


Abstract
mRNA 5′ cap recognition by eIF4F is a key element of eukaryotic translational control. Kinetic differences in eIF4F–mRNA interactions have long been proposed to mediate translation-efficiency differences between mRNAs, and recent transcriptome-wide studies have revealed significant heterogeneity in eIF4F engagement with differentially-translated mRNAs. However, detailed kinetic information exists only for eIF4F interactions with short model RNAs. We developed and applied single-molecule fluorescence approaches to directly observe real-time Saccharomyces cerevisiae eIF4F subunit interactions with full-length polyadenylated mRNAs. We found that eIF4E–mRNA association rates linearly anticorrelate with mRNA length. eIF4G–mRNA interaction accelerates eIF4E–mRNA association in proportion to mRNA length, as does an eIF4F-independent activity of eIF4A, though cap-proximal secondary structure still plays an important role in defining the final association rates. eIF4F–mRNA interactions remained dominated by effects of eIF4G, but were modulated to different extents for different mRNAs by the presence of eIF4A and ATP. We also found that eIF4A-catalyzed ATP hydrolysis ejects eIF4E, and likely eIF4E•eIF4G from the mRNA after initial eIF4F•mRNA complex formation, suggesting a mechanism to prepare the mRNA 5′ end for ribosome recruitment. Our results support a role for mRNA-specific, factor-driven eIF4F association rates in kinetically controlling translation.
INTRODUCTION
Protein synthesis is a key stage of gene expression, and is highly regulated to maintain cellular viability (1). In eukaryotes, translation is chiefly controlled during initiation. Translational control also allows gene expression to respond to changes in the cellular environment, and to external stimuli, on a timescale faster than transcriptional regulation (1).
Translation initiation on most eukaryotic mRNAs involves the 5′ m7G(5′)ppp(5′)N cap (where N is the transcript + 1 nucleotide) (2). Cap recognition by initiation factor eIF4F leads to recruitment of the small ribosomal subunit, in its 43S pre-initiation complex (PIC) (3). eIF4F is composed of three subunits. eIF4E binds the cap structure (1,4–7). eIF4G, a large, multidomain protein, binds eIF4E, mRNA, and eIF4A, a DEAD-box RNA helicase (8,9). eIF4G also directly contacts the 43S PIC (10). The resulting 48S PIC is thought to scan linearly through the mRNA 5′ leader to the start codon (3,11,12). Cap recognition, PIC recruitment and start-codon selection are each major targets for translational control mechanisms (10,13).
mRNAs encoding different proteins are translated with widely differing efficiencies (14,15). Early kinetic modeling studies implicated differing rates of cap recognition as kinetically controlling ‘discrimination’ between mRNAs competing for translation (16,17). Indeed, kinetics play an integral role throughout translation initiation, which is highly dynamic and proceeds through numerous intermediates with different molecular compositions and conformations on a seconds timescale (18,19). Understanding how eIF4F–mRNA coordination unfolds in real time during cap recognition is thus central to understanding molecular mechanisms of mRNA discrimination and translational control (20,21).
While the kinetics of eIF4F binding to capped RNA oligonucleotides have been elucidated (22–25), data are not available for interactions with full-length mRNAs, which present a much richer structural landscape that may impact eIF4F function. Several important questions therefore remain unanswered. For instance, in-vivo transcriptome-wide studies have indicated substantial heterogeneity in eIF4F•mRNA complex formation and utilization between mRNAs (26–31) These differences can be explained in part due to mRNA features, such as structures or sequences in the 5′ leader—frequently cap-proximal—that confer eIF4F dependence (1,7,15,32,33) However, the transcriptome-wide data also point to unanticipated mRNA features that may impact eIF4F function to define translational output. For instance, mRNA enrichment in eIF4E•eIF4G in vivo intriguingly appears to anticorrelate with coding-sequence length (26,34)—suggesting that mRNA elements beyond the cap-proximal region, or even beyond the leader, may be important determinants of eIF4F–mRNA interaction. Indeed, an mRNA ‘length sensing’ model has been proposed to contribute to variability in translation efficiency (34), though different potential mechanisms have been proposed to account for the phenomenon. Features beyond the 5′ leader have also been shown to contribute to the ability of eIF4A to promote PIC recruitment (35). There is consequently a need for a quantitative assessment of eIF4F dynamics on full-length mRNAs, and of the extent to which the dynamics may vary between transcripts.
Biochemical, genetic, and structural studies have also established that dynamic inter-subunit coordination is an important property of eIF4F function (3,8,11,13,20,21,36–43) However, a detailed kinetic framework for the molecular mechanisms of this coordination has not yet been established (21). Past kinetics studies of eIF4F–RNA interaction have utilized the intact heterotrimeric eIF4F complex purified from cell lysates, precluding isolation of the effects of individual subunits. Where complexes have been reconstituted from purified subunits, to isolate effects of individual subunits, kinetics were measured with capped oligonucleotides (25), or only partial eIF4G sequences were used that lack RNA-binding activity (44).
How kinetics underpin the coordinated biochemical functions of the eIF4F subunits thus remains poorly understood. For example, although eIF4G was shown to greatly enhance eIF4E–cap interaction in crosslinking and pulldown assays (45), through its RNA-binding activity (38), it is not known whether this is due to acceleration of initial eIF4E–mRNA encounter, to stabilization of the eIF4E•eIF4G•mRNA complex after initial binding, or to both effects. Likewise, whilst eIF4G–eIF4A interaction stimulates the eIF4A ATPase and helicase activities (46) by modulating eIF4A conformational cycling (41,42), and eIF4A globally promotes cap recognition (29), the extent to which eIF4A activity contributes to cap-recognition rates on different mRNAs is unknown. Moreover, the division of labor between eIF4F-bound eIF4A and the order-of-magnitude cellular excess of free eIF4A over the eIF4F-bound factor (47) is also not yet well understood, though the free eIF4A fraction is increasingly implicated as contributing to initiation (35,48) Indeed, although the function of eIF4A as an ATP-dependent RNA helicase is well established (49), the precise contributions of eIF4A ATP binding and hydrolysis to the dynamics of eIF4F–mRNA recognition remain to be fully determined (8), as again does their relative importance for eIF4F recognition of different mRNAs. More broadly beyond these examples for individual eIF4F subunits, the precise sequence of molecular events occurring in the eIF4F•mRNA complex prior to PIC recruitment is not fully known (8,20,21).
We developed a suite of single-molecule fluorescence assays to directly observe Saccharomyces cerevisiae eIF4F–mRNA recognition in real time. We dissected the contributions of the eIF4F subunits to cap recognition on individual, full-length yeast mRNAs that show varying binding to eIF4F in vivo, and varying translation-efficiency dependence on eIF4A. We also delineated the sequence of events that follows initial eIF4F–mRNA binding en route to recruitment of the 43S pre-initiation complex.
MATERIALS AND METHODS
In vitro transcription, RNA processing and labeling of oligonucleotides
DNA templates to be transcribed were PCR-amplified from Yeast Genomic DNA (EMD Millipore) with Phusion DNA polymerase (NEB) using standard procedures. Primers sequences are reported in Supplementary Table S12. A T7 promoter (TAATACGACTCACTATAGG) was incorporated into the PCR product through the forward primer, where the underlined bases become +1 and +2 nucleotides added to the transcript. The resulting templates were in-vitro transcribed to produce RNA using in-house purified or commercial (NEB) T7 RNA polymerase. Transcription reactions were typically carried out on a 40 μl scale, with ∼5 μg of DNA template, in a buffer consisting of 200 mM Tris–HCl, pH 7.9, 0.05% (v/v) Triton-X-100, 15 mM spermidine, 2 mM each NTP and 5 mM DTT. MgCl2 concentrations in this buffer were optimized for each transcript by titration, and were 15–30 mM. Unreacted nucleotides were removed from the transcription mixture with MicroBio-Spin gel filtration columns (P30, Bio-Rad), and the RNA product was then precipitated using 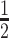 volumes of 7.5 M lithium chloride. The resulting RNA pellet was redissolved after three washes with 80% EtOH and run on a native 1% TBE–agarose gel, to check for integrity. The RNAs were then capped using the ScriptCap™ m7G Capping System (CellScript) with the following modifications to the manufacturer's protocol: the incubation time with the capping enzyme was increased to 2 h, and the volume of enzyme added was increased twofold. A poly(A) tail was added to the mRNA immediately after capping, using Escherichia coli poly(A) polymerase (NEB), following the manufacturer's guidelines. The capped and tailed mRNA was then re-purified by organic extraction with acidic phenol-chloroform, precipitated with 0.1 volumes of 3 M sodium acetate and two volumes of ethanol, and resuspended in RNase-free water. To assess the capping efficiency, the JJJ1, HXT2, and NCE102 RNAs were desalted using MicroBioSpin gel filtration columns (BioRad) after capping, and then treated with RNA 5′ polyphosphatase (Epicentre) and RNA 5’ terminator dependent exonuclease (Epicentre) for 1 h for each enzyme. Batches of the same RNA, either capped and uncapped, were treated separately, and RNA integrity was assessed on a 1.2% TAE–agarose gel immediately after treatment. To obtain an estimate of poly(A) tail lengths, RNAs were run on a 1.5% TAE–agarose gel for 20 min at 120 V after poly(A) tailing, loaded next to the same mRNA lacking a poly(A) tail, and staining with ethidium bromide.
volumes of 7.5 M lithium chloride. The resulting RNA pellet was redissolved after three washes with 80% EtOH and run on a native 1% TBE–agarose gel, to check for integrity. The RNAs were then capped using the ScriptCap™ m7G Capping System (CellScript) with the following modifications to the manufacturer's protocol: the incubation time with the capping enzyme was increased to 2 h, and the volume of enzyme added was increased twofold. A poly(A) tail was added to the mRNA immediately after capping, using Escherichia coli poly(A) polymerase (NEB), following the manufacturer's guidelines. The capped and tailed mRNA was then re-purified by organic extraction with acidic phenol-chloroform, precipitated with 0.1 volumes of 3 M sodium acetate and two volumes of ethanol, and resuspended in RNase-free water. To assess the capping efficiency, the JJJ1, HXT2, and NCE102 RNAs were desalted using MicroBioSpin gel filtration columns (BioRad) after capping, and then treated with RNA 5′ polyphosphatase (Epicentre) and RNA 5’ terminator dependent exonuclease (Epicentre) for 1 h for each enzyme. Batches of the same RNA, either capped and uncapped, were treated separately, and RNA integrity was assessed on a 1.2% TAE–agarose gel immediately after treatment. To obtain an estimate of poly(A) tail lengths, RNAs were run on a 1.5% TAE–agarose gel for 20 min at 120 V after poly(A) tailing, loaded next to the same mRNA lacking a poly(A) tail, and staining with ethidium bromide.
5′ biotinylated and 3′ amino-modified oligonucleotides purchased from IDT were reacted with a 1:8 molar ratio of oligonucleotide to NHS-ester derivatives of Cy5 (for intramolecular FRET experiments with dual labeled RNA), Cy3 and Cy3.5 in 0.1 M sodium bicarbonate for 4 h at room temperature, followed by four successive chloroform extractions to remove unreacted dye, and by buffer-exchange into ddH2O using MicroBio-Spin gel filtration columns (P30, Bio-Rad). Labeling efficiency was typically 75% as measured by UV/visible spectrophotometry. The labeled oligonucleotides were stored at –20°C and used without further purification.
Protein purification and labeling
Genes encoding translation factors were expressed from pET-28a(+) (Qiagen) or pTYB2 (New England Biolabs). Overexpression was carried out at 37°C in LB medium, in volumes ranging from 1 L to 12 L. E. coli BL21(DE3) CodonPlus RIL or BL21(DE3) cells expressing the target recombinant protein were grown to an OD600 of 0.5–1 at 37°C. Overexpression was then induced via addition of 0.5 mM IPTG, then the overexpression was allowed to proceed overnight at 16°C. For eIF4G, the induction was carried out at 37°C for 2–3 h. The resulting cells were harvested and stored at –80°C until purification.
His6-tagged yeast eIF4E(A124C) was purified as described previously (25) Briefly, cell lysate from the overexpression culture was loaded onto a gravity-flow Ni-NTA agarose column (Qiagen) pre-equilibrated with eIF4E buffer (50 mM HEPES–KOH, pH 7.4, 150 mM KCl, 2.5 mM TCEP). The column was then washed with 40 column volumes of eIF4E buffer containing 40 mM imidazole to remove nonspecifically-bound proteins, then eIF4E was eluted using eIF4E buffer containing 250 mM imidazole. Imidazole was removed from the protein eluate by desalting on a Bio-Rad 10-DG column equilibrated in containing 50 mM HEPES–KOH, pH 7.4, 150 mM KCl, 0.5 mM TCEP. The resulting protein was immediately labeled with a sulfonated Cyanine 5 maleimide (Lumiprobe) overnight at 6°C in darkness. Unreacted fluorophore was then removed by desalting on a BioRad 10-DG column. The labeled protein was purified by gel filtration using a Superdex 75 Increase column (GE healthcare), equilibrated in storage buffer (50 mM HEPES–KOH, pH 7.4, 150 mM KCl, 2.5 mM TCEP). The labeling efficiency was assessed by UV/visible spectrophotometry, and was typically ∼50%. The protein was stored at 6°C in darkness, and was prepared freshly every week as needed.
