

Abstract
Aberrant activation of CD4 T helper 2 (Th2) cells and excessive production of Th2 cytokines like interleukin (IL)-4 and IL-13 have been implicated in the pathogenesis of allergic diseases. Generally, IL-4 and IL-13 utilize JAK-STAT signaling pathways for induction of inflammatory gene expression and the effector functions associated with disease pathology in many allergic diseases. However, it is increasingly clear that JAK/STAT pathways activated by IL-4/IL-13 can themselves be modulated in the presence of other intracellular signaling programs, thereby changing the overall tone and/or magnitude of IL-4/IL-13 signaling. Apart from direct activation of the canonical JAK-STAT pathways, IL-4 and IL-13 also induce pro-inflammatory gene expression and effector functions through activation of additional signaling cascades. These alternative signaling cascades contribute to several specific aspects of IL-4/IL-13-associated cellular and molecular responses. A more complete understanding of IL-4/IL-13 signaling pathways, including the precise conditions under which non-canonical signaling pathways are activated, and the impact of these pathways on cellular and host level responses, will better allow us to design agents that target specific pathological outcomes, or tailor therapies for the treatment of uncommon disease endotypes.
Keywords: Allergic disease, cytokine signaling, IL-13, IL-4
Introduction:
Allergic diseases such as allergic asthma, atopic dermatitis, and allergic rhinitis collectively affect over 10–15% of the global population and their prevalence has doubled in last decade1. Allergic diseases are characterized by aberrant activation of CD4 T helper 2 (Th2) cells in response to innocuous environmental proteins2 - allergens - and subsequent production of Th2-derived cytokines such as interleukin (IL)-4, IL-5, and IL-13 at sites of allergic inflammation3, 4. Th2-derived cytokines are considered central to the pathology associated with allergic diseases, and as such excellent reviews on mechanisms driving Th2 development in the context of allergic diseases5, 6, and the mechanisms through which Th2 cytokines influence disease pathology7 are available. Accordingly, there has also been a great deal of interest in anti-Th2 cytokine biologicals for the treatment of these diseases8, 9. Moreover, as signaling pathways activated by Th2 cytokines typically use Janus kinase/Signal Transducer and Activator of Transcription (JAK/STAT) pathways for inducing expression of genes that contribute to their downstream effector functions, the use of JAK inhibitors is also being considered10. However, other understudied signaling intermediates have been reported to be activated in response to Th2 cytokines and recent work suggests that the intensity of Th2 cytokine signaling can be influenced by the presence of other inflammatory signals (i.e., Th17-associated cytokines). Given that the true power (and limitations) of targeting Th2-associated pathways in their entirety in the context of allergic diseases is still being deciphered, it is likely premature to speculate as to how and/or when specific pathways may represent unique and tractable targets for therapeutic intervention. Nonetheless, a more complete understanding of the multitude of pathways activated by disease relevant cytokines may lead to therapeutic interventions applicable to specific forms of allergic asthma (i.e., Th2 low or mixed Th2/Th17 responses9, 11), disease forms for which therapeutic options remain limited. Thus, the purpose of this article is to review evidence for activation of non-canonical IL-4/IL-13-induced signaling intermediates (such as MAPKs, PI3K/Akt, PKC and SRC), discuss negative and positive regulators of the IL-4/IL-13 signaling pathways, and describe interactions between IL-4/IL-13 signaling pathways and other pathways.
Role of IL-4 and IL-13 in allergic disease:
IL-4 and IL-13 are central to the pathology of many allergic disorders and have a variety of cellular targets that contribute to these responses. IL-4 serves as a key cytokine promoting the development of allergic disease through its ability to prime the differentiation of naïve T helper cells into Th2 cells12, 13 and its ability to promote immunoglobulin class switching to IgE in B cells14–16. In contrast, the role of IL-13 is primarily exerted in the effector phase via potent influences on structural cells such as epithelial cells, endothelial cells, fibroblasts, keratinocytes and smooth muscle cells17–22. On these cells, IL-13 induces a variety of effects, including induction of tissue remodeling23, regulation of barrier function24, 25, differentiation and proliferation of mucus-producing goblet cells26, and induction of smooth muscle hypertrophy19, 27. Although several agents targeting IL-4, IL-13 or their receptors are being explored for the treatment of allergic disease28–31, a complete understanding of the intracellular signaling cascades activated by these two allergic-disease relevant cytokines, and the biological processes controlled by various signaling pathways may inform potentially novel therapeutic options that can be tailored to control specific aspects of disease pathology.
IL-4 and IL-13 receptor complexes
IL-4 and IL-13 utilize shared receptors for activation of intracellular signaling cascades (Figure 1). The IL-4/IL-13 receptor family consists of two distinct heterodimeric receptors: the type I IL-4R (consisting of the IL-4Rα and the common gamma chain (γc)) and the type II IL-4R (consisting of IL-4Rα and the IL-13Rα1). Although IL-4 can bind to, and signal through, both type I and type II IL-4Rs, IL-13 binding and signaling is restricted to the type II IL-4 receptor23. Importantly, the cellular distribution of IL-4Rα and IL-13Rα1 is variable - although most cell types express at least low levels of IL-4Rα32, hematopoietic cells typically express higher levels of IL-4Rα, while non-hematopoietic cells express higher levels of IL-13Rα133. This may explain the particularly pronounced effects of IL-13 on non-hematopoietic cells (e.g. epithelial cells or fibroblasts) compared to lymphocytes (for more information on this aspect of type I/II IL-4R biology please see33, 34). Importantly, IL-13 (but not IL-4) can also bind to a unique receptor called IL-13Rα2. IL-13Rα2 binds to IL-13 with a higher affinity than IL-13Rα1, and because IL-13Rα2 has a short cytoplasmic tail (as well as a cleavage-induced, or alternatively spliced soluble forms), many consider IL-13Rα2 a decoy receptor35–37. However, recent reports suggest that IL-13Rα2 can activate alternative signaling pathways38, 39. The canonical and alternative pathways activated downstream of these receptors are discussed below.
Figure 1: Canonical and non-canonical IL-4 and IL-13 signaling pathways:
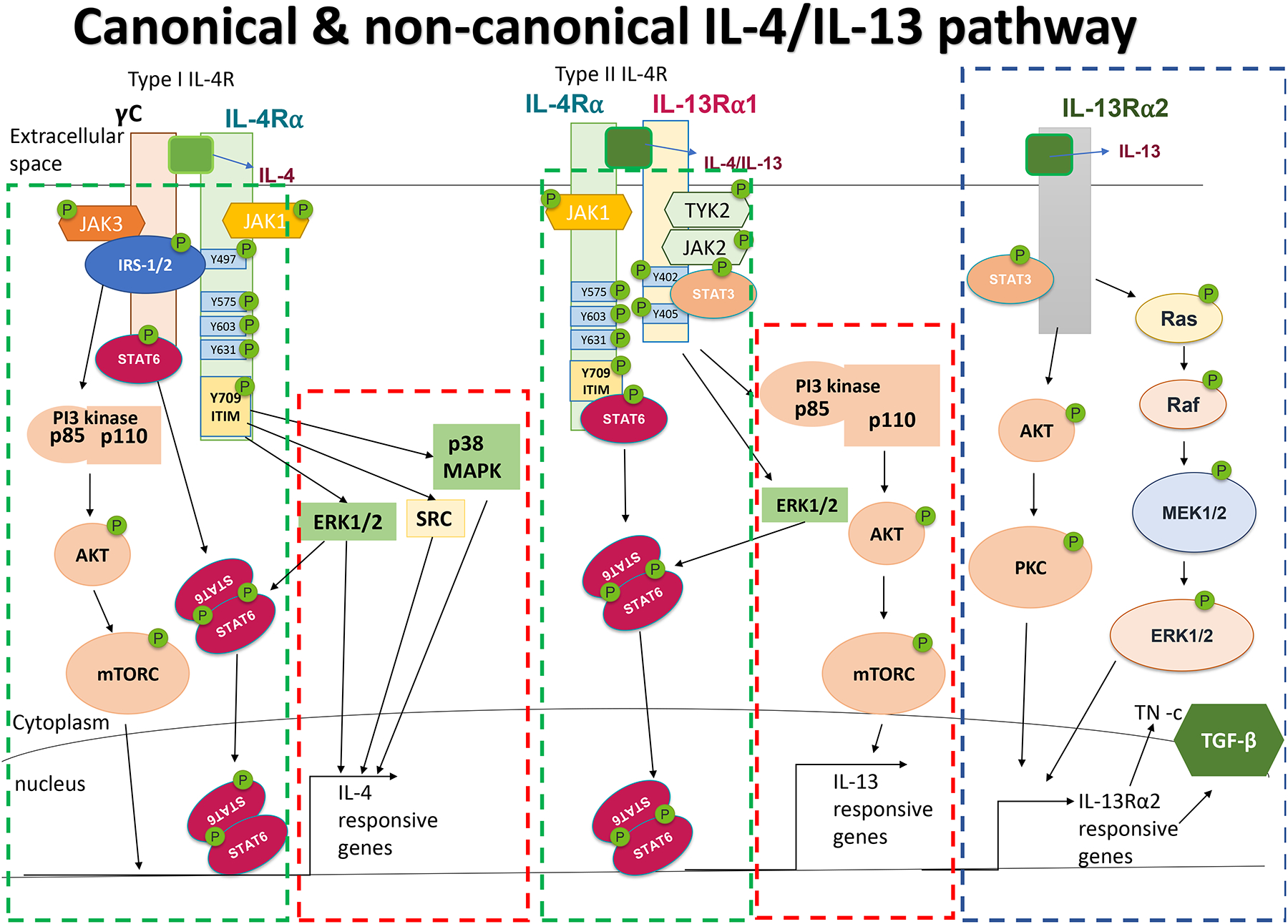
Canonical pathways (marked in dashed green box) activated in response to type I and type II IL-4R signaling involve JAK/STAT-mediated phosphorylation of STAT6. Activation of the type 1 IL-4R also leads to activation of IRS-2 and downstream activation of PI3K, Akt and mTOR. Non-canonical activation (marked in red dashed box) of SRC, ERK1/2 and p38 MAPK in response to IL-4, or ERK1/2 and PI3K in response to IL-13, have also been described to contribute important biological outcomes. As activation of PI3K/AKT/mTOR is directly attributable to γc/JAK3-driven phosphorylation of Y647 in IL-4Rα in the type 1 IL-4R complex, but IL-13Rα1/TYK2-driven phosphorylation of Y647 has not been described for the type II IL-4R, PI3K/AKT activation is considered canonical downstream of the type I IL-4R, but non-canonical downstream of the type II IL-4R complex. IL-13 can also signal through IL-13Rα2 (marked in dashed blue box) to activate/phosphorylate Akt/PKC and Ras/ERK1/2 pathway to induce TGFβ, Tenascin-c and other IL-13Rα2-associated genes.
