


Abstract
The RNA programmed non-specific (trans) nuclease activity of CRISPR-Cas Type V and VI systems has opened a new era in the field of nucleic acid-based detection. Here, we report on the enhancement of trans-cleavage activity of Cas12a enzymes using hairpin DNA sequences as FRET-based reporters. We discover faster rate of trans-cleavage activity of Cas12a due to its improved affinity (Km) for hairpin DNA structures, and provide mechanistic insights of our findings through Molecular Dynamics simulations. Using hairpin DNA probes we significantly enhance FRET-based signal transduction compared to the widely used linear single stranded DNA reporters. Our signal transduction enables faster detection of clinically relevant double stranded DNA targets with improved sensitivity and specificity either in the presence or in the absence of an upstream pre-amplification step.
Graphical Abstract
Graphical Abstract.
Here we report on the molecular basis for the enhancement of trans-cleavage activity of Cas12a enzyme using hairpin DNA as FRET reporters.
INTRODUCTION
The extensive use of CRISPR technology for diagnostic applications has been driven mainly by the discovery of the collateral trans-cleavage nuclease activities of CRISPR type V (Cas12) and type VI (Cas13) systems (1–5). Cas12 and Cas13 proteins are RNA-guided components of bacterial adaptive immune systems and represent an ideal toolbox for molecular diagnostics, as they integrate single- and double-stranded DNA (Cas12) or single-stranded RNA (Cas13) target recognition, signal transduction and amplification in one system (6–9). Their trans-cleavage nuclease activity (trans-activity) is activated only upon base-pairing of a nucleic acid target to a complementary crRNA guide. In the presence of a FRET-based single-stranded DNA (Cas12) or RNA (Cas13) reporter, consecutive probe degradation events result in an increase of the fluorescence, reflecting the presence of a specific nucleic acid target. So far, this peculiar RNA-programmed trans-activity towards single stranded DNA (i.e. Cas12) or RNA (i.e. Cas13) probes has been harnessed by DETECTR and SHERLOCK, respectively, to rapidly detect nucleic acid targets (10–15), and integrated in field-deployable devices for point-of-care use (13,16–20).
Although CRISPR-based technology has demonstrated vast potential for diagnostic applications, in the absence of a pre-amplification step the direct nucleic acid detection is limited to a detection limit in the picomolar range (10,21). This restricts the use of amplification-free CRISPR-based assays to the detection of few classes of nucleic acid targets (22–27). Therefore, most CRISPR-based platforms reported so far generally are combined with an isothermal pre-amplification step (e.g. LAMP, RPA, NASBA, etc.) of the nucleic acid target. However, the addition of this pre-amplification step demands for multiple handling steps and/or high-temperature incubation (55–65°C), procedures that cannot be easily translated in a Point of Care (PoC) test (28,29). In addition, the sensitivity of CRISPR-based detection intended as the ratio of the relative change in output to the relative change in input (the steepness of the input/output function) is relatively low, and this poses challenges to the absolute quantification of target also in the range where the cleavage activity directly correlates with target concentration (10–6–10–12 M). Recently, different strategies based on tandem CRISPR nucleases (e.g. Cas13 and Cmr-complex/Csm6) have been successful in achieving amplification-free detection of SARS-CoV-2 RNA (23,30–32). However, they have been reported only using combination of type III and VI CRISPR–Cas proteins and thus their application is limited to the direct detection of RNA targets. To the best of our knowledge, only one strategy has been reported being able to improve trans-activity of Cas12 enzyme until femtomolar limit of detection (33). It takes advantage of engineered crRNAs showing DNA extension of different length at 3′- or 5′-ends. However, enhancement of trans-cleavage activity comes at the cost of increased costs of chimeric DNA/RNA guide. Thus, new strategies to increase the speed and sensitivity of one-pot detection chemistries of Cas12-based diagnostic systems are required to further optimize its performance.
In this respect, the rational design of alternative DNA reporters with improved affinity for Cas12a proteins, and thus able to enhance the sensitivity and the trans-cleavage rate, has been largely neglected. In particular, it is well-recognized that short, single-stranded DNA probes are better performing FRET-based reporters of trans-cleavage activity than double stranded DNA (34,35). Some efforts have been also devoted to the study of different signal transductions,(36,37) but the vast majority of diagnostic assays still rely on the use of linear single-stranded DNA as signal transducers. However, the effect of specific DNA secondary structures (e.g. Hairpin; triplex, etc.) has not been fully investigated (38–40).
In response to the above considerations, we report here the rational design of single-stranded hairpin DNA reporters bearing a fluorophore and a quencher at the two ends of the self-complementary portions. We first demonstrate that FRET-based hairpin DNA reporters are able to generate faster and enhanced fluorescence signal change compared to linear single-stranded DNA (ssDNA) reporter. Second, we report that Cas12a proteins show higher trans-activity on hairpin DNA reporters, and we also provide mechanistic insights on the Cas12a interactions with hairpin substrates. Finally, we demonstrate that hairpin DNA reporters enhance the sensitivity and the specificity of Cas12a-based detection.
MATERIALS AND METHODS
Sodium chloride (NaCl), magnesium chloride (MgCl2), tris(hydroxymethyl)aminomethane hydrochloride (Tris–HCl), 2-amino-2-(hydroxymethyl)-1,3-propanediol (Tris base), boric acid, ethylenediamine tetraacetic acid (EDTA), acrylamide/bis-acrylamide 30% solution, ammonium persulfate (APS), N,N,N′,N′-tetramethyl ethylenediamine (TEMED), were purchased from Sigma-Aldrich, Italy and used without any further purifications. Orange DNA Loading Dye (6x) and O’Range Ruler 5 base pair DNA Ladder were purchased from Thermo Fisher Scientific Italia. EnGen® Lba Cas12a from Lachnospiraceae bacterium ND2006 was purchased from New England Biolabs Inc. (Ipswich, UK). EnGen Lba Cas12a is a native protein expressed as a N-terminal 6-His-tagged fusion in Escherichia coli. The enzyme has Simian virus 40 (SV40) T antigen nuclear localization sequences (NLSs) at both the N and C-termini of the protein. No mutations are present. Alt-R® A.s. Cas12a (Cpf1) V3 from Recombinant Acidaminococcus sp. BV3L6 (A.s.) nuclease, purified from an E. coli strain expressing Cas12a was purchased from Integrated DNA Technologies, Inc. (Coralville, UK). Alt-R® A.s. Cas12a (Cpf1) V3 nuclease is high purity, wild-type recombinant Acidaminococcus sp. Cas12a. The enzymes include NLSs and C-terminal 6-His tags. No mutations are present. FnCas12a from Francisella novocida bacterium was produced as reported in Heterologous expression and purification of FnCas12a.
Oligonucleotides
Oligonucleotides (HPLC purified) were purchased from Biosearch Technologies (Risskov, Denmark) and Metabion (Planegg, Germany). All oligonucleotides were dissolved in NE buffer (10 mM Tris–HCl/50 mM NaCl/10 mM MgCl2, pH 7.4) at a concentration of 100 μM and stored at –20°C. The concentration of the oligonucleotides was determined using Tecan Infinite M200pro (Männedorf, Switzerland) through NanoQuant Plate. Oligonucleotides were annealed before to use by thermocycler. Specifically, DNA oligonucleotides were incubated at 90°C for 2 min, RNA oligonucleotides at 65°C for 5 min, both slowly cooled to room temperature (1°C/min). The sequences of the oligonucleotides used are as follows.
FRET-based DNA reporters
See Table 1.
Table 1.
FRET-based DNA reporters
| Sequence name | Sequencea | Figures |
|---|---|---|
| Linear ss-DNA | 5′-(6-FAM)- TTATT – (BHQ1)- 3′ | 2-5, Supplementary Figures S1, S2, S3, S4, S5, S6, S7, S8, S9, S12, S13, S14 |
| Stem-loop #5T | 5′-(6-FAM)- CTCTCATTTTTAGAGAG– (BHQ1)- 3′ | 2, 3, Supplementary Figures S1, S2, S4, S6, S8 |
| Stem-loop #10T | 5′-(6-FAM)-CTCTCATTTTTTTTTTAGAGAG-(BHQ1)-3′ | 2-5, Supplementary Figures S1, S2, S3, S4, S5, S6, S7, S8, S9, S12, S13, S14 |
| Stem-loop #30T | 5′-(6-FAM)-CTCTCATTTTTTTTTTTTTTTTTTTTTTTTTTTT TTAGAGAG-(BHQ1)-3′ | 2, 3, Supplementary Figures S1, S2, S4, S6, S8 |
| Variant #1 | 5′-(6-FAM)-CTCTCTTTTTTTTTTTTGAGAG-(BHQ1)-3′ | Supplementary Figure S5 |
| Variant #2 | 5′-(6-FAM)-CTCTCATTATTATTATAGAGAG-(BHQ1)-3′ | Supplementary Figure S5 |
| Variant ss-DNA | 5′-(6-FAM)- TCGTCATTTTTTTTTTACTGAG-(BHQ1)-3′ | Supplementary Figure S9 |
aBold bases represent the stem portion, while the italic bases represent the loop portion.
Guide crRNA
See Table 2.
Table 2.
Guide crRNA
| Sequence name | Sequencea | Figures |
|---|---|---|
| crRNA | 5′-UAAUUUCUACUAAGUGUAGAUGUCUACA CAUGGCUAAAUCU-3′ | 2, 3, Supplementary Figures S3, S4, S5, S6, S7, S8, S9 |
| crRNA + 3′DNA7b | 5′-UAAUUUCUACUAAGUGUAGAUCUCAGGG CGGACUGGGUGCUTATTATT-3′ | Supplementary Figure S3 |
| crRNA- SARS-CoV-2c | 5′– UAAUUUCUACUAAGUGUAGAUAUUGUU ACUUUCCUUUACAA-3′ | 5a, 5b, Supplementary Figures S12, S13, S14 |
| crRNA-Salmonellad | 5′– UAAUUUCUACUAAGUGUAGAUACCG GCAUCGGCUUCAAUCA-3′ | 5c, 5d |
aUnderlined bases represent the portion complementary to the ssDNA or dsDNA activator.
bcrRNA + 3′DNA7 presents a 7-mer DNA extension to crRNA at the 3′-end that has been reported to enhance the sensitivity and specificity of LbCas12a-mediated nucleic acid detection (33) crRNA + 3′DNA7 was used in Supplementary Figure S3.
ccrRNA- SARS-CoV-2 has been designed for the recognition of the S gene SARS-CoV-2.
dcrRNA- Salmonella has been designed for the recognition of the invA gene.
Single- and double-stranded DNA activators
See Table 3.
Table 3.