The plasmid containing recombinant full-length eIF4G1 was a gift from Sarah Walker (also available from Addgene as plasmid #122248). This full-length eIF4G1 construct with a C-terminal chitin binding domain fusion was purified according to a published procedure (50), with the following modifications: the cells were lysed using a sonicator, and the final protein after elution was stored in 250 mM KCl instead of 250 mM KOAc, skipping the dialysis after the anion-exchange chromatography step in the procedure, and DTT for storage was substituted with 2.5 mM TCEP. Briefly, E. coli cells expressing full-length eIF4G1 were thawed, and lysed using a sonicator after resuspending in Intein Lysis Buffer (50 mM HEPES–KOH, pH 7.4, 500 mM KCl, 1 mM EDTA). The lysate was then clarified by centrifugation at 20 000 × g for 15 min. The clarified lysate was rocked with 4 ml of chitin resin (New England Biolabs) for 30 min at 4°C. The resin was then loaded into a gravity-flow column and washed with 100 ml of Intein Lysis Buffer. The column was then treated with micrococcal nuclease to remove nucleic acids from E. coli which co-purified with eIF4G. Briefly, the resin was first equilibrated with microccocal nuclease buffer (50 mM HEPES–KOH, pH 7.4, 100 mM KCl, 2 mM CaCl2). Then, a 3 ml solution containing 3 U/μl microccocal nuclease was passed through the column. The resin was then incubated for 30 min at 37°C. Following nuclease treatment, the column was washed with a further 50 ml of lysis buffer. The column was then flushed with 8 ml of lysis buffer containing 50 mM DTT, and 6 ml was allowed to pass through the column, which was then sealed and incubated overnight at 6°C. The following day, the cleaved protein was eluted with 10 ml of lysis buffer. The resulting protein solution was diluted to 100 mM KCl with lysis buffer lacking KCl, then manually loaded onto a Q HP column (1 ml; GE Healthcare Life Sciences) equilibrated in 50 mM HEPES–KOH, pH 7.4, 10% (v/v) glycerol, 2.5 mM TCEP, 100 mM KCl and washed with five column volumes of buffer containing 50 mM HEPES–KOH, pH 7.4, 10% (v/v) glycerol, 2.5 mM TCEP, 100 mM KCl. The column was then eluted manually with a step-gradient of 150, 200, 250 mM KCl in 50 mM HEPES–KOH, pH 7.4, 10% (v/v) glycerol, 2.5 mM TCEP (one column volume for each step). Eluate fractions were analyzed by SDS-PAGE; eIF4G typically eluted above ∼220 mM KCl. Single-use aliquots of purified eIF4G1 were prepared and were stored at –80°C. The His6-tagged eIF4G1–452 fragment (an NdeI fragment of the eIF4G1 CDS) was purified essentially as for eIF4E, but 1 M KCl was included in the purification buffers.
His6-tagged recombinant eIF4G83–452 was expressed in a pET-29b(+) vector using E. coli BL21(DE3) CodonPlus RIL cells, under dual selection with chloramphenicol and kanamycin. The resulting transformants were grown in a 10 ml starter culture at 37°C for 16 h. A 2 L LB medium flask was inoculated with this culture, and the culture was grown until an O.D.600 value of 1. Overexpression was then induced via addition of 1 mM IPTG, then the overexpression was allowed to proceed overnight at 16°C. Cells were harvested and resuspended in 30 ml of lysis buffer (50 mM HEPES–KOH, pH 7.4, 150 mM KCl, 2.5 mM TCEP, 40 mM imidazole). The resuspended cells were lysed using a sonicator, and the lysate was by centrifugation at 20 000 × g for 15 min. The lysate was then passed through a 0.2 μm syringe filter and applied to a 1 ml HisTrap column (Cytiva) equilibrated with 5 column volumes of lysis buffer. The column was washed with 5 ml of lysis buffer, then the protein was eluted using a lysis buffer containing 250 mM imidazole. The resulting eluate was diluted 3-fold using lysis buffer without salt or imidazole, and applied to a 1 ml HiTrap Heparin column (Cytiva) equilibrated in buffer containing 50 mM HEPES–KOH, pH 7.4, 10% (v/v) glycerol, 2.5 mM TCEP, 100 mM KCl. The column was washed with 5 column volumes of the same buffer, and bound protein was then eluted with the same buffer containing 250 mM KCl. The eluted protein was stored at −80°C.
For non-specific fluorescent labelling of full-length eIF4G, single-use aliquots of the eIF4G protein were treated with an equimolar concentration of Sulfo-Cy5.5 Maleimide (Lumiprobe), resuspended as a 2 mM stock in DMSO, for a total of 2 hours. The resulting fluorescent protein was then immediately used in the single molecule experiments.
His6-tagged recombinant eIF4A was purified as described previously (25). E. coli cells expressing recombinant eIF4A were first thawed and resuspended in eIF4A lysis buffer (50 mM HEPES–KOH, pH 7.4, 300 mM KCl, 2.5 mM TCEP). After sonication for cell lysis, the resulting lysate was clarified by spinning at 20 000 × g for 15 min. The clarified lysate was applied to Ni-NTA agarose (equilibrated in lysis buffer) as a first step, after filtering the lysate through a 0.22 μm syringe filter. The bound protein was eluted with lysis buffer containing 250 mM imidazole after washing with 10 column volumes of lysis buffer containing 40 mM Imidazole. The eluate was buffer-exchanged to Buffer A using a BioRad 10-DG column (50 mM HEPES–KOH, pH 7.4, 100 mM KCl, 2.5 mM TCEP) and subjected to anion-exchange chromatography using a 5 ml Q HP anion-exchange column (GE Healthcare). The column was eluted with a linear gradient of 0.1–1 M KCl. eIF4A typically eluted at 250 mM KCl. Fractions containing eIF4A were identified by SDS-PAGE analysis, then pooled, concentrated by centrifugal ultrafiltration, and further purified by gel filtration chromatography using a Superdex 200 column (GE healthcare) equilibrated in 50 mM HEPES–KOH, pH 7.4, 100 mM KOAc, 2.5 mM TCEP, 10% (v/v) glycerol. The final protein sample was divided into single-use aliquots and stored in storage buffer at –80°C.
For preparation of labeled eIF4A, a construct was designed that expresses the native eIF4A sequence with an N-terminal Met-Ala-(pAz)Phe tripeptide extension for unnatural amino acid incorporation. This plasmid was co-transformed into E. coli BL21(DE3) cells with the pEVOL-ps plasmid (a generous gift from Abhishek Chatterjee, Boston College) under dual selection with chloramphenicol and kanamycin. The resulting transformants were grown in a 10 ml starter culture overnight. Afterwards, a 1 l LB medium flask was inoculated with the starter culture and grown to an OD600 value of 0.5. 1 mM 4-azidophenylalanine was then added to the culture medium along with 2 mM arabinose to induce tRNA/aminoacyl-tRNA synthetase expression. Finally, 2 mM IPTG was added to induce expression of eIF4A. Overexpression was allowed to proceed for 5 h in darkness, to avoid photochemical damage to the unnatural amino acid. The cells were harvested and stored at –80°C until purification. MA(pAzF)-eIF4A was purified identically to unlabeled recombinant eIF4A, with the exception that after initial Ni-NTA purification the protein was treated with DBCO-Cy3 overnight to conjugate the fluorophore to the unnatural amino acid.
Electrophoretic mobility shift assays (EMSAs).
RNA (20 nM based on 260-nm absorbance) was incubated with eIF4G truncations (800 nM) or full-length eIF4G (400 nM) for 5 min in 1 × Assay Buffer (50 mM HEPES–KOH, pH 7.4, 3 mM Mg (OAc)2, 100 mM KOAc), and run on a 1.5% TAE-agarose gel at 6°C for 30 min at 80 V. Bound complexes were visualized using gels pre-cast with ethidium bromide.
Steady-state ATPase assay for eIF4A activity.
The NADH-coupled ATPase assay was carried out according to a published procedure (51) Briefly, reactions were assembled on ice and started by adding Mg-ATP. The reaction was set up with the KMg75 buffer (20 mM HEPES–KOH, pH 7.4, 75 mM KCl, 1 mM DTT, 5 mM MgCl2), 250 nM eIF4A or Cy3-eIF4A, 125 nM full-length eIF4G, 1 mM ATP, 1 mM (measured as concentration of bases) poly(U) RNA (Sigma), lactate dehydrogenase (20 U/ml final concentration), and pyruvate kinase (100 U/ml) Absorbance was recorded at 340 nM with a Shimadzu UV2600 UV-visible spectrophotometer, measuring the decrease of NADH absorbance with time. The slope of the absorbance versus time graph was converted to the rate of ATP hydrolysis using an extinction coefficient of 6220 M–1 cm–1 for NADH, and normalized to the eIF4A concentration to yield V/E0. Control reactions to establish the background rate of NADH oxidation included no eIF4A and no RNA.
Single-molecule experiments
The custom RS instrument was set up as described previously (52,53) The RNA to be immobilized was hybridized through its poly(A) tail to (dT)45 conjugated to biotin at its 5′ end and Cy3 or Cy3.5 at its 3′ end, to act as a FRET donor. Annealing was performed using a thermocycler, by heating 100 nM labeled oligonucleotide to 98°C for 2 min in the presence of 2- to 5-fold molar excess of mRNA, followed by cooling to 4°C at a ramp speed of 0.1°C s–1. The resulting mRNA:(dT)45 duplex was diluted to 3–10 nM fluorophore in smFRET assay buffer prior to immobilization on the ZMW using the assay buffer.
Zero-mode waveguides were set up as described previously (52) Briefly, the ZMW chip was hydrated with assay buffer (final concentrations of 50 mM HEPES–KOH, pH 7.4, 3 mM Mg(OAc)2, 100 mM KOAc) for 2 min, followed by incubation with 16 μM NeutrAvidin (Thermo Scientific) for 5 min to allow immobilization of biomolecules. The chip was then washed three times with the assay buffer, followed by addition of 10 nM mRNA:biotin–(dT)45–Cy3.5 duplex, which was allowed to immobilize for 20 min. The chip was then washed again three times to remove non-immobilized nucleic acids, and an imaging buffer containing PCA/PCD oxygen scavenging system and photostabilizer (TSY) (52,54) was added. Prior to imaging on the RS II, the chip was treated with 5% (v/v) each of BioLipidure 203 and 206, 1 mg/ml BSA and unlabeled eIF4E; this blocking step mitigates non-specific Cy5-eIF4E interactions with the surface. Inclusion of this step did not detectably alter the kinetics of eIF4E–mRNA interaction. After initiating the imaging on the RS II, between 4 and 30 nM Cy5-eIF4E were robotically injected onto the waveguide, starting the binding reaction. Where unlabeled eIF4G, and/or unlabelled or Cy3-eIF4A were included in experiments, they were co-delivered with Cy5-eIF4E at the concentrations indicated in the results section. Subunits were pre-incubated for ∼15 min prior to co-delivery. The ZMWs were imaged with 10-min movies acquired at 10 frames/second, at 0.7 μW/μm2 green (532 nm) laser power and 0.07 μW/μm2 red (642 nm) laser power (for dual illumination experiments).
Single-molecule data processing and analysis
Raw movie data were extracted and analyzed with an in-house MATLAB processing pipeline as described previously (25,52,53) Image files were first converted to fluorescence versus time traces. The locations of events in the traces were then manually assigned, resulting in distributions of event and inter-event durations (i.e. lifetimes and arrival times).
For the two-color FRET experiments, events showing anticorrelated bursts between the FRET donor and acceptor were manually assigned as FRET. For kinetic analysis, arrival-time or lifetime distributions for two-color smFRET experiments were constructed from analysis of events occurring on at least 100 mRNA molecules, which included at least 500 events, and typically >1000 events. Addition to the analysis of further molecules beyond this number neither significantly altered the kinetic parameters obtained, nor improved the quality of data fitting. Empirical cumulative distribution functions for unbinned distributions were fit in MATLAB, using nonlinear least-squares regression, to either single-exponential (1) or double-exponential (2) models, as appropriate:
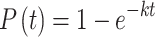 |
(1) |
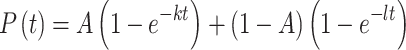 |
(2) |
where P(t) is the cumulative probability distribution function, A is an amplitude term that describes the contribution of each the two exponential phases to the cumulative probability in a double-exponential distribution, and k and l are first-order rate constants. For double-exponential arrival-time distributions, the fast-phase rate, which typically constituted at least 70% of the amplitude, was used for comparison of eIF4E binding between different mRNAs and conditions.
For goodness-of-fit evaluation, fits typically had an R2 value >0.99 (for all distributions generated from experiments with eIF4E-G, eIF4F) and >0.95 (for experiments containing only eIF4E and eIF4E-–eIF4A). Root-mean-squared errors of the fits for arrival-time distributions were typically 0.02 or a lower value; lifetime distributions showed more variable RMSE values with an upper limit of 0.1. RMSE values and 95% confidence intervals of the fits are provided in the Supplementary Tables.
For correlation of kinetic parameters with mRNA lengths (Figure 1H, I; Supplementary Figure S1E, F; Figure 2B) Pearson correlation coefficients (R) were calculated using GraphPad Prism software (Version 9.1.). Correlations with P < 0.05 based on a two-tailed Student's t-test were considered significant.
Figure 1.
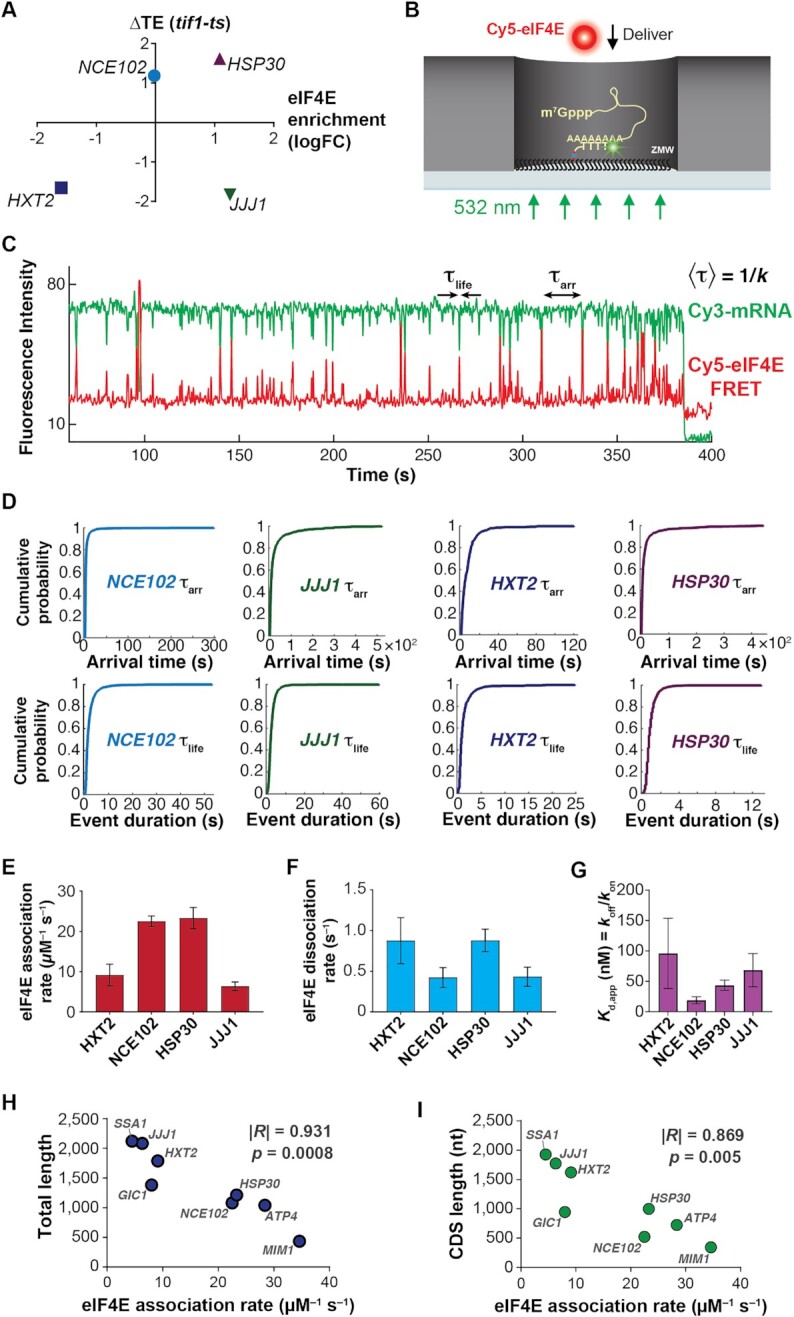
eIF4E interaction dynamics with full-length mRNAs depend on mRNA identity and length. (A) Selection of mRNAs with varying in-vivo enrichment in eIF4E•eIF4G and translation dependence on eIF4A, as measured by Costello et al. (2015), and Sen et al. (2015). (B) Schematic of single-molecule FRET experiment to detect binding of fluorescently-labeled eIF4E to surface-immobilized, fluorescently-labeled mRNA. (C) Sample smFRET trajectory showing eIF4E–mRNA interaction in the absence of other eIF4F components. (D) Representative cumulative distribution functions for eIF4E association with (top) and dissociation from (bottom) full-length mRNAs. (E) eIF4E–mRNA association rates quantified from exponential fitting of arrival-time and lifetime cumulative distribution functions. Error bars reflect the standard errors of the mean for three replicates of an experiment where the eIF4E–mRNA binding rate is measured across at least 100 mRNA molecules. (F) eIF4E–mRNA dissociation rates from the experiments in E. (G) eIF4E–mRNA equilibrium dissociation constants computed from the rates shown in E and F. (H) Dependence of eIF4E–mRNA association rate on mRNA length. The rates for JJJ1, HXT2, HSP30, NCE102, and MIM1 were measured in the present study. The rates for GIC1, SSA1 and ATP4, previously published in Çetin et al. (53), were added to include data points for correlation over a sufficient length range. (I) Dependence of eIF4E–mRNA association rate on coding-sequence length.
