

Abstract
The metabolism of acetone was investigated in the actinomycete Rhodococcus rhodochrous (formerly Nocardia corallina) B276. Suspensions of acetone- and isopropanol-grown R. rhodochrous readily metabolized acetone. In contrast, R. rhodochrous cells cultured with glucose as the carbon source lacked the ability to metabolize acetone at the onset of the assay but gained the ability to do so in a time-dependent fashion. Chloramphenicol and rifampin prevented the time-dependent increase in this activity. Acetone metabolism by R. rhodochrous was CO2 dependent, and 14CO2 fixation occurred concomitant with this process. A nucleotide-dependent acetone carboxylase was partially purified from cell extracts of acetone-grown R. rhodochrous by DEAE-Sepharose chromatography. Analysis by sodium dodecyl sulfate-polyacrylamide gel electrophoresis suggested that the acetone carboxylase was composed of three subunits with apparent molecular masses of 85, 74, and 16 kDa. Acetone metabolism by the partially purified enzyme was dependent on the presence of a divalent metal and a nucleoside triphosphate. GTP and ITP supported the highest rates of acetone carboxylation, while CTP, UTP, and XTP supported carboxylation at 10 to 50% of these rates. ATP did not support acetone carboxylation. Acetoacetate was determined to be the stoichiometric product of acetone carboxylation. The longer-chain ketones butanone, 2-pentanone, 3-pentanone, and 2-hexanone were substrates. This work has identified an acetone carboxylase with a novel nucleotide usage and broader substrate specificity compared to other such enzymes studied to date. These results strengthen the proposal that carboxylation is a common strategy used for acetone catabolism in aerobic acetone-oxidizing bacteria.
A variety of bacteria are capable of utilizing acetone as a growth-supporting substrate. Studies involving these bacteria have provided evidence leading to several proposed pathways for bacterial acetone metabolism (for a recent review, see reference 10). Collectively, these pathways have considered the initial conversion of acetone to occur by one of two strategies. For most aerobic bacteria, the initial step in acetone metabolism has been proposed to be catalyzed by an O2-dependent acetone monooxygenase to produce hydroxyacetone, although no acetone monooxygenase activity has been demonstrated in vitro (9, 16, 26, 27). For facultatively or strictly anaerobic bacteria, the initial step in acetone metabolism has been proposed to occur via a CO2-dependent carboxylation forming acetoacetate or an acetoacetyl derivative (7, 12, 14, 18–20, 22). Recently, a CO2-dependent pathway of acetone metabolism was identified in Xanthobacter strain Py2, an obligately aerobic bacterium (24). Thus, at present it is ambiguous which strategy is predominant in aerobic acetone-metabolizing bacteria. In light of this, it was of interest to investigate acetone metabolism in an obligately aerobic bacterium phylogenetically distinct from Xanthobacter strain Py2.
Rhodococcus rhodochrous (formerly Nocardia corallina) B-276 is a gram-positive obligate aerobe that is capable of growth on a variety of carbon sources, including alkanes and alkenes (11). In the present study, R. rhodochrous is reported for the first time to utilize acetone as a growth-supporting substrate. Evidence is presented that acetone is metabolized by R. rhodochrous in a CO2-dependent manner by a nucleotide-dependent acetone carboxylase. This enzyme appears to catalyze a reaction identical with that of the Xanthobacter strain Py2 and Rhodobacter capsulatus acetone carboxylases, previously studied in the purified and partially purified forms, respectively (6, 23). However, distinct differences are apparent, most notably in the nucleotide usage and substrate specificity of the R. rhodochrous acetone carboxylase. Presented here is an initial description of both the in vivo and in vitro metabolism of acetone in R. rhodochrous. This work is of interest because it provides further evidence that carboxylation may be a significant strategy for acetone metabolism by aerobic bacteria. Additionally, the demonstration of novel in vitro requirements by the R. rhodochrous acetone carboxylase helps to expand the very limited knowledge of bacterial acetone-degrading proteins and their catalytic requirements.
MATERIALS AND METHODS
Chemicals.
Nucleotides and antifoam 289 were purchased from Sigma Chemicals. NaH13CO3 (99% 13C atom) and all volatile organic compounds were purchased from Aldrich Chemicals. NaH14CO3 (specific activity, 54.4 mCi of 14C mmol−1) was purchased from ICN Radiochemicals, Irvine, Calif. Ascarite II was purchased from Thomas Scientific, Swedesboro, N.J. All other chemicals used were of analytical grade.
Bacteria and growth conditions.
Cultures of R. rhodochrous B276 (ATCC 31338) were grown at 30°C in either shake flasks or a carboy. The growth medium was a mineral salts medium (28) in which NaNO3 (2 g/liter) was substituted for NH4Cl as the primary nitrogen source. The carbon sources for growth were either glucose (1% [wt/vol]), isopropanol (40 mM), or acetone (40 mM). Shake flask cells were harvested by centrifugation (8,600 × g), resuspended into 50 mM phosphate buffer (pH 7.2), and frozen at −80°C for storage. Batch fermentation was carried out in a 45-liter glass carboy with forced aeration, containing 39 liters of minimal salts medium, antifoam 289 (0.1% [vol/vol]), and acetone (40 mM). Five liters of 48-h R. rhodochrous shake flask cultures grown on identical media served as the fermentor inoculum. Air was replenished at 24-h intervals, and acetone levels were monitored by gas chromatography. After reaching an A600 of approximately 2.5, cells were harvested by tangential-flow filtration with a Pellicon system (Millipore Corp.) followed by centrifugation (8,600 × g). Cell paste was drop frozen in liquid nitrogen and stored at −80°C.
Gas chromatography.
Gas chromatography (flame ionization detector) was performed by using a Shimadzu GC-8A interfaced with a Shimadzu CR601 integrator. Assays using 9- or 3-ml sealed serum vials involved injection of 100- or 30-μl gas-phase samples, respectively. Unless otherwise indicated, N2 was used as a carrier gas, the column packing material consisted of Porapak Q, and the injector temperature was 200°C. Conditions for assaying the following individual compounds are given in parentheses after the compound name(s): acetone (200 kPa; 0.3- by 49.5-cm column at 130°C), 2-butanone (100 kPa; 0.3- by 20.5-cm column at 130°C), 2-pentanone and 3-pentanone (100 kPa; 0.3- by 49.5-cm column at 150°C), and 2-hexanone (100 kPa; 0.3- by 49.5-cm column at 160°C). Liquid-phase sampling was performed for acetoacetate quantification by using the following parameters: carrier gas at 200 kPa, a 0.3- by 49.5-cm column at 120°C, and an injector temperature of 230°C.