Single- and double-stranded DNA activators
| Sequence namea | Sequenceb | crRNAs used for targeting | Figures |
|---|---|---|---|
| TS1 NTS1 |
5′-AGATTTAGCCATGTGTAGACTAAA-3′ 5′-TTTA GTCTACACATGGCTAAATCT-3′ |
crRNA | 2, 3, Supplementary Figures S3, S4, S5, S6, S7, S8, S9 |
| TS- crRNA + 3′DNA7 NTS- crRNA + 3′DNA7 |
5′-AGCACCCAGTCCGCCCTGAGTAAA-3′ 5′-TTTA CTCAGGGCGGACTGGGTGCT-3′ |
crRNA + 3′DNA7 | Supplementary Figure S3 |
| SARS-CoV-2 ds-DNA amplicon | 5′-GATCCTTGTAAAGGAAAGTAACAATTAAA ACCTTCAACACCATTACAAGGTGTGCTAGGATCCCCGGGTACCGAGCTCGAATTCACTGGCCGTCGTTTTACA-3′ 5′- TGTAAAACGACGGCCAGTGAATTCGA GCTCGGTACCCGGGGATCCTAGCACACCTTGTAATGGTGTTGAAGGTTTTAATTGTTACTTTCCTTTACAAGGATC-3′ |
crRNA- SARS-CoV-2 | 5a, 5b |
| PM MM1 MM4 MM8 MM12 MM16 MM20 MM1–2 MM4–5 MM8–9 MM12–13 MM16–17 MM19–20 NTS |
5′- TTGTAAAGGAAAGTAACAATTAAA-3′ 5′-TTGTAAAGGAAAGTAACAACTAAA-3′ 5′-TTGTAAAGGAAAGTAATAATTAAA-3′ 5′-TTGTAAAGGAAATTAACAATTAAA-3′ 5′-TTGTAAAGTAAAGTAACAATTAAA-3′ 5′-TTGTGAAGGAAAGTAACAATTAAA-3′ 5′-GTG TAA AGG AAA GTA ACA ATTAAA-3′ 5′-TTGTAAAGGAAAGTAACATCTAAA-3′ 5′-TTGTAAAGGAAAGTAGTAATTAAA-3′ 5′-TTGTAAAGGAAGTTAACAATTAAA-3′ 5′-TTGTAAACTAAAGTAACAATTAAA-3′ 5′-TTGGGAAGGAAAGTAACAATTAAA-3′ 5′-GAG TAA AGG AAA GTA ACA ATTAAA-3′ 5′-TTTA ATTGTTACTTTCCTTTACAA-3′ |
crRNA- SARS-CoV-2 | 5e, 5f, Supplementary Figures S12, S13, S14 |
| Salmonella ds-DNA amplicon | 5′-TGGCGATATTGGTGTTTATGGGGTCGTTCTACATTGACAGAATCCTCAGTTTTTCAACGTTTCCTGCGGTACTGTTAATTACCACGCTCTTTCGTCTGGCATTATCGATCAGTACCAGCCGTCTTATCTTGATTGAAGCCGATGCCGGTGAAATTATCGCCACGTTCGGGCAATTCGTTATTGGCGATAGCCTGGCGGTGGGTTTTGTTGTCTTCTCTATTGTCACCGTGGTCCAGTTTATCGTTATTAC-3′ | crRNA-Salmonella | 5c, 5d |
aTS represent the target single-stranded DNA, complementary to the RNA guide, whereas NTS represent the complementary non-target single-stranded DNA. PM stands for perfect match and MM for mismatch (reported in bold in the sequence). The numbers following ‘MM’ indicate the position of the mismatches respect to the PAM motif (i.e. MM1 indicates that mismatch is the first base following PAM motif, MM4 indicates that mismatch is the fourth base following PAM motif, etc).
bItalic bases represent the PAM motif; bold bases represent the mismatches.
Heterologous expression and purification of FnCas12a
The protocol provides by Mohanraju et al. was followed (41). The pDS015 plasmid, containing the Francisella novocida Cas12a gene was used for the heterologous expression of the protein in E. coli BL21(DE3) Rosetta™ 2 (Merck, Novagen), as fully described in the relative Bio-Protocol site (www.bio-protocol.org/e2842). Starting from 16 g of induced wet cells, we obtained ca. 10 mg of 95% purified protein, as confirmed by SDS-PAGE analysis. Aliquots of FnCas12a were finally stored at –20°C.
DNA preparation of plasmid containing the SARS-CoV-2 sequence (S gene)
The pUC19 plasmid was chosen as recipient vector to clone the sequence of the S gene of SARS-CoV-2 (42). The entire sequence was obtained from Eurofins Genomic (Germany) as oligonucleotides pairs flanked by a short sequence (underlined) which simulate, after oligonucleotide annealing, protruding ends after the BamH I digestion, for the subsequent cloning procedures.
NTS-Fwd: 5′-GATCCTAGCACACCTTGTAATGGTGTTGAAGGTTTTAATTGTTACTTTCCTTTACAAG-3′
NTS-Rev: 5′-GATCCTTGTAAAGGAAAGTAACAATTAAAACCTTCAACACCATTACAAGGTGT GCTAG-3′
The pUC19 plasmid was also digested with BamH I enzyme, and the resulting DNA fragment was gel-purified by using NucleoSpin® Gel and PCR Clean-up® kit from Macherey-Nagel GmbH (Germany). The annealed oligonucleotides were mixed with the digested plasmid and incubated in the ligation mixture with a plasmid:oligos 1:10 ratio. Afterwards, the mixture was used to transform E. coli DH5a competent cells and positive white colonies were selected on Ampicillin selective Luria-Bertani (LB) Agar plates supplemented with 5-bromo-4-chloro-3-indolyl-b-d-glucopyranoside (X-Glc) and Isopropyl β-d-1-thiogalactopyranoside (IPTG). One positive colony was used for the plasmid preparation, achieving the final pUC19-BZ construct.
Production of SARS-CoV-2 amplicons through PCR
In order to produce different amount of target SARS-CoV-2 amplicons for the CRISPR-based detection assay, six aliquots containing known concentrations of pUC19-BZ variant plasmid (from 20 nM to 20 aM) were amplified by PCR with M13-Fwd (5′-TGTAAAACGACGGCCAGT-3′)/NTS-Rev oligonucleotides, under the following conditions: an initial denaturation at 95°C for 5 min, 30 cycles of 30 s at 95°C, 30 s at 50°C and 30 s at 72°C, and a final extension of 5 min at 72°C. All amplified DNA products were cleaned by the NucleoSpin® Gel and PCR Clean-up® kit from Macherey-Nagel GmbH (Germany), in order to remove oligonucleotides, free nucleotides and the DNA polymerase enzyme.
Preparation and standardization of Salmonella strain
The sensitivity test was carried out using Salmonella Typhimurium ATCC 14028 (Oxoid Ltd, Basingstoke, UK). The strain was revitalized by incubation in 10 ml of Tryptone Soy Broth medium (Lab M Ltd, Heywood, UK) for 24 h at 37°C. Furthermore, the strain was standardized by determining the bacterial load with the method of serial dilutions and sowing by inclusion on Plate Count Agar (Lab M Ltd, Heywood, UK) that yielded about 108 CFU/ml. An aliquot (2 ml) of strain was centrifuged at 14 000 rpm for 10 minutes, the pellet was suspended in 1 ml of distilled water and centrifuged at 14 000 rpm for 10 min, and the pellet was suspended in 200 μl of 6% Chelex. The suspension was incubated for 8–10 min at 100°C, finally placed on ice for 1 minute. The suspension was then centrifuged (5 min at 14 000 rpm) to allow the Chelex, cell components, membrane residues and cell wall to settle to the bottom of the tube. 100 μl of the supernatant was carefully transferred to a new tube and used as a template in molecular methods.
PCR amplification of Salmonella samples
Briefly, the primer used amplify a highly specific segment of the region of invA gene, conserved in all Salmonella serotypes. The reaction mixture consists of a total of 50 μl divided as follows: 25 μl of master mix (QuantiTech Multiplex PCR Master Mix NoROX, QIAGEN), 1 μl (10 μM) of forward primer F3 (GGCGATATTGGTGTTTATGGGG), 1 μl (10 μM) of reverse primer B3 (5′-AACGATAAACTGGACCACGG-3′, Hara-Kudo et al., 2005), 19 μl of molecular biology water, 4 μl of DNA template. The reaction was run at 95°C for 15 min, followed by 40-cycle at 95°C for 30 s and 60°C for 30 s. All amplifications were performed using the GeneAmp PCR System 9700 instrument (Applied Biosystems). PCR products were analysed by 2% agarose gel electrophoresis using 5 μl of ethidium bromide (10 ng/mL) and run at 90 V for 60 min and finally visualized by UV transillumination. The sizes of the sample PCR fragment (244 bp) were determined comparing their position on the gel with those belonging to the standard DNA ladder (EZ Load 100 bp molecular ruler, Bio-Rad Life Science Research). Finally, the PCR products were purified with kit QIAquick® PCR Purification KIT (QIAGEN).
Melting assays and thermodynamic analysis
Fluorescence melting curves (Figure 2A and S1) were obtained by heating a solution containing 50 nM of DNA reporter in NE buffer (10 nM Tris–HCl, 50 mM NaCl, 10 mM MgCl2, pH 7.9) from 20°C to 90°C at a rate of 1°C min–1 and monitoring fluorescence at every 0.5°C, setting λex. = 488 nm and λem. = 520 nm. All the solutions were annealed prior to analysis by heating the solutions to 90°C and slowly cooling down at room temperature. The reported melting curves have been normalized through the use of the interpolation model that allows to estimate the melting temperature (Tm) for each experiment (Figure 2A and Supplementary Figure S1) (43). To estimate the switching equilibrium constant (Ks) of the hairpin DNA reporters, we assumed a two-state model and ΔH° independent of temperature (ΔCp = 0) (43,44). Firstly, we calculated the folded and unfolded fraction of the hairpin reporters as function of temperature according to the following equation:
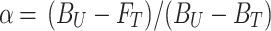 |
where α represents the folded fraction, BU and BF are the baseline fluorescence values of the unfolded and folded species, respectively and, FT is the fluorescence signal of the hairpin DNA reporter at a given temperature. Ks was then calculated as Ks = (1 – α)/α, by taking in accounts the α between 0.15 and 0.85. Van’t Hoff analysis was carried out by plotting ln(Ks) as a function of 1/T (Supplementary Figure S1) and data were fitted by using the following equation:
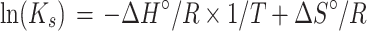 |
We assumed that all the variants by sharing the same stem portion (related to the enthalpy contribution) present the same average ΔH° value (44.1 ± 2.8 kcal/mol). Finally, Ks, was calculated as Ks = exp(–ΔG°/RT) at 37°C.
Fluorescence trans-cleavage assays
Double-stranded DNA activator was prepared by annealing 3-fold molar excess of the NTS to TS in buffer (10 nM Tris–HCl, 50 mM NaCl, 10 mM MgCl2, pH 7.9) heating at 90°C for 2 min, and slow-cooling on the benchtop. Trans-cleavage assays were performed as follow, unless otherwise stated. 4.5 μl of Cas12a/cRNA complex (200 nM) previously incubated with the single (ssDNA) or double stranded (dsDNA) DNA activator (10 nM) for 30 min at 37°C, are diluted into a final volume of 45 μl containing 100 nM DNA-reporter (final concentration) at 37°C. We performed all steady state fluorescence measurements using a Cary Eclipse Fluorimeter. We recorded emission spectra with excitation at 488 nm and acquisition from 500 to 600 nm. We fixed the excitation and emission bandwidths to 5 nm in all the experiments. Kinetics experiments were performed by fixing excitation at λexc = 488 nm and acquisition at λem = 520 nm, and setting the excitation and emission bandwidths to 5 nm in all the experiments. Background-corrected fluorescence values were calculated by subtracting fluorescence values obtained from reactions carried out in the absence of activator at the specific time of analysis.
