


Abstract
SUMO proteins are important regulators of many key cellular functions in part through their ability to form interactions with other proteins containing SUMO interacting motifs (SIMs). One characteristic feature of all SUMO proteins is the presence of a highly divergent intrinsically disordered region at their N-terminus. In this study, we examine the role of this N-terminal region of SUMO proteins in SUMO–SIM interactions required for the formation of nuclear bodies by the promyelocytic leukemia (PML) protein (PML-NBs). We demonstrate that the N-terminal region of SUMO1 functions in a paralog specific manner as an auto-inhibition domain by blocking its binding to the phosphorylated SIMs of PML and Daxx. Interestingly, we find that this auto-inhibition in SUMO1 is relieved by zinc, and structurally show that zinc stabilizes the complex between SUMO1 and a phospho-mimetic form of the SIM of PML. In addition, we demonstrate that increasing cellular zinc levels enhances PML-NB formation in senescent cells. Taken together, these results provide important insights into a paralog specific function of SUMO1, and suggest that zinc levels could play a crucial role in regulating SUMO1-SIM interactions required for PML-NB formation and function.
Graphical Abstract
Graphical Abstract.
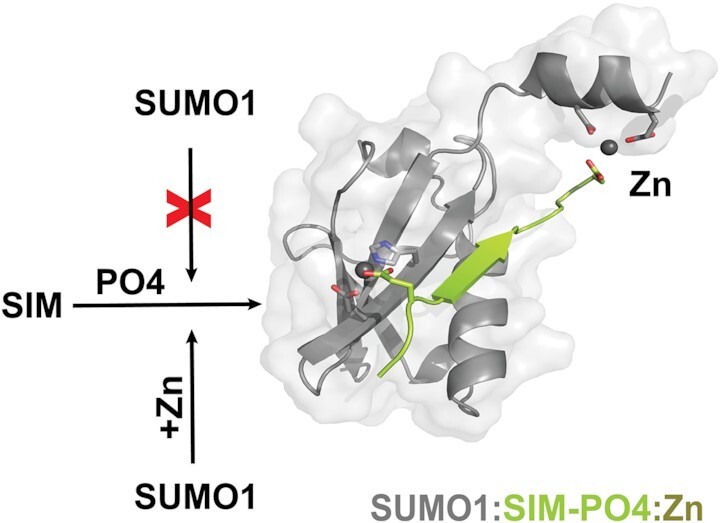
In the presence of zinc ions, the paralog specific auto-inhibition of SUMO1 is relieved to enhance binding to SUMO-interaction motifs (SIMs) in their phosphorylated forms.
INTRODUCTION
Post-translational modification with small ubiquitin-like modifier (SUMO) proteins occurs on a vast array of substrates and SUMOylation is a major regulatory mechanism that controls nearly all aspects of cell function (1). Like ubiquitination, SUMOylation involves the covalent attachment of SUMO-family proteins to a substrate through a series of enzymatic reactions that utilize a dimeric E1-activating enzyme, an E2-conjugating enzyme as well as in most cases an E3 ligase (2–5). The SUMOylation machinery in humans is significantly less diverse than the ubiquitination machinery as there is a single E1-activating enzyme (Aos1/Uba2), a single E2-conjugating enzyme (UBC9) and only a small group of E3-SUMO ligases characterized. SUMO proteins are conjugated to the side chain of Lys residues on substrates through an amide linkage and this posttranslational modification can be selectively reversed by SUMO proteases (6,7). Higher vertebrates express at least three different SUMO paralogs (SUMO1-3) ubiquitously in all cell types. SUMO2 and SUMO3 share 96% sequence identity between them and thus are often referred to as SUMO2/3, but they share only 53% and 52% sequence identity with SUMO1, respectively (6). In humans, two additional SUMO proteins (SUMO 4–5) have been recently identified (8,9). However, these proteins have been shown to be predominantly expressed in liver cells (9). The primary function of SUMO proteins appears to be their ability to control interactions with other partner proteins, and this can be accomplished either in the conjugated or unconjugated form (5,6). Such interactions occur with SUMO-binding domains (SBD) present within partner proteins, with the most common SBD being the SUMO-interacting motif (SIM) (5,6).
SIMs are characterized by the presence of a short hydrophobic core that contains the consensus sequence [V/I/L]-[V/I/L]-X-[V/I/L], and they are typically found in intrinsically disordered regions of proteins (5,10–12). SIMs bind in a β-strand configuration to a hydrophobic grove on the surface of SUMO proteins, and this binding can occur in either a parallel or an anti-parallel orientation relative to the second β-strand found in the core region of the SUMO protein (11,13). In addition, the hydrophobic core of many characterized SIMs is flanked by acidic residues (Asp/Glu) and/or phosphorylation sites (Ser/Thr) (14–17). These negatively charged residues present adjacent to the SIM typically enhance SUMO–SIM interactions, and it is generally thought that they dictate the binding orientation depending on whether they are located on the N-terminal (anti-parallel) or C-terminal (parallel) side of the hydrophobic core region (5,13,14,18). Some of the most thoroughly characterized examples of SUMO–SIM interactions are those that occur during the formation of nuclear bodies by the promyelocytic leukemia (PML) protein (17,19–21). PML-nuclear bodies (PML-NBs) are heterogeneous, dynamic structures where proteins involved in the regulation of diverse biological functions transit in and out of them (22,23). They are found in inter-chromosomal space and are tightly bound to the nuclear matrix (24), where they associate in a non-random manner with transcriptionally active gene-rich regions of the genome (25). Their number and size can be altered by controlling the transcription level of PML (26,27), and this occurs in response to cellular stresses such as viral infection, heat shock, exposure to heavy metals, transformation, or oxidative stress. (22,23). The PML protein is both SUMOylated at multiple sites and contains a SIM (17,21), and these two characteristics play a key role in regulating trafficking in and out of PML-NB through SUMO–SIM interactions with other proteins that share one or both of these same features (15,22,23,26,28–32). In the case of PML, the SIM is flanked by four Ser residues on the C-terminal side of the hydrophobic core, and phosphorylation of these four Ser residues by Casein Kinase II (CKII) has been shown to significantly enhance PML binding to SUMO proteins (17,19). Likewise, the Death-domain associated protein 6 (Daxx) protein is recruited to PML-NBs through interactions with a SIM motif located near the C-terminal end of the protein (20,30). Like the SIM of PML, phosphorylation of two Ser residues on the C-terminal side of the Daxx SIM by CKII enhances its binding to SUMO proteins in PML-NBs (14,20).
Despite a number of studies examining the details of SUMO–SIM interactions, there is still very little evidence of selectivity for the different SUMO paralogs for SIMs at the atomic level. This apparent lack of selectivity for most SUMO–SIM interactions is consistent with the fact that the core regions of the different SUMO paralogs, which contains the SIM-binding site, are highly homologous at both their sequence and structural level (18). The least conserved region between the various SUMO paralogs is found within the first 15–20 residues, and structural studies have shown that this N-terminal region forms an intrinsically disordered region that protrudes from the core ubiquitin-like domain (20,33–35). Although this N-terminal region is clearly a distinguishing feature of the various SUMO paralogs, its precise functional role is still not clearly defined despite the fact that this region appears to be post-translationally modified in a paralog specific fashion (36–38). For example, SUMO1 has been shown to be selectively phosphorylated at Ser2 and Ser9, whereas Lys11 of SUMO2/3 is a canonical site for the formation of poly-SUMO chains (36-38). In yeast, deletion of the N-terminal 17 amino acids of the lone SUMO-family protein (SMT3) has no apparent effect on either growth or response to stress, despite the fact that Lys15 is the major site required for poly-SUMO chain formation (39). In contrast, a similar N-terminal deletion in zebrafish SUMO2 leads to severe developmental defects and this suggests that there are paralog specific roles for this region in higher organisms (40). In addition, it has been shown that the N-terminal regions of both SUMO1 and SUMO2 prevents self-aggregation into amyloid-like fibrils and this anti-aggregation property was postulated to occurs through an intra-molecular shielding of hydrophobic residues in the β-strand 2 (β2) of the core domain (41,42).
Given that the β2 strand of SUMO-family proteins forms key interactions at the binding interface with SIMs, we were interested to determine whether the N-terminal region of SUMO1 and SUMO2 play an important role in defining paralog specific interactions. Using a combination of biochemical and structural studies, we have examined the role of the N-terminal regions of SUMO1 and SUMO2 in their binding to the SIMs of PML and Daxx. We establish that the N-terminal region of SUMO1 functions as an auto-inhibition domain for the binding to these two SIMs, but this is not the case for SUMO2. We also demonstrate that this auto-inhibition mechanism in SUMO1 was dependent on phosphorylation of serine residues adjacent to the hydrophobic core of the SIM of both Daxx and PML. In addition, biophysical and structural studies demonstrate that zinc can regulate the auto-inhibition by the N-terminal of SUMO1 through its ability to stabilize SUMO1-SIM interactions. The role of zinc in enhancing SUMO1-SIM interactions is supported by functional studies demonstrating that increases in zinc level enhance both the size and number of PML-NBs in cells under stress. Taken together, these results provide important insight into how the N-terminal region of SUMO1 functions in a paralog-specific manner to regulate SUMO1 binding to the SIMs of key components of PML nuclear bodies, and suggest that zinc levels could play a crucial role in forming PML-NBs in cells under stress.
MATERIALS AND METHODS
Expression vectors
The sequences encoding for the PML-SIM (residues 547–573 of human PML isoform I-V with a C-terminal Y residue for quantification) and the Daxx-SIM (residues 721–740 of human Daxx-1 with Cys to Ala substitution at position 728 and an N-terminal Y residue for quantification) were cloned into a modified pGEX-2T vector (Amersham) where the thrombin cleavage site was replaced with a Tobacco-Etch Virus (TEV) protease cleavage site as previously described (43). These constructs were also used to generate the PML-SIM-4SD (S/D substitutions at positions 560, 561, 562 and 565) and Daxx-SIM-2SD (S/D substitutions at positions 737 and 739) constructs by site directed mutagenesis. SUMO1 (residues 2–97 of human SUMO1 with a C52A substitution), SUMO1-E11Q (residues 2–97 of human SUMO1 with a E11Q and C52A substitution) and SUMO2 (residues 2–93 of human SUMO2 with a C48A substitution) cDNAs were cloned as BamHI/EcoRI fragments into the TEV modified pGEX-2T vector (16). The ΔN-SUMO1 (residues 17–97), ΔN-SUMO2 (residues 14–93) and UBC9 (residues 2–158 of human UBC9) cDNAs were generated through PCR amplification of the SUMO1 C52A and SUMO2 C48A as well as pGEX-2T-UBC9 (44) followed by their insertion as BamH1/EcoR1 into a modified pET-11b vector, where the thrombin cleavage site is replaced with a TEV protease cleavage site. The N2-SUMO1 sequence (residues 1–13 of human SUMO2 linked to residues 17–97 of human SUMO1 C52A) and the N1-S1 peptide sequence (residues 1–16 of SUMO1) were purchased as an oligonucleotide (BioBasic) with flanking BamHI/EcoRI restriction enzymes sites which were then 5′ phosphorylated, annealed and cloned into a TEV modified pGEX-2T vector. All constructs were verified by DNA sequencing.
Expression and protein purification
The PML and Daxx SIM peptides, the N1-S1 peptide as well as the SUMO1, SUMO1-E11Q, SUMO2 and N2-SUMO1 proteins were expressed as GST-fusion proteins in E. coli host strain TOPP2 (Bayou Biolabs) (19). The cells were grown at 37°C in Luria Broth (LB) media, and protein expression was induced for 4h at 30°C with 0.7 mM isopropyl-beta-d-galactopyranoside (IPTG; Inalco). The cells were harvested by centrifugation, suspended in lysis buffer (20 mM Tris–HCl pH 7.4, 1 M NaCl, 0.2 mM EDTA and 1 mM DTT), lysed with a French press and centrifuged at 105 000 × g for 1h at 4°C. The supernatant was collected and incubated with Glutathione Sepharose 4B (GSH) resin (GE Healthcare) for 1h at 4°C. Following incubation, the resin was collected by centrifugation, washed with lysis buffer and then with TEV buffer (25 mM Na2HPO4, 125 mM NaCl and 5 mM DTT). Removal of the GST tag was performed by incubating the resin for 2h with 100 units of TEV protease overnight at room temperature. The proteins were eluted with TEV buffer and dialysed into 10 mM Tris–HCl pH 7.5. The proteins were further purified over C4-reverse phase HPLC column (Vydac) and their identities confirmed by MALDI-TOF mass spectrometry (Regional Mass Spectrometry Centre of the University of Montreal, Montreal, Canada). Peptides were dialysed overnight into water, flash frozen, lyophilized and frozen at −80°C until being processed for ITC, fluorescence or X-ray crystallography experiments.