Induction of acetone-metabolizing activity.
R. rhodochrous cultures which had been grown for several generations on either glucose, isopropanol, or acetone were used to inoculate shake flasks with the respective carbon source. After reaching A600 values ranging from 1.25 to 1.96, cells were harvested by centrifugation (7,800 × g). Cell pellets were washed twice with 50 mM phosphate buffer (pH 7.2) and resuspended in the same buffer. Aliquots of these cell suspensions were added to sterile minimal salts medium with or without rifampin (0.2 mg ml−1) and chloramphenicol (0.4 mg ml−1). Assays were performed in 9-ml sealed serum vials with shaking (200 cycles min−1) in a 30°C water bath. Total volumes were 1 ml. Following a 10-min incubation, acetone was added, and the time-dependent consumption of acetone was monitored by gas chromatography.
CO2-dependent acetone metabolism in whole-cell suspensions of acetone-grown R. rhodochrous.
Assays were performed in 9-ml sealed serum vials with shaking (200 cycles min−1) in a 30°C water bath. All vials contained cell suspensions of acetone-grown R. rhodochrous in 50 mM phosphate buffer (pH 7.5). Vials enriched with carbonate species contained 4.5 mM NaHCO3 plus 5.5 mM CO2 gas. Vials were depleted of carbonate species by one of two CO2-absorbing traps: KOH or ascarite. Vials containing KOH traps used a cutoff 1.5-ml microcentrifuge tube with a Whatman glass microfiber filter (diameter, 1.8 cm) inserted into it and wetted with 200 μl of 6 M KOH. Four hundred microliters of 12 M HClO4 was added to the KOH trap at the indicated time in order to acidify the solution and thereby liberate CO2 trapped as K2CO3. Vials containing ascarite traps used a cutoff 1.5-ml microcentrifuge tube with 800 μl of ascarite II covered by loose cotton. All assays contained total volumes of 1 ml and were allowed to equilibrate 15 min prior to acetone addition. The time-dependent consumption of acetone was monitored by gas chromatography.
14C labeling of cell suspensions.
Assays were performed in 9-ml sealed serum vials with shaking (200 cycles min−1) in a 30°C water bath. Vials contained acetone-grown R. rhodochrous in 50 mM phosphate buffer (pH 7.2) with 50 mM carbonate species (added as 27.5 mM CO2 gas plus 22.5 mM NaHCO3 containing 59 μCi of 14C mmol−1) in a total volume of 1 ml. After incubation of the vials for 2 min, assays were initiated by addition of an organic substrate (glucose, acetone, propionaldehyde, or propylene oxide). At desired times, 25-μl liquid samples were removed and added to 200 μl of an ethanol-acetic acid mixture (95:5 [vol/vol]) in 500-μl microcentrifuge tubes. The quenched reaction mixtures were then dried at 50°C under a vacuum (0.67 kPa) overnight. Two hundred microliters of H2O was added to the dried samples, followed by incubation at room temperature for 30 min to solubilize dried material. Samples were then placed into scintillation vials (containing 10 ml of scintillation fluid), vortexed, and allowed to stand 20 min. The radioactivity of the samples was measured with a Beckman LS 6000 scintillation counter and compared to a standard curve relating disintegrations per minute to nanomoles of CO2 fixed, as described previously (24).
Partial purification of acetone carboxylase.
All steps were performed at 4°C unless otherwise indicated. Frozen cell paste of fermentor-grown R. rhodochrous was thawed at room temperature and resuspended in 50 mM phosphate buffer (pH 7.2), and cells were then centrifuged (8,600 × g). A second wash was performed in the same manner to further remove residual antifoam. The washed cell pellet was resuspended in 1.5 volumes of buffer (50 mM Tris-HCl [pH 8.0] containing 1 mM dithiothreitol, lysozyme at 0.4 mg ml−1, and DNAase I at 0.03 mg ml−1). After a 30-min incubation at room temperature, the cell suspension was passed through a French pressure cell three times at 125,000 kPa. The resulting extract was initially clarified by low-speed centrifugation (7,800 × g for 10 min) followed by ultracentrifugation (140,000 × g for 1 h). This clarified supernatant was referred to as cell extract, which was drop frozen in liquid nitrogen and stored at −80°C. For further purification, cell extracts (125 ml) were thawed and applied (linear flow rate of 7.3 cm h−1) to a 2.5- by 20.5-cm column of DEAE-Sepharose equilibrated in 50 mM Tris-HCl (pH 8.0) containing 1 mM dithiothreitol and 10% (vol/vol) glycerol (buffer A). After loading, the column was washed with 350 ml of buffer A. Bound proteins were eluted (linear flow rate, 12.2 cm h−1) with a 625-ml linear gradient of 0 to 500 mM KCl in buffer A. Fractions containing acetone carboxylase activity were pooled and concentrated 5.5-fold by ultrafiltration over a YM30 membrane (molecular mass cutoff, 30 kDa). The concentrate (15 ml) was then dialyzed (molecular mass cutoff, 6 to 8 kDa) for 12 h against 6 liters of buffer A with stirring. The partially purified acetone carboxylase was stored at −80°C.
Acetone carboxylase assays.
Assays were performed in 3-ml sealed serum vials with shaking (200 cycles min−1) in a 30°C water bath. All vials contained the standard assay mixture: buffer (50 mM Tris-HCl, pH 8.0), MgCl2 (5 mM), NH4Cl (100 mM), and ketone (1.5 mM), in a total volume of 335 μl. Additional components were included as indicated. Substrate degradation was monitored by gas chromatography.
13C NMR identification of the product of acetone carboxylation.
