


Abstract
Trypanosomatids cause the neglected tropical diseases, sleeping sickness, Chagas disease and the leishmaniases. Studies on these lethal parasites would be further facilitated by new and improved genetic technologies. Scalable precision editing methods, for example, could be used to improve our understanding of potential mutations associated with drug resistance, a current priority given that several new anti-trypanosomal drugs, with known targets, are currently in clinical development. We report the development of a simple oligo targeting method for rapid and precise editing of priority drug targets in otherwise wild type trypanosomatids. In Trypanosoma brucei, approx. 50-b single-stranded oligodeoxynucleotides were optimal, multiple base edits could be incorporated, and editing efficiency was substantially increased when mismatch repair was suppressed. Resistance-associated edits were introduced in T. brucei cyclin dependent kinase 12 (CRK12, L482F) or cleavage and polyadenylation specificity factor 3 (N232H), in the Trypanosoma cruzi proteasome β5 subunit (G208S), or in Leishmania donovani CRK12 (G572D). We further implemented oligo targeting for site saturation mutagenesis, targeting codon G492 in T. brucei CRK12. This approach, combined with amplicon sequencing for codon variant scoring, revealed fourteen resistance conferring G492 edits encoding six distinct amino acids. The outputs confirm on-target drug activity, reveal a variety of resistance-associated mutations, and facilitate rapid assessment of potential impacts on drug efficacy.
INTRODUCTION
The parasitic trypanosomatids cause a range of lethal and neglected tropical diseases. These protozoa include Trypanosoma brucei spp, Trypanosoma cruzi and Leishmania spp, which cause sleeping sickness, Chagas disease and leishmaniasis (both visceral and cutaneous) in humans, respectively, while T. brucei brucei causes nagana in livestock (1). All three human diseases have been targeted for elimination as part of the World Health Organisation's road map for neglected tropical diseases 2021–2030 (WHO/UCN/NTD/2020.01).
Encouragingly, several new drugs with known mechanisms of action and known primary targets are in clinical development, promising to deliver new, safe, affordable, efficacious and often, oral therapies. These new priority targets include cyclin dependent kinase 12 (CRK12) (2,3), cleavage and polyadenylation specificity factor 3 (CPSF3) (4) and the proteasome (5,6). Since drug resistance can undermine both the efficacy of new therapies and substantial investments in drug discovery and clinical development, there is an urgent need for further insights into resistance-associated mutations and structure–activity relationships.
A conventional approach often used to identify the target of an anti-parasitic compound involves exposing populations of cells to drug pressure, for up to 6 months, followed by sequencing the genomes of drug-resistant parasites. Indeed, this approach revealed mutations in the genes encoding Leishmania donovani CRK12 (2), T. cruzi proteasome β5 (5) and L. donovani proteasome β4 or β5 subunits (6), contributing towards identifying these proteins as drug targets. Notably though, these approaches typically reveal single point mutations, which can sample only a limited subset of alternative amino acids, contingent upon mutational space (7).
CRISPR Cas9 RNA-guided nucleases have been successfully employed for genome editing in trypanosomatids (8–13) and in other parasitic protozoa (14). Notwithstanding their broad utility, the associated DNA double-strand breaks are cytotoxic and can increase off-target mutation rates (15), by several hundred-fold in regions adjacent to a break (16). RNA-guided deaminases and base editors (17), and also prime editors (18), display much lower rates of off-target editing, but use of these editors has not yet been reported in trypanosomatids. In addition, a protospacer-adjacent motif (PAM) must typically be present close to a CRISPR Cas9 editing site. Oligo targeting is an alternative genome editing approach that has not previously been developed for parasitic protozoa.
Genome editing using oligo targeting in Saccharomyces cerevisiae involves incorporation of synthetic single-stranded DNA (ssDNA) oligodeoxynucleotides (ssODNs) on the lagging strand of the DNA replication fork, favoured by discontinuous replication on this strand (19). Consequently, the approach is not thought to create DNA breaks or nicks and does not require a proximal PAM. Previously deployed in bacteria, yeast, plant and mammalian cells, this approach facilitates targeted chromosome modification at single base-pair resolution. Indeed, the approach is scalable in Escherichia coli (20) and S. cerevisiae (19) and facilitates multiplex genome engineering in these cells. Given the simplicity and potential versatility of oligo targeting, we sought to develop this technology and to apply it to the parasitic trypanosomatids.
MATERIALS AND METHODS
Oligo targeting in T. brucei
Bloodstream form T. brucei Lister 427 wild type cells were grown in HMI-11 medium in a humidified incubator at 37°C with 5% CO2. Optimally, 2.5 × 107T. brucei cells were mixed with 100 μl of Nucleofector buffer (Amaxa T -cell kit) and then mixed with the appropriate ssODN (Thermo Fisher Scientific, except for LNA-modified ssODNs; Integrated DNA Technologies), 40 μg in 10 μL of 10 mM Tris–Cl, pH 8.5. The mixture was placed in a 0.2 cm gap electrocuvette and submitted to a single pulse using a Nucleofector (Amaxa) set on the Z-001 programme. The mixture was then transferred from the cuvette to 20 ml of prewarmed HMI-11 medium and placed in the incubator. After 6 h, the appropriate drug selection was applied, in excess of the previously determined EC50 and sufficient to eliminate mock-transfected cells; 10 nM unless stated otherwise in the case of cpd 2 (3) and 1 μM in the case of acoziborole (4). The cells were then distributed in multi-well plates for sub-cloning or were expanded in bulk-culture for amplicon-seq profiling, with drug selection maintained in each case. Estimates of oligo targeting efficiency were derived by counting positive wells in minimally diluted plates or, when efficiency was high, in serially diluted plates, 5–6 days after plating.