To determine the cleaved fraction of substrate over time (Figures 3B and Supplementary Figure S6), fluorescence kinetics were fitted to a single exponential decay curve (Kaleidagraph, Software), according to the following equation:
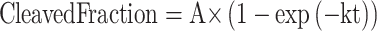 |
where A is the amplitude of the curve, k is the first-order rate constant and t is time. Calibration curves performed for sensitivity assays (Figure 5A) have been obtained after 1 h of incubation of different concentrations of ds-DNA amplicons in a solution containing LbCas12a/crRNA (20 nM, final concentration) and the Linear-ssDNA or the Stem-loop #10T DNA reporter (100 nM, final concentration), as previously reported. The detection of PCR-amplified amplicons (Figure 5B) have been performed by diluting the PCR products 500 times in NE buffer and then by transferring 5 μl to a vial containing 25 μl of 200 nM LbCas12a/crRNA-SARS-CoV-2. The resulting solutions were incubated for 30 min at 37°C. After that, 45 μl of DNA reporter (final concentration 100 nM) was added and incubated for 1 h at 37°C. Finally, the solutions were heated at 65°C for 10 min and cooled down to 37°C, in order to deactivate LbCas12a and trans-cleavage reaction. Detection of Salmonella (Figure 5C, D) was performed by adding 2.5 μl of 200 nM LbCas12a/crRNA (RNP) complex to the 25 ul of PCR products at different dilution factors in NE buffer containing a 500 nM final concentration of DNA reporter and kinetic were followed for 1 h at 37°C, by using a Tecan M200pro plate reader using top reading mode with black, flat bottom non-binding 96-well plates. Kinetic experiments performed for testing the specificity, were carried out by adding 4.5 μl of activator (final concentration 10 nM of TS or MM + 30 nM of NTS) in 45 μl of solution containing 100 nM of reporter and 20 nM of LbCas12a/ crRNA-SARS-CoV-2 (previously incubated for 30 min at 37°C at 200 nM concentration) (Figure 5e, 5f and S12).
Figure 3.

Kinetic analysis of LbCas12a trans-cleavage activity. (A) Time-dependent trans-cleavage activity of activated LbCas12a on FRET-labelled DNA reporters (250 nM). Substrate (e.g. DNA reporters, S) and degraded products (P) were resolved by polyacrylamide gel electrophoresis (PAGE). The intrinsic fluorescence background of the DNA reporters (lines 1) and signal change over time is consistent with the increased amount of degraded oligonucleotides over time (lines 3 to 8). (B) Fraction of DNA reporters cleaved by the active LbCas12a/crRNA/dsDNA complex (RNP complex, final concentration 20 nM/20 nM/1 nM) vs time. Fluorescence assays were performed at 37°C by adding 4.5 μl of a 10x concentrated RNP complex to a solution containing the DNA reporter (final concentration 100 nM). (C) Representative Michaelis-Menten plots for LbCas12a trans-cleavage activity on the different linear/hairpin DNA reporters using a dsDNA activator. Measured Km, kcat and kcat/Km values reported as mean ± SD, where n = 3.
Figure 5.
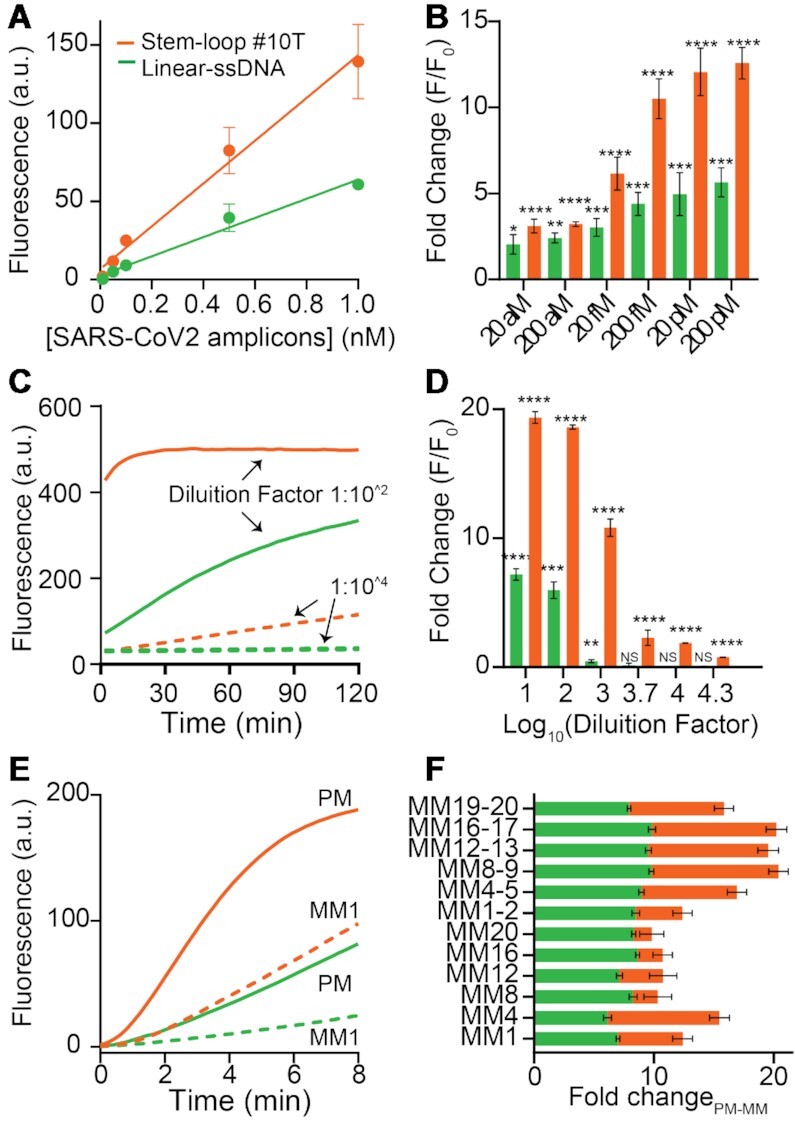
Sensitivity and specificity assays. (A) Amplification free, direct detection of SARS-CoV-2 ds-DNA amplicons. Calibration curves obtained after 1h of incubation of different concentrations of ds-DNA amplicons to a solution containing LbCas12a/crRNA (20 nM, final concentration) and the Linear-ssDNA or the Stem-loop #10T DNA reporter (100 nM). All the values reported show the mean ± SD, where n = 3 replicates. (B) Two-pot CRISPR-based detection of PCR amplified SARS-CoV-2 ds-DNA amplicons at known concentrations showing enhanced signal transduction using the Stem-loop #10T reporter. Pairwise comparisons of reactions containing target ds-DNA amplicons with the control (0 copies per μl of ds-DNA amplicon) were done using an unpaired t-test with Welch's correction (n = 5). The asterisks denote values levels of statistical significance: P< 0.0001 (****); P< 0.001 (***); P< 0.001 (**); p ≤ 0.005 (*). (C) Detection of PCR-amplified Salmonella Typhimurium samples, realized by adding diluted PCR-amplified Salmonella Typhimurium samples to a solution containing LbCas12a/crRNA (20 nM, final concentration) and the linear-ssDNA or the Stem-loop #10T DNA reporter (500 nM). Kinetic assays show enhanced signal transduction for stem-loop #30T reporter testing samples at different dilution factors (1:10^2 and 1:10^4). (D) Fold change of two-pot CRISPR-based detection of PCR-amplified Salmonella Typhimurium at different dilution factors. Pairwise comparisons of reactions containing target PCR-amplified Salmonella amplicons with the control (0 copies per μl of DNA amplicon) were done using an unpaired t-test with Welch's correction (n = 5). The asterisks denote values levels of statistical significance: P< 0.0001 (****); P< 0.001 (***); P< 0.001 (**); P ≤ 0.005 (*). Comparisons that were not significant (NS) have a P value of >0.05 or signal that was lower than the control (0 copies per μl of DNA amplicon). (E) Specificity test on single-nucleotide mismatch mutations (from MM1 to MM20) and double-nucleotide mismatch mutations (from MM1-2 to MM19-20) on synthetic SARS-CoV-2 dsDNA activator. Kinetic assays report the fluorescence difference between perfect match (PM) and mismatch 1 (MM1) target (right, t = 4 min). (F) Superimposed bar graphs showing the difference between the fold change related to the perfect match (PM) and to the mismatch (MM) target (right, 10 nM at t = 4 min). All the values reported show the mean ± SD, where n = 3 replicates.
Michaelis–Menten enzyme kinetic measurements and data analysis
Trans-cleavage reactions were measured by adding 4.5 μl of a 10× concentrated solution of active complex (10 nM of ds-DNA activator + 200 nM of LbCas12a/crRNA complex pre-incubated for 30 min at 37°C) to a solution (final volume of 45 μl) of FRET-based DNA reporters at different final concentrations (from 23 nM to 1.9 μM). Specifically, the final concentrations of LbCas12a/crRNA and ds-DNA activator are 20 and 1 nM, respectively, so that the concentration of the effective RNP complex is 1 nM. Reactions were carried out in three replicates and fluorescence measurements were acquired every 8 ms (λex:: 488 nm; λem: 520 nm). We calculated the initial reaction velocities (V0) for each concentration of reporters by fitting to a linear regression the data collected during the first 180 s. We converted the calculated reaction velocities from a.u./min to nM/min by assuming that fluorescence F(t) measured during trans-cleavage experiments is the sum of the fluorescence from cleaved and uncleaved reporters (21), as reported in the following equation:
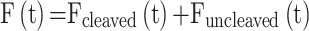 |
where Fcleaved(t) and Funcleaved(t) are the fluorescence from cleaved and uncleaved reporters, respectively. Fluorescence signal of fully cleaved reporters (Fcleaved) at the reported concentrations were obtained by incubating 45 μl of reporter for 24 h at 37°C in the same experimental conditions reported above by assuming the trans-cleavage reaction completed. Fluorescence signal of uncleaved reporters (Funcleaved) was obtained under the experimental procedure by using an inactive LbCas12a/crRNA complex (in the absence of the dsDNA target). We obtained calibrations curves of Fcleaved and Funcleavedversus increasing concentrations of reporters (from 24 nM to 1.9 μM) by fitting data to a linear regression (see Supplementary Figure S8).
