


Abstract
Although originally described as transcriptional activator, SPI1/PU.1, a major player in haematopoiesis whose alterations are associated with haematological malignancies, has the ability to repress transcription. Here, we investigated the mechanisms underlying gene repression in the erythroid lineage, in which SPI1 exerts an oncogenic function by blocking differentiation. We show that SPI1 represses genes by binding active enhancers that are located in intergenic or gene body regions. HDAC1 acts as a cooperative mediator of SPI1-induced transcriptional repression by deacetylating SPI1-bound enhancers in a subset of genes, including those involved in erythroid differentiation. Enhancer deacetylation impacts on promoter acetylation, chromatin accessibility and RNA pol II occupancy. In addition to the activities of HDAC1, polycomb repressive complex 2 (PRC2) reinforces gene repression by depositing H3K27me3 at promoter sequences when SPI1 is located at enhancer sequences. Moreover, our study identified a synergistic relationship between PRC2 and HDAC1 complexes in mediating the transcriptional repression activity of SPI1, ultimately inducing synergistic adverse effects on leukaemic cell survival. Our results highlight the importance of the mechanism underlying transcriptional repression in leukemic cells, involving complex functional connections between SPI1 and the epigenetic regulators PRC2 and HDAC1.
Graphical Abstract
Graphical Abstract.
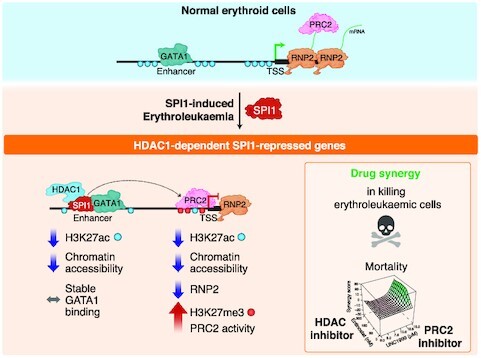
Abnormal high SPI1 expression leads to HDAC1-mediated transcriptional repression by deacetylating SPI1-bound enhancers that are GATA1 targets in erythropoiesis. PRC2 and HDAC1 synergize to mediate SPI1 repressive activity that enables leukaemic cell survival and expansion.
INTRODUCTION
Transcriptional regulation is a complex process that involves an array of protein activities in a specific chromatin context. Transcription factors (TFs) are major contributors to this process that act with partners, coactivators or epigenetic factors, some of which, known as pioneer TFs, are able to render chromatin structures permissive to coactivators and epigenetic factors. The epigenetic landscape plays a major role in haematopoietic homeostasis and the differentiation programme; therefore, it has been possible to construct a complete model of haematopoiesis from chromatin dynamics (1,2). Mutations of genes encoding epigenetic modifiers (TET2, IDH1/2, DNMT3A and ASXL1) are common in acute myeloid leukaemia (AML) patients, further indicating that this type of component plays a major role in driving AML development.
The TF SPI1/PU.1 belongs to the E26 transformation-specific (ETS) family and is a major contributor to haematopoietic control, playing an active role in myeloid and B lymphoid lineage specification and differentiation (3–5). SPI1 was originally described as a transcriptional activator and is considered a pioneer TF, since it has the ability to bind to or near closed nucleosome conformations and enable cofactors to bind the chromatin (6–9). For example, in macrophages, SPI1 activates the transcription of its target genes by binding to closed chromatin, where it evicts nucleosomes through recruitment of epigenetic modifiers, such as CBP/P300 or SWI/SNF complexes (6,7,10,11). This action gives instructions to create a new enhancer with monomethylation of lysine 4 of histone 3 (H3K4me1) and to recruit additional TFs at enhancer sites (6,7). The function of SPI1 in the control of transcriptional activation through epigenetic regulation has also been described in B lymphoid and osteoclast differentiation (12,13). Thus, in addition to playing well-known roles in controlling gene expression in coordination with lineage-determining cofactors, the effect of SPI1 on transcriptional activity is mediated in cooperation with epigenetic regulators. The role of SPI1 in repressing transcription has been recently reported in normal haematopoiesis, in controlling an appropriate neutrophil immune response (14), in early T cells (15,16) and in osteoclasts (12). Achieving a better understanding of how SPI1 represses gene expression is particularly relevant for such lineages as erythroid or T and B lymphoid cells, in which abnormally high SPI1 expression is oncogenic and whose transcriptional repression activity causes differentiation blockage (17–20).
SPI1 expression is fine-tuned during erythroid differentiation (21,22). A low level of SPI1 expression is required to maintain the pool of immature erythroid progenitors during foetal erythropoiesis and stress erythropoiesis in adults (21). Complete inhibition of the expression of SPI1 is required to enable terminal erythroid differentiation after the colony-forming-unit erythroid stage (CFU-E) (23,24). Maintaining elevated expression of SPI1 in mice leads to blockage of erythroid differentiation and favours survival of erythroid progenitors, initiating the erythroleukaemic process (17,23,24). Such high expression of transcriptional regulators, including SKI, ETO2 and ERG, was recently described in >25% of human acute erythroleukaemia (AEL) with a prominent ‘erythrocyte’ signature (25), thereby blocking erythroid differentiation. This finding emphasizes the need to understand their mode of action. All these TFs interfere with GATA1 activity, one of the major erythroid TFs. SPI1, interacting with GATA1 (26,27), creates a repressive histone context at GATA1 binding sites (28–30) or represses expression of GATA1 co-factors (31). Additionally, we showed that polycomb repressive complex 2 (PRC2), an epigenetic modifier complex that controls the repressive trimethylation of H3K27 (H3K27me3) (32,33), is involved in the transcriptional repression of the proapoptotic encoding gene Bcl2l11 by SPI1 in erythroleukaemia (23), providing further evidence of the action of SPI1 on the epigenetic landscape to repress genes.
In this study, we further investigated the mechanism underlying gene repression by SPI1. Specifically, we employed genome-wide analysis that combines characterization of transcriptional changes and the identification of SPI1 partners at the chromatin with epigenetic modifications of histones and chromatin accessibility. At least two types of SPI1-mediated repressive mechanisms have been highlighted: one involving modulation of histone acetylation at enhancer regions and another independent of such modulation. For the first one, we show that SPI1 cooperates with histone deacetylase 1 (HDAC1) to deacetylate the active enhancers it occupies, thereby reducing TSS acetylation, promoter–enhancer physical proximity, chromatin accessibility, RNA pol II occupancy and reducing transcription. Moreover, our results reveal a synergistic effect of HDAC1 and PRC2 in mediating SPI1-induced transcriptional repression and erythroid differentiation blockage, as well as a synergistic effect of inhibiting the two epigenetic modifiers in killing leukaemic cells. Taken together, our findings provide insight into the interplay between the SPI1 TF and two epigenetic regulators participating in the SPI1-induced transformation activity mediated by gene repression.