Acetone degradation assays were performed with the partially purified R. rhodochrous acetone carboxylase after desalting on a Sephadex G-25M PD10 column (Pharmacia Biotech) to remove glycerol. Assays were performed at 30°C with shaking (200 cycles min−1) in sealed 9-ml serum vials containing buffer (50 mM Tris-HCl, pH 8.0), MgCl2 (5 mM), NH4Cl (100 mM), partially purified acetone carboxylase (4.1 mg), and acetone (2 mM) in a total volume of 1 ml. When GTP was present, its concentration in assays was 30 mM. After complete degradation of acetone (or after 1 h, for assays lacking GTP), assay mixtures were centrifuged (5,000 × g for 30 min) in Microcon-30 microconcentrators (molecular mass cutoff, 30 kDa; Amicon, Inc.). The filtrates were analyzed for the 13C-carboxylation product by using 13C nuclear magnetic resonance (NMR) as described previously (24).
Quantification of acetoacetate formed from acetone carboxylation.
Assays were performed in a 30°C water bath with shaking (200 cycles min−1) in sealed 3-ml serum vials containing buffer (50 mM Tris-HCl, pH 8.0), GTP (20 mM), MgCl2 (5 mM), NH4Cl (100 mM), glycerol (1.5% [vol/vol]), partially purified acetone carboxylase (1.03 mg), and acetone (1.5 mM) in a total volume of 335 μl. At desired times, 1 μl of liquid was removed from assay vials and analyzed for acetoacetate as described previously (1).
SDS-PAGE analysis.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (12% total gel; 2.7% cross-linker running gel) was performed in a Mini-Protean II apparatus (Bio-Rad) by the Laemmli procedure (15). Electrophoresed proteins were visualized by Coomassie blue staining. Apparent molecular masses of polypeptides were determined by comparison of Rf values to those for molecular mass protein standards. The standards were myosin (200 kDa), β-galactosidase (116.25 kDa), phosphorylase b (97.4 kDa), bovine serum albumin (66.2 kDa), ovalbumin (45 kDa), carbonic anhydrase (31 kDa), soybean trypsin inhibitor (21.5 kDa), and lysozyme (14.4 kDa).
Protein determination.
Protein concentrations were determined by a modified biuret assay (8). The partially purified acetone carboxylase and cell extracts used in SDS-PAGE were precipitated with trichloroacetic acid (5% [wt/vol]) before protein determination. For whole-cell protein determination, cells were lysed by the addition of 3 M NaOH (1 h at 65°C) before protein determination. Bovine serum albumin was used as the protein standard.
RESULTS
Induction of acetone-degrading activity in R. rhodochrous.
R. rhodochrous B276 is capable of growth with short-chain aliphatic alkanes and alkenes as carbon sources, including propane (11). Since other propane oxidizers are capable of growth on isopropanol or acetone (5, 9), we investigated the possibility that these compounds could be utilized as carbon and energy sources for R. rhodochrous (5). Both isopropanol and acetone were found to support the growth of R. rhodochrous, with exponential-phase doubling times of approximately 3 h.
In order to determine whether the genes encoding the acetone-degrading enzyme of R. rhodochrous are expressed constitutively or induced during a specific condition, the ability of R. rhodochrous to metabolize acetone was investigated after growth with various carbon sources. As shown in Fig. 1, acetone- and isopropanol-grown R. rhodochrous readily consumed acetone from the onset of exposure, and this activity was not prevented by the inclusion of chloramphenicol and rifampin, which are protein and RNA synthesis inhibitors, respectively. In contrast to these results, R. rhodochrous cells cultured with glucose as the carbon source lacked the ability to consume acetone at the onset of the assay but gained the ability to do so in a time-dependent fashion (Fig. 1). The addition of chloramphenicol and rifampin to glucose-grown cells prevented this time-dependent increase in acetone-consuming activity (Fig. 1). These results suggest that the acetone-metabolizing enzyme in R. rhodochrous is not constitutively synthesized but instead is synthesized in response to growth on either acetone or isopropanol.
FIG. 1.

Effect of growth substrate on acetone metabolism by R. rhodochrous B276. Closed symbols, assays that contained rifampin and chloramphenicol; open symbols, assays without rifampin and chloramphenicol; squares, glucose-grown cells (0.48 mg of protein); circles, isopropanol-grown cells (0.38 mg of protein); triangles, acetone-grown cells (0.40 mg of protein). Each data point represents the average of duplicate assays.
CO2-dependent acetone metabolism and acetone-dependent 14CO2 fixation in cell suspensions of acetone-grown R. rhodochrous.
The study of acetone metabolism in the aerobic bacterium Xanthobacter strain Py2 had validated carboxylation as a potential strategy for aerobic acetone utilizers (23, 24). To investigate the possibility that acetone might be metabolized by carboxylation in R. rhodochrous, two experiments using whole-cell suspensions were performed. Together, these experiments investigated the effects of CO2 enrichment and depletion on acetone consumption, and the substrate-dependent fixation of 14CO2.
Assay vials containing cell suspensions supplemented with CO2 and NaHCO3 consumed acetone at rates 40% greater than those containing no added carbonate species (Fig. 2). When either of two CO2-trapping agents (6 M KOH or ascarite, a silicate carrier containing adsorbed NaOH) was added to microcentrifuge tubes placed inside assay vials, the consumption of acetone was almost completely prevented (Fig. 2). To verify that the decreased acetone consumption rates observed in the presence of the hydroxide-based traps were due to CO2 depletion, HClO4 was added to one of the traps (at 65 min) to liberate CO2 trapped as K2CO3. As shown in Fig. 2, the addition of HClO4 resulted in an immediate increase in the rate of acetone consumption, verifying the requirement of CO2 for acetone metabolism by R. rhodochrous.
FIG. 2.

Effects of CO2 enrichment and depletion on acetone metabolism by acetone-grown R. rhodochrous. Assays were performed with whole-cell suspensions (0.42 mg of protein) and 2,000 nmol of acetone. Symbols: □, boiled cells with no added CO2 or NaHCO3; ▴, cells enriched with CO2 and NaHCO3 (combined concentration, 10 mM); ■, no added CO2 or NaHCO3; ●, CO2 depleted via an ascarite trap; ⧫, CO2 depleted via a KOH trap (at 65 min HClO4 was added to the KOH trap to liberate CO2). Each data point represents the average of duplicate assays.
