

Abstract
Background:
An activated, pro-inflammatory endothelium is a key feature in the development of complications of obesity and type 2 diabetes and can be caused by insulin resistance in endothelial cells.
Methods:
We analyzed primary human endothelial cells by RNA sequencing to discover novel insulin-regulated genes and used endothelial cell culture and animal models to characterize signaling through CXCR4 in endothelial cells.
Results:
CXCR4 was one of the genes most potently regulated by insulin and this was mediated by phosphatidylinositol 3-kinase, likely through FoxO1 which bound to the CXCR4 promoter. CXCR4 mRNA in CD31+ cells was 77% higher in mice with diet-induced obesity compared to lean controls and 37% higher in db/db mice than db/+ controls, consistent with upregulation of CXCR4 in endothelial cell insulin resistance. Stromal derived factor-1 (SDF-1), the ligand for CXCR4, increased leukocyte adhesion to cultured endothelial cells. This effect was lost after deletion of CXCR4 by gene editing while 80% of the increase was prevented by treatment of endothelial cells with insulin. In vivo microscopy of mesenteric venules showed an increase in leukocyte rolling after intravenous injection of SDF-1 but most of this response was prevented in transgenic mice with endothelial overexpression of insulin receptor substrate-1 (IRS-1).
Conclusion:
Endothelial cell insulin signaling limits leukocyte-endothelial cell interaction induced by SDF-1 through downregulation of CXCR4. Improving insulin signaling in endothelial cells or inhibiting endothelial CXCR4 may reduce immune cell recruitment to the vascular wall or tissue parenchyma in insulin resistance and thereby help prevent several vascular complications.
Graphical Abstract
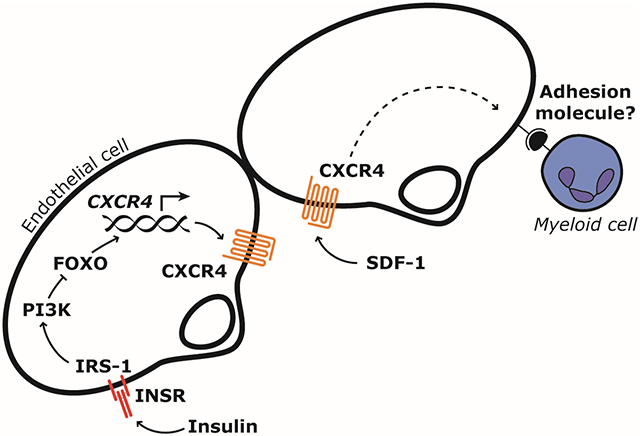
Introduction
An activated, pro-inflammatory endothelium is a key feature of obesity and type 2 diabetes1. It promotes recruitment of immune cells to tissues and contributes to accelerated atherosclerosis2-6, diabetic nephropathy7,8 and diabetic retinopathy9,10. It may also contribute to recruitment of macrophages11 or neutrophils12 into adipose tissue which in turn can cause insulin resistance in remote organs like liver and muscle. These mechanisms have important translational implications because blocking leukocyte-endothelial interaction in patients is sufficient to reduce tissue inflammation. For example, blocking α4 integrin, the leukocyte ligand for vascular cell adhesion molecule-1 (VCAM-1) and mucosal vascular addressin cell adhesion molecule-1 (MadCAM-1) expressed by endothelial cells, is efficacious in the treatment of multiple sclerosis13 and inflammatory bowel disease14. Further understanding the factors that causes activation of the endothelium in obesity and type 2 diabetes therefore has the potential to identify new therapeutic targets for several complications of these metabolic diseases.
We have previously demonstrated that insulin resistance in endothelial cells leads to an activated endothelium with increased leukocyte-endothelial cell interaction15,16. The ensuing immune cell recruitment can promote atherosclerosis15 or tumor formation16. These studies were performed in mice with knockout of the insulin receptor gene (Insr) in vascular endothelial cells which resulted in a 4-fold increase in adhesion to endothelium as observed by in vivo microscopy15. In atherosclerosis-prone mice with knockout of Apoe, endothelial Insr knockout increased atherosclerosis by up to 3-fold15 and in ApcMin/+ mice susceptible to intestinal tumor formation it increased tumor number by 42%16. Importantly, these pathologies occurred in the absence of changes in systemic insulin sensitivity15,16. Endothelial cell insulin resistance is present in animal models of obesity17,18 and develops before insulin resistance in many non-vascular tissues during high-fat feeding19,20. It is also present in humans with obesity or type 2 diabetes21-24.
Insulin increases nitric oxide (NO) production in endothelial cells and NO decreases leukocyte adhesion to endothelium25. Insulin can also decrease expression of VCAM-1 independently of NO15. However, we suspected that insulin action on endothelial cells could modify leukocyte adhesion through other mechanisms. In the current study we searched for targets of insulin signaling in endothelium and identified C-X-C motif chemokine receptor 4 (CXCR4) as a gene potently regulated by insulin and insulin-like growth factor-1 (IGF-1) in endothelial cells. We show that leukocyte interaction with endothelium in mesenteric venules in vivo is increased by stromal derived factor-1 (SDF-1), the main ligand for CXCR4, and that much of this increase is prevented in transgenic mice with gain of insulin signaling in endothelial cells. These results identify a new mechanism for increased leukocyte interaction with endothelial cells in obesity and type 2 diabetes and may have implications for prevention of leukocyte recruitment to tissues in insulin resistant states and consequent complications of metabolic disease.
