

Abstract
Naphthalene dioxygenase (NDO) is a multicomponent enzyme system that oxidizes naphthalene to (+)-cis-(1R,2S)-1,2-dihydroxy-1,2-dihydronaphthalene with consumption of O2 and two electrons from NAD(P)H. In the presence of benzene, NADH oxidation and O2 utilization were partially uncoupled from substrate oxidation. Approximately 40 to 50% of the consumed O2 was detected as hydrogen peroxide. The rate of benzene-dependent O2 consumption decreased with time, but it was partially increased by the addition of catalase in the course of the O2 consumption by NDO. Detailed experiments showed that the total amount of O2 consumed and the rate of benzene-induced O2 consumption increased in the presence of hydrogen peroxide-scavenging agents, and further addition of the terminal oxygenase component (ISPNAP) of NDO. Kinetic studies showed that ISPNAP was irreversibly inactivated in the reaction that contained benzene, but the inactivation was relieved to a high degree in the presence of catalase and partially relieved in the presence of 0.1 mM ferrous ion. Benzene- and naphthalene-reacted ISPNAP gave almost identical visible absorption spectra. In addition, hydrogen peroxide added at a range of 0.1 to 0.6 mM to the reaction mixtures inactivated the reduced ISPNAP containing mononuclear iron. These results show that hydrogen peroxide released during the uncoupling reaction acts both as an inhibitor of benzene-dependent O2 consumption and as an inactivator of ISPNAP. It is proposed that the irreversible inactivation of ISPNAP occurs by a Fenton-type reaction which forms a strong oxidizing agent, hydroxyl radicals (·OH), from the reaction of hydrogen peroxide with ferrous mononuclear iron at the active site. Furthermore, when [14C]benzene was used as the substrate, cis-benzene 1,2-dihydrodiol formed by NDO was detected. This result shows that NDO also couples a trace amount of benzene to both O2 consumption and NADH oxidation.
Dioxygenases which contain mononuclear iron and Rieske-type [2Fe-2S] clusters play a critical role in the bacterial aerobic degradation of aromatic hydrocarbons and many xenobiotic compounds. They are unique in terms of their ability to catalyze the enantiospecific addition of oxygen (O2) to substrates with π-electron systems to form cis-dihydrodiols (11). For example, naphthalene dioxygenase (NDO; EC 1.14.12.12) from Pseudomonas sp. strain NCIB 9816-4 catalyzes the addition of O2 to naphthalene to form (+)-cis-(1R,2S)-dihydroxy-1,2-dihydronaphthalene (cis-naphthalene dihydrodiol) in a reaction that requires NAD(P)H (20, 21). In addition, NDO has been shown to catalyze other diverse oxidative reactions, including monohydroxylation, desaturation, sulfoxidation, N and O dealkylation, and vinyl group dihydroxylation, depending on the substrate (reviewed in reference 43). It has also been shown that NDO catalyzes an alcohol oxidation reaction that requires NADH and O2 (28) and a partial uncoupling reaction with ethyl phenyl sulfide, methyl p-nitrophenyl sulfide, and acetophenone (27, 28). NDO is a multicomponent enzyme system (7) consisting of a 36.3-kDa iron-sulfur flavoprotein (reductaseNAP [RdNAP]) which contains flavin adenine dinucleotide and a chloroplast-type [2Fe-2S] redox center (16), a 13.6-kDa iron-sulfur protein (ferredoxinNAP [FdNAP]) which contains a single Rieske-type [2Fe-2S] redox center (15), and a terminal oxygenase component (ISPNAP) which has two different subunits (α = 55 kDa; β = 20 kDa) (6). Each α subunit contains a Rieske-type [2Fe-2S] redox center and a mononuclear iron binding site (22, 25, 50). Most preparations of ISPNAP require exogenous iron for maximum activity (50), suggesting that mononuclear iron plays an important role in the reaction catalyzed by NDO. The β subunit was shown to be essential for activity but to play no major role in determining the substrate specificity of NDO (39, 40). ISPNAP has been recently crystallized (30), and the crystal structure has been solved (25). It has been shown that ISPNAP has an α3β3 subunit structure and that mononuclear iron in the large subunit is coordinated by two histidine residues and one aspartate. The nucleotide sequences for all the structural genes of NDO have been determined (38, 46) and show that ligands are conserved throughout the family of NDO structural genes. The organization and electron transport of NDO are shown in Fig. 1. Several multicomponent enzyme systems that utilize oxygenase components similar to ISPNAP have been described previously (2, 33).
FIG. 1.

Proposed electron transport chain for NDO leading to the oxidation of naphthalene to cis-naphthalene dihydrodiol by the terminal oxygenase component, ISPNAP. e− represents the number of electrons transferred at a time.
Initial studies on the mechanism of action of cytochromes P-450 (P-450) have shown that certain substrates uncouple electron transfer from substrate oxygenation, and the resulting oxidase activity can lead to the formation of superoxide anion (47), hydrogen peroxide (12, 36), and water (1, 12). It has been reported in a few P-450-catalyzed reactions that hydrogen peroxide formed during the partial uncoupling reaction irreversibly inactivated the oxygenase components (23, 24). In addition, 4-methoxybenzoate monooxygenase is an oxygenase that contains [2Fe-2S] redox centers and mononuclear iron and produces hydrogen peroxide when incubated with different substrate analogs that can serve as partial or total uncoupling agents (54). However, the effects of hydrogen peroxide formed during the uncoupling reaction have not been determined.
In the present study, benzene is shown to be a partial uncoupling substrate for NDO, resulting in the formation of hydrogen peroxide and cis-benzene 1,2-dihydrodiol. Evidence also shows that hydrogen peroxide formed during the reaction acts both as an inhibitor of benzene-dependent O2 consumption and as an inactivator of ISPNAP.
(A preliminary report of this work has been published previously [29].)
MATERIALS AND METHODS
Materials.
Catalase (19,900 U/mg) from bovine liver, Mn-containing superoxide dismutase (SOD; 3,300 U/mg) from Escherichia coli, 30% solution of hydrogen peroxide, [14C]benzene (58.2 mCi/mmol), and [14C]naphthalene (49.8 mCi/mmol) were obtained from Sigma (St. Louis, Mo.). FerroZine [3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine-p,p′-disulfonic acid, monosodium salt hydrate, 97%] was obtained from Aldrich (Milwaukee, Wis.). Lactoperoxidase (72 U/mg) was obtained from Worthington Biochemical Co., Freehold, N.J. All commercially available chemicals were used without further purification. Deionized water and membrane-filtered (0.22-μm pore size) buffers were used for fast-performance liquid chromatography (FPLC; Pharmacia LKB).
Bacterial strains and growth conditions.