Since we know that at time (t) the overall fluorescence is the sum of Fcleaved(t) and Funcleaved(t) which is function of the reporter concentrations ([C]), we can assume that:
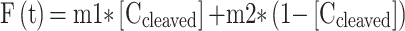 |
where m1 and m2 represent the calibration curve equations (i.e. angular coefficient) from Supplementary Figure S8c. The reaction velocities in nM/s is obtained by differentiating the above reported equation with respect to time. The calculated reaction velocities (nM/min) were plotted versus FRET-based DNA reporter (substrate) concentration and fitted to the Michaelis−Menten equation using GraphPad Prism software (GraphPad, CA, USA) to obtain Vmax and Km, according to the following equation: Y = (Vmax × X)/(Km + X), where X is the substrate concentration and Y is the enzyme velocity. Finally, turnover number (kcat) was calculated by using the following equation: kcat = Vmax/Et, where Et is the concentration of the active ribonucleoprotein complex = 1 nM.
Native polyacrylamide gel electrophoresis (PAGE) assays
Native PAGE experiments were obtained with 18% polyacrylamide (29:1 acrylamide/bisacrylamide) in TBE 10x buffer (1 M Tris, 0.9 M boric acid and 0.01 M EDTA), pH 8.3. Gel solution were prepared by mixing 5.7 ml of diH2O, 4.1 ml of TBE 10× buffer, 4.2 ml of 30% acrylamide/bisacrylamide solution, 75 μl of 10% APS, 14 μl of TEMED. A volume of 10 μl of each sample was mixed with 1 μl of 60% (w/w) Glycerol and then the mixture was added into the gel for the electrophoresis assay. O’Range Ruler 5 base pair DNA Ladder was used as the DNA standard. The samples were obtained following the same procedure reported for fluorescence trans-cleavage assay using 250 nM of FRET-based DNA reporter. The reactions were stopped at different times (0, 15, 30, 45, 60, 120 and 180 min), by heating the solutions for 10 min at 65°C in order to inactivate the Cas12a enzyme. The native PAGE was carried out in a Mini-PROTEAN Tetra cell electrophoresis unit (Bio-Rad) at room temperature, at a constant voltage of 120 V, using TBE 1× buffer (0.1 M Tris, 0.09 M boric acid and 0.001 M EDTA) at pH 8.3 for 80 min.
PAGE analysis of trans-cleavage digestion of FRET-based DNA reporters
Gel was scanned by a ChemiDoc Imaging System (Bio-Rad) that allows to image the fluorescent bands (FAM emission) associated to the cleaved and uncleaved FRET-based DNA reporters. However, the same gels were imaged also after 30 min of staining in SYBR gold (Invitrogen) dissolved in a TBE 1× buffer at pH 8.3 to visualize the ladder signal. We calculated the cleavage fraction (α) according to the following equation:
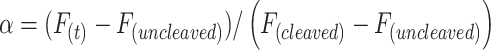 |
where Funcleaved and Fcleaved are the intensity of the bands associated to the reporter at reaction time equal to 0 s and 180 min, respectively, and F(t) is the fluorescence intensity associated to the cleaved reporter at a given time of analysis.
Structural models
Molecular simulations were based on the X-ray structure of the CRISPR-Cas12a system in complex with RNA and DNA (PDB ID: 5NFV),(45) as obtained from Francisella novicida at 2.5 Å resolution. The linear DNA was modelled from the DNA target strand of this X-ray structure, by mutating the sequence to 5′-CTCTCATTTTTAGAGAG-3′, following the experimental studies. The hairpin DNA was modelled from an NMR solved hairpin DNA (PDB ID: 1QE7),(46) by mutating the sequence same as the Linear-ssDNA mentioned above. The three systems were embedded in explicit waters, while Na+ ions were added to neutralize the total charge, leading to an orthorhombic periodic cell. The simulation cell for the CRISPR-Cas12a complex was of ∼120 × 130 × 150 Å containing a total of ∼210 000 atoms. The simulation cell of both the linear and stem loop DNA had a dimension of ∼90 × 60 × 60 Å3, containing ∼25 000 atoms.
Molecular simulations
All-atom molecular dynamics (MD) simulations were performed using the Amber ff12SB force field, which includes the ff99bsc0(46) corrections for DNA and the ff99bsc0+χOL3(46) corrections for RNA. The Allnér force field(46) was used for Mg2+ ions and the TIP3P model was used for water. These force field parameters and the simulation protocol are well-established for protein/nucleic acid complexes, as also shown in our recent studies of CRISPR-Cas9,(47–51) corroborated by NMR experiments (50) and quantum mechanical calculations (52). An integration time step of 2 fs was employed and all bond lengths involving hydrogen atoms were constrained using the SHAKE algorithm. Temperature control was performed at 300 K via Langevin dynamics(53) with a collision frequency of γ = 1/ps. Pressure control was accomplished by coupling the system to a Berendsen barostat (53), at a reference pressure of 1 atm and with a relaxation time of 2 ps. The systems were subjected to energy minimization to relax water molecules and counter ions, keeping the protein, RNA, DNA and Mg2+ ions fixed with harmonic position restraints of 300 kcal/mol·Å2. The systems were then heated from 0 to 100 K in the canonical ensemble (NVT), by running two simulations of 5 ps each while imposing position restraints of 100 kcal/mol·Å2. The temperature was further increased up to 200 K in a ∼100 ps MD run in the isothermal–isobaric ensemble (NPT), reducing the restraint to 25 kcal/mol·Å2. Subsequently, all restraints were released, and the temperature of the system was raised to 300 K in a single NPT simulation of 500 ps. After ∼1.1 ns of equilibration, a ∼10 ns of NPT run was carried out for the density of the system to stabilize at around 1.01 g/cm3. Production runs for the CRISPR-Cas12a system were carried out reaching ∼4 μs of MD simulations in two replicates, gaining ∼8 μs of collective ensemble. After these simulations, two structures, displaying an open conformation of the system, were extracted for further ∼200 ns of conventional MD to obtain an ensemble of conformations for molecular docking. The step loop DNA and linear DNA were also simulated for ∼200 ns of MD in two replicates, starting from different configurations and velocities. Molecular simulations have been carried out using the GPU-accelerated version of AMBER 19 (Case et al., Amber 2022, Univ. California, San Francisco). The most representative configurations were selected from the top three clusters, as obtained by performing k-means clustering on the collected ensemble of the CRISPR-Cas12a complex, stem loop DNA and linear DNA. These selected configurations of the DNA models and of the CRISPR-Cas12a system were object of docking calculations as described below.
Docking and binding energies
We performed rigid body docking of the stem loop and linear DNA models as ligands for the target CRISPR-Cas12a complex using HADDOCK2.4 (54) The catalytic core composed of the DED motif (D916, E1005, D1254) in Cas12a were treated as the ‘active’ residues for HADDOCK2.4 docking, while the solvent-exposed residues within 6.5 Å of the active residues were treated as ‘passive’ residues. The five dT-repeats in the DNA models were treated as active residues and passive residues were selected following the criterion mentioned above. Both the DNA models were restrained to preserve their structure while docking. For the all-atom representation, OPLSX non-bonded parameters are used both for the protein (55) and DNA (55). The most representative docked conformations from the top cluster, based on the electrostatic and Van der Waal energies, and HADDOCK2.4 score, were selected for further analyses. The docked structures were minimized and equilibrated as reported above (Molecular Simulations section) for subsequent ∼200 ns of conventional MD simulations. We calculated the binding free energy for the association of DNA with Cas12a using the Molecular Mechanics with Generalized Born and Surface Area (MM-GBSA) method (46). Binding free energy calculations where performed over the ∼200 ns ensemble, in windows of ∼20 ns. The GBn model described by Mongan, Simmerling, McCammon, Case and Onufriev was employed (46).
RESULTS AND DISCUSSION
Design of FRET-based hairpin DNA reporters
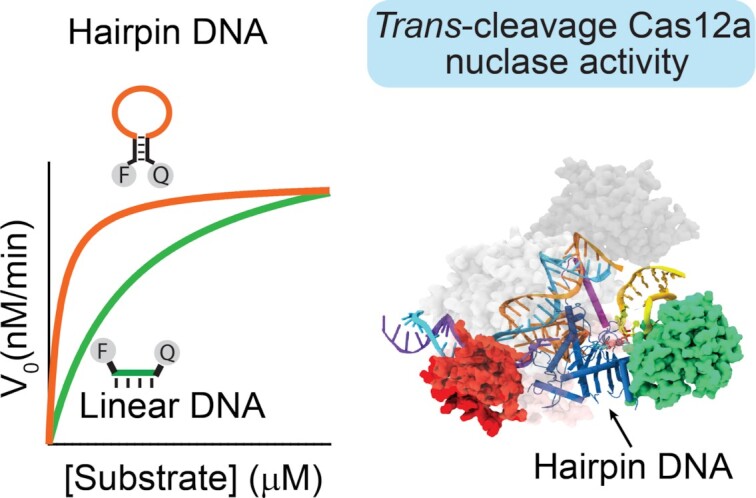
To enhance the trans-cleavage activity and the signal transduction of Cas12a, we have investigated the use of hairpin DNA molecules as fluorescent reporters. Consisting of a stem-loop DNA modified at the two self-complementary ends with a fluorophore/quencher pair, they are a class of synthetic biomolecular switches widely used for live-cell analysis of native RNA transcripts (56,57), in-vitro diagnostics (58–61) and intracellular analysis at the single-molecule level (62), and thus represent an ideal test bed for our studies. The simple hairpin structure indeed separates the single-stranded portion that preferentially can be cleaved by activated Cas12a proteins (the loop), from those that define the switching equilibrium and signal transduction (the stem). From the perspective of signal transduction, one crucial advantage of using stem-loop DNA reporters over conventional short ssDNA probes is the superior quenching efficiency and the relatively low fluorescence background due to the tight contact between the fluorophore and the quencher (63–65). In addition, we hypothesized that if Cas12a is able to cleave the single stranded loop portion, the short self-complementary stem should not be stable enough to maintain the non-emissive hairpin conformation, thus leading to a rapid increase of the fluorescence signal (Figure 1).
Figure 1.
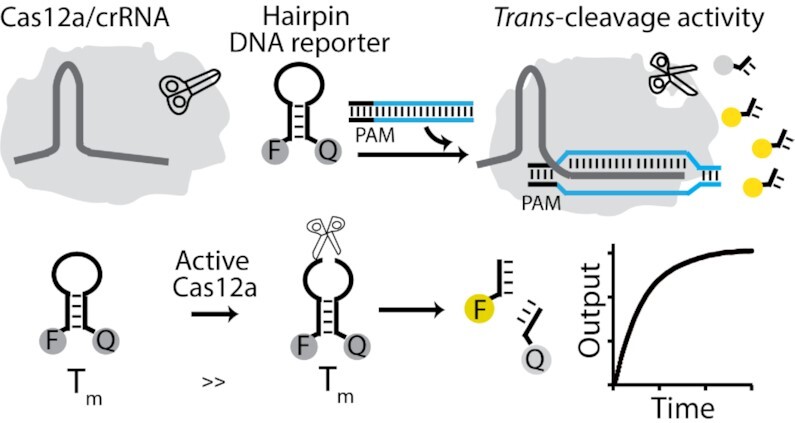
Schematic for Cas12a trans-cleavage activity based on hairpin DNA reporters. We explore the use of different FRET-based hairpin DNA reporters as signal transducers of the Cas12a-based trans-cleavage reaction. The hairpin structure separates the single-stranded portion that preferentially can be cleaved by activated Cas12a proteins (the loop) from those that define the switching equilibrium and signal transduction (the stem). Once the ternary complex of Cas12a/crRNA/dsDNA is formed, the trans-cleavage activity is activated and the hairpin DNA reporter can be cleaved, thus generating a fluorescence signal change.