Methods
A comprehensive description of methods can be found in the Supplemental Materials.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Results
Discovery of novel insulin-regulated genes in endothelial cells
We have shown that loss of insulin signaling in endothelial cells causes increased leukocyte rolling and adhesion to endothelial cells15,16, increased atherosclerotic lesions size in Apoe null mice15 and increased neutrophil recruitment to intestinal tumors in ApcMin/+ mice16. In a search for endothelial cell molecules responsible for insulin regulation of immune cell recruitment, we performed an unbiased screen of transcripts changed by insulin treatment in endothelial cell culture. RNA sequencing (RNAseq) analysis of endothelial cell lysate showed that 164 genes were significantly changed after 3 hours of insulin treatment (fig. 1A and the supplementary Data Set, which shows the regulation of all identified RNAs). The 30 most significantly enriched gene ontology terms are shown in Table S1. The most regulated gene, C10ORF10 (fig. 1A), was previously shown to be regulated by insulin, fasting and feeding in several tissues26 and by FOXO3 in endothelial cells27. The second-most regulated gene was CXCR4 which encodes C-X-C chemokine receptor type 4 (CXCR4) (fig. 1A). We validated the RNAseq result by measuring CXCR4 mRNA by real-time PCR in HUVEC exposed to increasing concentrations of insulin or IGF-1. Insulin and IGF-1 reduced CXCR4 mRNA by up to 45.5±9.7% and 53.2±15.3% respectively (fig. 1C).
Figure 1. Identification of insulin-regulated genes in human endothelial cells.

A, Human umbilical vein endothelial cells (HUVEC) were treated with 100 nM insulin for 3 hours or left untreated. Cell lysate from 5 cultures treated with insulin and 5 cultures left untreated was analyzed by RNAseq. Volcano plot of differentially regulated genes is shown. B-C, HUVEC were treated with insulin or IGF-1 for 3 hours or left untreated. Cell lysate was analyzed by TaqMan real-time PCR. Results are from 6 independent experiments. C, *, p=0.03 for all (Wilcoxon signed-rank test). D, *, p≤0.0025.
It is well-established that CXCR4 has a functional role in endothelial cells where it is critical to vasculogenesis during development28-32, pathological angiogenesis33,34 and reendothelialization35. CXCR4 is upregulated by hypoxia36,37 and downregulated by proinflammatory cytokines38. However, there is no information on regulation of CXCR4 by insulin or IGF-1 or dysregulation of CXCR4 in insulin resistant states. We therefore decided to study insulin regulation of CXCR4 in endothelial cells.
Insulin regulates CXCR4 via PI3K and FoxO1
In primary mouse endothelial cells, CXCR4 mRNA was downregulated as early as 1 hour after treatment with insulin (fig. 2A). In endothelial cells isolated from mice with double knockout of the insulin receptor and IGF-1 receptor genes, insulin receptor and IGF1R protein were barely detectable compared to control cells and insulin-stimulated Akt phosphorylation was reduced (fig. 2B). In these primary cell cultures, downregulation of CXCR4 mRNA by insulin or IGF-1 was completely abrogated (fig. 2C). Expression of CXCR4 protein at the cell surface, measured by flow cytometry, was also downregulated by insulin or IGF-1 in wildtype cells, but was unchanged in cells from mice with loss of endothelial cell insulin and IGF-1 receptors (fig. 2D-E).
Figure 2. Insulin regulates CXCR4 in primary endothelial cells through PI3K.

A, mRNA expression in primary mouse lung endothelial cells (MLEC) after treatment with 10 nM insulin for the time indicated. B-E, MLEC were isolated from mice with floxed mutations in the Insr and Igf1r gene with (“DKO”) or without (“lox”) a cre transgene controlled by the Cdh5 promoter causing knockout of the insulin and IGF-1 receptors in vascular endothelial cells. Serum-starved cultures were treated with 10 nM insulin or 10 nM IGF-1 for 4 hours. B, Western blots of MLEC lysate. C, Normalized Cxcr4 mRNA expression analyzed by real-time PCR. Results from 3 independent experiments are shown. *, p≤0.001. D, Analysis of cell surface CXCR4 expression in MLEC from lox and DKO mice using flow cytometry, representative result shown. E, Flow cytometry results from 6 independent experiment. F, MLEC were treated with 1 μM wortmannin, a PI3K inhibitor, or 10 μM U0126, a MEK1/2 inhibitor or left untreated. After 30 minutes, cultures were treated with 100 nM insulin for 4 hours or left untreated. CXCR4 mRNA was measured by real-time PCR. Results from 6 independent experiments are shown. *, p≤0.004. G-H, MLEC were isolated from Foxo1/3/4flox/flox mice and infected with adenovirus expressing GFP or cre. After 48 hours, cultures were lysed. G, Protein expression was measured by Western blotting. H, CXCR4 mRNA analyzed by real-time PCR. Results from 6 independent experiments are shown. *, p=0.007. I, MS1 endothelial cells were infected with an adenovirus expressing wild-type FOXO1 as described previously56. Cells were serum-starved overnight and chromatin immunoprecipitation (ChIP) was performed using FOXO1 antibody or rabbit IgG followed by real-time PCR using primers amplifying a candidate FoxO1 binding motifs in the Cxcr4 promoter or a sequence without such a motif in the Gck promoter.