Figure 2.

eIF4G1 accelerates eIF4E–mRNA binding in an mRNA-dependent manner and allows the interaction to persist on the translation initiation timescale. (A) eIF4E-mRNA association rates in the absence (grey) and presence (red) of full-length yeast eIF4G1. (B) Fold-stimulation of eIF4E–mRNA association rate by eIF4G1, as a function of mRNA length. (C) Kinetics of eIF4E–mRNA dissociation for transient binding events in the presence (blue) and absence (grey) of eIF4G1. (D) Representative single-molecule fluorescence trace for eIF4E–mRNA binding in the presence of eIF4G1. The inset shows representative transient and prolonged events on an expanded time axis. (E) Cumulative probability distributions of eIF4E–NCE102 mRNA event durations in the absence (grey) and presence (blue) of eIF4G1, showing appearance of slowly-dissociating events when eIF4G1 is present. (F) eIF4E–mRNA dissociation rates for long-lived binding events in the presence of eIF4G1. (G) Apparent equilibrium dissociation constants for the eIF4E–mRNA interaction in the presence of eIF4G1. (H) eIF4E–mRNA association rates in the presence (red) and absence (grey) of eIF4G11–452.
For the three-color FRET experiments, events showing Cy3 signal only were scored as free eIF4A binding. Events showing appearance of Cy3 fluorescence with a concomitant increase in Cy5 fluorescence significantly beyond the negligible expected bleedthrough (52), and showing apparent FRET efficiency changes during the ensuing binding event, were characterized as eIF4E–eIF4A FRET. A further type of binding event where eIF4E–eIF4A FRET disappeared while eIF4A stayed bound was also scored in both the number of occurrences and the dwell time of the initial FRET event. During these Cy3 fluorescence pulses, the dwell time of FRET when eIF4E–eIF4A FRET reappeared was also quantified. Rate constants were quantified by exponential fitting as described above.
RESULTS
eIF4E interaction with full-length mRNAs is highly dynamic and depends on mRNA length
From the ∼6000 S. cerevisiae protein-coding genes (55) we chose mRNAs spanning a range of eIF4E binding in cells (26). In vivo mRNA enrichments in eIF4E and eIF4G are broadly similar; therefore, our selection also reflects enrichment in eIF4G. We restricted mRNA candidates to those that span a range of changes in translation efficiency (‘TE’) on conditional knockdown of eIF4A, but where TE is not significantly dependent on the second translational helicase, Ded1p (29).
We arrived at four mRNAs that permute in vivo eIF4E binding and eIF4A translation dependence (Figure 1A)—with in-vivo eIF4E binding that is below average (HXT2), average (NCE102), or above average (JJJ1, HSP30), and where eIF4A knockdown either reduces translation efficiency (ΔTE < 0; JJJ1, HXT2), vs. where it remains unchanged or potentially increases (ΔTE > 0; NCE102, HSP30). We transcribed, capped and polyadenylated these mRNAs in vitro (Supplementary Table S1; Figure S1A). Analysis of the final transcripts indicated that they were near-quantitatively capped and that their poly(A) tails were consistently ∼100 nt in length (Supplementary Figures S1B, C).
We surface-immobilized the mRNAs for single-molecule fluorescence analysis of eIF4E binding, by hybridizing a fluorescently labeled, biotinylated (dT)45 oligonucleotide to their poly(A) tails (Figure 1B) (53). We then delivered Cy5-labeled eIF4E to the mRNAs (30–70 nM, depending on experiment; the normalized second-order rate constants did not vary with eIF4E concentration in this range) (Supplementary Figure S1D) (25). We chose these concentrations of eIF4E to balance between minimizing non-specific eIF4E–surface binding, which creates a background signal, and ensuring observation of a sufficient number of binding events to allow robust data analysis.
eIF4E delivery led to transient cycles of mRNA–eIF4E single-molecule FRET (Figure 1C), as observed previously for unidentified full-length yeast mRNAs and for capped oligoribonucleotides (25,53). FRET was observed uniformly for the present set of 0.4–2 kb-long mRNAs, suggesting eIF4E is within ∼7 nm of the poly(A) tail when bound to the cap. We previously showed that almost all eIF4E–mRNA binding events result in FRET (53). However, to ensure that mRNA end-to-end proximity was a consistent mRNA feature over a broad length range in our system, which is necessary to reliably interpret our signal, and also to test the possibility that eIF4E induced mRNA end-to-end proximity, we Cy5-labeled the 5′ triphosphate groups (56) of polyadenylated MIM1, NCE102, HXT2 and JJJ1 (∼0.4 kb to ∼2.1 kb), then immobilized them by poly(A) capture (Supplementary Figure S2A). Regardless of mRNA length across this 0.4–2.1 kb range, we observed persistent FRET between mRNA 5′ ends and poly(A) tails (Supplementary Figure S2B–E) for the majority of the Cy5 lifetime (Supplementary Figure S2F). These results are consistent with mRNA folding and tertiary compaction bringing the 5′ and 3′ ends within FRET distance (56–59). Thus, our smFRET signal reliably reports on eIF4E–mRNA interaction (Supplementary Discussion D1).
To quantify the extent to which eIF4E–mRNA kinetics differed between mRNAs, we determined eIF4E–mRNA binding and dissociation rates by exponential fitting of the distributions of waiting times between binding events, and of the event durations (Figure 1D) (53). For our four mRNAs chosen with variable in-vivo eIF4E enrichment and sole dependence on eIF4A, the dynamics differed substantially between mRNAs: association rates ranged over about four-fold, from 6.3 ± 1 μM–1 s–1 for JJJ1 to 23 ± 3 μM–1 s–1 for HSP30 (Figure 1E); dissociation rates depended less on mRNA identity (0.43 ± 0.08 s–1 to 0.9 ± 0.06 s–1; Figure 1F, Supplementary Table S2), as observed previously for populations of unidentified mRNAs from cells (53).
Assuming a two-state, on-off equilibrium-binding model (Kd = koff/kon), eIF4E–mRNA affinities spanned a Kd range of ∼18 ± 6 nM to ∼96 ± 58 nM (Figure 1G), a higher affinity than for the dinucleotide cap analog, and equal or significantly greater affinity than for capped oligonucleotides (25,60,61). However, whilst high-affinity, the bimolecular eIF4E–mRNA interaction is highly dynamic on the initiation timescale.
We did not observe a strong correlation between eIF4E–mRNA equilibrium dissociation constants and in-vivo RIP-seq enrichment; e.g. the JJJ1 mRNA is the most enriched (26) in eIF4E of our set of mRNAs in cells, but its measured equilibrium dissociation constant is indistinguishable within error from HXT2, which is the least enriched. However, mRNAs found to be more enriched in eIF4E in vivo tended on the whole to have faster eIF4E association rates in our experiments (Supplementary Figure S1E), though the trend did not reach the level of a statistically significant correlation. This observation would be consistent with eIF4E–mRNA enrichment being controlled kinetically, rather than thermodynamically in cells. An alternative explanation is that the eIF4E–mRNA interaction does equilibrate in vivo, but the position of equilibrium is dictated by additional translation components, e.g. eIF4G, the eIF4F complex, or other factors.
eIF4E–mRNA binding in vivo and translation dependence on eIF4G show an inverse relationship with mRNA and coding-sequence length (26,28,30). This has been interpreted in terms of more efficient formation of the ‘closed-loop’ mRNP, through enhanced eIF4G–poly(A) binding protein interaction owing to closer end-to-end proximity for shorter mRNAs (34). However, since our and others’ data suggest that equilibrium end-to-end proximity is relatively constant across mRNA lengths, we sought to test the alternative possibility that eIF4E– and or eIF4E•G–mRNA dynamics might intrinsically vary with mRNA length. To address this question, we correlated eIF4E–mRNA interaction kinetics with mRNA leader, coding-sequence, and total length. To provide a wider mRNA length range for this analysis, we supplemented our initial four mRNAs with data for four additional transcripts – ATP4, SSA1, GIC1 and MIM1. We previously measured (53) the eIF4E–mRNA association rates for SSA1, GIC1, and ATP4; here, to extend the range to shorter lengths we newly introduced MIM1 (434 nt). This combined set of mRNAs allowed coverage over a broad range of coding-sequence lengths (0.3–1.7 kb) and total mRNA lengths (0.4–2.1 kb).
We found no correlation of eIF4E association rates with mRNA leader length (Supplementary Figure S1F). However, remarkably, the association rates correlated significantly with total mRNA length (Pearson's R = –0.931, P = 0.0008; Figure 1H); i.e. mRNAs with longer coding sequences bound eIF4E more slowly. As the CDS accounts for 48–90% of the total length for these mRNAs, a similar but slightly less pronounced anticorrelation holds for CDS length (Pearson's R = –0.869, P = 0.005; Figure 1I).
Occluded-volume effects on eIF4E diffusion in zero-mode waveguides due to increased mRNA size are highly unlikely to decelerate eIF4E association, since the hydrodynamic radii of mRNAs in the 0.7–2.0 kb length range (7–12 nm) (62) are at most ∼8% of the zero-mode waveguide diameter (∼150 nm) (52,63). Thus, eIF4E–mRNA association rates differ by over four fold across a CDS length range that encompasses transcripts between the 10th and 80th percentiles of length in the yeast transcriptome (64).
These data suggest that at least one aspect of enhanced closed-loop mRNP formation on shorter mRNAs is their relative ease of binding eIF4E: faster eIF4E on-rates for shorter mRNAs would afford them an increased time-averaged occupancy of eIF4E•eIF4G at the 5’ end, allowing for more frequent interactions with the poly(A)-binding protein—essentially a kinetic formulation of the situation in classical thermodynamic coupling of linked equilibria. However, rather than the frequency of eIF4E•eIF4G–Pab1p encounters being driven by more frequent mRNA end collisions in short mRNAs (34), our data suggest it can be driven by more frequent eIF4E•eIF4G–cap association on those mRNAs. In turn, these findings raised the question of whether similar steric effects would operate for eIF4E•eIF4G or eIF4F.
Yeast eIF4G1 accelerates eIF4E–cap binding in proportion to mRNA length
We next co-delivered eIF4E (25 nM) to the immobilized mRNAs with full-length yeast eIF4G1 (250 nM). We chose this eIF4G1 concentration as the eIF4E•eIF4G equilibrium dissociation constant is ≤ 30 nM (65); eIF4E is thus expected to be quantitatively bound to eIF4G under these conditions. This expectation is consistent with the concentration dependence of the stimulation by eIF4G of Cy5-eIF4E association with capped oligoribonucleotides, which indicates a similar or higher Cy5-eIF4E•eIF4G affinity in our experimental system (25).
We examined the effects of eIF4G on eIF4E–mRNA association across an mRNA length range from MIM1 (434 nt) to SSA1 (2,122 nt). eIF4G (Supplementary Figure S1D) accelerated eIF4E–mRNA association for all mRNAs (Figure 2A). However, the extent of acceleration differed by mRNA, from almost eight-fold for the longest mRNAs (SSA1 and JJJ1) to just over two-fold for the shortest (MIM1), resulting in rates from 32 ± 4 to 89 ± 18 μM–1 s–1 (Supplementary Table S3). The degree of acceleration also correlated significantly with total mRNA and coding-sequence lengths (Figure 2B; Supplementary Figure S3A), again with a more significant correlation for total length than for coding-sequence length; shorter mRNAs showed less acceleration. Nevertheless, despite this accelerative effect eIF4E–mRNA association rates remained faster for shorter mRNAs, though the range of rates was narrower than for eIF4E alone.
The narrowed association-rate range we observed with eIF4G is consistent with cap accessibility being more similar between mRNAs in the presence of eIF4G. The accelerative effects could in principle result from structural changes in eIF4E induced by eIF4G binding, or from eIF4G–mRNA interactions. We found that an eIF4G1 fragment (eIF4G1–452) containing the N-terminal RNA-binding domain but truncated immediately N-terminal to the eIF4E-binding domain, also accelerated eIF4E association (Figure 2H), with the exception of the HSP30 mRNA. Therefore, the accelerative effect is not solely due to the eIF4E–eIF4G interaction. When the N-terminal RNA-binding domain was removed from this truncation (eIF4G83–452; Supplementary Figure S3B), which abolishes its RNA-binding activity (Supplementary Figure S3C), no stimulation of eIF4E–mRNA binding was observed (Supplementary Figure S3D). Thus, direct eIF4G–mRNA interactions play a deterministic role in accelerating eIF4E–cap binding.