In order to determine if CO2 is a cosubstrate of acetone metabolism, the abilities of acetone and other organic compounds to stimulate 14CO2 fixation into whole cells were investigated. Three isomeric compounds (acetone, propionaldehyde, and epoxypropane) and glucose were tested in this regard. Of the isomeric compounds, acetone and propionaldehyde were readily consumed by acetone-grown R. rhodochrous, while epoxypropane was not (Fig. 3). Glucose was also metabolized by acetone-grown cells, and at a rate comparable to those of acetone and propionaldehyde, as evidenced by the increased rate of O2 consumption observed when glucose was added to resting-cell suspensions (data not shown). As shown in Fig. 3, acetone consumption occurred concomitantly with the fixation of 14CO2 into acid-stable cell products. In contrast, the consumption of propionaldehyde and glucose (data not shown) did not support levels of 14CO2 fixation above the background rate.
FIG. 3.

Acetone-dependent 14CO2 fixation by R. rhodochrous. Assays contained acetone-grown R. rhodochrous (0.32 mg of protein). Closed symbols, 14CO2 fixation in the presence of substrate; open symbols, organic substrate remaining. Symbols: triangles, acetone; squares, epoxypropane; circles, propionaldehyde; diamonds, glucose. Each data point represents the average of duplicate assays.
Requirements for and optimization of in vitro acetone carboxylase activity.
In vitro acetone carboxylase activity has been demonstrated for two bacteria: Xanthobacter strain Py2 and R. capsulatus (6, 23, 24). In both cases acetone carboxylase activity required the addition of ATP. The carboxylation of acetone is an endergonic reaction (ΔG°′ = +17.1 kJ/mol), so it is not surprising that a source of energy would be required to support the reaction. For all other CO2-dependent acetone-degrading bacteria that have been studied, no in vitro acetone carboxylase activity has been successfully reconstituted (12, 20). It was therefore of interest to determine whether, and under what conditions, in vitro acetone degradation activity could be measured for R. rhodochrous.
No acetone degradation activity could be detected in cell extracts of R. rhodochrous in the absence of either CO2 or a nucleoside triphosphate. While low rates of CO2- and ATP-dependent acetone degradation activity (less than 0.2 mU mg−1) were observed in cell extracts, significantly higher rates (1.5 to 3.0 mU mg−1) were observed when GTP was included in the assay in place of ATP. The addition of the cations Mg2+ and NH4+ was found to have a stimulatory effect on activity, but they could not themselves replace the requirement of GTP or ATP. All of the acetone degradation activity was recovered in the soluble fraction after removal of membranes by centrifugation at 140,000 × g. The addition of membranes to the soluble fraction did not affect acetone degradation activity.
In order to further characterize the acetone carboxylase activity, cell extracts were fractionated by DEAE anion-exchange chromatography. Acetone carboxylase activity was recovered (>70% recovery) in the fractions eluting between 260 and 320 mM KCl (fourfold enrichment over cell extracts). Unfortunately, further attempts to purify the acetone carboxylase activity (e.g., by hydrophobic interaction, gel filtration, or Q-Sepharose anion-exchange chromatography) resulted in large losses of activity. These losses could not be prevented by the addition of stabilizing agents (glycerol, EDTA, or metal ions) or protease inhibitors. In addition, activity could not be restored by pooling together various fractions resolved by the chromatographic separations. Therefore, all subsequent studies of the acetone carboxylase activity made use of the active fractions partially purified by DEAE chromatography.
As observed for cell extracts, acetone carboxylation by the partially purified acetone carboxylase was CO2 and nucleotide dependent. Table 1 compares the acetone carboxylation rates supported by several nucleoside triphosphates. GTP and ITP supported the highest specific rates of acetone carboxylation, while ATP did not support detectable acetone carboxylase activity (Table 1). XTP, CTP, and UTP also supported acetone carboxylase activity, although at lower rates than either GTP or ITP (Table 1). No stimulation of activity was observed when ATP and GTP were added to assays simultaneously (i.e., ATP did not stimulate the GTP-dependent reaction). Acetone carboxylation was not supported by inorganic pyrophosphate or nucleoside diphosphates. As observed for cell extracts, activity was stimulated by the addition of Mg2+ and NH4+. No other factors, including biotin and the low-molecular-weight components of the cell extract resolved by using ultrafiltration versus a 30,000-Mr cutoff membrane, had any effect on acetone carboxylase activity. Avidin, a potent inactivator of biotin-dependent carboxylases, did not inhibit acetone carboxylase activity.
TABLE 1.
Effects of various nucleoside triphosphates on CO2-dependent acetone degradation by the partially purified acetone carboxylase from R. rhodochrous B276
| Nucleotide | Sp acta (mU · mg−1)
|
|
|---|---|---|
| No added CO2 | With CO2b | |
| None | 0 | 0 |
| ATP | 0 | 0.1 ± 0.1 |
| GTP | 1 ± 0.8 | 16 ± 1 |
| ITP | 2 ± 2 | 14 ± 1 |
| XTP | 0.2 ± 0.2 | 2 ± 0.2 |
| CTP | 2 ± 0.7 | 9 ± 1 |
| UTP | 0.2 ± 0.3 | 6 ± 0.4 |
All assays were performed in duplicate and contained a nucleoside triphosphate (20 mM), glycerol (1.5% [vol/vol]), and partially purified acetone carboxylase (1.03 mg) in addition to the standard assay mixture. One unit of activity is defined as 1 μmol of acetone degraded per min.
Vials contained 50 mM carbonate species, added as 43 mM KHCO3 plus 7 mM CO2.
Characterization of acetoacetate as the product of in vitro acetone carboxylation.
Acetoacetate has been shown to be the product of acetone carboxylation by the acetone carboxylase from Xanthobacter strain Py2 (24), and there is strong evidence that either acetoacetate or an acetoacetyl derivative is the product in anaerobic acetone-utilizing bacteria (10). Based on these precedents, we expected that acetoacetate would be the stoichiometric product of CO2-dependent acetone metabolism in R. rhodochrous. This prediction was verified by using 13C NMR spectroscopy and gas chromatography to identify and quantify acetoacetate produced from in vitro acetone metabolism. Figure 4A shows the 13C NMR spectrum obtained after incubation of the partially purified acetone carboxylase in an assay mixture containing GTP and NaH13CO3. This spectrum contains a resonance with a chemical shift at 174.7 ppm that is identical to that of the C-1 carbon of acetoacetate (24). This resonance is absent in an assay mixture lacking GTP (Fig. 4C). The identity of the resonance was confirmed by reducing the sample with sodium borohydride and then reanalyzing the reduced sample by NMR. As shown in Fig. 4B, the resonance at 174.7 ppm disappeared upon reduction, while a new resonance (180.3 ppm) appeared with a chemical shift identical to that of β-hydroxybutyrate, which is the product of acetoacetate reduction (24).