MSH2 knockdown by RNA interference
Initial analysis using msh2 null strains (21) consistently yielded cells that tolerated drug selection. Since we were concerned that many mutations may have accumulated in these cells due to the prolonged mismatch repair defect, we adopted a conditional knockdown strategy. PCR primers, for amplification of the T. brucei MSH2 (Tb927.10.11020) RNAi target fragment, were designed using RNAit (https://dag.compbio.dundee.ac.uk/RNAit/). Following PCR amplification, using T. brucei genomic DNA as template, the product was digested with BamHI and XmaI and ligated to similarly digested pRPaiSL plasmid (22). In the second step, additional PCR product was digested with HindIII and ApaI and ligated to similarly digested pRPaiSL plasmid from step 1. Correct assembly was confirmed by DNA sequencing. The T. brucei MSH2 RNAi strain was constructed by digesting the final pRPaMSH2-RNAi construct with AscI and electroporation into 2T1 T. brucei cells (23) as described above. After 6 h, phleomycin and hygromycin selection were applied, both at 2 μg/ml. The cells were then distributed in multi-well plates for sub-cloning. A puromycin-sensitive (2 μg/ml) sub-clone was selected for further analysis and subsequently maintained in phleomycin and hygromycin, both at 1 μg/ml. Knockdown by RNAi was induced by addition of tetracycline at 1 μg/ml to the growth medium. MSH2 knockdown was assessed using qRT-PCR. RNA was extracted using the RNeasy Mini Kit with an on-column DNase digest step (RNase-free DNase, Qiagen). RNA (1 μg) was reverse-transcribed to produce cDNA (M-MLV reverse transcriptase, Promega) and the equivalent of 25 ng of RNA was used in each qPCR reaction. The reactions were performed in technical triplicates on a QuantStudio3 system (Applied Biosystems) using the Luna® Universal qPCR Master Mix (NEB). TERT (Tb927.11.10190) and PFR (Tb927.8.5010) were used as reference genes (24) to calculate the 2^-deltaCt value for each sample.
Oligo targeting in T. cruzi
Oligo targeting was carried out as above for T. brucei, but with the following modifications. Wild type T. cruzi [MHOM/BR/78/Silvio; clone X10/7A] epimastigotes were grown in RTH medium [RPMI 1640 medium supplemented with trypticase, haemin and HEPES] plus 10% heat-inactivated FBS in a humidified incubator at 28°C with 5% CO2. 2.5 × 107T. cruzi cells were electroporated with the Nucleofector set on the X-014 programme. The mixture was then transferred from the cuvette to 10 mL of prewarmed medium and placed in the incubator. Cpd 7 (6) selection was applied at 350 nM after 24 h and thereafter. The cells were expanded in bulk-culture for 2–3 weeks, at which point no live cells were detectable in mock electroporated cultures (no oligo), and then distributed in multi-well plates for sub-cloning.
Oligo targeting in L. donovani
Oligo targeting was carried out as above for T. brucei, but with the following modifications. Wild type L. donovani BOB strain [from MHOM/SD/62/1S-CL2D] promastigotes were grown in LdBOBpro (M199) medium in a humidified incubator at 28°C with 5% CO2. 2.5 × 107L. donovani cells were electroporated with the Nucleofector set on the V-033 programme. The mixture was then transferred from the cuvette to 25 ml of prewarmed medium and placed in the incubator. Cpd 5 (2) selection was applied at 10 nM after 24 h, and cells were distributed in multi-well plates. Estimates of oligo targeting efficiency were derived by counting positive wells in minimally diluted plates, 14–16 days after plating. No drug-resistant cells were recovered from mock electroporation experiments (no oligo).
Confirmation of edits using DNA-sequencing
T. brucei DNA was extracted using a DNeasy blood and tissue Kit (Qiagen). Leishmania and T. cruzi cells were harvested and resuspended in lysis buffer [10 mM Tris–HCl, pH 8, 100 mM NaCl, 25 mM EDTA, 0.5% (w/v) SDS, 0.1 mg/ml proteinase K]. After overnight incubation at 56°C, DNA was extracted using the phenol:chloroform:isoamyl alcohol method. PCR was used to amplify an appropriate segment from each edited gene, which was subjected to Sanger DNA sequencing. PCR primer pairs are detailed in Supplementary Table S1. A minimum of two independent clones were sequenced in each case and all sequences revealed the expected edits.
Amplicon sequencing and analysis
T. brucei DNA was extracted as above; after 5 days of drug selection or after 24 h following mock transfection. A 176-bp PCR amplicon spanning the oligo targeted region was PCR-amplified using KOD Hot Start DNA Polymerase (Merck) and further processed using a PCR Purification Kit (Qiagen). Amplicon sequencing was then carried out using DNBseq at BGI Genomics. Briefly, rolling-circle amplification was used to produce DNA nanoballs followed by sequencing by combinatorial probe-anchor synthesis in 100 b paired-end mode on the BGISEQ-500 platform (The Beijing Genome Institute). Sequencing data were filtered using SOAPnuke software (BGI) with the following parameters: -n 0.001 -l 10 -q 0.4 -A 0.25 -Q 2 -G –cutAdaptor –minLen 100, to obtain between 17 and 30 million 100 b paired-end reads per sample. Reads were analysed with FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and filtered with fastp (https://github.com/OpenGene/fastp) using default options. The reference genome was assembled using v46 of the T. brucei reference genome, clone 427_2018, downloaded from TriTrypDB (25). Forward and reverse paired-end reads were aligned to the reference genome using Bowtie 2 (26), with the ‘very-sensitive-local’ pre-set alignment option. The alignments were converted to BAM format, reference sorted and indexed with SAMtools (27). The quality of alignments was evaluated with Qualimap 2 (28) using the bamqc and rnaseq options. The Qualimap2 output files, and the outputs of fastp, bowtie2, Picard Mark Duplicates (https://broadinstitute.github.io/picard/), SAMtools flagastat, SAMtools stats were aggregated with MultiQC (29) and inspected. Per-codon read counts were derived for ten codons at the 5′ and 3′ of the degenerate codon using a custom python script implemented in a jupyter notebook. Briefly, the script parses the aligned reads in the bam file to retrieve the codon counts. The genomic coordinates of the aligned reads are first used to extract the codons starts/ends and to reconstruct the codon sequence. Reads with a quality score <30 in the analysed region were discarded from the counts. The scripts to align the fastq files, qc control files and the scripts to extract the codon counts are available at https://github.com/mtinti/DH_Oligo-targeting and deposited in Zenodo (10.5281/zenodo.5139694).