E. coli JM109(DE3)(pDTG141) (49), which contains the cloned nahAaAbAcAd genes encoding the NDO components (RdNAP, FdNAP, and ISPNAP) from Pseudomonas sp. strain NCIB 9816-4 (5), was used for the purification of RdNAP. E. coli JM109(DE3)(pDTG135) (51), which contains the cloned nahAb genes encoding the FdNAP, was used for the purification of FdNAP. E. coli JM109(DE3)(pDTG121) (51), which contains the cloned nahAcAd genes which encode the large and small subunits of ISPNAP, was used for the purification of ISPNAP. Growth conditions for the recombinant E. coli strains and expression of the genes have been described previously (31).
Purification of NDO components.
Crude cell extracts were prepared as described previously (14). The buffer used for breakage was BTGD buffer (50 mM bis-Tris [pH 6.8], 5% glycerol, 1 mM dithiothreitol [DTT]) containing 0.1 mM phenylmethylsulfonyl fluoride and 1 μg of DNase/ml. All columns were operated with a Pharmacia FPLC system at 5°C. ISPNAP was purified to homogeneity as described previously (30). RdNAP and FdNAP were purified to homogeneity (26) by modifications of the methods described previously (15, 16). BTGD buffer was used for the purification of all three NDO components.
Biotransformation with whole cells and GC-MS analysis.
Biotransformation with isopropyl β-d-thiogalactopyranoside (IPTG)-induced cells of E. coli JM109(DE3)(pDTG141) has been described previously (31). Fifty microliters of benzene was used for a 100-ml reaction volume. Ethyl acetate extracts were prepared and analyzed by gas chromatography-mass spectrometry (GC-MS) as described previously (44).
Oxygen uptake studies.
The rates and stoichiometries of O2 consumption by NDO with benzene and naphthalene were determined polarographically with a Clark-type oxygen electrode (Rank Brothers, Cambridge, England) as described previously (27). Each reaction mixture contained in 1.0 ml of 50 mM 2-(N-morpholino)ethanesulfonate (MES) buffer (pH 6.8), NADH (0.35 μmol), RdNAP (10.5 μg of protein), FdNAP (48 μg of protein), and ISPNAP (40 μg of protein). Information about the addition of other components to the reaction mixtures is indicated in the figure legends. The initial slopes formed by the addition of each substrate were used to determine the initial rates of O2 consumption by NDO.
Time course of ISPNAP inactivation.
Time course of ISPNAP inactivation during reactions in the presence or absence of benzene was determined as follows. Reaction mixtures (0.5 ml) contained RdNAP (5.25 μg of protein), FdNAP (25.6 μg of protein), and ISPNAP (52.5 μg of protein) in 50 mM MES (pH 6.8). When appropriate, catalase (1,000 U), SOD (82.5 U), benzene (0.1 mM), and Fe(NH4)2(SO4)2 · 6H2O (0.1 mM) were added into the reaction mixtures. Reactions were initiated by the addition of 10 μl of 50 mM NADH to give a final concentration of 1 mM. Reactions were conducted in capped 1-dram (3.7-ml) tubes at 22°C and incubated horizontally with shaking (150 rpm). At appropriate time points, aliquots of 10 μl (each) were withdrawn from the reaction mixtures and rapidly suspended in the following assay mixtures to determine the remaining ISPNAP activity. The assay mixtures contained RdNAP (5.25 μg of protein), FdNAP (16 μg of protein), catalase (1,000 U), and Fe(NH4)2(SO4)2 · 6H2O (0.1 mM) in 50 mM MES (pH 6.8). The reactions were initiated by the addition of 20 μl of 5 mM [14C]naphthalene to give a final concentration of 0.25 mM (1.11 × 106 dpm/ml), followed by the addition of 2 μl of 50 mM NADH. The final reaction volume was 0.4 ml. Aliquots of 10 μl (each) were withdrawn from the assay mixtures after 1, 2, or 3 min and quickly mixed with 15 μl of a quenching solution containing 25 mM nonradioactive naphthalene dissolved in methanol in a 600-μl Eppendorf tube. Samples were applied to plastic-backed sheets of silica gel F254 (1 by 1.5 cm, 0.2-mm thickness; E. Merck Co.) and air dried in a fume hood for 30 min to remove volatile naphthalene. Radioactivity of nonvolatile cis- naphthalene dihydrodiol remaining in each plate was determined by scintillation counting with a Ready Safe cocktail solution (Beckman) as described previously (7). Initial NDO activity was determined from the linear portion of the reaction, and all experiments were conducted in duplicate.
Spectroscopic analysis.
Comparison of the UV/Vis spectra of ISPNAP after catalytic turnover with benzene and naphthalene was performed as follows. Reaction mixtures (0.4 ml) contained 0.5 mM NADH, substrate (8 μl of a 25 mM stock in methanol to yield a final concentration of 0.5 mM), RdNAP (4.2 μg of protein), FdNAP (22.4 μg of protein), and ISPNAP (520 μg of protein) in 50 mM MES buffer (pH 6.8). Reactions were conducted in capped 1-dram (3.7-ml) tubes and incubated horizontally with shaking (60 rpm) at 22°C. At the incubation time of 2 h, the protein was concentrated and desalted twice with 50 mM MES buffer (pH 6.8) containing 1 mM DTT by centrifugation over an Amicon YM100 Microcon at 5°C. The retentants were resuspended in BTGD buffer and subjected to the measurement of UV/Vis absorbance. The filtrates had no absorbance at 340 nm, indicating that NADH was completely oxidized in both reactions. The ISPNAP activity remaining in the retentants was also determined by oxygen uptake experiments as described in the paragraph entitled oxygen uptake studies. The reported results shown are the averages of duplicated measurements.
Removal of mononuclear iron from ISPNAP by FerroZine.
Removal of iron from ISPNAP by FerroZine was based on a modification of a published method (41). Briefly, purified ISPNAP (1,275 μg of protein) was added to a solution containing 5 mM FerroZine and 5 mM ascorbic acid in BTGD buffer (final preparation, 0.5 ml). The reaction mixture was incubated on ice for 63 h, and then the protein was concentrated and desalted three times with BTGD buffer by centrifugation over an Amicon YM100 Microcon. This preparation yielded 1,044 μg of ISPNAP.
Hydrogen peroxide-mediated ISPNAP inactivation.