ΔN-SUMO1, ΔN-SUMO2 and UBC9 were expressed as His-tagged fusion proteins in Escherichia coli strain BL21DE3 (Stratagene). Cells were grown overnight at 37°C and due to leaky expression of these proteins in this construct, induction was not necessary. Cells were harvested by centrifugation, suspended in lysis buffer (20 mM Tris–HCl pH 7.4, 1 M NaCl and 50 mM imidazole), lysed with a French press and centrifuged at 105 000 × g for 1h at 4°C. Supernatant was collected and incubated with nickel-charged chelating-sepharose resin (GE healthcare) for 1h at 4°C. Following incubation, the resin was thoroughly washed with lysis buffer and the proteins were eluted from the resin with elution buffer (20 mM Tris–HCl pH 7.4, 1 M NaCl and 500 mM imidazole). Eluted proteins were dialyzed against TEV buffer for 3h. Cleavage of the His-tag was performed by incubating the dialyzed protein with 100 units of TEV protease overnight at room temperature. The next day, the sample was dialysed into lysis buffer and incubated with nickel-charged chelating-sepharose resin in order to remove the remaining His-tag and TEV protease. Proteins were further purified over C4-reverse phase HPLC column (Vydac), dialysed overnight in water, flash frozen, lyophilized and stored at −80°C until being processed for ITC, fluorescent or X-ray crystallography experiments. Their identities were confirmed by MALDI-TOF mass spectrometry
In vitro phosphorylation
The Daxx-SIM-PO4 peptide was generated from the un-phosphorylated Daxx-SIM peptide by incubation with CKII (New England BioLabs) (16,43). The peptides (200–250 nmol) were resuspended in 1 ml of buffer (50 mM Tris–HCl pH 7.5, 10 mM MgCl2, 0.1 mM EDTA, 2 mM DTT, 0.01% Brij 35). 3000 units of CKII (NEB) were added to the solution, which was then incubated for 72 h at room temperature. Every 12h, the reaction was supplemented with fresh enzyme (500 units) and fresh ATP (2 μmol) for the entire reaction period. After 72h, acetic acid was added to a final concentration of 5% (v/v) to quench the reaction. The solution was then centrifuged 2 min at 13 000 rpm, filtered and purified by HPLC over a C4-reverse phase HPLC column (Vydac). The identity was confirmed by MALDI-TOF mass spectrometry.
ITC experiments
Proteins and peptides were dialysed overnight at 4°C against the experimental buffer (20 mM Tris–HCl, pH 7.4) and filtered before use. For experiments with zinc and calcium, the proteins and peptides were dialyzed into buffer containing zinc (20 mM Tris–HCl pH7.4, 50 mM NaCl, 1 mM ZnSO4) or calcium (20 mM Tris–HCl pH 7.4, 50 mM NaCl, 1 mM CaCl2) Titrations consisted of 25 injections of 12μL each with a delay time of 280 s and measurements were performed at 25°C using a VP-ITC calorimeter (GE Helthcare). All titration experiments were performed at least in duplicates and fit to a single-site interaction with 1:1 stoichiometry. The data were baseline-corrected and fit with Origin 7 software.
Fluorescence binding and competition assays
Binding of 4,4′-bis(1-anilinonaphtalene 8-sulfonate) dye (bis-ANS) to SUMO1 or ΔN-SUMO1 were measured at room temperature on a Cary Eclipse Fluorescence Spectrophotometer (Agilent). The fluorescence of bis-ANS was measured from 400–600 nm following excitation at 370 nm and all experiments were run in 20 mM Tris–HCl at pH 7.5. In standard binding studies, samples contained 25 μM of bis-ANS and 50 μM of either SUMO1 or ΔN-SUMO1. For the competition assay, fluorescence was measured at 487 nm following the addition of varying concentrations of either the PML-SIM-4SD or N1-S1 peptide as a lyophylized powder into a sample containing a mixture of 25 μM bis-ANS with 50 μM of ΔN-SUMO1.
Crystallization and data collection
SUMO1 was suspended in water at a final concentration of 1 mM. PML-SIM or PML-SIM-4SD peptides were added to the SUMO1 sample in order to obtain a molar ratio of 1:1.2 (SUMO1:SIM-peptide). Crystals were obtained by the hanging drop vapor diffusion method using a precipitant solution containing 100 mM HEPES pH 7.0, 19 to 31% (w/v) PEG 3350, and 1–5 mM zinc chloride. Crystals were cryo-protected by increasing the PEG 3350 to a final concentration of 35%, mounted on a loop and flashed-cooled in a stream of nitrogen gas at 100 K. Diffraction data were collected using either the F1 beamline at MacCHESS (Cornell University, Ithaca, NY) or the X-ray home source at the University of Victoria (UVic Facility- Victoria, BC Canada). For the X-ray structures, the model building was performed with Coot (45,46) and structural refinement with PHENIX (47). Statistics for the final models (Table 1), were obtained with PHENIX (47) and Molprobity (48). The structure figures were prepared with PyMOL.
Table 1.
Data collection and refinement statistics
| Dataset | SUMO1:PML-SIM:Zn | SUMO1:PML-SIM-4SD:Zn |
|---|---|---|
| Data collection | ||
| Beamline | F1, macChess | UVic Facility |
| Wavelength (Å) | 0.977 | 1.542 |
| Space group | P 21 21 21 | P 21 21 21 |
| Unit cell parameters |
a = 37.00, b = 47.16, c = 63.78 α = β = γ = 90 |
a = 37.78, b = 47.46, c = 63.63 α = β = γ = 90 |
| Resolution range (Å) | 29.11–1.97 (2.04–1.97) | 31.82–1.772 (1.835–1.772) |
| No. of unique reflections | 8027 (637) | 11 313 (972) |
| Multiplicity | 5.5 (3.2) | 3.0 (2.1) |
| Completeness (%) | 96.58 (74.91) | 97.23 (85.41) |
| R merge | 0.039 (0.338) | 0.079 (0.43) |
| CC1/2 | 0.982 (0.263) | 0.996 (0.75) |
| I/σ(I) | 5.34 (0.68) | 12.02 (1.98) |
| Refinement statistics | ||
| Reflections (total/test)a | 43 817/2030 | 34 004/2052 |
| Rwork/Rfree (%) | 23.44/25.51 | 21.71/24.88 |
| CCwork | 0.903 (0.532) | 0.930 (0.827) |
| CCfree | 0.913 (0.556) | 0.909 (0.768) |
| No. atoms (excluding hydrogens) | ||
| Protein | 734 | 854 |
| Water Zn | 60 3 | 128 4 |
| B-factors | ||
| Protein | 31.57 | 17.61 |
| Water Zn | 38.04 44.50 | 26.35 29.82 |
| Rmsd | ||
| Bond length (Å) | 0.006 | 0.014 |
| Bond angle (°) | 0.84 | 1.52 |
| Ramachandranb | ||
| Favored (%) | 100 | 98.99 |
| Outliers (%) | 0 | 1.01 |
Values in parentheses are for highest resolution shell. 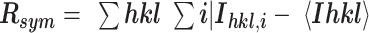 , where
, where 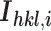 is the intensity of an individual measurement of the reflection with Miller indices
is the intensity of an individual measurement of the reflection with Miller indices 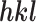 and
and 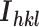 is the mean intensity of the reflection.
is the mean intensity of the reflection. 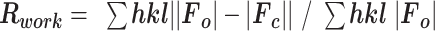 , where
, where 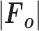 is the observed structure-factor amplitude and
is the observed structure-factor amplitude and 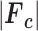 is the calculated structure-factor amplitude.
is the calculated structure-factor amplitude. 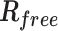 is the R factor based on at least 500 test reflections that were excluded from the refinement.
is the R factor based on at least 500 test reflections that were excluded from the refinement.
aReflection for Fo > 0.
bMolProbity analysis.
Analysis of PML-NBs in senescent cells
Normal human diploid fibroblasts IMR90 cells were purchased from Coriell Institute for Medical Research (Camden, NJ) and Phoenix Ampho packaging cells were a gift from Dr Scott W Lowe (Sloan Kettering Memorial). Cells were cultured in Dulbecco's modified Eagle medium (DMEM; Wisent) without pyruvate, supplemented with 10% high grade fetal bovine serum (FBS; Wisent) and 1% penicillin/streptomycin (Wisent). Retroviral control vector pBABE and Ras-activating vector pBABE-RAS-V12 were used as previously described to generate control and senescent IMR90 cells (27,49). Six days after introduction of the retroviral vectors, when the senescent phenotype is established, IMR90 cells were plated on coverslips to be treated the following day with either 5 μM pyrithione (PT; Toku-E) or 1 μM tetrakis(2-pyridinylmethyl)-1,2-ethanediamine (TPEN; Cayman), and fixed 24h later with 4% paraformaldehyde in phosphate buffered saline (PBS). For immuno-fluorescent analysis, the cells were washed with PBS containing 0.1 M glycine (2 × 5 min) and permeabilized using PBS containing 0.4% Triton X-100 and 0.1 M Glycine for 5 min a 4ºC. Following permeabilization, the cells were blocked using PBS with 3% bovine serum albumin (3 × 10 min) and treated overnight at 4ºC with a primary PML antibody (Bethyl; 301-167A) diluted 1/250 in blocking buffer. The next day, cells were washed and incubated with a secondary antibody (goat anti-rabbit Alex Fluor 568 diluted 1/1500) for 1h at room temperature in the dark. The cells were then washed with PBS (3 × 10 min) and mounted on a coverslip using Vectashield containing Dapi (Vector Laboratories H1200). All images were acquired sequentially on a Zeiss LSM 800 confocal microscope equipped with a spectral analysis detector. The data was acquired with a maximal of 2 Airy units. Data analysis were performed using the Image J data analysis software. An image with several regions of interest (ROIs; minimum of 50 cells) was taken with the intensity set between 10 000 and 65 550 for PML intensity. Images were converted into binary images and analyzed using the analyze particle function in image J with the parameter of 0.1 area to 5 and 0.3 to 1 circularity for PML bodies. To identify the number of PML per nucleus, images containing the nucleus were used to generate ROI and each ROI was counted individually with the aforementioned parameters. For nucleus, settings were set 20-infinity in area. For analysis of PML bodies larger than 1 mm2 analysis setting was set to 1 area to 5 and 0.3 to 1 circularity for PML bodies. For each condition, 50 cells were counted and statistics were performed using the GraphPad PRISM9 software. The experiments to characterize PML-NBs present in senescent cells were performed in duplicates and data points were shown as mean ± standard deviations.
For immunofluorescence of SUMO1 and SUMO2, cells were generated as described above until fixation. Prior to fixation cells were treated for 10 min with Cytoskeleton buffer (CSK-Buffer; 25 mM HEPES pH 7.8, 50 mM NaCl, 300 mM Sucrose, 1 mM EDTA, 3 mM MgCl2 and 0.5% Triton in distilled H2O with complete protease inhibitor), to permeabilize and remove non-bound cytosolic proteins. Cells were washed once with PBS followed by fixation for 10 min with 4% PFA in phosphate buffered saline (PBS). For immuno-fluorescent analysis, the cells were washed with PBS containing 0.1M Glycine (2 × 5 min) and blocked using PBS with 3% bovine serum albumin and 1% goat serum (3 × 10 min). For SUMO1, cells were then treated overnight at 4ºC with primary PML antibody (Bethyl; 301–167A, diluted 1/250 in blocking buffer) and primary SUMO1 (Invitrogen; 332400, diluted 1/100). The next day, cells were washed with PBS (3 × 10 min) and incubated with a secondary antibody (goat anti-rabbit Alexa Fluor 568 diluted 1/1000 for PML and goat anti-mouse Alexa Fluor 488 diluted 1/1000 for SUMO1 in PBS with 3% bovine serum albumin) for 1h at room temperature in the dark. For SUMO2, cells were then treated overnight at 4ºC with primary PML antibody (Santa Cruz; Sc-966, diluted 1/200 in blocking buffer) and primary SUMO2/3 (Abcam; AB3742, diluted 1/100). The next day, cells were washed with PBS (3 × 10 min) and incubated with a secondary antibody (goat anti-mouse Alexa Fluor 568 diluted 1/1000 for PML and goat anti-rabbit Alexa Fluor 488 diluted 1/1000 for SUMO2 in PBS with 3% bovine serum albumin) for 1h at room temperature in the dark. The cells were then washed with PBS (3 × 10 min) and mounted on a coverslip using Vectashield containing Dapi (Vector Laboratories H1200). Representative cells are shown and analysed for fluorescent intensity. To perform fluorescence intensity measurements of SUMO1 and PML or SUMO2/3, and PML, Image J with linear histogram intensity plugin was used as previously described (50). Briefly, a line was drawn through the image containing several PML/SUMO foci and then the intensity of fluorescent was extracted. Dotted lines in image show the intensity areas which were extracted for plot.