Dose-response assays
Half maximal effective drug concentrations (EC50) were measured by seeding mid-log cells in 96-well plates in a 2-fold dilution series of the appropriate drug; 103 cells/ml for T. brucei, 5 × 105 cells/ml for T. cruzi and 5 × 104 cells/ml for Leishmania. Resazurin sodium salt (Sigma) in PBS was then added to each well; 500 μM after 72 h growth for T. brucei, 500 μM after 96 h growth for T. cruzi and 50 μM after 72 h growth for Leishmania. The plates were incubated in resazurin prior to analysis; 6 h for T. brucei, 24 h for T. cruzi and 2–3 h for Leishmania. Fluorescence was then measured using an Infinite 200 pro plate reader (Tecan) at an excitation wavelength of 540 nm and an emission wavelength of 590 nm. Data were analysed using Prism (GraphPad).
RESULTS AND DISCUSSION
Establishing and optimising oligo targeting in T. brucei
While assessing a CRISPR Cas9 RNA-guided nuclease system to drive ssODN-templated editing in T. brucei, we consistently observed the rapid emergence of edited populations in control experiments combining wild type trypanosomes with a repair template but lacking CRISPR Cas9 components. This suggested, although not reported previously, that ssODN-directed mutagenesis, or oligo targeting could be an effective method for genetically manipulating these parasites.
The oligo targeting protocol (Figure 1A) that emerged employs wild type parasites mixed with a complementary but mismatched ssODN and subjected to a single pulse of electroporation. We first attempted to modify T. brucei CRK12 by introducing a L482F, CTC-TTC edit in the ATP binding site; this mutation preserves catalytic function and ATP-binding but confers resistance to compound 2 (3). We tested ssODNs of 21, 51 or 81 b in length and in either orientation, each with a centrally located mismatch. The drug was added six hours after electroporation and cells were distributed in multi-well plates, which were scored for clonal populations five to six days later. Selected clones were assessed for editing and drug resistance. This first set of six ssODNs tested yielded from ca. 3–94 drug-resistant clones. In terms of length, the 51 b ssODNs yielded the most clones (Figure 1B) and this was also the case compared to 41 or 61 b ssODNs (Supplementary Figure S1A), indicating a lower optimal ssODN length relative to E. coli and S. cerevisiae, where 90 b ssODNs are optimal (19,20). In terms of orientation, reverse ssODNs relative to the direction of transcription, and likely also DNA replication (30), yielded more clones (Figure 1B). This is consistent with insertion of complementary ssODNs on the lagging strand of the chromosomal DNA replication fork (19). DNA sequencing (Figure 1C) confirmed the desired heterozygous edit (trypanosomatids are diploid), which was associated with drug resistance (Figure 1D). We further validated the approach by introducing a N232H, AAT-CAT edit in T. brucei CPSF3 (Figure 1E), which creates a steric clash with acoziborole and confers resistance to this drug (4). No further base-changes were observed in the ssODN-complementary regions in either TbCRK12 or TbCPSF3 (Supplementary Figure S2A, B). These results indicate that oligo targeting is a precise gene-editing approach in T. brucei and that editing efficiency is both ssODN-length and ssODN-direction dependent.
Figure 1.
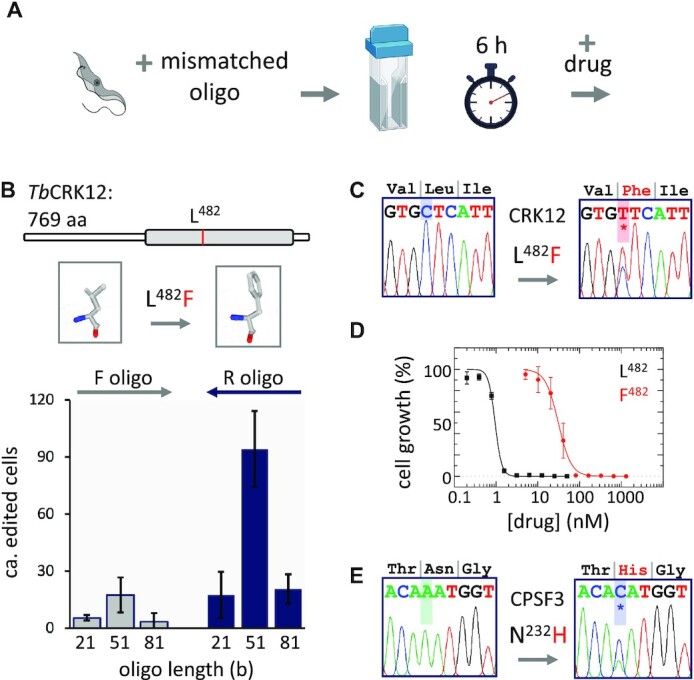
Establishing and optimising oligo targeting in T. brucei. (A) The schematic (created using BioRender.com) illustrates the oligo targeting protocol. (B) L482F, C-T editing in the kinase domain (grey bar) of TbCRK12. The plot shows editing efficiency in T. brucei using ssODNs of different lengths in the forward (F) or reverse (R) orientation. A control lacking an ssODN failed to yield any drug-resistant clones. Compound 2 selection was applied at 10 nM; n = 2 for each assay; error bars, SD. (C) DNA sequencing confirmed the desired edit (also see Supplementary Figure S2A). (D) The representative dose-response curves shows that edited cells are drug-resistant; two independent clones were assessed; error bars, SD. (E) N232H, A–C editing of TbCPSF3. DNA sequencing confirmed the desired edit (also see Supplementary Figure S2B).