MATERIALS AND METHODS
Cell culture and chemicals
Leukaemic cells overexpressing SPI1 were derived from bone marrow of Spi1 transgenic mice (TgSpi1) with erythroleukemia (17) (mice #763, #722 and #683) and cultured in alpha Eagle's minimal essential medium (αMEM, Gibco) supplemented with 5% Fetal Bovine Serum gold (FBS-gold, PAA, A15-151), l-glutamine (2 mM, Gibco), penicillin/streptomycin (100 μg/ml, Gibco) and 1 U/ml of erythropoietin (EPO). The erythroleukaemic cells from #763 and #722 mice were engineered to express anti-Spi1 shRNAs in the presence of doxycycline (dox, 100 ng/ml) as previously described (24). All the experiments using cells were performed with cells derived from mice #763 and, when indicated, with cells from #722 and #683 mice, as biological replicates. For HDAC inhibition, cells were incubated with indicated doses of Entinostat (MS-275, Santa Cruz), or TMP195 (M6176, AbMole). PRC2 inhibition was performed using the EZH1/2 inhibitor UNC1999 (M3107, AbMole). DMSO was used as control.
Human embryonic kidney (HEK293T) cells were grown in Dulbecco modified Eagle's medium (Gibco) supplemented with 10% FBS (Gibco), l-glutamine (2 mM, Gibco), penicillin/streptomycin (100 μg/ml, Gibco). KG1, K562 and A20 cells were grown in Roswell Park Memorial Institute medium (RPMI, Gibco) supplemented with 10% FBS (Gibco), l-glutamine (2 mM, Gibco), penicillin/streptomycin (100 μg/ml, Gibco).
CRISPR-Cas9 genome editing
Using the CRISPR design tools CHOPCHOP (34) or CRISPOR (35), we identified single-guide RNAs (sgRNAs) to target Alas2 or St3gal6-specific regions. The sgRNAs were synthesized by Synthego (see Supplementary materials). Cas9 ribonucleoprotein (RNP) complexes were assembled in vitro by incubating Cas9 (6pmol, Synthego) with sgRNAs (15 pmol; 500 ng, Synthego) and siGLO-green (31pmol, Dharmacon). To generate the genomic deletion in epigenetically-defined enhancer, two different pairs of sgRNAs for Alas2 and one for St3gal6 (see Supplementary materials) were electroporated with the Neon transfection system into 105 cells (1 pulse of 1450 V; pulse width: 10 ms). FITC-positive cells were sorted (BD FACSAriaIII cell sorter, BD biosciences) and clonally plated into 96-well plates at a limiting dilution of 0.5 cells per well to avoid mixed clones. After 7 days of clonal expansion, 200 000 cells were collected for DNA extraction using QuickExtract DNA extraction solution (Epicentre) and amplification of 2 μl DNA was performed by using the Platinum II Taq Hot-Start DNA polymerase (Invitrogen) for Alas2 enhancer or Platinum SuperFi II DNA polymerase (Invitrogen) for the large deletion in St3gal6 enhancer. Clones were screened for deletion in Alas2 or St3gal6 enhancer by PCR using primers as indicated in Supplementary materials. For Alas2, three clones (L1, L8, M2) were generated using the first pair of sgRNAs, while for two clones (P5, N3) the second pair was used. The FIMO tool from the MEME-suite was used to find individual occurrences of the SPI1 binding motif in the Alas2 enhancer region using a previously reported SPI1 position weight matrix (36). To mutate the SPI1 DNA motif sequence in the Alas2 enhancer, one sgRNA was used as reported in the in Supplementary materials and genomic DNA amplified by PCR was sequenced by Sanger method (Eurofins Genomics).
Methylcellulose culture and benzidine differentiation assay
Two thousand cells were seeded in 1 ml MethoCult (StemCell, M3334) following the manufacturer's instruction. Colonies were analyzed at day 5 by staining with a solution containing 0.2% benzidine (Sigma-Aldrich) in 10% H2O2 (Sigma-Aldrich) and 0.5 M glacial acetic acid (Sigma-Aldrich). The colonies were observed under the microscope (Motic AE2000 and Moticam 1080BMH).
Flow cytometry staining
For cell surface marker staining, 5 × 105 cells were collected and washed in cold 1× D-PBS and 2% BSA and were incubated for 30 min at 4°C in 100 μl of cold 1× D-PBS, 2% BSA and 2 ng/μl PE rat anti-mouse CD71 antibody (BD Pharmingen #553267). After washing, stained cells were then resuspended in FACS buffer supplemented with 0.5 μM DAPI and analyzed by BD LSRFortessa flow cytometer. Flow cytometry data have been analyzed using FlowJo v10.8.
HDAC1/3 and PRC2 inhibitors synergy assay
Synergy experiments were performed by treating leukaemic cells with 0, 50, 150 or 300 nM of Entinostat combined with 0, 5, 7.5, 10, 12.5, 15 μM of UNC1999 for 48 h. Number of viable cells was determined with DAPI labeling (0.5 μM) in 96-well plates by BD High Throughput Sampler (HTS) option from the BD LSRFortessa flow cytometer. FACS data were analysed using FlowJo v10.6.2 software (Tree Star). Synergy evaluation was performed using the zero-interaction potency (ZIP) model (37) with R v3.6.3 through the Bioconductor package synergyfinder v2.0.5 (38). Synergy effect is expressed as a synergy score, or delta score (δ), that corresponds to no interaction (δ = 0), synergy (δ > 0) or antagonism (δ < 0) between the combined drugs.
Generation of Avi-tagged HDAC1
Hdac1 was amplified with flanking AscI (5′-CCCTCACTCGGCGCGatggcgcagacgcagggcaccc-3′) and NsiI (5′-AAGAGGCAGAATGCATGCTCGAGTTAACggccaacttgacctcctcc-3′) restriction sites from the HDAC1 Flag plasmid (gift from Eric Verdin, Addgene plasmid #13820, (39)) using the KAPA HiFi HotStart ReadyMix (Roche). Using the In-Fusion HD Cloning Kit (Clontech) Hdac1 was cloned into the double digested pRRLs-Avi-MCS-iGFP-BirA (40) resulting into pRRLs-Avi-HDAC1-iGFP-BirA. Competent GT115 Escherichia coli (Invivogen) were transfected via heat-shock, recovered in SOC Outgrowth Medium (New England Biolabs), plated on LB-Amp plates and incubated overnight. The presence of HDAC1 was tested via colony PCR and sequencing (Eurofins Genomics) (primers: Fwd 5′-CTGCTTCTCGCTTCTGTTCG-3′, Rev 5′-CACACCGGCCTTATTCCAAG-3′).
