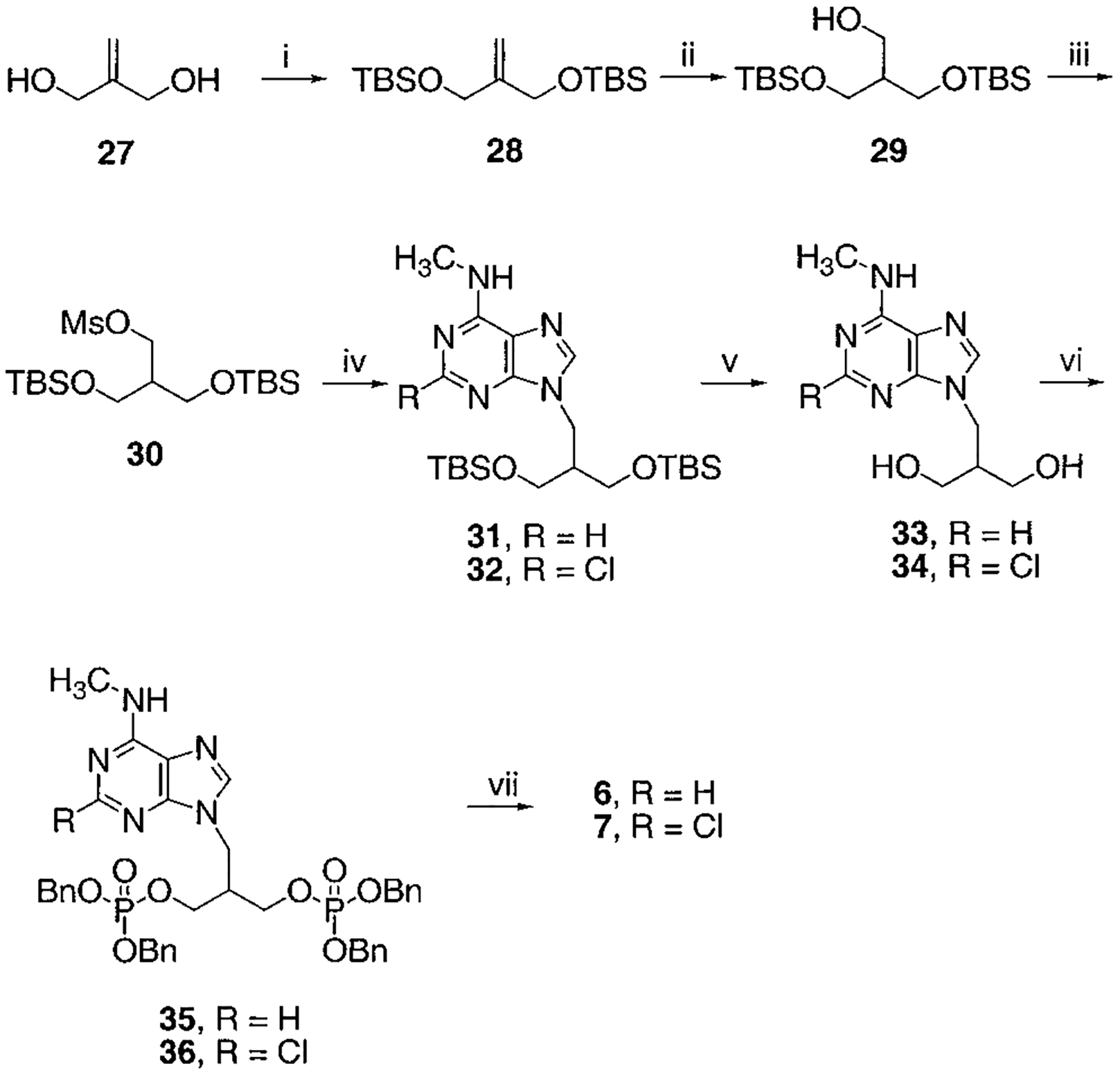
Synthesis of Isobutyl Bisphosphates, Compounds 6 and 7a
a Reaction conditions: (i) TBSCl, TEA, DMAP, CH2Cl2, rt, overnight, 99%; (ii) BH3, THF, rt, overnight, then H2O2, NaOH, 60%; (iii) MsCl, TEA, CH2Cl2, rt, 1 h; (iv) 2-chloro- N6-methylaminopurine (for 32) or N6-methylaminopurine (for 31), K2CO3, 18-crown-6, DMF, 60% two-step yield for 31, 72% two-step yield for 32; (v) AcOH:H2O:THF = 3:2:2, rt, 2 d, 79% for 33, 93% for 34; (vi) tetrabenzylpyrophosphate, NaH, THF, rt, 1 h, 81% for 35, 53% for 36; (vii) BCl3, CH2Cl2, 5 °C, 2 d, 75% for 6, 41% for 7.
Scheme 4.
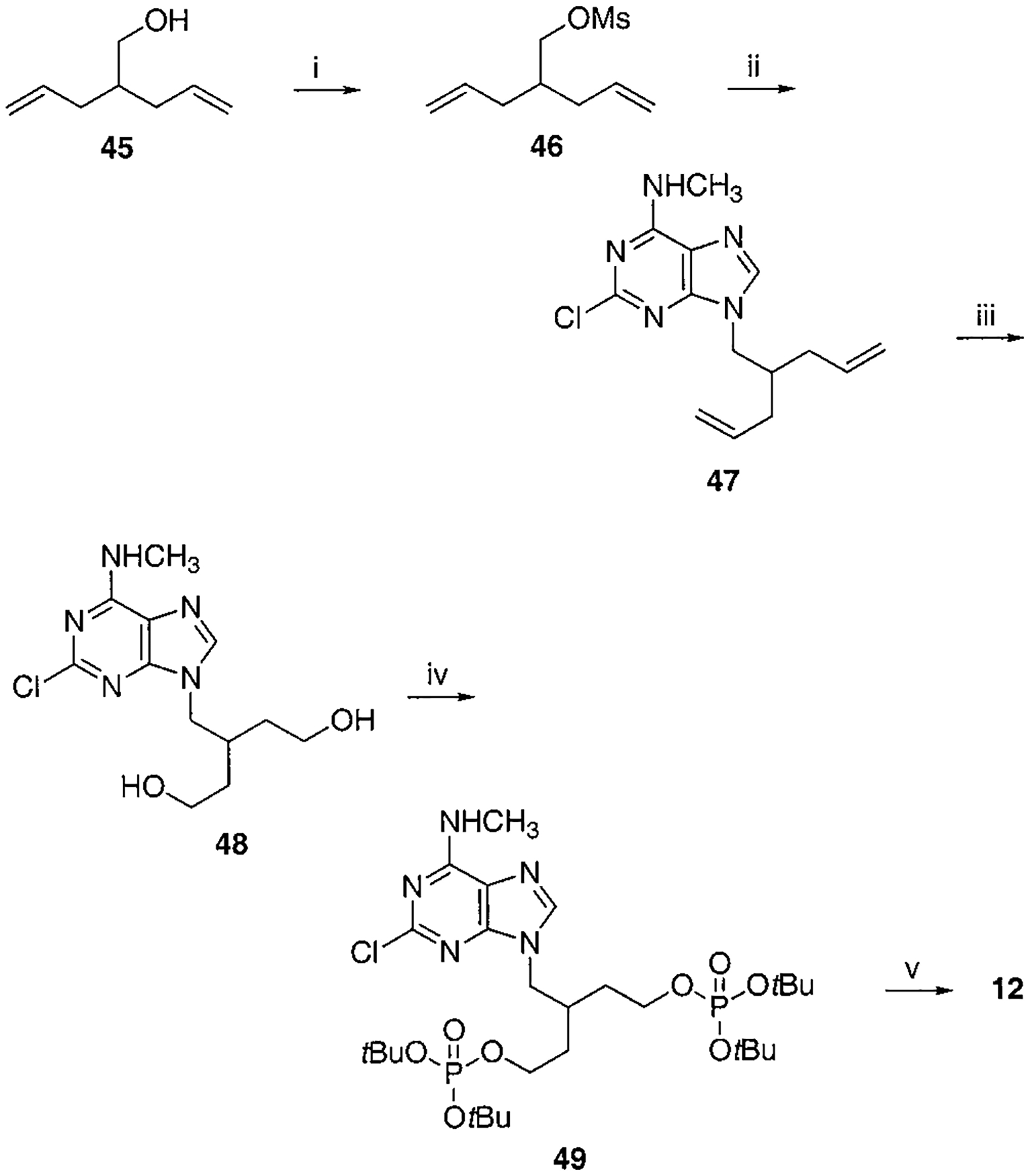
Synthesis of Compound 12a
a Reaction conditions: (i) MsCl, TEA, CH2Cl2, rt, 1 h; (ii) 2-chloro-N6-methylaminopurine, K2CO3, 18-crown-6, DMF, 60–70 °C, overnight, 60% two-step yield; (iii) (a) OsO4, NaIO4, acetone/H2O = 4:1, rt, 4 h, (b) NaBH4, EtOH, 0 °C, 2 h, 32% two-step yield; (iv) Et2NP(OtBu)2, tetrazole, THF, rt, 1 h then MCPBA, 73%; (v) 5% TFA in CH2Cl2, rt, 30 min, 69%.
Scheme 5.
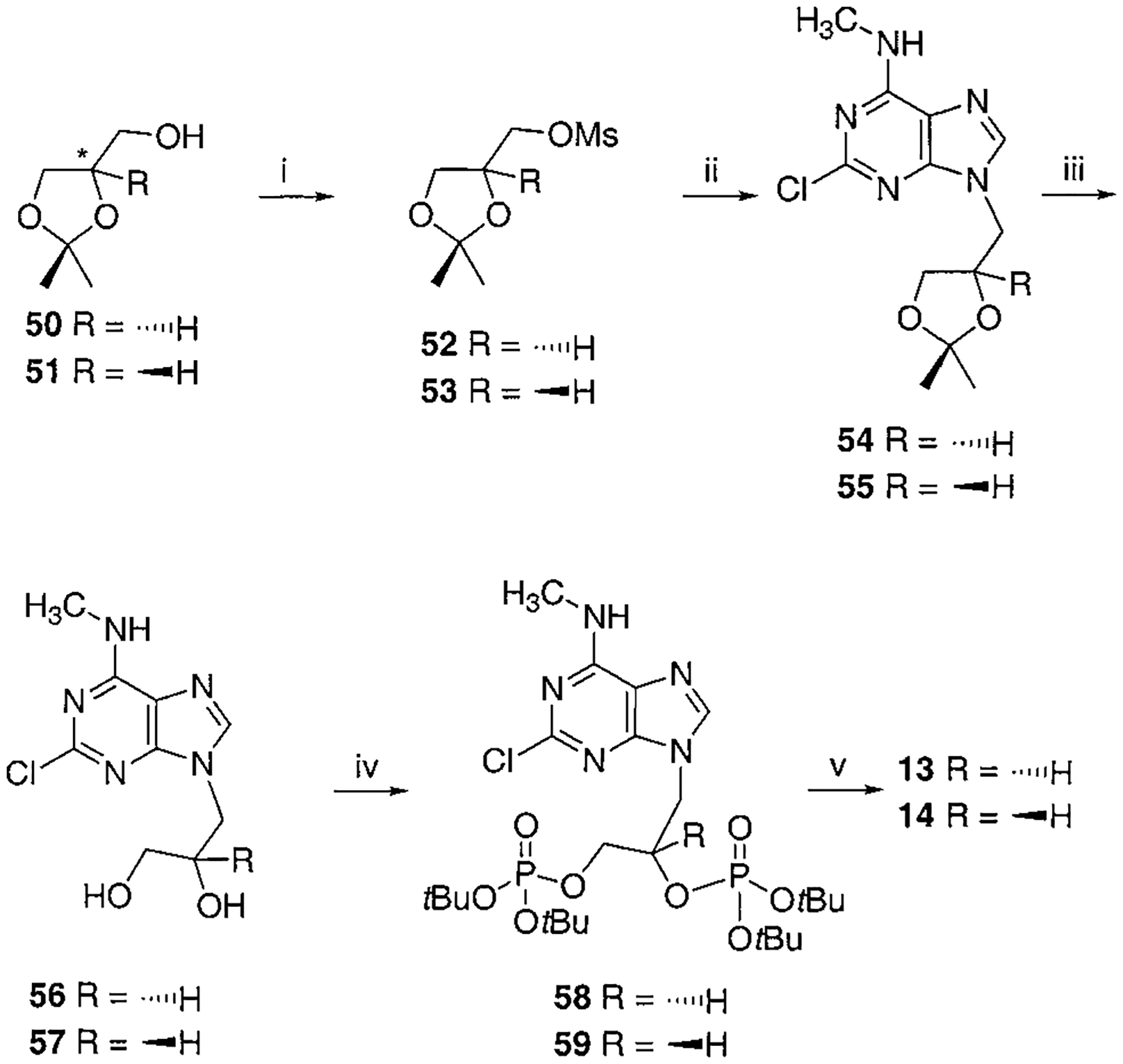
Synthesis of R- and S-Glyceryl Bisphosphate Derivatives, Compounds 13 and 14a
a Reaction conditions: (i) MsCl, TEA, CH2Cl2, rt, 1 h; (ii) 2-chloro-N6-methylaminopurine, K2CO3, 18-crown-6, DMF, 80–90 °C, 12 h, 50% two-steps yield for 54, 54% two-steps yield for 55; (iii) Dowex 50x-8 200, MeOH, 40 °C, 4 h, 88% for 56, 85% for 57; (iv) Et2NP(OtBu)2, tetrazole, THF, rt, overnight then MCPBA, 56% for 58, 69% for 59; (v) 5% TFA in CH2Cl2, rt, 1 h, 44% for 13, 55% for 14.
Scheme 6.
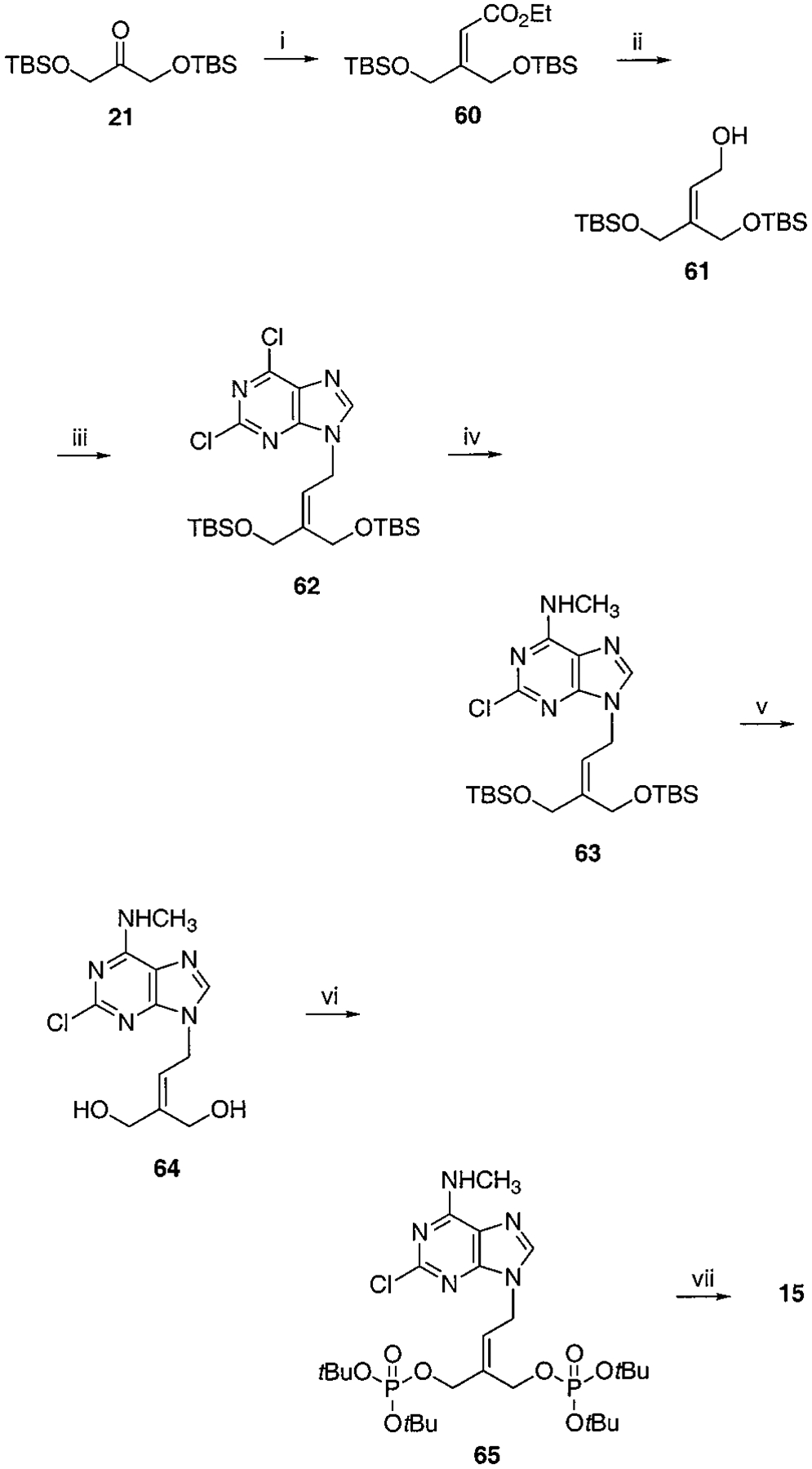
Synthesis of Isopentenyl Bisphosphate, Compound 10a
a Reaction conditions: (i) EtO2CCH2P(O)(OEt)2, NaH, THF, rt, 2 h, 85%; (ii) DIBAL, toluene, −78 °C to rt, 1 h, 74%; (iii) 2,6-chloropurine, DEAD, PPh3, THF, rt, 2 h, 57%; (iv) CH3NH2, THF, rt, 6 h, 85%; (v) AcOH:H2O:THF = 1:2:2, 40 °C, 5 h, 86%; (vi) Et2NP(OtBu)2, tetrazole, THF, rt, 1 h then MCPBA, 35%; (v) Dowex 50x-8 200, MeOH/H2O = 1:1, 50 °C, 3 h, 63%.
Scheme 7.