Multi-base editing and suppression by mismatch repair
We next asked whether the efficiency of oligo targeting could be improved in T. brucei and also whether multiple base changes could be incorporated in one step, again by introducing a CRK12–L482F edit. ssODNs with nuclease-resistant phosphorothioate internucleotide linkages (S-oligos) (31), also used for oligo targeting in E. coli (20), improved editing efficiency ∼2-fold (Figure 2A). A pair of ssODNs with two or three additional synonymous edits either 4 or 8 b apart, respectively, both yielded drug-resistant clones that incorporated all base-edits present in the ssODN; editing efficiency was reduced by 30 and 85%, respectively (Figure 2A). The protocol was next further optimised in terms of ssODN concentration and cell number (Supplementary Figure S1B). Given an electroporation survival rate of 44% (±5%, SEM) and an estimate of 342 edited clones per electroporation, we derive an optimal allele replacement frequency of 3 × 10–5 or 0.003% for these experiments. Finally, we asked whether terminal phosphate groups would impact oligo-targeting efficiency and found that these groups had only moderate impacts; a 5′-phosphate, which is required for ligation, reduced efficiency by ∼25%, whereas a 3′-phosphate, which can block extension by DNA polymerase, increased efficiency by ∼65% (Supplementary Figure S1C). These results suggested that neither direct ssODN ligation nor extension are required for oligo targeting. A 5′-phosphate may promote ssODN ligation, moderately limiting template molecule abundance and diffusion to target sites, while a 3′-phosphate may promote ssODN stability.
Figure 2.
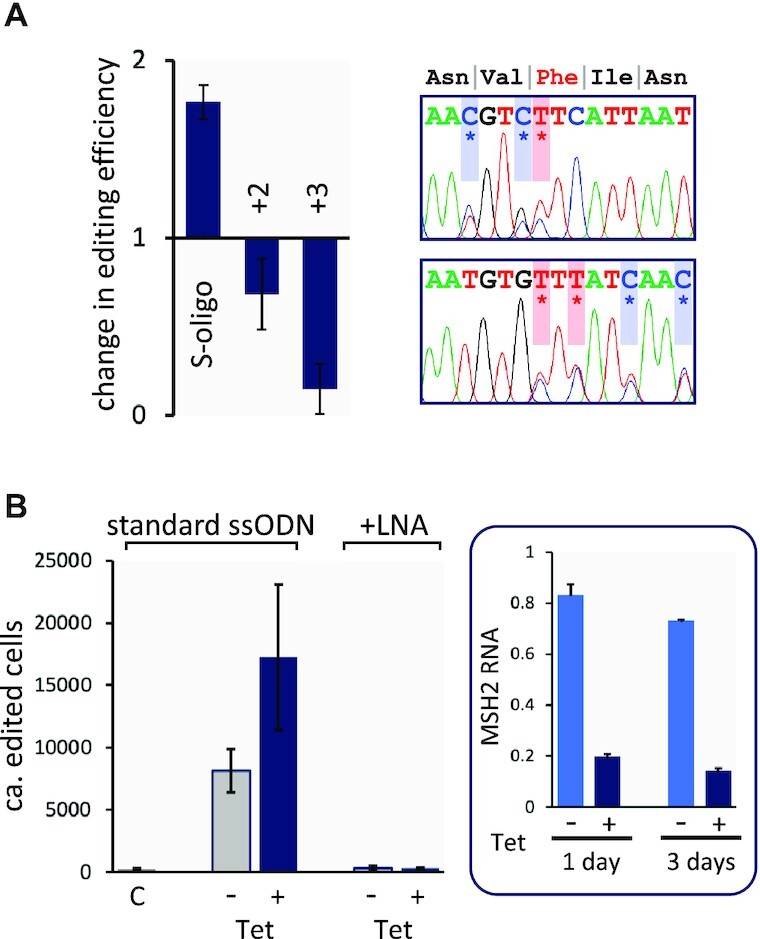
Multi-base editing and suppression by mismatch repair. (A) TbCRK12 L482F editing efficiency using a 51-b reverse S-oligo containing three phosphorothioate internucleotide linkages at each end or using standard ssODNs with 2–3 additional changes. DNA sequencing confirmed the desired edits (also see Supplementary Figure S2A). (B) Assessment of the impact of mismatch repair. TbCRK12 L482F editing efficiency following MSH2 knockdown using RNA interference; with either a standard 51-b reverse ssODN or with an equivalent ssODN containing a single locked nucleic acid (LNA) at the editing site. C, control; T. brucei cells lacking the RNAi construct. Another control following MSH2 knockdown but lacking the ssODN failed to yield any drug-resistant clones. The box on the right shows MSH2 knockdown relative to wild type cells as determined using qRT-PCR; 20–30% -Tet and >80% +Tet. Compound 2 selection was applied at 10 nM and n = 2 for each assay in A and B; error bars, SD.
Editing by oligo targeting is suppressed by the mismatch repair (MMR) machinery in E. coli (32), S. cerevisiae (33), and in embryonic stem cells (34). Indeed, MMR targets the newly replicated DNA strand (33,35). Using T. brucei cells in which the MMR component MSH2 (21) was knocked down, we observed a 68-fold increase in ssODN-dependent generation of drug-resistant clones relative to control cells (Figure 2B). The output was also increased by 32-fold in cells grown under non-inducing conditions, however, which was likely due to 20–30% ‘leaky’ MSH2 knockdown (Figure 2B, inset). Thus, editing by oligo targeting is also suppressed by MMR in T. brucei and an allele replacement frequency of 1.6 × 10–3 or 0.16% can be achieved following MSH2 knockdown.