We previously showed that the extent of cap-proximal secondary structure impedes eIF4E–mRNA association in the absence of eIF4G, in an approximately linear anti-correlation over a wide range of secondary-structural propensities (53). To investigate whether similar effects operate for eIF4E•eIF4G, we compared the eIF4E•eIF4G–mRNA association rates for mRNAs in our dataset that have well-defined PARS structural data, spanning the range of structuredness in their cap-proximal 30 nucleotides. NCE102, the mRNA with the least structured 5’ end (PARS score: –15.18), bound eIF4E•eIF4G with the highest rate: 89 ± 18 μM–1 s–1. SSA1, which has a highly structured 5’ end (PARS score of 45.7), bound eIF4E•eIF4G the slowest, at 32 ± 4 μM–1 s–1. An mRNA with intermediate cap-proximal secondary structure, HXT2 (PARS score –6.38), also showed an intermediate eIF4E•eIF4G–mRNA association rate of 41 ± 7 μM–1 s–1. Thus, cap-proximal structure that impedes eIF4E–mRNA association also impedes eIF4E•eIF4G–mRNA association.
More frequent eIF4E binding in the presence of eIF4G also raised the possibility that the additional events were due to eIF4E•eIF4G binding to internal, cap-proximal mRNA regions without eIF4E binding the cap. Experiments with uncapped (5′-triphosphate), polyadenylated NCE102 mRNA did result in transient eIF4E–mRNA FRET events (mean duration ∼1 ± 0.31 s), but at only ∼2% of the association rate relative to the capped mRNA (Supplementary Figure S3E–G). This is consistent with residual eIF4E•eIF4G binding either at the 5′ end, or at the 3′ end close to the FRET donor, and bolsters the conclusion that our FRET signal is specific for cap binding.
Taken together, our results imply that while eIF4E•eIF4G–mRNA association rates are dominated by eIF4G, the cap structure offers a point of attachment that biases stable accommodation of eIF4E•eIF4G at the 5′ end. The RNA-binding activity of eIF4G mitigates an mRNA length-dependent barrier to eIF4E–cap binding, but the final association rate still depends on the extent of cap-proximal secondary structure. The results are also consistent with previous results for mammalian eIF4F binding to RNAs (66), in that the affinity of eIF4F for RNA is driven dominantly by eIF4G.
Yeast eIF4G1 prolongs the eIF4E–cap interaction, allowing it to persist on the initiation timescale
Our data were surprising in that, for eIF4E alone, cap-binding events were short relative to the initiation timescale (67,68), contrasting with eIF4E remaining associated with the 48S ribosomal pre-initiation complex throughout scanning (69). Fast eIF4E dissociation would limit how long an intact eIF4F•mRNA complex is available for PIC recruitment. On the other hand, different models have been proposed in relation to whether eIF4E remains cap-bound as the mRNA 5′ end enters the PIC mRNA channel (70,71). eIF4E remaining cap-bound, or dissociating and rapidly rebinding, would favor maintaining eIF4G bound to the mRNA 5’ end, and thus looping of the leader as it moves through the PIC in search of the start site (39,70,72,73). We therefore assessed how eIF4G impacted the eIF4E–cap binding duration.
eIF4G slightly or moderately lengthened transient eIF4E–mRNA binding events for most mRNAs (koff = 0.35 ± 0.01 s–1 to 0.93 ± 0.25 s–1) (Figure 2C; Supplementary Table S3), resembling its effect on eIF4E binding to capped oligonucleotides (25). The effect varied between mRNAs, yielding a slightly narrower range of dissociation rates. However, the interactions remained transient on the initiation timescale.
Strikingly, though, a proportion of eIF4E-mRNA binding events lengthened by an order of magnitude in the presence of eIF4G (∼10–34%, depending on mRNA) (Figure 2D; Supplementary Table S3). The effect was observed as double-exponential behavior in the event-duration distribution (Figure 2E), and varied between mRNAs (koff = 0.03 ± 0.01 s–1 to 0.15 ± 0.01 s–1) (Figure 2F). These long events were not observed with the eIF4G1–452 fragment (Supplementary Figure S3H, I), implying they result from direct eIF4E–eIF4G interaction. Increasing the eIF4G concentration to 1 μM did not increase the relative proportion of the longer events (Supplementary Figure S3J, K), arguing that the remaining transient events are not due to eIF4E–mRNA binding without eIF4G. Similarly, titrating the eIF4G concentration from 75 to 250 nM did not alter the eIF4E–mRNA association rate, consistent with saturation of eIF4E in the eIF4E•eIF4G complex under our experimental conditions (Supplementary Figure S3L). As the Cy5 lifetime is >2 min in our illumination conditions (Supplementary Figure S2B–E), disappearance of FRET is unlikely to be due to Cy5 photobleaching.
Taken together, these results imply that the two dissociation events occur from two different states of the eIF4E•eIF4G•mRNA complex. The simplest possible interpretation for the identity of the second state is an alternative conformation in which one or more of the eIF4G RNA-binding domains have engaged the mRNA, leading to a higher energetic barrier for detachment of eIF4E from the eIF4E•eIF4G•mRNA complex, and thus the slower dissociation rate. Put otherwise, the data as a whole point to an accommodation mechanism where initial encounter of eIF4E•eIF4G with the mRNA through the cap structure precedes formation of more stable eIF4G–mRNA contacts.
These combined effects of eIF4G on eIF4E association and dissociation drastically enhanced the apparent eIF4E–cap affinity: Kd,app values for the interaction were reduced ∼4.8 – 15.8-fold (Kd range: 2.8 ± 0.9 to 23 ± 9 nM, calculated based on a weighted average of the two dissociation rate constants) (Figure 2G). As in the case of eIF4E alone, these equilibrium dissociation constants did not trend with the in-vivo RIP-seq eIF4E enrichments of the mRNAs, but, on the whole, the mRNAs enriched in eIF4E again showed faster association rates (Supplementary Figure S3M). This would also be consistent with eIF4E•eIF4G–mRNA association being kinetically, rather than thermodynamically controlled in vivo.
Taken together, our data show that the eIF4G RNA-binding activity is central to both accelerating cap recognition and allowing it to persist on the initiation timescale. While the accelerative effect is more pronounced on longer mRNAs, shorter mRNAs still bind eIF4E•eIF4G faster, at rates inversely dependent on cap-proximal secondary structure. Also, while eIF4G induces long eIF4E–mRNA binding events, the association rates still confer greater variability in eIF4E–mRNA interaction dynamics between mRNAs.
An eIF4F-independent function of eIF4A accelerates eIF4E–cap binding, depends on ATP binding but not hydrolysis, and correlates with in-vivo translation-efficiency dependence on eIF4A
Free eIF4A (i.e. not eIF4F-bound) is present at ∼20 μM in vivo (47,74,75), in large excess over eIF4E•eIF4G. Despite its abundance, cellular eIF4A appears to be rate-limiting for translation, at least in yeast—i.e. small reductions in concentration substantially reduce translational output (76). We recently reported that free yeast eIF4A (i.e. not bound to eIF4G or eIF4F) increased eIF4E association rates across mRNA populations (53). However, it remained unclear whether this effect operates to the same extent for all mRNAs, or whether they are affected differently. Moreover, the relationship of this phenomenon to translation dependence on eIF4A was unclear.
We co-delivered eIF4E to the immobilized mRNAs with eIF4A (2 μM) in the presence of 2.5 mM ATP. For these experiments we chose the HXT2, NCE102, HSP30 and JJJ1 mRNAs as their translation efficiency in vivo depends on eIF4A, but not the translational helicase Ded1p (29). We chose 2 μM as the eIF4A concentration to ensure a substantial excess of eIF4A over eIF4E, and where eIF4A–RNA equilibrium binding is expected to be substantially saturated (46).
eIF4A and ATP addition indeed accelerated eIF4E-cap association for each mRNA (Figure 3A,B). However, the fold-acceleration ranged from 1.2 ± 0.1 (marginal, if any, stimulation) to 4.2 ± 0.8-fold, yielding rates from 24.8 ± 0.1 to 52.8 ± 7.2 μM–1 s–1 (Supplementary Table S4). As observed for eIF4G, the fold-acceleration of eIF4E–mRNA binding induced by eIF4A again was greater for longer (JJJ1, HXT2) vs. shorter (NCE102, HSP30) mRNAs. The fastest association rate was again observed for the least 5′-structured mRNA (NCE102). No acceleration was induced by eIF4A in the absence of ATP (Supplementary Figure S4). However, acceleration was quantitatively indistinguishable when ATP was substituted by the slowly-hydrolyzable analog ATP-y-S (Figure 3C; Supplementary Table S5). We performed the ATP-less and ATP-y-S experiments with the JJJ1 and NCE102 mRNAs as they showed substantial eIF4A enhancement of eIF4E binding. We opted for ATP-y-S, rather than the non-hydrolyzable AMPPNP, because AMPPNP does not support PIC–mRNA recruitment (35). Nucleotide binding to eIF4A is thus sufficient for the accelerative effect. Meanwhile, eIF4E–mRNA binding events with free eIF4A and ATP remained essentially the same length relative to the eIF4E-only condition (Figure 3D).
Figure 3.
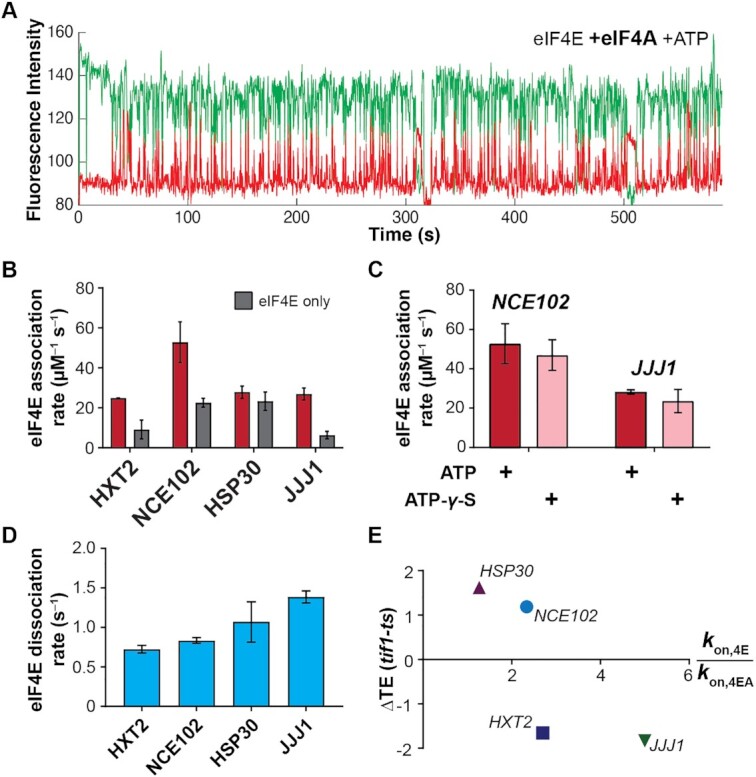
Free eIF4A with bound ATP stimulates eIF4E–mRNA association independently of eIF4G. (A) Representative single-molecule trace showing eIF4E–mRNA interaction in the presence of 2 μM eIF4A and 2.5 mM ATP. (B) eIF4E-mRNA association rates in the presence (red) and absence (grey) of eIF4A and ATP, compared with eIF4E-only rates. (C) eIF4E–mRNA association rates in the presence of eIF4A and ATP or ATP-γ-S, for the NCE102 and JJJ1 mRNAs. (D) eIF4E–mRNA dissociation rates in the presence of free eIF4A and ATP. (E) Relationship between translation-efficiency dependence on eIF4A and the fold-increase in eIF4E–mRNA association rate induced by free eIF4A and ATP.
This reduction in kinetic barrier height for eIF4E–mRNA binding caused by eIF4A could result from modulation of either steric (i.e. cap-accessibility) or electrostatic factors that determine the nature of the transition state for the binding reaction. Given that the electrostatics of eIF4E–cap interaction are thought to be dominated by elements common to all mRNAs (i.e. the m7G moiety and a portion of the cap-structure triphosphate bridge) (77), an eIF4A-mediated increase in cap accessibility most reasonably accounts for the accelerated binding. Since ATP hydrolysis is not required for the effect, it is unlikely to result from unwinding of secondary structures by eIF4A, although, as in the case of eIF4E•eIF4G, cap-proximal secondary structures clearly contribute to the final eIF4E–mRNA binding rate (53). Because the accelerative effect increased in proportion to mRNA length, we propose that it results from a weakening of tertiary or higher-order mRNA structures caused by formation of the eIF4A•mRNA complex—eIF4A ‘clamps’ the tertiary structure in a more open, decompacted conformation. Indeed, a number of DEAD-box helicase enzymes have been implicated in RNA-clamping roles, which in some cases promotes association of RNA-binding proteins (78). Along similar lines, eIF4A was recently shown to block intermolecular RNA–RNA interactions that form polymeric condensates in vitro and stress granules and P-bodies in vivo (79).
To probe the relevance of eIF4A-mediated acceleration of cap binding to translation in vivo, we correlated the eIF4E–mRNA association rates for each mRNA with publicly-available ribosome-profiling data for the mRNAs’ eIF4A–translation efficiency dependence (29). eIF4A indeed accelerates eIF4E binding to a greater degree for mRNAs that are hyperdependent (HXT2, JJJ1) on eIF4A compared to hypodependent mRNAs (HSP30, NCE102) (∼1.8-fold versus ∼3.6-fold) (Figure 3E). Thus, our results are consistent with ‘free’ eIF4A, outside the eIF4F complex, playing a physiologically relevant role in promoting cap-dependent translation initiation.
The full eIF4F complex modulates eIF4E–mRNA interaction dynamics in an ATP-dependent manner
We next assessed how formation of the full eIF4F complex modulated eIF4E–mRNA binding. We again included eIF4A (2 μM) and eIF4G (250 nM), reflecting their relative cellular concentrations and ensuring that eIF4E is near-quantitatively bound to both proteins in the eIF4F complex. eIF4E was included at 10–30 nM, as eIF4G potentiates nonspecific eIF4E interactions with the ZMW surface. Based on past thermodynamic analysis (65), under these conditions eIF4E is expected to be quantitatively bound in an eIF4F complex.