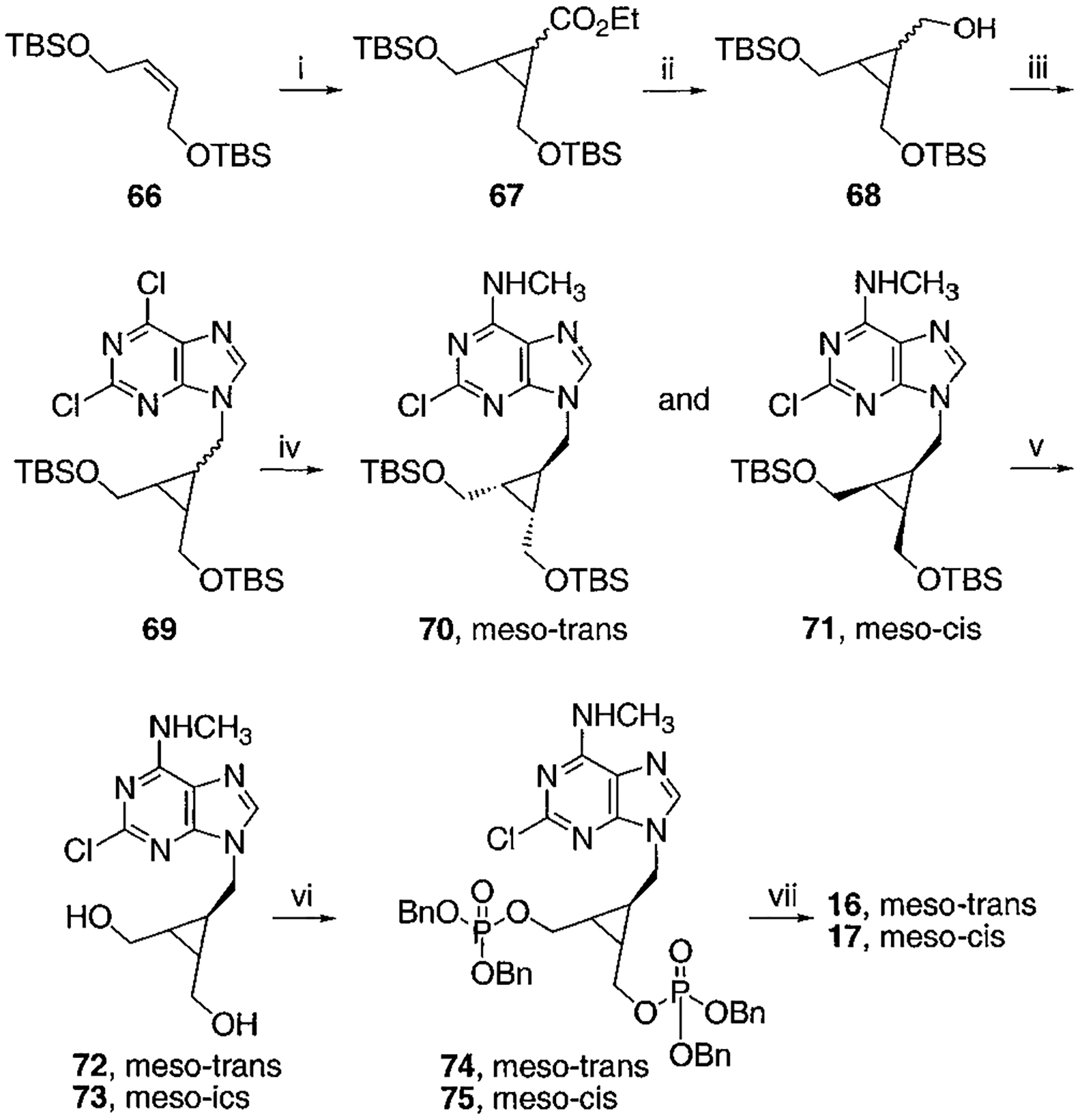
Synthesis of meso-cis- and meso-trans-Cyclopropyl Bisphosphate Derivatives, Compounds 16 and 17a
a Reaction conditions: (i) N2CH2CO2Et, Rh2(OAc)4, CH2Cl2, rt, 17 h, 61%; (ii) LiBH4, ethyl ether, toluene, 100 °C, 1 h, 93%; (iii) 2,6-chloropurine, DEAD, PPh3, THF, rt, overnight, 59%; (iv) CH3NH2, THF, rt, 5 min then separation, total 75%, 70:71 = 2.5: 1; (v) AcOH:THF:H2O = 3:1:1, rt, 4 h; (vi) ((BnO)2(O)P)2O, LDA, THF, −78 °C to rt, overnight, 46% two-step yield for 74, 33% two-step yield for 75; (vii) BCl3, CH2Cl2, −78 °C for 1 h and 6–7 °C for 2 d, 77% for 16, 57% for 17.
Subsequently, phosphorylation of each nucleoside analogue (yields in Table 2) was achieved in two steps. First, benzyl- or tert-butyl-protected phosphate groups were incorporated, thus facilitating product purification at this intermediate stage. Benzyl phosphates were introduced by the treatment of diols with NaH and tetrabenzylpyrophosphate (Schemes 2, 3B, and 7).20 The yields for this method of phosphorylation were variable, being limited by the solubility of the diols in THF. In such cases, use of DMF as solvent failed to give the desired product. Alternatively, bisphosphates were made via the tert-butyl phosphate esters,23 which were synthesized by treatment of diols with di-tert-butyl diethylphosphoramidite and tetrazole in THF, followed by oxidation with m-chloroperoxybenzoic acid (MCPBA, Schemes 1, 3A, 4–6, and 8–10). The two-step synthesis of tert-butyl phosphates had the following advantage over the tetrabenzylpyrophosphate method: limited solubility of the diols in THF could be overcome since these conditions allowed use of excess phosphoramidite and tetrazole reagents with overnight stirring, which either slowly or rapidly resulted in a homogeneous reaction mixture without significant side reactions.
Scheme 3.
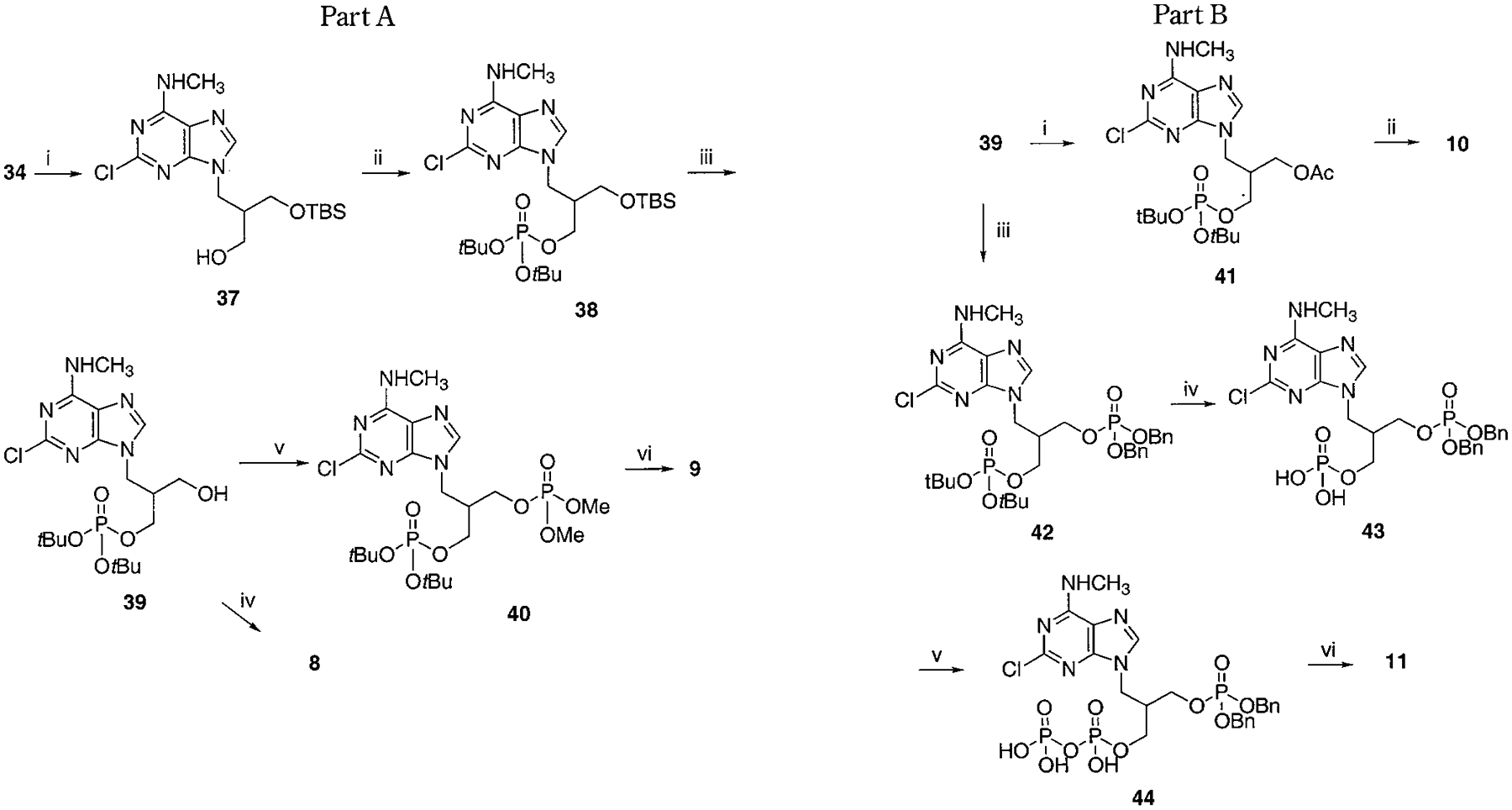
Synthesis of Asymmetrically Phosphorylated Isobutyl Derivatives, 8–11a
a Part A, reaction conditions: (i) TBSCl, imidazole, DMF, rt, overnight, 38%; (ii) Et2NP(OtBu)2, tetrazole, THF, rt, 20 min, then MCPBA, 83%; (iii) TBAF, THF, rt, 10 min, 76%; (iv) 5% TFA in CH2Cl2, rt, 2 h, 79%; (v) (MeO)2P(O)Cl, NaH, THF, rt, 1 h, 31%; (vi) 5% TFA in CH2Cl2, rt, 2 h, 76%. Part 3B, reaction conditions: (i) Ac2O, TEA, CH2Cl2, rt, 2 h, 96%; (ii) 5% TFA in CH2Cl2, rt, 30 min, 59%; (iii) iPr2NP(OBn)2, tetrazole, THF, rt, 20 min, then MCPBA, 45%; (iv) 5% TFA in CH2Cl2, rt, 30 min, 89%; (v) C(O)(imi)2, NEt3•H3PO4, DMF, rt, 3 d, 74%; (vi) TMSBr, rt, 10 min, 72%.
Scheme 8.
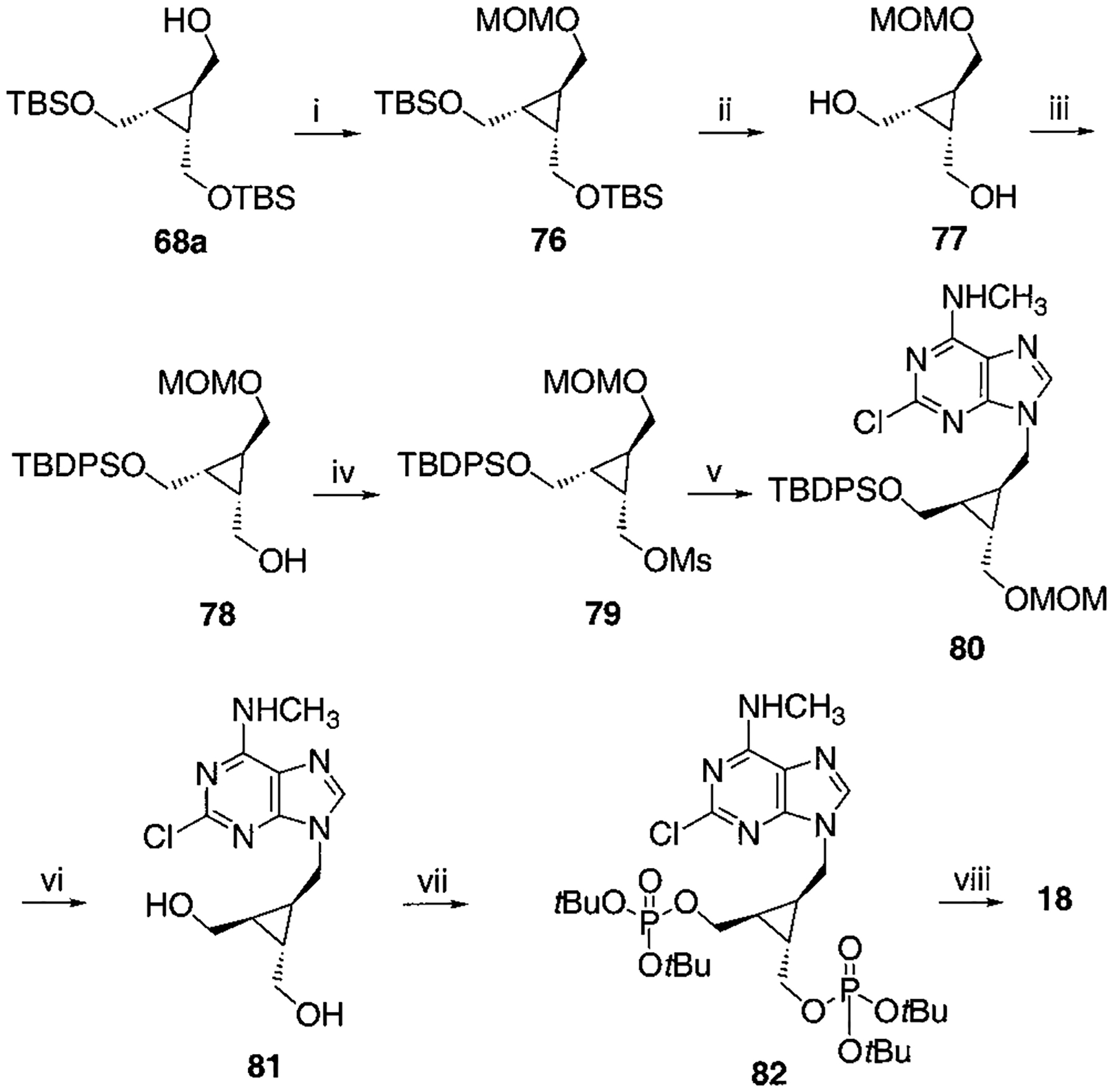
Synthesis of Racemic Cyclopropyl Bisphosphate Derivative, Compound 18a
a Reaction conditions: (i) CH3OCH2Cl, Hünig base, CH2Cl2, rt, 4 h, 93%; (ii) TBAF, THF, rt, 3 h, 95%; (iii) TBDPSCl (1 equiv), DMAP (1 equiv), CH2Cl2, rt, 3 d, 70%; (iv) MsCl, TEA, CH2Cl2, rt, 1 h, 95%; (v) 2-chloro-N6-methylaminopurine, K2CO3, DMF, 70 °C, overnight, 71%; (vi) c-HCl:MeOH = 1:3, 80 °C, 4 h, 65%; (vii) Et2NP(OtBu)2, tetrazole, THF, rt, 20 min then MCPBA, 62%; (viii) 5% TFA in CH2Cl2, rt, 30 min, 71%.
To synthesize compound 11, containing both monophosphate and diphosphate groups, it was necessary to distinguish between the two diols present. This was accomplished through the synthesis of 42, containing di-tert-butyl- and dibenzyl-protected phosphate groups (Schemes 3A,B), which was smoothly achieved in four steps from 34. The tert-butyl group was readily deprotected selectively in the presence of a dibenzyl phosphate ester by treatment with 5% trifluoroacetic acid in methylene chloride. The conversion of monophosphate of 43 to the diphosphate in moderate yield utilized a phosphoryl imidazolide intermediate, which reacted with triethylammonium phosphate.24 Preparation of analogues 16 and 17 (Scheme 7) required the synthesis of an intermediate, 68, using a modification of a literature procedure, which also permitted the assignment of the stereochemical identity of each isomer.25 meso-cis- (minor), 71, and meso-trans-cyclopropylmethyl (major), 70, isomers were separated after condensation of the mesylate with adenine moieties, using silica gel column chromatography. The preparation of a racemic cyclopropylmethyl moiety, leading to 18, was accomplished through a mesylate displacement (Scheme 8), utilizing TBDPS- (tert-butyl-diphenylsilyl) and MOM-(methoxymethyl) protected intermediates. Another cyclopropyl derivative, 19, was synthesized through displacement of a bromo diester, 83, with ring closure, followed by diester reduction to generate the diol group (Scheme 9).26
Scheme 9.
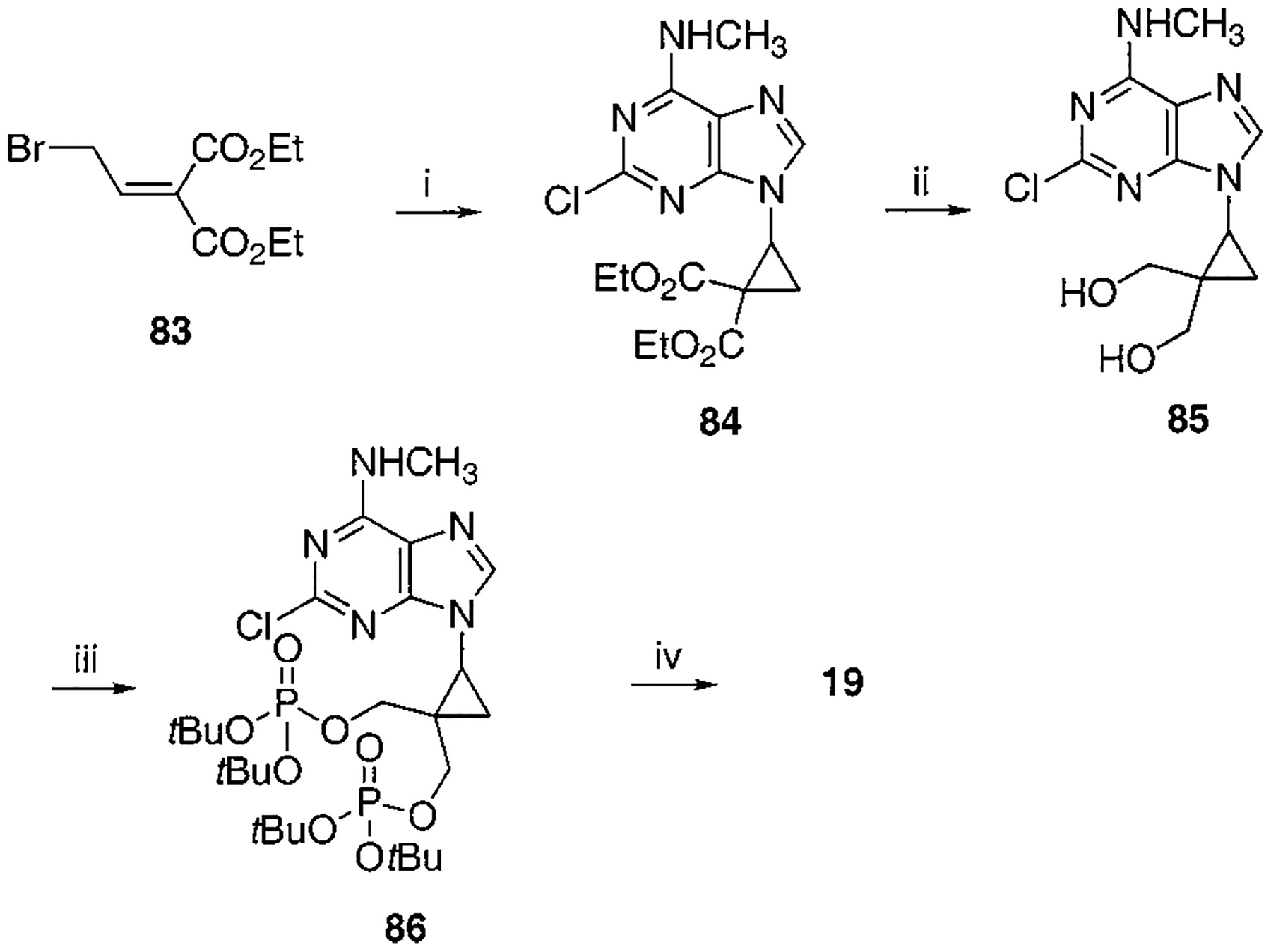
Synthesis of Cyclopropyl Bisphosphate Derivative, Compound 19a
a Reaction conditions: (i) 2-chloro-N6-methylaminopurine, K2CO3, DMF, 35 °C, 2 d, 41%; (ii) DIBAL, CH2Cl2, −78 °C to rt, 2 h, 70%; (iii) Et2NP(OtBu)2, tetrazole, THF, rt, 20 min then MCPBA, 85%; (iv) 5% TFA in CH2Cl2, rt, 20 min, 61%.