Lentiviral transduction
For lentiviral particles production, HEK293T were co-transfected with the Avi-GFP or HDAC1-Avi-GFP vectors carrying BirA biotin ligase with the lentiviral pMD2.G -VSVG and pCMV-Gag-Pol using jetPRIME reagent (Polyplus transfection). Lentiviral particles were harvested 48 h post-transfection and ultracentrifuged for 90 min at 80 000g. Leukaemic cells were transduced with lentiviral particles for 24 h before sorting on BD FACSAria III cell sorter. GFP positivity was checked before each experiment.
Immunoprecipitation, pull-down, protein extraction and immunoblotting
Avi-tagged protein technology is based on the biotinylation of Avi-Tag by the BirA biotin ligase and on the specific binding of streptavidin to biotin for pull-down.
40 × 106 cells for immunoprecipitation or 15 × 106 cells for Avi-tagged protein pull-down were lysed in 100 μl IP buffer per 107 cells (20 mM HEPES, 2 mM MgCl2, 0.5% NP-40, 0.1% Triton X-100) and incubated on ice for 5 min. NaCl was added at final concentration of 420 mM to the lysates, incubated on ice for 5 min and centrifuged for 15 min at 20 000g.
Supernatants were incubated 2 h with antibody of interest (Supplementary data, Table of antibodies). Then, for immunoprecipitation, 50 μl of magnetic beads were added for 1 h (Protein G coated Dynabeads, Invitrogen) or, for pull-down, 60 μl of streptavidin-coated beads (M-280 Dynabeads, Invitrogen) were added for 2 h. Beads were washed 5 times with IP-buffer and boiled for 10 min at 95°C in 25 μl Laemmli buffer 2× supplied with DTT.
Whole-cell extracts or immunoprecipitated/pulled-down proteins were resolved by SDS-PAGE and transferred to a nitrocellulose 0.22 μm membrane (BioRad). Membranes were blocked (1× D-PBS, 0.1% Tween20, 5% non-fat milk powder) and then incubated with primary antibody as described in Supplementary data (table of antibodies). Horse peroxidase-conjugated secondary antibody was used to detect proteins by LAS4000 digital imager (GE Healthcare Life Sciences) upon incubation with enhancer chemiluminescence kit (ThermoScientic). Stripping was performed in 1× stripping buffer (ST010, GeBa) according to manufacture instructions.
Recombinant proteins were visualized on acrylamide gels either by Fast Coomassie blue staining protocol as previously described (41) or by activation of Mini-PROTEAN 4–15% TGX Stain-Free precast gels (BioRad) by UltraViolet 302nm on GelDoc XR+ (BioRad) following manufacturer's instructions.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed using the ChIP assay kit (Millipore) following the manufacturer's protocol as previously described (36). ChIP samples were sonicated for 17 min using the Covaris S220 sonicator (peak power 220, duty factor 20 and cycle Burst 200) was used for sonication. For more details on antibodies and protein A/G coupled beads choice, see Supplementary data. ChIP were either sequenced or quantified by real-time quantitative PCR (RT-qPCR). ChIP experiments were repeated at least three times when analysed by quantitative PCR.
Quantitative PCR was performed on the 7500 RT-qPCR System (Applied Biosystems) using primers listed in Supplementary data and SYBR-green buffer (Applied Biosystems). Immunoprecipitated DNA is compared to input sample by the comparative Ct method. Immunoprecipitated DNA enrichment is expressed as % of input.
CUT&Tag assay, library preparation and sequencing
The HDAC1 CUT&Tag assay and library was performed using CUT&Tag-IT Assay Kit (Active Motif) following the manufacturer's protocol on 0.5 × 106 leukaemic cells. The libraries were sequenced on NovaSeq 6000 system to obtain 10 × 106 paired-end reads (50 bp) following Illumina's instructions.
In vitro PRC2 methyltransferase assay
Recombinant histone H3.1 (31894, Active Motif) was mixed in PRC2 buffer (50 mM Tris pH 8, 100 mM NaCl, 1 mM DTT, 0.01% Triton X-100) with or without 200 μM S-adenosyl methionine (SAM, Sigma Aldrich), PRC2 recombinant complex (31387, Active Motif) and SPI1 recombinant protein (MBS1265403, MyBioSource) at 1:1 or 5:1 SPI1:PRC2 molar ratios for 4 h. Recombinant PRC2 complex includes full length EZH2, SUZ12, EED and RBAP46/48 (accession numbers NP_001190176.1, NP_056170, NP_003788.2, NP_002884.1 and NP_005601.1, respectively). It was expressed in Sf9 and contains an N-terminal FLAG-Tag at the N-terminus of EZH2. Recombinant SPI1 was expressed in E. coli and contains ×6 N-terminal His-Tag (accession number NP_001074016.1). BSA (Sigma Aldrich) was used as a control protein. Combination of concentrations used are: (a) 500 ng H3.1, 73 nM PRC2 plus 73 nM SPI1 (1:1 molar ratio) or 365 nM SPI1 (5:1 molar ratio); (b) 250 ng H3.1, 36.5 nM PRC2 plus 36.5 nM SPI1 (1:1 molar ratio) or 182.5 nM SPI1 (5:1 molar ratio). Samples were separated by SDS-PAGE or directly dotblotted onto nitrocellulose membrane using Bio-Dot microfiltration apparatus (BioRad). Membranes were incubated with anti-H3K27me3 and HRP secondary antibody as described above. Specificity of the antibody used was verified with. Quantification of the signal intensity was performed by Fiji/ImageJ v2.1.0 software.
Reverse phase ultra-fast liquid chromatographic (RP-UFLC)-based separation and quantification of the fluorescein-labeled peptide H3K27me1
A 11-amino-acid peptide derived from the histone H3.1 protein centered around the lysine 27 residue was synthetized, conjugated to fluorescein amidite (FAM) on its N-terminus and modified by amidation (NH2) on its C-terminus (Proteogenix). A K27 mono-methylated form of this peptide (H3K27me1) was also synthesized and used as a standard. The peptide sequences used are: (a) H3K27: FAM-TKAARK27SAPAT-NH2; (b) H3K27me1: FAM-TKAARK27me1SAPAT-NH2.
