

Abstract
Rhodobacter capsulatus xanthine dehydrogenase (XDH) is composed of two subunits, XDHA and XDHB. Immediately downstream of xdhB, a third gene was identified, designated xdhC, which is cotranscribed with xdhAB. Interposon mutagenesis revealed that the xdhC gene product is required for XDH activity. However, XDHC is not a subunit of active XDH, which forms an α2β2 heterotetramer in R. capsulatus. It was shown that XDHC neither is a transcriptional regulator for xdh gene expression nor influences XDH stability. To analyze the function of XDHC for XDH in R. capsulatus, inactive XDH was purified from an xdhC mutant strain. Analysis of the molybdenum cofactor content of this enzyme demonstrated that in the absence of XDHC, no molybdopterin cofactor MPT is present in the XDHAB tetramer. In contrast, absorption spectra of inactive XDH isolated from the xdhC mutant revealed the presence of iron-sulfur clusters and flavin adenine dinucleotide, demonstrating that XDHC is not required for the insertion of these cofactors. The absence of MPT from XDH isolated from an xdhC mutant indicates that XDHC either acts as a specific MPT insertase or might be a specific chaperone facilitating the insertion of MPT and/or folding of XDH during or after cofactor insertion.
Xanthine dehydrogenase (XDH; EC 1.1.1.204) is a complex molybdo/iron-sulfur flavoprotein that catalyzes the oxidation of hypoxanthine to xanthine as well as the oxidation of xanthine to uric acid with concomitant reduction of NAD. Water is the ultimate source of the oxygen atom incorporated into the substrate (12). Eukaryotic XDHs are proteins with an Mr of 300,000 and are composed of two identical and independent subunits, containing one molybdopterin cofactor (MPT), two nonidentical [2Fe-2S] centers, and a flavin adenine dinucleotide (FAD) each (6). In contrast to XDH from eukaryotes, XDH from the phototrophic purple bacterium Rhodobacter capsulatus is a cytoplasmic enzyme with an α2β2 heterotetrameric structure and an Mr of 275,000 (21). DNA sequence analysis of genes encoding XDH in R. capsulatus revealed two open reading frames (ORFs), designated xdhA and xdhB. The overall arrangement of domains in R. capsulatus XDH, including binding sites for two [2Fe-2S] clusters, FAD, and the molybdenum cofactor (Moco), is similar to that in its eukaryotic counterparts. However, in R. capsulatus, the iron-sulfur and FAD cofactor binding domains, respectively, are located within the XDHA subunit polypeptide whereas the Moco binding domain is located within the XDHB polypeptide. In contrast, in eukaryotes all three cofactor binding domains reside within a single polypeptide chain. Analysis of Moco present in XDH from R. capsulatus revealed that this enzyme contains MPT, as found for all eukaryotic molybdenum enzymes. Therefore, R. capsulatus is an unique example of an organism that harbors three different kinds of molybdoenzymes: XDH containing MPT (21), dimethyl sulfoxide (DMSO) reductase containing the bis-molybdopterin guanine dinucleotide cofactor (bis-MGD) (32), and nitrogenase containing the iron-molybdenum cofactor (FeMoco) (23). Biosynthesis of Moco present in XDH and DMSO reductase of R. capsulatus might proceed in a similar manner to the Moco biosynthesis pathway in Escherichia coli (reviewed in reference 28), requiring the gene products of the moa, mod, moe, and mog loci. In contrast to the MPT cofactor of XDH, the MGD cofactor of DMSO reductase additionally requires the mobA gene product for the addition of the guanine dinucleotide.
This work describes the identification and characterization of an ORF, designated xdhC, which is located immediately downstream of xdhAB. Although XDHC is not a subunit of active XDH, it is required for XDH activity. XDH isolated from an xdhC mutant strain does not contain the MPT cofactor. Therefore, XDHC is suspected to be involved in MPT insertion or to act as a chaperone in proper folding of XDH during or after the insertion of the MPT cofactor.
MATERIALS AND METHODS
Bacterial strains, plasmids, media, and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. R. capsulatus strains were grown in RCV minimal medium as described previously (17). Methods for conjugational plasmid transfer between E. coli and R. capsulatus and the selection of mutants, anaerobic growth conditions, and antibiotic concentrations were as previously described (17, 18).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotype and/or characteristics | Reference or source |
|---|---|---|
| E. coli strains | ||
| S17-1 | RP4-2 (Tc::Mu) (Km::Tn7) integrated into the chromosome | 34 |
| JM83 | Host for pUC plasmids | 39 |
| R. capsulatus strains | ||
| B10S | Spontaneous Smr mutant of R. capsulatus B10 | 17 |
| KS36 | ΔnifHDK::Spc | 40 |
| SL42 | xdhB::Km, ΔnifHDK | 21 |
| SL72I/II | ORF1::Gm, ΔnifHDK | 21 |
| SL136I | xdhA::Gm, ΔnifHDK | 21 |
| SL138I/II | xdhC::Gm, ΔnifHDK | This work |
| Xan-5 | moeB::Tn5, ΔnifHDK | 21 |
| Xan-21 | moaE::Tn5, ΔnifHDK | 21 |
| Plasmids | ||
| pUC8 | Apr, Lac+ | 39 |
| pNF3 | nifH promoter plasmid | 26 |
| pML5 | Broad-host-range lacZ fusion vector | 19 |
| pPHU231 | Broad-host-range vector | 29 |
| pSL75 | pML5 carrying an xdhA-lacZ fusion | 21 |
| pSL144 | pPHU231 carrying xdhAB expressed from the nifH promoter derived from pNF3 | This work |
DNA biochemistry.