Biological Activity.
Adenine nucleotides activate a P2Y1 receptor in turkey erythrocyte membranes to markedly stimulate inositol lipid hydrolysis by phospholipase C.27–29 The analogues were screened for activation or inhibition of P2Y1 receptor promoted-effects on phospholipase C, as indicated by the accumulation of inositol phosphates in membranes isolated from [3H]inositol-labeled turkey erythrocytes (Table 1). As in our previous study,20 none of the acyclic adenosine bisphosphate nucleotide analogues displayed agonist activity at the P2Y1 receptor in turkey erythrocyte membranes. Concentration–response curves were obtained for each compound for inhibition of the effects of a fixed concentration (30 nM) of the agonist 2-MeSADP.
An acyclic 2-chloro-N6-methyladenine derivative, 7, containing a symmetric isobutyl bisphosphate moiety, was a potent P2Y1 receptor antagonist, with an IC50 value of 0.48 μM. The corresponding 2-H derivative, 6, displayed 2-fold weaker antagonist potency. Thus, antagonists 6 and 7 were roughly twice as potent as the next higher homologues, 3 and 4, respectively. The lower homologue, the isopropyl derivative, 5, was nearly an order of magnitude less potent than 7.
Replacement of one of the phosphate groups of 7 with an uncharged moiety (i.e., racemic alcohol, 8, phosphotriester, 9, or O-acetyl ester, 10) resulted in a 500- to 1000-fold loss of potency. Replacement of a phosphate group with diphosphate, 11, increased potency.
Methylene homologation at the phosphate ester positions of 7, resulting in compound 12, decreased potency at the P2Y1 receptor 4-fold. Removal of a phosphate-linked methylene group of 7, resulting in an enantiomeric pair of glycerol derivatives, 13 and 14, demonstrated a 5-fold stereoselectivity of binding at the P2Y1 receptor. The S-isomer, 14, was favored with an IC50 value of 1.47 μM. Introduction of an olefinic group, i.e., compound 15, decreased antagonist potency compared to 7 at the P2Y1 receptor 4-fold. meso-trans-, 16, and meso-cis-, 17, isomers of a cyclopropylmethyl derivative (two of the four possible stereoisomers) failed to display stereoselectivity of binding, and the IC50 values were approximately 2 μM. The racemate of the remaining two isomers, 18, as well as 19 and 20, had a similar potency. Thus, the antagonistic potency among cyclopropyl derivatives was nearly invariable.
Molecular Modeling.
To better understand the affinities of the acyclic bisphosphate adenine analogues and, in particular, the correspondence between the receptor bound conformers of these compounds and the rigid, high affinity analogues, we carried out molecular modeling of the ligand–receptor complexes. The human P2Y1 receptor model used for this purpose included the seven transmembrane helical domains (TMs) and the second extracellular loop (EL2) and was built in homology to the recently published X-ray structure of bovine rhodopsin.22 In this X-ray structure EL2, but not EL1 and 3, appears to interact directly with retinal.
Using this construct, docking experiments were carried out to establish a binding site for both prototypical agonists ATP or ADP and an antagonist, 2.19 These experiments were guided by our previous modeling experiments,30,31 in which parts of the binding pocket accommodating the adenine moiety have been identified, by mutagenesis studies,30 and by possible modes of superposition of the two ligand structures.
The energy-optimized structures of P2Y1 receptor complexes with ATP and 2 were practically superimposable with respect to the protein backbone atoms. The structural correspondence of the two ligands (Figure 2) indicated that in both cases the adenine moieties occupied the same binding subsite. According to this model, the 5′-phosphate of 2 overlapped the β-phosphate moiety of ATP, and the 3′-phosphate of 2 had no structural equivalent in the bound ATP structure. As in the previous model,30 neither the ribose ring of ATP nor the methanocarba ring of 2 participated in binding interactions with the receptor.
Figure 2.
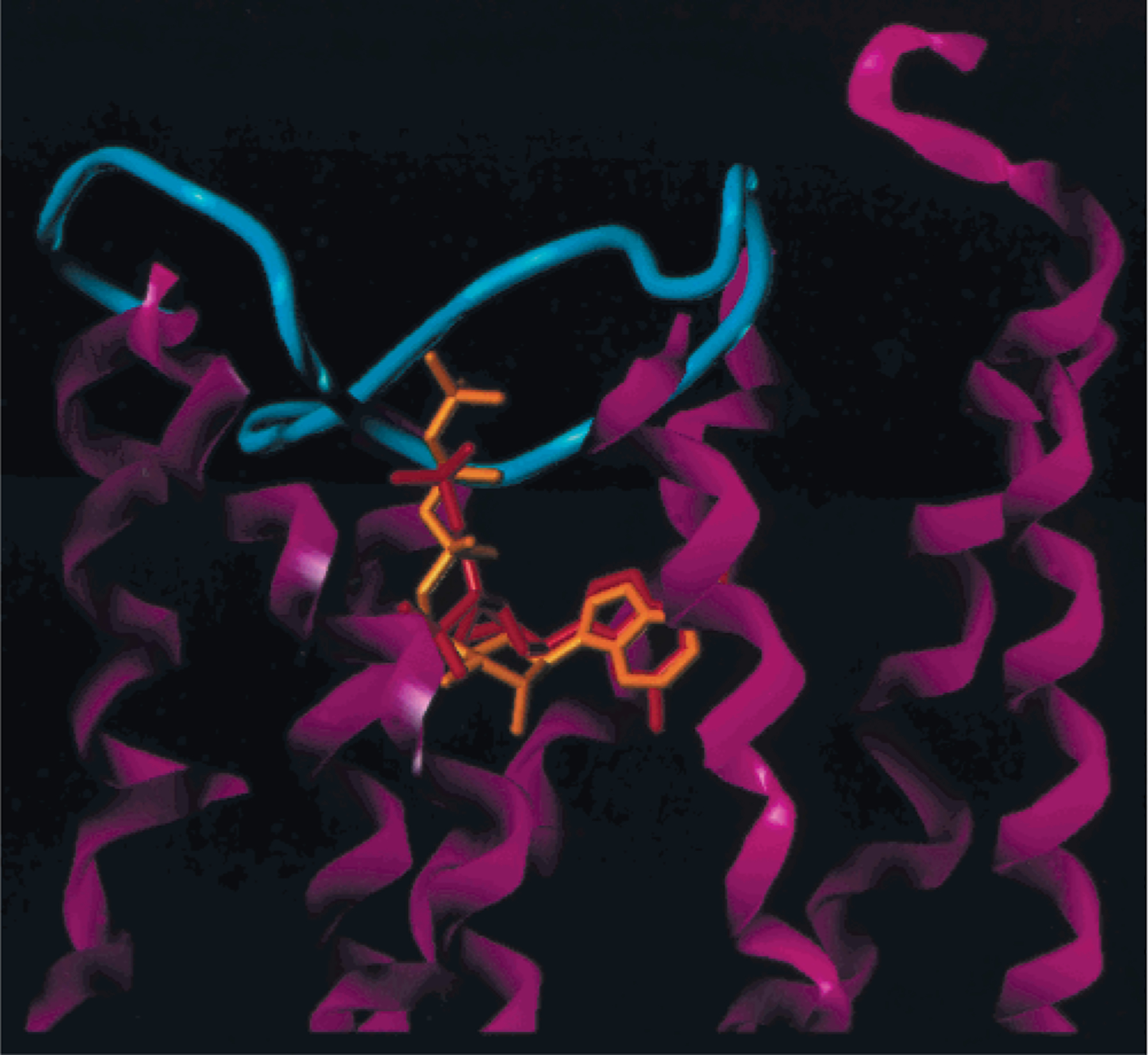
Structural correspondence of ATP (orange) and 2 (red), superimposed in the putative receptor binding site. TMs (magenta) and EL2 (cyan), joining TM4 and 5, are shown. The adenine moieties occupy the same binding subsite.
Using the receptor-2 complex as a template, the various acyclic bisphosphates were docked into the binding site and the models reoptimized. The structural correspondence between the receptor-bound conformer of 4 and that of 2 is shown in Figure 3. This figure also depicts the various residues involved in specific interactions with the ligands, including Lys280 and Arg310, which interacted with both phosphate groups of the ligands,30 and other residues. The residues Lys196, Tyr306, and Asp204 apparently were involved in polar interactions with the phosphate group corresponding to the 5′-phosphate of 2. The residues Phe226, Tyr273, and His277 formed a sterically restricted pocket to accommodate the 3′-phosphate of 2. The residues Tyr59, Tyr 273, and Ser314 interacted with the adjacent adenine nitrogen atoms (Figure 3). The similarity in positioning of the binding elements for 2 and 4 with respect to the modeled receptor manifold (see Figure 3) was consistent with the more potent inhibition of the P2Y1 receptor by the latter (see Table 1). Furthermore, ligands homologous to 4 (having 0 to 3 methylenes adjacent to adenine N-9), representing a >100-fold span of IC50 values, displayed varying degrees of correspondence to this pharmacophoric arrangement based on 2. An overall comparison of the bound conformations of 4 and the three homologues is shown in Figure 4, while specific deviations of atoms corresponding to 3′-P, 5′-P, and 6-N of 2 are listed in Table 3.
Figure 3.
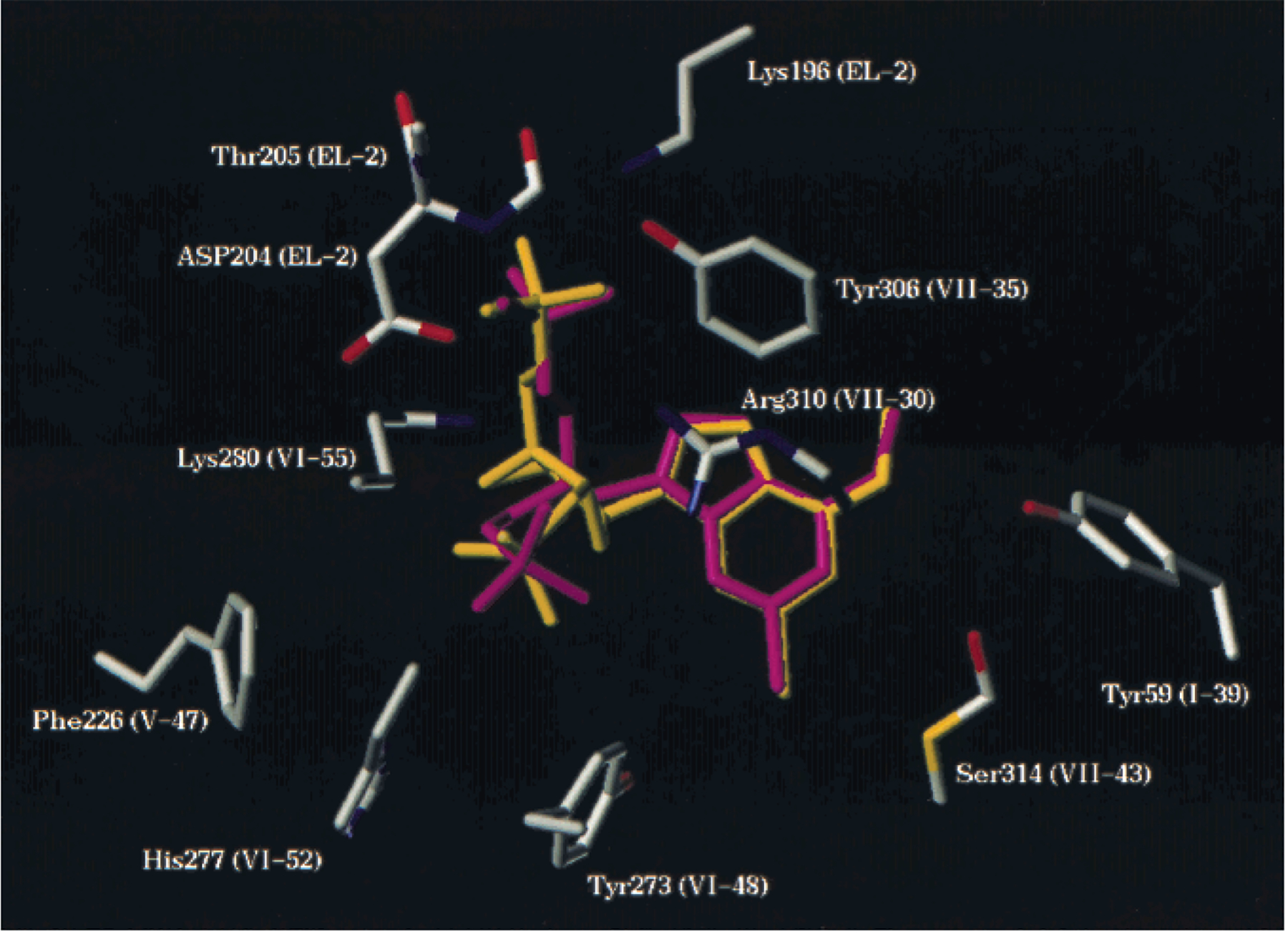
Structural correspondence between the receptor bound conformer of 4 (magenta) and that of 2 (yellow), depicting the residues involved in specific interactions with the ligands. Lys280 and Arg310 interact with both phosphate groups of the ligands.
Figure 4.
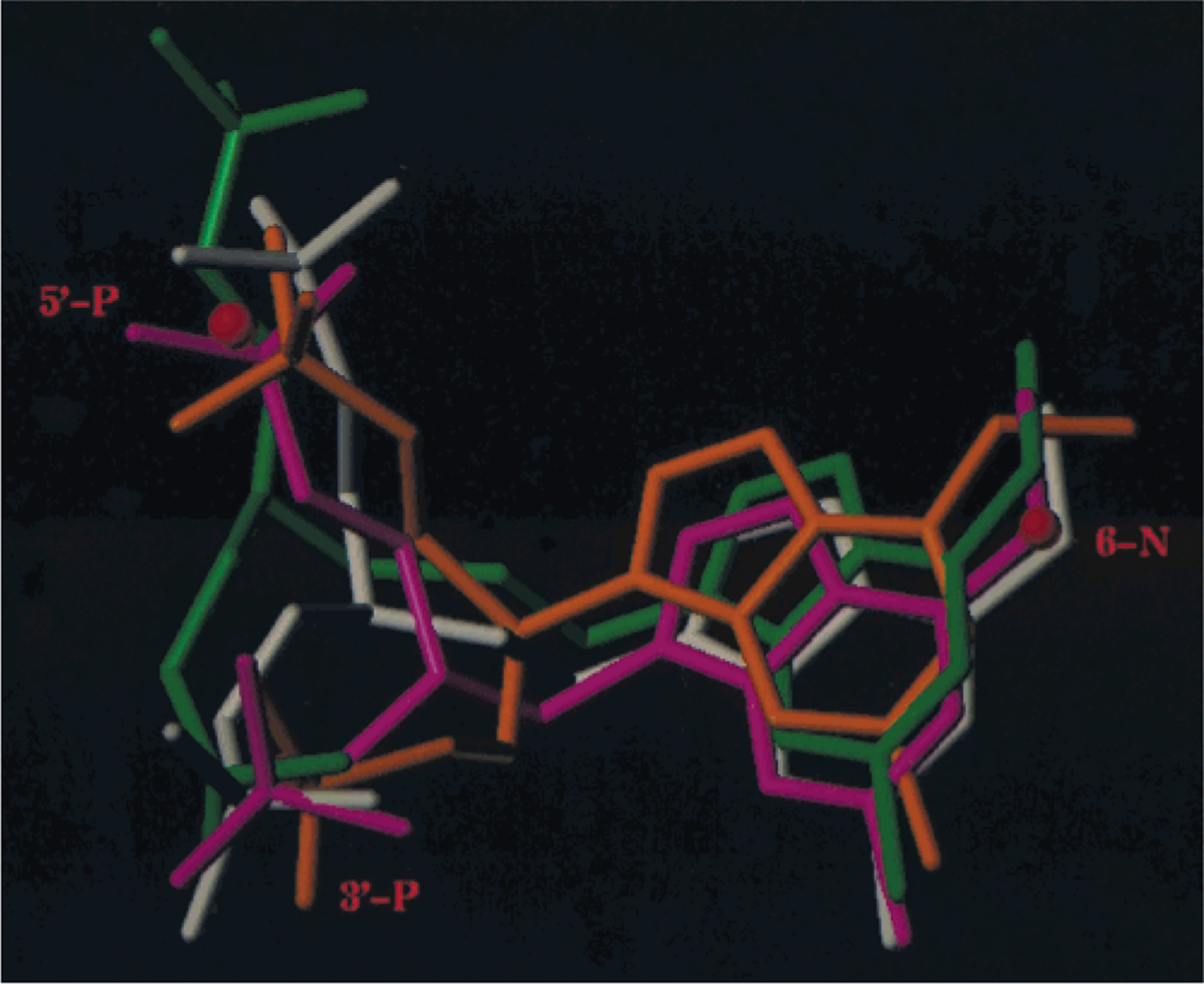
Comparison of the bound conformations of 4 (white) and three homologous derivatives, 5 (orange), 7 (magenta), and the analogue containing an n-propyl spacer 94 (green).
Table 3.
Distances in Angstroms between Pharmacophoric Points of P2Y1 Docked Antagonists and Template Compound 2a
| compd | 3′-P | 5′-P | 6-N | |
|---|---|---|---|---|
| 92 | 0.35 | 0.36 | 0.05 | |
| 93 | 0.28 | 1.88 | 0.20 | |
| 5 | 0.42 | 0.79 | 1.27 | |
| 7 | 0.38 | 0.50 | 0.27 | |
| 4 | 0.42 | 1.08 | 0.49 | |
| 94 | 0.33 | 2.22 | 0.7 | |
| 12 | 0.44 | 1.78 | 0.43 | |
| 13 | 0.45 | 0.83 | 1.24 | |
| 14 | 0.45 | 1.40 | 0.46 | |
| 15 | 0.16 | 0.42 | 0.25 | |
| 16 | 0.50 | 0.86 | 0.21 | |
| 17 | 0.27 | 1.60 | 0.30 | |
| 18 | isomer 1 | 0.78 | 0.72 | 0.50 |
| isomer 2 | 0.27 | 1.31 | 0.27 | |
| 19 | R-config | 0.10 | 0.81 | 0.15 |
| S-config | 0.15 | 0.83 | 0.21 | |
| 20 | R-config | 0.26 | 0.63 | 0.18 |
| S-config | 0.08 | 0.54 | 0.10 |
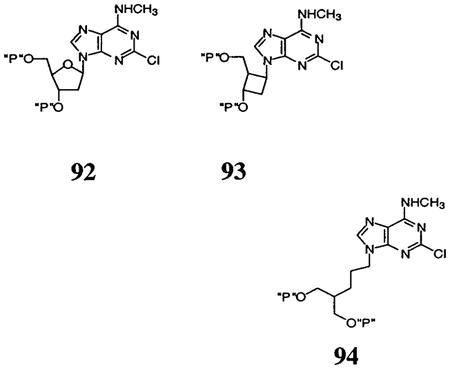
Compound 2 (MRS 2279) is (1R,2S,4S,5S)-1-[(phosphato)-methyl]-4-(2-chloro-6-methylaminopurin-9-yl)bicyclo [3.1.0]-hexane-2-phosphate. Compounds are grouped as cyclic template compounds (92, 93), acyclic bisphosphates of variable chain length (0–3 methylenes) (5, 7 (MRS 2298), 4, 94), other acyclic compounds (12-15), and cyclopropyl derivatives (16-20). Compounds 92 (2′-deoxy-2-chloro-N6-methyladenosine-3′,5′-bisphosphate (MRS 2216), IC50 0.206 μM), 93 (phosphoric acid mono-[3-(2-chloro-6-methylaminopurin-9-yl)-2-phosphonooxymethylcyclobutyl] ester (MRS 2264), IC50 0.805 μM), and 94 (2-[3-(6-methylaminopurin-9-yl)-propyl]propane-1,3-bisphosphate, IC50 >30 μM) were reported previously.19,20
Table 3 compares the receptor-bound conformations of rigid and acyclic P2Y1 antagonists. A relatively high affinity was maintained, as long as the position of a given antagonist did not deviate significantly with respect to 6-N and 3′-P of the template. In contrast, the binding pocket, including the relatively flexible EL2, provided significantly more room for the group bearing the 5′-phosphate. To further examine this suggestion, the docked conformations of three potent, cyclic P2Y1 antagonists,18,19 92 (2′-deoxy-2-chloro-N6-methyladeno-sine-3′,5′-bisphosphate), 2, and 93 (phosphoric acid mono-[3-(2-chloro-6-methylaminopurin-9-yl)-2-phosphonooxymethylcyclobutyl] ester), have been compared (Figure 5). Indeed, the requirements with respect to positioning the 5′-phosphate in the receptor appeared not to be very stringent, since the location of the corresponding phosphate of 93 deviated considerably from that of 2. Such a model is also consistent with the high activity of 11 (see Table 1), where the adenine and monophosphate binding elements could be expected to occupy receptor subsites analogous to those of 7. The flexible chain bearing the diphosphate moiety allows for its accommodation in a similar way to that of the α-and β-phosphates in ADP or ATP. The relative orientation of the three pharmacophoric elements in the template structure of 2 is presented (Figure 5). The resulting distances differ somewhat from those cited in our previous publication30 due to the refined receptor model used in the present study.