FIG. 4.

13C NMR identification of acetoacetate as the product of in vitro GTP-dependent acetone carboxylation. (A) Spectrum of the product of acetone carboxylation. The resonance at 174.7 ppm is identical in chemical shift to the resonance of the C-1 carbon atom of acetoacetate. (B) Spectrum of the product of acetone carboxylation after sodium borohydride reduction. The resonance at 180.3 ppm is identical in chemical shift to the resonance of the C-1 atom of β-hydroxybutyrate. (C) Spectrum of a sample prepared from an assay mixture containing all components except GTP. (D) Spectrum of the sample in panel C after sodium borohydride reduction. The resonance at 77.0 ppm in each spectrum is due to chloroform, which was used as the reference. The resonances at approximately 160 ppm (panel A, 160.1 ppm; panel B, 165.6 ppm; panel C, 160.4 ppm; panel D, 166.7 ppm) are due to the presence of NaH13CO3 in the samples. The shifts in position of the bicarbonate resonances from 160.1 and 160.4 ppm (in panels A and C, respectively) to 165.6 and 166.7 ppm (in panels B and D, respectively) are due to the change in pH resulting from the addition of sodium borohydride. The additional resonances at 170.9 ppm in panel B and 171.0 ppm in panel D are due to formate formed by the sodium borohydride reduction of a portion of the NaH13CO3 present in these samples.
The time course of acetone carboxylation and acetoacetate formation by the partially purified acetone carboxylase was analyzed quantitatively. Acetone carboxylation was correlated with the concomitant production of acetoacetate to near-stoichiometric levels. The ratio of acetoacetate formed to acetone degraded was 0.8:1 upon completion of the assay.
Substrate specificity of the partially purified R. rhodochrous acetone carboxylase.
R. rhodochrous acetone carboxylase was found to have a broad substrate specificity, catalyzing the CO2- and ATP-dependent consumption of longer-chain 2-ketones and 3-pentanone. 2-Butanone was consumed at a rate identical to that of acetone, while 2-pentanone, 3-pentanone, and 2-hexanone were degraded at rates that were 70, 40, and 42%, respectively, of the acetone-dependent rate. These results are dramatically different from those obtained for the Xanthobacter strain Py2 acetone carboxylase, which was unable to degrade 2-pentanone, 3-pentanone, or 2-hexanone (23). 2-Butanone was degraded by the Xanthobacter enzyme, but at a rate 60% lower than that of acetone (23).
Identification of inducible polypeptides in R. rhodochrous extracts and comparison of R. rhodochrous and Xanthobacter strain Py2 acetone carboxylase polypeptides.
SDS-PAGE analysis of cell extracts prepared from acetone- or isopropanol-grown R. rhodochrous cells shows the presence of two polypeptides produced at high levels, with apparent molecular masses of 74 and 85 kDa (Fig. 5, lanes 4 and 5). These polypeptides are not visible in extracts prepared from propylene- or glucose-grown cells, which lack acetone degradation activity (Fig. 5, lanes 2 and 3). The presence of acetone carboxylase activity in either Xanthobacter strain Py2 or R. capsulatus also leads to the high-level production of polypeptides with apparent molecular weights similar to those observed here for R. rhodochrous (6, 23). The acetone carboxylase from Xanthobacter strain Py2 has been purified to homogeneity and found to consist of these two polypeptides (apparent molecular masses of 68 and 79 kDa on SDS-PAGE; molecular masses of 78.3 and 85.3 kDa by mass spectrometry) in complex with a third polypeptide with an apparent molecular mass of 23 kDa on SDS-PAGE (19.6 kDa by mass spectrometry) (23) (see Fig. 5, lane 7). As shown in Fig. 5, lane 6, the fractionation of R. rhodochrous acetone carboxylase by DEAE-Sepharose chromatography resulted in the enrichment of the 74- and 85-kDa polypeptides seen in cell extracts, along with a third polypeptide with an apparent molecular mass of 16 kDa. Based on the staining intensities of these three polypeptides, they are present in a 1:1.2:1.2 ratio (in the order of largest to smallest Mr) and account for about 75% of the proteins present in the DEAE-Sepharose fraction exhibiting acetone carboxylase activity. These results suggest that the acetone carboxylase from R. rhodochrous is produced at high levels and has a three-subunit structure similar to that of the acetone carboxylase from Xanthobacter strain Py2.
FIG. 5.

Gel electrophoretic analysis of polypeptides in R. rhodochrous cell extracts and comparison to the partially purified R. rhodochrous and purified Xanthobacter strain Py2 acetone carboxylases. Lanes 1 and 8, molecular mass standards (1 μg per standard); lane 2, glucose-grown R. rhodochrous cell extracts (30 μg); lane 3, propylene-grown R. rhodochrous cell extracts (30 μg); lane 4, isopropanol-grown R. rhodochrous cell extracts (30 μg); lane 5, acetone-grown R. rhodochrous cell extracts (30 μg); lane 6, partially purified R. rhodochrous acetone carboxylase (5 μg); lane 7, purified Xanthobacter strain Py2 acetone carboxylase (2 μg).