The effect of hydrogen peroxide on the activity of either oxidized or reduced ISPNAP was determined with the following reaction mixtures. The reaction mixtures (0.25 ml in 1.5-ml Eppendorf tubes) used for the oxidized ISPNAP contained ISPNAP (26.2 μg of protein) and hydrogen peroxide at final concentrations of 0.1 to 0.6 mM in 50 mM MES (pH 6.8). Reaction mixtures that were performed with reduced ISPNAP contained RdNAP (2.62 μg of protein), FdNAP (12.8 μg of protein), and ISPNAP (26.2 μg of protein) with hydrogen peroxide in the same range as that used for the oxidized ISPNAP in 50 mM MES (pH 6.8). Stock solutions of hydrogen peroxide were made fresh prior to use. Actual concentrations of hydrogen peroxide were determined based on an extinction coefficient of 43.6 M−1 cm−1 at 240 nm (3). Reactions were initiated by the addition of 5 μl of 50 mM NADH to give a final concentration of 1 mM. The reaction mixtures were incubated at 22°C. At the incubation time of 10 min, aliquots of 20 μl were withdrawn from the reaction mixtures, and the remaining ISPNAP activity was determined as described above in the paragraph entitled time course of ISPNAP inactivation. The same activity assays were also conducted in the absence of 0.1 mM Fe(NH4)2(SO4)2 · 6H2O to determine the requirement of ferrous ion for the activity. The results shown are the averages of duplicated measurements.
Identification of cis-benzene 1,2-dihydrodiol.
In order to detect the benzene oxidation product formed by NDO, [14C]benzene was used as the substrate. Reaction mixtures (1 ml each) contained RdNAP (10.5 μg of protein), FdNAP (48 μg of protein), ISPNAP (40 μg of protein), and [14C]benzene (102.2 μM; 1.32 × 107 dpm/ml) in 50 mM MES buffer (pH 6.8). When appropriate, NADH (0.5 mM), Fe(NH4)2(SO4)2 · 6H2O (0.1 mM), and catalase (1,000 U) were added to the reaction mixtures. The same reaction was also conducted with corresponding amounts of NADH, [14C]benzene, Fe(NH4)2(SO4)2 · 6H2O, and purified toluene dioxygenase components from Pseudomonas putida F1 (27). Reactions were conducted in 7.4-ml screw-cap vials, with an agitation of 150 rpm at 22°C. After 1 h, aliquots of 20 μl were withdrawn and suspended in 10 μl of 25 mM cold benzene in methanol. The solutions were loaded onto silica gel plates (1.5 cm2; 0.2-mm thickness) and dried in the hood for 30 min. The amount of the radioactive polar product remaining on each plate was determined by scintillation counting. In addition, the reaction mixtures after incubation were extracted with 2 ml of NaOH-neutralized ethyl acetate, and each 1 ml of the extracts was concentrated over a stream of nitrogen to 25 μl. Aliquots (5 μl each) were subjected to thin-layer chromatography (TLC) on silica gel F254 plastic sheets with a developing solvent of chloroform-acetone (8:2), followed by autoradiography. X-ray films were exposed at −70°C for 5.5 days before development. The Rf value of the product was compared to those of phenol and cis-benzene 1,2-dihydrodiol formed by toluene dioxygenase (10).
RESULTS
Uncoupling of O2 consumption and formation of hydrogen peroxide in the presence of benzene.
NDO catalyzed the rapid and stoichiometric consumption of O2 when incubated with naphthalene (Fig. 2A). In addition, approximately identical O2 consumption rates were observed in the presence of 0.1 mM ferrous ion in the reaction mixture (Fig. 2B). Both had averages of 5.3 and 4.8 μmol of O2 consumed/min/mg of ISPNAP for the first and second additions of 0.1 mM naphthalene (Fig. 2A and B). GC-MS analysis of the reaction mixtures at the end of the experiment showed that cis-naphthalene dihydrodiol was the only product formed. In contrast, NDO catalyzed the steadily decreasing O2 consumption in the presence of benzene, with an initial rate of 0.96 μmol of O2 consumed/min/mg of ISPNAP (Fig. 2C). This reaction was tightly coupled to NADH oxidation and dependent on all three NDO components (data not shown). GC-MS analysis of the reaction mixtures did not reveal the presence of oxidation products. Since NDO did not incorporate a detectable amount of O2 into benzene, the reaction mixture was examined for reactive oxygen species that might account for the observed O2 consumption. Addition of catalase at the time shown in Fig. 2C increased the dissolved O2 level in the reaction mixture by 15.7 nmol, showing that part of the O2 consumed by NDO in the presence of benzene was converted to hydrogen peroxide with an uncoupling reaction (reaction 1) as shown below. According to the stoichiometry of reactions 1 and 2, the amount of O2 formed by the addition of catalase is equivalent to 50% of the O2 consumed by NDO during the reaction.
| 1 |
| 2 |
FIG. 2.

Oxygen consumption by NDO in the presence of naphthalene (Naph) and benzene (Benz). Additions of substrates (4 μl of 25 mM in methanol), catalase (1,000 U), and SOD (82.5 U) were made at the times indicated by arrows. Experimental details are described in Materials and Methods. Additions of 0.1 mM naphthalene (A); 0.1 mM naphthalene in the presence of 0.1 mM Fe(NH4)2(SO4)2 · 6H2O (B); 0.1 mM benzene, followed by additions of catalase, SOD, and 0.1 mM naphthalene (C); and 0.1 mM benzene in the presence of 0.1 mM Fe(NH4)2(SO4)2 · 6H2O (D).
The amount of hydrogen peroxide equivalent to 40 to 50% of the O2 consumed by NDO accumulated in the reaction mixture during the first 5 to 7 min of the reaction. The accumulation of hydrogen peroxide decreased over time (data not shown). Furthermore, the addition of increasing amounts of benzene to the reaction mixture showed that similar amounts of O2 were consumed by NDO as in the presence of 0.1 mM benzene, but the initial rates of O2 consumption increased slightly (data not shown). In spin trapping experiments with a hydroxyl radical (·OH) trapping agent, 5,5-dimethyl-1-pyrroline N-oxide (DMPO) (4), the electron spin resonance (ESR) spectrum of DMPO and ·OH adduct (DMPO-OH) was detected with sodium phosphate buffer under the conditions when hydrogen peroxide did not accumulate in the medium due to lesser amounts of NDO components (26). The ESR spectrum did not form in the presence of catalase, indicating that hydrogen peroxide released into the reaction medium decomposes to ·OH under the reaction conditions. Analogous experiments were also conducted in the presence of 1,3-diphenylisobenzofuran, a singlet oxygen acceptor (17). No decrease in absorbance at 415 nm due to the reaction of singlet oxygen with the acceptor was observed.
It should be further noted that the addition of catalase to the reaction mixture (Fig. 2C) increased the rate of O2 consumption from 0.11 to 0.22 μmol/min/mg of ISPNAP. The increase of NDO activity caused by the removal of hydrogen peroxide indicates that accumulated hydrogen peroxide acts as an inhibitor in benzene-dependent O2 consumption by NDO. In the time course of the O2 consumption, the addition of 0.1 mM naphthalene at the incubation time as indicated in Fig. 2C resulted in 1.18 μmol of O2 consumed/min/mg of ISPNAP, with a tight coupling to the amount of substrate added. The rate of O2 consumption was equivalent to 24.6% of second additions of 0.1 mM naphthalene in Fig. 2A and B. The addition of SOD to the reaction mixture in the presence of catalase did not affect the level of dissolved O2 in the reaction mixture (Fig. 2C), indicating that superoxide anion did not accumulate under the uncoupling reaction. Interestingly, the presence of 0.1 mM ferrous ion in the reaction mixture containing benzene increased the rate of O2 consumption (initial rate, 1.54 μmol/min/mg of ISPNAP) as well as the total amount of O2 consumed (Fig. 2D). This result suggests that ferrous ion may play another role in addition to supplementing iron ion at the active site of ISPNAP.