In the eIF4F complex without ATP, eIF4E–mRNA association accelerated on all mRNAs relative to eIF4E alone (Figure 4A,C), with rates between 43.3 ± 9.8 μM–1 s–1 for JJJ1 and 102.2 ± 0.5 μM–1 s–1 for HSP30 (Supplementary Table S6). This rate for HSP30 was the fastest measured in the present study. Acceleration again varied between mRNAs, from 6.8 ± 1.9 to 3.0 ± 0.4-fold. The net effect was to differentiate eIF4E–mRNA binding between mRNAs relative to the eIF4E•eIF4G condition, though, as with eIF4E and eIF4E•eIF4G, the shorter mRNAs (NCE102 and HSP30) retained faster association rates. Since the individual effects of eIF4G and eIF4A•ATP on eIF4E–mRNA cap association served to narrow the range of eIF4E–mRNA association rates relative to the eIF4E-only condition, this differentiation of rates observed with all components of the eIF4F complex present is reasonably ascribed to intersubunit coordination that leads to a different mode of mRNA engagement by the eIF4F heterotrimer than is afforded by the sum of the individual subunit activities. Based on the available structural and biochemical data (39,41,42,80,81), and supported by the results described below, we propose that the conformational changes induced in the eIF4F subunits on formation of the eIF4F complex enhance the ability of eIF4F to discriminate between mRNAs in cap recognition, relative to eIF4E•eIF4G.
Figure 4.

The eIF4F complex discriminates eIF4E–mRNA interaction dynamics in an ATP-dependent manner. (A) Representative single-molecule fluorescence trace for eIF4E–mRNA interaction in the eIF4F complex without added ATP, on NCE102. (B) Representative trace for eIF4E–mRNA binding in the eIF4F complex with ATP, on NCE102. (C) eIF4E–mRNA association rates for the eIF4F complex without ATP (red), compared with the rates in the presence of eIF4E only (grey). (D) Dissociation rates of transient eIF4E–mRNA interactions in the eIF4F complex without ATP. (E) Dissociation rates of long-lived eIF4E–mRNA interactions in the eIF4F complex without ATP. (F) eIF4E–mRNA association rates in the eIF4F complex with ATP (red), compared with the rates in the presence of eIF4E only. (G) Dissociation rates of transient eIF4E–mRNA interactions in the eIF4F complex with ATP. (H) Dissociation rates of long-lived eIF4E–mRNA interactions in the eIF4F complex with ATP.
Long and short eIF4E–mRNA binding events were also observed with eIF4F, and their relative incidence was unchanged within experimental error. Transient eIF4E dissociation occurred at around ∼0.3–0.6 s–1, similar to eIF4E•eIF4G (Figure 4D), while long events dissociated at between ∼0.05 and 0.10 s–1 (Figure 4E), slightly faster than for eIF4E•eIF4G. These results again place eIF4G as a dominant kinetic contributor to eIF4F–mRNA affinity, echoing thermodynamic data for human eIF4F (66).
However, addition of ATP led to both mRNA-specific and global changes in dynamics (Figure 4B, F–H). The HSP30 association rate was strikingly reduced, from being the fastest among the mRNAs, to being the slowest (32.8 ± 7.0 μM–1 s–1) with ATP present (Figure 4F; Supplementary Table S7). This almost entirely reversed the acceleration in eIF4E–mRNA binding afforded to HSP30 by eIF4F. On the other hand, the NCE102, HXT2 and JJJ1 mRNAs showed small or no reductions in association rate on ATP addition. Since the eIF4F-independent activity of eIF4A•ATP universally had the effect of accelerating eIF4E–mRNA association (Figure 3B), our data imply that the kinetic changes in eIF4E–cap binding observed on addition of ATP to eIF4F are mediated through eIF4A bound in the eIF4F complex.
As in the cases of eIF4E with added ‘free’ eIF4A(ATP), and eIF4E•eIF4G, the fold-acceleration of eIF4E–mRNA binding with eIF4F in the presence of ATP, relative to eIF4E alone, retained a length dependence—i.e. eIF4F(ATP) on the whole accelerated eIF4E binding to a greater extent for longer mRNAs (e.g. JJJ1 versus HSP30). Moreover, the net association rate following this acceleration was again fastest on the 5′-unstructured NCE102 mRNA.
ATP addition also shortened both the long and short eIF4E–mRNA binding events for the eIF4F complex, i.e. the complex became more dynamic (Figure 4B, G, H). For the long events, this effect ranged from a modest ∼50% for HSP30 to around two-fold for HXT2. Dissociation kinetics also became even more similar between mRNAs than in the other conditions, pointing to a common rate-limiting step for eIF4E dissociation from the eIF4F•mRNA complex. However, the identity of that step still remained unclear.
Because eIF4E–mRNA dynamics became much more similar between mRNAs with eIF4F in the presence of ATP, no trends were discernible with respect to the in vivo RIP-seq enrichment data. However, since yeast eIF4A does not co-purify with eIF4E•eIF4G (8), direct comparisons may not be possible between the RIP-seq experiment and our experimental conditions where eIF4E•eIF4G is exposed to constant and high eIF4A concentrations. Nevertheless, and as for eIF4E•eIF4G alone, our data indicate that the main source of mRNA-to-mRNA variability in eIF4E–mRNA interaction for the eIF4F(ATP) complex is the association rate.
We were initially surprised at the extent to which eIF4F(ATP) tended to equalize eIF4E association rates between mRNAs, with the exception of the NCE102 which had a higher association rate. A priori, this would run counter to the proposal that differential eIF4F interaction differentiates translation between mRNAs. However, inspection of publicly available published data revealed that the JJJ1, HXT2 and HSP30 mRNAs have quite similar ribosome occupancies and densities in vivo as measured by ribosome density mapping (82), and JJJ1 and HXT2 have similar translation efficiencies as measured by ribosome profiling (83) (the translation efficiency for HSP30 was not included in this ribosome profiling dataset). Meanwhile, NCE102 has significantly higher ribosome occupancies and densities, along with a higher translation efficiency than the other mRNAs (Supplementary Table S8). Thus, our data are consistent with a kinetic-control model for cap recognition in vivo, where differential eIF4F association rates limit translation differentially between mRNAs.
Simultaneous direct observation of eIF4A– and eIF4E–mRNA interaction reveals variable eIF4A and eIF4F dynamics on different mRNAs
To broaden our view of eIF4F intersubunit coordination during cap recognition, we performed three-color experiments that included fluorescent Cy3-eIF4A (15 nM) (Supplementary Figure S5A), co-delivered with Cy5-eIF4E (10 nM) and unlabelled eIF4G (250 nM) to surface-immobilized Cy3.5-mRNA (Figure 5A). Cy3-eIF4A RNA-dependent ATPase activity was indistinguishable from the unlabeled protein (Supplementary Figure S5B,C). The Cy3-eIF4A concentration was limited to 15 nM to prevent non-specific (i.e. RNA-independent) interactions with the surface at higher concentrations, which hinder data analysis. The concentrations of eIF4E and eIF4G were chosen for the reasons described previously. For these experiments we chose JJJ1, NCE102 and HXT2, which span the range of stimulation of eIF4E binding by eIF4A.
Figure 5.

Three-color smFRET to probe eIF4F- and eIF4A-mRNA interaction dynamics. (A) Schematic of the three-color smFRET experiment with two donors (on eIF4A and mRNA) and one acceptor (eIF4E). A FRET signal between Cy5-labeled eIF4E and the Cy3.5-labeled mRNA is tracked at the same time as a FRET signal between Cy3-eIF4A and Cy5-eIF4E. (B) Relative incidence of eIF4A-mRNA binding occurring with and without FRET to eIF4E. n is the number of molecules analyzed to enumerate the event types on each mRNA. (C) Reaction pathway and representative smFRET trace showing concomitant mRNA binding of eIF4E and eIF4A with eIF4E–eIF4A FRET, consistent with eIF4F–mRNA binding, on JJJ1. The eIF4A–mRNA lifetime measured in panel E is indicated. (D) Reaction pathway and representative single-molecule fluorescence trace for eIF4A–mRNA binding without eIF4E–eIF4A FRET on JJJ1. These events result both from ‘free’ eIF4A–mRNA interaction (‘eIF4A(–EG)’), and eIF4F–mRNA interaction where eIF4E is unlabeled (‘eIF4A(+EG)’. The Cy3 and Cy5 signals were manually corrected by linear subtraction to equalize their background values, for clarity of presentation. (E) eIF4A–mRNA dissociation rates following eIF4A–mRNA binding with eIF4E, i.e. with observable eIF4E–eIF4A FRET as shown in panel C. The Cy3 and Cy5 signals were manually corrected by linear subtraction to equalize their background values, for clarity of presentation. (F) eIF4A–mRNA dissociation rates following eIF4A–mRNA binding without FRET to eIF4E. (G) eIF4A–mRNA association rates across all binding event types.
We observed two types of eIF4A–mRNA binding event. In the first, eIF4A binding was accompanied by eIF4E–eIF4A FRET. (53%, 45% and 43% of all eIF4A binding events for HXT2, JJJ1 and NCE102, respectively) (Figure 5B, C). Since no direct eIF4E–eIF4A interaction is known, we interpret these events to represent assembly of an intact eIF4F complex.
In a second event type, eIF4A bound mRNA without FRET to eIF4E (47%, 55% and 57% for HXT2, JJJ1 and NCE102) (Figure 5D). This second class of events results from two separate processes: binding of an authentic eIF4F complex in which the eIF4E is non-fluorescent, and eIF4A–mRNA binding outside of eIF4F, i.e. ‘free’ eIF4A–mRNA interaction (approximately 10% of eIF4A is expected to be free—i.e. not bound to eIF4E•eIF4G—under our conditions) (65). Two types of this no-FRET eIF4A–mRNA binding mode were observed – transient and longer-lived, with the transient events constituting 25–90% of the eIF4A–mRNA encounters, depending on mRNA (Supplementary Figure S6; Table S9).
The eIF4A–mRNA dissociation rates in eIF4A–eIF4E co-binding events (i.e. authentic eIF4F–mRNA complex formation) were kinetically similar between mRNAs, and ranged from 0.027 s–1 ± 0.007 (JJJ1) to 0.041 ± 0.003 (HXT2) (Figure 5E; Supplementary Table S10). Conversely, the dynamics of eIF4A–RNA binding events lacking eIF4E–eIF4A FRET varied between mRNAs and also differed kinetically from events where eIF4E–eIF4A FRET was observed, consistent with a portion of them reporting on eIF4A–mRNA interactions outside the eIF4F complex. The dissociation rates for the dominant (higher-amplitude) eIF4A dissociation pathway in these no-FRET events also varied considerably between the mRNAs, from 0.095 ± 0.03 s–1 (NCE102) to 0.325 ± 0.003 s–1 (HXT2) (Figure 5F; Supplementary Table S9).
eIF4A–mRNA association rates were identical between mRNAs within experimental error (Figure 5G, Supplementary Table S9). Interestingly, then, and in contrast to eIF4E, variable affinity of free eIF4A for different mRNAs appears to result from differences in the lifetimes of the eIF4A•mRNA complexes. This echoes results that demonstrate different conformational dynamics of eIF4A in the presence of RNA oligonucleotides that differ in their duplex properties, as well as unwinding by eIF4F (46,80). Extrapolating our data to cellular concentrations of eIF4A, these results further implicate free eIF4A as a multifunctional ‘mRNA chaperone’ that maintains cap accessibility for eIF4F binding.
ATP hydrolysis ejects eIF4E from the cap after initial eIF4F–mRNA binding
Initial eIF4F•mRNA complex formation sets the stage for recruitment to the mRNA of the 43S ribosomal pre-initiation complex. A key question around cap recognition is how the mRNA 5′ end is transferred into its channel on the 40S subunit if the cap is bound by eIF4E/eIF4F. However, the sequence of events occurring in the eIF4F•mRNA complex immediately after its formation remains incompletely understood (8).
In our two-color smFRET experiments with eIF4F, we found that addition of ATP increased the rate of eIF4E–mRNA dissociation. Our three-color experiments now allowed us to directly follow the fates of eIF4E and eIF4A once bound to mRNA. In these three-color experiments, we also found that eIF4E fluorescence frequently disappeared before eIF4A fluorescence after formation of an eIF4F•mRNA complex detected by co-arrival of Cy5-eIF4E and Cy3-eIF4A fluorescence displaying Cy3-Cy5 FRET (Figure 6A). Within these eIF4F–mRNA events, eIF4E dissociation prior to eIF4A was the most common outcome, and occurred for ∼66% of eIF4F–mRNA binding events on NCE102, 51% on JJJ1 and 61% on HXT2 (Figure 6B). Simultaneous disappearance of eIF4E and eIF4A fluorescence thus occurred in 34%, 49%, and 39% of eIF4F–mRNA binding events on NCE102, JJJ1 and HXT2, respectively. We observed hardly any occurrences of eIF4A departing the mRNA before eIF4E. Thus, there is a preference for disrupting eIF4E–cap interaction whilst maintaining eIF4A–mRNA binding. This echoes findings for mammalian eIF4F where cap binding appears to reduce eIF4E affinity for eIF4F (8,84).
Figure 6.

Dynamic coordination within eIF4F after cap recognition. (A) Representative smFRET trajectory showing event with ejection of eIF4E prior to eIF4A, and fluctuations in the eIF4F•mRNA conformation on JJJ1. (B) Relative incidence of initial eIF4E dissociation vs. eIF4E/eIF4A co-dissociation from eIF4F•mRNA complexes. n is the number of molecules analyzed to enumerate the event types on each mRNA. (C) Reaction pathway and annotated representative single-molecule fluorescence trajectory for eIF4F•mRNA complex formation and dynamics, observed by dual red/green illumination which directly reports on the presence of both Cy3-eIF4A and Cy5-eIF4E. (D) Rates for the initial eIF4E–mRNA dissociation event after eIF4F–mRNA complex formation. (E) eIF4E–mRNA dissociation rates for events where eIF4E rebinds mRNA following initial dissociation from eIF4F•mRNA. (F) Reaction pathway and representative single-molecule fluorescence trajectory for a four-color experiment where eIF4G is non-specifically labeled with Cy5.5, allowing its simultaneous detection with Cy3-eIF4A and Cy5-eIF4E. eIF4E and eIF4G fluorescence co-depart the mRNA. (G) Reaction pathway and annotated representative single-molecule fluorescence trajectory for eIF4F•mRNA complex dynamics with ATP-γ-S. The Cy3 and Cy5 signals were manually corrected by linear subtraction to equalize their background values, for clarity of presentation. (H) Relative incidence of eIF4E or eIF4A dissociation, or co-dissociation from the eIF4F•mRNA complex in the presence of ATP-γ-S.