Inhibition of phosphatidylinositol 3-kinase (PI3K) with wortmannin blocked insulin-mediated downregulation of CXCR4 mRNA while inhibition of MEK1/2 with U0126 had no effect (fig. 2F). FoxO transcription factors are important mediators of insulin signaling downstream of PI3K. We isolated primary endothelial cells from animals carrying “floxed” alleles of FoxO1, 3 and 4 and expressed cre recombinase or a control construct by adenoviral vectors. FoxO1 and FoxO3 protein was reduced in endothelial cells infected with cre-expressing vectors compared to GFP-expressing vectors (fig. 2G). FoxO4 was not detectable in either condition, similar to what we have described previously for endothelial cells39. Loss of FoxO isoforms resulted in decreased expression of CXCR4 mRNA, demonstrating that FoxO isoforms maintains transcription of CXCR4 (fig. 2H).
To demonstrate whether CXCR4 is a direct target of FoxO1, we performed chromatin immunoprecipitation (ChIP) of FoxO1 followed by real-time PCR of sequences containing candidate FoxO-binding motifs. In cells overexpressing FoxO1, the amplification of a sequence containing the motif 5’-TTGTTGCC-3’ starting 9 nucleotides upstream of the translation initiation site was 13.8-fold greater after immunoprecipitation with a FoxO1 antibody compared with non-specific IgG (fig. 2I). Candidate FoxO binding motifs starting, relative to the translation initiation site, at −722, −618, −160, −130, −49, +971 and +1431 did not show significant enrichment in FoxO1 immunoprecipitates. A region in the Gck promoter, which does not contain FoxO binding motifs, also did not show significant enrichment (fig. 2I). Taken together, transcriptional regulation of CXCR4 by insulin is mediated by PI3K, but not by MAPK signaling, and may be mediated by inhibition of FoxO1, which we show binds directly to a FoxO response element in the proximal Cxcr4 promoter.
CXCR4 is upregulated in mouse models of insulin resistance and type 2 diabetes
Since insulin limits expression of CXCR4 in endothelial cells we expected increased expression of endothelial CXCR4 in insulin resistance. We tested this assumption in two different mouse models of obesity and insulin resistance: in mice fed a high-fat diet and in db/db mice. We dissociated cardiac tissue by enzymatic digestion, sorted cells by fluorescence-activated cell sorting (FACS) to separate endothelial cells for analysis and analyzed gene expression in sorted cells by real-time PCR. Endothelial cells were separated efficiently since expression of Pecam1 mRNA, the gene encoding CD31, was at least 24-fold higher higher in CD31+ cells than CD31− cells (fig. 3A), whereas expression of Kdr, the gene encoding vascular endothelial growth factor receptor-2, was at least 22-fold higher (fig. 3B). Endothelial CXCR4 mRNA was 77% higher in mice fed a high-fat diet than in mice fed a low-fat diet (fig. 3C) and 37% higher in db/db mice than db/+ controls (fig. 3D).
Figure 3. Regulation of CXCR4 in mouse models of insulin resistance.

A-D, Myocardial cells were dissociated enzymatically and sorted by FACS. CD31 sorting was performed after gating on PI– CD45– cells. mRNA in sorted cell lysate was analyzed by real-time PCR. A-C, Analysis of cells sorted from mice fed a high-fat diet (Hi-fat) or control mice fed a a low-fat diet (Lo-fat). C, *, p=0.04. D, Cells sorted from db/db mice or their db/+ controls. *, p=0.05 (Mann-Whitney test).
Insulin regulates SDF-1 dependent myeloid cell adhesion to endothelial cells
Treating cultured MS1 endothelial cells with SDF-1α increased subsequent THP-1 myeloid cell adhesion (fig. 4A). Adhesion was significantly increased 1 hour after SDF-1 treatment and was sustained after 2 through 8 hours (fig. 4A). Although we washed endothelial cells extensively after treatment with SDF-1, before adding myeloid cells to the endothelial cell monolayers, it is conceivable that residual SDF-1 could activate CXCR4 on myeloid cells. We therefore deleted the Cxcr4 gene in MS1 endothelial cells with CRISPR/Cas9 using 2 different gRNAs targeting distinct sequences in the Cxcr4 gene (Δ1 and Δ2 cell line). Almost all cells (99.4%) in control cultures expressed CXCR4 while in cultures with deletion of the Cxcr4 gene very few did (1.7% and 0.5% in the Δ1 and Δ2 cell line, respectively, fig. 4B). Downregulation of CXCR4 modestly changed expression of adhesion molecules, but not in the same direction. ICAM-1 was reduced by 54% and 51% in the Δ1 and Δ2 cell lines, respectively, compared to the control cells (fig. 4C & D). VCAM-1 was increased by 58% and 64%, respectively (fig. 4C & E). ICAM-1 or VCAM-1 did not change after treatment of cells with SDF-1 (fig. 4C-E). Myeloid cell adhesion increased by 42% (p=0.04) without SDF-1 treatment in the Δ1 endothelial cell line and by a non-significant 35% (p=0.20) in the Δ2 cell line (fig. 4F). Treatment of control cultures with SDF-1 increased subsequent myeloid cell adhesion by 2.5±0.5-fold but had no significant effect in the Δ1 and Δ2 cell lines (fig. 4F). These results show that increased myeloid cell adhesion to endothelial cells treated with SDF-1 is dependent on endothelial cell expression of CXCR4. In addition, they show that in quiescent endothelial cells in culture, loss of endothelial CXCR4 modestly changes ICAM-1 and VCAM-1 expression and causes a small increase in myeloid cell adhesion.
Figure 4. Endothelial insulin signaling regulates myeloid cell adhesion to endothelial cells induced by SDF-1.