RESULTS
The N-terminal region of SUMO1 inhibits the binding of the phospho-mimetic form of the PML SIM
The exact functional role of the N-terminal region of SUMO1 (residues 1–18 in human SUMO1) remains ambiguous despite the fact that its sequence is significantly different from the one found in SUMO2/3 (Supplemental Figure S1). Given its potential role in regulating protein–protein interactions, we were interested to determine whether the intrinsically disordered N-terminal region of SUMO1 has any influence on SUMO–SIM interactions. To investigate this possibility, we initially examined the binding of SUMO1 to the SIM of PML using isothermal titration calorimetry (ITC) studies (Figure 1 and Supplemental Table S1). For these experiments, we prepared SUMO1 and ΔN-SUMO1 (residues 17–97 of human SUMO1) as well as two peptides corresponding to the SIM of PML (residues 547–573 of human PML isoforms I-V). The two peptides contain the hydrophobic core region (V556, V557, V558, I559) of the SIM of PML as well as the four adjacent serine residues (560, 561, 562 and 565) that have been shown to be phosphorylated by CKII (Figure 1A). One peptide encompasses the native sequence (PML-SIM), whereas in the other peptide, S560, S561, S562 and S565 were substituted with Asp residues (PML-SIM-4SD) as a mimic of the tetra-phosphorylated form of the PML-SIM. The ITC experiments show that the PML-SIM-4SD peptide binds full-length SUMO1 and the ΔN-SUMO1 with dissociation constants values (KD’s) of 11 ± 2 μM and 0.52 ± 0.08 μM, respectively, under the experimental conditions (Figure 1B-C). In contrast, the PML-SIM peptide binds full-length SUMO1 and the ΔN-SUMO1 with KD’s of 44 ± 5 μM and 58 ± 2 μM, respectively under identical experimental conditions (Figure 1C). These ITC results demonstrate that ΔN-SUMO1 binds with roughly 20-fold higher affinity to the phospho-mimetic PML-SIM-4SD peptide in comparison to SUMO1, whereas the PML-SIM peptide binds SUMO1 and ΔN-SUMO1 with similar affinities. Based on these results, it appears that the N-terminal region of SUMO1 can function as an auto-inhibition domain for regulating the binding of the phospho-mimetic form of the SIM of PML to SUMO1.
Figure 1.

The N-terminal region of SUMO1 inhibits binding of phosphorylated SIMs. (A) Amino-acid sequence of SIMs from PML and Daxx used for ITC studies. Underlined residues represent the hydrophobic core region of the SIM and those in bold represent either the phospho-mimetic or phosphorylated serine residues. (B) Representative ITC thermograms for the interaction between PML-SIM-4SD and either SUMO1 (left panel) or ΔN-SUMO1 (right panel). (C) Comparison of the KD values (in μM) obtained in ITC studies for the binding of either SUMO1 or ΔN-SUMO1 to the different SIM peptides and UBC9. All experiments were conducted in 20 mM Tris–HCl, pH 7.4.
The N-terminal region of SUMO1 also inhibits the binding of the phosphoSIM of Daxx
Given that the auto-inhibition by the N-terminal region of SUMO1 appears to be targeting the SIM-binding region, we were interested in determining whether the phosphorylated SIM of other proteins that transit to PML-NB are subject to a similar type of regulation. In the case of Daxx, it has been shown that its C-terminal SIM is also phosphorylated by CKII at two serine residues (S737 and S739) and that phosphorylation is important for the regulation of a number of its biological functions, including its transit into PML-NBs (20). Given the similarities between the phosphoSIMs of Daxx and PML, ITC experiments were conducted in order to determine if the N-terminal region of SUMO1 also inhibits binding to the phosphoSIM of Daxx. For these experiments, two peptides corresponding to the phosphoSIM of Daxx (residues 721–740 of human Daxx) were prepared: a phospho-mimetic SIM (Daxx-SIM-2SD; S737D and S739D) and a di-phosphorylated SIM (Daxx-SIM-PO4; pS737 and pS739) (Figure 1C and Supplemental Table S1). The ITC experiments show that the Daxx-SIM-PO4 peptide binds 8.8-fold stronger (KD 0.88 ± 0.12 μM versus 0.10 ± 0.02 μM) and the Daxx-SIM-2SD binds 8.2-fold stronger (KD: 2.8 ± 0.6 μM versus 0.34 ± 0.05 μM) to ΔN-SUMO1 (Figure 1C). Consistent with the previous results with PML-SIM-4SD peptide, there is a significant increase in the binding of the phosphorylated and phospho-mimetic Daxx-SIM peptides to ΔN-SUMO1 in comparison to SUMO1 (Figure 1C). Taken together, these ITC results indicate that the auto-inhibition by the N-terminus of SUMO1 may be a general mechanism for regulating interactions with SIMs that contain phosphorylation sites adjacent to their hydrophobic core.
The N-terminal auto-inhibitory region does not alter UBC9 binding to SUMO1
In addition to the SIM-binding interface, SUMO-family proteins contain a second distinct binding surface for interaction with the E2 SUMO conjugating enzyme UBC9, which is located on the opposite face of the SUMO core domain with respect to the SIM-binding interface (51). This second interface is formed by the β1, β4 and β5 strands of SUMO1 and UBC9 forms a specific non-covalent interaction with this region. To verify that auto-inhibition by the N-terminal region of SUMO1 is specific to interactions with the SIM-binding interface, the binding affinity of UBC9 to SUMO1 and ΔN-SUMO1 were compared using ITC experiments (Figure 1C and Supplemental Table S1). In the ITC experiments, UBC9 binds to SUMO1 and ΔN-SUMO1with KD values of 29 ± 11 nM and 73 ± 15 nM, respectively. These KD values are similar to the 82 nM value that was previously reported for SUMO1 binding to UBC9 by ITC under slightly different conditions (52), and UBC9 binds roughly 2.5-fold stronger to SUMO1 in comparison to ΔN-SUMO1. Based on these results, the deletion of the N-terminal region does not significantly alter UBC9 binding to SUMO1, which strongly suggests that the auto-inhibition effect of the N-terminal region of SUMO1 specifically regulates interactions involving its SIM-binding interface.
The N-terminal region interacts with the SIM-binding interface of SUMO1
Auto-inhibition by an intrinsically disordered region in a protein has been reported for several other proteins and in some cases, this has been shown to be controlled by the phosphorylation status of the protein (53–55). In the case of the SUMO-family proteins, previous studies with SUMO2 have shown that the N-terminal region protects the protein from self-aggregation and it was suggested that this occurs via an unspecified intra-molecular interaction (41). To determine if the N-terminal region of SUMO1 behaves as an auto-inhibitory domain by forming an intra-molecular interaction, fluorescence spectroscopy studies were performed to compare the binding of SUMO1 and ΔN-SUMO1 to 4,4′-bis(1-anilinonaphthalene 8-sulfonate) (bis-ANS). Similar to what was previously observed with SUMO2 (41), bis-ANS gives a stronger fluorescence signal in the presence of ΔN-SUMO1 than in the presence of SUMO1 (Figure 2A). These results suggest that there are additional hydrophobic regions present in ΔN-SUMO1 that are accessible for binding to bis-ANS and implies that the N-terminal region of SUMO1 shields hydrophobic regions present on the surface of SUMO1.
Figure 2.
The N-terminal of SUMO1 inhibits interactions with the SIM-binding interface. (A) Fluorescence spectra of bis-ANS binding to either SUMO1 (full line) or ΔN-SUMO1 (dashed line). (B) Graphical plot of the bis-ANS fluorescence signal recorded at 487 nm in the presence of ΔN-SUMO1 following addition of various amounts (0–4 molar equivalence) of the PML-SIM-4SD peptide.
In order to determine whether the hydrophobic surface of SUMO1 shielded by its N-terminal region is involved in binding to the phosphorylated form of the SIM of PML, a competition assay was performed between bis-ANS and the PML-SIM-4SD peptide. In this assay, the change in fluorescence intensity was measured following the addition of increasing concentrations of the PML-SIM-4SD peptide to a sample containing bis-ANS ΔN-SUMO1 at a 1:2 molar ratio. In this experiment, the addition of the PML-SIM-4SD peptide significantly diminishes the bis-ANS fluorescent signal in a concentration dependent manner (Figure 2B). In addition, we performed a similar competition assay between bis-ANS and a peptide corresponding to the first 16 amino acids of SUMO1 (N1-S1 peptide) with bis-ANS and ΔN-SUMO1 at a 1:2 molar ratio. As seen with the PML-SIM-4SD peptide, the addition of the N1-S1 peptide significantly diminishes the bis-ANS fluorescent signal in a concentration dependent manner (Suppemental Figure S2), although less efficiently than the PML-SIM-4SD as expected given that it is now detached from ΔN-SUMO1. Based on these results, it appears that the PML-SIM-4SD peptide and bis-ANS compete for a common binding region present in SUMO1. Overall, these results support the notion that the N-terminal region of SUMO1 interacts with its SIM-binding region via an intra-molecular mechanism and competes with the phosphorylated SIM of PML for binding to SUMO1.
The N-terminal region of SUMO2 does not function as an auto-inhibitory domain
In some cases, SIMs have been reported to bind in a paralog specific manner to SUMO proteins (18,56), but the molecular determinants that underlie the ability of a SIM to recognize a specific SUMO paralog are still not well-defined. However, the high sequence divergence at the N-terminal regions suggests that this region could be important for defining paralog specificity in certain cases. Therefore, we attempted to determine whether the auto-inhibition by the N-terminal region of SUMO1 is also present in the N-terminal region of SUMO2. To test this, ITC experiments with the phospho-mimetic forms of the SIMs of PML and Daxx were performed with SUMO2 and ΔN-SUMO2 (residues 14–93). The ITC experiments show that the PML-SIM-4SD peptide binds with only 1.8-fold stronger (KD of 2.9 ± 0.8 μM versus 5.3 ± 1.0 μM) to ΔN-SUMO2 in comparison to SUMO2, and the Daxx-SIM-2SD binds only 1.9-fold stronger (KD of 0.51 ± 0.11 μM versus 1.0 ± 0.1 μM). (Figure 3 and Supplemental Table S1). These results show that the increase in binding observed with ΔN-SUMO2 is significantly less dramatic than the differences obtained with the ΔN-SUMO1 (over 20-fold with PML-SIM-4SD and over 8-fold with Daxx-SIM-2SD), which suggests that the auto-inhibition by the N-terminal region is specific to SUMO1 binding to phosphorylated SIMs.
Figure 3.

The N-terminal of SUMO2 does not inhibit binding of phosphorylated SIMs. (A) ITC thermograms of the titration of PML-SIM-4SD with either SUMO2 (left panel) and ΔN-SUMO2 (right panel). (B) Comparison of KD values (in μM) obtained in ITC studies for the binding of either SUMO2 or ΔN-SUMO2 to the different SIM peptides. All experiments were conducted in 20 mM Tris–HCl, pH 7.4.
To further verify that this auto-inhibition is specifically associated with the presence of the N-terminal region of SUMO1, a chimeric protein (N2-SUMO1) containing the N-terminal region of SUMO2 (residues 1–13) linked to the core region of SUMO1 (residues 17–97) was evaluated for binding to the PML-SIM-4SD by ITC. In the ITC experiments (Supplemental Figure S3 and Table S1), N2-SUMO-1 binds to the PML-SIM-4SD peptide with a KD of 1.4 ± 0.1 μM. This is a 7.9-fold higher affinity than PML-SIM-4SD binding to SUMO1, but only 2.7-fold weaker affinity than its binding to ΔN-SUMO1.