To demonstrate our hypothesis, we have designed a set of three hairpin DNA reporters that retain a common stem sequence (i.e. 3GC + 2AT) and fluorophore/quencher pair (6-FAM/BHQ-1), but present loop domains of different length (Figure 2A, left). In this way, it is possible to finely modulate the switching constant (Ks) associated to the equilibrium between the non-emitting hairpin configuration (OFF-state) and the emitting unfolded state (ON-state) of the DNA reporter (66–68). Specifically, by increasing the loop length (composed by thymine repetitions) from 5T (stem–loop #5T), to 10T (stem–loop #10T) and 30T (stem–loop #30T) we can rationally decrease the thermal stability of the hairpin DNA reporters (Figure 2A, right). Indeed, melting assays confirm that the melting temperature at which they unfold strongly correlates to the loop length (TM, stem–loop #5T = 72.4 ± 0.3°C; TM, stem–loop #10T = 67 ± 1°C; TM, stem–loop #30T = 53.3 ± 0.5°C, Figure 2A, right). From the thermodynamic analysis of the data we confirmed that a longer loop length corresponds to a higher switching constant (Ks, #30T = (2.9 ± 0.2) × 10–2), resulting in a destabilized non-emitting folded state and a lower signal gain due to the increased fluorescence background (Supplementary Figures S1 and S2). In contrast, a shorter loop produces an overly stabilized structure (Ks, #5T = (7 ± 2) × 10–4) and a higher signal change. As expected, the linear single-stranded FRET-based reporter used in DETECTR (Linear-ssDNA) does not show any fluorescence signal increase along with temperature, in agreement with the absence of secondary structures. Of note, we fixed the length of the stem because longer stems might lead to the formation of hairpin-hairpin dimers because of the self-complementary nature of the 5′- and 3′-ends.
Figure 2.

Design and characterization of different hairpin DNA reporters as FRET-based reporters of LbCas12a trans-cleavage activity. (A) We tested three different hairpin DNA reporters having same stem composition (3GC + 2AT) but different loop length and an increasing number of thymine (from 5 to 30). All the DNA reporters were functionalized with 6-FAM at 5′-end and with BHQ-1 at 3′-end. Melting curves indicate that by increasing the number of thymine in the loop we can rationally control the melting temperatures of the reporters and thus the stability of the hairpin structure. (B) LbCas12a trans nuclease activity on linear ssDNA and hairpin DNA reporters. The fluorescence kinetic analysis over time of LbCas12a trans-cleavage activity triggered by a fully complementary 20bp-long double stranded DNA activator show enhanced signal transduction using the Stem-loop #10T reporter. (C) Fluorescence fold change as a function of the temperature after 1h of LbCas12a trans-cleavage activity. (D) Effect of Mg2+ concentration on LbCas12a-based trans-cleavage activity. The assays were performed by adding 4.5 μl of LbCas12a/crRNA complex (200 nM) preassembled with the dsDNA activator (10 nM) to a solution containing the DNA reporter (100 nM). The trans-cleavage reactions in c) and d) were stopped after 1 h by heating the solutions at 65°C for 10 min. All bar graph shows the mean fold change calculated as F – F0/F0 (background-subtracted fluorescence divided by background fluorescence of the DNA reporter at time t = 0) ± SD, where n = 3 replicates.
Faster and enhanced signal change Using Hairpin DNA reporters
As our test bed for the study of signal transduction, we measured the a-specific nuclease activity of Lachnospiraceae bacterium Cas12a (LbCas12a) using the three FRET-based hairpin reporters and the linear-ssDNA. Consistent with our hypothesis, all DNA reporters display increased fluorescence upon trans-activity thus confirming that LbCas12a protein can degrade also DNA reporters folded in a hairpin structure. Notably, the Stem-loop #10T yields the highest fluorescence signal (∼200-fold change) compared to the other DNA reporters (Figure 2B). The lower fold signal change obtained using Stem-loop #30T reporter can be ascribed to the higher fluorescence background at 37°C, in agreement with the TM of melting of this reporter (Figure 2a and Supplementary FigureS2). We confirmed these results by testing multiple crRNAs/dsDNA activators (Supplementary Figure S3) and Cas12a orthologs, as from the Francisella novicida (FnCas12a) and the Acidaminococcus (AsCas12a; Supplementary Figure S4). In addition, the effect of temperature and Mg2+ concentration on the LbCas12a-based sensing assay has been investigated because of the synergistic effect on both Cas12a activity and stability of the hairpin DNA structures. The Stem-loop #10T reporter shows again the highest fluorescence increase at the temperatures of interest for diagnostic applications (25–45°C; Figure 2C) and optimal MgCl2 concentration is reported to be 10 mM (Figure 2D). As expected, at concentration of MgCl2 between 3 and 5 mM, the Stem-loop #10T and #5T reporters generate a lower but similar signal change, probably because the latter structure is more stable and less sensitive to the presence of Mg2+ ions. Of note, at these concentrations of Mg2+ the trans-activity is still detectable whereas the linear-ssDNA reporter does not provide any significant signal change. To properly compare trans-cleavage, we thus selected the experimental condition which is optimal for all the different FRET-based reporters.
Motivated by the enhanced signal transduction obtained using hairpin reporters, we sought to further explore the effect of DNA sequence content on the cleavage activity since previous publications reported a preferential trans-cleavage for AT-rich DNA reporters (33,34). Thus, we tested two variants of the Stem-loop #10T reporter containing different number of thymine in the loop region (Variant #1, loop composed of 12T; Variant #2, loop of 7T + 5A). Both the hairpin Variants confirm to better transduce the fluorescence signal compared to the Linear-ssDNA reporter (Supplementary Figure S5). This suggests that Cas12a trans-activity has no or very little preference for primary sequence content of the DNA reporter, and that probably the observed enhancement can be mainly ascribed to the presence of the secondary structure. However, it is very important to distinguish whether improved signal change from background can be ascribed to the real preference of Cas12a for hairpin DNA substrates, or to the intrinsic superior quenching efficiency of the FRET pair in this structure.
Hairpin reporters show enhanced rate of trans-cleavage
Motivated by the above considerations, we first carried out time-dependent gel electrophoresis assays of trans-activity on the DNA reporters. The trans-cleavage activity of the LbCas12a protein is a multi-turnover enzymatic reaction catalysed by the dsDNA target-activated ribonucleoprotein complex that degrades ssDNA probes in a RuvC- domain-dependent manner. To study the kinetics of LbCas12a trans-cleavage activity, we performed direct dsDNA detection without any pre-amplification step by preparing a 10X pre-incubated LbCas12a enzyme/crRNA/ds-DNA target complex (200, 200 and 10 nM, respectively) and then diluting the mix 10 times into a solution containing the DNA reporters (100 nM). This is necessary to assume that all target molecules have reacted and that the trans-cleavage reaction is the rate-limiting process. The results show faster trans-cleavage for hairpin probes compared to the linear one (Figure 3A). We quantified the PAGE data by FAM imaging in order to determine the cleavage fraction-time plot (see Supplementary Table S1) and then compared it to spectrofluorometric data. Fluorescence data are in agreement with gel assays confirming that all the three hairpin DNA reporters are degraded faster than the Linear-ssDNA reporter, regardless of whether LbCas12a is activated by dsDNA or ssDNA target sequences (Figure 3B and Supplementary Figure S6). We also carried out control experiments to evaluate if a-specific interactions between inactive RNP complex and reporters can affect the cleavage assays. Data obtained at saturating concentration of dsDNA activator (20 nM, equimolar to RNP complex) confirmed that a-specific interactions do not affect the assays (Supplementary Figure S7).
To study the LbCas12a enhanced activity, we performed a Michaelis-Menten kinetic study of the different DNA reporters. Different concentrations of DNA reporters were employed to determine the initial reaction velocities (V0, Supplementary Figure S8). Calibration curves were used to convert V0 from u.a./min to nM/min (see Supplementary Figure S8 and Material and Methods Section for the procedure). By plotting the measured reaction velocities versus DNA reporter (substrate) concentration and fitting the data using Michaelis−Menten equation we obtained kcat and Km values. The ratio between kcat/Km is ∼5-fold higher for the hairpin DNA reporters than for the linear-ssDNA (Supplementary Table S2). This difference can be ascribed to the decrease of Km values obtained for the three hairpin reporters (Kmlinear-ssDNA, = 250 ± 50 nM; Kmstem–loop #5T = 42 ± 5 nM; Kmstem–loop #10T = 34 ± 2 nM; Kmstem–loop #30T = 69 ± 5 nM) whereas kcat values are quite similar (kcat linear-ssDNA, = 0.30 ± 0.02 s–1; kcatstem–loop #5T = 0.24 ± 0.02 s–1; kcatstem–loop #10T = 0.23 ± 0.01 s–1; kcat stem–loop #30T = 0.28 ± 0.02 s–1). These data suggest that the enhanced binding affinity of LbCas12a for hairpin-forming substrates results in enhanced trans-cleavage as it is also confirmed through the comparison with previously reported systems (Supplementary Table S3). To exclude the possibility that enhanced binding affinity of stem-loop reporters can be due to an a-specific effect generated by the extra number of bases present in the sequence, we performed control assays using a 22 base long DNA reporter (i.e. variant-ssDNA reporter). It consists of single-stranded DNA terminally labelled with FAM/BHQ1 pair that presents the same sequence of the loop (i.e. 10 T, in the middle of the sequence) of the stem–loop #10T reporter and additional not self-complementary nucleotides at the 3′- and 5′-end in place of the stem in order to hinder any hairpin-like folding. PAGE analysis of trans-cleavage activity over time clearly indicates slower degradation of the variant-ssDNA reporter and, as expected, lower fluorescence signal transduction (Supplementary Figure S9) confirming the importance of the hairpin conformation to achieve enhanced trans-activity.
Mechanistic insight into the hairpin DNA reporter interactions with Cas12a
To elucidate the molecular basis for enhanced trans-cleavage, we performed molecular dynamics (MD) simulations and molecular docking studies. Molecular simulations were based on the X-ray structure of Cas12a obtained post non-target strand cleavage (PDB: 5NFV) (46). This structure contains a short dsDNA at the PAM distal site, which should be released from the protein after target strand cleavage to enable the access of ssDNA for trans-cleavage (69). We have removed this portion of dsDNA from the X-ray structure 5FNV, obtaining a state of Cas12a post cis-cleavage and thereby prone for trans-cleavage. This system has been an object of MD simulations, collecting ∼4 μs for two replicates. These simulations revealed a remarkable opening of the system (Figure 4A) that lead to the widening of the DNA-binding groove in the RuvC domain. Indeed, the Nuc domain significantly opens, as shown by an increase in the distance between the Rec2 and Nuc domains and an outward bending of Nuc, observed with respect to the RuvC domain (Figure 4B). This opens the room for the incoming ssDNA to bind for trans-cleavage (1). Notably, the observed opening of Nuc with respect to REC2 is in line with previous biophysical studies indicating concerted dynamics and high flexibility of the two domains (70,71). This open Nuc conformation provided us a suitable starting point to perform molecular docking of hairpin and linear DNA structures at the RuvC catalytic site. We selected the sequence of the Stem-loop #5T as our model hairpin structure. First, extensive MD simulations of the Stem-loop #5T and the Linear-ssDNA have been performed (i.e. ∼200 ns for each system in two replicates) to obtain an ensemble of conformations for molecular docking. Then, three representative conformations from the top three clusters were selected for molecular docking. Rigid body docking of the DNA models within the RuvC catalytic site was performed using HADDOCK2.4 (72).