Figure 5.
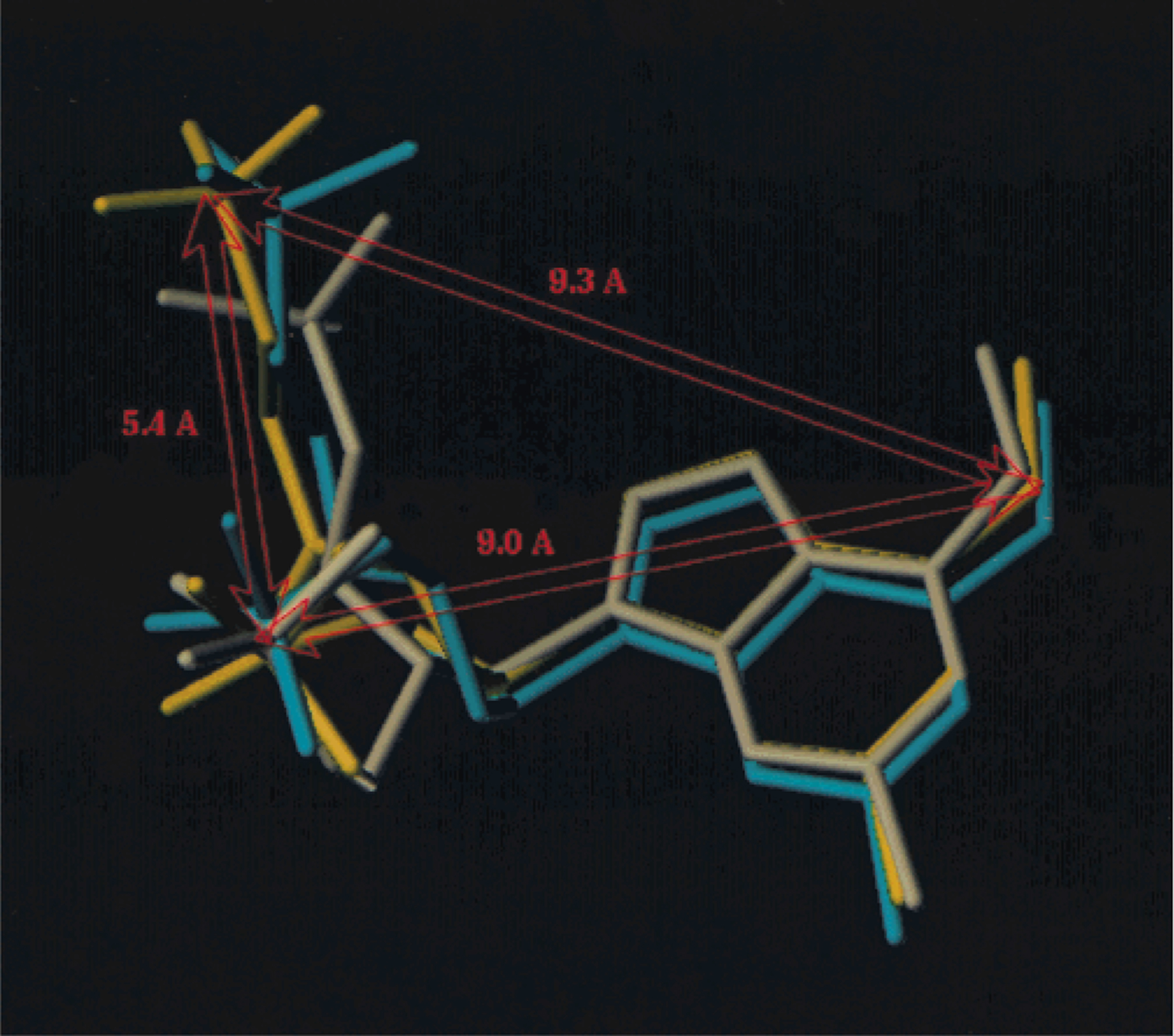
Docked conformations of three relatively rigid potent P2Y1 antagonists (see Table 3 for chemical names): 92 (cyan, a riboside), 2 (yellow), and 93 (white, a cyclobutyl analogue). Also shown is the relative orientation of the three pharmacophoric elements in the template structure of 2. The requirements with respect to positioning the 5′-phosphate in the receptor appeared not to be very stringent, since the location of the corresponding phosphate of 93 deviated considerably from that of 2.
Discussion
We have improved upon the affinity of 4 and found the optimal length of the spacer chain to be one methylene, i.e., 7. Numerous cyclopropyl analogues were designed to add an element of rigidity, by analogy to antagonists of metabotropic glutamate receptors in which selectivity was achieved specifically depending on the stereochemistry.32 However, all of the cyclopropyl analogues displayed Ki values in the 1–2 μM range. Although the lead bisphosphate structure, upon which these analogues were based, i.e., 4, was selective for the P2Y1 subtype,33 it will nevertheless be useful to examine the acyclic compounds as agonists and/or antagonists at other subtypes of P2 receptors.
We have refined our previous modeling of the P2Y1 receptor30 utilizing the high-resolution template structure of rhodopsin now available.22 In general, the two receptor models are similar with respect to the overall juxtaposition of the side chains forming the nucleotide binding pocket (Figure 3), while differing in the length of the individual helices and in their mutual orientation within the helical bundle. In addition, a portion of EL2 bridging TM4 and TM5 is deeply embedded in the TM-manifold, much like the β4 strand of the antiparallel β-sheet of EL2 in rhodopsin.22 Thus, while both P2Y1 receptor models30 predict proximity of EL2 to the path of approach of the ligand toward the TM binding site, the present model indicates participation of certain residues of this loop in coordination of the ligand bound within the TM domain (Figure 2).
Previous modeling studies19,30 suggested that agonists and antagonists may share a common binding site within the receptor. In particular cases, substitution of a single N6-methyl group converted a pure P2Y1 agonist into an antagonist, apparently without affecting affinity. The first modeling experiment carried out with this P2Y1 receptor model was the docking of the prototypical agonists ATP and ADP, aimed to rationalize the apparently superior binding of the latter to the receptor.31 The binding site model satisfied the following binding requirements: differentially accommodating (a) the conformationally constrained agonists ATP and ADP; (b) rigid antagonists such as 2 (Figure 2); (c) flexible, acyclic antagonists reported here (Figure 4).
Initially, basic residues needed to accommodate the phosphate moieties of the ligands were identified (Lys128, Lys196, Lys280, Arg310). The model suggested that the β-phosphate moieties of ATP and ADP, as well as the 5′-phosphate substituent of 2, may occupy a position adjacent to the antiparallel β-sheet of EL2. This interpretation was based on the mutual orientation of these basic side chains, as well as the juxtaposition of the main chain amide nitrogens of residues Asp204 and Thr205. In this position the 5′-phosphate moiety did not seem to be sterically constrained, and its main interactions were with the flexible lysine and arginine side chains. Interestingly, according to this ligand orientation, the γ-phosphate of ATP should not contribute to affinity due to its location above the β4 strand of the EL2 β-sheet.
In contrast to the relaxed steric requirements for accommodation of the 5′-phosphate moiety of 2, the receptor seemed to present a restricted binding pocket for the 3′-phosphate substituent. Consequently, it appeared that the ligand binding environment of the P2Y1 receptor consisted of rigidly defined binding subsites for the adenine and the 3′-phosphate moieties (using ligand 2 as structural template) while positioning of the 5′-phosphate was less stringent. Such notion is consistent with the affinities of the different P2Y1 ligands in which these three pharmacophoric elements are held together by various spacers, such as carbocyclic and heterocyclic rings or carbon chains. In all these cases, the ligand-bound conformation seemed to conform to the proposed model (Table 3). We note that compound 11, where the monophosphate and the diphosphate substituents mimic the 3′-substituent of 2 and the 5′-substituent of ADP, respectively, was the most potent of the acyclic P2Y1 antagonists reported here.
In conclusion, two binding pockets for monophosphate moieties were defined. The flexibility of the spacer chain linking bisphosphate groups to the adenine moiety, in general, allowed a relaxation of spatial requirements, although the more potent antagonists in the acyclic series conformed to a three-point model of the docked positions of the phosphates relative to the adenine N6. This was especially true for the series of symmetrically substituted homologues 3–7 and 94 (Table 3). Elongation of the ethylene group of 4 was previously shown to greatly decrease potency at the P2Y1 receptor.20 We have identified the optimal chain length for P2Y1 antagonism to be one methylene group, in 7. We have now achieved potency in an acyclic derivative comparable to the best antagonists in the ribose series.16 Thus, both approaches of rigidifying ribose19 and ring opening20 have proven successful design approaches for P2Y1 antagonists.
Experimental Section
Chemical Synthesis.
Materials and Instrumentation.
Nucleosides and synthetic reagents were purchased from Sigma Chemical Co. (St. Louis, MO) and Aldrich (Milwaukee, WI).
1H NMR spectra were obtained with a Varian Gemini-300 spectrometer using CDCl3, DMSO-d6, or D2O as a solvent. The chemical shifts are expressed as ppm downfield from tetramethylsilane or as relative ppm from DMSO (2.5 ppm) or HOD peaks (4.78 ppm). 31P NMR spectra were recorded at room temperature by use of Varian XL-300 spectrometer (121.42 MHz); orthophosphoric acid (85%) was used as an external standard. Low-resolution CI-NH3 (chemical ionization) mass spectra were carried out with a Finnigan 4600 mass spectrometer and high-resolution EI (electron impact) mass spectrometry with a VG7070F mass spectrometer at 6 kV. High-resolution FAB (fast atom bombardment) mass spectrometry was performed with JEOL SX102 spectrometer using 6-kV Xe atoms following desorption from a glycerol matrix.
The determinations of purity were performed with a Hewlett-Packard 1090 HPLC system using an SMT OD-5–60 C18 analytical column (250 mm × 4.6 mm, Separation Methods Technologies, Inc., Newark, DE) in two different linear gradient solvent systems. One solvent system (A) was 0.1 M triethylammonium acetate buffer:CH3CN in ratios of 95:5 to 40:60 for 20 min with flow rate 1 mL/min. The other (B) was 5 mM tetrabutylammonium phosphate buffer:CH3CN, 80:20 to 40:60, in 20 min with flow rate 1 mL/min. Peaks were detected by UV absorption using a diode array detector. All phosphate derivatives showed more than 95% purity as determined using HPLC.
1,3-Bis-(tert-butyldimethylsilanyloxy)propan-2-ol (22).
To a cooled (0 °C) and stirred solution of disilyl ketone 21 (1.20 g, 3.77 mmol) in ethyl alcohol (6 mL) was added a solid sodium borohydride (80 mg, 2.11 mmol) in one pot. After the mixture was stirred at 0 °C for 30 min, water (10 mL) was added, and solid ammonium bicarbonate (5 g) was added at the same temperature to quench excess residual sodium borohydride. The resulting mixture was partitioned between ethyl acetate (50 mL) and brine (20 mL). The organic layer was washed with brine/water (1:1, 10 mL × 2), dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo to give a crude disilyl alcohol 22 (1.10 g, 91%). 1H NMR (CDCl3) δ 0.07 (s, 12H), 0.90 (s, 18H), 3.54–3.72 (m, 5H).
Method A. Mitsunobu Reaction and Substitution of 6-Chloro Group by Methylamine To Introduce N6-Methylamino-2-chloro- (or hydro-)purine Moiety.
The alcohol 22 (250 mg, 0.78 mmol), triphenylphosphine (307 mg, 1.17 mmol), and 2,6-dichloropurine (189 mg, 0.858 mmol) were mixed and dried by high vacuum pump for about 30 min. The mixture was dissolved in anhydrous tetrahydrofuran (5 mL) and cooled to 0 °C. To this solution was dropwise added diethylazodicarboxylate (149 mg, 0.855 mmol) via syringe. After the mixture was stirred for 2–4 h at room temperature (the progress of the reaction was monitored by thin-layer chromatography, and if necessary it was overnight stirred), tetrahydrofuran was removed in vacuo. The residue was purified by silica gel column chromatography (hexanes/ethyl acetate = 5:1) to give a desired 2,6-dichloropurine analogue 23 (260 mg, 68%).
The 2,6-dichloropurine analogue 23 was treated with 2 M methylamine in tetrahydrofuran (5 mL, 10 mmol), stirred for 5 h, filtrated through silica gel (10 g), and washed with hexanes/ethyl acetate (1:2). Evaporation of the filtrate gave a desired 6-amino-2-chloropurine analogue 24 (225 mg, 88%).
9-[2-(tert-Butyldimethylsilanyloxy)-1-(tert-butyldimethylsilanyloxymethyl)ethyl]-2,6-dichloro-9H-purine (23).
1H NMR (CDCl3) δ 0.008 (s, 6H), 0.014 (s, 6H), 0.84 (s, 18H), 3.99 (dd, 2H, J = 5.2, 10.6 Hz), 4.08 (dd, 2H, J = 5.4, 10.6 Hz), 4.81 (m, 1H). 8.34 (s, 1H). MS (CI/NH3) m/z 491 (M + H+)+.
{9-[2-(tert-Butyldimethylsilanyloxy)-1-(tert-butyldimethylsilanyloxymethyl)ethyl]-2-chloro-9H-purin-6-yl}-methylamine (24).
1H NMR (CDCl3) δ 0.008 (s, 6H), 0.014 (s, 6H), 0.84 (s, 18H), 3.94 (dd, 2H, J = 5.4, 10.3 Hz), 4.01 (dd, 2H, J = 5.1, 10.3 Hz), 4.70 (m, 1H). 6.04 (bs, 1H), 7.91 (s, 1H). MS (CI/NH3) m/z 486 (M + H+)+.
Method B. Hydrolysis of TBS Group or Acetonide Group with Acidic Dowex Resin.
A mixture of disilyl ether 24 (215 mg, 0.442 mmol), Dowex 50x-8 200 (500 mg), and methanol (5 mL) was stirred overnight at refluxing temperature. The resin was removed by filtration of the resulting mixture through glass filter (10–20 μm, pore size). The filtrate was evaportated in vacuo to give diol 25 (85 mg, 75%). (This method was also used in deprotection of acetonide group of 54 and 55).
2-(2-Chloro-6-methylaminopurin-9-yl)propane-1,3-diol (25).
1H NMR (DMSO-d6) δ 2.91 (bs, 3H), 3.76 (dd, 2H, J = 5.4, 11.4 Hz), 3.83 (dd, 2H, J = 7.0, 11.4 Hz), 4.46 (m, 1H), 8.15 (s, 1H). MS (CI/NH3) m/z 258 (M + H+)+.
Method C. Preparation of Bis(di-tert-butyl phosphate) from Diol.
To a stirred suspension of diol 25 (22 mg, 0.0854 mmol) and diethyl di-tert-butylphosphoramidite (53 mg, 0.213 mmol) in anhydrous tetrahydrofuran (5 mL) at room temperature was added solid tetrazole (36 mg, 0.513 mmol) in one pot. The reaction mixture was stirred at room temperature for 20 min (sometimes overnight) and then cooled to −78 °C. To this cooled reaction mixture was added solid MCPBA (57–85%, calculated by 57%, 64 mg), and the resulting mixture was stirred at −78 °C for 30 min and at room temperature for 10 min. After removal of tetrahydrofuran in vacuo, the residue was directly purified by preparative thin-layer chromatography (CHCl3/MeOH = 20/1) to give 26 (40 mg, 73%).
Phosphoric Acid Di-tert-butyl Ester 2-(2-Chloro-6-methylaminopurin-9-yl)-3-(di-tert-butoxyphosphoryoxy)-propyl Ester (26).
1H NMR (CDCl3) δ 1.39 (s, 18H), 1.41 (s, 18H), 3.17 (bs, 3H), 4.34 (dd, 2H, J = 5.4, 11.0, 11.0 Hz), 4.47 (dd, 2H, J = 6.6, 11.0, 11.0 Hz), 5.00 (m, 1H), 6.16 (bs, 1H), 7.88 (s, 1H). MS (CI/NH3) m/z 642 (M + H)+, 659 (M + NH4+)+.
3-(tert-Butyldimethylsilanyloxy)-2-(tert-butyldimethylsilanyloxymethyl)propene (28).
To a stirred solution of propenediol 27 (3.0 g, 34.1 mmol), triethylamine (6.90 g, 68.2 mmol), and 4-(dimethylamino)pyridine (20 mg, 0.264 mmol) in methylene chloride (30 mL) at room temperature was added solid tert-butyldimethylsilyl chloride (10.7 g, 71.5 mmol). After being stirred overnight at room temperature, the resulting mixture was diluted with ethyl acetate (100 mL). The organic layer was washed with 1 N hydrochloric acid (20 mL), saturated aqueous sodium bicarbonate (30 mL × 2), brine/water (1:1, 20 mL × 2), dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo to dryness to give crude 28 (10.7 g, 99%). 1H NMR (CDCl3) δ 0.07 (s, 12H), 0.92 (s, 18H), 4.17 (t, 4H, J = 1.5 Hz), 5.09 (t, 2H, J = 1.5 Hz). MS (CI/NH3) m/z 317 (M + H+)+.
3-(tert-Butyldimethylsilanyloxy)-2-(tert-butyldimethylsilanyloxymethyl)propan-1-ol (29).
A 1 M solution of borane (in tetrahydrofuran, 34.2 mL, 34.2 mmol) was added dropwise to a cooled (0 °C), stirred solution of 28 (3.6 g, 11.37 mmol) in anhydrous tetrahydrofuran (10 mL). After being stirred at the same temperature, the reaction mixture was warmed to room temperature and stirred overnight. The resulting mixture was treated with potassium carbonate (10 g, 72.4 mmol) and 30% hydrogen peroxide (20 mL) at 0 °C, stirred at room temperature for 2 h, and partitioned between ethyl acetate (100 mL) and water (50 mL). The organic layer was separated, washed with brine/water (1:1, 20 mL × 3), dried over anhydrous sodium sulfate, filtered, concentrated in vacuo. The residue was purified by silica gel column chromatography (hexanes/ethyl acetate = 2:1) to give 29 (2.28 g, 60%). 1H NMR (CDCl3) δ 0.07 (s, 12H), 0.90 (s, 18H), 1.90 (m, 1H), 3.73 (d, 2H, J = 6.0 Hz), 3.75 (d, 2H, J = 6.0 Hz), 3.78 (d, 2H, J = 5.1 Hz). MS (CI/NH3) m/z 335 (M + H+)+.