Crystal structure of full length SUMO1 with PML-SIM-4SD and PML-SIM peptides
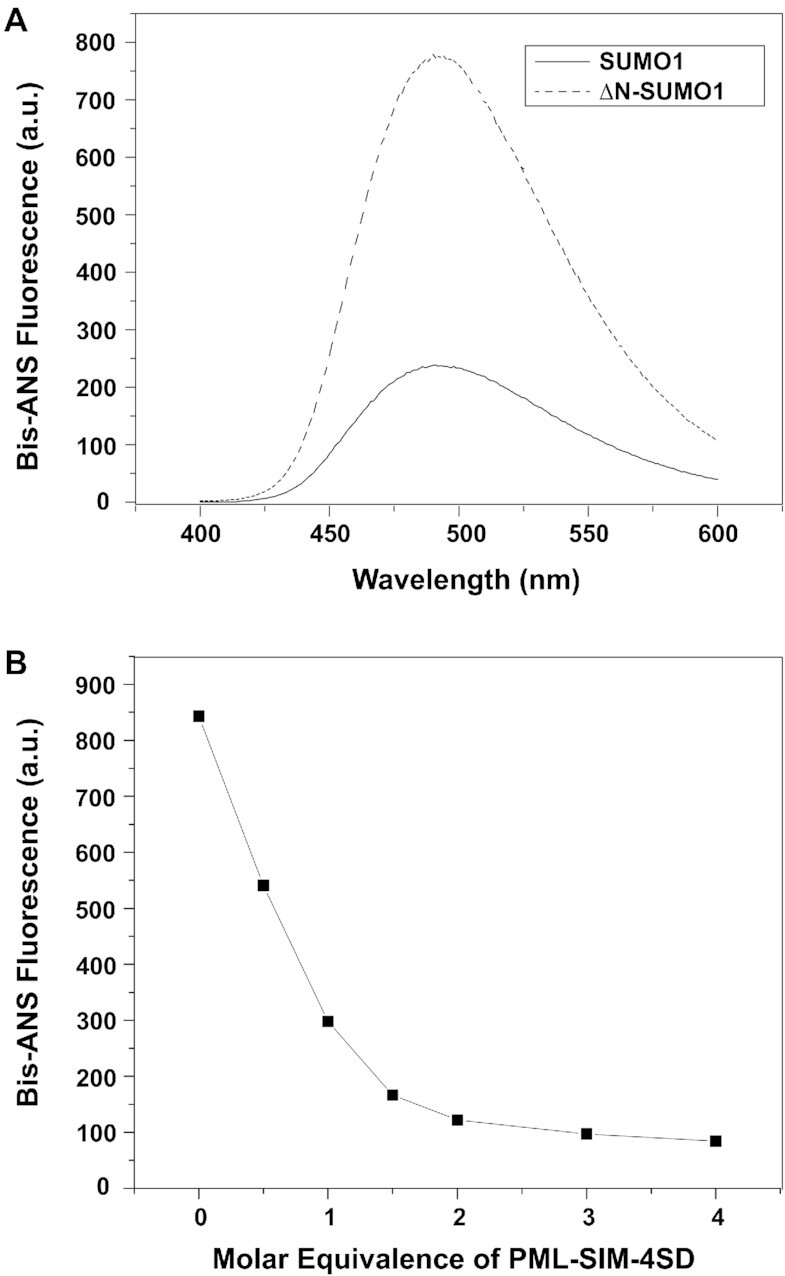
In previous structural studies with SUMO-family proteins, the N-terminal region of the protein has either been deleted (ΔN-SUMO1) or determined to exist in an intrinsically disordered conformation (5). Given that our ITC results indicate that the N-terminal can function as an auto-inhibition domain for the interaction between SUMO1 and the PML-SIM-4SD peptide, we attempted to crystallize the full-length SUMO1 protein alone or in the presence of either the PML-SIM-4SD or the PML-SIM peptides. Following an extensive screening of conditions, we were able to obtain crystals of SUMO1 in complex with both peptides, but not in its free form. The crystals with both peptides were obtained in similar conditions and in both cases, zinc was present in the buffer solution. The crystals obtained in the presence of zinc with the PML-SIM-4SD and the PML-SIM peptide are both of the P212121 space group and they diffract to a resolution of 1.77 and 1.97 Å, respectively (Table 1). In the crystals of the SUMO1:PML-SIM-4SD complex obtained in the presence of zinc (Figure 4), there is electron density for residues 9–93 of SUMO1 as well as for residues 552–564 of PML-SIM-4SD and the complex contains four bound zinc atoms. Two of the bound zinc atoms participate in the formation of the SUMO1:PML-SIM-4SD complex and the other two are associated with crystal packing. In the case of the SUMO1:PML-SIM complex in the presence of zinc (Figure 5), there is electron density for residues 18–97 of SUMO1 as well as for residues 552–560 of PML-SIM and the complex contains 3 bound zinc atoms. In this case, a single zinc atom is associated with the formation of the SUMO1:PML-SIM complex and again, the other two zinc atoms are associated with crystal packing
Figure 4.
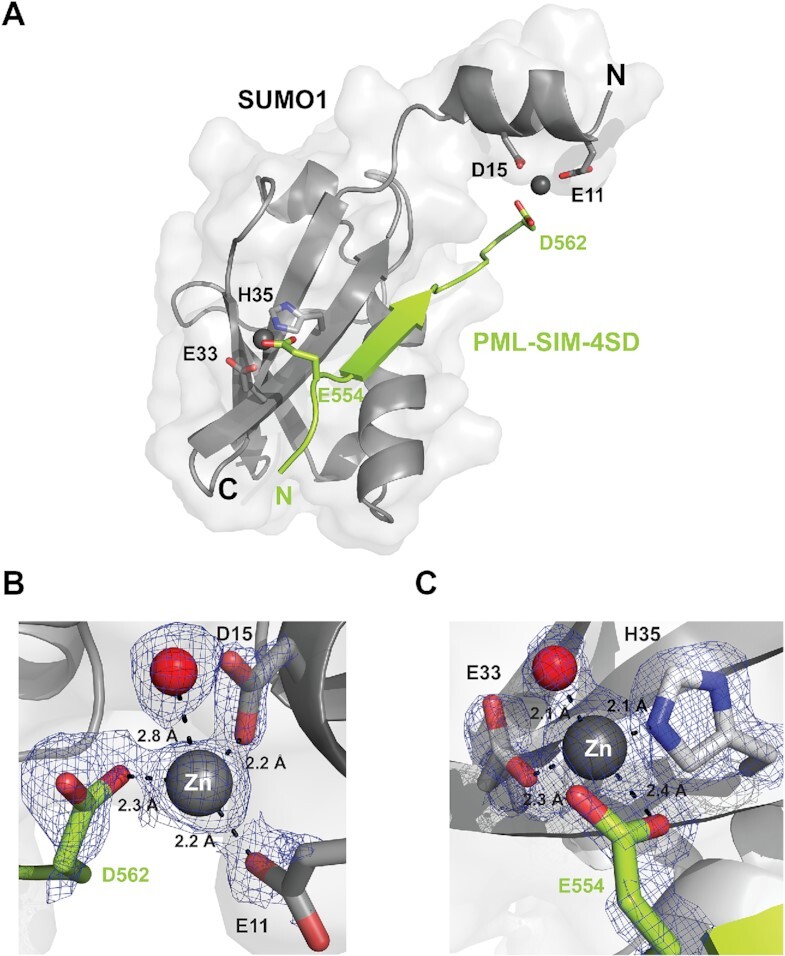
Structural characterization of a SUMO1:PML-SIM-4SD:Zn complex. (A) Cartoon representation of the structure of the SUMO1: PML-SIM-4SD complex in the presence of zinc highlighting the presence of an alpha helix in the N-terminal region of SUMO1. In the representation, SUMO1 is colored in gray and the PML-SIM-4SD peptide in lime green. The bound zinc atoms (Zn1 and Zn2) are represented as grey spheres and the side chains of the residues from SUMO1 and PML-SIM-4SD involved in chelating the zinc atoms are highlighted in stick representation. (B, C) Close-up and metrics (in Å) of Zn1 bound to the N-terminal region (B) and Zn2 bound to the core region (C) of SUMO1 and the PML-SIM-4SD peptide. The residues involved in coordinating the zinc atoms are in stick representation with their respective electron density contoured at 1.5σ cut-off from the 2FoFc map generated from the crystal diffraction data. The red spheres represent water molecules that are coordinated by the zinc atoms.
Figure 5.
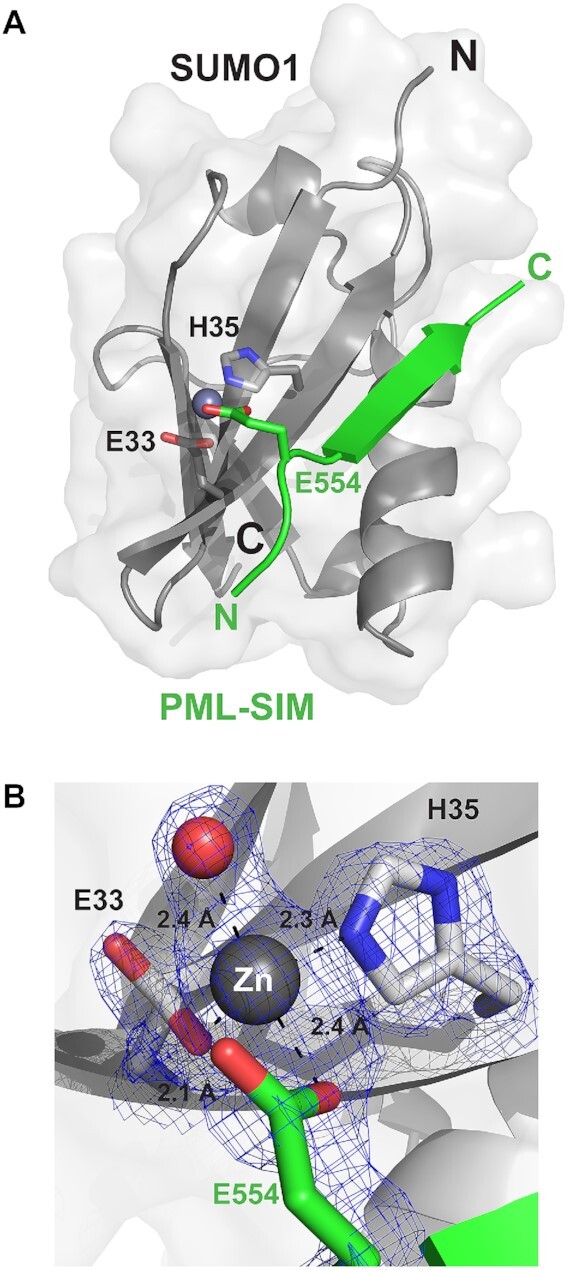
Structural characterization of a SUMO1:PML-SIM:Zn complex. (A) Overall structure of the SUMO1: PML-SIM complex in the presence of zinc highlighting the zinc bound between the core region of SUMO1 and the PML-SIM peptide. The proteins are in cartoon representation with SUMO1 in gray and PML-SIM peptide in green. The bound zinc atom is represented as a grey sphere and the side chains of the residues from SUMO1 and PML-SIM peptide involved in chelating the zinc atom is highlighted in stick representation. (B) Close-up and metrics (in Å) of the zinc atom bound to the core region of SUMO1 and the PML-SIM peptide. The residues involved in coordinating the zinc atom are in stick representation with their respective electron density contoured at 1.5σ cut-off from the 2FoFc map generated from the crystal diffraction data. The red sphere represents a water molecule that is coordinated by the zinc atom.
Like the previously reported crystal structures of complexes between ΔN-SUMO1 and the SIM of PML in either the tetra-phosphorylated or un-phosphorylated states, the core region of SUMO1 adopts a canonical ubiquitin-like fold whereas the PML-SIM-4SD and PML-SIM peptides adopt a parallel β-strand conformation with respect to the β2-strand of SUMO1. More specifically, the binding interface is stabilized by a network of hydrogen bonds between PML (residues 555–559) and SUMO1 (residues 35–37), and the side chains of V557 and I559 from the PML-SIM are buried in the hydrophobic groove formed between the α1 helix and the β2 strand of SUMO1. The most striking difference between these two new structures and all other structures of SUMO family proteins reported in the Protein Data Bank (PDB) occurs at the N-terminal region of SUMO1 and this occurs only in the SUMO1:PML-SIM-4SD complex. In this complex, SUMO1 adopts a 9-residue α-helix between residue 9–17 (Figure 4A), and this helix has not been previously observed in any other structure containing a SUMO-family protein including our current structure of SUMO1 in complex with the PML-SIM peptide in the presence of zinc.
The electron density map shows that this α-helix between residues 9–17 of SUMO1 is stabilized by a bound zinc atom (Zn1), which forms a bridge between the N-terminal region SUMO1 and the negatively charged D562 adjacent to the hydrophobic core in the PML-SIM-4SD peptide (Figure 4B and Supplemental Figure S4A). In the refined structure, Zn1 is coordinated to an oxygen atom from the side chains of E11 of SUMO1 (2.2 Å), an oxygen from the side chain of D15 of SUMO1 (2.1Å), an oxygen atom from the side chain of D562 of PML-SIM-4SD (2.3 Å) as well as a bound water molecule (2.8 Å) (Figure 4B and Supplemental Figure S4A). In addition, a second zinc atom (Zn2) involved in complex formation is coordinated to an oxygen from the side chain of E33 (2.3 Å) of SUMO1, a nitrogen atom from the side chain of H35 (2.1 Å) of SUMO1, an oxygen atom from the side chain of E554 of PML-SIM-4SD (2.4 Å) and an oxygen from a bound water molecule (2.1 Å) (Figure 4C and Supplemental Figure S4B).