Effect of the hydrogen peroxide-scavenging agents on benzene-induced O2 consumption by NDO.
The possible involvement of ferrous ion as the substrate of the Fenton reaction (reaction 3) was considered as an explanation for the increase of O2 consumption in the presence of benzene (Fig. 2D).
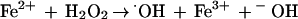 |
3 |
In reaction 3, the hydrogen peroxide formed could be removed from the reaction mixture by forming a strong oxidizing agent, ·OH, that could react with the components of the reaction mixture, such as MES in large excess, and even with the NDO components. Increases of the benzene-dependent O2 consumption in the presence of an enzyme such as catalase or the lactoperoxidase/KI system (34) that utilizes hydrogen peroxide as the substrate corroborated the proposed role of ferrous ion (Fig. 3). Benzene-induced O2 consumption of NDO increased slightly in the presence of ferric chloride and chelating agents such as diethyenetriaminepentaacetic acid (DETAPAC) and EDTA. DETAPAC and EDTA are known to enhance the Fenton reaction (56). Levels of hydrogen peroxide accumulation in the presence of hydrogen peroxide-removing agents were also determined by the addition of catalase (Fig. 3). The addition of the ·OH scavengers (17), including DMPO, thiourea, and mannitol, to the reaction mixture in a final concentration of 5 mM did not increase benzene-dependent O2 consumption.
FIG. 3.

Benzene-induced oxygen consumption by NDO in the presence of hydrogen peroxide-scavenging agents. Additions of benzene (Benz; final concentration, 0.1 mM) and catalase (1,000 U) were made at the times indicated by upward arrows. The reaction mixtures contained the same amounts of NADH and NDO components in 50 mM MES (pH 6.8) as described in Fig. 2. No addition (A) or addition of 0.1 mM FeCl3 · 6H2O (B), 1 mM DETAPAC (C), 1 mM EDTA (D), 10 mM KI and lactoperoxidase (9 U) (E), catalase (1,000 U) (F), and 0.1 mM Fe(NH4)2(SO4)2 · 6H2O (G).
Inactivation of ISPNAP during benzene-induced O2 consumption by NDO.
The decreasing rate of benzene-dependent O2 consumption by NDO with time was not completely reversible by the addition of catalase (Fig. 2C), indicating that NDO might be inactivated during the reaction. In order to determine the NDO component inactivated during the reaction, each NDO component was added back to the reaction mixture so that the stimulation of O2 consumption in the presence of catalase could be observed. Addition of RdNAP or/and FdNAP to the reaction mixture did not stimulate the reduced benzene-induced O2 consumption; however, the addition of ISPNAP resulted in increased O2 consumption (data not shown). This result shows that the ISPNAP component is inactivated in benzene-induced O2 consumption. Furthermore, the levels of naphthalene-dependent O2 consumption by NDO in the presence of different concentrations of hydrogen peroxide were determined to examine the effect of peroxide on NDO activity. The initial rates of the O2 consumption by NDO were steadily reduced in the presence of increasing amounts of hydrogen peroxide, and the O2 consumption activities were not stoichiometric in the presence of more than 0.5 mM hydrogen peroxide (data not shown). For instance, in the presence of 0.5 mM hydrogen peroxide the initial rate of naphthalene-dependent O2 consumption by NDO was 1.1 μmol/min/mg of ISPNAP. O2 consumption steadily decreased and ceased after 6 min. The addition of naphthalene, RdNAP, and FdNAP to the reaction mixture did not stimulate O2 consumption, but the addition of ISPNAP restarted O2 consumption. This result shows that hydrogen peroxide inactivates ISPNAP.
Spectroscopic studies.
Possible change of the Rieske [2Fe-2S] redox clusters of ISPNAP that reacted with benzene was determined by examining the change in the UV/Vis absorption spectrum ranging from 250 to 700 nm. As a control, the same reaction was also conducted in the presence of naphthalene. The observed differences between the two spectra were minimal (data not shown), and the R factors (A280/A450) of the naphthalene- and benzene-reacted ISPNAP were 17.6 and 17.0, respectively. However, the specific activities for naphthalene- and benzene-reacted ISPNAP were 2.7 and 0.88 μmol of O2 consumed/min/mg, respectively, by an assay in the presence of 0.1 mM ferrous ion. The specific activities for naphthalene- and benzene-reacted ISPNAP were 1.72 and 0.52 μmol of O2 consumed/min/mg, respectively, by an assay in the absence of ferrous ion.
Protective effect of hydrogen peroxide-scavenging agents on ISPNAP inactivation.
In order to determine the activity of each NDO component remaining during the uncoupling reaction, all three NDO components were incubated in the presence of NADH and benzene. After a 30-min incubation period, aliquots were withdrawn and assayed for the activities remaining for each individual component when there was an excess of the other two components. Under these conditions, RdNAP, FdNAP, and ISPNAP had 100, 94, and 20% of their original activities, respectively, indicating that benzene-dependent NDO inactivation was due primarily to the inactivation of the ISPNAP component as shown above. Figure 4 shows the time course of ISPNAP inactivation in the reaction mixtures containing benzene alone or benzene plus other hydrogen peroxide-scavenging agents. The presence of benzene resulted in a significant time-dependent inactivation of ISPNAP. For example, ISPNAP lost approximately 60% of the original activity when incubated with benzene for 10 min. ISPNAP activity was not lost under incubation conditions without either RdNAP or FdNAP (data not shown). Catalase greatly protected ISPNAP from inactivation. The same protection was also observed in the presence of both catalase and SOD (data not shown). This result shows that hydrogen peroxide released into the reaction medium during the reaction is largely responsible for ISPNAP inactivation. Partial relief of ISPNAP inactivation was also observed in the presence of 0.1 mM ferrous ion. Interestingly, in the absence of benzene, ISPNAP was also significantly inactivated in a prolonged incubation, but inactivation was fully prevented by the presence of catalase (data not shown). This result indicates that hydrogen peroxide released into the medium is also responsible for the inactivation of ISPNAP during very slow O2 consumption by NDO in the absence of substrate.
FIG. 4.

Time course of ISPNAP inactivation in NDO-catalyzed reaction mixtures containing benzene. Details of the experimental conditions are described in Materials and Methods. Reaction mixtures contained NADH and all three NDO components in 50 mM MES (pH 6.8) with added benzene (■), benzene and catalase (○), or benzene and Fe(NH4)2(SO4)2 · 6H2O (●) or with no addition (□).