Disappearance of eIF4E fluorescence could be due either to its complete dissociation from eIF4F•mRNA, or adoption of an extended eIF4F conformation that places eIF4E out of FRET range to both mRNA and eIF4A. To differentiate between these possibilities, we repeated the experiment with direct excitation of the Cy5-eIF4E fluorophore. In this illumination scheme, all eIF4E–mRNA interactions are detected, rather than only interactions that produce FRET—i.e. adoption of a no-FRET conformation would be reported by loss of FRET but persistence of the Cy5 signal. We found that disappearance of eIF4E–mRNA FRET following eIF4F–mRNA binding was due to complete dissociation of Cy5-eIF4E from mRNA for 90% of the FRET events on the JJJ1 mRNA (Figure 6C). Put otherwise, eIF4E is ejected from the eIF4F•mRNA complex shortly after cap recognition.
We also observed relatively frequent eIF4E rebinding after initial ejection (Figure 6C). However, while the first eIF4E dissociation event occurred at a rate of 0.07 – 0.09 s–1 across all mRNAs, dissociation during the subsequent rebinding events was slightly faster (koff ∼ 0.11–0.14 s–1) (Figure 6D, E; Supplementary Table S11). This raised the question of whether eIF4E was dissociating alone, or along with eIF4G. To directly address this question, we non-specifically labelled full-length eIF4G with Cy5.5, allowing eIF4E and eIF4G to be visualized simultaneously as they interact with mRNA. We then co-delivered labelled eIF4E, eIF4G and eIF4A to surface-immobilized mRNA. The most common behaviour observed after co-arrival of eIF4E, eIF4G and eIF4A (i.e. eIF4F•mRNA complex formation) was that the Cy5-eIF4E and Cy5.5-eIF4G fluorescence departed the mRNA simultaneously (Figure 6F). Thus, our data support a model where eIF4E•eIF4G is ejected from the cap as a unit shortly after eIF4F•mRNA complex formation, leaving eIF4A bound to mRNA. This model is also consistent with the finding that RIP-seq enrichments for eIF4E and eIF4G1 are highly similar in yeast (26); if eIF4E was frequently ejected on its own, RIP-seq enrichments would be expected to be higher for eIF4G than for eIF4E.
To establish the role of ATP hydrolysis in this ejection process, we substituted ATP with ATP-γ-S and monitored the dynamics of the corresponding eIF4F•mRNA complexes (Figure 6G). With the slowly-hydrolyzable analog, the relative incidence of eIF4E–eIF4A co-dissociation from RNA increased at the expense of eIF4E ejection (Figure 6H).
Thus, taken together our data indicate that eIF4A-catalyzed ATP hydrolysis occurring shortly after formation of the eIF4F•mRNA complex ejects eIF4E, and most likely eIF4E•eIF4G, from the mRNA 5′ end.
DISCUSSION
An mRNA-centric model for cap-recognition dynamics
Our results reveal that eIF4E interactions with full-length mRNAs are highly dynamic on the initiation timescale, and that the dynamics vary between mRNAs. This variability is dominantly due to differences in eIF4E–mRNA association rates, which are influenced by mRNA features and tuned by eIF4F subunits and the presence of ATP. For the mRNAs studied, we find that the eIF4E–mRNA association rate in the eIF4F•ATP complex largely accounts for relative translation efficiency in vivo. Our results thus provide experimental evidence supporting a model where translation initiation is kinetically controlled via the eIF4E–mRNA association rate, rather than the equilibrium affinity, echoing proposals from mathematical modelling of translational control (17).
Our results point to cap-proximal secondary structure as playing an important role in defining the cap-recognition rate. However, we find unexpectedly that increased mRNA length also impedes eIF4E–mRNA association in a manner that is overcome by eIF4G and eIF4A.
Taken together, our data support a model where a topographically condensed mRNA molecule initially has limited 5′ cap accessibility for eIF4E binding (Figure 7). This steric block results in a relatively wide variation of eIF4E–mRNA association rates, with slower rates for longer mRNAs where eIF4E access to the cap is sterically more demanding. The RNA-binding activities of eIF4G or free eIF4A(ATP) both separately overcome this steric block, and to a greater extent on longer mRNAs. In the case of free eIF4A, ATP binding but not hydrolysis is required to accelerate eIF4E–cap association. Because eIF4G and eIF4A bind RNA through different structural mechanisms, our findings point away from secondary structures as the main source of length-dependent steric hindrance, and rather to tertiary contacts in longer mRNAs that sterically oppose eIF4E–cap attachment.
Figure 7.

Mechanistic model and summary of kinetic data for eIF4F–mRNA interaction dynamics.
While detailed knowledge of mRNA structures in vivo remains incomplete, the available data suggests that cytosolic mRNAs in ribonucleoprotein assemblies prior to translation do not adopt an extended linear structure (57,85). Rather, intra-mRNA interactions promote secondary and tertiary structural contacts that contribute to establishing a ‘condensed’ or compacted state of the RNA polymer; active translation and polysome formation reverses this condensation by an order of magnitude (85,86). Even during active translation, electron microscopy of mammalian polysomes also suggests that mRNAs are not fully extended (87,88). In this manner, even if the mRNA ends are close, the overall structure of the mRNA can adopt more or less compacted forms depending on bound proteins or its translational status. Our finding that acceleration of eIF4E–mRNA binding by eIF4A and eIF4G is length-dependent is also consistent with such a model, as longer mRNAs are expected to have more opportunities to make intrastrand contacts. Our experimental conditions most closely resemble the situation for a pre-translational mRNA as it begins its first round of translation. Further studies will be required to directly assess the impact of active translation on eIF4F–mRNA dynamics.
Formation of the full eIF4F complex modulates the eIF4E–mRNA association rate relative to eIF4E•eIF4G alone, and addition of ATP further modulates the kinetics. This is consistent with allosteric structural crosstalk between eIF4A and eIF4G, and also with altered eIF4A structural dynamics in the presence of eIF4G and ATP (41,42,81). Nevertheless, our results still place eIF4G as playing a central role in defining the dynamics of cap recognition.
An eIF4F-independent role for eIF4A in translation
eIF4F- or eIF4G-independent acceleration of eIF4E–mRNA association by eIF4A was an unexpected finding. Past in-vitro studies have shown that eIF4A interaction with mRNA features beyond the 5′ leader accelerates loading of the first ribosome onto mRNA. This effect was attributed to relief of steric hindrance to pre-initiation complex recruitment due to transient, global intra-mRNA interactions (35). Our data indicate that this effect may at least partially be mediated by effects on cap recognition by eIF4F.
The relationship between this stimulatory effect of eIF4A and in-vivo translation dependence on the enzyme suggests that the activity is physiologically relevant. Nevertheless, in our experiments with eIF4F, excess eIF4A did not appear to accelerate cap binding relative to the eIF4E•eIF4G condition. This raises the question of whether or how the eIF4F-independent eIF4A activity might contribute to translation in cells. We suggest that this activity is functionally redundant with that of eIF4G in vitro, but is important in vivo. Along these lines, modest depletion of cellular eIF4A concentration reduces translational output in yeast cells, even though eIF4A is present in significant excess over eIF4E and eIF4G (76) and in vast excess of the equilibrium dissociation constant for eIF4A from eIF4E•eIF4G (65). In the same depletion experiments translation was more sensitive to eIF4A than eIF4E depletion, suggesting impact on an eIF4A function outside the eIF4F complex. Our single-molecule experimental approach constrains us by definition to using vast excesses of translation factors over mRNA, and thus does not fully recapitulate cellular conditions – where mRNA and eIF4E•eIF4G concentrations are quite similar, mRNAs compete for eIF4F (16,17), and eIF4E concentrations are limiting for translation. mRNA competition for eIF4F in vivo is expected to confer increased importance for the eIF4F-independent role of eIF4A. Moreover, this eIF4F-independent role of eIF4A echoes roles proposed for eIF4A in modulating RNA condensation and stress-granule formation (79). Additional RNA helicases (89), RNA-binding proteins targeted to specific mRNA features (90), or indeed general RNA-binding proteins (91), are also predicted to impact eIF4E–mRNA dynamics in this model, opening additional avenues for broad or targeted regulation of cap recognition.
mRNA length sensing during early initiation
Our results show that eIF4E– and eIF4F–mRNA dynamics are sensitive to coding-sequence and mRNA length, and that RNA-binding activity of individual initiation factors partially mitigates this sensitivity. These findings augment, or perhaps invert, the previously-proposed model that closed loop formation is more efficient on short mRNAs (34). Indeed, cryo-EM studies have found that polysomes remain circular for extended periods even without mRNA polyadenylation (88), even in actively translating cell lysates. In addition to, or perhaps instead of poly(A)-binding protein being more efficient at contacting eIF4E•eIF4G on shorter mRNAs, our results suggest that intrinsically faster eIF4E•eIF4G binding on shorter mRNAs allows more rapid formation of eIF4G–Pab1p interactions.
Importantly though, our data do not exclude a role for poly(A)-binding protein in modulating eIF4F•mRNA dynamics. PABP–eIF4G binding could alter eIF4F conformational dynamics to increase the likelihood of efficient binding of eIF4E to the mRNA cap. This would be consistent with the ability of poly(A)-binding protein to stimulate translation on non-polyadenylated mRNAs when provided poly(A) in trans (92). Moreover, several classes of mRNAs have been identified with differing relative enrichments in eIF4E•eIF4G and Pab1p (26), pointing to the potential for additional modes of crosstalk between these factors in cap recognition. Interestingly, this may differ in mammals, where the UTR regions comprise a greater portion of the mRNA length, and ORF length appears to have a reduced impact on translational efficiency (93).
That we observed a striking length dependence on a range of mRNAs picked arbitrarily based on size, and that the length dependence persists across conditions where the mRNA is exposed to different RNA-binding proteins (e.g. eIF4G, eIF4A), suggests that specific sequence features may be less important in determining eIF4F–mRNA interaction dynamics than simply the overall length. Put otherwise, the probability is rather low of obtaining a strong length-rate correlation if sequence effects were deterministic for the kinetics. Our results certainly do not exclude sequence elements in other mRNAs from further controlling eIF4F–mRNA dynamics; however, such effects are not apparent in our study. Similar to eIF4A and eIF4G, other RNA binding proteins may also act to modulate length-dependent eIF4E–mRNA interaction dynamics.
Dynamic eIF4F subunit coordination in the eIF4F•mRNA complex
Our results point to a dynamic progression of yeast eIF4F–mRNA interactions that unfold during cap recognition. Initial eIF4F–mRNA binding is kinetically biased to the cap structure through the eIF4E–cap interaction, which our data suggest precedes stable formation of eIF4G–mRNA contacts. The presence of eIF4A and ATP in the eIF4F complex further modulate this efficiency of eIF4E accessing the cap. After accommodation of eIF4F onto the mRNA, eIF4G–mRNA interactions maintain the eIF4E–cap interaction on the initiation timescale, though the complex is conformationally dynamic. eIF4A ATP hydrolysis then promotes eIF4E•eIF4G dissociation from the mRNA.
Each eIF4F subunit thus plays a role in establishing the net efficiency of cap recognition. mRNA identity—asserted through a combination of length and structural features – contributes more to variability in cap recognition toward the beginning of this sequence of events, while the factor activities dominate the later stages. As discussed below, such an arrangement is optimal for maximizing mRNA-to-mRNA discrimination through kinetic control of the recognition process.
Lack of structural information renders it challenging to assess the nature of conformational rearrangements in the eIF4F•mRNA complex. However, a structure of the human 48S PIC that included partial density for eIF4G indicated that the protein is highly flexible; likewise, a structure of the yeast 48S PIC did not identify the position of eIF4F, attributed to its dynamic nature (72,94), and pointing to this as one potential source of the conformational rearrangements. Similarly, eIF4G stabilizes a closed conformation of eIF4A (42), and this is associated with a reciprocal conformational change in eIF4G (81). Whatever their exact nature, our data suggest that conformational changes in the eIF4F•mRNA complex involve large-scale intersubunit coordination. Further experiments will be required to map out the topology and dynamics of this intricate molecular assembly.
Moving forward through the initiation process, our data also suggest a molecular mechanism for guiding the mRNA as it accommodates onto the 40S subunit during PIC recruitment. Our four-colour single-molecule fluorescence experiments strongly suggest that eIF4G is ejected from the cap at the same time as eIF4E (Figure 6F). In this model, ejection of eIF4E and eIF4G following eIF4A-catalyzed ATP hydrolysis frees the 5’ end whilst eIF4A remains mRNA-bound to act in contacting the PIC during its attachment to the mRNA (95,96). Release of eIF4E•eIF4G is consistent with evidence from genome-wide translation-complex profiling studies in yeast that scanning is cap-severed (70). While the 5’-proximal mRNA footprint of eIF4G has not been measured in yeast to our knowledge, crosslinking experiments in mammalian systems indicate a footprint of up to ∼12 nt from the 5′ end, with a broadly similar crosslinking profile for eIF4A (97). Yeast eIF4G has two additional mRNA binding domains over the mammalian protein; thus, the mammalian eIF4G footprint size might reasonably be expected to set a lower limit on how much of the 5’ end yeast eIF4G covers. Based on our additional data, the ejection of eIF4E•eIF4G would thus ‘free’ or uncover the mRNA 5’ end to a length no more than about half of the shortest footprint sizes measured for the yeast 48S pre-initiation complex (70). We are hesitant to speculate about the position of eIF4A relative to the mRNA 5′ end, though the observation of appreciable FRET between eIF4E and eIF4A shows that their respective fluorophores are relatively close (within ∼40–70 Å) in the cap-recognition complex. Nevertheless, the binding of yeast eIF4B to Rps20p, which promotes eIF4A interaction with the 43S pre-initiation complex that aid mRNA recruitment (98), argues that on arrival of the pre-initiation complex (PIC) eIF4A localises near the mRNA entry channel. This would contrast with the cryo-EM structure of the human 48S pre-initiation complex, where eIF4A was assigned to bind eIF3k and eIF3l; however, yeast eIF3 does not contain those subunits and eIF4B was omitted in the structure of the human PIC. Our proposal is thus consistent with the suggestion (72) that eIF4F positioning in the 48S pre-initiation complex may differ substantially between yeast and humans. The transient rebinding events we observe following initial eIF4E•eIF4G ejection may ensure that the eIF4F complex is recycled and retained at the mRNA 5’ end for the next round of PIC recruitment after scanning, or for efficient reinitiation. Further studies are required to understand the role of PIC-eIF4F•mRNA contacts in the dynamics of early initiation.