Method D. Mesylation and N-Alkylation To Introduce N6-Methylamino-2-chloro- (or 2-hydro)purine Moiety.
To a cooled (0 °C), stirred solution of alcohol 29 (1.47 g, 4.40 mmol) and triethylamine (1.34 g, 13.2 mmol) in methylene chloride (10 mL) was dropwise and neatly added methanesulfonyl chloride (756 mg, 6.60 mmol). After being stirred at 0 °C for 10 min and at room temperature for 1 h, the resulting mixture was partitioned between ethyl acetate (50 mL) and brine (20 mL). The organic layer was separated and washed with saturated sodium bicarbonate (20 mL) and brine/water (1:1, 10 mL × 2), dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo to give a crude mesylate 30 (1.90 g). The mesylate 30 was used in the following N-alkylation without further purification.
A mixture of crude mesylate 30 (1.9 g), 2-chloro-6-methylaminopurine (808 mg, 4.40 mmol), potassium carbonate (912 mg, 6.60 mmol), 18-crown-6 (10 mg, mmol), and dimethylformamide (10 mL) was stirred overnight at 40–60 °C. The resulting mixture was partitioned between ethyl acetate (150 mL) and brine/water (40 mL:20 mL). The organic layer was separated, washed with brine (20 mL × 4), and dried over anhydrous sodium sulfate, filtered, concentrated to dryness. The residue was purified by silica gel column chromatography (hexanes/ethyl acetate = 1:1) to give 32 (1.47 g, 72% two steps yield).
Methanesulfonic Acid 3-(tert-Butyldimethylsilanyloxy)-2-(tert-butyldimethylsilanyloxymethyl)propyl Ester (30).
1H NMR (CDCl3) δ 0.06 (s, 12H), 0.89 (s, 18H), 2.10 (m, 1H), 3.00 (s, 3H), 3.63 (dd, 2H, J = 5.2, 10.2 Hz), 3.68 (dd, 2H, J = 5.8, 10.2 Hz), 4.30 (d, 2H, J = 5.8 Hz). MS (CI/NH3) m/z 413 (M + H+)+.
{9-[3-(tert-Butyldimethylsilanyloxy)-1-(tert-butyldimethylsilanyloxymethyl)-propyl]-9H-purin-6-yl}-methylamine (31). Method D.
Compound 31 (280 mg, 60% two steps yield) was obtained from 30 (410 mg, 0.993 mmol). 1H NMR (CDCl3) δ 0.04 (s, 12H), 0.90 (s, 18H), 2.32 (m, 1H), 3.22 (bs, 3H), 3.54 (dd, 2H, J = 5.9, 10.2 Hz), 3.65 (dd, 2H, J = 4.8, 10.2 Hz), 4.22 (d, 2H, J = 7.2 Hz), 5.79 (bs, 1H), 7.75 (s, 1H), 8.41 (s, 1H). MS (CI/NH3) m/z 466 (M + H+)+.
{9-[3-(tert-Butyldimethylsilanyloxy)-1-(tert-butyldimethylsilanyloxymethyl)propyl]-2-chloro-9H-purin-6-yl}-methylamine (32). Method D.
1H NMR (CDCl3) δ 0.04 (s, 12H), 0.90 (s, 18H), 2.29 (m, 1H), 3.20 (bs, 3H), 3.52 (dd, 2H, J = 5.9, 10.2 Hz), 3.64 (dd, 2H, J = 5.0, 10.2 Hz), 4.19 (d, 2H, J = 7.0 Hz), 5.93 (bs, 1H), 7.71 (s, 1H). MS (CI/NH3) m/z 500 (M + H+)+.
Method E. Deprotection of Disilyl Ether and Acetonide Groups with Acetic Acid/THF/Water.
A solution of disilyl ether 32 (300 mg, 0.644 mmol) in acetic acid/tetrahydofuran/water (3 mL:2 mL:2 mL) was stirred at 50 °C for 1 day. All volatile material was removed in vacuo, and the residue was washed with hexanes/ethyl ether (1:1) to give diol 34 (163 mg, 93%).
2-(6-Methylaminopurin-9-ylmethyl)propane-1,3-diol (33). Method E.
Compound 33 (106 mg, 79%) was obtained from 31 (260 mg, 0.993 mmol). 1H NMR (CD3OD) δ 2.23 (m, 1H), 3.12 (bs, 3H), 3.54 (m, 4H), 4.33 (d, 2H, J = 6.7 Hz), 8.05 (s, 1H), 8.26 (s, 1H). MS (CI/NH3) m/z 238 (M + H+)+.
2-(2-Chloro-6-methylaminopurin-9-ylmethyl)propane-1,3-diol (34). Method E.
1H NMR (CD3OD) δ 2.23 (m, 1H), 3.06 (bs, 3H), 3.54 (m, 4H), 4.27 (d, 2H, J = 6.7 Hz), 8.00 (s, 1H). MS (CI/NH3) m/z 272 (M + H+)+.
Method F. Preparation of Tetrabenzylbisphosphates from Diol.
To a cooled (0 °C), stirred of diol 34 (50 mg, 0.184 mmol) and tetrabenzylpyrophosphate (247 mg, 0.423 mmol) in anhydrous tetrahydrofuran (5 mL) was added solid sodium hydride (80 mg, 50% in mineral oil, ~1.67 mmol) in one pot. The reaction mixture was stirred at 0 °C for 10 min and at room temperature for 30 min. The resulting mixture was poured into ice–water (20 mL) solution, and the aqueous layer was extracted with ethyl acetate (20 mL × 3). The combined organic phase was washed with brine (10 mL), dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (ethyl acetate/methanol = 15:1) to afford a desired bis(dibenzyl phosphate) 36 (82 mg, 53%). (In the case of cyclopropanediols 73 and 72, lithium diethylamide was used as a base as substitute for sodium hydride.)
Phosphoric Acid Dibenzyl Ester 3-(Bisbenzyloxyphosphoyloxy)-2-(6-methylaminopurin-9-ylmethyl)propyl Ester (35). Method F.
Compound 35 (59 mg, 81%) was obtained from 33 (23 mg, 0.097 mmol). 1H NMR (CDCl3) δ 2.57 (m, 1H), 3.19 (bs, 3H), 3.90 (m, 4H), 4.03 (d, 2H, J = 6.9 Hz), 5.01 (m, 8H), 5.99 (q, 1H, J = 5.1 Hz), 7.22–7.38 (m, 20H), 7.61 (s, 1H), 8.34 (s, 1H). 31P NMR (D2O) δ −22.363. MS (positive-ion FAB) m/z 758 (M + H+)+.
Phosphoric Acid Dibenzyl Ester 3-(Bisbenzyloxyphosphoyloxy)-2-(2-chloro-6-methylaminopurin-9-ylmethyl)-propyl Ester (36). Method F.
1H NMR (CDCl3) δ 2.49 (m, 1H), 3.17 (bs, 3H), 3.86 (m, 4H), 3.97 (d, 2H, J = 7.0 Hz), 4.89–5.08 (m, 8H), 5.99 (bs, 1H), 7.15–7.0.38 (m, 20H), 7.57 (s, 1H). MS (CI/NH3) m/z 792 (M + H+)+.
(±)-3-(tert-Butyldimethylsilanyloxy)-2-(2-chloro-6-methylaminopurin-9-ylmethyl)propan-1-ol ((±)-37).
To a stirred solution of diol 34 (370 mg, 1.34 mmol), imidazole (140 mg, 2.06 mmol), in dimethylformamide at room temperature was added solid tert-butyldimethylsilyl chloride (226 mg, 1.50 mmol) in one pot. The reaction mixture was overnight stirred at room temperature. Dimethylformamide was removed in vacuo, and the residue was purified by preparative thin-layer chromatography (chloroform/methanol = 10:1) to give monosilyl derivative, 37 (200 mg, 38%). 1H NMR (CDCl3) δ 0.05 (s, 6H), 0.89 (s, 9H), 2.22 (m, 1H), 3.15 (bs, 3H), 3.35–3.52 (m, 2H), 3.51 (dd, 1H, J = 7.1, 10.4 Hz), 3.61 (dd, 1H, J = 5.0, 10.4 Hz), 4.32 (m, 2H), 6.60 (bs, 1H), 7.71 (s, 1H). MS (positive-ion FAB) 386 (M + H+)+.
(±)-Phosphoric Acid Di-tert-butyl Ester 3-(tert-Butyldimethylsilanyloxy)-2-(2-chloro-6-methylaminopurin-9-ylmethyl)propyl Ester (38). Method C.
Compound 38 (250 mg, 83%) was obtained from 37 (200 mg, 0.52 mmol). 1H NMR (CDCl3) δ 0.04 (s, 6H), 0.90 (s, 9H), 1.49 (s, 18H), 2.49 (m, 1H), 3.19 (bs, 3H), 3.56 (dd, 1H, J = 5.4, 10.5 Hz), 3.61 (dd, 1H, J = 4.8, 10.5 Hz), 3.88–4.06 (m, 2H), 4.24 (d, 2H, J = 6.9 Hz), 6.05 (bs, 1H), 7.77 (s, 1H). MS (positive-ion FAB) 578 (M + H+)+.
(±)-Phosphoric Acid Di-tert-butyl Ester 2-(2-Chloro-6-methylaminopurin-9-ylmethyl)-3-hydroxy-propyl Ester (39).
A solution of silyl phosphate 38 (250 mg, 0.432 mmol) in tetrahydrofuran (10 mL) at room temperature was treated with tetrabutylammonium fluoride (0.52 mL, 0.52 mmol, 1 M solution in tetrahydrofuran). The reaction mixture was stirred for 10 min and applied directly to preparative thin-layer chromatography (chloroform/methanol = 10:1) to give 39 (190 mg, 76%). 1H NMR (CDCl3) δ 1.51 (s, 9H), 1.52 (s, 9H), 2.41 (m, 1H), 3.19 (bs, 3H), 3.40–3.56 (m, 2H), 3.84–4.11 (m, 2H), 4.23–4.40 (m, 2H), 6.34 (bs, 1H), 7.81 (s, 1H). MS (positive-ion FAB) 464 (M + H+)+.
(±)-Phosphoric Acid Di-tert-butyl Ester 2-(2-Chloro-6-methylaminopurin-9-ylmethyl)-3-(dimethyoxyphosphoryloxy)propyl Ester (40).
To a stirred solution of 39 (10 mg, 0.0216 mmol) and dimethylphosphoryl chloride (12 mg, 0.083 mmol) in anhydrous tetrahydrofuran at room temperature was added solid sodium hydride (8.6 mg, 0. 215 mmol, 60% in mineral oil) in one pot. The reaction mixture was stirred for 1 h at the same temperature, followed by quenching with water under water-ice bath. The mixture was applied to prepatative thin-layer chromatography (chloroform/methanol = 10:1) to give 40 (3.8 mg, 31%). 1H NMR (CDCl3) δ 1.497 (s, 9H), 1.500 (s, 9H), 2.73 (m, 1H), 3.18 (bs, 3H), 3.80 (d, 3H, J = 11.1 Hz), 3.82 (d, 3H, J = 11.1 Hz), 3.86–4.19 (m, 4H), 4.21–4.38 (m, 2H), 6.17 (bs, 1H), 7.85 (s, 1H). MS (positive-ion FAB) (M + H +)+.
(±)-Acetic Acid 2-(2-Chloro-6-methylaminopurin-9-ylmethyl)-3-(di-tert-butoxyphosphoryloxy)propyl Ester (41).
A mixture of 39 (11.5 mg, 0.0248 mmol), triethylamine (9.4 mg, 0.0933 mmol), and acetic anhydride (3.80 mg, 0.0372 mmol) was stirred for 2 h at room temperature. The resulting mixture was purified by preparative thin-layer chromatography (chloroform/methanol = 15:1) to give 41 (12 mg, 96%). 1H NMR (CDCl3) δ 1.497 (s, 9H), 1.502 (s, 9H), 2.07 (s, 3H), 2.70 (m, 1H), 3.18 (bs, 3H), 3.86–4.05 (m, 2H), 4.09 (d, 2H, J = 5.9 Hz), 4.29 (d, 2H, J = 7.0 Hz), 6.08 (bs, 1H), 7.79 (s, 1H). MS (positive-ion FAB) 506 (M + H+)+.
(±)-Phosphoric Acid Dibenzyl Ester 2-(2-Chloro-6-methylaminopurin-9-ylmethyl)-3-(di-tert-butoxyphosphoryloxy)propyl Ester (42).
To a stirred suspension of 39 (13 mg, 0.028 mmol) and diisopropyl dibenzylphosphoramidite (29 mg, 0.084 mmol) in anhydrous tetrahydrofuran (2 mL) at room temperature was added solid tetrazole (11.7 mg, 0.167 mmol) in one pot. The reaction mixture was stirred at room temperature for 20 min and cooled to −78 °C. To this cooled reaction mixture was added solid MCPBA (57–85%, calculated by 57%, 25 mg), and the resulting mixture was stirred at −78 °C for 30 min and at room temperature for 10 min. After removal of tetrahydrofuran in vacuo, the residue was directly purified by preparative thin-layer chromatography (CHCl3/MeOH = 20/1) to give 42 (9.2 mg, 45%). 1H NMR (CDCl3) δ 1.46 (s, 18H), 2.61 (m, 1H), 3.18 (bs, 3H), 3.79–4.08 (m, 4H), 4.14 (d, 2H, J = 7.0 Hz), 4.97–5.13 (m, 4H), 6.04 (bs, 1H), 7.28–7.42 (m, 10H), 7.74 (s, 1H). MS (positive-ion FAB) 724 (M + H+)+.
(±)-Phosphoric Acid Dibenzyl Ester 2-(2-Chloro-6-methylaminopurin-9-ylmethyl)-3-phosphonooxypropyl Ester (43). Method I.
Compound 43 (8 mg, 89%) was obtained from 42 (9.1 mg, 0.0126 mmol) as a triethylammonium salt. 1H NMR (CD3OD) δ 2.65 (m, 1H), 3.07 (bs, 3H), 3.93–4.13 (m, 4H), 4.24 (d, 2H, J = 7.0 Hz), 5.03 (d, 4H, J = 8.9 Hz), 7.34 (s, 10H). 8.07 (s, 1H). HPLC 16.372 min (purity >98%) in solvent system A, 13.449 min in system B.
(±)-Phosphoric Acid Dibenzyl Ester 2-(2-Chloro-6-methylaminopurin-9-ylmethyl)-3-phosphonophosphonooxypropyl Ester (44).
Monotriethylamine salt 43 (8 mg, 0.0112 mmol) was dissolved in anhydrous dimethylformamide (1 mL), followed by an addition of carbonyldiimidazole (5.46 mg, 0.0337 mmol). The mixture was stirred for 20 min at room temperature and then triethylammonium phosphate (13.4 mg, 0.067 mmol) in anhydrous dimethylformamide (0.5 mL) was added. The reaction mixture was stirred for 3 days at room temperature. All volatile material was removed by nitrogen stream. The residue was purified with an ion-exchange column chromatography using Sephadex-DEAE-25 resin with a linear gradient (0.01 to 1.0 M) of 1.0 M ammonium bicarbonate as the mobile phase to give phosphate 44 (6 mg, 74%) as ammonium salt. 1H NMR (D2O) δ 2.66 (m, 1H), 2.87 (bs, 3H), 3.91–4.19 (m, 4H), 4.20–4.41 (m, 2H), 4.65–4.90 (m, 2H), 7.06–7.25 (m, 5H), 7.30–7.41 (m, 5H). 8.08 (s, 1H). High-resolution MS (negative-ion FAB) calcd for C24H28O11N5ClP3 [M − H+]− 690.0687, found 690.0684.
Methanesulfonic Acid 2-Allylpent-4-enyl Ester (46) Method D.
1H NMR (CDCl3) δ 1.96 (m, 1H), 2.17 (t, 4H, J = 7.0 Hz), 3.15 (s, 3H), 4.14 (d, 2H, J = 5.5 Hz), 5.03–5.16 (m, 4H), 5.76 (m 2H).
[9-(2-Allyl-pent-4-enyl)-2-chloro-9H-purin-6-yl]methylamine (47). Method D.
Compound 47 (69 mg, 60% two steps yield) obtained from 45 (50 mg, 0.396 mmol). 1H NMR (CDCl3) δ 2.07 (t, 4H, J = 6.6 Hz), 2.23 (m, 1H), 3.18 (s, 3H), 4.06 (d, 2H, J = 7.2 Hz), 5.01–5.11 (m, 4H), 5.75 (m 2H). MS (positive-ion FAB) 292 (M + H+)+.
3-(2-Chloro-6-methylaminopurin-9-ylmethyl)pentane-1,5-diol (48).
A mixture of diene 47 (39 mg, 0.134 mmol), osmium tetroxide (0.3 mL of stock solution, 1 g OsO4/100 mL H2O), sodium metaperiodate (340 mg, 1.59 mmol), and a mixed solvent (5 mL, acetone/water = 4:1) was stirred at room temperature for 4 h. All volatile materials were removed in vacuo to dryness. The residue was stirred in a mixed solvent (ethyl acetate/methanol = 10:1) and filtrated through a short column packed with silica gel (20 g). The filtrate was collected and evaporated, and the residue was dissolved in ethyl alcohol in an ice–water bath. Solid sodium borohydride was added to this solution, and the reaction mixture was stirred for 2 h at room temperature, followed by quenching excess sodium borohydride with 1 N hydrochloric acid. After removal of all volatile materials in vacuo to dryness, the residue was purified by preparative thin-layer chromatography (chloroform/methanol = 10:1) to give 48 (13 mg, 32%). 1H NMR (CD3OD) δ 1.43 – 1.65 (m, 4H), 2.27 (m, 1H), 3.07 (bs, 3H), 3.51–3.71 (m, 4H), 4.18 (d, 2H, J = 7.2 Hz), 8.05 (s, 1H). MS (positive-ion FAB) 300 (M + H+)+.
Phosphoric Acid Di-tert-butyl Ester 3-(2-Chloro-6-methylaminopurin-9-ylmethyl)-5-(di-tert-butoxyphosphoryloxy)-pentyl Ester (49). Method C.
Compound 49 (20 mg, 73%) obtained from 48 (12 mg, 0.040 mmol). 1H NMR (CDCl3) δ 1.47 (s, 36H), 1.65–1.75 (m, 4H), 2.29 (m, 1H), 3.18 (bs, 3H), 3.93–4.16 (m, 4H), 4.21 (d, 2H, J = 6.7 Hz), 6.12 (bs, 1H), 7.84 (s, 1H). MS (positive-ion FAB) 684 (M + H+)+.
(4S)-Methanesulfonic Acid 2,2-Dimethyl-[1,3]dioxolan-4-ylmethyl Ester (52) (from R-Isomer Alcohol 50). Method D.
Crude 52 (700 mg) was obtained from 50 (500 mg, 3.78 mmol).1H NMR (CDCl3) δ 1.36 (s, 3H), 1.44 (s, 3H), 3.06 (s, 3H), 3.82 (dd, 1H, J = 5.5, 8.8 Hz), 4.10 (dd, 1H, J = 6.5, 8.8 Hz), 4.22 (d, 2H, J = 5.2 Hz), 4.37 (m, 1H). MS (CI/NH3) m/z 211 (M + H+)+, 228 (M + NH4+)+.