Type I IL-4R signaling pathway:
IL-4 binds with high affinity to IL-4Rα (KD = 0.1 nM)40, 41 and formation of the IL-4/IL-4Rα complex facilitates the recruitment of γc to complete the formation of the type I IL-4R system (Figure 1). Assembly of this receptor complex allows phosphorylation and activation of JAK family members that are constitutively associated with IL-4Rα and γc: JAK1 and JAK3 respectively42. Once phosphorylated, JAK1 phosphorylates five tyrosine residues (Y497, Y575, Y603, Y631, and Y713) within the IL-4Rα cytoplasmic tail. These phospho-tyrosine residues subsequently act as docking sites for downstream phospho-tyrosine binding (PTB) signaling molecules43, 44. Specifically, pY497 residues form an insulin/interleukin-4 receptor (I4R) motif that interacts with PTB domains on insulin receptor substrate-2 (IRS-2)45 and allows γc-dependent phosphorylation of IRS-246. Phosphorylated IRS-2 binds to and activates the p85 domain of phosphatidylinositol-3-kinase (PI3K) leading to activation of Akt, PDK1/2 and p70S6 kinases47. In monocytes, IRS-2 activation is important for skewing towards an alternatively activated macrophage phenotype and expression of genes such as Arg1, Chi3l (Ym1), and Chi3l1 (YKL-40)46, 48. IL-4 is the major inducer of alternatively activated macrophages which have been shown to enhance Th2 responses, elevate eosinophilic inflammation, promote tissue remodeling and fibrosis and exacerbate AHR49–54. Phosphorylation of Y575, Y603, and Y631 of IL-4Rα enables the recruitment of STAT6. Once recruited to the receptor, JAK1 and JAK3 phosphorylate the C-terminal tyrosine residue of STAT6 (Y631) leading to homodimerization of STAT6. Dimerized STAT6 translocates to the nucleus and activates the expression of many IL-4-responsive genes. In contrast to the activating role of the other tyrosine residues, tyrosine Y713 on the IL-4Rα is part of an immune-tyrosine-based inhibitory motif (ITIM) and helps in negative regulation of IL-4 signaling responses through recruitment of SHP1/255, important negative regulators of IL-4 signaling (see below).
Type II IL-4R signaling pathway:
In contrast to the type I IL-4R, which binds only IL-4, the heterodimeric type II IL-4R binds both IL-4 and IL-13 (Figure 1). Although IL-4 has higher binding affinity towards IL-4Rα (ΚD = 0.1 nM)41 than that of IL-13 to IL-13Rα1 (ΚD = 1.7 nM)56, IL-13 is typically produced in higher quantities than IL-4. Binding of IL-4/IL-13 to type II IL-4R activates JAK1, which is constitutively associated with IL-4Rα, and tyrosine kinase 2 (TYK2), which is associated with IL-13Rα1. Activated JAK1 and TYK2 phosphorylate residues Y575, Y603, Y631, Y709 in the IL-4Rα leading to recruitment and activation of STAT6, as in the Type I IL-4R. Importantly, as γc is not present in type II IL-4R system, IRS-2 is not activated46. On the other hand, the cytoplasmic domain of IL-13Rα1 contains two tyrosine residues, Y402 and Y405, which have been proposed to act as a docking site for STAT355. Unfortunately, the physiological consequences of potential STAT3 activation in this context are not well understood.
IL-13Rα2:
IL-13Rα2 was initially described as a decoy receptor capable of limiting IL-13-induced pathology. This was based on several properties, including a higher binding affinity of IL-13 for IL-13Rα2 than IL-13Rα1 (ΚD < 10−15 M vs ΚD = 1.7 nM)56, 57, and the observation that the cytoplasmic tail of IL-13Rα2 (17 amino acids58) is much shorter than that of IL-13Rα1 and IL-4Rα (60 & 785 amino acids respectively) and lacks the conserved JAK binding sites essential for signal transduction56, 59. The concept of IL-13Rα2 serving an inhibitory function is supported by studies demonstrating that 1) endogenous expression of IL-13Rα2 attenuated IL-13-induced gene expression in lung fibrosis and atopic dermatitis models37, 38; 2) endogenous expression of IL-13Rα2 decreased the sensitivity of the type II IL-4R to IL-1337; 3) antibodies blocking interactions between IL-13 and type II IL-4R in IL-13Rα2-sufficient mice did not induce IL-13 relevant gene expression37; 4) mice lacking IL-13Rα2 demonstrated increased IL-13-mediated cutaneous inflammation, IgG1 levels, and trans-epidermal water loss in a mouse model of atopic dermatitis compared to wild-type mice36; and 5) IL-13Rα2 null mice had increased IL-13-mediated STAT6 activation compared to wild type mice36. Importantly, IL-13Rα2 expression is induced by IL-13 in multiple cell types (primary human keratinocytes, keratinocyte cell lines60,36,61), suggesting that expression of IL-13Rα2 may represent a negative feedback loop which can attenuate IL-13 signaling.
Although these studies suggest a negative role for IL-13Rα2 in regulation of IL-13 signaling, contradicting observations exist – we39 and others62 have demonstrated that IL-13Rα2 KO mice display reduced airway hyperresponsiveness (AHR), mucus production, and IL-13-induced gene expression in the lung compared to wild type mice in both HDM-challenge and IL-13-challenge models (Figure 1). Moreover, IL-13Rα2 overexpression in lung epithelial cells reconstituted airway inflammation comparable to HDM-challenged wild type mice, suggesting that pulmonary epithelial cell expression of IL-13Rα2 was the mediator of these effects62. Mechanistically, IL-13 interaction with IL-13Rα2 has been shown to activate the transcription factor activator protein-1 (AP-1), which then induced TGFβ production63–65. Other studies show that IL-13 binds to IL-13Rα2 and induces extracellular signal-regulated kinases 1/2 (ERK1/2), and the downstream AP-1-related gene C-Jun in human nasal epithelial cells65. Another recent study examining potential IL-13Rα2 signaling demonstrated that IL-13 binding to IL-13Rα2 increased epidermal growth factor receptor vIII (EGFRvIII) tyrosine kinase activities, leading to increased RAS/RAF/MEK/ERK and STAT3 activation in a human glioblastoma cell line66. Although signaling via the IL-13Rα2 has been implicated in many tumor models67–70, its role in allergic diseases remains understudied. Nonetheless, these studies suggest a complex role for IL-13Rα2 in regulating allergic diseases.
IL-4 and IL-13 mediated activation of non JAK/STAT-mediated pathways:
Apart from direct activation of above-described JAK-STAT pathways, IL-4 and IL-13 also induce pro-inflammatory gene expression and effector functions through activation of additional signaling cascades, including AKT/phosphoinositide 3-kinase (PI3K)/mammalian target of rapamycin (mTOR), mitogen activated protein kinases (MAPK), or SRC, through STAT6-dependent and - independent signaling pathways (Figure 1). These alternative signaling cascades contribute to important aspects of IL-4/IL-13-associated cellular and molecular responses.
PI3K activation:
IL-4 and IL-13 have been shown to induce activation of a PI3K/AKT/mTOR cascade, which has profound effects on cell biology. PI3K is a lipid kinase that is typically activated by receptor tyrosine kinases (RTKs) binding to the P85 subunit of PI3K71. Activated PI3K then phosphorylates the plasma membrane lipid phosphatidylinositol (4,5)-bisphosphate (PIP2) to generate phosphatidylinositol (3,4,5)-trisphosphate (PIP3), which is responsible for further activation and initiation of signaling cascades such as protein serine/threonine kinases, AKT, and phosphoinositide-dependent kinase (PDK)71. Activated AKT initiates downstream gene expression that contributes to cell growth, proliferation, and anti-apoptotic activity through engagement of mTOR signaling72. Studies have shown that IL-4-induced IRS-2 activates PI3K and the downstream protein serine/threonine AKT46 and that this contributes to IL-4-induced changes such as alternatively activated macrophage differentiation and enhanced vascular remodeling and muscularization54, 73–75. Interestingly, although IL-13 signals exclusively through the type 2 IL-4R complex, and thus does not activate IRS-2 (as it utilizes IL13Rα1/TYK2 rather than γc/JAK3, which are required for phosphorylation of Y647 and docking of IRS-2 to the IL-4Rα), IL-13 has also been shown to activate PI3K (via the p110 subunit), which then phosphorylates AKT in murine smooth muscle tracheal cells27, 76, 77. IL-13-driven activation of a PI3K/AKT/Protein Kinase C (PKC) cascade contributes to induction of Tenascin-C expression in a mouse bleomycin-induced fibrosis model77. Interestingly, Tenascin-C is a glycoprotein found to be upregulated in bronchial tissue of asthmatic patients78, suggesting that this pathway may contribute to IL-13-induced lung remodeling in the context of asthma. The mechanisms through which IL-13 supports activation of PI3K, remain unclear.
MAPK activation:
Both IL-4 and IL-13 also activate members of the MAPK family - a family of serine threonine kinases that induce expression of inflammatory-, growth factor- and osmotic stress-related genes79. MAPKs are classified into 3 subfamilies: 1) extracellular signal related protein kinase 1/2 (ERK1/2), 2) Jun N-terminal kinases (JNKs) and 3) p38 MAPK80–82. Activation of these signaling intermediates contributes to the increased activity of transcriptional regulators including nuclear factor of activated T-cells (NFAT), cyclic AMP-response element binding protein (CREB), and AP-1, and have been described to mediate specific aspects of IL-4/IL-13 responses. IL-4-mediated induction of p38 MAPK stabilized IL-6 mRNA in human keratinocyte cell line83, suggesting that IL-4 can regulate innate inflammatory responses in structural cells. IL-4-mediated activation of ERK has been reported in a human T cell line, and inhibition of ERK was found to inhibit IL-4-driven STAT6 activation and subsequent Th2 differentiation84, suggesting an important role for IL-4-induced MAPK activation in Th2 skewing. Using IL-13-transgenic mice, it was demonstrated that IL-13 induces phosphorylation of ERK1/2 (but not JNK and p38) through a STAT6-independent pathway85. Interestingly, chemokine production via canonical STAT6 signaling versus ERK1/2 activation varied by chemokine, with some chemokines demonstrating complete dependence upon STAT6 (CCL11, CCL2, CCL6), complete dependence on ERK1/2 (CXCL1) or equivalent induction by STAT6 and ERK1/2 (CCL3, CCL4, CCL5)85. In a human keratinocyte cell line, inhibition of ERK1/2 completely abrogated IL-4- and IL-13-mediated induction of IL-13Rα279, suggesting ERK1/2 is also important for IL-4/IL-13-mediated IL-13Rα2 expression. Thus, specific inhibition of IL-13 or IL-4 driven ERK may have selective effects on IL-4 and IL-13-induced cellular recruitment and disease pathology.