Figure 4.

Mechanistic insights from molecular simulations. (A) Long timescale molecular dynamics (MD) simulations reporting a large-scale opening of the Nuc (indicated by the arrow in green). (B) Outward bending of Nuc along ∼4 μs MD trajectories (in two replicates), shown through the time evolution of the center of mass distance between the REC2 and Nuc domains (top), and the angle of Nuc with respect to the RuvC domain (bottom). (C) Comparison of the binding free energy between the association of the stem loop (green) and linear ssDNA (purple) with Cas12a using the MM-GBSA solvation method.
The most representative docked conformations, based on the electrostatic and Van der Waal energies, and HADDOCK score, were selected for further molecular simulations and analyses. This approach using MD before and after molecular docking allowed us to reduce the possible biases arising from the docking calculations of single stranded DNA (73). To compute the binding free energy, ∼200 ns of unconstrained MD simulations were carried out for both the stem-loop DNA and linear DNA in complex with Cas12a. This was obtained using the Molecular Mechanics with Generalized Born and Surface Area (MM-GBSA) solvation method (46). This method gave us an appreciable comparison of the binding energies between the two systems. We observed a difference in free energy of ∼8.24 kcal/mol in favor of the stem–loop #5T, compared to the Linear-ssDNA (Figure 4c and S10). This suggests that the Hairpin DNA model promotes stronger binding with the Cas12a catalytic pocket. Analysis of the interactions further reveals that the stem loop DNA interacts more extensively with the Nuc domain than the linear DNA (Supplementary Figure S11). Accordingly, the stem loop DNA binds more stably within the Cas12a core, as an effect of increased interactions with Nuc, when compared to the linear DNA. A more stable location of the substrate within the active site can favor the catalysis, ultimately resulting in an increase in the catalytic efficiency. This is in agreement with our experimental observations, reporting a lower Km and higher cleavage activity for the hairpin DNA reporter when compared with Linear-ssDNA. Moreover, these simulations provide a mechanistic understanding of the binding of hairpin and linear DNA. These findings also take us a step further toward understanding the binding of single stranded DNA (both hairpin and linear) at Cas12a for trans-cleavage.
Enhanced sensitivity and specificity using hairpin DNA reporter
To better characterize the advantages of using a hairpin DNA reporter as fluorescence reporter, we selected the best-performing FRET-based DNA reporter (i.e. Stem-loop #10T) and compared its analytical features with those of the Linear-ssDNA. To do this, we designed an LbCas12a-based detection assays to detect synthetically generated dsDNA amplicons of the S gene of SARS-CoV-2 (74). Although no clinical samples were tested, we performed quantitative detection of synthetic SARS-CoV-2 dsDNA amplicons (102 bp). The results clearly indicate that the Stem-loop #10T reporter consistently has a higher sensitivity (slope stem–loop #10T = 137.0 a.u./nM) and lower limit of detection (LOD = 10 pM) for detecting SARS-CoV-2 as compared to the linear-ssDNA reporter (slope linear-ssDNA = 61.3 a.u./nM; LOD = 150 pM; Figure 5A), and exhibit an average of 2-fold signal improvement between 1 nM and 10 pM of target SARS-CoV-2 dsDNA amplicons. In addition, when samples at known concentration (from 20 aM to 200 pM, see Figure 5B) were pre-amplified by PCR (30 cycles, two-pot PCR-CRISPR reaction), the Stem-loop #10T demonstrated higher sensitivity intended as the upstroke of the input/output response in a two-pot reaction assay (i.e. by diluting PCR samples 500 times before the incubation with the LbCas12a/crRNA complex). To demonstrate the generalizability of the results for the detection of bacteria strains, the amplification of nucleotide sequences within the invA gene of Salmonella Typhimurium was evaluated as a means to detect Salmonella serotypes. Salmonella is an important food-borne pathogen responsible for human disease outbreaks worldwide. The reference method for its detection in food requires up to 5–6 days to produce final results, as it involves pre-enrichment, selective enrichment, isolation on selective agar, biochemical characterisation of presumptive colonies and final serological confirmation. (75). Therefore, there is a need for new methods capable of detecting small amounts of Salmonella in food samples in a short time. To this end, several molecular methods for the detection of Salmonella in food have been developed (76,77) and we also report here preliminary results for an alternative and more sensitive detection of this pathogen. The specific PCR product was a 244 bp DNA amplicon which was analysed in CRIPSR-Cas12a assays through serial dilution obtained from the same PCR product. Both the kinetic analysis and the fold change associated to different dilution factors clearly confirmed that enhanced sensitivity and more rapid signal transduction using the stem–loop #10T reporter (Figure 5C and D).
We evaluated also the specificity of LbCas12a-based detection in discriminating point mutations across dsDNA amplicons using both the Stem-loop #10T and the Linear-ssDNA reporters. We mutated either a single or two consecutive nucleotides at different points of the dsDNA amplicons of SARS-CoV-2. Since stem–loop #10T reporter show enhanced and faster cleavage rate compared to linear-ssDNA, we hypothesized that by performing short-time fluorescence analysis of trans-cleavage activity (t = 4 min), discrimination between perfect match (PM) and mismatch (MM) targets could be improved (Figure 5E and Supplementary Figure S12). As expected, the single-point mutations were more easily tolerated than the double-point mutations by LbCas12a. Nevertheless, it is noteworthy that the difference of fold signal change between PM and MM is significantly higher for the stem–loop #10T compared to Linear-ssDNA reporter at different ds-DNA target concentration (Figure 5F and Supplementary Figure S13). Cleavage PAGE assays over time using mismatches confirmed that the significant difference of trans-cleavage rate between the two reporter systems allows to better discriminate the presence of MMs using Stem-loop #10T (Supplementary Figure S14).
In this work, we have studied a set of hairpin DNA reporters with loop domains of different length as FRET-based reporters of the trans-activity of Cas12a enzymes. We have first confirmed that the loop length can be used to finely regulate the switching mechanism between the folded (ON)/unfolded (OFF) state of the reporters, and this also provides a means to rationally control the overall fluorescence signal change produced by the trans-activity of Cas12a. Then, kinetic analysis of the trans-activity showed that all the hairpin DNA reporters have faster Cas12a-based cleavage rate compared to that observed using short linear ssDNA probes. In addition, the Michaelis-Menten analysis performed on the different DNA reporters indicated an enhanced affinity (lower Km values) of Cas12a for the hairpin DNA reporters. Molecular dynamics simulations of Cas12a showed a remarkable opening of the system that lead to the widening of the DNA-binding groove in the RuvC domain, and docking studies confirmed the stronger binding of stem-loop DNA to the Cas12a catalytic pocket. Finally, we tested the best performing hairpin DNA reporter (Stem-loop #10T) for the detection of synthetic ds-DNA amplicons related to S gene of SARS-CoV-2 and Salmonella strains. The use of the stem–loop #10T resulted in a sensing platform with improved sensitivity and enhanced specificity in discriminating single and double point mutations across dsDNA amplicons. These data suggest that the enhanced affinity of Cas12a for hairpin probes, combined to the well-established capacity of FRET-based stem-loop DNA to improve fluorescence signal transduction, can significantly ehnance the sensitivity and the specificity of CRISPR-Cas12-based sensing platforms. The faster signal transduction and the ability to discriminate among different DNA sequences offers the opportunity to use hairpin-based transducer of trans-activity in a variety of high-throughput applications, from metagenomics to biomedicine.
Our findings represent a step toward a better understanding of Cas12a nuclease activity that can potentially assist in the development of improved diagnostic platforms for the diagnosis of various diseases, including the COVID-19. Beyond the diagnostic applications, this work paves the way for exploring evolutionary and mechanistic aspects of CRISPR type V enzyme that govern the affinity of CRISPR Type V systems for hairpin-like structures. Further studies are currently in progress to understand whether other CRISPR Type systems (Cas13) showing trans-activity show a similar behaviour.
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: A.P., G.P. and G.P. conceived the idea and supervised the research; M.R., J.v.O., A.P., G.P. and G.P. designed the experiments; F.R. and R.I. designed the probes; M.R. and N.B. performed the experiments and analysed the data; R.M. and A.V. produced FnCas12a and SARS-CoV-2 sequence and amplicons; D.M., I.T. and E.D. prepared Salmonella samples; A.S. and P.A. performed molecular simulations; M. R., G.P., G.P., and A. P. wrote the manuscript.
Contributor Information
Marianna Rossetti, Department of Chemistry, University of Rome, Tor Vergata, Via della Ricerca Scientifica 00133, Rome, Italy.
Rosa Merlo, Institute of Biosciences and BioResources, National Research Council of Italy, Via Pietro Castellino 111, 80131 Naples, Italy.
Neda Bagheri, Department of Chemistry, University of Rome, Tor Vergata, Via della Ricerca Scientifica 00133, Rome, Italy.
Danila Moscone, Department of Chemistry, University of Rome, Tor Vergata, Via della Ricerca Scientifica 00133, Rome, Italy.
Anna Valenti, Institute of Biosciences and BioResources, National Research Council of Italy, Via Pietro Castellino 111, 80131 Naples, Italy.
Aakash Saha, Department of Bioengineering and Department of Chemistry, University of California Riverside, 900 University Avenue, Riverside, CA 52512 USA.
Pablo R Arantes, Department of Bioengineering and Department of Chemistry, University of California Riverside, 900 University Avenue, Riverside, CA 52512 USA.
Rudy Ippodrino, Ulisse BioMed S.r.l. Area Science Park, 34149 Trieste, Italy.
Francesco Ricci, Department of Chemistry, University of Rome, Tor Vergata, Via della Ricerca Scientifica 00133, Rome, Italy.
Ida Treglia, Department of Food Safety, Nutrition and Veterinary Public Health, Istituto Superiore di Sanità, Viale Regina Elena 299, Rome, Italy.
Elisabetta Delibato, Department of Food Safety, Nutrition and Veterinary Public Health, Istituto Superiore di Sanità, Viale Regina Elena 299, Rome, Italy.
John van der Oost, Laboratory of Microbiology, Wageningen University, Stippeneng 4, 6708 WE Wageningen, The Netherlands.
Giulia Palermo, Department of Bioengineering and Department of Chemistry, University of California Riverside, 900 University Avenue, Riverside, CA 52512 USA.