(4R)-Methanesulfonic Acid 2,2-Dimethyl-[1,3]dioxolan-4-ylmethyl Ester (53) (from S-Isomer Alcohol 51). Method D.
Crude 53 (850 mg) was obtained from 51 (500 mg, 3.78 mmol), and it showed the identical proton NMR spectrum to 52. MS (CI/NH3) m/z 228 (M + NH4)+.
(R)-[2-Chloro-9-(2,2-dimethyl-[1,3]dioxolan-4-ylmethyl)-9H-purin-6-yl]-methyl-amine (54) (Originated from R-Isomer 50). Method D.
Compound 54 (567 mg, 50% two steps yield) was obtained from 50 (500 mg, 3.78 mmol). 1H NMR (CDCl3) δ 1.33 (s, 3H), 1.39 (s, 3H), 3.19 (bs, 3H), 3.69 (dd, 1H, J = 5.8, 8.8 Hz), 4.14 (dd, 1H, J = 6.6, 8.8 Hz), 4.20 (dd, 1H, J = 6.4, 14.4 Hz), 4.27 (dd, 1H, J = 3.2, 14.4 Hz), 4.46 (m, 1H). MS (CI/NH3) 298 m/z (M + H+)+.
(S)-[2-Chloro-9-(2,2-dimethyl-[1,3]dioxolan-4-ylmethyl)-9H-purin-6-yl]-methyl-amine (55) (Originated from S-Isomer 51). Method D.
Compound 55 (611 mg, 54% two steps yield) was obtained from 51 (500 mg, 3.78 mmol), and it showed the identical proton NMR spectrum and mass spectrum to 54. MS (CI/NH3) 298 m/z (M + H+)+.
(R)-3-(2-Chloro-6-methylaminopurin-9-yl)propane-1,2-diol (56) (Originated from R-Isomer 50). Method B.
Compound 56 (114 mg, 88%) was obtained from 54 (150 mg, 0.504 mmol).1H NMR (DMSO-d6) δ (d, 3H, J = 3.6 Hz), 3.32 (dd, 1H, J = 6.2, 11.1 Hz), 3.40 (dd, 1H, J = 5.1, 11.1 Hz), 3.82 (m, 1H), 3.95 (dd, 1H, J = 8.5, 13.9 Hz), 4.25 (dd, 1H, J = 3.4, 13.9 Hz), 8.04 (s, 1H), 8.16 (m, 1H). MS (CI/NH3) m/z 258 (M + H+)+.
(S)-3-(2-Chloro-6-methylaminopurin-9-yl)propane-1,2-diol (57) (Originated from S-Isomer 51) Method B.
Compound 57 (110 mg, 85%) was obtained from 55 (150 mg, 0.504 mmol) and it showed the identical proton NMR spectrum and mass spectrum to 56.
(R)-Phosphoric Acid Di-tert-butyl Ester 1-(2-Chloro-6-methylaminopurin-9-ylmethyl)-2-(di-tert-butoxyphosphoryloxy)ethyl Ester (58) (Originated from R-Isomer 50). Method C.
Compound 58 (42 mg, 56%) was obtained from 56 (30 mg, 0.116 mmol). 1H NMR (CDCl3) δ 1.34 (s, 9H), 1.44 (s, 9H), 1.507 (s, 9H), 1.513 (s, 9H), 3.18 (bs, 3H), 4.04–4.27 (m, 2H), 4.34–4.55 (m, 2H), 4.78 (m, 1H), 6.50 (bs, 1H), 7.88 (s, 1H). MS (CI/NH3) m/z 642 (M + H+)+.
(S)-Phosphoric Acid Di-tert-butyl Ester 1-(2-Chloro-6-methylaminopurin-9-ylmethyl)-2-(di-tert-butoxyphosphoryloxy)ethyl Ester (59) (Originated from R-Isomer 50). Method C.
Compound 59 (43 mg, 69%) was obtained from 57 (25 mg, 0.097 mmol). 1H NMR (CDCl3) δ 1.34 (s, 9H), 1.44 (s, 9H), 1.507 (s, 9H), 1.513 (s, 9H), 3.18 (bs, 3H), 4.04–4.27 (m, 2H), 4.34–4.55 (m, 2H), 4.78 (m, 1H), 6.21 (bs, 1H), 7.85 (s, 1H). MS (CI/NH3) m/z 642 (M + H+)+.
4-(tert-Butyldimethylsilanyloxy)-3-(tert-butyldimethylsilanyloxylmethyl)but-2-enoic Acid Ethyl Ester (60).
Neat ethyl (diethoxy)phosphoacetate (3.10 g, 13.8 mmol) was added dropwise to a cooled (0 °C), stirred suspension of sodium hydride (50%, 662 mg, 13.8 mmol) in anhydrous tetrahydrofuran (60 mL). After being stirred at room temperature for 1 h until the reaction mixture became homogeneous, the reaction mixture was cooled to 0 °C, and to this solution was added a solution of 21 (4.0 g, 12.6 mmol) in anhydrous tetrahydrofuran (10 mL) at the same temperature. The reaction mixture was stirred at room temperature for 2 h and poured into ethyl acetate (100 mL) and water (50 mL). The organic layer was separated, washed with brine/water (1:1, 20 mL × 3), dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexanes/ethyl acetate = 15:1) to give 60 (4.17 g, 85%). 1H NMR (CDCl3) δ 0.07 (s, 6H), 0.10 (s, 6H), 0.90 (s, 9H), 0.93 (s, 9H), 1.29 (t, 3H, J = 7.2 Hz), 4.16 (q, 2H, J = 7.2 Hz), 4.44 (m, 2H), 4.87 (m, 2H), 5.98 (m, 1H). MS (CI/NH3) m/z 389 (M + H+)+.
4-(tert-Butyldimethylsilanyloxy)-3-(tert-butyldimethylsilanyloxylmethyl)but-2-en-1-ol (61).
To a cooled (−78 °C), stirred solution of 60 (1.3 g, 3.34 mmol) in toluene (20 mL) was added a 1 M solution of DIBAL (in tetrahydrofuran, 8.0 mL, 8.0 mmol). After being stirred for 2 h at −20 °C, the mixture was warmed to room temperature, stirred for 1 h, and cooled to 0 °C, followed by an addition of methanol (7 mL). The resulting mixture was partitioned between 10% Rochelle salt solution (100 mL) and ethyl acetate (100 mL). This two-phase solution was stirred until the two phases were clearly separated with more addition of Rochelle salt. The organic layer was separated, washed with brine/water (1:1, 20 mL × 2), dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexanes/ethyl acetate = 1:1) to give 61 (860 mg, 74%). 1H NMR (CDCl3) δ 0.08 (s, 6H), 0.09 (s, 6H), 0.91 (s, 9H), 0.92 (s, 9H), 4.10–4.28 (m, 6H), 5.82 (m, 1H). MS (CI/NH3) m/z 347 (M + H+)+.
9-[4-(tert-Butyldimethylsilanyloxy)-3-(tert-butyldimethylsilanyloxylmethyl)but-2-enyl]-2,6-dichloro-9H-purine (62). Method A.
Crude 62 (230 mg, 57%) was obtained from 61 (270 mg, 0.779 mmol). 1H NMR (CDCl3) δ 0.05 (s, 6H), 0.11 (s, 6H), 0.89 (s, 9H), 0.91(s, 9H), 4.19 (s, 2H), 4.25 (s, 2H), 5.03 (d, 2H, J = 7.5 Hz), 5.78 (t, 1H, J = 7.5 Hz), 8.17 (s, 1H).
{9-[4-(tert-Butyldimethylsilanyloxy)-3-(tert-butyldimethylsilanyloxylmethyl)but-2-enyl]-2-chloro-9H-purin-6-yl}-methylamine (63). Method A.
Compound 63 (168 mg, 85%) was obtained from crude 62 (200 mg, 0.386 mmol). 1H NMR (CDCl3) δ 0.05 (s, 6H), 0.09 (s, 6H), 0.89 (s, 9H), 0.90 (s, 9H), 3.19 (bs, 3H), 4.20 (s, 2H), 4.33 (s, 2H), 4.89 (d, 2H, J = 7.4 Hz), 5.76 (t, 1H, J = 7.4 Hz), 7.74 (s, 1H). MS (CI/NH3) m/z 512 (M + H+)+.
2-[2-(2-Chloro-6-methylaminopurin-9-yl)-ethylidene]-propane-1,3-diol (64). Method E.
Compound 64 (100 mg, 86%) was obtained from 63 (210 mg, 0.410 mmol). 1H NMR (DMSO-d6) δ 2.91 (s, 3H), 3.96 (s, 2H), 4.12 (s, 2H), 4.85 (d, 2H, J = 7.4 Hz), 5.60 (t, 1H, J = 7.4 Hz), 8.11 (s, 1H), 8.20 (bs, 1H). MS (CI/NH3) m/z 284 (M + H+)+.
Phosphoric Acid Di-tert-butyl Ester 4-(2-Chloro-6-methylaminopurin-9-yl)-2-(di-tert-butoxyphosphoryloxymethyl)but-2-enyl Ester (65). Method C.
Compound 65 (40 mg, 34%) was obtained from 64 (50 mg, 0.176 mmol). 1H NMR (CDCl3) δ 1.45 (s, 18H), 1.49 (s, 18H), 3.18 (bs, 3H), 4.52 (d, 2H, J = 6.7 Hz), 4.70 (d, 2H, J = 9.1 Hz), 4.96 (d, 2H, J = 7.1 Hz), 5.95 (t, 1H, J = 7.3 Hz), 6.17 (bs, 1H), 7.87 (s, 1H). MS (CI/NH3) m/z 668 (M + H+)+.
2,3-Bis-(tert-butyldimethylsilanyloxymethyl)cyclopropanecarboxylic Acid Ethyl Ester (67).
A solution of ethyl diazoacetate (containing <10% methylene chloride, 9 mL) at room temperature was added dropwise over 18 h, using a syringe pump, to a stirred solution of (Z)-1,4-bis[(tertbutyldimethylsilyl)oxy]-2-butene 66 (25 g, 78.95 mmol) and rhodium-(II) acetate dimer (410 mg, 0.928 mmol) in methylene chloride (50 mL). After the mixture was stirred for an additional 6 h, the solvent was removed in vacuo, and the residue was chromatographed on silica gel eluting 0.5–2% ethyl acetate in hexanes to give a mixture 67 of meso-cis and meso-trans isomers (19.27 g, 61%, meso-cis:meso-trans = 7.36 g: 11.91 g). 67: 1H NMR (CDCl3) δ 0.01–0.08 (m, 12H), 0.80–0.99 (m, 18H), 1.20–1.35 (m, 3H), 1.52–1.88 (m, 3H), 3.65–4.26 (m, 6H).
[2,3-Bis-(tert-butyldimethylsilanyloxymethyl)cyclopropyl]methanol (68).
To a stirred solution of the mixture 67 (400 mg, 0.99 mmol) in ether (5 mL) and toluene (2.7 mL) at room temperature was added solid lithium borohydride (22 mg, 1.01 mmol), and the reaction mixture was stirred at 100 °C with continuous removal of ethyl ether. More lithium borohydride (40 mg, 2.02 mmol) was added. After the mixture was stirred for 1 h at the same temperature, ice (20 g) was added in one pot with a rigorous stirring to decompose excess lithium borohydride, and the resulting mixture was stirred for 30 min. The aqueous layer was extracted with ethyl acetate (50 mL), and the organic layer was washed with water (30 mL × 2), dried over anhydrous sodium sulfate, filtered, concentrated in vacuo to give a crude mixture of meso-cis and meso-trans cyclopropanol isomers 68 (320 mg, 1:1.6, 93%). 1H NMR (CDCl3) δ 0.02–0.11 (m, 12H), 0.87–0.95 (m, 18H), 1.01–1.54 (m, 3H), 3.46–3.95 (m, 6H). MS (CI/NH3) m/z 361 (M + H+)+.
9-[2,3-Bis-(tert-butyldimethylsilanyloxymethyl)cyclopropylmethyl]-2,6-dichloro-9H-purine (69). Method A.
A mixture of two isomers 69 (147 mg, 59%) was obtained from a mixture 68 (160 mg, 0.464 mmol). 1H NMR (CDCl3) δ 0.01–0.20 (m, 12H), 0.61–0.89 (m, 18H), 1.42–1.55 (m, 3H), 3.54–4.48 (m, 6H). 8.59 and 8.61 (two singlets, 1H).
meso-1,2-trans-{9-[2,3-Bis-(tert-butyldimethylsilanyloxymethyl)cyclopropylmethyl]-2-chloro-9H-purin-6-yl}-methylamine (70) (meso-trans). Method A.
Compound 70 (78 mg) and 71 (31 mg) were obtained from 69 (147 mg, 0.276 mmol) and separated by silica gel column chromatography. 1H NMR (CDCl3) δ 0.03 (s, 12H), 0.87 (s, 18H), 1.20–1.35 (m, 3H), 3.18 (bs, 3H), 3.62 (dd, 2H, J = 5.9, 10.3 Hz), 3.78 (dd, 2H, J = 4.4, 10.3 Hz), 4.05 (d, 2H, J = 7.3 Hz), 6.11 (bs, 1H), 8.19 (s, 1H). MS (CI/NH3) m/z 526 (M + H+)+.
meso-1,2-cis-{9-[2,3-Bis-(tert-butyldimethylsilanyloxymethyl)cyclopropylmethyl]-2-chloro-9H-purin-6-yl}-methylamine (71) (meso-cis). Method A.
Compound 70 (78 mg) and 71 (31 mg) were obtained from 69 (147 mg, 0.276 mmol) and separated by silica gel column chromatography. 1H NMR (CDCl3) δ 0.06 (s, 6H), 0.07 (s, 6H), 0.90 (s, 18H), 1.35–1.55 (m, 3H), 3.19 (bs, 3H), 3.78 (dd, 2H, J = 8.8, 11.7 Hz), 3.92 (dd, 2H, J = 5.9, 11.7 Hz), 4.32 (d, 2H, J = 5.9 Hz), 6.07 (bs, 1H), 8.18 (s, 1H). MS (CI/NH3) m/z 526 (M + H+)+.
meso-1,3-trans-[2-(2-Chloro-6-methylaminopurin-9-ylmethyl)-3-hydroxymethylcyclopropyl]methanol (72) (meso-trans). Method E.
Crude 72 (50 mg) was obtained from 70 (78 mg, 0.148 mmol). 1H NMR (CDCl3) δ 1.24 (m, 1H), 1.38(m, 2H), 3.05 (bs, 3H), 3.48–3.71 (m, 2H), 4.08 (d, 2H, J = 6.9 Hz), 8.13 (s, 1H). MS (CI/NH3) m/z 298 (M + H+)+.
meso-1,3-cis-[2-(2-Chloro-6-methylaminopurin-9-ylmethyl)-3-hydroxymethylcyclopropyl]methanol (73) (meso-cis). Method E.
Crude 73 (20 mg) was obtained from 71 (31 mg, 0.059 mmol). 1H NMR (CD3OD) δ 1.38–1.63 (m, 3H), 3.07 (bs, 3H), 3.72–3.93 (m, 4H), 4.36 (d, 2H, J = 7.1 Hz), 8.16 (s, 1H). MS (CI/NH3) m/z 298 (M + H+)+.
meso-1,3-trans-Phosphoric Acid Dibenzyl Ester 2-(bisbenzyloxyphosphoyloxymethyl)-3-(2-chloro-6-methylaminopurin-9-ylmethyl)cyclopropylmethyl Ester (74) (meso-trans). Method F.
Compound 74 (56 mg, 46% two steps yield from 70) was obtained from crude 72 (50 mg).1H NMR (CDCl3) δ 1.14–1.43 (m, 3H), 3.10 (bs, 3H), 3.82–3.97 (m, 6H), 4.90–5.06 (m, 8H), 5.99 (bs, 1H), 7.21–7.39(m, 20H), 7.68 (bs, 1H). 31P NMR (CDCl3) δ −22.458. MS (CI/NH3) m/z 818 (M + H) +. High-resolution MS (positive-ion FAB) calcd for C40H42N5O8- ClP2Cs [M2− + H+ + Cs2+]+ 950.1252, found 950.1229.
meso-1,3-cis-Phosphoric Acid Dibenzyl Ester 2-(bisbenzyloxyphosphoyloxymethyl)-3-(2-chloro-6-methylaminopurin-9-ylmethyl)cyclopropylmethyl Ester (75) (meso-cis). Method F.
Compound 75 (17 mg, 33% two steps yield from 71) was obtained from crude 73 (20 mg). 1H NMR (CDCl3) δ 1.46 (m, 2H), 1.61 (m, 2H), 3.15 (bs, 3H), 3.97 (dd, 2H, J = 7.8, 10.8 Hz), 4.04 (d, 2H, J = 6.9 Hz), 4.18 (dd, 2H, J = 6.6, 10.8 Hz), 4.90–5.19 (m, 8H), 5.99 (bs, 1H), 7.23–7.40(m, 20H), 7.70 (bs, 1H). 31P NMR (CDCl3) δ −22.338. MS (CI/NH3) m/z 818 (M + H+)+. High-resolution MS (positive-ion FAB) calcd for C40H42N5O8ClP2Cs [M2− + H+ + Cs2+]+ 950.1252, found 950.1298.
1,2-Bis-(tert-butyldimethylsilanyloxymethyl)-3-methoxymethoxymethylcyclopropane (76).
Chloromethylmethyl ether (234 mg, 2.90 mmol) was added dropwise by syringe over 2 min to a stirred solution of cyclopropyl alcohol 68a (1.0 g, 2.90 mmol) and diisopropylethylamine (563 mg, 4.36 mmol) in methylene chloride (15 mL) cooled in an ice-water bath. The reaction mixture was stirred for 1 h at the same temperature, for 4 h at room temperature. Ice (10 g) was added, the mixture was stirred for 10 min, and water (30 mL) was added. The aqueous layer was extracted with ethyl acetate (150 mL), and the organic layer was washed with brine/water (30 mL × 2, 1/1), dried over sodium sulfate, filtered, and concentrated to give 76 (1.05 g, 93%). 1H NMR (CDCl3) δ 0.05 (s, 6H), 0.90 (s, 9H), 0.94–1.16 (m, 3H), 3.36 (s, 3H), 3.45 (d, 1H, J = 5.9 Hz), 3.66–3.76 (m 4H), 4.66 (s, 2H). MS (CI/NH3) m/z 405 (M + H+)+
(2-Hydroxymethyl-3-methoxymethoxymethylcyclopropyl)methanol (77).
A mixture of disilyl ether 76 (640 mg, 1.65 mmol), tetrabutylammonium fluoride (2.47 mL, 2.47 mmol, 1 M solution in tetrahydrofuran), and tetrahydrofuran (5 mL) was stirred for 3 h at room temperature. After removal of volatile materials in vacuo, the residue was purified by column chromatography (ethyl acetate/methanol = 10:1) to give diol 77 (275 mg, 95%). MS (CI/NH3) m/z 194 (M + NH4+)+.
(±)-1,2-cis-1,3-trans-[2-(tert-Butyldiphenylsilanyloxymethyl)-3-methoxymethoxymethylcyclopropyl]methanol ((±)-78).