DISCUSSION
Acetone is a toxic molecule formed through both biological and industrial processes. While diverse microorganisms have been shown to grow using acetone as a source of carbon and energy, the microbial pathways of acetone metabolism and the properties of acetone-degrading enzymes have not been fully characterized. In vitro acetone degradation activity has been demonstrated for only two bacteria: Xanthobacter strain Py2, an obligate aerobe (24), and R. capsulatus, a purple nonsulfur photosynthetic bacterium (6). In both cases, in vitro acetone degradation is ATP dependent and occurs via carboxylation to acetoacetate, observations that support the CO2-dependent pathways of acetone metabolism proposed in earlier work (7, 13, 14, 18, 19, 21, 22). To date, only the acetone carboxylase from Xanthobacter strain Py2 has been purified to homogeneity (23). This enzyme, which is expressed at high levels (∼20% of cell protein) in acetone- or isopropanol-grown cells, requires ATP and exhibits a specific activity of 225 nmol · min−1 · mg−1 for acetone carboxylation (23). The products of ATP hydrolysis formed during the course of the reaction are AMP and inorganic phosphate, as shown in reaction 1:
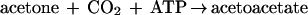 |
1 |
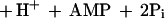 |
The present work expands our base of knowledge concerning bacterial acetone metabolism by demonstrating that an aerobic hydrocarbon-oxidizing actinomycete, R. rhodochrous B276 (formally N. corallina), metabolizes acetone via a CO2-dependent process analogous to those described for Xanthobacter strain Py2 and anaerobic acetone-utilizing bacteria. This finding supports the idea that carboxylation may be a common pathway for acetone metabolism in both aerobic and anaerobic bacteria. Previous studies of acetone metabolism in Mycobacterium vaccae JOB5 (27) and four gram-positive enrichment cultures (26) led to the hypothesis that aerobic acetone metabolism involves an initial monooxygenase-catalyzed hydroxylation producing acetol (hydroxyacetone). Acetol is proposed to undergo further oxidation to pyruvate or cleavage to acetaldehyde and formaldehyde (26, 27). However, acetone monooxygenase activity has not been demonstrated for any aerobic acetone-oxidizing bacteria, and this proposed route remains speculative.
The metabolism of acetone by R. rhodochrous B276 may be relevant to the pathway of propane metabolism, and possibly to that of longer-chain saturated-hydrocarbon metabolism, in this bacterium, although this has not been investigated in the present work. Propane catabolism in hydrocarbon-oxidizing Mycobacterium strains has been shown to proceed by oxidation to isopropanol and then acetone (16, 27). Isopropanol and acetone are potential intermediates in the pathway of propane metabolism in R. rhodochrous, based on the present work showing that they support the growth of the bacterium. Interestingly, R. rhodochrous B276 is capable of growth using short-chain unsaturated hydrocarbons (e.g., propylene and 1-butylene) as carbon sources as well. The pathway of propylene metabolism has been characterized for R. rhodochrous B276 (2) and Xanthobacter strain Py2 (1, 25) and shown to proceed through epoxypropane and acetoacetate as intermediates, as shown in reactions 2 and 3:
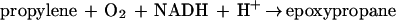 |
2 |
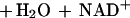 |
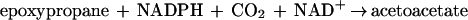 |
3 |
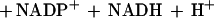 |
The isomeric compounds epoxypropane and acetone, intermediates of unsaturated and saturated C3 hydrocarbon metabolism, respectively, are thus metabolized by carboxylation reactions that produce the common central intermediate acetoacetate.
While the carboxylation of epoxypropane and acetone form the same product, the reactions are catalyzed by distinct enzymes induced under different growth conditions. In addition, the cofactor requirements for the two reactions are dramatically different. In both R. rhodochrous and Xanthobacter strain Py2, epoxypropane carboxylation is catalyzed by a complex four-component enzyme system that requires NADPH and NAD+ as cofactors (3, 4). In contrast, acetone carboxylation requires energy input in the form of nucleoside triphosphate hydrolysis, as illustrated by the stoichiometry of reaction 1 for the Xanthobacter strain Py2 acetone carboxylase.
The initial characterization of acetone carboxylase from R. rhodochrous highlights similarities and differences with regard to the corresponding enzymes studied in Xanthobacter strain Py2 and R. capsulatus. With regard to similarities, in all three cases acetone carboxylase activity correlates with the inducible, high-level production (10 to 20% of cell protein) of two polypeptides with apparent molecular masses of 70 to 75 and 80 to 85 kDa on SDS-PAGE (Fig. 5) (6, 23). A polypeptide with a molecular mass of 21 kDa constitutes the third subunit of the Xanthobacter acetone carboxylase, which has an α2β2γ2 quaternary structure (23). A polypeptide of similar molecular weight, present in a stoichiometric ratio to the larger polypeptides, can be observed in the partially purified acetone carboxylase of R. rhodochrous (Fig. 5). While a corresponding small peptide was not specifically noted for the partially purified acetone carboxylase of R. capsulatus, there appears to be a significant amount of protein at the dye front, present just under the 20-kDa marker protein, in the SDS-PAGE system of Birks and Kelly (see Fig. 4 in reference 6). If the acetone carboxylase of R. capsulatus does indeed contain a similar peptide, it would indicate that the three acetone carboxylases of diverse bacteria contain a conserved subunit structure of (αβγ)n.
With regard to differences between the three acetone carboxylase systems studied in vitro, the nucleoside triphosphate specificity of the R. rhodochrous enzyme is significantly different from those of the other two (Table 1). For the purified acetone carboxylase from Xanthobacter, no activity could be observed when GTP, CTP, UTP, or TTP replaced ATP in the assay (23). As noted earlier, ATP supported low rates of acetone carboxylase activity in cell extracts and for the partially purified enzyme from R. capsulatus (6). However, it is not known whether other nucleoside triphosphates were tested for their ability to support activity.
If GTP is indeed the physiologically relevant cofactor for acetone carboxylation in R. rhodochrous, it would be a new role without precedent in prokaryotic metabolism. In fact, there are few examples of the direct utilization of GTP as a cofactor and energy source in either prokaryotic or eukaryotic pathways for the assimilation or breakdown of carbon-containing compounds. The only characterized example is that of phosphoenolpyruvate carboxykinase (PEPCK), a gluconeogenic enzyme that catalyzes the reversible decarboxylation and nucleoside triphosphate-dependent phosphorylation of oxaloacetate (OAA) to form phosphoenolpyruvate (PEP), as shown in reaction 4 (17):
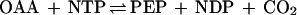 |
4 |
where NTP is a nucleoside triphosphate. The nature of the nucleoside triphosphate supporting the above reaction varies depending on the organism: ATP is used by the PEPCK enzymes from bacteria, yeast, and plants, while GTP is used by the PEPCK enzymes in mammals and some other eukaryotes (17). A characteristic feature of the GTP-dependent PEPCK enzymes is their ability to substitute ITP for GTP in the reaction (17); in light of this, it is intriguing that ITP supported R. rhodochrous acetone carboxylase activity at a rate nearly identical to that of GTP (Table 1). It is unclear why GTP would have been selected as the cofactor for acetone carboxylation in R. rhodochrous, a gram-positive actinomycete, while ATP was selected for Xanthobacter and R. capsulatus, both gram-negative eubacteria. It is also not known at present whether the stoichiometry of nucleoside triphosphate hydrolysis supporting the R. rhodochrous reaction is the same as, or different from, that found for the Xanthobacter enzyme (reaction 1).