The methyltransferase reaction was carried out overnight in PRC2 buffer (50 mM Tris pH 8, 100 mM NaCl, 1 mM DTT, 0.01% Triton X-100) containing 75 μM of H3K27 peptide, 36.5 nM of PRC2 recombinant complex (acm31387, Active Motif), 100 μM of S-adenosyl methionine (SAM, Sigma Aldrich) and different molar ratios of SPI1 to PRC2 (MBS1265403, MyBioSource) or BSA (Sigma Aldrich). The reaction was stopped with perchloric acid (HClO4) (15% v/v in H2O). Samples were separated by RP-UFLC (Shimadzu) using Kromasil 100-5-C18 column, 5μm particle size at 40°C. The mobile phase used for the separation consisted of a mix of solvent A containing water with 0.1% perchloric acid (HClO4) and solvent B containing acetonitrile with 0.12% trifluoacetic acid (TFA). Separation was performed by an isocratic flow as followed: 80% solvent A + 20% solvent B, rate of 1 ml/min, run time of 30 min. H3K27 peptide and its methylated product were monitored by fluorescence emission (λ = 530 nm) after excitation at λ = 485 nm and quantified by integration of the peak absorbance area.
RNA extraction and quantification by real-time quantitative PCR (RT-qPCR)
Total RNA was isolated from cells using RNeasy Plus Mini Kit (Qiagen) and 1 μg was reverse-transcribed using Superscript IV Reverse Transcriptase (Invitrogen). RT-qPCR was performed using TaqMan Gene Expression Assays (Applied Biosystems) on the 7500 RT-qPCR System (Applied Biosystems) with the probes listed in Supplementary data. Changes in mRNA expression levels were calculated relative to untreated control condition and normalized to Polr2a mRNA level using the 2–ΔΔCt formula.
RNA library preparation and sequencing
RNA quality was evaluated using an Agilent Fragment Analyser apparatus as described in the manufacturer's procedure (Agilent Technologies, Basel, Switzerland). RNA-seq libraries were generated from 300 ng of total RNA Illumina TruSeq RNA Sample Preparation Kit v2 (Part Number RS-122-2001). Libraries were sequenced on Illumina HiSeq 2500 or 4000 as 100 bp paired-end reads following Illumina's instructions. Sequencing was performed to obtain at least 5.5 × 107 reads for each sample. Sequence reads were mapped onto mm10/GRCm38 assembly of mouse genome using STAR v2.5.3a (42).
ChIP library preparation and sequencing
ChIP samples were purified using Agencourt AMPure XP beads (Beckman Coulter). ChIP-seq libraries were prepared from 5 ng of double-stranded purified DNA using the MicroPlex Library Preparation kit v2 (C05010014, Diagenode s.a.).
The libraries were sequenced on an Illumina Hiseq2500 or Hiseq4000 sequencer as 100 bp (for SPI1, RNA-pol II and H3K27ac upon Entinostat treatment) or 50 bp (for H3K27me3, H3K4me3, H3K4me1 and H3K27ac upon dox treatment) Paired-End reads following Illumina's instructions. Image analysis and base calling were performed using RTA v2.7.7 and bcl2fastq v2.17.1.14. Adapter dimer reads were removed using DimerRemover. Reads were aligned to the mouse reference genome (GRCm38/mm10) using Burrows-Wheeler Aligner (BWA) (bwa-mem algorithm) (version: 0.7.17-r1188) (43). Peak calling was performed using HMCan (44). Signals from two comparative conditions have been normalized by CHIPIN v0.1.0. (45). More details are described in Supplemental data. Two independent experiments of H3K27ac and H3K27me3 were performed.
ATAC assay, library preparation and sequencing
5 × 104 cells frozen in culture media containing FBS and 10% DMSO were sent to Active Motif to perform the ATAC-seq assays. Cells were washed with cold PBS, and tagmented using the Omni-ATAC protocol described in (46), using the enzyme and buffer provided in the Nextera Library Prep Kit (Illumina). Tagmented DNA was purified using the MinElute PCR purification kit (Qiagen), amplified with 10 cycles of PCR, and purified using Agencourt AMPure SPRI beads (Beckman Coulter). Resulting material was quantified using the KAPA Library Quantification Kit for Illumina platforms (KAPA Biosystems), and sequenced with PE42 sequencing on the NextSeq 500 sequencer (Illumina). Two independent experiments were performed.
Bioinformatic data analysis
Details for bioinformatic analyses are described in Supplemental data.
RESULTS
SPI1 represses gene transcription through binding to active enhancers
To characterize the process of gene repression mediated by SPI1 in erythroleukaemia, we performed RNA-seq on erythroleukaemic cells derived from a sick TgSpi1 mouse (#763) and secondarily engineered to produce inducible shRNA against Spi1 after dox treatment (24). Forty-eight hours of dox treatment reduced the SPI1 protein expression level by up to 90% (Figure 1A), and SPI1 was no longer detected on the chromatin (Figure 1B). We have previously characterized the SPI1 oncogenic functions in the murine erythroleukaemic process and shown that decrease of SPI1 expression in those leukemic cells re-instates erythroid differentiation and reduces expansion of leukemic population by inducing apoptosis, establishing that SPI1 initiates leukemia by blocking differentiation and cellular expansion (17,23,24).
Figure 1.

SPI1 represses gene transcription by binding to active enhancers in leukaemic cells. (A) SPI1 expression was determined by immunoblotting of lysates of leukaemic cells (#763) treated for 48 or 72 h with or without doxycycline (dox) to induce the expression of Spi1 shRNA. HSC70 was used as loading control. (B) Heatmap of SPI1-normalized ChIP-seq reads performed in leukaemic cells treated (SPI1–) or not (SPI1+) with dox for 48 h, sorted according to the decreasing signal intensity in SPI1+ cells. (C) SPI1 ChIP-seq data were crossed with RNA-seq data to identify the percentage of genes that are bound by SPI1 among activated (gene expression relative to SPI1– cells ≥1.5, P < 0.05), repressed (gene expression relative to SPI1– cells ≤ 0.67, P < 0.05), or NoResp (0.9 ≤ gene expression relative to SPI1– cells ≤ 1.1, P ≥ 0.05) genes. (D, E) Top 10 enriched biological processes identified by DAVID functional analysis (P < 0.05) and ranked by P-value (red scale) using repressed (D) or activated (E) genes that are bound (upper panel) or not (lower panel) by SPI1. Gray scale indicates the relative enrichment of a corresponding biological process. (F) Enrichment of transcriptional categories in each genomic region compared to the total number of SPI1-bound genes in the indicated genomic region for repressed or NoResp genes using Fisher's test. * P < 0.05, ** P < 0.01. (G) UpSet plot representing the number of genes with SPI1 peaks at single or multiple genomic regions as indicated by the black dots. Horizontal stack plots show the number of genes with one peak in a specific region that are associated with additional peaks in other genomic regions. (H) Doughnut chart showing the percentage of active enhancers (with H3K27ac and H3K4me1 marks), inactive enhancers (only H3K4me1 mark) or Not enhancers (without H3K27ac or H3K4me1 marks) at SPI1-bound intergenic and gene body regions of SPI1-repressed genes. (I) Top: Alas2 genomic locus with SPI1, H3K27ac, H3K4me1 ChIP-seq tracks, the sgRNA target site (scissors and gray bar) and putative SPI1-binding DNA sequences at Alas2 enhancer of Control (Ctrl) and mutant clones. PAM: Protospacer Adjacent Motif. Bottom-left: SPI1 ChIP-qPCR in the controls (Ctrl1 and 2) and 2 clones (MC2 and MC8) deleted for the SPI1 binding site at Alas2 enhancer. Ah1 enhancer site is an untargeted positive control bound by SPI1. Mean % Input ± SEM of 4 independent experiments is shown. Bottom-right: RT-qPCR analysis of the Alas2 and Spi1 transcript expression in the two controls and two independent clones. Data represent the mean ± SEM of expression relative to Ctrl1 normalized to Hprt1 and Polr2a gene expression of three independent experiments. * P < 0.05, *** P < 0.001, ns: not significant, two-tailed Student's t-test.