Inactivation of ISPNAP in the presence of hydrogen peroxide.
Experiments were conducted to determine whether hydrogen peroxide inactivation is specific for oxidized or reduced ISPNAP. As shown in Fig. 5, oxidized ISPNAP was resistant to the range of hydrogen peroxide tested. However, in the presence of excess hydrogen peroxide (10 mM), oxidized ISPNAP had 57% of the original activity under the same incubation conditions. In contrast, reduced ISPNAP was easily inactivated by hydrogen peroxide. For example, reduced ISPNAP had no activity after incubation with 0.4 mM hydrogen peroxide for 10 min by an assay in the absence of added ferrous ion. However, the same incubation had 35% of the original activity by an assay with 0.1 mM ferrous ion. This result indicates that reduced ISPNAP containing the mononuclear iron lost the activity by the treatment of hydrogen peroxide and that reduced ISPNAP without the mononuclear iron which was intact during the incubation gained activity because of the supply of ferrous ion in the assay mixture. The role of mononuclear iron at the active site in the inactivation was further examined with partially mononuclear iron-depleted (FerroZine-treated) ISPNAP, whose specific activities were 1.0 and 4.1 μmol of O2 consumed/min/mg of ISPNAP by assay without and with 0.1 mM ferrous ion, respectively. As shown in Fig. 5, partially mononuclear iron-depleted ISPNAP was more resistant to hydrogen peroxide than original ISPNAP by an assay of the activity in the presence of 0.1 mM ferrous ion. The effect of FerroZine-treated ISPNAP is illustrated by the fact that ca. 25% of the enzyme that contained mononuclear iron became inactive in a typical manner, while the apoprotein could be reconstituted to achieve near full activity when ferrous ion was supplied in the assay. This result supports the inactivation of reduced ISPNAP containing mononuclear iron by hydrogen peroxide. In addition, the [2Fe-2S] redox centers of FerroZine-treated ISPNAP were also fully reduced in the presence of NADH, RdNAP, and FdNAP (data not shown).
FIG. 5.

ISPNAP inactivation in the presence of hydrogen peroxide. Details of the experimental conditions are described in Materials and Methods. Reaction mixtures contained oxidized ISPNAP and the amount of hydrogen peroxide indicated (circles); NADH, all three NDO components, and the amount of hydrogen peroxide indicated (triangles); or NADH, RdNAP, FdNAP, and FerroZine-treated ISPNAP and the amount of hydrogen peroxide indicated (squares). The activity of treated ISPNAP was determined under normal assay conditions in the presence of RdNAP and FdNAP as described in Materials and Methods. Open and filled symbols represent percentages of remaining ISPNAP activity assayed in the presence and absence of 0.1 mM added Fe(NH4)2(SO4)2 · 6H2O, respectively.
Formation of cis-benzene 1,2-dihydrodiol by NDO.
Since no benzene oxidation product was detected by GC-MS from concentrated ethyl acetate extracts obtained through purified NDO and IPTG-induced E. coli JM109(DE3)(pDTG141) biotransformations, the possibility of the formation of a trace amount of product was examined with [14C]benzene. Surprisingly, NDO oxidized benzene to a polar product that stuck on the silica gel plate. To identify the product, the reaction mixtures were further extracted with ethyl acetate, concentrated about 20-fold, and subjected to TLC. The only polar product formed by NDO had an Rf of 0.11, identical to that of cis-benzene 1,2-dihydrodiol. The amounts of cis-benzene 1,2-dihydrodiol formed by NDO under various reaction conditions are listed in Table 1. The results show that the formation of cis-benzene 1,2-dihydrodiol by NDO is highly dependent on the presence of ferrous ion or catalase. For example, in the presence of 0.1 mM ferrous ion, a maximal accumulation of 1.31 μM (equivalent to a turnover ratio of 2.59 [product formed/active site]) was observed for 1 h of incubation.
TABLE 1.
Formation of cis-benzene 1,2-dihydrodiol by NDOa
| Reaction condition | Amt (nmol) of cis-benzene 1,2-di-hydrodiol formed/mlb | Turnoverc |
|---|---|---|
| NADH + benzene | 0.23 | 0.45 |
| NADH + benzene + Fe2+ | 1.31 | 2.59 |
| NADH + benzene + catalase | 0.90 | 1.78 |
| NADH + benzene + catalase + Fe2+ | 1.10 | 2.17 |
| Benzene + Fe2+ + catalase | —d | — |
Reactions were conducted for 1 h. Details of the experimental conditions and product analysis are described in Materials and Methods.
Values are the averages of two different experiments with two different assays. Standard deviations were less than 5% of the values given.
Ratio of cis-benzene 1,2-dihydrodiol to the α and β subunits of ISPNAP.
Not detected.
DISCUSSION
Purified NDO has been shown to oxidize toluene sequentially through benzyl alcohol to benzaldehyde by reactions involving benzylic monooxygenation and alcohol oxidation, respectively (28). In this study the reactions catalyzed by NDO with a smaller substrate, benzene, were investigated to examine the factors involved in the uncoupling of substrate oxidation. As shown in Fig. 2, 40 to 50% of the O2 consumed by NDO in the presence of benzene was reduced to hydrogen peroxide by an uncoupling reaction. In addition, a trace amount of cis-benzene 1,2-dihydrodiol was formed by a coupling reaction (Table 1). These results indicate that in the presence of benzene, NDO catalyzes a partial uncoupling reaction, resulting in the formation of hydrogen peroxide and cis-benzene 1,2-dihydrodiol. The sum of the O2 utilized to form hydrogen peroxide and cis-benzene 1,2-dihydrodiol is insufficient to account for the O2 consumption during the reaction, indicating that some intermediates formed from the consumed O2 disappeared during the reaction. However, since O2 consumption was tightly coupled to NADH oxidation, the formation of H2O resulting from a four-electron reduction reaction (12) was not considered during the uncoupling reaction. Under the experimental conditions, hydrogen peroxide spontaneously decomposed to hydroxyl radicals, interfering with the precise measurement of accumulated hydrogen peroxide. A two-component 2-oxo-1,2-dihydroquinoline 8-monooxygenase which contains Rieske-type [2Fe-2S] clusters and additional iron in its oxygenase component has been shown to consume O2 in the presence of pseudosubstrates without the oxidation of substrate (45). The decay under the experimental conditions and the present results suggest that hydrogen peroxide decomposes spontaneously under certain reaction conditions.