A, Adhesion of THP-1 myeloid cells to MS1 endothelial cells. Endothelial cells were treated with 100 ng/ml SDF-1 for the time indicated or left untreated, then washed before adding THP-1 cells, labeled with Vybrant CFDA SE, for 30 minutes. Non-adhering cells were washed away. B-F, The Cxcr4 gene was targeted by CRISPR/Cas9 in MS1 cells using two independent gRNAs (Cxcr4Δ1 and Cxcr4Δ2, respectively, abbreviated Δ1 and Δ2) and compared to wildtype cells (wt) cells. B, Fraction of CXCR4+ cells, measured by flow cytometry. C-F, Cells were treated with 100 ng/ml SDF-1 for 4 hours. C, Representative Western blots. D, ICAM-1 relative to actin densitometry from 5 independent experiments. a, p=0.06; b, p=0.06 (Wilcoxon signed-rank test). E, Western blot results from 6 independent experiments. a, p=0.02 and b, p=0.04. F-G, Adhesion of THP-1 myeloid cells to MS1 endothelial cells as in A. F, Results from 5-6 independent experiments using the wt, Δ1 and Δ2 cell lines. *, p=0.01. G, Serum-starved, “wildtype” MS1 endothelial cells were treated with 100 nM insulin for 12 hours or left untreated, then treated with 100 ng/ml SDF-1 for 4 hours or left untreated, with or without AMD3100, an inhibitor of CXCR4. Results from 4 independent experiments. a, p=0.007; b, p=0.01.
Since insulin downregulates endothelial CXCR4, we then studied how insulin affected myeloid adhesion to endothelial cells treated with SDF-1. Adding insulin to endothelial cells 12 hours prior to addition of SDF-1α prevented 80% of the SDF-1-induced increase in myeloid cell adhesion (fig. 4G and Supplemental fig. S1). SDF-1 had no effect after treating endothelial cells with AMD3100, a small-molecule inhibitor of CXCR4 known in clinical medicine as plerixafor (fig. 4G and S1).
Endothelial cell IRS-1 regulates leukocyte/endothelial cell interaction in vivo
We next wished to study how CXCR4, SDF-1 and insulin/IGF-1 signaling influences leukocyte/endothelial cell interaction in vivo by observing leukocyte interaction with endothelium of mesenteric venules during intravital microscopy in mice.
We first used Cxcr4iΔEC mice40 with deletion of CXCR4 targeted to endothelial cells. CXCR4 deletion in these mice was efficient as evidenced by expression of CXCR4 in primary cultures of endothelial cells cultured from these animals, measured by flow cytometry (4.2% CXCR4+ cells in in cultures from Cxcr4iΔEC mice compared to 34.4% in cultures from control animals, fig. 5A). Leukocyte rolling on mesenteric venules was similar in mice with or without endothelial cell CXCR4 knockout (fig. 5B). Therefore, the modest change in expression of adhesion molecules and adhesion of myeloid cells in cultured cells after deletion of CXCR4 (fig. 4C-F) is not paralleled by changes in leukocyte/endothelial cell interaction after deletion of CXCR4 in vivo, at least not in the vascular bed of mesenteric venules.
Figure 5. Endothelial insulin signaling regulates leukocyte-endothelial cell interaction in vivo.

A-B, Mice with deletion of CXCR4 targeted to endothelial cells (cre Cxcr4lox/lox mice abbreviated Cxcr4iΔEC) were compared to Cxcr4lox/lox mice (flox). A, The fraction of CXCR4+ cells in primary cultures of lung endothelial cells was measured by flow cytometry. B, Intravital microscopy of leukocyte interaction with mesenteric venules was performed in 3-4 week old mice. The frequency of rolling leukocytes in 7 pair of animals was analyzed. C-D, Tg(Irs1) (“Tg”) mice, with overexpression of IRS-1 in endothelial cells, or their wildtype controls wildtype (“wt”) were studied. C, The fraction of CXCR4+ cells in primary cultures of lung endothelial cells was measured by flow cytometry. A representative result is shown. D, Flow cytometry was performed in 3 wt mice and 3 Tg mice. *, p=0.008. E-G, 3-4 week old wildtype (“wt”) or Tg(Irs1) (“Tg”) mice were studied 4 hours after intravenous injection of 1 μg SDF-1α or saline. E, Leukocyte interaction with mesenteric venules was observed during intravital microscopy. Arrows point to rolling leukocytes. Scalebar, 50 μm. F, Quantification of leukocyte rolling. *, p=0.03 (Mann-Whitney test).
To study the interaction of SDF-1 action and endothelial cell insulin/IGF-1 signaling in vivo, we observed leukocyte rolling and adhesion during intravital microscopy in mice with overexpression of IRS-1 in vascular endothelial cells under control of the vascular endothelial-cadherin (VE-cadherin or Cdh5) promoter (Tg(Irs1) mice). These mice have been characterized previously41,42. In primary lung endothelial cells isolated from wildtype or Tg(Irs1) mice, the fraction of CXCR4+ cells was 69±13% and 33±2%, respectively (fig. 5C-D, p=0.008). No difference in CXCR4+ cells was observed in CD11b+ Ly-6G+ neutrophils or CD11b+ CD115+ monocytes (fig. S2). Among CD11b+ Ly-6G+ cells (marking neutrophils), 99.8 and 99.7% were CXCR4+, respectively (fig. S2, p=0.50) with the median intensity of the CXCR4 label among CXCR4+ cells in Tg(Irs1) mice at 106% of the level in wildtype controls (data not shown, p=0.74). In CD11b+ CD115+ cells (marking monocytes), 44.6 and 39.3% were CXCR4+, respectively (fig. S2 , p=0.29).