DNA isolation, restriction enzyme analysis, agarose gel electrophoresis, and cloning procedures were performed by standard methods (31). Restriction endonucleases and T4 DNA ligase were purchased from MBI Fermentas and used as recommended by the supplier.
DNA sequencing.
DNA sequence analysis was carried out by the Eurogentec DNA-sequencing service. The Staden programs were used for editing and translating DNA sequences as well as for statistical and structural predictions (37). For homology searches in sequence databases, the BLAST algorithms (Basic Local Alignment Search Tool [1]) were used. Sequence alignments were performed with the CLUSTAL W program (38).
Construction of R. capsulatus xdhC mutant strains.
For the construction of R. capsulatus xdhC interposon mutants, a 2.5-kb BamHI fragment from the xdh gene region was cloned by standard methods (31) into mobilizable vector plasmids. A 519-bp XhoI fragment (Fig. 1A) from the xdhC coding region was replaced by a gentamicin resistance gene (13). The resulting hybrid plasmids were mobilized from E. coli S17-1 into R. capsulatus KS36 (40) by filter mating (24). Mutants were selected for the interposon-encoded resistance, and marker rescue was identified by the loss of the vector-encoded resistance.
FIG. 1.

Physical and genetic map of the R. capsulatus xdh gene region. (A) The localization of ORFs is shown as arrows carrying their respective gene designations. The DNA region sequenced in this study is marked by a heavy line. The locations of interposon insertions are shown below the restriction map. The directions of transcription of interposon resistance genes are symbolized by arrows in boxes (open arrows, gentamicin resistance gene; solid arrows, kanamycin resistance gene), indicating polar and nonpolar insertions. The ability of the corresponding R. capsulatus mutant strains to grow with xanthine as the sole nitrogen source is indicated by + or −. (B) Construction of a transcriptional xdhA-lacZ fusion. A HindIII-BamHI fragment was fused to the reporter gene lacZ in the broad-host-range vector plasmid pML5, resulting in plasmid pSL75. Abbreviations: B, BamHI; E, EcoRI; H, HindIII; S, SalI; Sm, SmaI; X; XhoI.
Construction of an xdhA-lacZ fusion plasmid.
To create a transcriptional xdhA-lacZ fusion, a 562-bp HindIII-BamHI fragment (Fig. 1B) carrying the 5′ part of R. capsulatus xdhA was cloned into the polylinker of pML5 (19). The resulting plasmid was designated pSL75.
β-Galactosidase assays.
To determine the β-galactosidase activities of R. capsulatus strains carrying lacZ fusions on the broad-host-range vector plasmid pSL75 (21), the corresponding strains were grown in RCV medium supplemented with tetracycline (0.25 μg/ml). Purines were added at a final concentration of 1 mM. Following growth in the respective media to late exponential phase, β-galactosidase activities of R. capsulatus strains were determined by the sodium dodecyl sulfate (SDS)-chloroform method described previously (14, 25).
Enzyme assays and spectroscopic methods.
XDH activities were assayed as described by Leimkühler et al. (21), with NAD as the electron acceptor. Absorption spectra were recorded at ambient temperature on a Pharmacia LKB Biochrom4060 spectrophotometer by using 50 mM Tris–1 mM EDTA (pH 8.0).
Enzyme purification.
To purify XDH from the R. capsulatus xdhC mutant strain SL138I, a plasmid overexpressing the xdhAB genes was constructed. To uncouple xdh expression from its native promoter, the nifH promoter region derived from pNF3 (26) was cloned in front of xdhAB. To fuse the ATG start codon of nifH to xdhA, an NdeI site was introduced in front of xdhA by PCR mutagenesis. Subsequently, a SacI-XhoI fragment carrying the structural genes of XDH (xdhAB) expressed from the nifH promoter was cloned into the polylinker of the broad-host-range plasmid pPHU231 (29). The resulting plasmid was designated pSL144. This plasmid was introduced into R. capsulatus xdhC mutant strain SL138I. A 10-liter volume of the corresponding strain was grown under anaerobic, phototrophic conditions in five 2-liter bottles containing RCV minimal medium with 10 mM serine as the nitrogen source. Cells were harvested by centrifugation at late log phase after 40 h of growth, when the absorbance at 660 nm reached 0.8. The cells were washed with a buffer containing 50 mM Tris-HCl (pH 8.0), 1 mM EDTA, and 2.5 mM dithiothreitol (buffer 1). Unless otherwise stated, all purification steps were carried out at 4°C and in buffer 1. Cell lysis was achieved by repeated passages through a French pressure cell (9,000 lb/in2); the suspension was centrifuged at 16,000 × g for 30 min, and the supernatant was used as a crude extract for enzyme purification. The crude extract was centrifuged at 100,000 × g in an ultracentrifuge for 1 h. The resulting supernatant was brought to 45% ammonium sulfate saturation at 4°C, and the suspension was centrifuged at 16,000 × g for 30 min. The pellet was dissolved in the minimal volume of buffer 1 and dialyzed against the same buffer to remove ammonium sulfate. Ion-exchange chromatography was performed on DEAE-Sepharose Fast Flow (Pharmacia). Bound protein was eluted with a linear 0 to 250 mM NaCl gradient in buffer 1. Fractions containing inactive XDH were detected on native polyacrylamide gels with purified, active XDH as reference. The corresponding fractions were pooled and electrophoresed by means of the model 491 Prep Cell system (Bio-Rad) in a gel column (28 mm long) consisting of 4 ml of stacking gel (4% acrylamide) and 10 ml of separating gel (4% acrylamide). Electrophoresis was carried out at 12 W (constant power) and 4°C. Fractions were collected in buffer 1, and those containing XDH were pooled and concentrated by ultrafiltration. By this method, inactive XDH from the xdhC mutant could be purified with a yield of approximately 6 mg. Protein concentrations were determined by the method of Smith et al. (35).