In contrast to what is observed in the complex with the phospho-mimetic PML-SIM-4SD, no electron density is observed for the N-terminal region (residues 1–17) of SUMO1 in the complex with the unphosphorylated PML-SIM in the presence of zinc (Figure 5A). However, this complex does contain electron density for a single zinc atom bound between SUMO1 and PML-SIM. This zinc atom is coordinated in a similar manner as Zn2 in the SUMO1:PML-SIM-4SD complex by an oxygen from the carboxylic group of E33 (2.1 Å) of SUMO1, a nitrogen atom from the imidazole ring of H35 (2.3 Å) of SUMO1, an oxygen atom from the carboxylic group of E554 of PML-SIM (2.4 Å) and an oxygen from a bound water molecule (2.4 Å) (Figure 5B and Supplemental Figure S4C). Taken together, these structures demonstrate that zinc binding stabilizes the formation of an α-helix in the N-terminal region of SUMO1 when bound to the phospho-mimetic PML-SIM-4SD peptide and suggests that zinc ions have the capacity to serve as a regulator for the auto-inhibition by the N-terminal region of SUMO1.
Zinc regulates the auto-inhibitory activity of the SUMO1 N-terminal region
Given the presence of two bound zinc atoms binding between SUMO1 the PML-SIM-4SD peptide in the structure of the complex and the fact that one of these zinc atoms was bound by two residues in the helix in the N-terminal region of SUMO1, we were interested to determine whether or not zinc plays a role in the auto-inhibition mechanism observed in SUMO1. To test this, ITC experiment were performed to determine the affinity of the PML-SIM and the PML-SIM-4SD peptides for SUMO1 in the presence of 1mM zinc sulfate (ZnSO4). Under the experimental conditions for these studies, PML-SIM-4SD binds to SUMO1 with a KD = 0.57 ± 0.08 μM in the presence of zinc, but with a KD = 22 ± 1 μM in the absence of zinc (Figure 6 and Supplemental Table S2), and the PML-SIM binds to SUMO1 with a KD = 42 ± 8.0 μM in the presence of zinc, but with a KD = 160 ± 10 μM in the absence of zinc. Thus, the binding of the phospho-mimetic PML-SIM-4SD to SUMO1 increases by 38-fold in the presence of zinc, whereas the binding of the unphosphoylated PML-SIM to SUMO1 increases by only 4-fold in the presence of zinc. To verify that this increase in binding is specific to zinc, additional ITC experiments were performed under identical conditions, but in the presence of 1 mM calcium chloride (CaCl2). In contrast to what was observed in the presence of zinc, there is essentially no difference observed in SUMO1 binding to the PML-SIM-4SD peptide in the presence of calcium (KD = 22 ± 3 μM) compared to its binding in the absence of calcium (KD = 22 ± 1 μM) (Figure 6B and Supplemental Figure S5). In addition, we tested whether or not the increased binding in the presence of zinc is specific to SUMO1. In these ITC experiments, we compared the binding of SUMO2 to the PML-SIM-4SD peptide in the presence and absence of zinc. Under our experimental conditions, PML-SIM-4SD binds to SUMO2 with a KD = 6.5 ± 1.2 μM in the presence of zinc, but with a KD = 12 ± 1 μM in the absence of zinc (Figure 6C). Thus, the binding of the PML-SIM-4SD to SUMO2 increases by only 2-fold in the presence of zinc as compared to the 38-fold difference seen with SUMO1. To verify the key role of E11 of SUMO1 in bridging the binding to zinc, we examined the binding of SUMO1 containing a Gln substitution for E11 (SUMO1-E11Q) to the PML-SIM-4SD peptide in the presence and absence of zinc. Under our ITC experimental conditions, PML-SIM-4SD binds to SUMO1-E11Q with a KD = 4.2 ± 1.3 μM in the presence of zinc, but with a KD = 8.7 ± 1.4 μM in the absence of zinc (Figure 6B). Thus, the binding of the PML-SIM-4SD to SUMO1-E11Q increases by only 2-fold in the presence of zinc, which is similar to what was observed with SUMO2, but significantly less than the 38-fold difference seen with SUMO1. Taken together, the results demonstrate that zinc ions have the ability to enhance binding to SUMO1 to the PML-SIM-4SD in a paralog specific manner. In addition, these results suggest that both bound zinc atoms in the structure of the complex help enhance the binding of SUMO1 to the SIM of PML, but the zinc atom that bridges between the N-terminal of SUMO1 and the phospho-mimetic PML-SIM-4SD plays a more significant role in enhancing the affinity.
Figure 6.

Zinc ions specifically enhance binding of the phospho-mimetic SIM of PML to SUMO1. (A) ITC thermograms from the titration of the PML-SIM-4SD peptide with SUMO1 either in the absence (left panel) or the presence of zinc sulfate (right panel). (B) Comparison of the KD values (in μM) obtained in ITC studies for SUMO1 binding to PML-SIM and PML-SIM-4SD and SUMO1E11Q binding to PML-SIM-4SD in the presence and absence of zinc as well as the binding to PML-SIM-4SD in the presence of calcium. (C) Comparison of the KD values (in μM) obtained in ITC studies for PML-SIM-4SD binding to SUMO1 and SUMO2 in the presence and absence of zinc. All experiments were conducted in 20 mM Tris–HCl pH7.4, 50 mM NaCl in the absence or presence of 1 mM ZnSO4.
Changes in cellular zinc levels modulate PML-NB formation in senescent cells
Given that the results from the biophysical and structural studies demonstrate that zinc has the capacity to enhance the interaction of SUMO1 with the phospho-mimetic SIM of PML, we were interested to determine whether increases in cellular zinc levels alter PML-NB formation which are formed in part by SUMO–SIM interaction (11,22,23). Cellular zinc levels are tightly regulated (57), but several small molecules known as zinc ionophores have the capacity to bind zinc and enhance its passage into cells leading to increases in cellular zinc levels (58,59). To test whether PML-NB formation is altered by varying cellular zinc levels, we selected the model of oncogene-induced senescence of IMR90 normal human fibroblasts expressing the activated Ras oncogene, where an increase in PML-NB formation occurs (27,60,61). Next, the cells were treated for 24h with either the zinc ionophore pyrithione (5 μM) to increase cellular zinc levels or with the zinc specific chelator tetrakis(2-pyridinylmethyl)-1,2-ethanediamine (TPEN; 1 μM) to decrease cellular zinc levels. Immunofluorescence analysis using an antibody specific for PML demonstrates that the pyrithione treatment induces significantly the formation of PML-NBs in the Ras-expressing cells, but only a marginal increase in the vector-control cells (Figure 7). Actually, in the Ras-expressing cells, there is an increase in both the total number of PML-NBs formed (Figure 8A) as well as the size of the PML-NBs (Figure 8B) with pyrithione treatment. In contrast, treatment of TPEN results in a decrease is both the total number and size of the PML-NBs in the Ras-senescent cells, but these decreases are less significant than the increases associated with the pyrithione treatment. In addition, immunofluorescence analysis using specific antibodies for either SUMO1 or SUMO2/3 demonstrates that their levels in PML-NBs increase significantly in senescent cells treated with pyrithione, but not with TPEN (Supplemental Figure S6). The increase in SUMO proteins in parallel to the increase in PML-NBs we observe in response to zinc could be explained by an increase in SUMO–SIM interactions in response to zinc as suggested by our structural model. Taken together, the results demonstrate that increases in cellular zinc levels can significantly increase PML-NB formation as well as the levels of SUMO proteins in PML-NBs in Ras-induced senescence cells.
Figure 7.
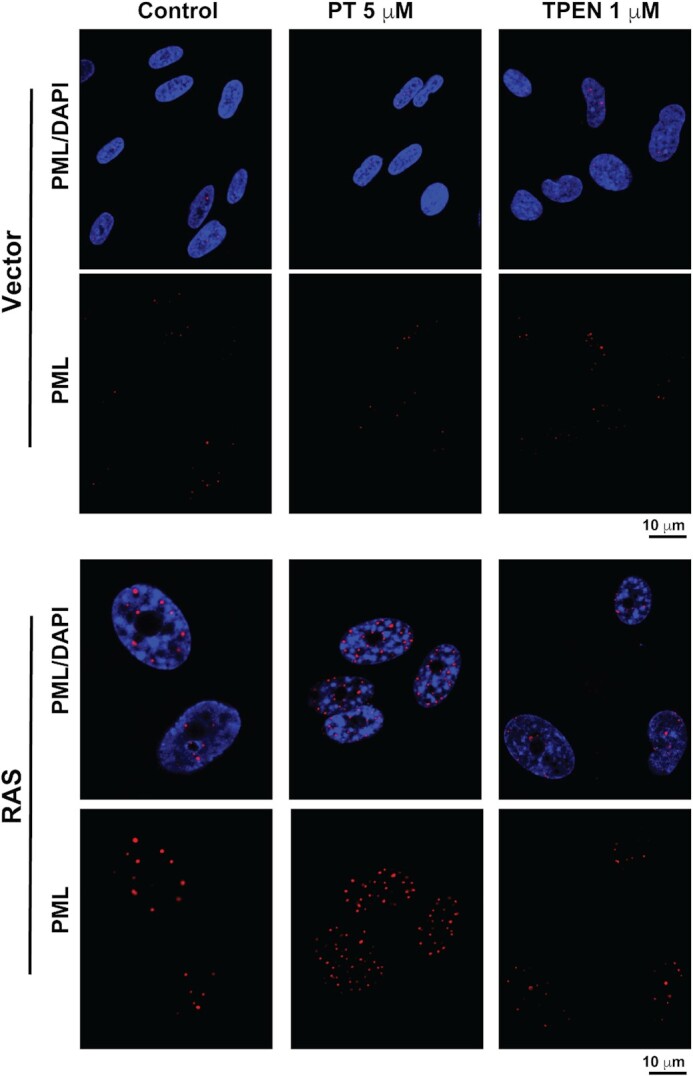
Increasing cellular zinc levels in senescent cells enhances PML-NB formation. Normal human fibroblasts IMR90 cells expressing a control vector or the activated RAS oncogene, to induce senescence were treated for 24h with either control vehicle, 5 μM PT (pyrithione) or 1 μM TPEN prior to fixation and indirect immunofluorescence with a specific anti-PML antibody (Bethyl # A301-167A) and with DAPI to stain DNA in the nucleus.
Figure 8.
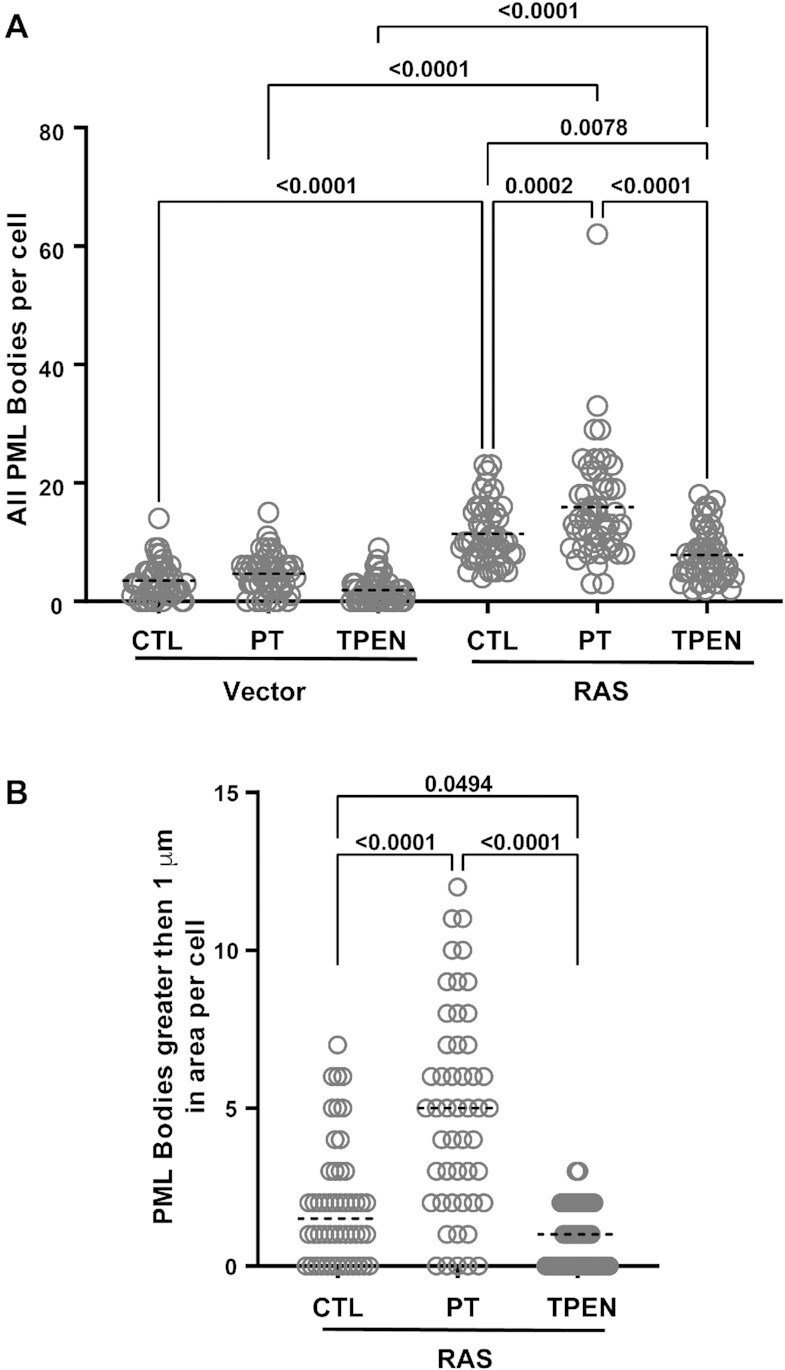
Increasing cellular zinc levels in senescent cells enhances PML-NB formation. Normal human fibroblasts IMR90 cells expressing a control vector or the activated RAS oncogene, to induce senescence, were treated for 24 h with either control vehicle, 5 μM pyrithione (PT) or 1 μM TPEN prior to fixation and quantitative analysis for the overall number of PML-NBs per cell (A) and the number of PML-NBs greater than 1 μm in size (B). For each condition, 50 cells from two independent experiments were counted and statistics were performed using the PRISM9 software.