We used these mice to observe leukocyte interaction with mesenteric venules 4 hours after intravenous injection of SDF-1α. In wild-type mice, injection of SDF-1α increased leukocyte rolling by 3.1-fold (fig. 5E-F). However, this increase in rolling was mostly prevented in Tg(Irs1) mice (fig. 5E-F). These observations show that SDF-1 potently increases peripheral leukocyte adhesion in vivo and that this effect is largely prevented by gain of endothelial cell insulin/IGF-1 signaling, likely due to the effect of these hormones on CXCR4 expression.
Discussion
Low-grade chronic inflammation is an important component in the development of insulin resistance, type 2 diabetes and cardiovascular disease43,44. Insulin resistance in endothelial cells causes an activated, pro-adhesive endothelium which may contribute to increased immune cell recruitment to the vascular wall and surrounding tissue. In the current study, we have identified CXCR4 as an insulin-regulated gene and described a new mechanism for increased leukocyte interaction with insulin resistant endothelial cells, caused by upregulation of endothelial CXCR4. The availability of clinically safe antagonists of CXCR4 readily gives this finding translational value.
Endothelial dysfunction is associated with a range of cardiovascular risk factors. Insulin resistance in endothelial cells represents a cause of endothelial dysfunction that is uniquely pertinent to obesity and type 2 diabetes. In humans, insulin resistance in endothelium can be demonstrated as impaired insulin-stimulated vasodilation21, which is dependent on endothelium-derived NO45, or an impaired effect of insulin to potentiate endothelial-dependent vasodilation stimulated with acetylcholine or its analogues22,23. It can also be demonstrated as impaired insulin signaling and eNOS phosphorylation in endothelial cells24. However, insulin action on endothelial cells regulates a range of signaling targets which are only beginning to be characterized. For example, we have shown that insulin, through suppression of FoxO activity, regulates several genes involved in angiogenesis, including the transcriptional repressor CBP/p300-interacting transactivator with ED-rich tail 2 (CITED2)39. The current study identified a range of novel transcriptional targets of insulin signaling and we characterized the functional consequence of CXCR4 regulation by insulin.
CXCR4 is a G-protein-coupled chemokine receptor and its main ligand is SDF-1. It is expressed in many cell types but its function is best characterized in hematopoietic stem and progenitor cells (HSPC) and immune cells. HSPC are retained in bone marrow niches by responding to SDF-1 released by endothelial or perivascular cells46,47. Consequently, HSPC are mobilized from the bone marrow by CXCR4 antagonists, including AMD3100, which block the chemotactic signal induced by SDF-1 in HSPC48. Homing of HSPC to peripheral tissues expressing SDF-1 is also mediated by CXCR4 expressed by circulating HSPC49. CXCR4 also regulates homing of differentiated immune cells. This is perhaps best described for neutrophils for which CXCR4 inhibits mobilization from the bone marrow50 or from a neutrophil pool in the lungs51 whereas senescent neutrophils are cleared in the bone marrow after upregulation of CXCR452.
CXCR4 expressed by endothelial cells is critical for vasculogenesis during development, including in the intestine28,29, coronary arteries30,31 and renal vasculature32. CXCR4 also mediates neoangiogenesis, including tumor angiogenesis 33 or coronary collateral formation in response to injury34. The phenotype resulting from deletion of SDF-1 has been described as very similar to endothelial deletion of CXCR4 in vasculogenesis and CXCR4 likely works as a mediator of chemotactic signals in this context. The mechanism by which endothelial CXCR4 confers a pro-adhesive function to endothelium may be quite different since, unlike mobile hematopoietic cells, quiescent endothelial cells reside in a fixed position on the basal membrane.
We show that SDF-1 can act on endothelial cells in a CXCR4-dependent manner and increase myeloid cell adhesion to cultured endothelial cells and to endothelium of mesenteric venules in vivo. In turn, insulin decreases SDF-1-stimulated leukocyte interaction with endothelium by downregulating endothelial cell CXCR4. Although deletion of CXCR4 in endothelial cell culture modestly perturbed expression of ICAM-1 and VCAM-1 and caused a small increase in baseline adhesion of myeloid cells, we found no change in leukocyte adhesion to mesenteric venules in mice with endothelial cell deletion of CXCR4. To our knowledge, SDF-1-induced leukocyte adhesion through endothelial CXCR4 has not been described previously. Important work by Christian Weber’s group showed that deletion of CXCR4 in arterial endothelial cells in Apoe null mice caused increased adhesion of myeloid cells to the aorta after topical application of TNF-α53. This mechanism, elegantly described by Weber et al., involved endothelial damage with breakdown of endothelial barrier function53. It is therefore possible that endothelial cell CXCR4 affects leukocyte adhesion differently in quiescent versus activated endothelium, in arterial versus venous endothelium, or in endothelium responding to SDF-1. We plan to use Cxcr4iΔEC mice to study the role of CXCR4-dependent signaling in vascular endothelium in tissue affected by inflammation and hypoxia with the expected induction of inflammatory cytokines and SDF-1 in these conditions.