PAGE.
Analytical polyacrylamide gel electrophoresis (PAGE) was carried out in a discontinuous gel system (20). For nondenaturing PAGE, 4% acrylamide stacking gels and 6% acrylamide separating gels were used; for denaturing SDS-PAGE, 4% acrylamide stacking gels and 12% acrylamide separating gels were used. Protein staining was performed with Coomassie brilliant blue. Molecular mass standards were obtained from Bio-Rad.
Western blot analysis.
Immunoblot analysis of XDH from crude extracts of different R. capsulatus strains was performed after electrophoresis on nondenaturating 6% polyacrylamide gels. The proteins were transferred onto polyvinylidene difluoride membranes (Bio-Rad) by wet electrotransfer in carbonate-methanol buffer. After blocking with 3% bovine serum albumin, XDH was detected with rabbit anti-XDH antisera (1:10,000) raised against native XDH (Bioscience, Göttingen, Germany) and horseradish peroxidase-labelled second antibodies (goat antibodies 1:5,000) purchased from Bio-Rad. Horseradish peroxidase was detected with the enhanced chemiluminescence reagent (Amersham) as instructed by the manufacturer.
MPT analysis.
To analyze MPT present in XDH, the purified protein was subjected to a procedure which converts MPT to its oxidized fluorescent degradation product (Form A) after the enzyme was boiled at pH 2.5 in the presence of iodine, as originally described by Johnson et al. (15). After treatment with alkaline phosphatase, dephospho-Form A was further purified and desalted on 0.5-ml QAE-Sephadex columns. The columns were each washed with 10 ml of water. Dephospho-Form A was subsequently eluted with 10 mM acetic acid and immediately analyzed by high-pressure liquid chromatography (HPLC). HPLC was performed at room temperature with a Perkin-Elmer C18 reverse-phase column as described by Schwarz et al. (33). Comparable amounts of proteins were used for these analysis. Dephospho-Form A was detected by its fluorescence, which was monitored with excitation at 370 nm and emission at 450 nm. The presence of Form A in the respective fractions was verified by its absorption spectrum, with a an absorbance maximum at 380 nm.
Nucleotide sequence accession number.
The nucleotide sequence of a DNA fragment encompassing the xdhC coding region has been submitted to the EMBL nucleotide sequence database under accession no. AJ131529.
RESULTS
Identification of an ORF downstream of the xdhAB gene region, which is essential for XDH activity.
To determine whether the DNA fragment located immediately downstream of xdhAB, encoding the two subunits of XDH, is also involved in xanthine degradation, defined mutant strains were constructed. An interposon encoding gentamicin resistance was used to replace a 519-bp XhoI fragment downstream of xdhB (Fig. 1A). The corresponding R. capsulatus mutant strains (SL138I and SL138II) were tested for their ability to use xanthine as the sole nitrogen source. As shown in Fig. 1A, the XDH phenotype of SL138I and SL138II was compared to the XDH phenotype of mutant strains SL136I (xdhA, XDH−), SL42 (xdhB, XDH−), and SL72I/II (ORF1−, XDH+). Mutants SL138I and SL138II were unable to grow with xanthine as the sole source of nitrogen, demonstrating that the DNA region immediately downstream of xdhAB is essential for XDH activity. DNA sequence analysis of a 1,247-bp XhoI-HindIII fragment (Fig. 1A) downstream of the R. capsulatus xdhAB gene region revealed the presence of an ORF preceded by a typical ribosome binding site, which is transcribed in the same direction as xdhAB. The start codon of this ORF overlaps the 3′ end of xdhB, indicating translational coupling with xdhB. The corresponding gene was termed xdhC, since the xdhC gene product is required for XDH activity. The xdhC gene encodes a putative protein of 312 amino acid residues with a deduced molecular mass of 33,414 Da. XDHC shows weak similarity (21% identity) only to an ORF in E. coli with unknown function (AE000136-3) (5). However, the corresponding gene in E. coli is located downstream of three genes coding for a putative XDH (AE000136-6, AE000136-5, and AE000136-4) (21).
Immediately downstream of xdhC, another ORF was identified, which is transcribed in the same direction as xdhABC (ORF1; Fig. 1A). The predicted GTG start codon of ORF1 overlaps the stop codon of xdhC. In contrast to xdhC, interposon insertions into the HindIII site of ORF1 (SL72I/II) had no influence on xanthine utilization (Fig. 1A) (21). ORF1 shows a high degree of identity in its 157 N-terminal amino acids to ATP binding proteins of ATP binding cassette (ABC) transport systems.