Neat tert-butyldiphenylsilyl chloride (498 mg, 1.56 mmol) was added to a stirred solution of diol 77 (275 mg, 1.56 mmol) and (dimethylamino)pyridine (190 mg, 1.56 mmol) in methylene chloride (7 mL). The reaction mixture was stirred for 3 days at room temperature. Methylene chloride was removed in vacuo, and the residue was purified by column chromatography (hexanes/ethyl acetate = 1:1) to give 78 (479 mg, 70%). 1H NMR (CDCl3) δ 0.93 (m, 1H), 1.07 (s, 9H), 1.21 (m, 1H), 1.41 (m, 1H), 3.30 (s, 3H), 3.34–3.49 (m, 4H), 4.02 (dd, 1H, J = 5.4, 12.0 Hz), 4.11 (dd, 1H, J = 5.4, 11.5 Hz), 4.57 (s, 2H), 7.36–7.50 (m, 6H), 7.65–7.76 (m, 4H). MS (CI/NH3) m/z 415 (M + H+)+.
(±)-1,2-cis-1,3-trans-Methanesulfonic Acid 2-(tert-Butyldiphenylsilanyloxymethyl)-3-methoxymethoxymethylcyclopropylmethyl Ester ((±)-79). Method D.
Crude (±)-79 (538 mg, 95%) was obtained from (±)-78 (479 mg, 1.15 mmol).
(±)-1,2-cis-1,3-trans-{9-[2-(tert-Butyldiphenylsilanyloxymethyl)-3-methoxymethoxymethyl-cyclopropylmethyl]-2-chloro-9H-purin-6-yl}-methylamine ((±)-80). Method D.
Compound (±)-80 (430 mg, 71%) was obtained from (±)-79 (520 mg, 1.05 mmol). 1H NMR (CDCl3) δ 1.05 (s, 9H), 1.04–1.46 (m, 3H), 3.18 (bs, 3H), 3.28 (s, 3H), 3.24–3.45 (m, 2H), 3.50 (dd, 1H, J = 6.0, 10.4 Hz), 3.70 (dd, 1H, J = 8.0, 11.4 Hz), 3.98 (dd, 1H, J = 5.1, 11.4 Hz), 4.28 (dd, 1H, J = 7.0, 14.6 Hz), 4.54 (m, 2H), 6.18 (bs, 1H), 7.33–7.49 (m, 6H), 7.61–7.74 (m, 4H), 8.05 (s, 1H). MS (positive-ion FAB) 580 (M + H+)+.
(±)-1,2-cis-1,3-trans-[2-(2-Chloro-6-methylaminopurin-9-ylmethyl)-3-hydroxymethylcyclopropyl]methanol ((±)-81).
A mixture of silyl ether (±)-80 (230 mg, 0.467 mmol), c-HCl (2 mL), and methanol (6 mL) was stirred at 80 °C for 4 h. All volatile materials were removed by nitrogen purging. The residue was dissolved in methanol (20 mL) and treated with potassium carbonate (100 mg). The mixture was stirred at room temperature for 4 h, filtered, and evaporated. The residue was purified by PTLC (chloroform/methanol = 4:1) to give diol (±)-81 (90 mg, 65%). 1H NMR (CD3OD) δ 1.07–1.43 (m, 3H), 3.08 (bs, 3H), 3.32–3.66 (m, 3H), 3.92 (dd, 1H, J = 5.8, 11.8 Hz), 4.20–4.34 (m, 2H), 8.20 (s, 1H). MS (positive-ion FAB) 298 (M + H+)+.
(±)-1,2-cis-1,3-trans-Phosphoric Acid Di-tert-butyl Ester 2-(2-Chloro-6-methylaminopurin9-yl)-3-(di-tert-butoxyphosphoryloxymethyl)cyclopropylmethyl Ester ((±)-82). Method C.
Compound (±)-82 (46 mg, 62%) was obtained from diol (±)-81 (32 mg, 0.108 mmol). 1H NMR (CDCl3) δ 1.43 (s, 9H), 1.45 (s, 9H), 1.49 (s, 18H), 3.18 (s, 3H), 3.73 3.98 (m, 3H), 4.15–4.42 (m, 3H), 6.19 (bs. 1H), 7.93 (s, 1H). MS (positive-ion FAB) 682 (M + H+)+.
(±)-2-(2-Chloro-6-methylaminopurin-9-yl)cyclopropane-1,1-dicarboxylic Acid Diethyl Ester ((±)-84).
A mixture of allyl bromide 83 (115 mg, 0.435 mmol), potassium carbonate (71 mg, 0.52 mmol), N6-methyl-2-chloropurine (80 mg, 0.435 mmol), and dimethylformamide (3 mL) was stirred for 2 days at 30–35 °C. After the usual aqueous workup using ethyl acetate (40 mL) and 5% NaCl solution (20 mL × 3), the organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The residue was purified by column chromatography (hexanes/ethyl acetate = 1:5) to give (±)-84 (68 mg, 41%). 1H NMR (CDCl3) δ 1.01 (t, 3H, J = 7.1 Hz), 1.32 (t, 3H, J = 7.1 Hz), 2.06 (dd, 1H, J = 6.6, 8.2 Hz), 2.74 (t, 1H, J = 6.3 Hz), 3.15 (bs, 3H), 3.87–4.06 (m, 2H), 4.19–4.39 (m, 3H), 6.19 (bd, 1H, J = 3.6 Hz), 7.68 (s, 1H). MS (positive-ion FAB) 368 (M + H+)+.
(±)-[2-(2-Chloro-6-methylaminopurin-9-yl)-1-hydroxymethylcyclopropyl]methanol ((±)-85).
To a cooled (−78 °C), stirred solution of diester (±)-84 (33 mg, 0.0860 mmol) in methylene chloride was added 1 M DIBAL (in hexanes, 0.52 mL, 0.52 mmol). The reaction mixture was stirred for 20 min at the same temperature and for 1 h at room temperature. After quenching with methanol (0.5 mL), the resulting solution was directly purified by PTLC (chloroform/methanol = 4:1) to give (±)-85 (17 mg, 70%). 1H NMR (CDCl3 0.5 mL + CD3OD 30 μL) δ 1.14 (dd, 1H, J = 4.5, 6.8 Hz), 1.32 (t, 1H, J = 6.3, 6.7 Hz), 3.07 (bs, 3H), 3.11 (d, 1H, J = 12.4 Hz), 3.29 (dd, 1H, J = 4.3, 7.7 Hz), 3.60 (d, 1H, J = 12.2 Hz), 3.70 (ABq, 1H, J = 12.0 Hz), 7.09 (bs, 1H), 7.74 (s, 1H). MS (positive-ion FAB) 284 (M + H+)+.
(±)-Phosphoric Acid Di-tert-butyl Ester 2-(2-Chloro-6-methylaminopurin-9-yl)-1-(di-tert-butoxyphosphoryloxymethyl)cyclopropylmethyl Ester ((±)-86). Method C.
(±)-86 (30 mg, 85%) was obtained from (±)-85 (15 mg, 0.053 mmol). 1H NMR (CDCl3) δ 1.39 (s, 9H), 1.41 (s, 9H), 1.51 (s, 9H), 1.52 (s, 9H), 1.63 (t, 1H, J = 7.3 Hz), 1.78 (dd, 1H, J = 4.9, 7.0 Hz), 3.17 (bs, 3H), 3.58 (dd, 1H, J = 5.0, 7.7 Hz), 3.74–3.87 (m, 2H), 4.07–4.21 (m, 2H), 6.10 (bs, 1H), 7.79 (s, 1H). MS (positive-ion FAB) 668 (M + H+)+.
(±)-[2,2-Bis-(tert-butyldimethylsilanyloxymethyl)-cyclopropyl]methanol ((±)-87).
To a stirred, cooled (0 °C) solution of allyl alcohol 61 (220 mg, 0.635 mmol) in methylene chloride (8 mL) was added diethylzinc (1 M in hexane, 4.45 mL, 4.45 mmol), followed by an addition of diiodomethane (510 mg, 1.91 mmol). After 20 min at room temperature, the mixture was allowed to reach rt and stirred overnight. Aqueous saturated ammonium chloride was carefully added, and the resulting mixture was extracted with ether (30 mL), washed successively with saturated sodium bicarbonate (20 mL), brine (20 mL), dried over sodium sulfate, filtered, evaporated. The residue was purified by silica gel column chromatography (hexanes/ethyl acetate = 4:1) to give 87 (220 mg, 96%). 1H NMR (CDCl3) δ 0.04 (s, 6H), 0.11 (s, 3H), 0.12 (s, 3H), 0.35 (t, 1H, J = 4.1 Hz), 0.70 (dd, 1H, J = 5.0, 8.4 Hz), 0.90 (s, 9H), 0.92 (s, 9H), 1.19 (m, 1H), 2.87 (d, 1H, J = 10.3 Hz), 3.25 (d, 1H, J = 11.0 Hz), 3.26 (t, 1H, J = 12.1 Hz), 3.96 (dd, 1H, J = 5.4, 12.1 Hz), 4.13 (d, 1H, J = 10.3 Hz), 4.29 (d, 1H, J = 11.0 Hz). MS (positive-ion FAB) 361 (M + H+)+.
(±)-Methanesulfonic Acid 2,2-bis-(tert-butyldimethylsilanyloxymethyl)cyclopropylmethyl Ester ((±)-88). Method D.
Crude (±)-88 (250 mg, 93%) was obtained from (±)-87 (220 mg, 0.610 mmol).
(±)-{9-[2,2-Bis-(tert-butyldimethylsilanyloxymethyl)-cyclopropylmethyl]-2-chloro-9H-purin-6-yl}methylamine ((±)-89). Method D.
Compound (±)-89 (185 mg, 62%) was obtained from crude (±)-88 (250 mg, 0.570 mmol). 1H NMR (CDCl3) δ 0.02 (s, 6H), 0.04 (s, 3H), 0.06 (s, 3H), 0.0.54 (t, 1H, J = 5.0 Hz), 0.79 (dd, 1H, J = 5.0, 8.7 Hz), 0.88 (s, 9H), 0.89 (s, 9H), 1.31 (m, 1H), 3.20 (bs, 3H), 3.42 (d, 1H, J = 10.2 Hz), 3.62 (d, 1H, J = 11.1 Hz), 3.66 (d, 1H, J = 10.3 Hz), 3.97 (d, 1H, J = 11.0 Hz), 4.04 (dd, 1H, J = 9.1, 14.3 Hz), 4.42 (dd, 1H, J = 6.0, 14.4 Hz), 8.08 (s, 1H). MS (positive-ion FAB) 526 (M + H+)+.
(±)-[2-(2-Chloro-6-methylaminopurin-9-ylmethyl)-1-hydroxymethylcyclopropyl]methanol ((±)-90). Method E.
Compound (±)-90 (56 mg, 55%) was obtained from (±)-89 (180 mg, 0.342 mmol). 1H NMR (CD3OD) δ 0.60 (t, 1H, J = 5.4 Hz), 0.77 (dd, 1H, J = 5.1, 8.7 Hz), 1.37 (m, 1H), 3.08 (bs, 3H), 3.41 (d, 1H, J = 11.4 Hz), 3.62 (d, 1H, J = 11.1 Hz), 3.65 (d, 1H, J = 11.8 Hz), 3.99 (d, 1H, J = 12.0 Hz), 4.28 (d, 2H, J = 7.3 Hz), 8.18 (s, 1H). MS (positive-ion FAB) 298 (M + H+)+.
(±)-Phosphoric Acid Di-tert-butyl Ester 2-(2-chloro-6-methylaminopurin-9-ylmethyl)-1-(di-tert-butoxyphosphoryloxymethyl)cyclopropylmethyl Ester ((±)-91). Method C.
Compound (±)-91 (23 mg, 40%) was obtained from (±)-90 (25 mg, 0.0840 mmol). 1H NMR (CDCl3) δ 0.77 (t, 1H, J = 5.7 Hz), 1.03 (dd, 1H, J = 5.7, 8.4 Hz), 1.21–1.41 (m, 1H), 1.44 (s, 9H), 1.45 (s, 9H), 1.49 (s, 18H), 3.17 (bs, 3H), 3.74 (dd, 1H, J = 5.9, 10.4 Hz), 3.95 (dd, 1H, J = 5.4, 11.5 Hz), 4.04 (dd, 1H, J = 6.6, 10.6 Hz), 4.27 (d, 2H, J = 6.8 Hz), 4.38 (dd, 1H, J = 4.5, 11.4 Hz), 6.66 (bs, 1H), 7.95 (s, 1H). MS (positive-ion FAB) 682 (M + H+)+.
Method G. Deprotection of tert-Butylphosphates with Dowex 50 Resin.
A mixture of bis(di-tert-butyl phosphate) 26 (20 mg, mmol) and Dowex 50 resin (100 mg) and methanol/water (1:1, 5 mL) was stirred at 80 °C for 16 h. The resulting mixture was filtered off through glass filter and washed with water (5 mL × 2), the combined filtrate was lyophilized, and the residue was purified with an ion-exchange column chromatography using Sephadex-DEAE A-25 resin with a linear gradient (0.01 to 0.5 M) of 0.5 M ammonium bicarbonate as the mobile phase to give bisphosphate 5 (9.0 mg, 63%) as an ammonium salt.
Phosphoric Acid Mono-[2-(2-chloro-6-methylaminopurin-9-yl)-3-phosphonooxypropyl] Ester, Ammonium Salt (5).
1H NMR (D2O) δ 3.03 (s, 3H), 4.35 (m, 2H), 4.46 (m, 2H), 5.16 (m, 1H), 8.97 (s, 1H). 31P NMR (D2O) 4.107. MS (negative-ion FAB) 416 (M − H+)−.
Method H. Deprotection of Benzyl Phosphates with Boron Trichloride.
Bis(dibenzyl phosphate) 36 (37 mg, 0.0442 mmol) was dissolved in anhydrous methylene chloride (3 mL) and cooled to 0 °C. To this solution was added 1 M boron trichloride (in dichloromethane, 0.442 mL, 0.442 mmol), and the reaction mixture was stirred at 5 °C for 2 d (the progress of the reaction was monitored by HPLC). After the deprotection was done, the resulting mixture was quenched by 1 M triethylammonium acetate (5 mL), followed by addition of water (5 mL). This mixture was lyophilized, and the residue was purified with an ion-exchange column chromatography using Sephadex-DEAE-25 resin with a linear gradient (0.01 to 0.5 M) of 0.5 M ammonium bicarbonate as the mobile phase to give bisphosphate 7 (9.6 mg, 41%).
Phosphoric Acid Mono-[2-(6-methylaminopurin-9-ylmethyl)-3-phosphonooxy-propyl] Ester, Ammonium Salt (6). Method H.
Compound 6 (12.0 mg, 70%) was obtained from 35 (28 mg, 0.369 mmol). 1H NMR (D2O) δ 2.57 (m, 1H), 3.11 (bs, 3H), 3.87 (m, 4H), 4.36 (d, 2H, J = 7.1 Hz), 8.17 (s, 1H), 8.22 (s, 1H). 31P NMR (D2O) −21.005. MS (negative-ion FAB) 396 (M − H+)−.
Phosphoric Acid Mono-[2-(2-chloro-6-methylaminopurin-9-ylmethyl)-3-phosphonooxypropyl] Ester, Ammonium Salt (7). Method H.
Compound 7 (9.6 mg, 41%) was obtained from 36 (37 mg, 0.0467 mmol). 1H NMR (D2O) δ 2.56 (m, 1H), 3.04 (bs, 3H), 3.87 (m, 4H), 4.28 (d, 2H, J = 7.3 Hz), 8.09 (s, 1H). 31P NMR (D2O) −20.975 (s).
Method I. Deprotection of tert-Butylphosphates with Trifluoroacetic Acid.
A solution of tert-butylphosphate 39 (5 mg, 0.011 mmol) in 5% trifluoroacetic acid in methylene chloride was stirred for 2 h at room temperature. All volatile material was removed in vacuo. The residue was purified using ion-exchange column chromatography on Sephadex-DEAE-25 resin and a linear gradient (0.01 to 0.5 M) of 0.5 M ammonium bicarbonate as the mobile phase to give monophosphate 8 (3.3 mg, 79%) as an ammonium salt.
(±)-Phosphoric Acid Mono-[2-(2-chloro-6-methylaminopurin-9-ylmethyl)-3-hydroxypropyl] Ester, Ammonium Salt ((±)-8). Method I.
Compound 8 (3.3 mg, 79%) was obtained from (5.0 mg, 0.0107 mmol). 1H NMR (D2O) δ 2.55 (m, 1H), 3.11 (s, 3H), 3.69 (d, 2H, J = 5.9 Hz), 3.82–4.02 (m, 2H), 4.44 (d, 2H, J = 7.8 Hz), 8.87 (s, 1H). 31P NMR (D2O) 1.292 (s).
(±)-Phosphoric Acid Mono-[2-(2-chloro-6-methylami nopurin-9-ylmethyl)-3-(dimethoxyphosphoryloxy)propyl] Ester, Ammonium Salt ((±)-9). Method I.
Compound 9 (2.5 mg, 76%) was obtained from 40 (3.8 mg, 0.00664 mmol). 1H NMR (D2O) δ 2.73 (m, 1H), 3.09 (bs, 3H), 3.67 (d, 3H, J = 3.2 Hz), 3.71 (d, 3H, J = 3.2 Hz), 3.96 (m, 2H), 4.16 (m, 2H), 4.38 (d, 2H, J = 6.0 Hz), 8.14 (s, 1H). 31P NMR (D2O) 0.664 (bs), 1.538 (s).
(±)-Acetic Acid 2-(2-Chloro-6-methylaminopurin-9-ylmethyl)-3-phosphoryloxypropyl Ester, Ammonium Salt ((±)-10). Method I.
Compound 10 (6 mg, 59%) obtained from 41 (12 mg, 0.0237 mmol). 1H NMR (D2O) δ 1.99 (s, 3H), 2.73 (m, 1H), 3.09 (bs, 3H), 3.98 (m, 2H), 4.12 (d, 2H, J = 5.1 Hz), 4.44 (d, 2H, J = 6.3 Hz), 8.60 (s, 1H). 31P NMR (D2O) 0.352 (s).
(±)-Diphosphoric Acid Mono-[2-(2-chloro-6-methylami nopurin-9-ylmethyl)-3-phosphonooxypropyl] Ester, Ammonium Salt ((±)-11).
Compound 44 (6 mg, 0.00808 mmol) was dissolved in methylene chloride (1 mL), and trimethylsilyl bromide (60 μL, 0.455 mmol) was added. The reaction mixture was stirred at room temperature for 10 min, followed by quenching with 0.1 M triethylammonium bicarbonate (5 mL). Methylene chloride was removed in vacuo, and the residue was lyophilized and purified using ion-exchange column chromatography on Sephadex-DEAE-25 resin with a linear gradient (0.01–1.0 M) of 1.0 M ammonium bicarbonate as the mobile phase to give bisphosphate 11 (3.5 mg, 72%) as an ammonium salt. 1H NMR (D2O) δ 2.59 (m, 1H), 3.05 (bs, 3H), 3.74–4.09 (m, 4H), 4.31 (d, 2H, J = 7.1 Hz), 8.12 (s, 1H). 31P NMR (D2O) −10.457 (bs), 0.774 (s).
Phosphoric Acid Mono-[3-(2-chloro-6-methylaminopurin-9-ylmethyl)-5-phosphonooxypentyl] Ester, Ammonium Salt (12). Method I.
Compound 12 (8.0 mg, 69%) was obtained from 49 (15 mg, 0.0219 mmol) as an ammonium salt. 1H NMR (D2O) δ 1.61–1.85 (m, 4H), 2.41 (m, 1H), 3.08 (s, 3H), 3.80–4.09 (m, 4H), 4.33 (d, 2H, J = 7.3 Hz), 8.95 (s, 1H). 31P NMR (D2O) 0.689 (s).