SRC Activation:
A final signaling intermediate implicated in IL-4/IL-13 signaling is the proto-oncogene tyrosine-protein kinase SRC. SRC signaling has been implicated in cell proliferation, growth, and survival86 as well as downstream activation of various STAT proteins86–89. Consistent with reports that SRC can promote activation of some STAT proteins86, overexpression of v-SRC in mouse fibroblasts induced constitutive STAT6 phosphorylation and activation, while fibroblasts lacking SRC displayed reduced IL-4-driven activation of STAT6, suggesting that SRC might be involved upstream of STAT690. Interestingly, although IL-4 and IL-13 both utilize STAT6 as an important signaling intermediate, the activation and involvement of SRC in IL-13-induced signaling pathways has not been explored. Nonetheless, these studies implicate SRC as an important upstream activator of STAT6 in response to IL-4. Given the interest in development of therapeutics based on targeting SRC91, it would be important to further delineate the relationship between IL-4/IL-13 signaling and SRC activity.
Collectively these studies suggest that IL-4 and IL-13 have the potential to activate several signaling pathways beyond their well described influence on JAK/STAT6 signaling. Confirming the importance of these pathways, identifying situations where they are preferentially induced, and delineating the specific molecular processes that are activated in response to their signaling will be important to fully understand the molecular mechanisms driving disease in situations where excessive IL-4/IL-13 production are expected to play a role.
Regulators of IL-4 and IL-13 signaling:
As described above, IL-4 and IL-13 signaling utilizes multiple signaling pathways. These pathways can themselves be modulated in the presence of other intracellular signaling programs, thereby changing the overall tone and magnitude of IL-4/IL-13 signaling. Both positive and negative regulators of IL-4/IL-13-induced responses have been described. Below we discuss these positive and negative regulators of IL-4/IL-13 signaling, the mechanisms they utilize to alter these signaling pathways, and the expected influence of these pathways on the allergic response.
Negative regulators of IL-4 and IL-13 signaling:
The key negative regulators of JAK-STAT signaling pathways are Protein Tyrosine Phosphatases (PTPs), Src homology 2-containing protein tyrosine phosphatase (SHP-1), and Suppressor of Cytokine Signaling (SOCS) proteins (Figure 2).
Figure 2: Negative and positive regulators of IL-4 and IL-13 signaling:
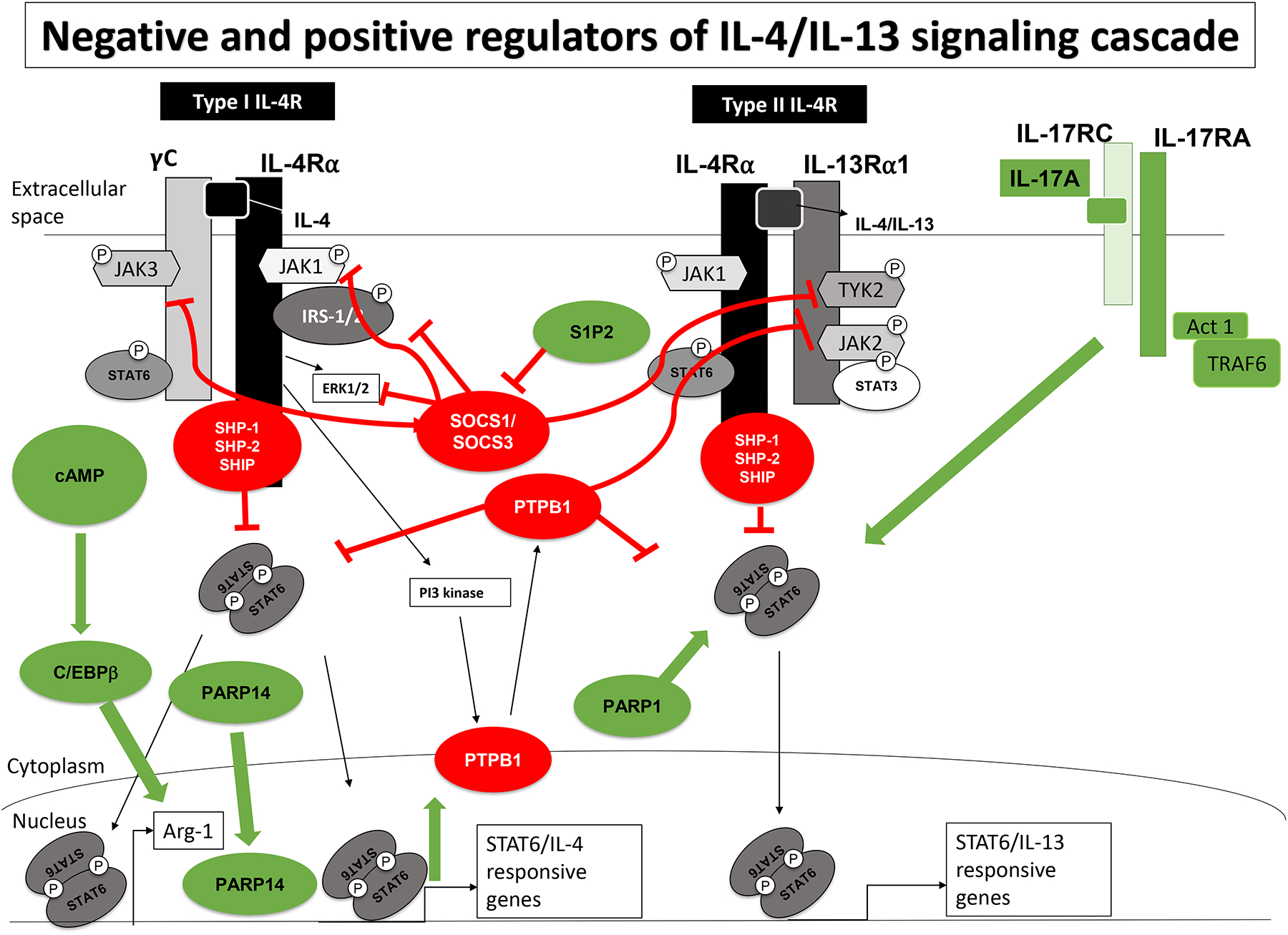
IL-13 and IL-4 signaling intermediates and receptors are marked in black and grey. Negative regulators (marked in red circles and red lines) of IL-4/13 signaling (SOCS proteins, protein tyrosine phosphatases) function by inhibiting JAK kinase activity, targeting activated signaling intermediates for proteolysis, and removing phosphate groups from activated signaling intermediates. Positive regulators (marked in green circles and green arrows) of IL-4/13 signaling pathway (PARP proteins, cAMP, S1P2 and IL-17A) facilitate efficient STAT6 DNA binding, protect STAT6 from proteolytic degradation, enhance STAT6 phosphorylation, enhance IL-4-induced gene expression through recruitment of C/EBPβ, and suppress SOCS3-mediated inhibition.
Protein Tyrosine phosphatases (PTPs):
Some of the most widely recognized negative regulators of IL-4/IL-13 signaling are the Src homology 2-containing phosphatases, including tyrosine-protein phosphatase non-receptor type 6 (PTPN6/SHP-1)92–94, PTPN11 (SHP-2)95, 96, and SH2-containing inositol phosphatase (SHIP)97–99. The 5th tyrosine residue (Y709) of the IL-4Rα cytoplasmic domain serves as a docking site for regulatory phosphatases including SHP-1, SHP-2 and SHIP, and as such, this region of the IL-4Rα has been termed the Immunoreceptor Tyrosine-based Inhibitory Motif (ITIM)100. Once recruited to the IL-4 receptor complexes, these phosphatases have the capacity to attenuate or terminate these signaling pathways by removing phosphate groups on activated signaling intermediates owing to their intrinsic phosphatase capacity. In support of their role as a negative regulator of IL-4 and IL-13, transgenic expression of SHP-1 in mouse fibroblasts (NIH3T3) significantly reduced IL-4-induced STAT6 phosphorylation and STAT6 responsive gene expression compared to control cell line94. Additionally, bone marrow derived macrophages (BMDM) from mice with a spontaneous mutation rendering SHP-1 inactive (“motheaten”, or mev mice) demonstrated enhanced IL-4-mediated phosphorylation of STAT6 compared to cells from wild type mice94. Similarly macrophages from SHIP deficient mice demonstrated more substantial IL-4-driven skewing towards an alternatively activated phenotype with enhanced Arg1 activity101. Finally, ITIM or SHP-1 deletion in mice led to a dramatic decrease in dephosphorylation of IL-4/IL-13-induced STAT692, 102–104. In vivo studies also suggest these PTPs negatively regulate allergic inflammation. Mev mutant mice had increased eosinophils, elevated IgE levels, and higher AHR compared to wild type mice in OVA-induced allergic asthma models102, 103, 105–107. Likewise, SHP-2 deletion in myeloid cells decreased OVA-induced eosinophil recruitment and AHR compared to wild type mice108. Finally, mice in which the ITIM on the IL-4Rα chain is mutated to be resistant to phosphorylation (IL-4Rα Y709F mice) and thus unable to activate SHP-1/SHP-2 during IL-4/IL-13 signaling demonstrate increased Th2-driven inflammation, such as increased STAT6 phosphorylation, increased activation of alternative macrophages, elevated eosinophil recruitment, and exacerbated AHR compared to WT mice in an OVA-induced allergic asthma model102, 109, 110.
PTP1B, another PTP, had also been reported to modulate cytokine signaling by dephosphorylating JAK2 and TYK2111. Overexpression of PTP1B led to dephosphorylation of STAT6 and decreased STAT6 transcriptional activity, while PTP1B deficiency led to prolonged STAT6 phosphorylation112. Co-precipitation studies demonstrated a direct interaction between PTP1B and both JAK1 and STAT6, suggesting multiple different pathways through which PTP1B might influence IL-4 signaling. Importantly, IL-4 also induces expression of PTP1B through a STAT6-independent, PI3K-dependent signaling pathway112. As IL-4 and IL-13 both utilize JAK1 and STAT6, PTP1B is likely to inhibit IL-13-induced JAK and STAT6 phosphorylation as well, although such regulation has not been formally tested.
SOCS-mediated inhibition of IL-4/IL-13 signaling:
SOCS proteins are negative regulators of JAK-STAT signaling113. The SOCS family of proteins contains 8 unique members (SOCS1 – 7 and Cytokine Inducible Sh2 protein (CIS)). This family is defined by the presence of a C-terminal SH2 domain, and a SOCS box which contains both kinase inhibitory and ubiquitin ligase functions114. This domain also allows association with elongin BC, an adaptor protein which helps recruit E3 ubiquitin ligase scaffold (cullin5)115. Patterns, and cell type-specific expression of SOCS proteins varies amongst cell types, but expression is typically of an inducible nature, and often can be induced by STAT transcription factors, suggesting that these represent feedback inhibitors of inflammatory cytokine signaling that prevent prolonged STAT signaling (recently reviewed in116). SOCS proteins regulate JAK-STAT activation through direct inhibition of JAK activity via a unique short motif within the SOCS box called the kinase inhibitory region (KIR)117, 118. Following interaction with activated kinases (via SH2-mediated phosphotyrosine binding), SOCS proteins promote ubiquitination of associated targets and subsequent proteasomal degradation73,119.