We identified 1579 differentially expressed genes (DEGs) between dox-untreated (SPI1+) and dox-treated cells (SPI1–) for 48 h; 972 genes were activated and 607 were repressed in the presence of SPI1 (Supplementary Figure S1A and Supplementary Table S1). Using SPI1 ChIP-seq results obtained from TgSpi1 erythroleukaemic cells (36), we assigned to genes peaks within 50 kb upstream of the transcriptional start site (TSS) and 3 kb downstream of the transcription end site (TES) (Supplementary Table S2). The fraction of genes bound by SPI1 was higher among SPI1-repressed (57%) and SPI1-activated (66%) genes than among NoResp genes (25%) (Figure 1C). DAVID gene ontology enrichment analyses for repressed genes with SPI1 bound to chromatin demonstrated transcriptional misregulation in erythroid differentiation processes, but this phenomenon was not observed for genes not bound by SPI1 (Figure 1D and E).
Taken together, these results indicate that similar to gene activation, SPI1 chromatin occupancy is associated with repression of gene expression.
To characterize how SPI1 bound to chromatin reduces gene expression, we first investigated whether the repression capacity of SPI1 was associated with the nature of the interacting subgenomic region. Peak annotation was performed using ChIPseeker across the six following genomic subregions of gencode-annotated genes: intergenic regions (−50 kb; −2 kb), the distal promoter (−2 kb; −1 kb), the immediate upstream promoter (−1 kb; TSS), the immediate downstream promoter (TSS; +1 kb), the gene body (+1 kb; TES), and the downstream region (TES; TES + 3 kb) (Figure 1F). By computing the odds ratios and significance using Fisher's test, we evaluated the association between two events, that are, the transcriptional status (repressed or NoResp) and the SPI1-occupied genomic subregion. SPI1-repressed genes exhibited a strong and significant enrichment for SPI1 peaks located in intergenic regions (×1.5), in the distal promoter (×1.8) and inside the gene body (×2.5), with no enrichment being observed close to TSS (from −1 kb to +1 kb). Considering the number of SPI1 peaks in each subregion of repressed genes, we also found that the large majority of peaks were located in intergenic regions and gene body regions (Supplementary Figure S1B). A total of 226 out of the 325 SPI1-bound genes contained a peak in the gene body, of which 72% (163/226) displayed no other peak elsewhere and 13% (30/226) showed another peak in the intergenic region (Figure 1G). Sixty-nine genes displayed a SPI1 peak in the intergenic region, of which 45% (35/69) displayed no other peak elsewhere and 43% (30/69) showed another peak in the gene body. These findings indicate that in the majority of repressed genes, SPI1 binds only to one location. Furthermore, in the repressed genes, the peaks located in intergenic regions were distributed near the promoter, while the peaks located inside the gene body were more homogenously distributed across this region (Supplementary Figure S1C). Due to the low number of genes repressed and bound by SPI1 in the distal promoter region (14/325) (Supplementary Figure S1B), we excluded this category for further study.
These results suggest that the gene body and intergenic regions are two regions that play key roles in the transcriptional repressing effect exerted by SPI1.
Next, we explored whether the gene body and intergenic regions bound by SPI1 in repressed genes are potential distal cis-regulatory elements (DREs) using the presence of two chromatin marks, histone 3 lysine 27 acetylation (H3K27ac) and lysine 4 mono-methylation (H3K4me1), identified by ChIP-seq in two erythroleukaemic cells derived from sick mice (#722 and #763). Using ChromHMM, we established the three following chromatin statuses: regions presenting only H3K4me1 marks are defined as ‘inactive enhancers’, regions presenting both H3K4me1 and H3K27ac are ‘active enhancers’, and regions with neither of the two are called ‘non-enhancers’. A total of 90% of the SPI1 peaks located in intergenic and gene body regions of repressed genes, contained H3K4me1 peaks compatible with enhancer functions, for both #763 and #722 leukaemic cells, SPI1 peaks of which 53% and 58%, respectively, were active (Figure 1H and Supplementary Figure S1D). In agreement with our annotation, the region bound by SPI1 in the intron 1 of the Alas2 gene that we defined to be active enhancer on the basis of epigenetic marks was previously described to be indeed a functional cis-regulatory region of the Alas2 gene by CRISPR/Cas9 deletion in normal murine proerythroblast-like cells (47). Deleting this enhancer region in the TgSpi1 erythroleukemic blasts using CRISPR-Cas9 system strongly reduced Alas2 gene expression, validating the functionality of this enhancer region in the leukemic cells (Supplementary Figure S1E and Supplementary materials). Cas9-mediated deletion of the St3gal6 region identified as enhancer with H3K4me1 and H3K27ac and bound by SPI1 was also used to examine the functionality of the annotated enhancer region (Supplementary Figure S1F and Supplementary materials). The reduction of St3gal6 mRNA expression associated with the deletion of this genomic region confirmed that this region was an active enhancer. These data validated our approach of defining active enhancers based on epigenetic information in erythroid cells.