Another distinguishing feature of the R. rhodochrous acetone carboxylase is its ability to use other ketones (butanone, 2-pentanone, 3-pentanone, and 2-hexanone) as substrates at rates comparable to that with acetone. By comparison, only acetone and butanone (46% of the acetone consumption rate) were found to be substrates for the Xanthobacter acetone carboxylase (23). Birks and Kelly have reported that butanone was not a substrate for the acetone carboxylase activity of R. capsulatus (6). Thus, the R. rhodochrous acetone carboxylase has a broader substrate specificity than the two other acetone carboxylases studied.
It is intriguing that 3-pentanone is a substrate for the acetone carboxylase, due to the lack of a terminal carbon alpha to the carbonyl, a feature which was thought to be required for carboxylation. Instead, the symmetrical 3-pentanone contains two methylene carbon atoms flanking the carbonyl, both of which are sites representing greater steric hindrance to carboxylation relative to a terminal position. Nonetheless, 3-pentanone was metabolized at rates 40% of that observed for acetone. It is not known at present whether 3-pentanone or the other longer-chain ketones will support the growth of R. rhodochrous.
It should be noted that the specific activities measured in cell extracts and for the partially purified preparation of R. rhodochrous acetone carboxylase are significantly lower than the rates observed in whole-cell suspensions. Typical rates for acetone degradation in whole-cell suspensions were in the range of 70 to 150 nmol of acetone degraded min−1 mg−1, while the rates in cell extracts were in the range of 1.5 to 3.0 nmol min−1 mg−1. The in vitro rates are thus only about 2% of the maximal rates observed in cell suspensions. At present it is not known why the in vitro rates are so much lower. Possible explanations include inactivation during and after cell lysis, nonoptimal assay conditions, absence or limitation of an unidentified cofactor, and/or the presence of inhibitory components in the assay (e.g., phosphatases or nucleotide hydrolysis products). In light of the low activity, some caution must be exercised in definitively ascribing a physiological role to GTP in the acetone carboxylase assay. While acetone carboxylase activity was recovered at a good yield and with the expected fold purification (about fourfold) after a single DEAE-Sepharose column, further attempts to purify the enzyme resulted in low recoveries and decreases in the specific activity of the enzyme. Again, it is not known whether this loss of activity is due to inherent instability of the protein, the loss of a required cofactor, or some other phenomenon. Studies of other acetone-metabolizing bacteria have been similarly hampered by the complete or partial loss of acetone-degrading activity upon cell lysis. For example, no acetone carboxylase activity has to date been successfully reconstituted in cell extracts of acetone-grown sulfate reducers, denitrifiers, or fermentative enrichment cultures (12, 20). Schink and coworkers have, however, successfully measured ADP-dependent acetoacetate decarboxylase activity in cell extracts of an acetone-grown denitrifying bacterium (20). In addition, they have demonstrated ADP-dependent 14CO2-acetoacetate exchange in cell extracts from the same bacterium (12). Strong evidence was provided that both the decarboxylase activity and the exchange activity were catalyzed by the acetone carboxylase (12, 20). As noted earlier, ATP-dependent acetone carboxylase activity has been measured in vitro for R. capsulatus. However, as for R. rhodochrous, the specific activities in cell extracts of R. capsulatus were dramatically lower (100- to 1,000-fold) than those in whole-cell suspensions (6). The only acetone carboxylase that can be reconstituted in vitro at physiologically relevant rates is the enzyme from Xanthobacter strain Py2: this enzyme exhibits a specific activity in cell extracts equivalent to that in whole cells and can be purified in a fully active state (23). The only cofactors required by the Xanthobacter enzyme are ATP and Mg2+, while K+ exerts a stimulatory, but not essential, effect on activity (23). Further avenues need to be explored in order to optimize the in vitro activity of the R. rhodochrous acetone carboxylase, with the hope of purifying the enzyme to homogeneity in an active state.
In summary, evidence has been provided for the presence of a nucleotide-dependent acetone carboxylase in the gram-positive actinomycete R. rhodochrous. The occurrence of acetone carboxylases in two distantly related aerobic bacteria such as R. rhodochrous, a gram-positive actinomycete, and Xanthobacter strain Py2, a gram-negative eubacterium, provides further evidence that carboxylation may be a significant strategy of acetone metabolism in aerobic bacteria. The novel nucleotide usage and substrate specificity of the R. rhodochrous acetone carboxylase set it apart from the Xanthobacter strain Py2 and R. capsulatus acetone carboxylases previously studied. This work provides additional insights into the novelty of bacterial acetone-degrading enzymes and their catalytic requirements.
ACKNOWLEDGMENTS
This work was supported by National Science Foundation grant MCB9630081.
We thank Miriam Sluis for helpful discussions and technical assistance.