Among SOCS proteins, SOCS1 has been demonstrated to have direct inhibitory activity relevant to IL-4/IL-13 signaling. SOCS1 directly limits JAK phosphorylation and facilitates the ubiquitination of IRS-2 following activation of the type I IL-4R. In monocytes, IRS-2 tyrosine phosphorylation is important for alternative macrophage activation through induction of IL-4-dependent gene expression. As SOCS1 suppresses IRS-2 tyrosine phosphorylation by ubiquitinating IRS-2 and targeting IRS-2 for proteasomal degradation120, this directly limits alternative macrophage activation. In B cells, SOCS1 negatively regulates IL-4- and IL-13-induced STAT6 activity by direct inhibition of JAK1121, suggesting that SOCS1 can also regulate IgE production. Not surprisingly, the role of SOCS1 as a negative regulator of IL-4 and IL-13 signaling is supported by many studies114, 120–124. In an OVA-induced allergic asthma model, mice expressing SOCS1 that is unable to enter the nucleus (which spares them from the early lethal phenotype observed in complete SOCS1-deficient animals125) had enhanced Th2 inflammation, such as elevated eosinophils, airway epithelial cell remodeling, and increased Th2 cytokines in response to OVA-albumin exposure compared to wildtype mice126. Similarly, human nasal epithelial cells taken from individuals with severe eosinophilic asthma had reduced SOCS1 expression levels compared to healthy controls122.
Like SOCS1, SOCS3 directly binds to and inhibits the catalytic domain of JAK1, JAK2 and TYK2 through its KIR motif. Apart from inhibiting JAK kinase activity, SOCS3 also inhibits JAK1, IRS-2 through E3 ubiquitination and proteasomal degradation127. E3 ubiquitin ligase activity of SOCS3 was particularly important for inhibition of IL-4-induced ERK phosphorylation and the C-Jun128. Not surprisingly, overexpression of SOCS3 in a rat mast cell line reduced calcium-induced ERK1/2 phosphorylation and C-Jun transcription factor activity resulting in reduced IL-4-induced gene expression. Conversely, knockdown of SOCS3 triggered higher ERK1/2 phosphorylation and C-Jun transcription factor expression, suggesting SOCS3 negatively regulates IL-4-induced outcomes by inhibiting ERK signaling128.
Collectively, these studies provide a rationale for use of PTP or SOCS-activating therapeutics for treatment of allergic disease. Interestingly, transcellular liposomal delivery of SOCS3 in lung epithelial cells inhibited the allergen-induced IL-4/IL-13-mediated phosphorylation of STAT6 and STAT3129, revealing the therapeutic potential of such an approach.
Positive modulators of IL-4 and IL-13 signaling:
In addition to the negative regulators of IL-4/IL-13 signaling, factors which positively influence the intracellular signaling pathways activated by these cytokines have been described, including post-translational modification of STAT6 (via PARP-ylation), cyclic AMP (cAMP), sphingosine receptor signaling and IL-17A signaling (Figure 2).
PARP-mediated modulation of IL-4/IL-13 induced STAT6 activation
Poly (ADP-ribose) polymerase (PARP) is a nuclear enzyme that mediates a unique post-translational modification by attaching ADP-ribose polymer chains to target proteins. Classically, PARP proteins have been implicated in DNA repair as many PARP targets include enzymes important in these processes (e.g. DNA ligase III). Recent studies on asthma suggest PARP activity levels are enhanced in asthma patients’ lungs, possibly as a result of reactive oxygen species (ROS)-mediated DNA damage in pulmonary epithelial cells130. However, beyond its role in DNA repair, PARP proteins have been reported to contribute to asthma pathology by influencing cellular capacity to respond to, and produce, Th2 cytokines131, 132. Interestingly, PARP1 deficiency results in markedly reduced levels of STAT6 in spleen cells, despite comparable levels of STAT6 mRNA expression, suggesting that PARP1 is an important promoter of STAT6 stability133. Although the mechanism behind PARP1-mediated regulation of STAT6 integrity remain unclear, it is possible that PARP-ylation of STAT6 inhibits proteolytic cleavage of STAT6 induced after activation. In support of this possibility, cellular exposure to a calpain inhibitor (but not a proteosome inhibitor) reversed the rapid degradation of STAT6 seen in cytokine stimulated PARP1−/− cells133.
Like PARP1, PARP14 was implicated in the progression of asthma pathogenesis. PARP14 has been described as a transcriptional factor switch for STAT6-associated gene expression: when IL-4 is absent, PARP14 is present at the promoters of IL-4 responsive genes and recruits HDAC2/3 to limit the expression of these genes134. However, upon IL-4 signaling, PARP14 PARP-ylates itself as well as HDAC2/3, resulting in the loss of HDAC2/3 at these sites and simultaneous recruitment of transcriptional activators with HAT activity. These transcriptional activators facilitate more efficient STAT6 binding to promoters of IL-4 responsive genes and increase IL-4-dependent gene epression134. PARP14 was found to directly PARP-ylate STAT6 itself, suggesting an additional means whereby PARP14 might regulate STAT6 activity134. Not surprisingly then, overexpression of PARP14 promotes IL-4-induced Th2 differentiation and gene expression, while PARP14 deficient mice had evidence of decreased OVA-induced airway inflammation due to decreased Th2 differentiation135, 136. These findings suggest that blocking PARP activity could act as potential therapeutic for Th2-mediated allergic disease.
cAMP in Th2 immunopathology:
3’,5’-cyclic-adenosine monophosphate (cAMP) is a secondary messenger that plays diverse signaling roles in cells137. Intracellular cAMP levels are regulated through the balance of adenylyl cyclase (AC) activity, which creates cAMP, and phosphodiesterase 4 (PDE4), which converts cAMP to 5’-AMP138. Importantly, regulation of intracellular cAMP levels has roles in asthma: activation of Gαs-linked G protein coupled receptors (GPCRs – e.g. β2-adrenergic receptor139) lead to smooth muscle relaxation140, while Gαi-linked GPCRs (e.g. Muscarinic Acetylcholine receptor 2) lead to activation of PDE4 and inhibition of smooth muscle relaxation141, 142. cAMP has been shown to regulate gene expression downstream of IL-4 - particularly the expression of Arginase I (Arg1), a key protein associated with alternatively activated macrophages. Specifically, cAMP143,144 and IL-4 synergistically induce arginase I expression in RAW264.7 macrophages and primary BMDM. The effects of cAMP on arginase I expression were found to be dependent on increased nuclear localization of C/EBPβ (independent of C-EBPβ expression levels), which in turn facilitated increased STAT6 binding to the Arg1 promoter143, 144. Collectively, these findings suggest cAMP could function as a both positive and negative regulator of allergic inflammation. However, cAMP acts as a positive regulator of Th2 signaling.
Role of Sphingosine 1-phosphate in Th2 cytokine signaling:
Sphingosine 1-phosphate (S1P) is a bioactive lipid known to activate 5 unique GPCRs, S1P1 through S1P5145. S1P binds to receptors on multiple cell types involved in allergic diseases to mediate changes such as stimulation of airway smooth muscle cell proliferation146 and FcεRI-mediated mast cell activation147 which disrupts the integrity of airway epithelium148. Recently, S1P receptor 2 (S1P2) was found to be implicated in exacerbating allergic diseases through interactions with IL-4 and IL-13 downstream signaling intermediates. In a bleomycin-induced, IL-13-mediated model of lung fibrosis, S1P2 deficient mice demonstrated decreased fibrosis following administration of bleomycin149. Further examination showed reduced activation of STAT6 in the lungs compared to controls despite equivalent levels of IL-13 protein production and IL-13 receptor expression149, suggesting that responsiveness to IL-13 was limited in the absence of S1P2 signaling. Supporting this possibility, knocking out S1P2 from PMA-differentiated THP-1 cells in vitro, or inhibiting the Rho kinase pathway (a pathway strongly activated by S1P2 signaling150), reduced the IL-4/IL-13-mediated phosphorylation of STAT6, JAK1, and JAK3151. Decreased IL-4/IL-13-induced STAT6 activation observed in the context of limited S1P2 signaling was associated with increased SOCS3 expression128 – and although not formally tested, the authors suggest that the S1P2 signaling limits SOCS3 expression, thereby enhancing IL-4/IL-13 signaling. Together, this evidence suggests that S1P2-Rho pathway is necessary for optimal activation of STAT6.
Effects Th17 and Th1 cytokines on IL-13 signaling cascades:
Recent studies on the severe asthma endotype identified elevated levels of mixed Th2/Th17 cells in bronchioalveolar lavage fluid (BALF) and lungs of more severe asthmatic patients152–154 and that IL-17A can augment IL-13-induced pathology in mouse models39. Our group has explored mechanisms behind these observations by examining the effects of IL-17A on IL-13-induced signaling cascades. We demonstrated that, even though IL-17A utilizes a completely JAK/STAT-independent signaling pathway, the presence of IL-17A enhanced IL-13-mediated STAT6 phosphorylation and expression of downstream genes in both human and mouse cells of various types (hematopoietic, non-hematopoietic). The effects of IL-17A on IL-13 signaling are very rapid (occurring in <5 minutes of stimulation), do not require additional protein expression, and require concurrent IL-13 and IL-17A signaling in the same cells. Interestingly, in contrast to the synergistic effects of IL-17A on IL-13 induced pathways, IL-13 downregulated IL-17A-related gene expression, suggesting reciprocal coregulation of IL-13 and IL-17A39. Although another IL-17A family member, IL-17E (IL-25) has clear pro-Th2 supporting activities, these appear to be through direct activity on immune initiating cells (ILC2s, Th2 cells) rather than by modulating signaling in response to other Th2 cytokines155. These findings suggest that IL-17A (but not IL-17E) modulates IL-13-mediated intracellular signaling pathways through direct effects on signaling intermediates activated in response to IL-13.
Although Th1-derived cytokines are known to counter the development of Th2 cells156, 157, and thus can negatively regulate production of both IL-4 and IL-13, Th1 cytokines like IL-12 and IFNγ can directly antagonize IL-4R signaling. Pre-treatment of both epithelial cells158 and monocytes159, 160 with IFNγ prior to IL-4 stimulation reduced IL-4-induced STAT6 activation. In monocytes, IFNγ pre-treatment triggered inhibition of STAT6 activation was a result of increased SOCS1 expression159, while in epithelial cells it was associated with increased expression of both SOCS1 and SOCS3158. Collectively, these observations are consistent with SOCS proteins acting as inducible, negative regulators of IL-4/IL-13 cytokine signaling. However, our group has shown a direct inhibitory effect of IL-12 on IL-13-mediated STAT6 phosphorylation and IL-13-induced gene expression in murine BMDCs and primary murine tracheal epithelial cells161. Importantly, the IL-12-mediated inhibition of IL-13-induced STAT6 activation occurred when IL-12 and IL-13 were added simultaneously to the culture (i.e., not in pre-treated cells) and within 15 minutes - too rapidly to be due to de novo IFNγ synthesis – suggesting that IL-12 signaling was directly antagonizing IL-13-induced STAT6 activation. The mechanisms responsible for this remain unclear.