Our study identifies endothelial CXCR4 as an insulin-regulated receptor that mediates immune cell adhesion to endothelium and has found that CXCR4 is upregulated in mouse models of obesity or type 2 diabetes, as would be expected when downregulation by insulin is impaired by insulin resistance. Future research should seek to determine whether this translates into homing of immune cells to the vascular wall or tissue parenchyma, whether this mechanism preferentially operates in certain vascular beds and whether it primarily affects certain types of immune cells. Results from such studies could provide a clear rationale to block CXCR4 in order to reduce complications of obesity and type 2 diabetes. Several CXCR4 antagonists are in clinical trial54,55, including AMD3100 (plerixafor) which is approved for routine clinical use, so the translational value of this principle is readily testable. More generally, future studies will establish how improving insulin resistance in endothelial cells or targeting insulin-regulated molecules may help prevent complications in metabolic disease where insulin resistance is a central feature.
Supplementary Material
Higlights.
Insulin downregulates the chemokine receptor CXCR4 in vascular endothelial cells
Endothelial CXCR4 is upregulated in animal models of insulin resistance
The CXCR4 ligand SDF-1 increases myeloid cell adhesion to endothelial cells through endothelial CXCR4
Sources of Funding
This work was supported by NIH grant R21CA185196 (to CRM) and NIH grant R01DK053105 (to GKL). BK was a recipient of the NIH training grant T32DK007260. CRM also recieved research support from the Novo Nordisk R & D Science Talent Attraction and Recruitment (STAR) Programme. Flow cytometry and real-time PCR was done using cores in the Diabetes Research Center, supported by NIH grant 5P30DK036836 and S10OD021740, at Joslin Diabetes Center. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Nonstandard Abbreviations and Acronyms
- ChIP
chromatin immunoprecipitation
- CITED2
CBP/p300-interacting transactivator with ED-rich tail 2
- CXCR4
C-X-C motif chemokine receptor 4
- HUVEC
human umbilical vein endothelial cells
- IGF-1
insulin-like growth factor-1
- MadCAM-1
mucosal vascular addressin cell adhesion molecule-1
- NO
nitric oxide
- PI3K
phosphatidylinositol 3-kinase
- SDF-1
stromal derived factor-1
- VCAM-1
vascular cell adhesion molecule-1
Footnotes
Disclosures
Thomas Rathjen was an employee of Novo Nordisk, Inc. as a participant in the STAR Programme (see above) for postdoc training. He is currently an employee of Bayer AG. Hanni Willenbrock, Grith Skytte Olsen, Gro Klitgaard Povlsen are employees of Novo Nordisk, Inc. Christian Rask-Madsen is currently on a leave of absence from Joslin Diabetes Center and an employee of Sanofi, Inc. The remaining authors disclose no financial, professional, or personal conflict of interest.
References
- 1.Rask-Madsen C, King GL. Vascular complications of diabetes: mechanisms of injury and protective factors. Cell Metab. 2013;17:20–33. doi: 10.1016/j.cmet.2012.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nageh MF, Sandberg ET, Marotti KR, Lin AH, Melchior EP, Bullard DC, Beaudet AL. Deficiency of inflammatory cell adhesion molecules protects against atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 1997;17:1517–1520. [DOI] [PubMed] [Google Scholar]
- 3.Huo Y, Hafezi-Moghadam A, Ley K. Role of vascular cell adhesion molecule-1 and fibronectin connecting segment-1 in monocyte rolling and adhesion on early atherosclerotic lesions. Circ Res. 2000;87:153–159. [DOI] [PubMed] [Google Scholar]
- 4.Dong ZM, Brown AA, Wagner DD. Prominent role of P-selectin in the development of advanced atherosclerosis in ApoE-deficient mice. Circulation. 2000;101:2290–2295. [DOI] [PubMed] [Google Scholar]
- 5.Collins RG, Velji R, Guevara NV, Hicks MJ, Chan L, Beaudet AL. P-Selectin or intercellular adhesion molecule (ICAM)-1 deficiency substantially protects against atherosclerosis in apolipoprotein E-deficient mice. J Exp Med. 2000;191:189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW, Milstone DS. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107:1255–1262. doi: 10.1172/JCI11871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galkina E, Ley K. Leukocyte recruitment and vascular injury in diabetic nephropathy. J Am Soc Nephrol. 2006;17:368–377. [DOI] [PubMed] [Google Scholar]
- 8.Pichler R, Afkarian M, Dieter BP, Tuttle KR. Immunity and inflammation in diabetic kidney disease: translating mechanisms to biomarkers and treatment targets. Am J Physiol Renal Physiol. 2017;312:F716–F731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joussen AM, Poulaki V, Le ML, Koizumi K, Esser C, Janicki H, Schraermeyer U, Kociok N, Fauser S, Kirchhof B, Kern TS, Adamis AP. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004;18:1450–1452. [DOI] [PubMed] [Google Scholar]
- 10.Roy S, Kern TS, Song B, Stuebe C. Mechanistic Insights into Pathological Changes in the Diabetic Retina: Implications for Targeting Diabetic Retinopathy. Am J Pathol. 2017;187:9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y, Ofrecio J, Lin M, Brenner MB, Olefsky JM. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med. 2012;18:1407–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chataway J, Miller DH. Natalizumab therapy for multiple sclerosis. Neurotherapeutics. 2013;10:19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Targan SR, Feagan BG, Fedorak RN, Lashner BA, Panaccione R, Present DH, Spehlmann ME, Rutgeerts PJ, Tulassay Z, Volfova M, Wolf DC, Hernandez C, Bornstein J, Sandborn WJ. Natalizumab for the treatment of active Crohn's disease: results of the ENCORE Trial. Gastroenterology. 2007;132:1672–1683. [DOI] [PubMed] [Google Scholar]