Influence of xdhC mutations on xdhA gene expression.
As described by Leimkühler et al. (21), active XDH purified from R. capsulatus forms an α2β2 heterotetramer with a molecular mass of 275 kDa. SDS-PAGE revealed two noncovalently bound subunits with molecular masses of 50 kDa for the α-subunit, encoded by xdhA, and 85 kDa for the β-subunit, encoded by xdhB. A subunit corresponding to the molecular mass of XDHC (33,414 Da) was not identified in native XDH. Therefore, XDHC, which is required for XDH activity, is not part of the active enzyme.
Previously it was shown that transcription of the xdhAB genes is induced threefold in the presence of the substrates xanthine or hypoxanthine (21). To test whether the xdhC gene product is a transcriptional regulator of xdh gene expression, an xdhA-lacZ reporter plasmid (pSL75; Fig. 1B) was introduced into a strain carrying a mutation in xdhC (SL138I) and into a strain carrying a nonpolar mutant defective in xdhA (SL136I). The expression pattern of the xdhA-lacZ transcriptional fusion revealed no significant differences between an xdhC mutant and a mutant defective in xdhA (data not shown), assuming that the xdhC gene product has no influence on the expression of the xdhABC operon. The inducibility by hypoxanthine was severely reduced in both mutant strains, whereas the inducibility by xanthine was retained. As discussed previously (21), a putative activator system responsible for the induction of xdh gene expression seems to sense mainly the concentration of xanthine and not that of hypoxanthine. The presence of the same expression patterns of xdhA-lacZ in a mutant defective in xdhC and in a mutant defective in xdhA, which are both unable to convert hypoxanthine to xanthine, strongly suggests that XDHC is not a transcriptional activator protein for xdh gene expression.
Immunodetection of XDH in crude extracts of R. capsulatus wild type, different xdh mutant strains, and mutants defective in Moco biosynthesis.
To determine whether XDHC has an influence on XDH stability, immunodetection of XDH with antisera raised against purified XDH was carried out. Figure 2A shows the result of Western blot analysis comparing crude extracts of the R. capsulatus parental strain KS36 to xdhA, xdhB, and xdhC mutant strains on native polyacrylamide gels. The results revealed that inactive XDH can be detected in an xdhC mutant strain. Although the protein exhibited a slightly reduced electrophoretic mobility in the xdhC mutant compared to active protein from KS36 extracts (compare lane 4 with lane 1 in Fig. 2A), it still retained its ability to form a tetramer. In contrast, XDHB or XDHA dimers or monomers could not be detected in xdhA or xdhB mutant strains, respectively (Fig. 2A, lanes 2 and 3). In these mutants, the XDHA or XDHB subunits, without the corresponding counterpart, are suspected to be degraded immediately.
FIG. 2.

Western blot analysis of XDH accumulated in different R. capsulatus mutant strains. Preparation and electrophoresis of R. capsulatus crude extracts from different mutant strains in 6% native polyacrylamide gels were performed as described in Materials and Methods. Cells were grown in the presence of 10 mM NH4+ and 1 mM hypoxanthine to induce xdhABC gene expression. Equal amounts of total protein (20 μg) were subjected to electrophoresis and analyzed by Western blotting with antisera against purified XDH. (A) Lanes: 1, KS36 (parental strain); 2, SL136I (xdhA); 3, SL42 (xdhB); 4, SL138I (xdhC); 5, 1 μg of purified XDH. (B) Lanes: 1, SL138I (xdhC); 2, R507 (moeA) grown in the absence of added molybdate; 3, R507 (moeA) supplemented with 1 mM molybdate in the medium; 4, Xan-5 (moeB); 5, Xan-21 (moaE); 6, KS36 (parental strain).
To test whether the reduced electrophoretic mobility of inactive XDH from xdhC mutants compared to active XDH was due to absence of MPT, XDH was analyzed in crude extracts of different Moco biosynthesis mutants (Fig. 2B). XDH is inactive in a moeA mutant strain, but an active XDH can be obtained from this mutant strain after growth on medium supplemented with 1 mM molybdate (21a). In addition, crude extracts of moaE and moeB mutant strains were tested. The Western blot analysis in Fig. 2B indicated that inactive XDH from moeA, moaE, or moeB mutants exhibited the same electrophoretic mobility as did XDH from an xdhC mutant (compare lanes 2, 4, and 5 with lane 1). In contrast, active XDH from a moeA mutant strain supplemented with 1 mM molybdate had the same electrophoretic mobility as did XDH from extracts of the parental strain (lanes 3 and 6). Since inactive XDH isolated either from mutants defective in Moco biosynthesis or from xdhC mutants exhibited comparable changes in electrophoretic mobility, it could be assumed that XDH from xdhC mutant strains might be devoid of MPT (see below).
Purification of XDH from an xdhC mutant strain.