Further identification of disease endotypes where enhancing pathways are activated to potentially amplify IL-4/IL-13 signaling may present novel therapeutic options for the treatment of disease. In support of this idea, therapeutic inhibition of PARP1 with Olaparib was found to ameliorate HDM/OVA-induced airway inflammation162. In addition, our group demonstrated that combination therapy of anti-IL-13 and anti-IL-17A (at levels too low to individually impact IL-13- or IL-17A-induced disease processes) ameliorated HDM-induced allergic asthma in a model of Th2/Th17-driven allergic asthma163.
Conclusions:
IL-4 and IL-13 can both signal through the type II IL-4R, which leads to the activation of STAT6 and expression of STAT6-dependent genes. Despite this similarity, these two cytokines have been ascribed different functions in the context of allergic disease. These differences may arise due to unique activation of additional signaling pathways by IL-4 signaling through the type I IL-4R (IRS-2) and differential expression of the type I and type II IL-4R on hematopoietic, verses non-hematopoietic cells. Moreover, additional inputs provided by either positive or negative regulators of IL-4/IL-13 signaling pathways can selectively fine tune responses to these cytokines, thereby subtly altering the biological outcomes of local signaling in response to these molecules. However, it is also clear that additional signaling pathways may be activated by these cytokines (MAPK, PI3K, SRC), and that activation of these unique pathways may in turn influence IRS-2/STAT6 activation or promote altered biological outcomes in response to these paradigmatic Th2 cytokines. Although a great deal of attention has been paid to the effects of IL-4/IL-13-driven activation of IRS-2 and STAT6, the role of these additional signaling pathways is poorly understood. It is our belief that a more complete understanding of IL-4/IL-13 signaling pathways, including the precise conditions under which non-canonical signaling pathways are activated, and the impact of these pathways on cellular and host level responses, will better allow us to design agents that target specific pathological outcomes, or tailor therapies for the treatment of uncommon disease endotypes.
Funding:
This work was funded by R01HL149366 (IPL).
Abbreviations used:
- Th2
CD4 T helper 2
- IL
Interleukin
- JAK
Janus kinase
- STAT
Signal Transducer and Activator of Transcription
- γc
common gamma chain
- ΚD
Dissociation constant
- PTB
phospho-tyrosine binding
- IRS-2
insulin receptor substrate-2
- PI3K
phosphatidylinositol-3-kinase
- MAPK
mitogen activated protein kinases
- Akt
serine threonine protein kinase
- PKC
protein kinase C
- SRC
proto-oncogene tyrosine-protein kinase
- I4R
insulin/interleukin-4 receptor
- TYK2
tyrosine kinase 2
- mTOR
mammalian target of rapamycin
- RTK
receptor tyrosine kinase
- PIP2
phosphatidylinositol (4,5)-bisphosphate
- PIP3
phosphatidylinositol (3,4,5)-trisphosphate
- PDK
phosphoinositide-dependent kinase
- EGFRvIII
epidermal growth factor receptor vIII
- ERK
extracellular signal-regulated kinases
- JNK
Jun N-terminal Kinase
- NFAT
nuclear factor of activated T-cells
- CREB
cyclic AMP-response element binding protein
- AP-1
activator protein-1
- mRNA
messenger ribonucleic acid
- PTPs
Protein Tyrosine phosphatases
- SOCS
Suppressor of Cytokine Signaling
- SHP-1
Src homology 2-containing protein tyrosine phosphatase
- SHIP
SH2-containing inositol phosphatase
- ITIM
immune-tyrosine-based inhibitory motif
- BMDM
bone marrow derived macrophages
- KIR
kinase inhibitory region
- AHR
airway hyper responsiveness
- IgG
immunoglobulin G
- IgE
immunoglobulin E
- CIS
cytokine Inducible Sh2 protein
- ADP-ribose
adenosine diphosphate ribose
- PARP
poly (ADP-ribose) polymerase
- DNA
deoxy ribonucleic acid
- cAMP
cyclic AMP
- S1P
Sphingosine 1-phosphate
- ROS
reactive oxygen species
- HDAC
histone deacetylase
- HAT
histone acetyltransferase
- OVA
ovalbumin
- HDM
house dust mite
- BALF
bronchoalveolar lavage fluid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: All authors have nothing to disclose.
References:
- 1.Pawankar R Allergic diseases and asthma: a global public health concern and a call to action. World Allergy Organ J 2014; 7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim HY, DeKruyff RH, Umetsu DT. The many paths to asthma: phenotype shaped by innate and adaptive immunity. Nat Immunol 2010; 11:577–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chung KF, Barnes PJ. Cytokines in asthma. Thorax 1999; 54:825–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu W, Liu S, Verma M, Zafar I, Good JT, Rollins D, et al. Mechanism of TH2/TH17-predominant and neutrophilic TH2/TH17-low subtypes of asthma. J Allergy Clin Immunol 2017; 139:1548–58 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wallrapp A, Riesenfeld SJ, Burkett PR, Kuchroo VK. Type 2 innate lymphoid cells in the induction and resolution of tissue inflammation. Immunol Rev 2018; 286:53–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakayama T, Hirahara K, Onodera A, Endo Y, Hosokawa H, Shinoda K, et al. Th2 Cells in Health and Disease. Annu Rev Immunol 2017; 35:53–84. [DOI] [PubMed] [Google Scholar]
- 7.Hammad H, Debeuf N, Aegerter H, Brown AS, Lambrecht BN. Emerging Paradigms in Type 2 Immunity. Annu Rev Immunol 2022; 40:443–67. [DOI] [PubMed] [Google Scholar]
- 8.van de Veen W, Akdis M. The use of biologics for immune modulation in allergic disease. J Clin Invest 2019; 129:1452–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leon B, Ballesteros-Tato A. Modulating Th2 Cell Immunity for the Treatment of Asthma. Front Immunol 2021; 12:637948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Georas SN, Donohue P, Connolly M, Wechsler ME. JAK inhibitors for asthma. J Allergy Clin Immunol 2021; 148:953–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Q, Nian S, Ye Y, Liu D, Yu H, Xiong H, et al. The Emerging Roles of T Helper Cell Subsets and Cytokines in Severe Neutrophilic Asthma. Inflammation 2022;45:1007–22. [DOI] [PubMed] [Google Scholar]
- 12.Forbes E, van Panhuys N, Min B, Le Gros G. Differential requirements for IL-4/STAT6 signalling in CD4 T-cell fate determination and Th2-immune effector responses. Immunol Cell Biol 2010; 88:240–3. [DOI] [PubMed] [Google Scholar]
- 13.Seder RA, Paul WE, Davis MM, Fazekas de St Groth B. The presence of interleukin 4 during in vitro priming determines the lymphokine-producing potential of CD4+ T cells from T cell receptor transgenic mice. J Exp Med 1992; 176:1091–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finkelman FD, Katona IM, Urban JF Jr., Holmes J, Ohara J, Tung AS, et al. IL-4 is required to generate and sustain in vivo IgE responses. J Immunol 1988; 141:2335–41. [PubMed] [Google Scholar]
- 15.Chen L, Grabowski KA, Xin JP, Coleman J, Huang Z, Espiritu B, et al. IL-4 induces differentiation and expansion of Th2 cytokine-producing eosinophils. J Immunol 2004; 172:2059–66. [DOI] [PubMed] [Google Scholar]
- 16.Ho IC, Miaw SC. Regulation of IL-4 Expression in Immunity and Diseases. Adv Exp Med Biol 2016; 941:31–77. [DOI] [PubMed] [Google Scholar]
- 17.Booth BW, Sandifer T, Martin EL, Martin LD. IL-13-induced proliferation of airway epithelial cells: mediation by intracellular growth factor mobilization and ADAM17. Respir Res 2007; 8:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Risse PA, Jo T, Suarez F, Hirota N, Tolloczko B, Ferraro P, et al. Interleukin-13 inhibits proliferation and enhances contractility of human airway smooth muscle cells without change in contractile phenotype. Am J Physiol Lung Cell Mol Physiol 2011; 300:L958–66. [DOI] [PubMed] [Google Scholar]
- 19.Chiba Y, Nakazawa S, Todoroki M, Shinozaki K, Sakai H, Misawa M. Interleukin-13 augments bronchial smooth muscle contractility with an up-regulation of RhoA protein. Am J Respir Cell Mol Biol 2009; 40:159–67. [DOI] [PubMed] [Google Scholar]
- 20.Doucet C, Brouty-Boye D, Pottin-Clemenceau C, Canonica GW, Jasmin C, Azzarone B. Interleukin (IL) 4 and IL-13 act on human lung fibroblasts. Implication in asthma. J Clin Invest 1998; 101:2129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Purwar R, Werfel T, Wittmann M. IL-13-stimulated human keratinocytes preferentially attract CD4+CCR4+ T cells: possible role in atopic dermatitis. J Invest Dermatol 2006; 126:1043–51. [DOI] [PubMed] [Google Scholar]
- 22.Wongpiyabovorn J, Suto H, Ushio H, Izuhara K, Mitsuishi K, Ikeda S, et al. Up-regulation of interleukin-13 receptor alpha1 on human keratinocytes in the skin of psoriasis and atopic dermatitis. J Dermatol Sci 2003; 33:31–40. [DOI] [PubMed] [Google Scholar]
- 23.Wills-Karp M IL-12/IL-13 axis in allergic asthma. J Allergy Clin Immunol 2001; 107:9–18. [DOI] [PubMed] [Google Scholar]
- 24.Huang ZQ, Liu J, Ong HH, Yuan T, Zhou XM, Wang J, et al. Interleukin-13 Alters Tight Junction Proteins Expression Thereby Compromising Barrier Function and Dampens Rhinovirus Induced Immune Responses in Nasal Epithelium. Front Cell Dev Biol 2020; 8:572749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dalessandri T, Crawford G, Hayes M, Castro Seoane R, Strid J. IL-13 from intraepithelial lymphocytes regulates tissue homeostasis and protects against carcinogenesis in the skin. Nat Commun 2016; 7:12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tukler Henriksson J, Coursey TG, Corry DB, De Paiva CS, Pflugfelder SC. IL-13 Stimulates Proliferation and Expression of Mucin and Immunomodulatory Genes in Cultured Conjunctival Goblet Cells. Invest Ophthalmol Vis Sci 2015; 56:4186–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farghaly HS, Blagbrough IS, Medina-Tato DA, Watson ML. Interleukin 13 increases contractility of murine tracheal smooth muscle by a phosphoinositide 3-kinase p110delta-dependent mechanism. Mol Pharmacol 2008; 73:1530–7. [DOI] [PubMed] [Google Scholar]
- 28.Chan R, Stewart K, Misirovs R, Lipworth BJ. Targeting Downstream Type 2 Cytokines or Upstream Epithelial Alarmins for Severe Asthma. J Allergy Clin Immunol Pract 2022;10:1497–505 [DOI] [PubMed] [Google Scholar]