We also tested an ssODN containing a locked nucleic acid, which can preclude MMR protein binding in E. coli (36). The results indicated that oligo targeting efficiency is indeed unaffected by MMR when using this modified ssODN in T. brucei (Figure 2B). However, this ssODN yielded relatively low efficiency editing, both before and after MSH2 knockdown, suggesting that the locked nucleic acid also interferes with incorporation at the replication fork in T. brucei.
We concluded that oligo targeting in T. brucei was optimal when combining 25 million cells with 40 μg of a 50–55-b reverse or ‘antisense’ ssODN, stabilized with phosphorothioate bonds, and with 1–3 centrally located and closely clustered base-edits. Editing efficiency can be further increased by perturbing the native mismatch repair machinery.
Establishing oligo targeting in T. cruzi and Leishmania
Having established and optimised oligo targeting in T. brucei, we next sought to apply the technology to other trypanosomatid parasites. In T. cruzi, we targeted the proteasome β5 subunit and introduced a G208S, GGC-AGC edit, not reported previously but equivalent to the G197S mutation in the ligand binding pocket reported to produce a steric clash with, and resistance to, compound 7 in L. donovani (6). DNA sequencing confirmed the desired edit (Figure 3A), which was associated with drug resistance (Figure 3B), as well as the ability to introduce further adjacent edits (Figure 3A, Supplementary Figure S2C).
Figure 3.
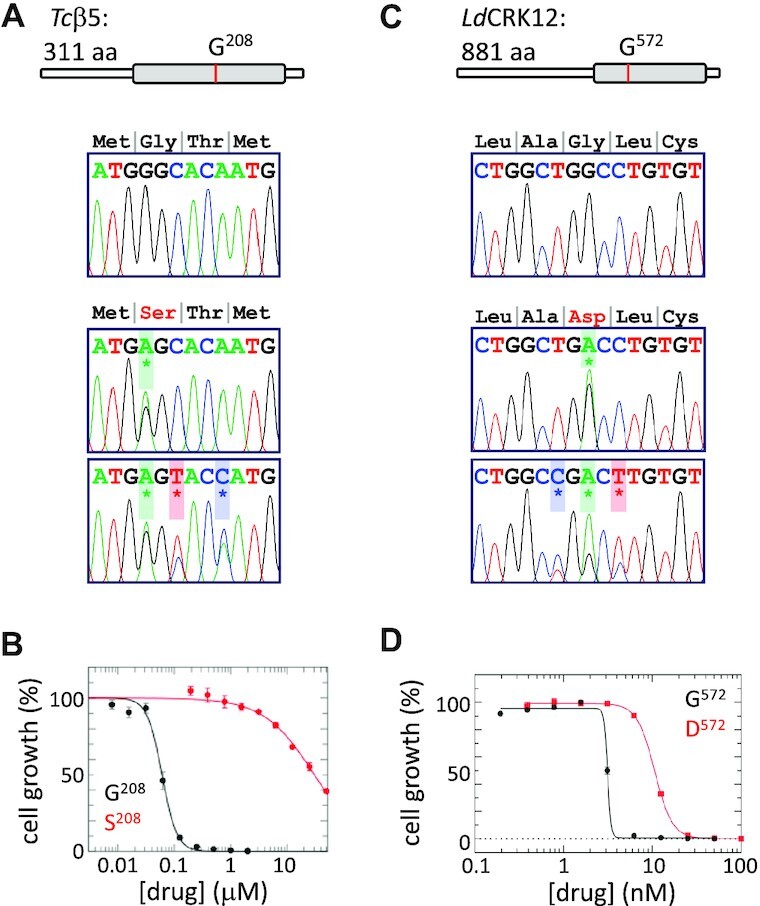
Establishing oligo targeting in T. cruzi and Leishmania. (A) G208S editing in the core domain (grey bar) of the T. cruzi proteasome β5 subunit, using either a 51-b reverse ssODN with a single G-A edit or a 56-b reverse ssODN with two additional edits. Compound 7 selection was applied at 350 nM. DNA sequencing confirmed the desired edits, n = 2 (also see Supplementary Figure S2C). A control lacking an ssODN failed to yield any drug-resistant clones. (B) The representative dose-response curves show that T. cruzi with the proteasome β5 G208S edit are drug-resistant; data are shown for one representative clone from three independent clones analysed; error bars, SD from two technical replicates. (C) As in A but showing G572D editing in the L. donovani CRK12 kinase domain (grey bar). In this case, a 55-b reverse ssODN was used to incorporate two additional synonymous edits. Compound 5 selection was applied at 10 nM (also see Supplementary Figure S2D). (D) As in B but for L. donovani CRK12 G572D editing; data are shown for one representative clone from two independent clones analysed; error bars, SD from two technical replicates. Similar results were obtained using S-oligos with a single mismatch.
In L. donovani, we targeted CRK12 and introduced a G572D, GGC-GAC edit, which conferred resistance to compound 5, either when the native gene was mutated or when an ectopic mutant copy was overexpressed (2). Again, DNA-sequencing confirmed the desired edit (Figure 3C), which was associated with drug resistance (Figure 3D), and the ability to introduce further synonymous edits (Figure 3C, Supplementary Figure S2D). From these experiments, we were able to estimate an allele replacement frequency of ∼10–5 for Leishmania. Indeed, all transfections, using either standard ssODNs or S-oligos, yielded drug-resistant populations for both Leishmania and T. cruzi. Using the assays described above, we found that editing and drug resistance were always ssODN-dependent, edits were always heterozygous, and drug resistance was always similarly increased in two or more independent edited clones. These results revealed the versatility and utility of oligo targeting in the trypanosomatid parasites associated with major neglected tropical diseases.