DISCUSSION
The interaction of the different SUMO paralogs with proteins containing SIMs are important for the formation of PML-NBs, and these interactions are regulated by a number of different post-translational modifications, including SUMOylation, phosphorylation and acetylation (19,32,62). Although the different SUMO paralogs display many overlapping functions, they also appear to have specific functions and previous studies have shown that they can have varying affinities for select SIMs (18,56,63–65). From a sequence/structure point of view, all SUMO paralogs possess a conserved ubiquitin-like core domain that contains the SIM-binding region. However, there is a much greater variability when comparing the sequences of their intrinsically disordered N-terminal region. This suggests that the N-terminal region of the different SUMO proteins could potentially play an important role in distinguishing paralog specificity in SUMO–SIM interactions. In this work, we investigated the role of the N-terminal region of SUMO1 in its binding to SIMs from proteins that help regulate PML-NB formation. Using a combination of biophysical, structural and functional experiments, we demonstrate that the N-terminal region of SUMO1 functions as an auto-inhibition domain for regulating the binding to the SIMs of PML and Daxx. This auto-inhibition mechanism is specific for SUMO1 binding to the phosphorylated forms of the SIMs, as we were not able to observe similar effects with either the unphosphorylated SIMs binding to SUMO1 or in experiments with the SIMs binding to SUMO2. Structural characterization of crystals of a SUMO:PML-SIM-4SD complex in the presence of zinc demonstrates that a bound zinc atom bridges the N-terminal of SUMO1 and negatively charged residues from the PML-SIM-4SD. Importantly, this zinc atom stabilizes the formation of a 9-residue helix within the normally disordered N-terminal region of SUMO1. In support of an important role for zinc ions in SUMO1-SIM interactions, ITC studies showed that SUMO1 binding to PML-SIM-4SD is significantly enhanced in the presence of zinc, and enhancing cellular zinc levels with the zinc ionophore pyrithione leads to increases in formation of PML-NBs in senescent cells.
The presence of an auto-inhibition domain within the N-terminal region of SUMO1, but not in SUMO2, is somewhat surprising. It was previously shown that the N-terminal regions of both SUMO1 and SUMO2 possess an anti-aggregation activity, which was postulated to occur through an in trans interaction within the two proteins (41). More specifically, it was suggested that the N-terminal region of both of these SUMO paralogs interacts with the hydrophobic SIM-binding region within the core of the proteins, and this prevents protein aggregation. Our ITC and fluorescent studies support this notion that the presence of the N-terminal of SUMO1 alters access to the hydrophobic SIM-binding region, and this serves to inhibit interactions with SIMs of PML and Daxx in their phosphorylated form. However, we did not observe a similar inhibition for SUMO2 binding to either of the same two SIMs in their phosphorylated forms. One explanation for the difference between the two paralogs is that the N-terminal region of SUMO2 lacks key amino acids residues present in SUMO1, which are required to interfere with the ability of the SIM-binding region to interact with other proteins containing SIMs. This would be consistent with the fact that there is very little sequence conservation between these two SUMO paralogs in the N-terminal region and is further supported by our experiments demonstrating that swapping the N-terminal region of SUMO2 onto SUMO1 eliminates a significant portion of the auto-inhibition effect towards binding the phosphorylated SIM of PML. Alignment of SUMO1 sequences from different animal species (Supplemental Figure S7) shows that the two negatively charged amino acids in the N-terminal that bind to the zinc atom (E11 and D15) in the crystal structure of the complex are conserved. However, this is not the case for the five human SUMO paralogs, as these two residues are conserved in SUMO5, but not in SUMO2, SUMO3 or SUMO4 (Supplemental Figure S1B). This suggests that the regulation of auto-inhibitory activity of SUMO1 by zinc is very likely conserved in many other animal species, but not in the other human paralogs with the possible exception of SUMO5.
It is well documented that the non-biological metals arsenic, antimony and cadmium can all significantly alter the dynamics of PML-NBs in cells (66–75). Thus, it is not surprising that a biological metal, such as zinc, is playing an important role in regulating SUMO–SIM interactions since they play such a crucial role in controlling PML-NB dynamics (11,19,22,23,62,76,77). Arsenic, antimony, zinc and cadmium are all known to associated with sulfur atoms present in cysteine residues in proteins (74,78–81), and As2O3 is an effective treatment for Acute Promyelocytic Leukemia (APL) associated with a translocation in the PML gene (69,82–84). Mechanistically, arsenic has been shown to bind to cysteine residues within the zinc-binding domains of PML, and it has been proposed that arsenic has the capacity to selectively displace zinc from PML in comparison to other proteins containing zinc-binding domains (68,74,85). Arsenic binding to the cysteine residues was shown to result in a local misfolding of PML and this results in the formation of PML oligomers (68). The oligomerization leads to the formation of larger PML-NBs almost immediately following exposure to arsenic (68,70,86,87). However, the PML in these larger bodies is rapidly degraded over the next few hours (87). The degradation of PML is due to the fact that the partially unfolded PML binds more readily to UBC9 (68), which results in increased formation of poly-SUMOylated PML that is degraded by the SUMO-dependent ubiquitin ligase RNF4 (67–71,83,87,88). Given its similarity to arsenic, antimony treatment has also been shown to enhance formation of poly-SUMOylated PML, which is then degraded by RNF4 (74,75). In contrast, increases in cellular levels of cadmium enhance disruption of PML-NBs into microstructures (66,72,73,86,89–91). Although the exact mechanism by which cadmium disrupts PML-NB dynamics has not been identified at the molecular level, cadmium also has the capacity to bind cysteine residues in proteins (77). However, it does not appear to have the ability to induce RNF4 dependent degradation of PML. Therefore, it appears that cadmium, like arsenic and antimony, is also binding to cysteine residues in the zinc-binding domains of PML and alters its folding by displacing zinc (92), but without inducing PML poly-SUMOylation. Based on our results, it appears that zinc availability alters the dynamics of PML-NB formation in a dramatically different manner as to what is observed following exposure to these non-biological metals. Our results demonstrate that altering zinc levels in cells with the zinc ionophore pyrithione leads to a significant increase both in the size and the number of PML-NBs that are present even 24h after the treatment is initiated and these increases can be blocked by the addition of the zinc specific chelator TPEN. Thus, zinc does not appear to enhance the degradation of PML by RNF4 in these larger bodies, as seen following treatment with arsenic and antimony. This may be due to the fact that increases in zinc levels help stabilize the native fold of PML, which has lower affinity for UBC9 than the partially unfolded form adopted following treatment with arsenic (68). In addition, our results suggest that zinc can also enhance SUMO–SIM interactions with phosphorylated SIMs required for the formation of the higher order structure of PML-NBs (23).
The role of zinc could also be linked to the functions of PML-NBs during the stress response (93,94). PML-NBs share common properties with many other liquid-liquid phase separation processes that play important roles in regulating cellular responses to stress (32,95–98). In particular, PML condenses into droplets using weak interactions between its modular domains, and it has been shown that key factors regulating the biogenesis of these droplets include the affinity, stoichiometry and valency of SUMO–SIM interactions (76,99,100). Therefore, it is possible that zinc plays a key role in regulating the phase separation processes associated with PML-NB by enhancing SUMO1 interaction with multiple different proteins containing phosphorylated SIMs (20,30). Several proteins that locate to PML-NBs contain a SIM with either a negatively charged amino acid residue or a phosphorylation site immediately agent to the hydrophobic core residues in the same position as D562 of PML-SIM-4SD, including Daxx, PIAS1-4, RNF4 and SP100. Therefore, it is possible that zinc could regulate the interaction of SUMO1 with a plethora of SIM-containing proteins that transit within PML-NBs. However, more detailed studies in cells are required to determine the exact mechanism by which increased cellular zinc levels enhance formation of PML-NBs in senescence cells. Still, the effect of increased cellular zinc levels on PML-NB formation is an intriguing observation, since stress granule formation is also enhanced by increasing cellular zinc levels (101) and SUMO proteins also play an important role in stress granule formation (102,103). In future studies, it will be important to determine the cellular details of interactions within PML-NBs regulated by enhanced zinc levels as well as to investigate whether zinc is required for the formation of other membrane-less organelles where SUMO proteins have been shown to play a key role (104), through its ability to regulate SUMO1–SIM interactions.
DATA AVAILABIILITY
The atomic coordinates and structure factors have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/). The PDB Accession codes are: 8DJI for the SUMO1:PML-SIM:Zn complex and 8DJH for the SUMO1:PML-SIM-4SD-Zn complex.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Dr Alisdair Borastan for usage of the diffractometer at the University of Victoria, Dr. Nicolas Stifani for help with the data analysis of PML-NBs and to Dr Stephen Michnick for useful discussion.
Author contributions: M.L.P., G.F., L.C. and J.G.O. designed experiments; M.L.P., H.W., X.H.M., S.I., V.B., C.G. and L.C. conducted experiments; M.L.P., X.H.M., J.G.O., S.I., V.B., L.C., C.G. and H.W. analyzed the results; M.L.P. and J.G.O. wrote the paper with critical revisions by X.H.M., G.F., L.C. and H.W.
Notes
Present address: Laurent Cappadocia, Département de Chimie, Université du Québec à Montréal, Montréal, QC, Canada.
Contributor Information
Mathieu Lussier-Price, Département de Biochimie et Médicine Moléculaire, Université de Montréal, Montréal, QC, Canada.
Haytham M Wahba, Département de Biochimie et Médicine Moléculaire, Université de Montréal, Montréal, QC, Canada; Department of Biochemistry, Beni-Suef University, Beni-Suef, Egypt.
Xavier H Mascle, Département de Biochimie et Médicine Moléculaire, Université de Montréal, Montréal, QC, Canada.
Laurent Cappadocia, Département de Biochimie et Médicine Moléculaire, Université de Montréal, Montréal, QC, Canada.
Veronique Bourdeau, Département de Biochimie et Médicine Moléculaire, Université de Montréal, Montréal, QC, Canada.
Christina Gagnon, Département de Biochimie et Médicine Moléculaire, Université de Montréal, Montréal, QC, Canada.
Sebastian Igelmann, Département de Biochimie et Médicine Moléculaire, Université de Montréal, Montréal, QC, Canada.
Kazuyasu Sakaguchi, Department of Chemistry, Faculty of Science, Hokkaido University, Sapporo, Japan.
Gerardo Ferbeyre, Département de Biochimie et Médicine Moléculaire, Université de Montréal, Montréal, QC, Canada.
James G Omichinski, Département de Biochimie et Médicine Moléculaire, Université de Montréal, Montréal, QC, Canada.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Canadian Institute for Health Research [70439 and 130414 to J.G.O., 166104 to G.F.]; Natural Science and Engineering Research Council (to J.G.O.); Photo-excitonix Project at Hokkaido University (to K.S.); Macromolecular Diffraction Facility at Cornell High Energy Synchrotron Source (MacCHESS), which is supported by the NSF [DMR-1332208] (in part); NIH/NIGMS [GM-103485]; Canadian Light Source (CLS) which is supported by the Natural Sciences and Engineering Research Council of Canada; National Research Council Canada; Canadian Institutes of Health Research; Province of Saskatchewan; Western Economic Diversification Canada; University of Saskatchewan. Funding for open access charge: Canadian Institutes of Health Research.
Conflict of interest statement. None declared.