- 29.Lawrence MG, Steinke JW, Borish L. Cytokine-targeting biologics for allergic diseases. Ann Allergy Asthma Immunol 2018; 120:376–81. [DOI] [PubMed] [Google Scholar]
- 30.Guilleminault L, Conde E, Reber LL. Pharmacological approaches to target type 2 cytokines in asthma. Pharmacol Ther 2022; 237:108167. [DOI] [PubMed] [Google Scholar]
- 31.Romeo MJ, Agrawal R, Pomes A, Woodfolk JA. A molecular perspective on TH2-promoting cytokine receptors in patients with allergic disease. J Allergy Clin Immunol 2014; 133:952–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKnight CG, Potter C, Finkelman FD. IL-4Ralpha expression by airway epithelium and smooth muscle accounts for nearly all airway hyperresponsiveness in murine allergic airway disease. Mucosal Immunol 2020; 13:283–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Junttila IS. Tuning the Cytokine Responses: An Update on Interleukin (IL)-4 and IL-13 Receptor Complexes. Front Immunol 2018; 9:888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCormick SM, Heller NM. Commentary: IL-4 and IL-13 receptors and signaling. Cytokine 2015; 75:38–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rahaman SO, Sharma P, Harbor PC, Aman MJ, Vogelbaum MA, Haque SJ. IL-13R(alpha)2, a decoy receptor for IL-13 acts as an inhibitor of IL-4-dependent signal transduction in glioblastoma cells. Cancer Res 2002; 62:1103–9. [PubMed] [Google Scholar]
- 36.Sivaprasad U, Warrier MR, Gibson AM, Chen W, Tabata Y, Bass SA, et al. IL-13Ralpha2 has a protective role in a mouse model of cutaneous inflammation. J Immunol 2010; 185:6802–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chandriani S, DePianto DJ, N’Diaye EN, Abbas AR, Jackman J, Bevers J, 3rd, et al. Endogenously expressed IL-13Ralpha2 attenuates IL-13-mediated responses but does not activate signaling in human lung fibroblasts. J Immunol 2014; 193:111–9. [DOI] [PubMed] [Google Scholar]
- 38.Ulzii D, Kido-Nakahara M, Nakahara T, Tsuji G, Furue K, Hashimoto-Hachiya A, et al. Scratching Counteracts IL-13 Signaling by Upregulating the Decoy Receptor IL-13Ralpha2 in Keratinocytes. Int J Mol Sci 2019; 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hall SL, Baker T, Lajoie S, Richgels PK, Yang Y, McAlees JW, et al. IL-17A enhances IL-13 activity by enhancing IL-13-induced signal transducer and activator of transcription 6 activation. J Allergy Clin Immunol 2017; 139:462–71 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim JE, Jung K, Kim JA, Kim SH, Park HS, Kim YS. Engineering of anti-human interleukin-4 receptor alpha antibodies with potent antagonistic activity. Sci Rep 2019; 9:7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kraich M, Klein M, Patino E, Harrer H, Nickel J, Sebald W, et al. A modular interface of IL-4 allows for scalable affinity without affecting specificity for the IL-4 receptor. BMC Biol 2006; 4:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kelly-Welch AE, Hanson EM, Boothby MR, Keegan AD. Interleukin-4 and interleukin-13 signaling connections maps. Science 2003; 300:1527–8. [DOI] [PubMed] [Google Scholar]
- 43.Keegan AD, Nelms K, White M, Wang LM, Pierce JH, Paul WE. An IL-4 receptor region containing an insulin receptor motif is important for IL-4-mediated IRS-1 phosphorylation and cell growth. Cell 1994; 76:811–20. [DOI] [PubMed] [Google Scholar]
- 44.Wang HY, Paul WE, Keegan AD. IL-4 function can be transferred to the IL-2 receptor by tyrosine containing sequences found in the IL-4 receptor alpha chain. Immunity 1996; 4:113–21. [DOI] [PubMed] [Google Scholar]
- 45.Wang HY, Zamorano J, Keegan AD. A role for the insulin-interleukin (IL)-4 receptor motif of the IL-4 receptor alpha-chain in regulating activation of the insulin receptor substrate 2 and signal transducer and activator of transcription 6 pathways. Analysis by mutagenesis. J Biol Chem 1998; 273:9898–905. [DOI] [PubMed] [Google Scholar]
- 46.Heller NM, Qi X, Junttila IS, Shirey KA, Vogel SN, Paul WE, et al. Type I IL-4Rs selectively activate IRS-2 to induce target gene expression in macrophages. Sci Signal 2008; 1:ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franke TF. PI3K/Akt: getting it right matters. Oncogene 2008; 27:6473–88. [DOI] [PubMed] [Google Scholar]
- 48.Kreider T, Anthony RM, Urban JF Jr., Gause WC. Alternatively activated macrophages in helminth infections. Curr Opin Immunol 2007; 19:448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kelly-Welch AE, Melo ME, Smith E, Ford AQ, Haudenschild C, Noben-Trauth N, et al. Complex role of the IL-4 receptor alpha in a murine model of airway inflammation: expression of the IL-4 receptor alpha on nonlymphoid cells of bone marrow origin contributes to severity of inflammation. J Immunol 2004; 172:4545–55. [DOI] [PubMed] [Google Scholar]
- 50.Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol 2009; 183:6469–77. [DOI] [PubMed] [Google Scholar]
- 51.Subrata LS, Bizzintino J, Mamessier E, Bosco A, McKenna KL, Wikstrom ME, et al. Interactions between innate antiviral and atopic immunoinflammatory pathways precipitate and sustain asthma exacerbations in children. J Immunol 2009; 183:2793–800. [DOI] [PubMed] [Google Scholar]
- 52.Holt PG, Strickland DH. Interactions between innate and adaptive immunity in asthma pathogenesis: new perspectives from studies on acute exacerbations. J Allergy Clin Immunol 2010; 125:963–72; quiz 73–4. [DOI] [PubMed] [Google Scholar]
- 53.Melgert BN, ten Hacken NH, Rutgers B, Timens W, Postma DS, Hylkema MN. More alternative activation of macrophages in lungs of asthmatic patients. J Allergy Clin Immunol 2011; 127:831–3. [DOI] [PubMed] [Google Scholar]
- 54.Ford AQ, Dasgupta P, Mikhailenko I, Smith EM, Noben-Trauth N, Keegan AD. Adoptive transfer of IL-4Ralpha+ macrophages is sufficient to enhance eosinophilic inflammation in a mouse model of allergic lung inflammation. BMC Immunol 2012; 13:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Umeshita-Suyama R, Sugimoto R, Akaiwa M, Arima K, Yu B, Wada M, et al. Characterization of IL-4 and IL-13 signals dependent on the human IL-13 receptor alpha chain 1: redundancy of requirement of tyrosine residue for STAT3 activation. Int Immunol 2000; 12:1499–509. [DOI] [PubMed] [Google Scholar]
- 56.Lupardus PJ, Birnbaum ME, Garcia KC. Molecular basis for shared cytokine recognition revealed in the structure of an unusually high affinity complex between IL-13 and IL 13-Ralpha2. Structure 2010; 18:332–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mueller TD, Zhang JL, Sebald W, Duschl A. Structure, binding, and antagonists in the IL-4/IL-13 receptor system. Biochim Biophys Acta 2002; 1592:237–50. [DOI] [PubMed] [Google Scholar]
- 58.Arima K, Sato K, Tanaka G, Kanaji S, Terada T, Honjo E, et al. Characterization of the interaction between interleukin-13 and interleukin-13 receptors. J Biol Chem 2005; 280:24915–22. [DOI] [PubMed] [Google Scholar]
- 59.Konstantinidis AK, Puddicombe SM, Mochizuki A, Sheth PD, Yang IA, Yoshisue H, et al. Cellular localization of interleukin 13 receptor alpha2 in human primary bronchial epithelial cells and fibroblasts. J Investig Allergol Clin Immunol 2008; 18:174–80. [PubMed] [Google Scholar]
- 60.Liu J, Li YY, Andiappan AK, Yan Y, Tan KS, Ong HH, et al. Role of IL-13Ralpha2 in modulating IL-13-induced MUC5AC and ciliary changes in healthy and CRSwNP mucosa. Allergy 2018; 73:1673–85. [DOI] [PubMed] [Google Scholar]
- 61.Penke LR, Ouchi H, Speth JM, Lugogo N, Huang YJ, Huang SK, et al. Transcriptional regulation of the IL-13Ralpha2 gene in human lung fibroblasts. Sci Rep 2020; 10:1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen W, Sivaprasad U, Gibson AM, Ericksen MB, Cunningham CM, Bass SA, et al. IL-13 receptor alpha2 contributes to development of experimental allergic asthma. J Allergy Clin Immunol 2013; 132:951–8 e1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fujisawa T, Shimamura T, Goto K, Nakagawa R, Muroyama R, Ino Y, et al. A Novel Role of Interleukin 13 Receptor alpha2 in Perineural Invasion and its Association with Poor Prognosis of Patients with Pancreatic Ductal Adenocarcinoma. Cancers (Basel) 2020; 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.He CH, Lee CG, Dela Cruz CS, Lee CM, Zhou Y, Ahangari F, et al. Chitinase 3-like 1 regulates cellular and tissue responses via IL-13 receptor alpha2. Cell Rep 2013; 4:830–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med 2006; 12:99–106. [DOI] [PubMed] [Google Scholar]
- 66.Newman JP, Wang GY, Arima K, Guan SP, Waters MR, Cavenee WK, et al. Interleukin-13 receptor alpha 2 cooperates with EGFRvIII signaling to promote glioblastoma multiforme. Nat Commun 2017; 8:1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hsi LC, Kundu S, Palomo J, Xu B, Ficco R, Vogelbaum MA, et al. Silencing IL-13Ralpha2 promotes glioblastoma cell death via endogenous signaling. Mol Cancer Ther 2011; 10:1149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fujisawa T, Joshi BH, Takahashi S, Takasaki Y, Suzuki A, Ito K, et al. IL-13Ralpha2 Is a Biomarker of Diagnosis and Therapeutic Response in Human Pancreatic Cancer. Diagnostics (Basel) 2021; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao Z, Wang L, Xu W. IL-13Ralpha2 mediates PNR-induced migration and metastasis in ERalpha-negative breast cancer. Oncogene 2015; 34:1596–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kwon HJ, Choi JE, Bae YK. Interleukin-13 receptor alpha 2 expression in tumor cells is associated with reduced disease-free survival in patients with luminal subtype invasive breast cancer. Tumour Biol 2018; 40:1010428318783657. [DOI] [PubMed] [Google Scholar]
- 71.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov 2009; 8:627–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017; 168:960–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Keegan AD, Zamorano J, Keselman A, Heller NM. IL-4 and IL-13 Receptor Signaling From 4PS to Insulin Receptor Substrate 2: There and Back Again, a Historical View. Front Immunol 2018; 9:1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dasgupta P, Keegan AD. Contribution of alternatively activated macrophages to allergic lung inflammation: a tale of mice and men. J Innate Immun 2012; 4:478–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim EY, Battaile JT, Patel AC, You Y, Agapov E, Grayson MH, et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nat Med 2008; 14:633–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu H, Li Q, Kolosov VP, Perelman JM, Zhou X. Interleukin-13 induces mucin 5AC production involving STAT6/SPDEF in human airway epithelial cells. Cell Commun Adhes 2010; 17:83–92. [DOI] [PubMed] [Google Scholar]