Oligo targeting for site saturation mutagenesis
Our maximal editing efficiency observed using oligo targeting, following MSH2 knockdown, remains 50-fold lower than the efficiency obtained previously using Cas9-based editing (10). Consequently, edited clones generated by oligo targeting are not readily isolated from populations in the absence of selection. The approach could be used to profile the variety of mutant amino acids and codons at a specific site that impact drug resistance, however. For such a site saturation mutagenesis approach, wild type T. brucei were mixed with an S-oligo containing a fully (64-fold) degenerate CRK12 G492 codon; G492 is adjacent to a conserved motif at the ATP binding site and mutation of this residue is a common route to kinase inhibitor resistance, including compound 2 resistance in T. brucei (3). Electroporated populations were grown in two different drug concentrations for five days, after which we used high-throughput amplicon sequencing to quantify those CRK12 edits that allowed the cells to tolerate drug pressure. The analysis revealed fourteen distinct resistance-conferring G492 codon edits encoding six distinct amino acids (Figure 4A). Following 5 nM drug selection, G492N, G492C, G492Q, G492I, G492M and G492T edits were observed (all possible codons in every case). Following 20 nM drug selection, the abundance of all edits encoding amino acids with polar neutral sidechains was diminished by >500-fold, while edits encoding amino acids with hydrophobic sidechains, G492I and G492M, persisted. Notably, editing was restricted to the target codon and all three nucleotides were edited in many of the new codons (Figure 4A). Thus, oligo targeting can be used to sample all possible codons at a given position, revealing edits and novel mutants with distinct drug resistance profiles, even following only a single electroporation (Figure 4B). Finally, we calculated how many clones likely represent each edited codon in these populations. A yield of 69 ± 32 resistant clones per electroporation, and fourteen among a possible sixty-four distinct codons observed following 5 nM drug selection, suggested an average of five clones representing each edited codon, or 20 when four electroporated populations were pooled.
Figure 4.
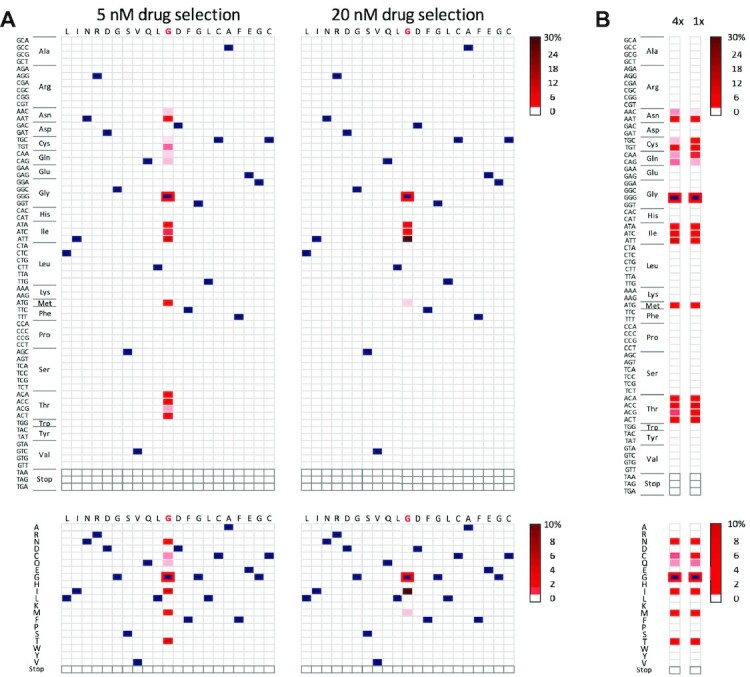
Oligo targeting for site saturation mutagenesis. (A) Amplicon sequencing and codon variant profiling following TbCRK12 editing in T. brucei using a 53-b S-oligo with a centrally located degenerate G492 codon. The heatmaps show average proportions of edited codons within the targeted region following compound 2 selection, relative to mock-transfected control cells (no ssODN). The lower panels show averages for edited codons representing each amino acid. Dark blue indicates the wild type codons or amino acids while the unedited GGG/G492 allele is indicated by red outlining. Drug at either 5 nM or 20 nM was applied to two independent populations. (B) Data from panel A (5 nM drug selection), derived by averaging two pools of cells from four electroporations (4×), are compared to data derived in parallel but using a single population derived from a single electroporation (1×). Other details as in A except only the targeted codon/amino acid position is shown in this case.
Our findings demonstrate the utility of oligo targeting to generate focussed genetic diversity within practical timescales in the parasitic trypanosomatids (see Table 1). The approach can be used to confirm on-target activity and can facilitate the assessment of mutations that impact drug efficacy. This relatively simple, rapid, precise, versatile, scalable and low-cost mutagenesis approach can be used to further develop our understanding of drug resistance-associated mutations and structure-activity relationships, which should facilitate the rational optimization of drug efficacy and durability.
Table 1.
Edited targets
| Parasite | Target name | GeneID | Edit (aa, nt) | WT EC50 (RΔ) | Drug |
|---|---|---|---|---|---|
| T. brucei | CRK12 | Tb927.11.12310 | L482F, CTC-TTC | 0.96 nM (33) | cpd 2 (3) |
| T. brucei | CRK12 | Tb927.11.12310 | G492x, GGG-NNN | 0.96 nM (ND) | cpd 2 (3) |
| T. brucei | CPSF3 | Tb927.4.1340 | N232H, AAT-CAT | 310 nM (4.8)* | acoziborole (4) |
| T. cruzi | proteasome β5 | TCSYLVIO_004939 | G208S, GGC-AGC | 63.9 nM (666) | cpd 7 (6) |
| L. donovani | CRK12 | LdBPK_090270 | G572D, GGC-GAC | 2.2 nM (3.3) | cpd 5 (2) |
EC50, Half maximal effective concentrations; RΔ, fold-increase in EC50 in edited and resistant cells; cpd, compound; * EC50 and RΔ determined previously (4).