To rigorously test whether SPI1 represses gene expression by directly binding to DNA, we used CRIPSR-Cas9 to generate erythroleukemic cells lacking a SPI1 DNA motif in the Alas2 enhancer bound by SPI1 (Figure 1I). Individual occurrences of SPI1 binding motif in the Alas2 enhancer were identified with FIMO (Figure 1I). We selected two clones that have a deletion of a SPI1 binding motif (Table in Supplementary materials and Figure 1I). The SPI1 motif deletion reduced SPI1 occupancy at Alas2 enhancer in the two cell lines compared to the control cells. Such a decrease in SPI1 binding was associated with an increase in Alas2 mRNA expression relative to controls. This result, which links the efficiency of SPI1 binding to an enhancer with the level of gene repression, provides strong evidence, at least for a portion of genes, that SPI1 represses gene transcription through direct binding to DNA.
Taken together, these results suggest that SPI1 represses gene expression primarily by binding distal regulatory elements, many of which are active enhancers in the intergenic or gene body regions of the repressed genes.
SPI1 and HDAC1 cooperate to repress SPI1 target genes
The SPI1 protein does not contain any transcriptional repression domain (48) but interacts with several cofactors that may mediate its repressive activity. To identify putative SPI1 corepressors acting when SPI1 is bound to DNA, we performed SPI1 ChIP experiments followed by mass spectrometry (RIME) (49). We identified 58 proteins that were reproducibly immunoprecipitated with SPI1 and not in the IgG control (Supplementary Figure S2A). In particular, FUS/TLS, a well-described interacting factor involved in SPI1 splicing activity (50,51), and the SWI/SNF complex, which is also known to interact with SPI1 (52), were recovered, validating the SPI1-RIME approach. Notably, we found peptides for two epigenetic factors involved in transcriptional repression, chromodomain helicase DNA binding protein 4 (CHD4) and histone deacetylase 1 (HDAC1). HDAC1 plays a catalytic role as a deacetylase in 4 different complexes, namely, NuRD (including CHD4), SIN3A, CoREST and SHIP complexes (53).
We validated the interaction of SPI1 with HDAC1 in leukaemic cells engineered to produce Avi-tagged HDAC1 (Supplementary Figure S2B) and with endogenous HDAC1 (Figure 2A). Moreover, to verify that the interaction between SPI1 and HDAC1 was efficient at the chromatin, we performed SPI1 ChIP followed by western blotting coupled with HDAC1 or SPI1 antibodies and showed that they indeed formed a complex at chromatin (Figure 2B). SPI1, SIN3A and HDAC1 seem to belong to the same complex as deduced from their co-immunoprecipitations with SIN3A and HDAC1 (Figure 2A). SPI1 also interacts with CHD4 belonging to the NuRD complex in the erythroleukaemic TgSpi1 cells (Supplementary Figure S2C). We ensured that HDAC1 expression was not affected by the level of SPI1 expression in erythroleukaemic cells at the RNA (Supplementary Figure S2D) or protein levels (Figure 2C).
Figure 2.

SPI1 interacts with HDAC1 to repress gene expression. (A) Protein lysates were immunoprecipitated (IP) with antibodies against HDAC1, SIN3A and IgG (negative control) and immunoblotted with antibodies against SIN3A, HDAC1, SPI1 and HSP90β (negative control). The membrane of the input samples was cut for ECL detection, as indicated by the vertical bar. (B) ChIP performed using SPI1 antibody was immunoblotted with antibodies against HDAC1, SPI1 and STAT3 (negative control). The HDAC1 input signal was from a lower exposure than IP, as indicated by the vertical bar. (C) Leukaemic cells were treated for 48 h with or without dox and for additional 6 h with HDAC1/3 inhibitor (Entinostat, 3 μM) or DMSO. Whole-cell extracts were analyzed by immunoblotting with antibodies against HDAC1, SPI1, H3K27ac and H4 (loading control). (D, I) Venn diagram showing the overlapping genes bound by SPI1 at enhancers that are repressed (D) or activated (I) by SPI1 (blue) or by HDAC1 (green). RNA expression was measured by RNA-seq (3 independent experiments). (E, F) Full (•) and empty (○) circles indicate the presence or absence (dox treatment), respectively, of SPI1 expression or HDAC1/3 activity (Entinostat treatment). (E) Violin plots showing the distribution of changes in mRNA expression by SPI1 depletion (○) in the presence of normal HDAC1/3 activity (blue) or in the absence of HDAC1/3 activity (purple) of the genes repressed and bound by SPI1 at enhancers. (F) Violin plots showing the distribution of changes in mRNA expression by HDAC1/3 inhibition (○) in the presence of SPI1 overexpression (green) or in the absence of SPI1 (violet) of the genes repressed and bound by SPI1 at enhancers. (G) Mean of relative expression ± SEM of RNA-seq of three experiments for the same comparison as those described in (E and F) illustrated for four SPI1-repressed genes bound at enhancers. (H) Violin plots showing the distribution of changes in mRNA expression by SPI1 depletion (blue) or by HDAC1/3 inhibition (green) of the genes activated and bound by SPI1 at enhancers.
HDAC1 plays a critical role in the suppression of gene transcription but can also upregulate specific gene transcription (54). To define whether HDAC1 is a mediator of SPI1 repressive activity on transcription, we analysed the consequences of inhibiting HDAC1 on the transcription of SPI1 targets. The leukaemic cells producing dox-inducible Spi1-shRNA were treated with dox for 48 h alone and with 6 h of treatment concomitant with a pharmacological inhibitor of HDAC1, entinostat (Figure 2C). The efficiency of entinostat was validated by the increase in H3K27 acetylation (Figure 2C). Importantly, the expression of HDAC1 was unaltered by entinostat treatment. RNA-seq was performed for the four following conditions: untreated leukaemic cells (SPI1+DMSO), leukaemic cells inhibited for HDAC1 activity (SPI1+Entinostat+), leukaemic cells downregulated for SPI1 (SPI1–DMSO), and leukaemic cells downregulated for SPI1 and inhibited for HDAC activity (SPI1–Entinostat+). The 60% of genes repressed and bound at enhancers by SPI1 were also repressed by HDAC1 (159 out of 264 SPI1-repressed genes), as deduced from the Venn diagram (Figure 2D and Supplementary Table S3) with a strong association between HDAC1 and SPI1 repressed genes (odds ratio of 6, Fisher exact test P ≃ 3.5 × 10–35). These results are consistent with coregulation by HDAC1 and SPI1 on a high fraction of repressed genes. Moreover, the derepression of transcription by knockdown of SPI1 was stronger when HDAC1 was active than inactive (Figure 2E), and the derepression by HDAC1 inhibitor was more efficient when SPI1 was overexpressed (Figure 2F). In other words, reducing the expression level of SPI1 and the absence of HDAC1 activity had little or no greater effect than when only one of the two proteins was inhibited. The Figure 2G presents this result for four representative repressed genes, Sox6, Alas2, Nprl3 and St3gal6. These results, showing that inhibition of HDAC1 activity alleviates the ability of SPI1 to repress genes on which it binds, suggested an epistatic interaction between HDAC1 and SPI1. In contrast, for SPI1-activated genes, SPI1 knockdown and reduction of HDAC1 activity triggered primarily opposite effects on gene regulation (Figure 2H), with only 8% of SPI1-activated genes also being activated by HDAC1 (Figure 2I), showing that SPI1 gene activation is largely HDAC1-independent. Interestingly, only 22% of the genes occupied by SPI1 but which are not transcriptionally regulated by SPI1 were also repressed by HDAC1 (Supplementary Figure S2E), supporting that the presence of HDAC1 is critical for the repression of a large part of genes by SPI1. The TMP195 inhibitor, which is efficient on HDAC4, −5, −7 and −9, had no consequence on gene expression in the presence or absence of SPI1 (Supplementary Figure S3).