Implications for cellular translation and translational control
Our results provide a rationale for how eIF4E/eIF4F concentration can be leveraged to mediate differences in mRNA activation for translation, even though eIF4F–mRNA binding is saturated in an equilibrium sense under cellular conditions, where eIF4E and total mRNA are present at similar concentrations in yeast cells—∼1 μM (74,75)—in great excess over eIF4F•mRNA Kd values. Based on the eIF4E–mRNA binding rates we measured for the eIF4F complex, at these concentrations eIF4E will be substantially bound to mRNAs in cells, thus limiting the pool of eIF4F available for de-novo translation initiation (7,48). In this regime, the rate of the eIF4E–mRNA interaction that begins de-novo cap recognition becomes an important determinant of initiation efficiency. Differences in this rate between mRNAs differentiate the mRNAs’ ability to compete for initiation, in accord with mathematical modeling (17).
Cellular mRNAs begin de-novo translation in an environment replete with RNA-binding proteins, and are subsequently incorporated into polysomes. While our experiments do not contain the cellular complement of RNA-binding proteins, the two-color experiments containing eIF4A (Figures 3 and 4) include it at concentrations that fully saturate its double-stranded RNA binding and significantly saturate single-stranded RNA binding (46). Thus, the mRNAs in these experiments are expected to exist in an eIF4A mRNP. Our data (Figure 3) then suggest that mRNP formation accelerates eIF4F–cap association in a length-dependent manner, and that the acceleration is physiologically relevant.
While our study does not probe eIF4F–mRNA dynamics in polysomes, polysome formation is expected to disrupt both secondary and tertiary structures in mRNA. This is consistent with the finding that very long mRNAs engaged in translation show end-to-end separation that greatly reduces when ribosomes are released by puromycin treatment (86). Since our data suggest that disruption of mRNA structure leads to enhanced cap recognition, our model predicts that cap recognition may become more efficient as polysomes form and begin to grow in size. Interestingly this could also, in principle, permit a cooperative effect on cap-/eIF4F-dependent pre-initiation complex loading rates during the earlier rounds of translation. As higher-order polysomes form, eIF4F-dependence of initiation may become less prominent, due both to efficient re-initiation of terminating ribosomes and also enhanced mRNA compaction observed in very heavy polysomes (99).
Given our finding that mRNA length impacts the eIF4E association rate, these results highlight how information encoded along the mRNA length intrinsically contributes to the efficiency of early initiation, and offers one explanation for why longer mRNAs are often translated less efficiently and with higher eIF4F dependence (29,30,34). Moreover, reduction in the availability or activity of any one eIF4F subunit is expected to impact different mRNAs differently in this model. Short mRNAs and/or less structured mRNAs that effectively compete for eIF4F–cap binding are predicted to be less sensitive to depletion of active eIF4F. mRNAs that associate faster with eIF4F might be expected to have an advantage in terms of translational efficiency under conditions such as stress where eIF4F activity is downregulated, provided they do not accumulate in stress granules or P bodies.
A second aspect of this kinetic scheme is that eIF4G, by inducing long eIF4E–mRNA binding events, has the effect of rendering cap recognition a non-equilibrium process on the initiation timescale. PIC–mRNA recruitment rates measured in vitro are in the ∼0.001–0.01 s–1 range at 30 nM 40S subunits (35,65). Extrapolating to a ∼1 μM lower limit for the cellular concentration of free 40S subunits thus implies a minimum recruitment rate of ∼0.05–0.4 s–1 at physiological PIC concentrations. This places recruitment occurring at rates close to or much faster than slow eIF4E–cap dissociation in the presence of eIF4G. With this kinetic balance, the overall efficiency of all steps from initial cap recognition to PIC–mRNA attachment is partially or fully limited by the eIF4E–mRNA association rate if PICs are sufficiently abundant. In contrast, for mRNAs that associate rapidly with eIF4E, PIC availability is predicted to limit initiation, suggesting a rationale for why translation of different subsets of mRNAs are affected differently by regulation directed at eIF4F and the PIC (100). Kinetic control of this type is consistent with our observation that eIF4E– and eIF4E•eIF4G–mRNA association rates show evidence for a discernible trend (if short of a statistically-significant correlation) with in-vivo RIP-seq enrichment, while the corresponding equilibrium dissociation constants do not, and our observation that the eIF4E–mRNA on-rate for eIF4F in the presence of ATP mirrors the translation efficiency of mRNAs measured by ribosome-density mapping and ribosome profiling (82,83).
Interestingly, mammalian eIF4F differs from yeast eIF4F in several aspects, particularly in terms of eIF4G1 architecture and eIF4A helicase activity. Moreover, mammalian mRNAs are generally longer and more structurally complex due to their higher GC content. Hence, it will be important to separately characterize these dynamics for mammalian eIF4F in future experiments.
In summary, we find that an intricate interplay of mRNA identity with factor activities facilitates mRNA-to-mRNA discrimination in cap recognition. Within this, ATP binding and hydrolysis also play important and distinct roles. The dynamics of the eIF4F•mRNA complex suggest a mechanism for coordinating eIF4F–cap recognition with PIC recruitment, and for conferring mRNA sensitivity to distinct translational control pathways.
DATA AVAILABILITY
All data generated and analysed in this study are included in the manuscript and Supplementary Information.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Jin Chen (UT Southwestern) and Gregor Blaha (UC Riverside) for insightful discussions on the manuscript.
Contributor Information
Burak Çetin, Graduate Program in Cell, Molecular, and Developmental Biology, University of California Riverside, Riverside, CA 92521, USA.
Seán E O’Leary, Graduate Program in Cell, Molecular, and Developmental Biology, University of California Riverside, Riverside, CA 92521, USA; Department of Biochemistry, University of California Riverside, Riverside, CA 92521, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [GM111858, GM138939, GM139056 to S.O’L.]; UC Riverside (Regents’ Faculty Fellowship award to S.O’L.). Funding for open access charge: National Institutes of Health [GM139056].
Conflict of interest statement. None declared.
REFERENCES
- 1. Hershey J.W.B., Sonenberg N., Mathews M.B.. Principles of translational control: an overview. Cold Spring Harb. Perspect. Biol. 2012; 4:a011528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Furuichi Y., Shatkin A.J.. Viral and cellular mRNA capping: past and prospects. Adv. Virus Res. 2000; 55:135–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fraser C.S. Quantitative studies of mRNA recruitment to the eukaryotic ribosome. Biochimie. 2015; 114:58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marcotrigiano J., Gingras A.C., Sonenberg N., Burley S.K.. Cocrystal structure of the messenger RNA 5’ cap-binding protein (eIF4E) bound to 7-methyl-GDP. Cell. 1997; 89:951–961. [DOI] [PubMed] [Google Scholar]
- 5. Siddiqui N., Sonenberg N.. Signalling to eIF4E in cancer. Biochem. Soc. Trans. 2015; 43:763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. von Der Haar T., Ball P.D., McCarthy J.E.. Stabilization of eukaryotic initiation factor 4E binding to the mRNA 5’-Cap by domains of eIF4G. J. Biol. Chem. 2000; 275:30551–30555. [DOI] [PubMed] [Google Scholar]
- 7. Pelletier J., Graff J., Ruggero D., Sonenberg N.. Targeting the eIF4F translation initiation complex: a critical nexus for cancer development. Cancer Res. 2015; 75:250–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Merrick W.C. eIF4F: a retrospective. J. Biol. Chem. 2015; 290:24091–24099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Altmann M., Linder P.. Power of yeast for analysis of eukaryotic translation initiation. J. Biol. Chem. 2010; 285:31907–31912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sonenberg N., Hinnebusch A.G.. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009; 136:731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kozak M., Shatkin A.J.. Migration of 40 S ribosomal subunits on messenger RNA in the presence of edeine. J. Biol. Chem. 1978; 253:6568–6577. [PubMed] [Google Scholar]
- 12. Hinnebusch A.G., Lorsch J.R.. The mechanism of eukaryotic translation initiation: new insights and challenges. Cold Spring Harb. Perspect. Biol. 2012; 4:a011544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jackson R.J., Hellen C.U.T., Pestova T.V.. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010; 11:113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sonneveld S., Verhagen B.M.P., Tanenbaum M.E.. Heterogeneity in mRNA translation. Trends Cell Biol. 2020; 30:606–618. [DOI] [PubMed] [Google Scholar]
- 15. Genuth N.R., Barna M.. Heterogeneity and specialized functions of translation machinery: from genes to organisms. Nat. Rev. Genet. 2018; 19:431–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Temple G., Lodish H.F.. Competition between α and β globin messenger RNA. Biochem. Biophys. Res. Commun. 1975; 63:971–979. [DOI] [PubMed] [Google Scholar]
- 17. Godefroy-Colburn T., Thach R.E.. The role of mRNA competition in regulating translation. IV. Kinetic model. J. Biol. Chem. 1981; 256:11762–11773. [PubMed] [Google Scholar]
- 18. Prabhakar A., Puglisi E.V., Puglisi J.D.. Single-Molecule fluorescence applied to translation. Cold Spring Harb. Perspect. Biol. 2019; 11:a032714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang J., Johnson A.G., Lapointe C.P., Choi J., Prabhakar A., Chen D.-H., Petrov A.N., Puglisi J.D.. eIF5B gates the transition from translation initiation to elongation. Nature. 2019; 573:605–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pelletier J., Sonenberg N.. The organizing principles of eukaryotic ribosome recruitment. Annu. Rev. Biochem. 2019; 88:307–335. [DOI] [PubMed] [Google Scholar]
- 21. Sokabe M., Fraser C.S.. Toward a kinetic understanding of eukaryotic translation. Cold Spring Harb. Perspect. Biol. 2019; 11:a032706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khan M., Yumak H., Goss D.J.. Kinetic mechanism for the binding of eIF4F and tobacco etch virus internal ribosome entry site RNA. J. Biol. Chem. 2009; 284:35461–35470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Khan M., Ma J., Walden W.E., Merrick W.C., Theil E.C., Goss D.J.. Rapid kinetics of iron responsive element (IRE) RNA/iron regulatory protein 1 and IRE-RNA/eIF4F complexes respond differently to metal ions. Nucleic Acids Res. 2014; 42:6567–6577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Khan M.A., Domashevskiy A.V.. Iron enhances the binding rates and translational efficiency of iron responsive elements (IREs) mRNA with initiation factor eIF4F. PLoS One. 2021; 16:e02500374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. O’Leary S.E., Petrov A., Chen J., Puglisi J.D.. Dynamic recognition of the mRNA cap by saccharomyces cerevisiae eIF4E. Structure. 2013; 21:2197–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Costello J., Castelli L.M., Rowe W., Kershaw C.J., Talavera D., Mohammad-Qureshi S.S., Sims P.F.G., Grant C.M., Pavitt G.D., Hubbard S.J.et al.. Global mRNA selection mechanisms for translation initiation. Genome Biol. 2015; 16:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Costello J.L., Kershaw C.J., Castelli L.M., Talavera D., Rowe W., Sims P.F.G., Ashe M.P., Grant C.M., Hubbard S.J., Pavitt G.D.. Dynamic changes in eIF4F–mRNA interactions revealed by global analyses of environmental stress responses. Genome Biol. 2017; 18:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Park E.-H., Zhang F., Warringer J., Sunnerhagen P., Hinnebusch A.G.. Depletion of eIF4G from yeast cells narrows the range of translational efficiencies genome-wide. BMC Genomics. 2011; 12:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sen N.D., Zhou F., Ingolia N.T., Hinnebusch A.G.. Genome-wide analysis of translational efficiency reveals distinct but overlapping functions of yeast DEAD-box RNA helicases ded1 and eIF4A. Genome Res. 2015; 25:1196–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sen N.D., Zhou F., Harris M.S., Ingolia N.T., Hinnebusch A.G.. eIF4B stimulates translation of long mRNAs with structured 5’ UTRs and low closed-loop potential but weak dependence on eIF4G. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:10464–10472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jensen K.B., Dredge B.K., Toubia J., Jin X., Iadevaia V., Goodall G.J, Proud C.G.. capCLIP: a new tool to probe translational control in human cells through capture and identification of the eIF4E–mRNA interactome. Nucleic Acids Res. 2021; 49:e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Avni D., Biberman Y., Meyuhas O.. The 5′ terminal oligopyrimidine tract confers translational control on TOP mRNAs in a cell-type and sequence context-dependent manner. Nucleic Acids Res. 1997; 25:995–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Svitkin Y.V., Pause A., Haghighat A., Pyrronet S., Witherell G., Belsham G.J., Sonenberg N.. The requirement for eukaryotic initiation factor 4A (eIF4A) in translation is in direct proportion to the degree of mRNA 5′ secondary structure. RNA. 2001; 7:382–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thompson M.K., Gilbert W.V.. mRNA length-sensing in eukaryotic translation: reconsidering the ‘closed loop’ and its implications for translational control. Curr. Genet. 2017; 63:613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yourik P., Aitken C.E., Zhou F., Gupta N., Hinnebusch A.G., Lorsch J.R.. Yeast eIF4A enhances recruitment of mRNAs regardless of their structural complexity. Elife. 2017; 6:e31476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gross J.D., Moerke N.J., von der Haar T., Lugovskoy A.A., Sachs A.B., McCarthy J.E.G., Wagner G.. Ribosome loading onto the mRNA cap is driven by conformational coupling between eIF4G and eIF4E. Cell. 2003; 115:739–750. [DOI] [PubMed] [Google Scholar]
- 37. Grüner S., Peter D., Weber R., Wohlbold L., Chung M.-Y., Weichenrieder O., Valkov E., Igreja C., Izaurralde E.. The structure of eIF4E–eIF4G complexes reveal an extended interface to regulate translation initiation. Mol. Cell. 2016; 64:467–469. [DOI] [PubMed] [Google Scholar]