REFERENCES
- 1.Allen J R, Ensign S A. Carboxylation of epoxides to β-keto acids in cell extracts of Xanthobacter strain Py2. J Bacteriol. 1996;178:1469–1472. doi: 10.1128/jb.178.5.1469-1472.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen J R, Ensign S A. Identification and characterization of epoxide carboxylase activity in cell extracts of Nocardia corallina B276. J Bacteriol. 1998;180:2072–2078. doi: 10.1128/jb.180.8.2072-2078.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen J R, Ensign S A. Purification to homogeneity and reconstitution of the individual components of the epoxide carboxylase multiprotein enzyme complex from Xanthobacter strain Py2. J Biol Chem. 1997;272:32121–32128. doi: 10.1074/jbc.272.51.32121. [DOI] [PubMed] [Google Scholar]
- 4.Allen J R, Ensign S A. Two short-chain dehydrogenases confer stereoselectivity for enantiomers of epoxypropane in the multiprotein epoxide carboxylating systems of Xanthobacter strain Py2 and Nocardia corallina B276. Biochemistry. 1999;38:247–256. doi: 10.1021/bi982114h. [DOI] [PubMed] [Google Scholar]
- 5.Ashraf W, Mihdhir A, Murrell J C. Bacterial oxidation of propane. FEMS Microbiol Lett. 1994;122:1–6. doi: 10.1111/j.1574-6968.1994.tb07134.x. [DOI] [PubMed] [Google Scholar]
- 6.Birks S J, Kelly D J. Assay and properties of acetone carboxylase, a novel enzyme involved in acetone-dependent growth and CO2 fixation in Rhodobacter capsulatus and other photosynthetic and denitrifying bacteria. Microbiology. 1997;143:755–766. doi: 10.1099/00221287-143-3-755. [DOI] [PubMed] [Google Scholar]
- 7.Bonnet-Smits E M, Robertson L A, Van Dijken J P, Senior E, Kuenen J G. Carbon dioxide fixation as the initial step in the metabolism of acetone by Thiosphaera pantotropha. J Gen Microbiol. 1988;134:2281–2289. [Google Scholar]
- 8.Chromy V, Fischer J, Kulhanek V. Re-evaluation of EDTA-chelated biuret reagent. Clin Chem. 1974;20:1362–1363. [PubMed] [Google Scholar]
- 9.Coleman J P, Perry J J. Fate of the C1 product of propane dissimilation in Mycobacterium vaccae. J Bacteriol. 1984;160:1163–1164. doi: 10.1128/jb.160.3.1163-1164.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ensign S A, Small F J, Allen J R, Sluis M K. New roles for CO2 in the microbial metabolism of aliphatic epoxides and ketones. Arch Microbiol. 1998;169:179–187. doi: 10.1007/s002030050558. [DOI] [PubMed] [Google Scholar]
- 11.Furuhashi K, Taoka A, Uchida S, Karube I, Suzuki S. Production of 1,2-epoxyalkanes from 1-alkenes by Nocardia corallina B-276. Eur J Appl Microbiol Biotechnol. 1981;12:39–45. [Google Scholar]
- 12.Janssen P H, Schink B. 14CO2 exchange with acetoacetate catalyzed by dialyzed cell-free extracts of the bacterial strain BunN grown with acetone and nitrate. Eur J Biochem. 1995;228:677–682. doi: 10.1111/j.1432-1033.1995.0677m.x. [DOI] [PubMed] [Google Scholar]
- 13.Janssen P H, Schink B. Catabolic and anabolic enzyme activities and energetics of acetone metabolism of the sulfate-reducing bacterium Desulfococcus biacutus. J Bacteriol. 1995;177:277–282. doi: 10.1128/jb.177.2.277-282.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janssen P H, Schink B. Metabolic pathways and energetics of the acetone-oxidizing, sulfate-reducing bacterium, Desulfobacterium cetonicum. Arch Microbiol. 1995;163:188–194. doi: 10.1007/BF00305352. [DOI] [PubMed] [Google Scholar]
- 15.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 16.Lukins H H, Foster J W. Methyl ketone metabolism in hydrocarbon-utilizing mycobacteria. J Bacteriol. 1963;85:1074–1087. doi: 10.1128/jb.85.5.1074-1087.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matte A, Tari L W, Goldie H, Delbaere L T J. Minireview: structure and mechanism of phosphoenolpyruvate carboxykinase. J Biol Chem. 1997;272:8105–8108. doi: 10.1074/jbc.272.13.8105. [DOI] [PubMed] [Google Scholar]
- 18.Platen H, Janssen P H, Schink B. Fermentative degradation of acetone by an enrichment culture in membrane-separated culture devices and in cell suspensions. FEMS Microbiol Lett. 1994;122:27–32. doi: 10.1111/j.1574-6968.1994.tb07138.x. [DOI] [PubMed] [Google Scholar]
- 19.Platen H, Schink B. Anaerobic degradation of acetone and higher ketones by newly isolated denitrifying bacteria. J Gen Microbiol. 1989;135:883–891. doi: 10.1099/00221287-135-4-883. [DOI] [PubMed] [Google Scholar]
- 20.Platen H, Schink B. Enzymes involved in anaerobic degradation of acetone by a denitrifying bacterium. Biodegradation. 1990;1:243–251. doi: 10.1007/BF00119761. [DOI] [PubMed] [Google Scholar]
- 21.Platen H, Temmes A, Schink B. Anaerobic degradation of acetone by Desulfococcus biacutus spec. nov. Arch Microbiol. 1990;154:355–361. doi: 10.1007/BF00276531. [DOI] [PubMed] [Google Scholar]
- 22.Siegel J M. The metabolism of acetone by the photosynthetic bacterium Rhodopseudomonas gelatinosa. J Bacteriol. 1950;60:595–606. doi: 10.1128/jb.60.5.595-606.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sluis M K, Ensign S A. Purification and characterization of acetone carboxylase from Xanthobacter strain Py2. Proc Natl Acad Sci USA. 1997;94:8456–8461. doi: 10.1073/pnas.94.16.8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sluis M K, Small F J, Allen J R, Ensign S A. Involvement of an ATP-dependent carboxylase in a CO2-dependent pathway of acetone metabolism by Xanthobacter strain Py2. J Bacteriol. 1996;178:4020–4026. doi: 10.1128/jb.178.14.4020-4026.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Small F J, Ensign S A. Carbon dioxide fixation in the metabolism of propylene and propylene oxide by Xanthobacter strain Py2. J Bacteriol. 1995;177:6170–6175. doi: 10.1128/jb.177.21.6170-6175.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor D G, Trudgill P W, Cripps R E, Harris P R. The microbial metabolism of acetone. J Gen Microbiol. 1980;118:159–170. [Google Scholar]
- 27.Vestal J R, Perry J J. Divergent metabolic pathways for propane and propionate utilization by a soil isolate. J Bacteriol. 1969;99:216–221. doi: 10.1128/jb.99.1.216-221.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weigant W W, deBont J A M. A new route for ethylene glycol metabolism in Mycobacterium E44. J Gen Microbiol. 1980;120:325–331. [Google Scholar]