Taken together, these results demonstrate that SPI1 interacts with HDAC1 at the chromatin and indicate that both factors may cooperate to repress SPI1 target genes.
SPI1 and HDAC1 induce histone deacetylation of active enhancers at SPI1 binding sites
We previously showed that SPI1 requires HDAC1 to repress a large portion of its target genes and that SPI1 represses genes primarily by binding to active enhancers. The next step of our study was to define whether SPI1 acts through HDAC1 at active enhancers of repressed genes to induce local deacetylation. We performed ChIP-seq of the H3K27ac histone mark in leukaemic cells treated or not with dox. To evaluate the ChIP efficiency and sensitivity, we verified that the H3K27ac ChIP-seq signal at the TSS of the genes in leukaemic cells was positively correlated with their expression level, as previously demonstrated (Supplementary Figure S4) (55).
To investigate whether the absence of H3K27ac at enhancers in repressed genes (inactive, Figure 1H) was attributable to the binding of SPI1, we compared SPI1-bound enhancer statuses between leukaemic cells (SPI1+, dox-untreated) and SPI1 knockdown cells (SPI1–, dox-treated) using ChromHMM. Most of the inactive enhancers were not changed by SPI1 depletion (Supplementary Figure S5A), indicating that in most cases, SPI1 did not repress gene expression by completely inhibiting enhancers. This result was confirmed by the absence of H3K27ac signal around SPI1 peaks in SPI1+ leukaemic cells and SPI1– cells (Figure 3A). Regarding active enhancers, the intensity of the H3K27ac signal was lower in the presence of SPI1 than when SPI1 was depleted (Figure 3A). This result was observed for both regulatory regions, that is, gene bodies and intergenic regions (Supplementary Figure S5B and C). In contrast, for SPI1-activated genes, H3K27ac profiles were opposite, exhibiting an increase in the H3K27ac intensity at TSS and at SPI1-bound enhancers in the presence of SPI1 (Supplementary Figure S5D).
Figure 3.

SPI1 represses gene expression by decreasing H3K27 acetylation at SPI1-bound enhancers that are targets of HDAC1. (A and B) Density plot profiles of H3K27ac signals in SPI1+ (untreated, red) or SPI1– (54 h dox treated, blue) cells for SPI1-repressed genes centered on SPI1 peak summits at active or inactive enhancers (A) or in two separated subgroups of genes according to H3K27ac signal in dox untreated relative to dox treated cells at enhancers bound by SPI1. (B) Profiles of H3K27ac signal at H3K27acDown or H3K27acNotDown enhancers bound by SPI1 and at the TSS of the same genes are shown as illustrated. n = number of SPI1 peaks or of TSSs. (C) Density plot profiles of H3K27ac normalized signal in untreated (DMSO, red) or Entinostat-treated (for 6 h at 3 μM, green) cells for SPI1-repressed genes whose enhancers are bound by SPI1. Profiles are centered on SPI1 peak summits at enhancers or at the TSS for the same groups of genes as in (B). (D) The H3K27ac, H3K4me1 and SPI1 binding patterns are shown at the St3gal6 and Alas2 loci for SPI1+ or SPI1– and untreated (DMSO) or Entinostat-treated cells. Sites of SPI1 binding at enhancers and around TSSs of the same genes are highlighted in yellow. Representative results from one experiment. (E) SIN3A (ChIP) in MEL cells (56), the HDAC1 signal profile (CUT&Tag) and the SPI1 binding pattern in TgSpi1 erythroleukaemic cells are shown at the St3gal6 and Alas2 loci. Sites of SPI1 binding at enhancers and TSSs are highlighted in yellow.
To better identify active enhancers at which the presence of SPI1 may restrain acetylation, we measured the H3K27ac signal intensity in untreated (SPI1+) relative to dox-treated (SPI1–) cells at each SPI1-bound enhancer. We divided the repressed genes into two categories according to the modulation of H3K27ac by SPI1 at bound enhancers: enhancers with reduction of H3K27ac signal intensity by SPI1, H3K27acDown (RatioH3K27ac SPI1+/SPI1– ≤ 0.67) and enhancers with no reduction of H3K27ac signal intensity, H3K27acNotDown, including enhancers unchanged and with increased H3K27ac signal by SPI1 (RatioH3K27ac SPI1+/SPI1– ≥ 1; see methods for more details) (Figure 3B, D and Supplementary Figures S5E, G). While acetylation of SPI1-bound enhancers was differently modified for the two groups, at TSS, H3K27 acetylation was reduced by SPI1 for both groups, consistent with reduced promoter activity in repressed genes (Figure 3B, D and Supplementary Figures S5E, G).
Investigations were also performed in the leukaemic cells #722 in which SPI1 was knocked-down by dox treatment (Supplementary Figure S6A). Changes in gene expression level evaluated by comparing RNA-seq between leukaemic cells (SPI1+, dox-untreated) and SPI1 knockdown cells (SPI1–, dox-treated) were highly correlated between #722 and #763 cells (Pearson's correlation coefficient: 0.84) (Supplementary Figure S6B). Similar epigenetic H3K27ac and/or H3K4me1 marks were detected and depletion of SPI1 showed also similar impact on the status of enhancers in the #763 and #722 leukaemic cells (Supplementary Figure S6C–G).
We observed that enhancers whose H3K27ac signal level was increased by SPI1 depletion (H3K27acDown group) were also increased by HDAC1 inhibition, supporting that HDAC1 plays a role in the deacetylation of the SPI1-bound enhancers of those genes (Figure 3B–D and Supplementary Figure S5F, G). In contrast, the highest acetylation in the presence of SPI1 for the H3K27acNotDown enhancers, argues against HDAC1 deacetylation activity at those SPI1 binding sites of this subset of repressed genes (Figure 3B and C).