REFERENCES
- 1. Kerscher O. SUMO junction-what's your function? New insights through SUMO-interacting motifs. EMBO Rep. 2007; 8:550–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Desterro J.M., Rodriguez M.S., Kemp G.D., Hay R.T.. Identification of the enzyme required for activation of the small ubiquitin-like protein SUMO-1. J. Biol. Chem. 1999; 274:10618–10624. [DOI] [PubMed] [Google Scholar]
- 3. Pichler A., Gast A., Seeler J.S., Dejean A., Melchior F.. The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell. 2002; 108:109–120. [DOI] [PubMed] [Google Scholar]
- 4. Schmidt D., Muller S.. PIAS/SUMO: new partners in transcriptional regulation. Cell. Mol. Life Sci. 2003; 60:2561–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cappadocia L., Lima C.D.. Ubiquitin-like protein conjugation: structures, chemistry, and mechanism. Chem. Rev. 2018; 118:889–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Geiss-Friedlander R., Melchior F.. Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol. 2007; 8:947–956. [DOI] [PubMed] [Google Scholar]
- 7. Hickey C.M., Wilson N.R., Hochstrasser M.. Function and regulation of SUMO proteases. Nat. Rev. Mol. Cell Biol. 2012; 13:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guo D., Li M., Zhang Y., Yang P., Eckenrode S., Hopkins D., Zheng W., Purohit S., Podolsky R.H., Muir A.et al.. A functional variant of SUMO4, a new I kappa B alpha modifier, is associated with type 1 diabetes. Nat. Genet. 2004; 36:837–841. [DOI] [PubMed] [Google Scholar]
- 9. Liang Y.C., Lee C.C., Yao Y.L., Lai C.C., Schmitz M.L., Yang W.M.. SUMO5, a novel Poly-SUMO isoform, regulates PML nuclear bodies. Sci. Rep. 2016; 6:26509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Song J., Durrin L.K., Wilkinson T.A., Krontiris T.G., Chen Y.. Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:14373–14378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gareau J.R., Lima C.D.. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 2010; 11:861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beauclair G., Bridier-Nahmias A., Zagury J.F., Saib A., Zamborlini A.. JASSA: a comprehensive tool for prediction of SUMOylation sites and SIMs. Bioinformatics. 2015; 31:3483–3491. [DOI] [PubMed] [Google Scholar]
- 13. Song J., Zhang Z., Hu W., Chen Y.. Small ubiquitin-like modifier (SUMO) recognition of a SUMO binding motif: a reversal of the bound orientation. J. Biol. Chem. 2005; 280:40122–40129. [DOI] [PubMed] [Google Scholar]
- 14. Stehmeier P., Muller S.. Phospho-regulated SUMO interaction modules connect the SUMO system to CK2 signaling. Mol. Cell. 2009; 33:400–409. [DOI] [PubMed] [Google Scholar]
- 15. Sung K.S., Lee Y.A., Kim E.T., Lee S.R., Ahn J.H., Choi C.Y.. Role of the SUMO-interacting motif in HIPK2 targeting to the PML nuclear bodies and regulation of p53. Exp. Cell. Res. 2011; 317:1060–1070. [DOI] [PubMed] [Google Scholar]
- 16. Lussier-Price M., Mascle X.H., Cappadocia L., Kamada R., Sakaguchi K., Wahba H.M., Omichinski J.G.. Characterization of a C-terminal SUMO-interacting motif present in select PIAS-family proteins. Structure. 2020; 28:573–585. [DOI] [PubMed] [Google Scholar]
- 17. Scaglioni P.P., Yung T.M., Cai L.F., Erdjument-Bromage H., Kaufman A.J., Singh B., Teruya-Feldstein J., Tempst P., Pandolfi P.P.. A CK2-dependent mechanism for degradation of the PML tumor suppressor. Cell. 2006; 126:269–283. [DOI] [PubMed] [Google Scholar]
- 18. Hecker C.M., Rabiller M., Haglund K., Bayer P., Dikic I.. Specification of SUMO1- and SUMO2-interacting motifs. J. Biol. Chem. 2006; 281:16117–16127. [DOI] [PubMed] [Google Scholar]
- 19. Cappadocia L., Mascle X.H., Bourdeau V., Tremblay-Belzile S., Chaker-Margot M., Lussier-Price M., Wada J., Sakaguchi K., Aubry M., Ferbeyre G.et al.. Structural and functional characterization of the phosphorylation-dependent interaction between PML and SUMO1. Structure. 2015; 23:126–138. [DOI] [PubMed] [Google Scholar]
- 20. Chang C.C., Naik M.T., Huang Y.S., Jeng J.C., Liao P.H., Kuo H.Y., Ho C.C., Hsieh Y.L., Lin C.H., Huang N.J.et al.. Structural and functional roles of Daxx SIM phosphorylation in SUMO paralog-selective binding and apoptosis modulation. Mol. Cell. 2011; 42:62–74. [DOI] [PubMed] [Google Scholar]
- 21. Shen T.H., Lin H.K., Scaglioni P.P., Yung T.M., Pandolfi P.P.. The mechanisms of PML-Nuclear body formation. Mol. Cell. 2006; 24:805. [DOI] [PubMed] [Google Scholar]
- 22. Lallemand-Breitenbach V., de The H.. PML nuclear bodies. Cold Spring Harb. Perspect. Biol. 2010; 2:a000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lallemand-Breitenbach V., de The H.. PML nuclear bodies: from architecture to function. Curr. Opin. Cell Biol. 2018; 52:154–161. [DOI] [PubMed] [Google Scholar]
- 24. Stuurman N., de Graaf A., Floore A., Josso A., Humbel B., de Jong L., van Driel R.. A monoclonal antibody recognizing nuclear matrix-associated nuclear bodies. J. Cell Sci. 1992; 101:773–784. [DOI] [PubMed] [Google Scholar]
- 25. Wang J., Shiels C., Sasieni P., Wu P.J., Islam S.A., Freemont P.S., Sheer D. Promyelocytic leukemia nuclear bodies associate with transcriptionally active genomic regions. J. Cell Biol. 2004; 164:515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ishov A.M., Sotnikov A.G., Negorev D., Vladimirova O.V., Neff N., Kamitani T., Yeh E.T., Strauss J.F. 3rd, Maul G.G.. PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol. 1999; 147:221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ferbeyre G., de Stanchina E., Querido E., Baptiste N., Prives C., Lowe S.W.. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev. 2000; 14:2015–2027. [PMC free article] [PubMed] [Google Scholar]
- 28. Maroui M.A., Kheddache-Atmane S., El Asmi F., Dianoux L., Aubry M., Chelbi-Alix M.K.. Requirement of PML SUMO interacting motif for RNF4- or arsenic trioxide-induced degradation of nuclear PML isoforms. PLoS One. 2012; 7:e44949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cho G., Lim Y., Golden J.A.. SUMO interaction motifs in Sizn1 are required for promyelocytic leukemia protein nuclear body localization and for transcriptional activation. J. Biol. Chem. 2009; 284:19592–19600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lin D.Y., Huang Y.S., Jeng J.C., Kuo H.Y., Chang C.C., Chao T.T., Ho C.C., Chen Y.C., Lin T.P., Fang H.I.et al.. Role of SUMO-interacting motif in Daxx SUMO modification, subnuclear localization, and repression of sumoylated transcription factors. Mol. Cell. 2006; 24:341–354. [DOI] [PubMed] [Google Scholar]
- 31. Rasheed Z.A., Saleem A., Ravee Y., Pandolfi P.P., Rubin E.H.. The topoisomerase I-binding RING protein, topors, is associated with promyelocytic leukemia nuclear bodies. Exp. Cell. Res. 2002; 277:152–160. [DOI] [PubMed] [Google Scholar]
- 32. Corpet A., Kleijwegt C., Roubille S., Juillard F., Jacquet K., Texier P., Lomonte P.. PML nuclear bodies and chromatin dynamics: catch me if you can!. Nucleic. Acids. Res. 2020; 48:11890–11912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bayer P., Arndt A., Metzger S., Mahajan R., Melchior F., Jaenicke R., Becker J.. Structure determination of the small ubiquitin-related modifier SUMO-1. J. Mol. Biol. 1998; 280:275–286. [DOI] [PubMed] [Google Scholar]
- 34. Anamika, Spyracopoulos L. Molecular basis for Phosphorylation-dependent SUMO recognition by the DNA repair protein RAP80. J. Biol. Chem. 2016; 291:4417–4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Namanja A.T., Li Y.J., Su Y., Wong S., Lu J., Colson L.T., Wu C., Li S.S., Chen Y.. Insights into high affinity small ubiquitin-like modifier (SUMO) recognition by SUMO-interacting motifs (SIMs) revealed by a combination of NMR and peptide array analysis. J. Biol. Chem. 2012; 287:3231–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matic I., Macek B., Hilger M., Walther T.C., Mann M.. Phosphorylation of SUMO-1 occurs in vivo and is conserved through evolution. J. Proteome Res. 2008; 7:4050–4057. [DOI] [PubMed] [Google Scholar]
- 37. Gartner A., Wagner K., Holper S., Kunz K., Rodriguez M.S., Muller S.. Acetylation of SUMO2 at lysine 11 favors the formation of non-canonical SUMO chains. EMBO Rep. 2018; 19:e46117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tatham M.H., Jaffray E., Vaughan O.A., Desterro J.M., Botting C.H., Naismith J.H., Hay R.T.. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 2001; 276:35368–35374. [DOI] [PubMed] [Google Scholar]
- 39. Newman H.A., Meluh P.B., Lu J., Vidal J., Carson C., Lagesse E., Gray J.J., Boeke J.D., Matunis M.J.. A high throughput mutagenic analysis of yeast sumo structure and function. PLoS Genet. 2017; 13:e1006612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yuan H., Zhou J., Deng M., Liu X., Le Bras M., de The H., Chen S.J., Chen Z., Liu T.X., Zhu J.. Small ubiquitin-related modifier paralogs are indispensable but functionally redundant during early development of zebrafish. Cell Res. 2010; 20:185–196. [DOI] [PubMed] [Google Scholar]
- 41. Grana-Montes R., Marinelli P., Reverter D., Ventura S.. N-terminal protein tails act as aggregation protective entropic bristles: the SUMO case. Biomacromolecules. 2014; 15:1194–1203. [DOI] [PubMed] [Google Scholar]
- 42. Sabate R., Espargaro A., Grana-Montes R., Reverter D., Ventura S.. Native structure protects SUMO proteins from aggregation into amyloid fibrils. Biomacromolecules. 2012; 13:1916–1926. [DOI] [PubMed] [Google Scholar]
- 43. Mascle X.H., Gagnon C., Wahba H.M., Lussier-Price M., Cappadocia L., Sakaguchi K., Omichinski J.G.. Acetylation of SUMO1 alters interactions with the SIMs of PML and Daxx in a protein-specific manner. Structure. 2020; 28:157–168. [DOI] [PubMed] [Google Scholar]
- 44. Mascle X.H., Lussier-Price M., Cappadocia L., Estephan P., Raiola L., Omichinski J.G., Aubry M.. Identification of a non-covalent ternary complex formed by PIAS1, SUMO1, and UBC9 proteins involved in transcriptional regulation. J. Biol. Chem. 2013; 288:36312–36327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta. Crystallogr. D Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 46. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta. Crystallogr. D Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W.et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta. Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen V.B., Arendall W.B., Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lessard F., Igelmann S., Trahan C., Huot G., Saint-Germain E., Mignacca L., Del Toro N., Lopes-Paciencia S., Le Calvé B., Montero M.et al.. Senescence-associated ribosome biogenesis defects contributes to cell cycle arrest through the Rb pathway. Nat. Cell Biol. 2018; 20:789–799. [DOI] [PubMed] [Google Scholar]
- 50. Igelmann S., Lessard F., Uchenunu O., Bouchard J., Fernandez-Ruiz A., Rowell M.C., Lopes-Paciencia S., Papadopoli D., Fouillen A., Ponce K.J.et al.. A hydride transfer complex reprograms NAD metabolism and bypasses senescence. Mol. Cell. 2021; 81:3848–3865. [DOI] [PubMed] [Google Scholar]
- 51. Capili A.D., Lima C.D.. Structure and analysis of a complex between SUMO and Ubc9 illustrates features of a conserved E2-Ubl interaction. J. Mol. Biol. 2007; 369:608–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Knipscheer P., van Dijk W.J., Olsen J.V., Mann M., Sixma T.K.. Noncovalent interaction between Ubc9 and SUMO promotes SUMO chain formation. EMBO J. 2007; 26:2797–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kasahara K., Shiina M., Higo J., Ogata K., Nakamura H.. Phosphorylation of an intrinsically disordered region of Ets1 shifts a multi-modal interaction ensemble to an auto-inhibitory state. Nucleic Acids Res. 2018; 46:2243–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bah A., Forman-Kay J.D.. Modulation of intrinsically disordered protein function by Post-translational modifications. J. Biol. Chem. 2016; 291:6696–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bah A., Vernon R.M., Siddiqui Z., Krzeminski M., Muhandiram R., Zhao C., Sonenberg N., Kay L.E., Forman-Kay J.D.. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature. 2015; 519:106–109. [DOI] [PubMed] [Google Scholar]