Since the rate of the benzene-dependent O2 consumption by NDO decreased with time, the biochemical background for the observation was further studied. The presence of hydrogen peroxide-scavenging agents such as catalase, ferrous ion, lactoperoxidase, EDTA, and DETAPAC increased O2 consumption (Fig. 3), leading to the conclusion that the hydrogen peroxide released was responsible for the decrease of O2 consumption in the presence of benzene. Hydrogen peroxide was found to function as an inhibitor (Fig. 2) of O2 consumption and as an irreversible inactivator of ISPNAP. In the reconstituted system, ISPNAP was shown to be largely protected from benzene-dependent inactivation in the presence of catalase (Fig. 4). A similar result was obtained in the formation of cis-benzene 1,2-dihydrodiol (Table 1). ISPNAP inactivation was not related to the destruction of Rieske [2Fe-2S] clusters and was heavily dependent on the reduced form of ISPNAP containing mononuclear iron (Fig. 5). These results suggest that residues at the mononuclear iron-containing active site are the primary reaction target for hydrogen peroxide-dependent ISPNAP inactivation. Some purified (oxidized) ISPNAP is known to contain ferrous ion, based on the ESR spectrum of a Fe2+-NO complex giving S = 3/2 spins (55). It has also been shown in the related phthalate dioxygenase that the mononuclear iron of the active enzyme is ferrous (9). The presence of ferrous mononuclear iron at the active site of the oxidized ISPNAP may be the reason part of the ISPNAP is inactivated in the presence of excess hydrogen peroxide. Since the reduced form of ISPNAP is more sensitive to hydrogen peroxide, it may play another role in reactivity to hydrogen peroxide. Many enzymes are reported to be inactivated in the presence of hydrogen peroxide and ferrous ion (8). In most cases, enzyme inactivation was proposed to occur via a Fenton-type reaction (reaction 3), in which the strong oxidizing agent ⋅OH reacts with an amino acid(s) at or near the active site (48). This type of irreversible inactivation resulting from the formation of reactive oxygen species is distinct from oxygenase inactivation resulting from the formation of reactive substrate intermediates, capable of formation of covalent adducts at the functional groups of the enzymes (19, 35, 37). Since only the former type of enzyme inactivation is inhibited by catalase, these two types of enzyme inactivation can be differentiated. ISPNAP inactivation during NDO catalysis in the presence of benzene was also partially prevented when ferrous ion was present in the reaction mixture (Fig. 4). This effect appears to be contradictory to the proposed mechanism of ISPNAP inactivation by a Fenton-type reaction. However, ferrous ion in the reaction mixture seems to participate in the breakdown of hydrogen peroxide that is released and enters into the active site. Hydroxyl radicals formed in the medium can be easily quenched by reacting with excess MES buffer. This explanation can also be applied to the increase in O2 consumption in the presence of ferrous ion and chelating agents such as EDTA and DETAPAC that help break down hydrogen peroxide by accelerating the Fenton reaction (56) (Fig. 3). In addition, the possibility cannot be excluded that added ferrous ion plays an extra role in protecting ISPNAP from inactivation by hydrogen peroxide.
With respect to the electron transport reaction catalyzed by NDO, a role for hydrogen peroxide formed during the benzene-dependent uncoupling reaction can be proposed. It has been proposed and shown in many studies with iron-containing oxygenases that the reduction of O2 by NAD(P)H involves the release of hydrogen peroxide at the ferric state in the form of [FeO2]+ or Fe3+(O22−) (13, 32, 42). This is reasonable in terms of a known chemistry in which a ferrous peroxo form [FeO2]0 can undergo a Fenton-type reaction, causing suicide inactivation of the oxygenase in the absence of substrate (17). With a continuous supply of two reducing equivalents from NAD(P)H, O2 bound at the mononuclear iron site of ISPNAP can be reduced to a peroxide level to be released as hydrogen peroxide by the benzene-dependent uncoupling reaction catalyzed by NDO. During the electron transfer cycle, mononuclear iron undergoes ferric and ferrous states. Released hydrogen peroxide could reversibly bind to ferric ion at the active site to block O2 binding or could react with ferrous ion at the active site of reduced ISPNAP formed during the catalytic cycle. This could result in inhibition of O2 consumption by ISPNAP and in ISPNAP inactivation via a Fenton-type reaction, respectively. From this study it can be concluded that a ferric peroxide is an intermediate for NDO-dependent O2 activation as proposed earlier for a similar oxygenase, 4-methoxybenzoate monooxygenase (52, 54). A ferric peroxide per se, however, cannot participate in all of the reactions catalyzed by NDO (18). In addition, the heterolytic cleavage of an O-O bond from a ferric peroxide intermediate proposed for monooxygenase-type P-450 (13, 42) and methane monooxygenase (32) cannot be a part of the dihydroxylation reaction, which requires both atoms of O2 incorporated into the substrate. Thus, the mechanism for further activation of the intermediate in NDO catalysis remains to be elucidated; this mechanism should account for dioxygenation, monooxygenation, and radical reactions (43, 53).
ACKNOWLEDGMENTS
I am indebted to David T. Gibson for his generous permission to publish these results and for his stimulating discussions and help. I thank John D. Lipscomb and Matt D. Wolfe (University of Minnesota) for providing unpublished data, G. Buettner for DMPO-OH ESR spectral analysis, and Rebecca E. Parales, Sol M. Resnick, Juanito V. Parales, Haiyan Jiang, and Julie R. Nealson for their help and discussions. I greatly acknowledge Matt D. Wolfe for critically reading the manuscript and for suggestions. I also appreciate the constructive comments of the anonymous reviewers and their suggestions for improving the manuscript.