To test if inactive XDH from xdhC mutants lacks MPT, XDH was purified from an xdhC mutant strain. For this purpose, an xdhAB-overexpressing hybrid plasmid (pSL144) was constructed as described in Materials and Methods. Plasmid pSL144 carries the structural genes for XDH (xdhAB) under the control of the strong nifH promoter, which is inducible under nitrogen-limiting conditions. Plasmid pSL144 was introduced into the R. capsulatus xdhC mutant strain SL138I. The resulting strain was grown under anaerobic, phototrophic conditions in RCV minimal medium containing serine as the nitrogen source. These growth conditions result in induction of the nifH promoter and overproduction of XDH. Inactive XDH was purified by ion-exchange chromatography and preparative gel electrophoresis as described in Materials and Methods. The purity of the inactive enzyme was demonstrated by detection on Coomassie blue-stained native polyacrylamide gels and on denaturing SDS gels with purified XDH from wild-type R. capsulatus as the control (Fig. 3). Although the apparent molecular masses of the native proteins appeared to be slightly different (Fig. 3A), the subunit compositions of the proteins were identical on SDS gels; i.e., the proteins consisted of two subunits with molecular masses of 50 and 85 kDa, (Fig. 3B). By this method, inactive XDH from a strain lacking XDHC could be partially purified.
FIG. 3.
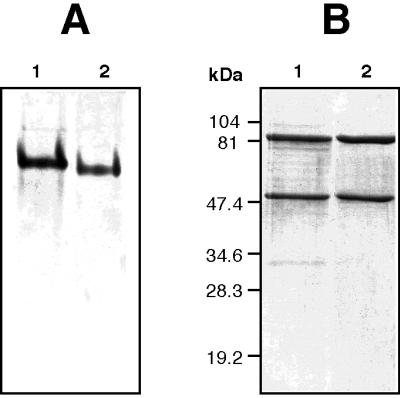
Analysis of active and inactive XDH of R. capsulatus by PAGE. (A) Purified XDH was electrophoresed under nondenaturing conditions in 6% polyacrylamide gels, and proteins were stained with Coomassie brilliant blue. Lanes: 1, 5 μg of inactive XDH purified from R. capsulatus SL138I (xdhC); 2, 4 μg of active XDH purified from wild-type R. capsulatus. (B) SDS-PAGE on 12% polyacrylamide gels. Lanes: 1, 8 μg of inactive XDH purified from R. capsulatus SL138I (xdhC); 2, 7 μg of active XDH purified from wild-type R. capsulatus.
Absorption spectra of XDH isolated from wild-type R. capsulatus and an xdhC mutant strain.
To identify the cofactors present in inactive XDH purified from an xdhC mutant strain, the visible absorption spectrum of this protein was recorded and compared to the absorption spectrum of active XDH purified by the method described by Leimkühler et al. (21). Air-oxidized XDH isolated from an xdhC mutant strain as well as from the wild type was brownish, and its absorption spectrum is shown in Fig. 4. The absorption spectra of active and inactive XDH from R. capsulatus are similar to absorption spectra studied for xanthine-oxidizing enzymes from other sources (9, 11). The visible spectrum of the inactive enzyme under oxic conditions was characterized by a pronounced absorption maximum at 460 nm and a broad shoulder centered around 550 nm, which can be attributed to the presence of FAD and iron-sulfur centers, respectively. The ratio of E460/E550 is 3.0, which corresponds to a 1:4 ratio of FAD to Fe-S (9). Both active and inactive XDH were completely reduced by dithionite, resulting in a complete reduction of FAD and iron-sulfur centers. In contrast to the wild-type enzyme, which was partially reduced by hypoxanthine, no reduction by hypoxanthine was observed for the inactive enzyme isolated from the xdhC mutant. These results indicated that MPT, which is involved in the reduction of hypoxanthine followed by a reductive electron transfer to the iron-sulfur centers and FAD, is not present in inactive XDH isolated from a mutant strain lacking XDHC. Therefore, XDHC might be involved in the insertion of MPT into XDH but not play a role in the insertion of FAD and iron-sulfur clusters into the enzyme.
FIG. 4.

Absorption spectra of R. capsulatus XDH. The spectra of 2 mg of XDH isolated from R. capsulatus SL138I (xdhC) in 50 mM Tris buffer–1 mM EDTA (pH 8.0) were recorded with respect to a buffer blank. The inset shows the absorption spectra of 460 μg of XDH isolated from wild-type R. capsulatus by the method described by Leimkühler et al. (21). Curve 1, air-oxidized enzyme; curve 2, enzyme reduced with 1 μmol of hypoxanthine; curve 3, reduced enzyme after addition of 50 μl of an aqueous solution, saturated with sodium dithionite.
Fluorescence analysis of molybdopterin derivatives isolated from active and inactive XDH.
To prove that XDH isolated from an xdhC mutant strain does not contain MPT, analysis of the cofactor present in active and inactive XDH was carried out as described by Leimkühler et al. (21). MPT was released from the active and inactive enzymes by heat treatment and converted to its oxidized fluorescent degradation product, Form A, by treatment with acidic iodine. As described in Materials and Methods, dephospho-Form A obtained from both enzymes was eluted with 10 mM acetic acid from QAE-Sephadex and analyzed by HPLC. As shown in Fig. 5A, large amounts of dephospho-Form A were obtained from wild-type XDH. In contrast, the amount of dephospho-Form A obtained from inactive XDH isolated from the xdhC mutant strain corresponded only to 3% of the amount of dephospho-Form A obtained from active XDH (Fig. 5B). These results clearly demonstrated that the inactivity of XDH in the R. capsulatus xdhC mutant is due to the lack of MPT. Although XDH from an xdhC mutant contained residual amounts of MPT, no enzyme activity could be detected (data not shown), which is in line with the inability of an xdhC mutant to grow with xanthine as the sole source of nitrogen.