- 15.Rask-Madsen C, Li Q, Freund B, et al. Loss of insulin signaling in vascular endothelial cells accelerates atherosclerosis in apolipoprotein E null mice. Cell Metab. 2010;11:379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Haring MF, Rathjen T, Lockhart SM, Sorensen D, Ussar S, Rasmussen LM, Bertagnolli MM, Kahn CR, Rask-Madsen C. Insulin resistance in vascular endothelial cells promotes intestinal tumour formation. Oncogene. 2017;36:4987–4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang ZY, Lin YW, Clemont A, Feener EP, Hein KD, Igarashi M, Yamauchi T, White MF, King GL. Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats. J Clin Invest. 1999;104:447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kubota T, Kubota N, Kumagai H, et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab. 2011;13:294–307. [DOI] [PubMed] [Google Scholar]
- 19.Kim F, Pham M, Maloney E, Rizzo NO, Morton GJ, Wisse BE, Kirk EA, Chait A, Schwartz MW. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler Thromb Vasc Biol. 2008;28:1982–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao L, Fu Z, Wu J, Aylor KW, Barrett EJ, Cao W, Liu Z. Inflammation-induced microvascular insulin resistance is an early event in diet-induced obesity. Clin Sci (Lond). 2015;129:1025–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laakso M, Edelman SV, Brechtel G, Baron AD. Impaired insulin-mediated skeletal muscle blood flow in patients with NIDDM. Diabetes. 1992;41:1076–1083. [DOI] [PubMed] [Google Scholar]
- 22.Steinberg HO, Chaker H, Leaming R, Johnson A, Brechtel G, Baron AD. Obesity/insulin resistance is associated with endothelial dysfunction. Implications for the syndrome of insulin resistance. J Clin Invest. 1996;97:2601–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rask-Madsen C, Ihlemann N, Krarup T, Christiansen E, Kober L, Nervil Kistorp C, Torp-Pedersen C. Insulin therapy improves insulin-stimulated endothelial function in patients with type 2 diabetes and ischemic heart disease. Diabetes. 2001;50:2611–2618. doi: 10.2337/diabetes.50.11.2611 [DOI] [PubMed] [Google Scholar]
- 24.Tabit CE, Shenouda SM, Holbrook M, et al. Protein kinase C-beta contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Circulation. 2013;127:86–95. doi: 10.1161/CIRCULATIONAHA.112.127514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA Jr., Shin WS, Liao JK. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96:60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuroda Y, Kuriyama H, Kihara S, Kishida K, Maeda N, Hibuse T, Nishizawa H, Matsuda M, Funahashi T, Shimomura I. Insulin-mediated regulation of decidual protein induced by progesterone (DEPP) in adipose tissue and liver. Hormone and metabolic research = Hormonund Stoffwechselforschung = Hormones et metabolisme. 2010;42:173–177. [DOI] [PubMed] [Google Scholar]
- 27.Chen S, Gai J, Wang Y, Li H. FoxO regulates expression of decidual protein induced by progesterone (DEPP) in human endothelial cells. FEBS Lett. 2011;585:1796–1800. doi: 10.1016/j.febslet.2011.04.024 [DOI] [PubMed] [Google Scholar]
- 28.Tachibana K, Hirota S, Iizasa H, Yoshida H, Kawabata K, Kataoka Y, Kitamura Y, Matsushima K, Yoshida N, Nishikawa S, Kishimoto T, Nagasawa T. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature. 1998;393:591–594. [DOI] [PubMed] [Google Scholar]
- 29.Ara T, Tokoyoda K, Okamoto R, Koni PA, Nagasawa T. The role of CXCL12 in the organspecific process of artery formation. Blood. 2005;105:3155–3161. [DOI] [PubMed] [Google Scholar]
- 30.Harrison MR, Bussmann J, Huang Y, Zhao L, Osorio A, Burns CG, Burns CE, Sucov HM, Siekmann AF, Lien CL. Chemokine-guided angiogenesis directs coronary vasculature formation in zebrafish. Dev Cell. 2015;33:442–454. doi: 10.1016/j.devcel.2015.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ivins S, Chappell J, Vernay B, Suntharalingham J, Martineau A, Mohun TJ, Scambler PJ. The CXCL12/CXCR4 Axis Plays a Critical Role in Coronary Artery Development. Dev Cell. 2015;33:455–468. doi: 10.1016/j.devcel.2015.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takabatake Y, Sugiyama T, Kohara H, Matsusaka T, Kurihara H, Koni PA, Nagasawa Y, Hamano T, Matsui I, Kawada N, Imai E, Nagasawa T, Rakugi H, Isaka Y. The CXCL12 (SDF-1)/CXCR4 axis is essential for the development of renal vasculature. J Am Soc Nephrol. 2009;20:1714–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guleng B, Tateishi K, Ohta M, et al. Blockade of the stromal cell-derived factor-1/CXCR4 axis attenuates in vivo tumor growth by inhibiting angiogenesis in a vascular endothelial growth factor-independent manner. Cancer Res. 2005;65:5864–5871. doi: 10.1158/0008-5472.CAN-04-3833 [DOI] [PubMed] [Google Scholar]
- 34.Das S, Goldstone AB, Wang H, et al. A Unique Collateral Artery Development Program Promotes Neonatal Heart Regeneration. Cell. 2019;176:1128–1142 e1118. doi: 10.1016/j.cell.2018.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noels H, Zhou B, Tilstam PV, et al. Deficiency of endothelial CXCR4 reduces reendothelialization and enhances neointimal hyperplasia after vascular injury in atherosclerosis-prone mice. Arterioscler Thromb Vasc Biol. 2014;34:1209–1220. [DOI] [PubMed] [Google Scholar]