- 77.Moriya C, Jinnin M, Yamane K, Maruo K, Muchemwa FC, Igata T, et al. Expression of matrix metalloproteinase-13 is controlled by IL-13 via PI3K/Akt3 and PKC-delta in normal human dermal fibroblasts. J Invest Dermatol 2011; 131:655–61. [DOI] [PubMed] [Google Scholar]
- 78.Yasuda M, Harada N, Harada S, Ishimori A, Katsura Y, Itoigawa Y, et al. Characterization of tenascin-C as a novel biomarker for asthma: utility of tenascin-C in combination with periostin or immunoglobulin E. Allergy Asthma Clin Immunol 2018; 14:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.David M, Ford D, Bertoglio J, Maizel AL, Pierre J. Induction of the IL-13 receptor alpha2-chain by IL-4 and IL-13 in human keratinocytes: involvement of STAT6, ERK and p38 MAPK pathways. Oncogene 2001; 20:6660–8. [DOI] [PubMed] [Google Scholar]
- 80.Owens DM, Keyse SM. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 2007; 26:3203–13. [DOI] [PubMed] [Google Scholar]
- 81.Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene 2007; 26:3100–12. [DOI] [PubMed] [Google Scholar]
- 82.Zhang Y, Dong C. Regulatory mechanisms of mitogen-activated kinase signaling. Cell Mol Life Sci 2007; 64:2771–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wery-Zennaro S, Zugaza JL, Letourneur M, Bertoglio J, Pierre J. IL-4 regulation of IL-6 production involves Rac/Cdc42- and p38 MAPK-dependent pathways in keratinocytes. Oncogene 2000; 19:1596–604. [DOI] [PubMed] [Google Scholar]
- 84.So EY, Oh J, Jang JY, Kim JH, Lee CE. Ras/Erk pathway positively regulates Jak1/STAT6 activity and IL-4 gene expression in Jurkat T cells. Mol Immunol 2007; 44:3416–26. [DOI] [PubMed] [Google Scholar]
- 85.Lee PJ, Zhang X, Shan P, Ma B, Lee CG, Homer RJ, et al. ERK1/2 mitogen-activated protein kinase selectively mediates IL-13-induced lung inflammation and remodeling in vivo. J Clin Invest 2006; 116:163–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Haura EB. SRC and STAT pathways. J Thorac Oncol 2006; 1:403–5. [PubMed] [Google Scholar]
- 87.Amata I, Maffei M, Pons M. Phosphorylation of unique domains of Src family kinases. Front Genet 2014; 5:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Roskoski R Jr., Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol Res 2015; 94:9–25. [DOI] [PubMed] [Google Scholar]
- 89.Cortes JR, Perez GM, Rivas MD, Zamorano J. Kaempferol inhibits IL-4-induced STAT6 activation by specifically targeting JAK3. J Immunol 2007; 179:3881–7. [DOI] [PubMed] [Google Scholar]
- 90.Perez GM, Melo M, Keegan AD, Zamorano J. Aspirin and salicylates inhibit the IL-4- and IL-13-induced activation of STAT6. J Immunol 2002; 168:1428–34. [DOI] [PubMed] [Google Scholar]
- 91.Rivera-Torres J, San Jose E. Src Tyrosine Kinase Inhibitors: New Perspectives on Their Immune, Antiviral, and Senotherapeutic Potential. Front Pharmacol 2019; 10:1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kamata T, Yamashita M, Kimura M, Murata K, Inami M, Shimizu C, et al. src homology 2 domain-containing tyrosine phosphatase SHP-1 controls the development of allergic airway inflammation. J Clin Invest 2003; 111:109–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huang Z, Coleman JM, Su Y, Mann M, Ryan J, Shultz LD, et al. SHP-1 regulates STAT6 phosphorylation and IL-4-mediated function in a cell type-specific manner. Cytokine 2005; 29:118–24. [DOI] [PubMed] [Google Scholar]
- 94.Haque SJ, Harbor P, Tabrizi M, Yi T, Williams BR. Protein-tyrosine phosphatase Shp-1 is a negative regulator of IL-4- and IL-13-dependent signal transduction. J Biol Chem 1998; 273:33893–6. [DOI] [PubMed] [Google Scholar]
- 95.Shi L, Kidder K, Bian Z, Chiang SKT, Ouellette C, Liu Y. SIRPalpha sequesters SHP-2 to promote IL-4 and IL-13 signaling and the alternative activation of macrophages. Sci Signal 2021; 14:eabb3966. [DOI] [PubMed] [Google Scholar]
- 96.Spalinger MR, Crawford M, Bobardt SD, Li J, Sayoc-Becerra A, Santos AN, et al. Loss of protein tyrosine phosphatase non-receptor type 2 reduces IL-4-driven alternative macrophage activation. Mucosal Immunol 2022; 15:74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lioubin MN, Algate PA, Tsai S, Carlberg K, Aebersold A, Rohrschneider LR. p150Ship, a signal transduction molecule with inositol polyphosphate-5-phosphatase activity. Genes Dev 1996; 10:1084–95. [DOI] [PubMed] [Google Scholar]
- 98.Damen JE, Liu L, Rosten P, Humphries RK, Jefferson AB, Majerus PW, et al. The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-triphosphate 5-phosphatase. Proc Natl Acad Sci U S A 1996; 93:1689–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zamorano J, Keegan AD. Regulation of apoptosis by tyrosine-containing domains of IL-4R alpha: Y497 and Y713, but not the STAT6-docking tyrosines, signal protection from apoptosis. J Immunol 1998; 161:859–67. [PubMed] [Google Scholar]
- 100.Giallourakis C, Kashiwada M, Pan PY, Danial N, Jiang H, Cambier J, et al. Positive regulation of interleukin-4-mediated proliferation by the SH2-containing inositol-5’-phosphatase. J Biol Chem 2000; 275:29275–82. [DOI] [PubMed] [Google Scholar]
- 101.Weisser SB, McLarren KW, Voglmaier N, van Netten-Thomas CJ, Antov A, Flavell RA, et al. Alternative activation of macrophages by IL-4 requires SHIP degradation. Eur J Immunol 2011; 41:1742–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tachdjian R, Al Khatib S, Schwinglshackl A, Kim HS, Chen A, Blasioli J, et al. In vivo regulation of the allergic response by the IL-4 receptor alpha chain immunoreceptor tyrosine-based inhibitory motif. J Allergy Clin Immunol 2010; 125:1128–36 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Oh SY, Zheng T, Kim YK, Cohn L, Homer RJ, McKenzie AN, et al. A critical role of SHP-1 in regulation of type 2 inflammation in the lung. Am J Respir Cell Mol Biol 2009; 40:568–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Varone A, Mariggio S, Patheja M, Maione V, Varriale A, Vessichelli M, et al. A signalling cascade involving receptor-activated phospholipase A2, glycerophosphoinositol 4-phosphate, Shp1 and Src in the activation of cell motility. Cell Commun Signal 2019; 17:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhu Z, Oh SY, Cho YS, Zhang L, Kim YK, Zheng T. Tyrosine phosphatase SHP-1 in allergic and anaphylactic inflammation. Immunol Res 2010; 47:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhou L, Oh SY, Zhou Y, Yuan B, Wu F, Oh MH, et al. SHP-1 regulation of mast cell function in allergic inflammation and anaphylaxis. PLoS One 2013; 8:e55763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang Y, Zhu Z, Church TD, Lugogo NL, Que LG, Francisco D, et al. SHP-1 as a critical regulator of Mycoplasma pneumoniae-induced inflammation in human asthmatic airway epithelial cells. J Immunol 2012; 188:3371–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xu C, Wu X, Lu M, Tang L, Yao H, Wang J, et al. Protein tyrosine phosphatase 11 acts through RhoA/ROCK to regulate eosinophil accumulation in the allergic airway. FASEB J 2019; 33:11706–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tomar S, Ganesan V, Sharma A, Zeng C, Waggoner L, Smith A, et al. IL-4-BATF signaling directly modulates IL-9 producing mucosal mast cell (MMC9) function in experimental food allergy. J Allergy Clin Immunol 2021; 147:280–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Keegan AD, Leonard WJ, Zhu J. Recent advances in understanding the role of IL-4 signaling. Fac Rev 2021; 10:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Myers MP, Andersen JN, Cheng A, Tremblay ML, Horvath CM, Parisien JP, et al. TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. J Biol Chem 2001; 276:47771–4. [DOI] [PubMed] [Google Scholar]
- 112.Lu X, Malumbres R, Shields B, Jiang X, Sarosiek KA, Natkunam Y, et al. PTP1B is a negative regulator of interleukin 4-induced STAT6 signaling. Blood 2008; 112:4098–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yoshimura A, Nishinakamura H, Matsumura Y, Hanada T. Negative regulation of cytokine signaling and immune responses by SOCS proteins. Arthritis Res Ther 2005; 7:100–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol 2007; 7:454–65. [DOI] [PubMed] [Google Scholar]
- 115.Okumura F, Matsuzaki M, Nakatsukasa K, Kamura T. The Role of Elongin BC-Containing Ubiquitin Ligases. Front Oncol 2012; 2:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sobah ML, Liongue C, Ward AC. SOCS Proteins in Immunity, Inflammatory Diseases, and Immune-Related Cancer. Front Med (Lausanne) 2021; 8:727987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Liau NPD, Laktyushin A, Lucet IS, Murphy JM, Yao S, Whitlock E, et al. The molecular basis of JAK/STAT inhibition by SOCS1. Nat Commun 2018; 9:1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kershaw NJ, Murphy JM, Liau NP, Varghese LN, Laktyushin A, Whitlock EL, et al. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol 2013; 20:469–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol 2008; 19:414–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.McCormick SM, Gowda N, Fang JX, Heller NM. Suppressor of Cytokine Signaling (SOCS)1 Regulates Interleukin-4 (IL-4)-activated Insulin Receptor Substrate (IRS)-2 Tyrosine Phosphorylation in Monocytes and Macrophages via the Proteasome. J Biol Chem 2016; 291:20574–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ritz O, Guiter C, Dorsch K, Dusanter-Fourt I, Wegener S, Jouault H, et al. STAT6 activity is regulated by SOCS-1 and modulates BCL-XL expression in primary mediastinal B-cell lymphoma. Leukemia 2008; 22:2106–10. [DOI] [PubMed] [Google Scholar]