DATA AVAILABILITY
Fastq files for the amplicon-seq analysis in this study have been deposited in the Short Read Archive (SRA) at https://www.ncbi.nlm.nih.gov/sra/ under BioProject ID PRJNA749253. The scripts to align the fastq files, qc control files and the scripts to extract the codon counts are available at https://github.com/mtinti/DH_Oligo-targeting and deposited in Zenodo (https://doi.org/10.5281/zenodo.5139694).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the chemistry teams within the Drug Discovery Unit, University of Dundee, for providing compounds used in this study. We also thank Richard McCulloch (University of Glasgow) for providing msh2 null strains.
Author contributions: S.A. Conceptualization, Formal analysis, Investigation, Methodology, Writing—review and editing; E.R. Conceptualization, Investigation, Methodology; S.C. Formal analysis, Investigation, Writing—review and editing; M.R. Formal analysis, Investigation, Writing—review and editing; A.T. Investigation, Supervision; H.D. Investigation; M.T. Data curation, visualisation, and analysis; S.W. Conceptualization, Funding acquisition, Supervision; D.H. Conceptualization, Formal analysis, Funding acquisition, Supervision, Writing—original draft.
Contributor Information
Simone Altmann, The Wellcome Trust Centre for Anti-Infectives Research, School of Life Sciences, University of Dundee, Dow Street, Dundee DD1 5EH, UK.
Eva Rico, The Wellcome Trust Centre for Anti-Infectives Research, School of Life Sciences, University of Dundee, Dow Street, Dundee DD1 5EH, UK.
Sandra Carvalho, The Wellcome Trust Centre for Anti-Infectives Research, School of Life Sciences, University of Dundee, Dow Street, Dundee DD1 5EH, UK.
Melanie Ridgway, The Wellcome Trust Centre for Anti-Infectives Research, School of Life Sciences, University of Dundee, Dow Street, Dundee DD1 5EH, UK.
Anna Trenaman, The Wellcome Trust Centre for Anti-Infectives Research, School of Life Sciences, University of Dundee, Dow Street, Dundee DD1 5EH, UK.
Hannah Donnelly, The Wellcome Trust Centre for Anti-Infectives Research, School of Life Sciences, University of Dundee, Dow Street, Dundee DD1 5EH, UK.
Michele Tinti, The Wellcome Trust Centre for Anti-Infectives Research, School of Life Sciences, University of Dundee, Dow Street, Dundee DD1 5EH, UK.
Susan Wyllie, The Wellcome Trust Centre for Anti-Infectives Research, School of Life Sciences, University of Dundee, Dow Street, Dundee DD1 5EH, UK.
David Horn, The Wellcome Trust Centre for Anti-Infectives Research, School of Life Sciences, University of Dundee, Dow Street, Dundee DD1 5EH, UK.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Investigator Award [217105/Z/19/Z to D.H.]; Centre Award [203134/Z/16/Z]; Innovations Award [218448/Z/19/Z to S.W. and I.H.G.] all from the Wellcome Trust; M.T. is also supported by a Wellcome Trust Investigator Award [101842/Z13/Z to M.A.J.F.]. Funding for open access charge: Wellcome Trust.
Conflict of interest statement. None declared.
REFERENCES
- 1. Field M.C., Horn D., Fairlamb A.H., Ferguson M.A., Gray D.W., Read K.D., De Rycker M., Torrie L.S., Wyatt P.G., Wyllie S.et al.. Anti-trypanosomatid drug discovery: an ongoing challenge and a continuing need. Nat. Rev. Microbiol. 2017; 15:217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wyllie S., Thomas M., Patterson S., Crouch S., De Rycker M., Lowe R., Gresham S., Urbaniak M.D., Otto T.D., Stojanovski L.et al.. Cyclin-dependent kinase 12 is a drug target for visceral leishmaniasis. Nature. 2018; 560:192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Smith A., Wall R.J., Patterson S., Rowan T., Rico Vidal E., Stojanovski L., Huggett M., Hampton S.E., Thomas M.G., Corpas Lopez V.et al.. Repositioning of a diaminothiazole series confirmed to target the cyclin-dependent kinase CRK12 for use in the treatment of African animal trypanosomiasis. J. Med. Chem. 2022; 65:5606–5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wall R.J., Rico E., Lukac I., Zuccotto F., Elg S., Gilbert I.H., Freund Y., Alley M.R.K., Field M.C., Wyllie S.et al.. Clinical and veterinary trypanocidal benzoxaboroles target CPSF3. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:9616–9621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Khare S., Nagle A.S., Biggart A., Lai Y.H., Liang F., Davis L.C., Barnes S.W., Mathison C.J., Myburgh E., Gao M.Y.et al.. Proteasome inhibition for treatment of leishmaniasis, chagas disease and sleeping sickness. Nature. 2016; 537:229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wyllie S., Brand S., Thomas M., De Rycker M., Chung C.W., Pena I., Bingham R.P., Bueren-Calabuig J.A., Cantizani J., Cebrian D.et al.. Preclinical candidate for the treatment of visceral leishmaniasis that acts through proteasome inhibition. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:9318–9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Creixell P., Schoof E.M., Tan C.S., Linding R.. Mutational properties of amino acid residues: implications for evolvability of phosphorylatable residues. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012; 367:2584–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hefnawy A., Negreira G., Jara M., Cotton J.A., Maes I., D’Haenens E., Imamura H., Cuypers B., Monsieurs P., Mouchtoglou C.et al.. Genomic and phenotypic characterization of experimentally selected resistant Leishmaniadonovani reveals a role for dynamin-1-like protein in the mechanism of resistance to a novel antileishmanial compound. Mbio. 2022; 13:e0326421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Potvin J.E., Leprohon P., Queffeulou M., Sundar S., Ouellette M.. Mutations in an aquaglyceroporin as a proven marker of antimony clinical resistance in the parasite leishmaniadonovani. Clin. Infect. Dis. 2021; 72:e526–e532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rico E., Jeacock L., Kovarova J., Horn D. Inducible high-efficiency CRISPR-Cas9-targeted gene editing and precision base editing in african trypanosomes. Sci. Rep. 2018; 8:7960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vasquez J.J., Wedel C., Cosentino R.O., Siegel T.N.. Exploiting CRISPR-Cas9 technology to investigate individual histone modifications. Nucleic Acids Res. 2018; 46:e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vergnes B., Gazanion E., Mariac C., Du Manoir M., Sollelis L., Lopez-Rubio J.J., Sterkers Y., Banuls A.L.. A single amino acid substitution (H451Y) in leishmania calcium-dependent kinase SCAMK confers high tolerance and resistance to antimony. J. Antimicrob. Chemother. 2019; 74:3231–3239. [DOI] [PubMed] [Google Scholar]