- 56. Gareau J.R., Reverter D., Lima C.D.. Determinants of small ubiquitin-like modifier 1 (SUMO1) protein specificity, E3 ligase, and SUMO-RanGAP1 binding activities of nucleoporin RanBP2. J. Biol. Chem. 2012; 287:4740–4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kambe T., Tsuji T., Hashimoto A., Itsumura N.. The physiological, biochemical, and molecular roles of zinc transporters in zinc homeostasis and metabolism. Physiol. Rev. 2015; 95:749–784. [DOI] [PubMed] [Google Scholar]
- 58. Ding W.Q., Lind S.E.. Metal ionophores - an emerging class of anticancer drugs. IUBMB Life. 2009; 61:1013–1018. [DOI] [PubMed] [Google Scholar]
- 59. Weekley C.M., He C.. Developing drugs targeting transition metal homeostasis. Curr. Opin. Chem. Biol. 2017; 37:26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pearson M., Carbone R., Sebastiani C., Cioce M., Fagioli M., Saito S., Higashimoto Y., Appella E., Minucci S., Pandolfi P.P.et al.. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000; 406:207–210. [DOI] [PubMed] [Google Scholar]
- 61. Vernier M., Bourdeau V., Gaumont-Leclerc M.F., Moiseeva O., Begin V., Saad F., Mes-Masson A.M., Ferbeyre G.. Regulation of E2Fs and senescence by PML nuclear bodies. Genes Dev. 2011; 25:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ullmann R., Chien C.D., Avantaggiati M.L., Muller S.. An acetylation switch regulates SUMO-dependent protein interaction networks. Mol. Cell. 2012; 46:759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cai Q., Cai S., Zhu C., Verma S.C., Choi J.Y., Robertson E.S.. A unique SUMO-2-interacting motif within LANA is essential for KSHV latency. PLoS Pathog. 2013; 9:e1003750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ouyang J., Shi Y., Valin A., Xuan Y., Gill G.. Direct binding of CoREST1 to SUMO-2/3 contributes to gene-specific repression by the LSD1/CoREST1/HDAC complex. Mol. Cell. 2009; 34:145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sriramachandran A.M., Meyer-Teschendorf K., Pabst S., Ulrich H.D., Gehring N.H., Hofmann K., Praefcke G.J.K., Dohmen R.J.. Arkadia/RNF111 is a SUMO-targeted ubiquitin ligase with preference for substrates marked with SUMO1-capped SUMO2/3 chain. Nat. Commun. 2019; 10:3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Topisirovic I., Capili A.D., Borden K.L.. Gamma interferon and cadmium treatments modulate eukaryotic initiation factor 4E-dependent mRNA transport of cyclin D1 in a PML-dependent manner. Mol. Cell. Biol. 2002; 22:6183–6198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hands K.J., Cuchet-Lourenco D., Everett R.D., Hay R.T.. PML isoforms in response to arsenic: high-resolution analysis of PML body structure and degradation. J. Cell Sci. 2014; 127:365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang X.W., Yan X.J., Zhou Z.R., Yang F.F., Wu Z.Y., Sun H.B., Liang W.X., Song A.X., Lallemand-Breitenbach V., Jeanne M.et al.. Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science. 2010; 328:240–243. [DOI] [PubMed] [Google Scholar]
- 69. Zhu J., Koken M.H., Quignon F., Chelbi-Alix M.K., Degos L., Wang Z.Y., Chen Z., de Thé H.. Arsenic-induced PML targeting onto nuclear bodies: implications for the treatment of acute promyelocytic leukemia. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:3978–3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lallemand-Breitenbach V., Jeanne M., Benhenda S., Nasr R., Lei M., Peres L., Zhou J., Zhu J., Raught B., de The H.. Arsenic degrades PML or PML-RARalpha through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Biol. 2008; 10:547–555. [DOI] [PubMed] [Google Scholar]
- 71. Tatham M.H., Geoffroy M.C., Shen L., Plechanovova A., Hattersley N., Jaffray E.G., Palvimo J.J., Hay R.T.. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell Biol. 2008; 10:538–546. [DOI] [PubMed] [Google Scholar]
- 72. Maul G.G., Yu E., Ishov A.M., Epstein A.L.. Nuclear domain 10 (ND10) associated proteins are also present in nuclear bodies and redistribute to hundreds of nuclear sites after stress. J. Cell. Biochem. 1995; 59:498–513. [DOI] [PubMed] [Google Scholar]
- 73. Korioth F., Gieffers C., Maul G.G., Frey J.. Molecular characterization of NDP52, a novel protein of the nuclear domain 10, which is redistributed upon virus infection and interferon treatment. J. Cell Biol. 1995; 130:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yang C., Hao R., Lan Y.F., Chen Y.J., Wang C., Bu N., Wang Q.Q., Hussain L., Ma L.Y., Maimaitiyiming Y.et al.. Integrity of zinc finger motifs in PML protein is necessary for inducing its degradation by antimony. Metallomics. 2019; 11:1419–1429. [DOI] [PubMed] [Google Scholar]
- 75. Müller S., Miller W.H. Jr, Dejean A. Trivalent antimonials induce degradation of the PML-RAR oncoprotein and reorganization of the promyelocytic leukemia nuclear bodies in acute promyelocytic leukemia NB4 cells. Blood. 1998; 92:4308–4316. [PubMed] [Google Scholar]
- 76. Banani S.F., Rice A.M., Peeples W.B., Lin Y., Jain S., Parker R., Rosen M.K.. Compositional control of phase-separated cellular bodies. Cell. 2016; 166:651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Friedman R. Structural and computational insights into the versatility of cadmium binding to proteins. Dalton Trans. 2014; 43:2878–2887. [DOI] [PubMed] [Google Scholar]
- 78. Jacobson K.B., Turner J.E.. The interaction of cadmium and certain other metal ions with proteins and nucleic acids. Toxicology. 1980; 16:1–37. [DOI] [PubMed] [Google Scholar]
- 79. Shen S., Li X.F., Cullen W.R., Weinfeld M., Le X.C.. Arsenic binding to proteins. Chem. Rev. 2013; 113:7769–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jiang G., Gong Z., Li X.F., Cullen W.R., Le X.C.. Interaction of trivalent arsenicals with metallothionein. Chem. Res. Toxicol. 2003; 16:873–880. [DOI] [PubMed] [Google Scholar]
- 81. Andreini C., Banci L., Bertini I., Rosato A.. Zinc through the three domains of life. J. Proteome Res. 2006; 5:3173–3178. [DOI] [PubMed] [Google Scholar]
- 82. Ghavamzadeh A., Alimoghaddam K., Ghaffari S.H., Rostami S., Jahani M., Hosseini R., Mossavi A., Baybordi E., Khodabadeh A., Iravani M.et al.. Treatment of acute promyelocytic leukemia with arsenic trioxide without ATRA and/or chemotherapy. Ann. Oncol. 2006; 17:131–134. [DOI] [PubMed] [Google Scholar]
- 83. Nasr R., Guillemin M.C., Ferhi O., Soilihi H., Peres L., Berthier C., Rousselot P., Robledo-Sarmiento M., Lallemand-Breitenbach V., Gourmel B.et al.. Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nat. Med. 2008; 14:1333–1342. [DOI] [PubMed] [Google Scholar]
- 84. Mathews V., George B., Lakshmi K.M., Viswabandya A., Bajel A., Balasubramanian P., Shaji R.V., Srivastava V.M., Srivastava A., Chandy M.. Single-agent arsenic trioxide in the treatment of newly diagnosed acute promyelocytic leukemia: durable remissions with minimal toxicity. Blood. 2006; 107:2627–2632. [DOI] [PubMed] [Google Scholar]
- 85. Kaiming C., Sheng Y., Zheng S., Yuan S., Huang G., Liu Y.. Arsenic trioxide preferentially binds to the ring finger protein PML: understanding target selection of the drug. Metallomics. 2018; 10:1564–1569. [DOI] [PubMed] [Google Scholar]
- 86. Jeanne M., Lallemand-Breitenbach V., Ferhi O., Koken M., Le Bras M., Duffort S., Peres L., Berthier C., Soilihi H., Raught B.et al.. PML/RARA oxidation and arsenic binding initiate the antileukemia response of As2O3. Cancer Cell. 2010; 18:88–98. [DOI] [PubMed] [Google Scholar]
- 87. Geoffroy M.C., Jaffray E.G., Walker K.J., Hay R.T.. Arsenic-induced SUMO-dependent recruitment of RNF4 into PML nuclear bodies. Mol. Biol. Cell. 2010; 21:4227–4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Percherancier Y., Germain-Desprez D., Galisson F., Mascle X.H., Dianoux L., Estephan P., Chelbi-Alix M.K., Aubry M.. Role of SUMO in RNF4-mediated promyelocytic leukemia protein (PML) degradation: sumoylation of PML and phospho-switch control of its SUMO binding domain dissected in living cells. J. Biol. Chem. 2009; 284:16595–16608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Eskiw C.H., Dellaire G., Mymryk J.S., Bazett-Jones D.P.. Size, position and dynamic behavior of PML nuclear bodies following cell stress as a paradigm for supramolecular trafficking and assembly. J. Cell Sci. 2003; 116:4455–4466. [DOI] [PubMed] [Google Scholar]
- 90. Nefkens I., Negorev D.G., Ishov A.M., Michaelson J.S., Yeh E.T., Tanguay R.M., Müller W.E., Maul G.G.. Heat shock and Cd2+ exposure regulate PML and Daxx release from ND10 by independent mechanisms that modify the induction of heat-shock proteins 70 and 25 differently. J. Cell Sci. 2003; 116:513–524. [DOI] [PubMed] [Google Scholar]
- 91. Janer A., Martin E., Muriel M.P., Latouche M., Fujigasaki H., Ruberg M., Brice A., Trottier Y., Sittler A.. PML clastosomes prevent nuclear accumulation of mutant ataxin-7 and other polyglutamine proteins. J. Cell Biol. 2006; 174:65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Petering D.H. Reactions of the Zn proteome with Cd(2+) and other xenobiotics: trafficking and toxicity. Chem. Res. Toxicol. 2017; 30:189–202. [DOI] [PubMed] [Google Scholar]
- 93. Sahin U., de The H., Lallemand-Breitenbach V.. PML nuclear bodies: assembly and oxidative stress-sensitive sumoylation. Nucleus. 2014; 5:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sahin U., Ferhi O., Jeanne M., Benhenda S., Berthier C., Jollivet F., Niwa-Kawakita M., Faklaris O., Setterblad N., de The H.et al.. Oxidative stress-induced assembly of PML nuclear bodies controls sumoylation of partner proteins. J. Cell Biol. 2014; 204:931–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kato M., Han T.W., Xie S., Shi K., Du X., Wu L.C., Mirzaei H., Goldsmith E.J., Longgood J., Pei J.et al.. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012; 149:753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mateju D., Franzmann T.M., Patel A., Kopach A., Boczek E.E., Maharana S., Lee H.O., Carra S., Hyman A.A., Alberti S.. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017; 36:1669–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Weber S.C., Brangwynne C.P.. Inverse size scaling of the nucleolus by a concentration-dependent phase transition. Curr. Biol. 2015; 25:641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Murray D.T., Kato M., Lin Y., Thurber K.R., Hung I., McKnight S.L., Tycko R.. Structure of FUS protein fibrils and its relevance to self-assembly and phase separation of low-complexity domains. Cell. 2017; 171:615–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Ditlev J.A., Case L.B., Rosen M.K.. Who's in and who's out-compositional control of biomolecular condensates. J. Mol. Biol. 2018; 430:4666–4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Banani S.F., Lee H.O., Hyman A.A., Rosen M.K.. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017; 18:285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Rayman J.B., Karl K.A., Kandel E.R.. TIA-1 self-multimerization, phase separation, and recruitment into stress granules are dynamically regulated by Zn(2). Cell Rep. 2018; 22:59–71. [DOI] [PubMed] [Google Scholar]
- 102. Keiten-Schmitz J., Wagner K., Piller T., Kaulich M., Alberti S., Müller S.. The nuclear SUMO-targeted ubiquitin quality control network regulates the dynamics of cytoplasmic stress granules. Mol. Cell. 2020; 79:54–67. [DOI] [PubMed] [Google Scholar]
- 103. Marmor-Kollet H., Siany A., Kedersha N., Knafo N., Rivkin N., Danino Y.M., Moens T.G., Olender T., Sheban D., Cohen N.et al.. Spatiotemporal proteomic analysis of stress granule disassembly using APEX reveals regulation by SUMOylation and links to ALS pathogenesis. Mol. Cell. 2020; 80:876–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Keiten-Schmitz J., Röder L., Hornstein E., Müller-McNicoll M., Müller S.. SUMO: glue or solvent for phase-separated ribonucleoprotein complexes and molecular condensates?. Front. Mol. Biosci. 2021; 8:673038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.