Giuseppe Perugino, Institute of Biosciences and BioResources, National Research Council of Italy, Via Pietro Castellino 111, 80131 Naples, Italy; Department of Biology, University of Naples “Federico II”, Complesso Universitario di Monte Sant’Angelo, Ed. 7, Via Cintia 26, 80126 Naples, Italy.
Alessandro Porchetta, Department of Chemistry, University of Rome, Tor Vergata, Via della Ricerca Scientifica 00133, Rome, Italy.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement ‘Nano-Oligo Med’ [778133]; Italian Ministry of University and Research Project of National Interest, PRIN, 2017Y2PAB8_004 through the project ‘Cutting Edge Analytical Chemistry Methodologies and Bio-Tools to Boost Precision Medicine in Hormone-Related Diseases’; M. Rossetti was supported from a Fondazione Umberto Veronesi postdoctoral fellowship; computational studies are based upon work supported by the National Institutes of Health [R01GM141329 to G.P.]; National Science Foundation [CHE-2144823 to G.P.]; COVID-19 HPC Consortium (https://covid19-hpc-consortium.org/), which is a unique private-public effort to bring together government, industry, and academic leaders who are volunteering free compute time and resources in support of COVID-19 research [MCB200150 to G.P.]; computer time has also been awarded by XSEDE [TG-MCB160059]; NERSC [M3807 to G.P.]. Funding for open access charge: NanoOligo Med.
Conflict of interest statement. None declared.
REFERENCES
- 1. Chen J.S., Ma E., Harrington L.B., Da Costa M., Tian X., Palefsky J.M., Doudna J.A.. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science. 2018; 360:436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kaminski M.M., Abudayyeh O.O., Gootenberg J.S., Zhang F., Collins J.J.. CRISPR-based diagnostics. Nat. Biomed. Eng. 2021; 5:643–656. [DOI] [PubMed] [Google Scholar]
- 3. Li S.Y., Cheng Q.X., Li X.Y., Zhang Z.L., Gao S., Cao R.B., Zhao G.P., Wang J., Wang J.M.. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 2018; 4:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li S.Y., Cheng Q.X., Liu J.K., Nie X.Q., Zhao G.P., Wang J.. CRISPR-Cas12a has both cis- and trans-cleavage activities on single-stranded DNA. Cell Res. 2018; 28:491–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abudayyeh O.O., Gootenberg J.S., Konermann S., Joung J., Slaymaker I.M., Cox D.B.T., Shmakov S., Makarova K.S., Semenova E., Minakhin L.et al.. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016; 353:aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Makarova K.S., Wolf Y.I., Alkhnbashi O.S., Costa F., Shah S.A., Saunders S.J., Barrangou R., Brouns S.J.J., Charpentier E., Haft D.H.et al.. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015; 13:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. East-Seletsky A., O’Connell M.R., Knight S.C., Burstein D., Cate J.H.D., Tjian R., Doudna J.A.. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature. 2016; 538:270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yan W.X., Hunnewell P., Alfonse L.E., Carte J.M., Keston-Smith E., Sothiselvam S., Garrity A.J., Chong S., Makarova K.S., Koonin E.V.et al.. Functionally diverse type v CRISPR-Cas systems. Science. 2019; 363:88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tang Y., Gao L., Feng W., Guo C., Yang Q., Li F., Le X.C.. The CRISPR-Cas toolbox for analytical and diagnostic assay development. Chem. Soc. Rev. 2021; 50:11844–11869. [DOI] [PubMed] [Google Scholar]
- 10. Kellner M.J., Koob J.G., Gootenberg J.S., Abudayyeh O.O., Zhang F.. SHERLOCK: nucleic acid detection with CRISPR nucleases. Nat. Protoc. 2019; 14:2986–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. De Puig H., Lee R.A., Najjar D., Tan X., Soekensen L.R., Angenent-Mari N.M., Donghia N.M., Weckman N.E., Ory A., Ng C.F.et al.. Minimally instrumented SHERLOCK (miSHERLOCK) for CRISPR-based point-of-care diagnosis of SARS-CoV-2 and emerging variants. Sci. Adv. 2021; 7:eabh2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gootenberg J.S., Abudayyeh O.O., Lee J.W., Essletzbichler P., Dy A.J., Joung J., Verdine V., Donghia N., Daringer N.M., Freije C.A.et al.. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. 2017; 356:438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Broughton J.P., Deng X., Yu G., Fasching C.L., Servellita V., Singh J., Miao X., Streithorst J.A., Granados A., Sotomayor-Gonzalez A.et al.. CRISPR-Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 2020; 38:870–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gootenberg J.S., Abudayyeh O.O., Kellner M.J., Joung J., Collins J.J., Zhang F.. Multiplexed and portable nucleic acid detection platform with cas13, cas12a, and csm6. Science. 2018; 360:439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chertow D.S. Next-generation diagnostics with CRISPR. Science. 2018; 360:381–382. [DOI] [PubMed] [Google Scholar]
- 16. Myhrvold C., Freije C.A., Gootenberg J.S., Abudayyeh O.O., Metsky H.C., Durbin A.F., Kellner M.J., Tan A.L., Paul L.M., Parham L.A.et al.. Field-deployable viral diagnostics using CRISPR-Cas13. Science. 2018; 360:444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barnes K.G., Lachenauer A.E., Nitido A., Siddiqui S., Gross R., Beitzel B., Siddle K.J., Freije C.A., Dighero-Kemp B., Mehta S.B.et al.. Deployable CRISPR-Cas13a diagnostic tools to detect and report ebola and lassa virus cases in real-time. Nat. Commun. 2020; 11:4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xiong E., Jiang L., Tian T., Hu M., Yue H., Huang M., Lin W., Jiang Y., Zhu D., Zhou X.. Simultaneous dual-gene diagnosis of SARS-CoV-2 based on CRISPR/Cas9-Mediated lateral flow assay. Angew. Chem. Int. Ed. 2021; 60:5307–5315. [DOI] [PubMed] [Google Scholar]
- 19. Mukama O., Wu J., Li Z., Liang Q., Yi Z., Lu X., Liu Y., Liu Y., Hussain M., Makafe G.G.et al.. An ultrasensitive and specific point-of-care CRISPR/Cas12 based lateral flow biosensor for the rapid detection of nucleic acids. Biosens. Bioelectron. 2020; 159:112143. [DOI] [PubMed] [Google Scholar]
- 20. Wang X., Ji P., Fan H., Dang L., Wan W., Liu S., Li Y., Yu W., Li X., Ma X.et al.. CRISPR/Cas12a technology combined with immunochromatographic strips for portable detection of african swine fever virus. Commun. Biol. 2020; 3:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ramachandran A., Santiago J.G.. CRISPR enzyme kinetics for molecular diagnostics. Anal. Chem. 2021; 93:7456–7464. [DOI] [PubMed] [Google Scholar]
- 22. Choi J.-H., Lim J., Shin M., Paek S.-H., Choi J.-W.. CRISPR-Cas12a-Based nucleic acid amplification-free DNA biosensor via au nanoparticle-assisted metal-enhanced fluorescence and colorimetric analysis. Nano Lett. 2021; 21:693–699. [DOI] [PubMed] [Google Scholar]
- 23. Fozouni P., Son S., Díaz de León Derby M., Knott G.J., Gray C.N., D’Ambrosio M.V., Zhao C., Switz N.A., Kumar G.R., Stephens S.I.et al.. Amplification-free detection of SARS-CoV-2 with CRISPR-Cas13a and mobile phone microscopy. Cell. 2021; 184:323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tian T., Shu B., Jiang Y., Ye M., Liu L., Guo Z., Han Z., Wang Z., Zhou X.. An ultralocalized cas13a assay enables universal and nucleic acid amplification-free single-molecule RNA diagnostics. ACS Nano. 2021; 15:1167–1178. [DOI] [PubMed] [Google Scholar]
- 25. Bruch R., Johnston M., Kling A., Mattmüller T., Baaske J., Partel S., Madlener S., Weber W., Urban G.A., Dincer C.. CRISPR-powered electrochemical microfluidic multiplexed biosensor for target amplification-free miRNA diagnostics. Biosens. Bioelectron. 2021; 177:112887. [DOI] [PubMed] [Google Scholar]
- 26. Bao M., Chen Q., Xu Z., Jensen E.C., Liu C., Waitkus J.T., Yuan X., He Q., Qin P., Du K.. Challenges and opportunities for clustered regularly interspaced short palindromic repeats based molecular biosensing. ACS Sensors. 2021; 6:2497–2522. [DOI] [PubMed] [Google Scholar]
- 27. Bruch R., Baaske J., Chatelle C., Meirich M., Madlener S., Weber W., Dincer C., Urban G.A.. CRISPR/Cas13a-Powered electrochemical microfluidic biosensor for nucleic acid amplification-free miRNA diagnostics. Adv. Mater. 2019; 31:1905311. [DOI] [PubMed] [Google Scholar]
- 28. Lillis L., Siverson J., Lee A., Cantera J., Parker M., Piepenburg O., Lehman D.A., Boyle D.S.. Factors influencing recombinase polymerase amplification (RPA) assay outcomes at point of care. Mol. Cell. Probes. 2016; 30:74–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li L., Li S., Wu N., Wu J., Wang G., Zhao G., Wang J.. HOLMESv2: a CRISPR-Cas12b-Assisted platform for nucleic acid detection and DNA methylation quantitation. ACS Synth. Biol. 2019; 8:2228–2237. [DOI] [PubMed] [Google Scholar]
- 30. Steens J.A., Zhu Y., Taylor D.W., Bravo J.P.K., Prinsen S.H.P., Schoen C.D., Keijser B.J.F., Ossendrijver M., Hofstra L.M., Brouns S.J.J.et al.. SCOPE enables type III CRISPR-Cas diagnostics using flexible targeting and stringent CARF ribonuclease activation. Nat. Commun. 2021; 12:5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Santiago-Frangos A., Hall L.N., Nemudraia A., Nemudryi A., Krishna P., Wiegand T., Wilkinson R.A., Snyder D.T., Hedges J.F., Cicha C.et al.. Intrinsic signal amplification by type III CRISPR-Cas systems provides a sequence-specific SARS-CoV-2 diagnostic. Cell Reports Med. 2021; 2:100319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu T.Y., Knott G.J., Smock D.C.J., Desmarais J.J., Son S., Bhuiya A., Jakhanwal S., Prywes N., Agrawal S., Díaz de León Derby M.et al.. Accelerated RNA detection using tandem CRISPR nucleases. Nat. Chem. Biol. 2021; 17:982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nguyen L.T., Smith B.M., Jain P.K.. Enhancement of trans-cleavage activity of cas12a with engineered crRNA enables amplified nucleic acid detection. Nat. Commun. 2020; 11:4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fuchs R.T., Curcuru J., Mabuchi M., Yourik P., Robb G.B.. Cas12a trans-cleavage can be modulated in vitro and is active on ssDNA, dsDNA, and RNA. 2019; bioRxiv doi:08 April 2019, preprint: not peer reviewed 10.1101/600890. [DOI]