- 13. Zhang W.W., Matlashewski G.. CRISPR-Cas9-mediated genome editing in Leishmaniadonovani. Mbio. 2015; 6:e00861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bryant J.M., Baumgarten S., Glover L., Hutchinson S., Rachidi N.. CRISPR in parasitology: not exactly cut and dried. Trends Parasitol. 2019; 35:409–422. [DOI] [PubMed] [Google Scholar]
- 15. Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D.. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013; 31:822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gallagher D.N., Pham N., Tsai A.M., Janto A.N., Choi J., Ira G., Haber J.E.. A Rad51-independent pathway promotes single-strand template repair in gene editing. PLoS Genet. 2020; 16:e1008689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim D., Luk K., Wolfe S.A., Kim J.-S. Evaluating and enhancing target specificity of gene-editing nucleases and deaminases. Annu. Rev. Biochem. 2019; 88:191–220. [DOI] [PubMed] [Google Scholar]
- 18. Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A.et al.. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019; 576:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barbieri E.M., Muir P., Akhuetie-Oni B.O., Yellman C.M., Isaacs F.J.. Precise editing at DNA replication forks enables multiplex genome engineering in eukaryotes. Cell. 2017; 171:1453–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang H.H., Isaacs F.J., Carr P.A., Sun Z.Z., Xu G., Forest C.R., Church G.M.. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009; 460:894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bell J.S., McCulloch R.. Mismatch repair regulates homologous recombination, but has little influence on antigenic variation, in Trypanosomabrucei. J. Biol. Chem. 2003; 278:45182–45188. [DOI] [PubMed] [Google Scholar]
- 22. Alsford S., Horn D. Single-locus targeting constructs for reliable regulated RNAi and transgene expression in Trypanosomabrucei. Mol. Biochem. Parasitol. 2008; 161:76–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alsford S., Kawahara T., Glover L., Horn D. Tagging a T. brucei RRNA locus improves stable transfection efficiency and circumvents inducible expression position effects. Mol. Biochem. Parasitol. 2005; 144:142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brenndorfer M., Boshart M.. Selection of reference genes for mRNA quantification in trypanosomabrucei. Mol. Biochem. Parasitol. 2010; 172:52–55. [DOI] [PubMed] [Google Scholar]
- 25. Aslett M., Aurrecoechea C., Berriman M., Brestelli J., Brunk B.P., Carrington M., Depledge D.P., Fischer S., Gajria B., Gao X.et al.. TriTrypDB: a functional genomic resource for the trypanosomatidae. Nucleic Acids Res. 2010; 38:D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Langmead B., Salzberg S.L.. Fast gapped-read alignment with bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.Genome Project Data Processing, S. . The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Okonechnikov K., Conesa A., Garcia-Alcalde F.. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 2016; 32:292–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ewels P., Magnusson M., Lundin S., Kaller M.. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016; 32:3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tiengwe C., Marcello L., Farr H., Dickens N., Kelly S., Swiderski M., Vaughan D., Gull K., Barry J.D., Bell S.D.et al.. Genome-wide analysis reveals extensive functional interaction between DNA replication initiation and transcription in the genome of Trypanosomabrucei. Cell Rep. 2012; 2:185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Putney S.D., Benkovic S.J., Schimmel P.R.. A DNA fragment with an alpha-phosphorothioate nucleotide at one end is asymmetrically blocked from digestion by exonuclease III and can be replicated in vivo. Proc. Natl. Acad. Sci. U.S.A. 1981; 78:7350–7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Costantino N., Court D.L.. Enhanced levels of lambda Red-mediated recombinants in mismatch repair mutants. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:15748–15753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kow Y.W., Bao G., Reeves J.W., Jinks-Robertson S., Crouse G.F.. Oligonucleotide transformation of yeast reveals mismatch repair complexes to be differentially active on DNA replication strands. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:11352–11357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dekker M., Brouwers C., te Riele H.. Targeted gene modification in mismatch-repair-deficient embryonic stem cells by single-stranded DNA oligonucleotides. Nucleic Acids Res. 2003; 31:e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Reyes G.X., Kolodziejczak A., Devakumar L., Kubota T., Kolodner R.D., Putnam C.D., Hombauer H.. Ligation of newly replicated DNA controls the timing of DNA mismatch repair. Curr. Biol. 2021; 31:1268–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Ravesteyn T.W., Dekker M., Fish A., Sixma T.K., Wolters A., Dekker R.J., Te Riele H.P.. LNA modification of single-stranded DNA oligonucleotides allows subtle gene modification in mismatch-repair-proficient cells. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:4122–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Fastq files for the amplicon-seq analysis in this study have been deposited in the Short Read Archive (SRA) at https://www.ncbi.nlm.nih.gov/sra/ under BioProject ID PRJNA749253. The scripts to align the fastq files, qc control files and the scripts to extract the codon counts are available at https://github.com/mtinti/DH_Oligo-targeting and deposited in Zenodo (https://doi.org/10.5281/zenodo.5139694).