FIG. 5.

Analysis of fluorescent derivatives of MPT from R. capsulatus XDH. HPLC elution profiles of dephospho-Form A isolated from MPT of XDH from wild-type (WT) R. capsulatus (A), and R. capsulatus SL138I (xdhC) (B). MPT were converted into Form A by oxidation with iodine at room temperature. Dephospho-Form A was eluted from a QAE-Sephadex column with 10 mM acetic acid and analyzed by HPLC. Fluorescence was monitored with excitation at 370 nm and emission at 450 nm. Active and inactive XDH at 0.25 μM were used for these analyses.
DISCUSSION
The xdhC gene was identified as the third gene in the xdhABC operon. The overlapping start codon of xdhC with the 3′ end of xdhB indicated translational coupling of these genes, ensuring the synthesis of equimolar amounts of the corresponding proteins. Downstream of xdhC, another open reading frame (ORF1) was identified, which is probably also cotranscribed with xdhABC. Interposon mutagenesis demonstrated that ORF1 is not required for XDH activity (21). The N-terminal amino acid sequence of ORF1 shows high similarity to ATP binding proteins from ABC transport systems. However, it remains speculative whether ORF1 belongs to an ABC transport system involved in purine uptake. In all systems studied so far, purines are imported into the cell via permeases and not by ABC transport systems (7, 8, 10). At least under the conditions of high xanthine or hypoxanthine concentrations used in this study, ORF1 is not essential for purine uptake in R. capsulatus.
It was shown that XDHC is required for XDH activity, although the protein is not a subunit of the active protein. Furthermore, XDHC neither constitutes a transcriptional regulator of xdh gene expression nor influences XDH stability. Based on the finding that XDH purified from an xdhC mutant strain contained iron-sulfur clusters as well as the FAD cofactor, but not MPT, a possible function of XDHC in MPT insertion into XDH has to be assumed.
The crystal structures of several molybdoenzymes revealed (reviewed in references 16 and 30) that Moco is deeply buried within the protein, at the end of a funnel-shaped passage giving access only to the substrate. Therefore, it seems unlikely that Moco is inserted into the protein after the assembly and correct folding of the protein. Thus, it has to be assumed that during Moco biosynthesis, until MPT is completed, the apo-XDHAB tetramer has to stay in a suitable open conformation which enables XDHB to bind mature MPT (Fig. 6). This is in line with the observation that XDH isolated from Moco biosynthesis mutants exhibited a change in electrophoretic mobility in native polyacrylamide gels, indicating a different folding of XDH in these mutant strains. Additionally, XDH isolated from xdhC mutants, which was shown to be devoid of MPT, exhibited the same electrophoretic mobility as XDH in crude extracts of Moco biosynthesis mutants. Thus, the role of XDHC could be as a molybdopterin carrier protein, an MPT insertase, or a chaperone involved in proper folding of XDH during or after the insertion of MPT.
FIG. 6.

Model for the assembly of XDH in R. capsulatus. MPT for XDH is synthesized in a series of reactions catalyzed by the moa, mod, moe, and mog gene products (28). Finally, MPT might be inserted into XDH by the XDHC protein. The role of XDHC could either be a molybdopterin carrier protein, an MPT insertase, or a chaperone involved in proper folding of XDH during or after the insertion of MPT. In contrast to MPT, two [2Fe-2S] clusters and FAD are inserted into the XDHA subunit of XDH without involvement of XDHC.
As mentioned above, R. capsulatus XDHC is not a subunit of XDH but is essential for XDH activity. Similarly, E. coli NarJ does not belong to the nitrate reductase complex but is also required for nitrate reductase activity (2, 3, 36). E. coli nitrate reductase A is a membrane-bound molybdoenzyme composed of three subunits, NarG, NarH, and NarI, harboring MGD. It has been suggested that NarJ functions as a chaperone in nitrate reductase maturation, being involved in MGD insertion into NarG (4, 22). After the insertion of MGD a conformational change in nitrate reductase results in dissociation of NarJ from the protein (4). Taking into account similar functions in Moco insertion of XDHC for XDH to those of NarJ for nitrate reductase, it can be assumed that XDHC might also act as a specific chaperone for XDH. However, XDHC could not be copurified with XDHAB in the absence of MPT.
The presence of specific chaperones for molybdoenzymes in prokaryotes seems to be widespread, since a chaperone for trimethylamine N-oxide reductase (TMAO reductase) in E. coli has recently been identified (TorD) (27). TorD homologous proteins were also identified for DMSO reductases of R. sphaeroides and R. capsulatus (DmsB and DorD, respectively), suggesting similar functions of these proteins for DMSO and TMAO reductases (27). Additionally, an ORF with weak similarity to xdhC could be identified in the E. coli genome (AE000136-3) (5), which is located immediately downstream of three ORFs with homology to XDH (AE000136-6, AE000136-5, and AE000136-4) (21).
In conclusion, it seems likely that each prokaryotic molybdoenzyme has its own system-specific chaperone that plays a special role in Moco insertion and target protein folding, which cannot be replaced by another protein. This might explain why none of these proteins have structural features in common, which might imply general functional homologies.
ACKNOWLEDGMENTS
We thank G. Schwarz and R. Mendel (Braunschweig) for help in cofactor analysis and B. Masepohl and K.-U. Riedel (Bochum) for critical reading of the manuscript.