- 36.Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature. 2003;425:307–311. doi: 10.1038/nature01874 [DOI] [PubMed] [Google Scholar]
- 37.Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, Bernasconi S, Saccani S, Nebuloni M, Vago L, Mantovani A, Melillo G, Sica A. Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med. 2003;198:1391–1402. doi: 10.1084/jem.20030267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gupta SK, Lysko PG, Pillarisetti K, Ohlstein E, Stadel JM. Chemokine receptors in human endothelial cells. Functional expression of CXCR4 and its transcriptional regulation by inflammatory cytokines. J Biol Chem. 1998;273:4282–4287. [DOI] [PubMed] [Google Scholar]
- 39.Wang X, Lockhart SM, Rathjen T, Albadawi H, Sorensen D, O'Neill BT, Dwivedi N, Preil SR, Beck HC, Dunwoodie SL, Watkins MT, Rasmussen LM, Rask-Madsen C. Insulin downregulates the transcriptional coregulator CITED2, an inhibitor of proangiogenic function in endothelial cells. Diabetes. 2016;65:3680–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ding BS, Cao Z, Lis R, Nolan DJ, Guo P, Simons M, Penfold ME, Shido K, Rabbany SY, Rafii S. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature. 2014;505:97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park K, Mima A, Li Q, et al. Insulin decreases atherosclerosis by inducing endothelin receptor B expression. JCI Insight. 2016;1. doi: 10.1172/jci.insight.86574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Katagiri S, Park K, Maeda Y, Rao TN, Khamaisi M, Li Q, Yokomizo H, Mima A, Lancerotto L, Wagers A, Orgill DP, King GL. Overexpressing IRS1 in endothelial cells enhances angioblast differentiation and wound healing in diabetes and insulin resistance. Diabetes. 2016;65:2760–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goldfine AB, Shoelson SE. Therapeutic approaches targeting inflammation for diabetes and associated cardiovascular risk. J Clin Invest. 2017;127:83–93. doi: 10.1172/JCI88884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Libby P Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045–2051. doi: 10.1161/ATVBAHA.108.179705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steinberg HO, Brechtel G, Johnson A, Fineberg N, Baron AD. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. J Clin Invest. 1994;94:1172–1179. doi: 10.1172/JCI117433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crane GM, Jeffery E, Morrison SJ. Adult haematopoietic stem cell niches. Nat Rev Immunol. 2017;17:573–590. doi: 10.1038/nri.2017.53 [DOI] [PubMed] [Google Scholar]
- 47.Hoggatt J, Kfoury Y, Scadden DT. Hematopoietic Stem Cell Niche in Health and Disease. Annual review of pathology. 2016;11:555–581. doi: 10.1146/annurev-pathol-012615-044414 [DOI] [PubMed] [Google Scholar]
- 48.Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, Liles WC, Li X, Graham-Evans B, Campbell TB, Calandra G, Bridger G, Dale DC, Srour EF. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. 2005;201:1307–1318. doi: 10.1084/jem.20041385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng M, Huang K, Zhou J, Yan D, Tang YL, Zhao TC, Miller RJ, Kishore R, Losordo DW, Qin G. A critical role of Src family kinase in SDF-1/CXCR4-mediated bone-marrow progenitor cell recruitment to the ischemic heart. J Mol Cell Cardiol. 2015;81:49–53. doi: 10.1016/j.yjmcc.2015.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eash KJ, Means JM, White DW, Link DC. CXCR4 is a key regulator of neutrophil release from the bone marrow under basal and stress granulopoiesis conditions. Blood. 2009;113:4711–4719. doi: 10.1182/blood-2008-09-177287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Devi S, Wang Y, Chew WK, et al. Neutrophil mobilization via plerixafor-mediated CXCR4 inhibition arises from lung demargination and blockade of neutrophil homing to the bone marrow. J Exp Med. 2013;210:2321–2336. doi: 10.1084/jem.20130056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang J, Hossain M, Thanabalasuriar A, Gunzer M, Meininger C, Kubes P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science. 2017;358:111–116. doi: 10.1126/science.aam9690 [DOI] [PubMed] [Google Scholar]
- 53.Doring Y, Noels H, van der Vorst EPC, et al. Vascular CXCR4 Limits Atherosclerosis by Maintaining Arterial Integrity: Evidence From Mouse and Human Studies. Circulation. 2017;136:388–403. doi: 10.1161/CIRCULATIONAHA.117.027646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duda DG, Kozin SV, Kirkpatrick ND, Xu L, Fukumura D, Jain RK. CXCL12 (SDF1alpha)-CXCR4/CXCR7 pathway inhibition: an emerging sensitizer for anticancer therapies? Clin Cancer Res. 2011;17:2074–2080. doi: 10.1158/1078-0432.CCR-10-2636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scala S. Molecular Pathways: Targeting the CXCR4-CXCL12 Axis--Untapped Potential in the Tumor Microenvironment. Clin Cancer Res. 2015;21:4278–4285. doi: 10.1158/1078-0432.CCR-14-0914 [DOI] [PubMed] [Google Scholar]
- 56.Wang X, Lockhart SM, Rathjen T, Albadawi H, Sorensen D, O'Neill BT, Dwivedi N, Preil SR, Beck HC, Dunwoodie SL, Watkins MT, Rasmussen LM, Rask-Madsen C. Insulin Downregulates the Transcriptional Coregulator CITED2, an Inhibitor of Proangiogenic Function in Endothelial Cells. Diabetes. 2016;65:3680–3690. doi: 10.2337/db16-0001 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.