(R)-Phosphoric Acid Mono-[1-(2-chloro-6-methylaminopurin-9-ylmethyl)-2-phosphonooxyethyl] Ester, Ammonium Salt (13). Method I.
Compound 13 (5.0 mg, 44%) was obtained from 58 (15 mg, 0.0234 mmol) as an ammonium salt. 1H NMR (D2O) δ 3.07 (bs, 3H), 3.97 (m, 2H), 4.32–4.60 (m, 3H), 8.10 (s, 1H). 31P NMR (D2O) −0.446, −1.673.
(S)-Phosphoric Acid Mono-[1-(2-chloro-6-methylaminopurin-9-ylmethyl)-2-phosphonooxyethyl] Ester, Ammonium Salt (14). Method I.
Compound 14 (8.3 mg, 55%) was obtained from 59 (20 mg, 0.0311 mmol) as an ammonium salt. 1H NMR (D2O) δ 3.06 (bs, 3H), 3.78 (t, 2H, J = 4.5 Hz), 4.30–4.55 (m, 3H), 8.17 (s, 1H). 31P NMR (D2O) 3.851, 2.745. MS (negative-ion FAB) 416 (M − H+)−.
Phosphoric Acid 4-(2-Chloro-6-methylaminopurin-9-yl)-2-phosphonooxymethylbut-2-enyl Ester, Ammonium Salt (15). Method I.
Compound 15 (9.7 mg, 63%) was obtained from 65 (20 mg, 0.0299 mmol) as an ammonium salt. 1H NMR (D2O) δ 3.03 (bs, 3H), 4.53 (d, 2H, J = 7.0 Hz), 4.67 (d, 2H, J = 7.0 Hz), 5.01 (d, 2H, J = 6.6 Hz), 6.03 (m, 1H), 8.61 (s, 1H). 31P NMR (D2O) 0.342.
meso-1,3-trans-Phosphoric Acid Mono-[2-(2-chloro-6-methylaminopurin-9-ylmethyl)-3-phosphonooxymethylcyclopropylmethyl] Ester, Ammonium Salt (16). Method H.
Compound 16 (28.0 mg, 77%) was obtained from 74 (56 mg, 0.068 mmol) as an ammonium salt.1H NMR (D2O) δ 1.42–1.52 (m, 3H), 3.05 (bs, 3H), 3.93 (m, 4H), 4.13 (d, 2H, J = 6.6 Hz), 8.32 (s, 1H). 31P NMR (D2O) −21.217. MS (positive-ion FAB) 458 (M + H+)+.
meso-1,3-cis-Phosphoric Acid Mono-[2-(2-chloro-6-methylaminopurin-9-ylmethyl)-3-phosphonooxymethylcyclopropylmethyl] Ester, Ammonium Salt (17). Method H.
Compound 17 (6.2 mg, 57%) was obtained from 75 (17.0 mg, 0.0208 mmol) as an ammonium salt. 1H NMR (D2O) δ 1.67 (m, 2H), 1.82 (m, 1H), 3.07 (bs, 3H), 4.05 (m, 2H), 4.22 (m, 2H), 4.40 (d, 2H, J = 6.9 Hz), 8.28 (s, 1H). 31P NMR (D2O) −21.287. MS (positive-ion FAB) 458 (M + H+) +.
(±)-1,2-cis-1,3-trans-Phosphoric Acid Mono-[2-(2-chloro-6-methylaminopurin-9-ylmethyl)-3-phosphonooxymethylcyclopropylmethyl] Ester, Ammonium Salt ((±)-18). Method H.
Compound ((±)-18) (11.0 mg, 71%) was obtained from (±)-82 (20 mg, 0.0293 mmol) as an ammonium salt. 1H NMR (D2O) δ 1.41 (m, 1H), 1.49–1.67 (m, 2H), 2.99 (bs, 3H), 3.62–3.97 (m, 4H), 4.15–4.28 (m, 4H), 8.48 (s, 1H). 31P NMR (D2O) 0.583.
(±)-Phosphoric Acid Mono-[2-(2-chloro-6-methylaminopurin-9-yl)-1-phosphonooxymethylcyclopropylmethyl] Ester ((±)-19). Method I.
Compound (±)-19 (7.0 mg, 61%) was obtained from (±)-86 (15 mg, 0.0225 mmol) as an ammonium salt. 1H NMR (D2O) δ 1.69 (t, 1H, J = 7.8 Hz), 1.77 (dd, 1H, J = 4.6, 7.2 Hz), 3.06 (bs, 3H), 3.49 (dd, 1H, J = 4.4, 11.3 Hz), 3.79 (dd, 1H, J = 4.6, 7.8 Hz), 3.91 (dd, 1H, J = 6.4, 11.0 Hz), 4.10 (dd, 1H, J = 4.4, 11.3 Hz), 4.35 (dd, 1H, J = 6.7, 11.0 Hz), 8.67 (s, 1H). 31P NMR (D2O) 0.183, 0.644 (s).
(±)-Phosphoric Acid Mono-[2-(2-chloro-6-methylaminopurin-9-ylmethyl)-1-phosphonooxymethylcyclopropylmethyl] Ester ((±)-20). Method I.
Compound (±)-20 (5.0 mg, 55%) was obtained from (±)-91 (12 mg, 0.0176 mmol) as an ammonium salt. 1H NMR (D2O) δ 0.85 (t, 1H, J = 5.5 Hz), 1.06 (dd, 1H, J = 5.8, 8.7 Hz), 1.63 (m, 1H), 3.08 (bs, 3H), 3.67 (dd, 1H, J = 4.8, 10.5 Hz), 3.86 (dd, 1H, J = 3.6, 11.3 Hz), 4.01 (dd, 1H, J = 5.5, 10.6 Hz), 4.20–4.40 (m, 3H), 8.39 (s, 1H). 31P NMR (D2O) 0.548 (s).
Pharmacological Analyses.
P2Y1 receptor promoted stimulation of inositol phosphate formation by adenine nucleotide analogues was measured in turkey erythrocyte membranes as previously described.27,28 The K0.5 values were averaged from 3 to 8 independently determined concentration-effect curves for each compound. Briefly, 1 mL of washed turkey erythrocytes was incubated in inositol-free medium (DMEM; Gibco, Gaithersburg MD) with 0.5 mCi of 2-[3H]myo-inositol (20 Ci/mmol; American Radiolabeled Chemicals, Inc., St. Louis MO) for 18–24 h in a humidified atmosphere of 95% air/5% CO2 at 37 °C. Erythrocyte ghosts were prepared by rapid lysis in hypotonic buffer (5 mM sodium phosphate, pH 7.4, 5 mM MgCl2, 1 mM EGTA) as described.27 Phospholipase C activity was measured in 25 μL of [3H]inositol-labeled ghosts (approximately 175 μg of protein, 200–500,000 cpm/assay) in a medium containing 424 μM CaCl2, 0.91 mM MgSO4, 2 mM EGTA, 115 mM KCl, 5 mM KH2PO4, and 10 mM Hepes, pH 7.0. Assays (200 μL final volume) contained 1 μM GTPγS, and the indicated concentrations of nucleotide analogues. Ghosts were incubated at 30 °C for 5 min, and total [3H]inositol phosphates were quantitated by anion exchange chromatography as previously described.27,28
Data Analysis.
Agonist potencies were calculated using a four-parameter logistic equation and the GraphPad software package (GraphPad, San Diego, CA). EC50 values (mean ± standard error) represent the concentration at which 50% of the maximal effect is achieved. Relative efficacies (%) were determined by comparison with the effect produced by a maximal effective concentration of 2-MeSADP in the same experiment. Antagonist IC50 values (mean ± standard error) represent the concentration needed to inhibit by 50% the effect elicited by 30 nM 2-MeSADP. The percent of maximal inhibition is equal to 100 minus the residual fraction of stimulation at the highest antagonist concentration. All concentration-effect curves were replicated in at least three separate experiments, carried out with different membrane preparations using duplicate or triplicate assays.
Computational Methods.
A molecular model of the human P2Y1 receptor was built and optimized using Sybyl 6.634 modeling package, following the homology modeling approach described in our previous studies30,35 and using the recently reported X-ray structure of bovine rhodopsin22 as a structural template. All calculations were performed on a Silicon Graphics Octane R12000 workstation. Briefly, sequences of the P2Y1 transmembrane domains, identified in our previously published model, were amended by comparison to the corresponding domains of rhodopsin, according to a published sequence alignment.36 Transmembrane P2Y1 helices were built from these sequences, in homology to the corresponding helices of rhodopsin, and minimized individually. The minimized helices were then grouped by adding one at a time until a helical bundle (TM region), matching the overall characteristics of the crystallographic structure of rhodopsin, was obtained. The TM region was further modified by adding the amino acid residues 189–213, which comprise EL2. The conformation of EL2 was initially modeled according to the corresponding domain in rhodopsin including the Cys124–Cys202 disulfide bond. At each step the structures were minimized using the Tripos force field with Amber37 all-atom force parameters until the rms value of the conjugate gradient (CG) was <0.1 kcal/mol/Å. A fixed dielectric constant = 4.0 was used throughout these calculations. A model of adenosine 5′-triphosphate (ATP) was constructed from the crystallographic coordinates of ATP-phosphoglycerate kinase.38 Models of 92 and other nucleotides were constructed from ATP using the “Sketch Molecule” module of Sybyl. The ligands were minimized in Sybyl (using MOPAC calculated partial atomic charges) and were rigidly docked into the helical bundle using graphical manipulation coupled to continuous energy monitoring (Dock module of Sybyl). Whenever a final position was reached, consistent with a local energy minimum, the complexes of receptor and ligand were subjected to an additional CG minimization run of up to 1500 steps.
Acknowledgment.
This work was supported by USPHS Grants GM38213 and HL54889. D.B. is grateful for financial support from Gilead Sciences (Foster City, CA).
References
- (1).Fredholm BB; Abbracchio MP; Burnstock G; Dubyak GR; Harden TK; Jacobson KA; Schwabe U; Williams M Toward a revised nomenclature for P1 and P2 receptors. Trends Pharm. Sci 1997, 18, 79–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).North RA; Barnard EA Nucleotide receptors. Curr. Opin. Neurobiol 1997, 7, 346–357. [DOI] [PubMed] [Google Scholar]
- (3).Burnstock G; Williams M P2 Purinergic Receptors: Modulation of cell function and therapeutic potential. J. Pharmacol. Exp. Ther 2000, 295, 862–869. [PubMed] [Google Scholar]
- (4).Webb TE; Simon J; Krishek BJ; Bateson AN; Smart TG; King BF; Burnstock G; Barnard EA Cloning and functional expression of a brain G-protein-coupled ATP receptor. FEBS Lett. 1993, 324, 219–225. [DOI] [PubMed] [Google Scholar]
- (5).Schachter JB; Li Q; Boyer JL; Nicholas RA; Harden TK Second messenger cascade specificity and pharmacological selectivity of the human P2Y1-purinoceptor. Br. J. Pharmacol 1996, 118, 167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).King BF; Townsend-Nicholson A; Burnstock G Metabotropic receptors for ATP and UTP: exploring the correspondence between native and recombinant nucleotide receptors. Trends Pharmacol. Sci 1998, 19, 506–514. [DOI] [PubMed] [Google Scholar]
- (7).Jin J; Daniel JL; Kunapuli SP Molecular basis for ADP-induced platelet activation. II. The P2Y1 receptor mediates ADP-induced intracellular calcium mobilization and shape change in platelets. J. Biol. Chem 1998, 273, 2030–2034. [DOI] [PubMed] [Google Scholar]
- (8).Hechler B; Leon C; Vial C; Vigne P; Frelin C; Cazenave JP; Gachet C The P2Y1 receptor is necessary for adenosine 5′-diphosphate-induced platelet aggregation. Blood 1998, 92, 152–159. [PubMed] [Google Scholar]
- (9).Jarvis GE; Humphries RG; Robertson MJ; Leff P ADP can induce aggregation of human platelets via both P2Y1 and P2T receptors. Br. J. Pharmacol 2000, 129, 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hollopeter G; Jantzen H-M; Vincent D; Li G; England L; Ramakrishnan V; Yang R-B; Nurden P; Nurden A; Julius D; Conley P Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature 2001, 409, 202–207. [DOI] [PubMed] [Google Scholar]
- (11).Boyer JL; Adams M; Jang SY; Ravi RG; Jacobson KA; Harden TK Development of P2Y1 receptor antagonists. Drug Dev. Res (Abstract S14–02) 2000, 50, 21. [Google Scholar]
- (12).Gachet C The role of ADP receptors in experimental thrombosis and haematostasis. Drug Dev. Res (Abstract S11–03) 2000, 50, 19. [Google Scholar]
- (13).Leon C; Hechler B; Freund M; Eckly A; Vial C; Ohlmann P; Dierich A; LeMeur M; Cazenave JP; Gachet C Defective platelet aggregation and increased resistance to thrombosis in purinergic P2Y1 receptor-null mice. J. Clin. Invest 1999, 104, 1731–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Fabre JE; Nguyen M; Latour A; Keifer JA; Audoly LP; Coffman TM; Koller BH Decreased platelet aggregation, increased bleeding time and resistance to thromboembolism in P2Y1-deficient mice. Nat. Med 1999, 5, 199–202. [DOI] [PubMed] [Google Scholar]
- (15).Boyer JL; Romero-Avila T; Schachter JB; Harden TK Identification of competitive antagonists of the P2Y1 receptor. Mol. Pharmacol 1996, 50, 1323–1329. [PubMed] [Google Scholar]
- (16).Camaioni E; Boyer JL; Mohanram A; Harden TK; Jacobson KA Deoxyadenosine-bisphosphate derivatives as potent antagonists at P2Y1 receptors. J. Med. Chem 1998, 41, 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Boyer JL; Mohanram A; Camaioni E; Jacobson KA; Harden TK Competitive and selective antagonism of P2Y1 receptors by N6-methyl 2′-deoxyadenosine 3′,5′-bisphosphate. Br. J. Pharmacol 1998, 124, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Nandanan E; Camaioni E; Jang SY; Kim Y-C; Cristalli G; Herdewijn P; Secrist JA; Tiwari KN; Mohanram A; Harden TK; Boyer JL; Jacobson KA Structure activity relationships of bisphosphate nucleotide derivatives as P2Y1 receptor antagonists and partial agonists. J. Med. Chem 1999, 42, 1625–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Nandanan E; Jang SY; Moro S; Kim H; Siddiqi MA; Russ P; Marquez V; Busson R; Herdewijn P; Harden TK; Boyer JL; Jacobson KA Synthesis, biological activity, and molecular modeling of ribose-modified adenosine bisphosphate analogues as P2Y1 receptor ligands. J. Med. Chem 2000, 43, 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kim YC; Gallo-Rodriguez C; Jang SY; Nandanan E; Adams M; Harden TK; Boyer JL; Jacobson KA Acyclic analogues of deoxyadenosine 3′,5′-bisphosphates as P2Y(1) receptor antagonists. J. Med. Chem 2000, 43, 746–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Brown SG; King BF; Kim Y-C; Burnstock G; Jacobson KA Activity of novel adenine nucleotide derivatives as agonists and antagonists at recombinant rat P2X receptors. Drug Dev. Res 2000, 49, 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Palczewski K; Kumasaka T; Hori T; Behnke CA; Motoshima H; Fox BA; Trong IL; Teller DC; Okada T; Stenkamp RE; Yamamoto M; Miyano M Crystal structure of rhodopsin: a G protein-coupled receptor. Science 2000, 289, 739–745. [DOI] [PubMed] [Google Scholar]
- (23).Perich JW; Johns RB Di-tert-butyl N, N-diethylphosphoramidite. A new phosphitylating agent for the efficient phosphorylation of alcohols. Synthesis 1988, 2, 142–144. [Google Scholar]
- (24).Zatorski A; Goldstein BM; Colby TD; Jones JP; Pankiewicz KW Potent inhibitors of human inosine monophosphate dehydrogenase Type II. Fluorine-substituted analogues of thialzole-4-carboxamide adenine dinucleotide. J. Med. Chem 1995, 38, 1098–1105. [DOI] [PubMed] [Google Scholar]
- (25).Sekiyama T; Hatsuya S; Tanaka Y; Uchiyama M; Ono N Synthesis and antiviral activity of novel acyclic nucleosides: Discovery of a cyclopropyl nucleoside with potent inhibitory activity against Herpes viruses. J. Med. Chem 1998, 41, 1284–1298. [DOI] [PubMed] [Google Scholar]
- (26).Green GR; Kincey PM; Choudary BM Regiospecific Michael additions with 2-aminopurines. Tetrahedron Lett. 1992, 33, 4609–4612. [Google Scholar]
- (27).Boyer JL; Downes CP; Harden TK Kinetics of activation of phospholipase C by P2Y purinergic receptor agonists and guanine nucleotides. J. Biol. Chem 1989, 264, 884–890. [PubMed] [Google Scholar]
- (28).Harden TK; Hawkins PT; Stephens L; Boyer JL; Downes P Phosphoinositide hydrolysis by guanosine 5′-(gamma-thio]triphosphate-activated phospholipase C of turkey erythrocyte membranes. Biochem. J 1988, 252, 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Filtz TM; Li Q; Boyer JL; Nicholas RA; Harden TK Expression of a cloned P2Y purinergic receptor that couples to phospholipase C. Mol. Pharmacol 1994, 46, 8–14. [PubMed] [Google Scholar]
- (30).Moro S; Guo D; Camaioni E; Boyer JL; Harden TK; Jacobson KA Human P2Y1 receptor: molecular modeling and site-directed mutagenesis as tools to identify agonist and antagonist recognition sites. J. Med. Chem 1998, 41, 1456–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Moro S; Hoffmann C; Jacobson KA Role of the extracellular loops of G protein-coupled receptors in ligand recognition: A molecular modeling study of the human P2Y1 receptor. Biochemistry 1999, 38, 3498–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Pellicciari R; Marinozzi M; Costantino G; Natalini B; Moroni F; Pellegrini-Giampietro D (2R,1′S,2′R,3′S)-2-(2′-Carboxy-3′-phenylcyclopropyl)glycine (PCCG-13), the first potent and selective competitive antagonist of phospholipase D-coupled metabotropic glutamate receptors: Asymmetric synthesis and preliminary biological properties. J. Med. Chem 1999, 42, 2716–2720. [DOI] [PubMed] [Google Scholar]
- (33).Palmer RK; Boyer JL; Schachter JB; Nicholas RA; Harden TK Agonist action of adenosine triphosphates at the human P2Y1 receptor. Mol. Pharmacol 1998, 54, 1118–1123. [PubMed] [Google Scholar]
- (34).The program SYBYL 6.3 is available from TRIPOS Associates, St. Louis, MO, 1993. [Google Scholar]
- (35).van Rhee AM; Fischer B; van Galen PJM; Jacobson KA Modelling the P2Y purinoceptor using rhodopsin as template. Drug Des. Discovery 1995, 13, 133–154. [PMC free article] [PubMed] [Google Scholar]
- (36).Horn F; Weare J; Beukers MW; Hörsch S; Bairoch A; Chen W; Edvardsen Ø; Campagne F; Vriend G GPCRDB: an information system for G protein-coupled receptors. Nucleic Acids Res. 1998, 26, 277–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Weiner SJ; Kollman PA; Nguyen DT; Case DA An all-atom force field for simulation of protein and nucleic acids. J. Comput. Chem 1986, 7, 230–252. [DOI] [PubMed] [Google Scholar]
- (38).Watson HC; Walker NP; Shaw PJ; Bryant TN; Wendell PL; Fothergill LA; Perkins RE; Conroy SC; Dobson MJ; Tuite MF; et al. Sequence and structure of yeast phosphoglycerate kinase. EMBO J. 1982, 1, 1635–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]