This work was supported by financial grants from Fonds der Chemischen Industrie and Deutsche Forschungsgemeinschaft (DFG).
REFERENCES
- 1.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 2.Blasco F, Iobbi C, Giordano G, Chippaux M, Bonnefoy V. Nitrate reductase of Escherichia coli: completion of the nucleotide sequence of the nar operon and reassessment of the role of the α and β subunits in iron binding and electron transfer. Mol Gen Genet. 1989;218:249–256. doi: 10.1007/BF00331275. [DOI] [PubMed] [Google Scholar]
- 3.Blasco F, Pommier J, Augier V, Chippaux M, Giordano G. Involvement of the narJ or narW gene product in the formation of active nitrate reductase in Escherichia coli. Mol Microbiol. 1992;6:221–230. doi: 10.1111/j.1365-2958.1992.tb02003.x. [DOI] [PubMed] [Google Scholar]
- 4.Blasco F, Dos Santos J-P, Magalon A, Frixon C, Guigliarelli B, Santini C-L, Giordano G. NarJ is a specific chaperone required for molybdenum cofactor assembly in nitrate reductase A of Escherichia coli. Mol Microbiol. 1998;28:435–447. doi: 10.1046/j.1365-2958.1998.00795.x. [DOI] [PubMed] [Google Scholar]
- 5.Blattner F R, Plunkett III G, Bloch C A, Perna N T, Burland V, Riley M, Collado-Vides J, Glasner J D, Rode C K, Mayhew G F, Gregor J, Davis N W, Kirkpatrick H A, Goeden M A, Rose D J, Mau B, Shao Y. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 6.Bray R C, Bennett B, Burke J F, Chovnick A, Doyle W A, Howes B D, Lowe D J, Richards R L, Turner N A, Ventom A, Whittle J R S. Recent studies on xanthine oxidase and related enzymes. Biochem Soc Trans. 1996;24:99–105. doi: 10.1042/bst0240099. [DOI] [PubMed] [Google Scholar]
- 7.Burton K. Transport of nucleic acid bases into Escherichia coli. J Gen Microbiol. 1983;129:3505–3513. doi: 10.1099/00221287-129-11-3505. [DOI] [PubMed] [Google Scholar]
- 8.Christiansen L C, Schou S, Nygaard P, Saxild H H. Xanthine metabolism in Bacillus subtilis: characterization of the xpt-pbuX operon and evidence for purine- and nitrogen-controlled expression of genes involved in xanthine salvage and catabolism. J Bacteriol. 1997;179:2540–2550. doi: 10.1128/jb.179.8.2540-2550.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coughlan M P. Molybdenum and molybdenum-containing enzymes. New York, N.Y: Pergamon Press; 1980. pp. 119–185. [Google Scholar]
- 10.Diallinas G, Gorfinkiel L, Arst H N, Cecchetto G, Scazzocchio C. Genetic and molecular characterization of a gene encoding a wide specificity purine permease of Aspergillus nidulans reveals a novel family of transporters conserved in prokaryotes and eukaryotes. J Biol Chem. 1995;270:8610–8622. doi: 10.1074/jbc.270.15.8610. [DOI] [PubMed] [Google Scholar]
- 11.Hille R. The mononuclear molybdenum enzymes. Chem Rev. 1996;96:2757–2816. doi: 10.1021/cr950061t. [DOI] [PubMed] [Google Scholar]
- 12.Hille R, Sprecher H. On the mechanism of action of xanthine oxidase. J Biol Chem. 1987;262:10914–10917. [PubMed] [Google Scholar]
- 13.Hirsch P R, Beringer J E. A physical map of pPH1JI and pJB4JI. Plasmid. 1984;12:139–141. doi: 10.1016/0147-619x(84)90059-3. [DOI] [PubMed] [Google Scholar]
- 14.Hübner P, Willison J C, Vignais P M, Bickle T A. Expression of regulatory nif genes in Rhodobacter capsulatus. J Bacteriol. 1991;173:2993–2999. doi: 10.1128/jb.173.9.2993-2999.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson J L, Hainline B E, Rajagopalan K V, Arison B H. The pterin component of the molybdenum cofactor. Structural characterization of two fluorescent derivatives. J Biol Chem. 1984;259:5414–5422. [PubMed] [Google Scholar]
- 16.Kisker C, Schindelin H, Rees D C. Molybdenum-cofactor-containing enzymes: structure and mechanism. Annu Rev Biochem. 1997;66:233–267. doi: 10.1146/annurev.biochem.66.1.233. [DOI] [PubMed] [Google Scholar]
- 17.Klipp W, Masepohl B, Pühler A. Identification and mapping of nitrogen fixation genes of Rhodobacter capsulatus: duplication of a nifA-nifB region. J Bacteriol. 1988;170:693–699. doi: 10.1128/jb.170.2.693-699.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kutsche M, Leimkühler S, Angermüller S, Klipp W. Promoters controlling expression of the alternative nitrogenase and the molybdenum uptake system in Rhodobacter capsulatus are activated by NtrC, independent of ς54, and repressed by molybdenum. J Bacteriol. 1996;178:2010–2017. doi: 10.1128/jb.178.7.2010-2017.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Labes M, Pühler A, Simon R. A new family of RSF1010-derived expression and lac-fusion broad-host-range vectors for Gram-negative bacteria. Gene. 1990;89:37–46. doi: 10.1016/0378-1119(90)90203-4. [DOI] [PubMed] [Google Scholar]
- 20.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 21.Leimkühler S, Kern M, Solomon P S, McEwan A G, Schwarz G, Mendel R R, Klipp W. Xanthine dehydrogenase from the phototrophic purple bacterium Rhodobacter capsulatus is more similar to its eukaryotic counterparts than to prokaryotic molybdenum enzymes. Mol Microbiol. 1998;27:853–869. doi: 10.1046/j.1365-2958.1998.00733.x. [DOI] [PubMed] [Google Scholar]
- 21a.Leimkühler, S., and W. Klipp. Unpublished results.