REFERENCES
- 1.Atkins W M, Sligar S G. Metabolic switching in cytochrome P-450cam: deuterium isotope effects on regiospecificity and monooxygenase/oxidase ratio. J Am Chem Soc. 1987;109:3754–3760. [Google Scholar]
- 2.Batie C J, Ballou D P, Corell C C. Phthalate dioxygenase reductase and related flavin-iron-sulfur containing electron transferases. In: Müller F, editor. Chemistry and biochemistry of flavoenzymes. Boca Raton, Fla: CRC Press; 1991. pp. 543–556. [Google Scholar]
- 3.Beers R F, Sizer I W. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J Biol Chem. 1952;195:133–140. [PubMed] [Google Scholar]
- 4.Buettner G R. Spin trapping: ESR parameters of spin adducts. Free Radic Biol Med. 1987;3:259–303. doi: 10.1016/s0891-5849(87)80033-3. [DOI] [PubMed] [Google Scholar]
- 5.Davies J I, Evans W C. Oxidative metabolism of naphthalene by soil pseudomonads: the ring-fission mechanism. Biochem J. 1964;91:251–261. doi: 10.1042/bj0910251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ensley B D, Gibson D T. Naphthalene dioxygenase: purification and properties of a terminal oxygenase component. J Bacteriol. 1983;155:505–511. doi: 10.1128/jb.155.2.505-511.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ensley B D, Gibson D T, Laborde A L. Oxidation of naphthalene by a multicomponent enzyme system from Pseudomonas sp. strain NCIB 9816. J Bacteriol. 1982;149:948–954. doi: 10.1128/jb.149.3.948-954.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fucci L, Oliver C N, Coon M J, Stadtman E R. Inactivation of key metabolic enzymes by mixed-function oxidation reactions: possible implication in protein turnover and ageing. Proc Natl Acad Sci USA. 1983;80:1521–1525. doi: 10.1073/pnas.80.6.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gassner G T, Ballou D P, Landrum G A, Whittaker J W. Magnetic circular dichroism studies on the mononuclear ferrous active site of phthalate dioxygenase from Pseudomonas cepacia show a change of ligation state on substrate binding. Biochemistry. 1993;32:4820–4825. doi: 10.1021/bi00069a017. [DOI] [PubMed] [Google Scholar]
- 10.Gibson D T, Cardini G E, Masales F C, Kallio R E. Incorporation of oxygen-18 into benzene by Pseudomonas putida. Biochemistry. 1970;9:1631–1635. doi: 10.1021/bi00809a024. [DOI] [PubMed] [Google Scholar]
- 11.Gibson D T, Subramanian V. Microbial degradation of aromatic hydrocarbons. In: Gibson D T, editor. Microbial degradation of organic compounds. New York, N.Y: Marcel Dekker Inc.; 1984. pp. 181–251. [Google Scholar]
- 12.Gorsky L D, Koop D R, Coon M J. On the stoichiomerty of the oxidase and monooxygenase reactions catalyzed by liver microsomal cytochrome P-450. J Biol Chem. 1984;259:6812–6817. [PubMed] [Google Scholar]
- 13.Guengerich F P. Reactions and significance of cytochrome P-450 enzymes. J Biol Chem. 1991;266:10019–10022. [PubMed] [Google Scholar]
- 14.Haddock J D, Nadim L M, Gibson D T. Oxidation of biphenyl by a multicomponent enzyme system from Pseudomonas sp. strain LB400. J Bacteriol. 1993;175:395–400. doi: 10.1128/jb.175.2.395-400.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haigler B E, Gibson D T. Purification and properties of ferredoxinNAP, a component of naphthalene dioxygenase from Pseudomonas sp. strain NCIB 9816. J Bacteriol. 1990;172:465–468. doi: 10.1128/jb.172.1.465-468.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haigler B E, Gibson D T. Purification and properties of NADH-ferredoxinNAP reductase, a component of naphthalene dioxygenase from Pseudomonas sp. strain NCIB 9816. J Bacteriol. 1990;172:457–464. doi: 10.1128/jb.172.1.457-464.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halliwell B, Gutteridge J M C. Free radicals in biology and medicine. 2nd ed. New York, N.Y: Oxford University Press; 1989. [Google Scholar]
- 18.Hamilton G A. Chemical models and mechanisms for oxygenases. In: Hayaishi O, editor. Molecular mechanisms of oxygen activation. New York, N.Y: Academic Press; 1974. pp. 405–451. [Google Scholar]
- 19.Hyman M R, Arp D J. 14C2H2- and 14CO-labelling studies of the de novo synthesis of polypeptides by Nitrosomonas europaea during recovery from acetylene and light inactivation of ammonia monooxygenase. J Biol Chem. 1992;267:1534–1545. [PubMed] [Google Scholar]
- 20.Jeffrey A M, Yeh H J C, Jerina D M, Patel T R, Davey J F, Gibson D T. Initial reactions in the oxidation of naphthalene by Pseudomonas putida. Biochemistry. 1975;14:575–583. doi: 10.1021/bi00674a018. [DOI] [PubMed] [Google Scholar]
- 21.Jerina D M, Daly J W, Jeffrey A M, Gibson D T. cis-1,2-Dihydroxy-1,2-dihydronaphthalene: a bacterial metabolite from naphthalene. Arch Biochem Biophys. 1971;142:394–396. doi: 10.1016/0003-9861(71)90298-0. [DOI] [PubMed] [Google Scholar]
- 22.Jiang H, Parales R E, Lynch N A, Gibson D T. Site-directed mutagenesis of conserved amino acids in the alpha subunit of toluene dioxygenase: potential mononuclear non-heme iron coordination sites. J Bacteriol. 1996;178:3133–3139. doi: 10.1128/jb.178.11.3133-3139.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karuzina I I, Archakov A I. Hydrogen peroxide-mediated inactivation of microsomal cytochrome P450 during monooxygenase reactions. Free Radic Biol Med. 1994;17:557–567. doi: 10.1016/0891-5849(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 24.Karuzina I I, Archakov A I. The oxidative inactivation of cytochrome P450 in monooxygenase reactions. Free Radic Biol Med. 1994;16:73–97. doi: 10.1016/0891-5849(94)90245-3. [DOI] [PubMed] [Google Scholar]
- 25.Kauppi B, Lee K, Carredano E, Parales R E, Gibson D T, Eklund H, Ramaswamy S. Structure of an aromatic ring-hydroxylating dioxygenase—naphthalene 1,2-dioxygenase. Structure. 1998;6:571–586. doi: 10.1016/s0969-2126(98)00059-8. [DOI] [PubMed] [Google Scholar]
- 26.Lee K. Ph.D. thesis. Iowa City: University of Iowa; 1995. [Google Scholar]
- 27.Lee K, Brand J M, Gibson D T. Stereospecific sulfoxidation by toluene and naphthalene dioxygenases. Biochem Biophys Res Commun. 1995;212:9–15. doi: 10.1006/bbrc.1995.1928. [DOI] [PubMed] [Google Scholar]
- 28.Lee K, Gibson D T. Toluene and ethylbenzene oxidation by purified naphthalene dioxygenase from Pseudomonas sp. strain NCIB 9816-4. Appl Environ Microbiol. 1996;62:3101–3106. doi: 10.1128/aem.62.9.3101-3106.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee K, Gibson D T. Abstracts of the 97th General Meeting of the American Society for Microbiology 1997. Washington, D.C: American Society for Microbiology; 1997. Naphthalene dioxygenase: factors involved in the uncoupling of substrate oxidation by benzene, abstr. K-31; p. 347. [Google Scholar]