- 35. Nalefski E.A., Patel N., Leung P.J.Y., Islam Z., Kooistra R.M., Parikh I., Marion E., Knott G.J., Doudna J.A., Le Ny A.-L.M.et al.. Kinetic analysis of cas12a and cas13a RNA-Guided nucleases for development of improved CRISPR-Based diagnostics. Iscience. 2021; 24:102996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fu X., Shi Y., Peng F., Zhou M., Yin Y., Tan Y., Chen M., Yin X., Ke G., Zhang X.-B.. Exploring the trans-cleavage activity of CRISPR/Cas12a on gold nanoparticles for stable and sensitive biosensing. Anal. Chem. 2021; 93:4967–4974. [DOI] [PubMed] [Google Scholar]
- 37. Dai Y., Somoza R.A., Wang L., Welter J.F., Li Y., Caplan A.I., Liu C.C.. Exploring the trans-cleavage activity of CRISPR-Cas12a (cpf1) for the development of a universal electrochemical biosensor. Angew. Chem. Int. Ed. 2019; 58:17399–17405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li Y., Li T., Liu B.-F., Hu R., Zhu J., He T., Zhou X., Li C., Yang Y., Liu M.. CRISPR-Cas12a: trans-cleaves DNA G-quadruplexes. Chem. Commun. 2020; 56:12526–12529. [DOI] [PubMed] [Google Scholar]
- 39. Smith C.W., Nandu N., Kachwala M.J., Chen Y.-S., Uyar T.B., Yigit M.V.. Probing CRISPR-Cas12a nuclease activity using double-stranded DNA-Templated fluorescent substrates. Biochemistry. 2020; 59:1474–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang D., Yan Y., Que H., Yang T., Cheng X., Ding S., Zhang X., Cheng W.. CRISPR/Cas12a-mediated interfacial cleaving of hairpin DNA reporter for electrochemical nucleic acid sensing. ACS Sensors. 2020; 5:557–562. [DOI] [PubMed] [Google Scholar]
- 41. Mohanraju P., Oost J., Jinek M., Swarts d. Heterologous expression and purification of the CRISPR-Cas12a/Cpf1 protein. Bio-protocol. 2018; 8:e2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marques M.-C., Ruiz R., Montagud-Martinez R., Marquez-Costa R., Albert S., Domingo-Calap P., Rodrigo G.. CRISPR-Cas12a-Based detection of SARS-CoV-2 harboring the E484K mutation. ACS Synth. Biol. 2021; 10:3595–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mergny J.L., Lacroix L.. Analysis of thermal melting curves. Oligonucleotides. 2003; 13:515–537. [DOI] [PubMed] [Google Scholar]
- 44. Idili A., Ricci F., Vallée-Bélisle A.. Determining the folding and binding free energy of DNA-based nanodevices and nanoswitches using urea titration curves. Nucleic Acids Res. 2017; 45:7571–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Swarts D.C., van der Oost J., Jinek M.. Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR-Cas12a. Mol. Cell. 2017; 66:221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mongan J., Simmerling C., McCammon J.A., Case D.A., Onufriev A.. Generalized born model with a simple, robust molecular volume correction. J. Chem. Theory Comput. 2007; 3:156–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Palermo G., Miao Y., Walker R.C., Jinek M., McCammon J.A.. CRISPR-Cas9 conformational activation as elucidated from enhanced molecular simulations. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:7260–7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Palermo G., Ricci C.G., Fernando A., Basak R., Jinek M., Rivalta I., Batista V.S., McCammon J.A.. Protospacer adjacent motif-induced allostery activates CRISPR-Cas9. J. Am. Chem. Soc. 2017; 139:16028–16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Palermo G., Miao Y., Walker R.C., Jinek M., McCammon J.A.. Striking plasticity of CRISPR-Cas9 and key role of non-target DNA, as revealed by molecular simulations. ACS Cent. Sci. 2016; 2:756–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. East K.W., Newton J.C., Morzan U.N., Narkhede Y.B., Acharya A., Skeens E., Jogl G., Batista V.S., Palermo G., Lisi G.P.. Allosteric motions of the CRISPR-Cas9 HNH nuclease probed by NMR and molecular dynamics. J. Am. Chem. Soc. 2020; 142:1348–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Palermo G., Chen J.S., Ricci C.G., Rivalta I., Jinek M., Batista V.S., Doudna J.A., McCammon J.A.. Key role of the REC lobe during CRISPR-Cas9 activation by ‘sensing’, ‘regulating’, and ‘locking’ the catalytic HNH domain. Q. Rev. Biophys. 2018; 51:e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Casalino L., Nierzwicki L., Jinek M., Palermo G.. Catalytic mechanism of non-target DNA cleavage in CRISPR-Cas9 revealed by ab initio molecular dynamics. ACS Catal. 2020; 10:13596–13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Turq P., Lantelme F., Friedman H.L.. Brownian dynamics: its application to ionic solutions. J. Chem. Phys. 1976; 66:3039–3044. [Google Scholar]
- 54. Van Zundert G.C.P., Rodrigues J.P.G.L.M., Trellet M., Schmitz C., Kastritis P.L., Karaca E., Melquiond A.S.J., Van Dijk M., De Vries S.J., Bonvin A.M.J.J.. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016; 428:720–725. [DOI] [PubMed] [Google Scholar]
- 55. Lippert R.A., Predescu C., Ierardi D.J., Mackenzie K.M., Eastwood M.P., Dror R.O., Shaw D.E.. Accurate and efficient integration for molecular dynamics simulations at constant temperature and pressure. J. Chem. Phys. 2013; 139:164106. [DOI] [PubMed] [Google Scholar]
- 56. Braselmann E., Rathbun C., Richards E.M., Palmer A.E.. Illuminating RNA biology: tools for imaging RNA in live mammalian cells. Cell Chem. Biol. 2020; 27:891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tyagi S. Imaging intracellular RNA distribution and dynamics in living cells. Nat. Methods. 2009; 6:331–338. [DOI] [PubMed] [Google Scholar]
- 58. Kim Y., Sohn D., Tan W.. Molecular beacons in biomedical detection and clinical diagnosis. Int. J. Clin. Exp. Pathol. 2008; 1:105–116. [PMC free article] [PubMed] [Google Scholar]
- 59. Zanut A., Rossetti M., Marcaccio M., Ricci F., Paolucci F., Porchetta A., Valenti G.. DNA-Based nanoswitches: insights into electrochemiluminescence signal enhancement. Anal. Chem. 2021; 93:10397–10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rossetti M., Ippodrino R., Marini B., Palleschi G., Porchetta A.. Antibody-Mediated small molecule detection using programmable DNA-Switches. Anal. Chem. 2018; 90:8196–8201. [DOI] [PubMed] [Google Scholar]
- 61. Porchetta A., Ippodrino R., Marini B., Caruso A., Caccuri F., Ricci F.. Programmable nucleic acid nanoswitches for the rapid, single-step detection of antibodies in bodily fluids. J. Am. Chem. Soc. 2018; 140:947–953. [DOI] [PubMed] [Google Scholar]
- 62. Mao S., Ying Y., Wu R., Chen A.K.. Recent advances in the molecular beacon technology for live-cell single-molecule imaging. Iscience. 2020; 23:101801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vallée-Bélisle A., Ricci F., Plaxco K.W.. Thermodynamic basis for the optimization of binding-induced biomolecular switches and structure-switching biosensors. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:13802–13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Farag N., Mattossovich R., Merlo R., Nierzwicki Ł., Palermo G., Porchetta A., Perugino G., Ricci F.. Folding-upon-Repair DNA nanoswitches for monitoring the activity of DNA repair enzymes. Angew. Chem. Int. Ed. 2021; 60:7283–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mocenigo M., Porchetta A., Rossetti M., Brass E., Tonini L., Puzzi L., Tagliabue E., Triulzi T., Marini B., Ricci F.et al.. Rapid, cost-effective peptide/nucleic acid-based platform for therapeutic antibody monitoring in clinical samples. ACS Sensors. 2020; 5:3109–3115. [DOI] [PubMed] [Google Scholar]
- 66. Mariottini D., Idili A., Nijenhuis M.A.D., De Greef T.F.A., Ricci F.. DNA-Based nanodevices controlled by purely entropic linker domains. J. Am. Chem. Soc. 2018; 140:14725–14734. [DOI] [PubMed] [Google Scholar]
- 67. Mariottini D., Idili A., Nijenhuis M.A.D., Ercolani G., Ricci F.. Entropy-Based rational modulation of the pKa of a synthetic pH-Dependent nanoswitch. J. Am. Chem. Soc. 2019; 141:11367–11371. [DOI] [PubMed] [Google Scholar]
- 68. Idili A., Ricci F., Vallée-Bélisle A.. Determining the folding and binding free energy of DNA-based nanodevices and nanoswitches using urea titration curves. Nucleic Acids Res. 2017; 45:7571–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Swarts D.C., Jinek M.. Mechanistic insights into the cis- and trans-Acting DNase activities of cas12a. Mol. Cell. 2019; 73:589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Stella S., Mesa P., Thomsen J., Paul B., Alcón P., Jensen S.B., Saligram B., Moses M.E., Hatzakis N.S., Montoya G.. Conformational activation promotes CRISPR-Cas12a catalysis and resetting of the endonuclease activity. Cell. 2018; 175:1856–1871. [DOI] [PubMed] [Google Scholar]
- 71. Saha A., Arantes P.R., Hsu R.V., Narkhede Y.B., Jinek M., Palermo G.. Molecular dynamics reveals a DNA-Induced dynamic switch triggering activation of CRISPR-Cas12a. J. Chem. Inf. Model. 2020; 60:6427–6437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Van Zundert G.C.P., Rodrigues J.P.G.L.M., Trellet M., Schmitz C., Kastritis P.L., Karaca E., Melquiond A.S.J., Van Dijk M., De Vries S.J., Bonvin A.M.J.J.. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016; 428:720–725. [DOI] [PubMed] [Google Scholar]
- 73. Rodrigues J.P.G.L.M., Bonvin A.M.J.J.. Integrative computational modeling of protein interactions. FEBS J. 2014; 281:1988–2003. [DOI] [PubMed] [Google Scholar]
- 74. Hacisuleyman E., Hale C., Saito Y., Blachere N.E., Bergh M., Conlon E.G., Schaefer-Babajew D.J., DaSilva J., Muecksch F., Gaebler C.et al.. Vaccine breakthrough infections with SARS-COV-2 variants. N. Engl. J. Med. 2021; 384:2212–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bell R.L., Jarvis K.G., Ottesen A.R., Mcfarland M.A., Brown E.W.. Recent and emerging innovations in Salmonella detection: a food and environmental perspective. Microb. Biotechnol. 2016; 9:279–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rodriguez-Lazaro D., Gonzalez-García P., Delibato E., De Medici D., García-Gimeno R.M., Valero A., Hernandez M.. Next day Salmonella Spp. detection method based on real-time PCR for meat, dairy and vegetable food products. Int. J. Food Microbiol. 2014; 184:113–120. [DOI] [PubMed] [Google Scholar]
- 77. Malorny B., Paccassoni E., Fach P., Bunge C., Martin A., Helmuth R.. Diagnostic real-time PCR for detection of Salmonella in food. Appl. Environ. Microbiol. 2004; 70:7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.