- 22.Liu X, DeMoss J A. Characterization of NarJ, a system-specific chaperone required for nitrate reductase biogenesis in Escherichia coli. J Biol Chem. 1997;272:24266–24271. doi: 10.1074/jbc.272.39.24266. [DOI] [PubMed] [Google Scholar]
- 23.Masepohl B, Klipp W. Organization and regulation of genes encoding the molybdenum nitrogenase and the alternative nitrogenase in Rhodobacter capsulatus. Arch Microbiol. 1996;165:80–90. [Google Scholar]
- 24.Masepohl B, Klipp W, Pühler A. Genetic characterization and sequence analysis of the duplicated nifA/nifB gene region of Rhodobacter capsulatus. Mol Gen Genet. 1988;212:27–37. doi: 10.1007/BF00322441. [DOI] [PubMed] [Google Scholar]
- 25.Miller J H. Experiments in molecular genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- 26.Pollock D, Bauer C E, Scolnik P A. Transcription of the Rhodobacter capsulatus nifHDK operon is modulated by the nitrogen source. Construction of plasmid expression vectors based on the nifHDK promoter. Gene. 1988;65:269–275. doi: 10.1016/0378-1119(88)90463-5. [DOI] [PubMed] [Google Scholar]
- 27.Pommier J, Méjean V, Giordano G, Iobbi-Nivol C. TorD, a cytoplasmic chaperone interacts with unfolded trimethylamine N-oxide reductase enzyme (TorA) in Escherichia coli. J Biol Chem. 1998;273:16615–16620. doi: 10.1074/jbc.273.26.16615. [DOI] [PubMed] [Google Scholar]
- 28.Rajagopalan K V. Biosynthesis of the molybdenum cofactor. In: Neidhardt F C, Curtiss III R, Ingraham J L, Lin E C C, Low K B, Magasanik B, Reznikoff W S, Riley M, Schaechter M, Umbarger H E, editors. Escherichia coli and Salmonella: cellular and molecular biology. 2nd ed. Washington, D.C: ASM Press; 1996. pp. 674–679. [Google Scholar]
- 29.Reyes F, Roldan M D, Klipp W, Castillo F, Moreno-Vivian C. Isolation of periplasmic nitrate reductase genes from Rhodobacter sphaeroides DSM 158: structural and functional differences among prokaryotic nitrate reductases. Mol Microbiol. 1996;19:1307–1318. doi: 10.1111/j.1365-2958.1996.tb02475.x. [DOI] [PubMed] [Google Scholar]
- 30.Romão M J, Knäblein J, Huber R, Moura J J G. Structure and function of molybdopterin containing enzymes. Prog Biophys Mol Biol. 1997;68:121–144. doi: 10.1016/s0079-6107(97)00022-9. [DOI] [PubMed] [Google Scholar]
- 31.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 32.Schneider F, Löwe J, Huber R, Schindelin H, Kisker C, Knäblein J. Crystal structure of dimethyl sulfoxide reductase from Rhodobacter capsulatus at 1.88 Å resolution. J Mol Biol. 1996;263:53–69. doi: 10.1006/jmbi.1996.0555. [DOI] [PubMed] [Google Scholar]
- 33.Schwarz G, Boxer D H, Mendel R R. Molybdenum cofactor biosynthesis. The plant protein Cnx1 binds molybdopterin with high affinity. J Biol Chem. 1997;272:26811–26814. doi: 10.1074/jbc.272.43.26811. [DOI] [PubMed] [Google Scholar]
- 34.Simon R, Priefer U, Pühler A. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Bio/Technology. 1983;1:784–791. [Google Scholar]
- 35.Smith P K, Krohn R I, Hermanson G T, Mallia A K, Gartner F H, Provenzano M D, Fujimoto E K, Goeke N M, Olson B J, Klenk D C. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 36.Sodergren E J, DeMoss J A. narI region of the Escherichia coli nitrate reductase (nar) operon contains two genes. J Bacteriol. 1988;170:1721–1729. doi: 10.1128/jb.170.4.1721-1729.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Staden R. The current status and portability of our sequence handling software. Nucleic Acids Res. 1986;14:217–232. doi: 10.1093/nar/14.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thompson J D, Higgins D G, Gibson T J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vieira J, Messing J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 1982;19:259–268. doi: 10.1016/0378-1119(82)90015-4. [DOI] [PubMed] [Google Scholar]
- 40.Wang G, Angermüller S, Klipp W. Characterization of Rhodobacter capsulatus genes encoding a molybdenum transport system and putative molybdenum-pterin-binding proteins. J Bacteriol. 1993;175:3031–3042. doi: 10.1128/jb.175.10.3031-3042.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]