- 38. Yanagiya A., Svitkin Y.V., Shibata S., Mikami S., Imataka H., Sonenberg N.. Requirement of RNA binding of mammalian eukaryotic translation initiation factor 4GI (eIF4GI) for efficient interaction of eIF4E with the mRNA cap. Mol. Cell Biol. 2009; 29:1661–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marintchev A., Edmonds K.A., Marintcheva B., Hendrickson E., Oberer M., Suzuki C., Herdy B., Sonenberg N., Wagner G.. Topology and regulation of the human eIF4A/4G/4H helicase complex in translation initiation. Cell. 2009; 136:447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Feoktistova K., Tuvshintogs E., Do A., Fraser C.S.. Human eIF4E promotes mRNA restructuring by stimulating eIF4A helicase activity. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:13339–13344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Andreou A.Z., Klostermeier D.. eIF4B and eIF4G jointly stimulate eIF4A ATPase and unwinding activities by modulation of the eIF4A condormational cycle. J. Mol. Biol. 2014; 426:51–61. [DOI] [PubMed] [Google Scholar]
- 42. Harms U., Andreou A.Z., Gubaev A., Klostermeier D.. eIF4B, eIF4G and RNA regulate eIF4A activity in translation initiation by modulating the eIF4A conformational cycle. Nucleic Acids Res. 2014; 42:7911–7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gingras A.C., Raught B., Sonenberg N.. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 1999; 68:913–963. [DOI] [PubMed] [Google Scholar]
- 44. Slepenkov S.V., Korneeva N.L., Rhoads R.E.. Kinetic mechanism for assembly of the m7GpppG·eIF4E·eIF4G complex. J.Biol. Chem. 2008; 283:25227–25237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Haghighat A., Sonenberg N.. eIF4G dramatically enhances the binding of eIF4E to the mRNA 5′-cap structure. J. Biol. Chem. 1997; 272:21677–21680. [DOI] [PubMed] [Google Scholar]
- 46. Rajagopal V., Park E.-H., Hinnebusch A.G., Lorsch J.R.. Specific domains in yeast translation initiation factor eIF4G strongly bias RNA unwinding activity of the eIF4F complex toward duplexes with 5’-overhangs. J. Biol. Chem. 2012; 287:20301–20312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. von der Haar T., McCarthy J.E.G.. Intracellular translation initiation factor levels in saccharomyces cerevisiae and their role in cap-complex function. Mol. Microbiol. 2002; 46:531–544. [DOI] [PubMed] [Google Scholar]
- 48. Chu J., Zhang W., Cencic R., O’Connor P.B.F., Robert F., Devine W.G., Selznick A., Henkel T., Merrick W.C., Brown L.E.et al.. Rocaglates induce Gain-of-Function alterations to eIF4A and eIF4F. Cell Rep. 2020; 30:2481–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Blum S., Schmid S.R., Pause A., Buser P., Linder P., Sonenberg N., Trachsel H.. ATP hydrolysis by initiation factor 4A is required for translation initiation in saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:7664–7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liu X., Schuessler P.J., Sahoo A., Walker S.E.. Reconstitution and analyses of RNA interactions with eukaryotic translation initiation factors and ribosomal preinitiation complexes. Methods. 2019; 162-163:42–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bradley M.J., De La Cruz E.M.. Analyzing ATP utilization by DEAD-Box RNA helicases using kinetic and equilibrium methods. Methods Enzymol. 2012; 511:29–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen J., Dalal R.V., Petrov A.N., Tsai A., O’Leary S.E., Chapin K., Cheng J., Ewan M., Hsiung P.L., Lundquist P.et al.. High-throughput platform for real-time monitoring of biological processes by multicolour single-molecule fluorescence. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:664–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Çetin B., Song G.J., O’Leary S.E.. Heterogeneous dynamics of protein–RNA interactions across transcriptome-derived messenger RNA populations. J. Am. Chem. Soc. 2020; 142:21249–21253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Aitken C.E., Marshall R.A., Puglisi J.D.. An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments. Biophys. J. 2008; 94:1826–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hirschman J.E., Balakrishnan R., Christie K.R., Costanzo M.C., Dwight S.S., Engel S.R., Fisk D.G., Hong E.L., Livstone M.S., Nash R.et al.. Genome snapshot: a new resource at the saccharomyces genome database (SGD) presenting an overview of the saccharomyces cerevisiae genome. Nucleic Acids Res. 2006; 34:D442–D445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lai W.-J.C., Kayedkhordeh M., Cornell E.V., Farah E., Bellaousov S., Rietmeijer R., Salsi E., Mathews D.H., Ermolenko D.N.. mRNAs and lncRNAs intrinsically form secondary structures with short end-to-end distances. Nat. Commun. 2018; 9:4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Khong A., Parker R.. The landscape of eukaryotic mRNPs. RNA. 2020; 26:229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vicens Q., Kieft J.S., Rissland O.S.. Revisiting the closed-loop model and the nature of mRNA 5’-3' communication. Mol. Cell. 2018; 72:805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yoffe A.M., Prinsen P., Gelbart W.M., Ben-Shaul A.. The ends of a large RNA molecule are necessarily close. Nucleic Acids Res. 2011; 39:292–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Carberry S.E., Rhoads R.E., Goss D.J.. A spectroscopic study of the binding of m7GTP and m7GpppG to human protein synthesis initiation factor 4E. Biochemistry. 1989; 28:8078–8083. [DOI] [PubMed] [Google Scholar]
- 61. Ueda H., Maruyama H., Doi M., Inoue M., Ishida T., Morioka H., Tanaka T., Nishikawa S., Uesugi S.. Expression of a synthetic gene for human cap binding protein (Human IF-4E) in esherichia coli and fluorescence studies on interaction with mRNA cap structure analogues. J. Biochem. 1991; 109:882–889. [DOI] [PubMed] [Google Scholar]
- 62. Borodavka A., Singaram S.W., Stockley P.G., Gelbart W.M., Ben-Shaul A., Tuma R.. Sizes of long RNA molecules are determined by the branching patterns of their secondary structures. Biophys. J. 2016; 111:2077–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Moran-Mirabal J.M., Craighead H.G.. Zero-mode waveguides: sub-wavelength nanostructures for single molecule studies at high concentrations. Methods. 2008; 46:11–17. [DOI] [PubMed] [Google Scholar]
- 64. Tuller T., Ruppin E., Kupiec M.. Properties of untranslated regions of the s. cerevisiae genome. BMC Genomics. 2009; 10:391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mitchell S.F., Walker S.E., Algire M.A., Park E.-H., Hinnebusch A.G., Lorsch J.R.. The 5′-7-methylguanosine cap on eukaryotic mRNAs serves both to stimulate canonical translation initiation and to block an alternative pathway. Mol. Cell. 2010; 39:950–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kaye N.M., Emmett K.J., Merrick W.C., Jankowsky E.. Intrinsic RNA binding by the eukaryotic initiation factor 4F depends on a minimal RNA length but not on the m7G cap. J. Biol. Chem. 2009; 284:17742–17750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Acker M.G., Shin B.S., Nanda J.S., Saini A.K., Dever T.E., Lorsch J.R.. Kinetic analysis of late steps of eukaryotic translation initiation. J. Mol. Biol. 2009; 385:491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Palmiter R.D. Quantitation of parameters that determine the rate of ovalbumin synthesis. Cell. 1975; 4:189. [DOI] [PubMed] [Google Scholar]
- 69. Bohlen J., Fenzl K., Kramer G., Bukau B., Teleman A.A.. Selective 40S footprinting reveals cap-tethered ribosome scanning in human cells. Mol. Cell. 2020; 79:561–574. [DOI] [PubMed] [Google Scholar]
- 70. Archer S.K., Shirokikh N.E., Beilharz T.H., Preiss T.. Dynamics of ribosome scanning and recycling revealed by translation complex profiling. Nature. 2016; 535:570–574. [DOI] [PubMed] [Google Scholar]
- 71. Shirokikh N.E., Preiss T.. Translation initiation by cap-dependent ribosome recruitment: recent insights and open questions. Wiley Interdiscip. Rev. RNA. 2018; 9:e1473. [DOI] [PubMed] [Google Scholar]
- 72. Querido J.B., Sokabe M., Kraatz S., Gordiyenko Y., Skehel M., Fraser C., Ramakrishnan V.. Structure of a human 48S translational initiation complex. Science. 2020; 369:1220–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Paek K.Y., Hong K.Y., Ryu I., Park S.M., Keum S.J., Kwon O.S., Jang S.K.. Translation initiation mediated by RNA looping. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:1041–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ghaemmaghami S., Huh W.-K., Bower K., Howson R.W., Belle A., Dephoure N., O’Shea E.K., Weissman J.S.. Global analysis of protein expression in yeast. Nature. 2003; 425:737–741. [DOI] [PubMed] [Google Scholar]
- 75. Lu P., Vogel C., Wang R., Yao X., Marcotte E.M.. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol. 2007; 25:117–124. [DOI] [PubMed] [Google Scholar]
- 76. Firczuk H., Kannambath S., Pahle J., Claydon A., Beynon R., Duncan J., Westerhoff H., Mendes P., McCarthy J.E.G.. An in vivo control map for the eukaryotic mRNA translation machinery. Mol. Syst. Biol. 2013; 9:635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Niedzwiecka A., Darzynkiewicz E., Stolarski R.. Thermodynamics of mRNA 5′ cap binding by eukaryotic translation initiation factor eIF4E. Biochemistry. 2004; 43:13305–13317. [DOI] [PubMed] [Google Scholar]
- 78. Linder P., Jankowsky E.. From unwinding to clamping — the DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 2011; 12:505–516. [DOI] [PubMed] [Google Scholar]
- 79. Tauber D., Tauber G., Khong A., Van Treeck B., Pelletier J., Parker R.. Modulation of RNA condensation by the DEAD-Box protein eIF4A. Cell. 2020; 180:411–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Andreou A.Z., Harms U., Klostermeier d. Single-stranded regions modulate conformational dynamics and ATPase activity of eIF4A to optimize 5’-UTR unwinding. Nucleic Acids Res. 2019; 47:5260–5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Schütz P., Bumann M., Oberholzer A.E., Bieniossek C., Trachsel H., Altmann M., Baumann U.. Crystal structure of the yeast eIF4A-eIF4G complex: an RNA-helicase controlled by protein-protein interactions. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:9564–9569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Arava Y., Boas F.E., Brown P.O., Herschlag D.. Dissecting eukaryotic translation and its control by ribosome density mapping. Nucleic Acids Res. 2005; 33:2421–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zinshteyn B., Rojas-Duran M.F., Gilbert W.V.. Translation initiation factor eIF4G1 preferentially binds yeast transcript leaders containing conserved oligo-uridine motifs. RNA. 2017; 23:1365–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ray B.K., Lawson T.G., Kramer J.C., Cladaras M.H., Grifo J.A., Abramson R.D., Merrick W.C., Thach R.E.. ATP-dependent unwinding of messenger RNA structure by eukaryotic initiation factors. J. Biol. Chem. 1985; 260:7651–7658. [PubMed] [Google Scholar]
- 85. Adivarahan S., Livingston N., Nicholson B., Rahman S., Wu B., Rissland O.S., Zenklusen D.. Spatial organization of single mRNPs at different stages of the gene expression pathway. Mol. Cell. 2018; 72:727–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Khong A., Parker R.. mRNP architecture in translating and stress conditions reveals an ordered pathway of mRNP compaction. J. Cell Biol. 2018; 217:4124–4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kopeina G.S., Afonina Z.A., Gromova K.V., Shirokov V.A., Vasiliev V.D., Spirin A.S.. Step-wise formation of eukaryotic double-row polyribosomes and circular translation of polysomal mRNA. Nucleic Acids Res. 2008; 36:2476–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Afonina Z.A, Myasnikov A.G., Shirokov V.A., Klaholz B.P., Spirin A.S.. Formation of circular polyribosomes on eukaryotic mRNA without cap-structure and poly(A)-tail: a cryo-electron tomography study. Nucleic Acids Res. 2014; 42:9461–9469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gao Z., Putnam A.A., Bowers H.A., Guenther U.-P., Ye X., Kindsfather A., Hilliker A.K., Jankowsky E.. Coupling between the DEAD-box RNA helicases ded1p and eIF4A. Elife. 2016; 5:e16408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hentze M.W., Castello A., Schwarzl T., Preiss T.. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018; 19:327–341. [DOI] [PubMed] [Google Scholar]
- 91. Svitkin Y.V., Evdokimova V.M., Brasey A., Pestova T.V., Fantus D., Yanagiya A., Imataka H., Skabkin M.A., Ovchinnikov L.P., Merrick W.C.et al.. General RNA-binding proteins have a function in poly(A)-binding protein-dependent translation. EMBO J. 2009; 28:58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Munroe D., Jacobson A.. mRNA poly(A) tail, a 3’ enhancer of translational initiation. Mol. Cell. Biol. 1990; 10:3441–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Riba A., Di Nanni N., Mittal N., Arhné E., Schmidt A., Zavolan M.. Protein synthesis rates and ribosome occupancies reveal determinants of translation elongation rates. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:15023–15032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Llácer J.L., Hussain T., Saini A.K., Nanda J.S., Kaur S., Gordiyenko Y., Kumar R., Hinnebusch A.G., Lorsch J.R., Ramakrishnan V.. Translational initiation factor eIF5 replaces eIF1 on the 40S ribosomal subunit to promote start-codon recognition. Elife. 2018; 7:e39273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. He H., von der Haar T., Singh C.R., Ii M., Li B., Hinnebusch A.G., McCarthy J.E.G., Asano K.. The yeast eukaryotic initiation factor 4G (eIF4G) HEAT domain interacts with eIF1 and eIF5 and is involved in stringent AUG selection. Mol. Cell. Biol. 2003; 23:5431–5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yamamoto Y., Singh C.R., Marintchev A., Hall N.S., Hannig E.M., Wagner G., Asano K.. The eukaryotic initiation factor (eIF) 5 HEAT domain mediates multifactor assembly and scanning with distinct interfaces to eIF1, eIF2, eIF3, and eIF4G. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:16164–16169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lindqvist L., Imataka H., Pelletier J.. Cap-dependent eukaryotic initiation factor–mRNA interactions probed by cross-linking. RNA. 2008; 14:960–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Walker S.E., Zhou F., Mitchell S.F., Larson V.S., Valasek L., Hinnebusch A.G., Lorsch J.R.. Yeast eIF4B binds to the head of the 40S ribosomal subunit and promotes mRNA recruitment through its N-terminal and internal repeat domains. RNA. 2013; 19:191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Afonina Z.A., Myasnikov A.G., Shirokov V.A., Klaholz B., Spirin A.S.. Conformational transitions of eukaryotic polyribosomes during multi-round translation. Nucleic Acids Res. 2015; 43:618–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gebauer F., Hentze M.W.. Molecular mechanisms of translational control. Nat. Rev. Mol. Cell Biol. 2004; 5:827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated and analysed in this study are included in the manuscript and Supplementary Information.