- 122.Doran E, Choy DF, Shikotra A, Butler CA, O’Rourke DM, Johnston JA, et al. Reduced epithelial suppressor of cytokine signalling 1 in severe eosinophilic asthma. Eur Respir J 2016; 48:715–25. [DOI] [PubMed] [Google Scholar]
- 123.Hebenstreit D, Luft P, Schmiedlechner A, Regl G, Frischauf AM, Aberger F, et al. IL-4 and IL-13 induce SOCS-1 gene expression in A549 cells by three functional STAT6-binding motifs located upstream of the transcription initiation site. J Immunol 2003; 171:5901–7. [DOI] [PubMed] [Google Scholar]
- 124.Hebenstreit D, Luft P, Schmiedlechner A, Duschl A, Horejs-Hoeck J. SOCS-1 and SOCS-3 inhibit IL-4 and IL-13 induced activation of Eotaxin-3/CCL26 gene expression in HEK293 cells. Mol Immunol 2005; 42:295–303. [DOI] [PubMed] [Google Scholar]
- 125.Marine JC, Topham DJ, McKay C, Wang D, Parganas E, Stravopodis D, et al. SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell 1999; 98:609–16. [DOI] [PubMed] [Google Scholar]
- 126.Zimmer J, Weitnauer M, Boutin S, Kublbeck G, Thiele S, Walker P, et al. Nuclear Localization of Suppressor of Cytokine Signaling-1 Regulates Local Immunity in the Lung. Front Immunol 2016; 7:514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Williams JJ, Munro KM, Palmer TM. Role of Ubiquitylation in Controlling Suppressor of Cytokine Signalling 3 (SOCS3) Function and Expression. Cells 2014; 3:546–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kim D, Kim SH, Cho SH, Shin K, Kim S. SOCS3 suppresses the expression of IL-4 cytokine by inhibiting the phosphorylation of c-Jun through the ERK signaling pathway in rat mast cell line RBL-2H3. Mol Immunol 2011; 48:776–81. [DOI] [PubMed] [Google Scholar]
- 129.Draijer C, Speth JM, Penke LRK, Zaslona Z, Bazzill JD, Lugogo N, et al. Resident alveolar macrophage-derived vesicular SOCS3 dampens allergic airway inflammation. FASEB J 2020; 34:4718–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Andreadis AA, Hazen SL, Comhair SA, Erzurum SC. Oxidative and nitrosative events in asthma. Free Radic Biol Med 2003; 35:213–25. [DOI] [PubMed] [Google Scholar]
- 131.Oumouna M, Datta R, Oumouna-Benachour K, Suzuki Y, Hans C, Matthews K, et al. Poly(ADP-ribose) polymerase-1 inhibition prevents eosinophil recruitment by modulating Th2 cytokines in a murine model of allergic airway inflammation: a potential specific effect on IL-5. J Immunol 2006; 177:6489–96. [DOI] [PubMed] [Google Scholar]
- 132.Havranek T, Aujla PK, Nickola TJ, Rose MC, Scavo LM. Increased poly(ADP-ribose) polymerase (PARP)-1 expression and activity are associated with inflammation but not goblet cell metaplasia in murine models of allergen-induced airway inflammation. Exp Lung Res 2010; 36:381–9. [DOI] [PubMed] [Google Scholar]
- 133.Datta R, Naura AS, Zerfaoui M, Errami Y, Oumouna M, Kim H, et al. PARP-1 deficiency blocks IL-5 expression through calpain-dependent degradation of STAT-6 in a murine asthma model. Allergy 2011; 66:853–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Iwata H, Goettsch C, Sharma A, Ricchiuto P, Goh WW, Halu A, et al. PARP9 and PARP14 cross-regulate macrophage activation via STAT1 ADP-ribosylation. Nat Commun 2016; 7:12849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Riley JP, Kulkarni A, Mehrotra P, Koh B, Perumal NB, Kaplan MH, et al. PARP-14 binds specific DNA sequences to promote Th2 cell gene expression. PLoS One 2013; 8:e83127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mehrotra P, Hollenbeck A, Riley JP, Li F, Patel RJ, Akhtar N, et al. Poly (ADP-ribose) polymerase 14 and its enzyme activity regulates T(H)2 differentiation and allergic airway disease. J Allergy Clin Immunol 2013; 131:521–31 e1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mathiesen JM, Vedel L, Brauner-Osborne H. cAMP biosensors applied in molecular pharmacological studies of G protein-coupled receptors. Methods Enzymol 2013; 522:191–207. [DOI] [PubMed] [Google Scholar]
- 138.Tavares LP, Negreiros-Lima GL, Lima KM, PMR ES, Pinho V, Teixeira MM, et al. Blame the signaling: Role of cAMP for the resolution of inflammation. Pharmacol Res 2020; 159:105030. [DOI] [PubMed] [Google Scholar]
- 139.Davis DJ, Dattel BJ, Ballard PL, Roberts JM. Beta-adrenergic receptors and cyclic adenosine monophosphate generation in human fetal lung. Pediatr Res 1987; 21:142–7. [DOI] [PubMed] [Google Scholar]
- 140.Lincoln TM, Cornwell TL. Towards an understanding of the mechanism of action of cyclic AMP and cyclic GMP in smooth muscle relaxation. Blood Vessels 1991; 28:129–37. [DOI] [PubMed] [Google Scholar]
- 141.Hansen G, Jin S, Umetsu DT, Conti M. Absence of muscarinic cholinergic airway responses in mice deficient in the cyclic nucleotide phosphodiesterase PDE4D. Proc Natl Acad Sci U S A 2000; 97:6751–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Torphy TJ, Undem BJ, Cieslinski LB, Luttmann MA, Reeves ML, Hay DW. Identification, characterization and functional role of phosphodiesterase isozymes in human airway smooth muscle. J Pharmacol Exp Ther 1993; 265:1213–23. [PubMed] [Google Scholar]
- 143.Sheldon KE, Shandilya H, Kepka-Lenhart D, Poljakovic M, Ghosh A, Morris SM Jr., Shaping the murine macrophage phenotype: IL-4 and cyclic AMP synergistically activate the arginase I promoter. J Immunol 2013; 191:2290–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Polumuri S, Perkins DJ, Vogel SN. cAMP levels regulate macrophage alternative activation marker expression. Innate Immun 2021; 27:133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Proia RL, Hla T. Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J Clin Invest 2015; 125:1379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fuerst E, Foster HR, Ward JP, Corrigan CJ, Cousins DJ, Woszczek G. Sphingosine-1-phosphate induces pro-remodelling response in airway smooth muscle cells. Allergy 2014; 69:1531–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Saluja R, Kumar A, Jain M, Goel SK, Jain A. Role of Sphingosine-1-Phosphate in Mast Cell Functions and Asthma and Its Regulation by Non-Coding RNA. Front Immunol 2017; 8:587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gon Y, Wood MR, Kiosses WB, Jo E, Sanna MG, Chun J, et al. S1P3 receptor-induced reorganization of epithelial tight junctions compromises lung barrier integrity and is potentiated by TNF. Proc Natl Acad Sci U S A 2005; 102:9270–5. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 149.Zhao J, Okamoto Y, Asano Y, Ishimaru K, Aki S, Yoshioka K, et al. Sphingosine-1-phosphate receptor-2 facilitates pulmonary fibrosis through potentiating IL-13 pathway in macrophages. PLoS One 2018; 13:e0197604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tani M, Kawakami A, Nagai M, Shimokado K, Kondo K, Yoshida M. Sphingosine 1-phosphate (S1P) inhibits monocyte-endothelial cell interaction by regulating of RhoA activity. FEBS Lett 2007; 581:4621–6. [DOI] [PubMed] [Google Scholar]
- 151.Okamoto Y, Kitakaze K, Takenouchi Y, Yamamoto S, Ishimaru H, Tsuboi K. Sphingosine 1-phosphate receptor type 2 positively regulates interleukin (IL)-4/IL-13-induced STAT6 phosphorylation. Cell Signal 2021; 88:110156. [DOI] [PubMed] [Google Scholar]
- 152.Wang YH, Voo KS, Liu B, Chen CY, Uygungil B, Spoede W, et al. A novel subset of CD4(+) T(H)2 memory/effector cells that produce inflammatory IL-17 cytokine and promote the exacerbation of chronic allergic asthma. J Exp Med 2010; 207:2479–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Irvin C, Zafar I, Good J, Rollins D, Christianson C, Gorska MM, et al. Increased frequency of dual-positive TH2/TH17 cells in bronchoalveolar lavage fluid characterizes a population of patients with severe asthma. J Allergy Clin Immunol 2014; 134:1175–86 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Al-Ramli W, Prefontaine D, Chouiali F, Martin JG, Olivenstein R, Lemiere C, et al. T(H)17-associated cytokines (IL-17A and IL-17F) in severe asthma. J Allergy Clin Immunol 2009; 123:1185–7. [DOI] [PubMed] [Google Scholar]
- 155.Deng C, Peng N, Tang Y, Yu N, Wang C, Cai X, et al. Roles of IL-25 in Type 2 Inflammation and Autoimmune Pathogenesis. Front Immunol 2021; 12:691559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gajewski TF, Fitch FW. Anti-proliferative effect of IFN-gamma in immune regulation. I. IFN-gamma inhibits the proliferation of Th2 but not Th1 murine helper T lymphocyte clones. J Immunol 1988; 140:4245–52. [PubMed] [Google Scholar]
- 157.Oriss TB, McCarthy SA, Morel BF, Campana MA, Morel PA. Crossregulation between T helper cell (Th)1 and Th2: inhibition of Th2 proliferation by IFN-gamma involves interference with IL-1. J Immunol 1997; 158:3666–72. [PubMed] [Google Scholar]
- 158.Heller NM, Matsukura S, Georas SN, Boothby MR, Rothman PB, Stellato C, et al. Interferon-gamma inhibits STAT6 signal transduction and gene expression in human airway epithelial cells. Am J Respir Cell Mol Biol 2004; 31:573–82. [DOI] [PubMed] [Google Scholar]
- 159.Dickensheets HL, Venkataraman C, Schindler U, Donnelly RP. Interferons inhibit activation of STAT6 by interleukin 4 in human monocytes by inducing SOCS-1 gene expression. Proc Natl Acad Sci U S A 1999; 96:10800–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bonder CS, Davies KV, Hosszu EK, Finlay-Jones JJ, Hart PH. IFN-gamma downregulates interleukin-4 functional activity on monocytes by multiple mechanisms. J Interferon Cytokine Res 2002; 22:287–93. [DOI] [PubMed] [Google Scholar]
- 161.Lewkowich IP, Lajoie S, Stoffers SL, Suzuki Y, Richgels PK, Dienger K, et al. PD-L2 modulates asthma severity by directly decreasing dendritic cell IL-12 production. Mucosal Immunol 2013; 6:728–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sethi GS, Sharma S, Naura AS. PARP inhibition by olaparib alleviates chronic asthma-associated remodeling features via modulating inflammasome signaling in mice. IUBMB Life 2019; 71:1003–13. [DOI] [PubMed] [Google Scholar]
- 163.Kim D, McAlees JW, Bischoff LJ, Kaur D, Houshel LK, Gray J, et al. Combined administration of anti-IL-13 and anti-IL-17A at individually sub-therapeutic doses limits asthma-like symptoms in a mouse model of Th2/Th17 high asthma. Clin Exp Allergy 2019; 49:317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]