- 30.Lee K, Kauppi B, Parales R E, Gibson D T, Ramaswamy S. Purification and crystallization of the oxygenase component of naphthalene dioxygenase in native and selenomethionine-derivatized forms. Biochem Biophys Res Commun. 1997;241:553–557. doi: 10.1006/bbrc.1997.7863. [DOI] [PubMed] [Google Scholar]
- 31.Lee K, Resnick S M, Gibson D T. Stereospecific oxidation of (R)- and (S)-indanol by naphthalene dioxygenase from Pseudomonas sp. strain NCIB 9816-4. Appl Environ Microbiol. 1997;63:2067–2070. doi: 10.1128/aem.63.5.2067-2070.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lipscomb J D. Biochemistry of the soluble methane monooxygenase. Annu Rev Microbiol. 1994;48:371–399. doi: 10.1146/annurev.mi.48.100194.002103. [DOI] [PubMed] [Google Scholar]
- 33.Mason J R, Cammack R. The electron-transport proteins of hydroxylating bacterial dioxygenases. Annu Rev Microbiol. 1992;46:277–305. doi: 10.1146/annurev.mi.46.100192.001425. [DOI] [PubMed] [Google Scholar]
- 34.Morrison M. Iodination of tyrosine: isolation of lactoperoxidase (bovine) Methods Enzymol. 1970;17A:653–657. [Google Scholar]
- 35.Newman L M, Wackett L P. Trichloroethylene oxidation by purified toluene 2-monooxygenase: products, kinetics, and turnover-dependent inactivation. J Bacteriol. 1997;179:90–96. doi: 10.1128/jb.179.1.90-96.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nordblom G D, Coon M J. Hydrogen peroxide formation and stoichiometry of hydroxylation reactions catalyzed by highly purified liver microsomal cytochrome P450. Arch Biochem Biophys. 1977;180:343–347. doi: 10.1016/0003-9861(77)90047-9. [DOI] [PubMed] [Google Scholar]
- 37.Ortiz de Montellano P R. Cytochrome P-450 catalysis: radical intermediates and dehydrogenation reactions. Trends Pharmacol Sci. 1989;10:354–359. doi: 10.1016/0165-6147(89)90007-2. [DOI] [PubMed] [Google Scholar]
- 38.Parales J V, Kumar A, Parales R E, Gibson D T. Cloning and sequencing of the genes encoding 2-nitrotoluene dioxygenase from Pseudomonas sp. JS42. Gene. 1996;181:57–61. doi: 10.1016/s0378-1119(96)00462-3. [DOI] [PubMed] [Google Scholar]
- 39.Parales J V, Parales R E, Resnick S M, Gibson D T. Enzyme specificity of 2-nitrotoluene 2,3-dioxygenase from Pseudomonas sp. strain JS42 is determined by the C-terminal region of the α subunit of the oxygenase component. J Bacteriol. 1998;180:1194–1199. doi: 10.1128/jb.180.5.1194-1199.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parales R E, Emig M D, Lynch N A, Gibson D T. Substrate specificities of hybrid naphthalene and 2,4-dinitrotoluene dioxygenase enzyme systems. J Bacteriol. 1998;180:2337–2344. doi: 10.1128/jb.180.9.2337-2344.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Percival M D. Human 5-lipoxygenase contains an essential iron. J Biol Chem. 1991;266:10058–10061. [PubMed] [Google Scholar]
- 42.Porter T D, Coon M J. Cytochrome P-450: multiplicity of isoforms, substrates and catalytic and regulatory mechanisms. J Biol Chem. 1991;266:13469–13472. [PubMed] [Google Scholar]
- 43.Resnick S M, Lee K, Gibson D T. Diverse reactions catalyzed by naphthalene dioxygenase from Pseudomonas sp. strain NCIB 9816. J Ind Microbiol. 1996;17:438–457. [Google Scholar]
- 44.Resnick S M, Torok D S, Lee K, Brand J M, Gibson D T. Regiospecific and stereoselective hydroxylation of 1-indanone and 2-indanone by naphthalene dioxygenase and toluene dioxygenase. Appl Environ Microbiol. 1994;60:3323–3328. doi: 10.1128/aem.60.9.3323-3328.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosche B, Tshisuaka B, Fetzner S, Lingens F. 2-Oxo-1,2-dihydroquinoline 8-monooxygenase, a two-component enzyme system from Pseudomonas putida 86. J Biol Chem. 1995;270:17836–17842. doi: 10.1074/jbc.270.30.17836. [DOI] [PubMed] [Google Scholar]
- 46.Simon M J, Osslund T D, Saunders R, Ensley B D, Suggs S, Harcourt A, Suen W-C, Cruden D L, Gibson D T, Zylstra G J. Sequences of genes encoding naphthalene dioxygenase in Pseudomonas putida strains G7 and NCIB 9816-4. Gene. 1993;127:31–37. doi: 10.1016/0378-1119(93)90613-8. [DOI] [PubMed] [Google Scholar]
- 47.Sligar S G, Lipscomb J D, Debrunner P G, Gunsalus I C. Superoxide anion production by the autoxidation of cytochrome P450cam. Biochem Biophys Res Commun. 1974;61:290–296. doi: 10.1016/0006-291x(74)90565-8. [DOI] [PubMed] [Google Scholar]
- 48.Stadtman E R. Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal catalyzed reactions. Annu Rev Biochem. 1993;62:797–821. doi: 10.1146/annurev.bi.62.070193.004053. [DOI] [PubMed] [Google Scholar]
- 49.Suen W-C. Ph.D. thesis. Iowa City: University of Iowa; 1991. [Google Scholar]
- 50.Suen W-C, Gibson D T. Isolation and preliminary characterization of the subunits of the terminal component of naphthalene dioxygenase from Pseudomonas putida NCIB 9816-4. J Bacteriol. 1993;175:5877–5881. doi: 10.1128/jb.175.18.5877-5881.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suen W-C, Gibson D T. Recombinant Escherichia coli strains synthesize active forms of naphthalene dioxygenase and its individual α and β subunits. Gene. 1994;143:67–71. doi: 10.1016/0378-1119(94)90606-8. [DOI] [PubMed] [Google Scholar]
- 52.Twilfer H, Bernhardt F-H, Gersonde K. Dioxygen-activating iron center in putidamonooxin: electron spin resonance investigation of the nitrosylated putidamonooxin. Eur J Biochem. 1985;147:171–176. doi: 10.1111/j.1432-1033.1985.tb08733.x. [DOI] [PubMed] [Google Scholar]
- 53.Wackett L P, Kwart L D, Gibson D T. Benzylic monooxygenation catalyzed by toluene dioxygenase from Pseudomonas putida. Biochemistry. 1988;27:1360–1367. doi: 10.1021/bi00404a041. [DOI] [PubMed] [Google Scholar]
- 54.Wende P, Bernhardt F-H, Pfleger K. Substrate-modulated reactions of putidamonooxin: the nature of the active oxygen species formed and its reaction mechanism. Eur J Biochem. 1989;181:189–197. doi: 10.1111/j.1432-1033.1989.tb14710.x. [DOI] [PubMed] [Google Scholar]
- 55.Wolfe, M. D., and J. D. Lipscomb. Personal communication.
- 56.Yamazaki I, Piette H. ESR spin-trapping studies on the reaction of Fe2+ ions with H2O2-reactive species in oxygen toxicity in biology. J Biol Chem. 1990;265:13589–13594. [PubMed] [Google Scholar]