Importantly, HDAC1 occupied active enhancers of repressed genes that were deacetylated in the presence of SPI1, as shown by CUT&Tag experiments performed using a HDAC1 antibody (Figure 3E and Supplementary Figure S5G). Notably, the HDAC1 profile in TgSpi1 erythroleukaemic cells proportionally colocalized with the SIN3A profile but not with the histone acetylase P300 (previously published datasets, (56)) profile in erythroleukaemic cells (Supplementary Figure S5H), consistent with their opposite roles in transcription.
Taken together, these results suggest that in a subset of genes, SPI1 cooperates with HDAC1 to deacetylate active enhancers located at intergenic and gene body regions, thereby reducing TSS acetylation and inhibiting gene transcription.
SPI1 represses gene transcription by reducing chromatin opening at enhancer and TSS and RNA pol II occupancy
Lysine acetylation is believed to control chromatin accessibility by changing histone charge and, consequently, loosening the binding of histones with DNA and reducing the contact between histones in adjacent nucleosomes (57). Lysine acetylation is also a binding target for proteins bearing bromodomains, which are mainly proteins stimulating transcriptional machinery (58–60). To evaluate whether the presence of SPI1 at enhancers impacts chromatin accessibility and/or the transcriptional machinery at promoters, we performed a comparative analysis of transposable-accessible chromatin (ATAC)-seq and RNA pol II ChIP-seq between SPI1+ leukaemic cells and SPI1– cells. The quality and sensitivity of ATAC-seq and RNA pol II ChIP-seq were evaluated by the correlation of the signal at the TSS with the level of gene expression in leukaemic cells (61). As expected, ATAC signal intensity was lower for genes with low expression than for genes with high expression but was barely able to differentiate genes with intermediate expression from genes with high expression levels (Supplementary Figure S7A). RNA pol II was positively proportional to mRNA quantity. Notably, even though SPI1 strongly reduced H3K27ac intensity (H3K27acDown), SPI1 binding at enhancers only slightly diminished local chromatin accessibility, exhibiting a narrowing of chromatin accessibility in the presence of SPI1 (Figure 4A and Supplementary Figure S7B). A stronger reduction in chromatin accessibility by SPI1 was observed at the TSS of those genes. Figure 4B presents the differences in ATAC density signals between SPI1– and SPI1+ cells at each SPI1 peak and confirms that chromatin accessibility was reduced by SPI1 at a majority of SPI1 binding sites and associated TSSs. This reduction is illustrated by the signal track at the St3gal6 and Alas2 loci (Figure 4C). Interestingly, SPI1 repressed RNA pol II occupancy in leukaemic cells (Figure 4D). The images were different for the H3K27acNotDown group of repressed genes in which enhancers were not deacetylated by SPI1. Indeed, enhancer regions were opened up by SPI1, the TSS region was significantly closed down by SPI1, and the level of RNA pol II at the TSS was not modified (Figure 4A, B and D). The features of chromatin accessibility described in this group of genes were surprising, as they resembled those of the activated genes (Figure 4E). These findings again support that SPI1 acts in different manners to repress transcription of the H3K27acDown and H3K27acNotDown groups of genes.
Figure 4.

SPI1 reduces chromatin opening and RNA pol II occupancy at the TSS without impacting GATA1 accessibility to chromatin. (A) Density plot profiles of ATAC signals in SPI1+ (untreated, red) or SPI1– (54 h dox treated, blue) cells for SPI1-repressed genes bound at H3K27acDown or H3K27acNotDown enhancers. Profiles of ATAC signal are centered on SPI1 peak summits at bound enhancers or at the TSS of the same genes. (B) Differences between SPI1– and SPI1+ cells of ATAC signals within regions ±1 kb from the SPI1 peak summit at active enhancers or within regions ±1 kb from the TSS of the same genes. The dots represent the signal difference value over each SPI1 peak at enhancers or each TSS. Blue and red dots represent ATAC signal intensities higher or lower in SPI1– cells, respectively. (C) The H3K27ac, H3K4me1 and SPI1 binding patterns for SPI1+ cells and ATAC signal for SPI1+ and SPI1– cells are shown at the St3gal6 and Alas2 loci. Representative results of one experiment. (D) Density plot profiles of RNA pol II signals in SPI1+ or SPI1– cells for SPI1-repressed genes bound at H3K27acDown or H3K27acNotDown enhancers or for SPI1-activated genes whose enhancers are bound by SPI1. Profiles of ATAC signal are centered on SPI1 peak summits at enhancers or on TSSs of the same genes. (E) Density plot profiles of ATAC signals in SPI1+ or SPI1– cells for SPI1-activated genes whose enhancers are bound by SPI1. Profiles are centered on SPI1 peak summits at bound enhancers or on TSS of the same genes. (F) Top 25 motifs obtained by motif enrichment analysis at ±150 bp from the SPI1 peak summit position at H3K27acDown or H3K27acNotDown enhancers for repressed genes or at active enhancers bound by SPI1 for activated genes. Transcription factor motifs are ranked by increasing adjusted P-value. Only motives corresponding to expressed genes in leukaemic cells are labelled. (G) Density plot profiles of ATAC signal in SPI1+ or SPI1– cells at H3K27acDown enhancers co-bound by SPI1 and GATA1 for repressed genes. Profiles are centered on SPI1 peak summits at bound enhancers or on TSS of the same genes
Recent data indicate that histone acetylation dynamics can modulate enhancer-promoter interactions and associated gene expression in cancer cells (62). To investigate if the SPI1-associated reduction of acetylation at enhancer and promoter is accompanied by a change in promoter–enhancer interaction, we performed circularized chromosome conformation capture sequencing (4C-seq) experiments and compared the behaviour of the two regions at Alas2 and St3gal6 loci between cells overexpressing SPI1 and cells depleted for SPI1 (#763 and #722). We used the enhancer as view-point for Alas2 and the TSS as view-point for St3gal6 (Supplementary Figure S8A and S8B, respectively). We observed that the 4C-seq signal was slightly reduced in the presence of SPI1 at the promoter of Alas2 or at the enhancer of St3gal6, suggesting a modest reduction of the enhancer-promoter contact as an additional consequence of the presence of SPI1 (Supplementary Figure S8).
Taken together, our findings revealed at least two mechanisms by which SPI1 inhibits transcription. The first mechanism involves deacetylation of active enhancers mediated by HDAC1, with a reduction of chromatin accessibility locally at SPI1 binding sites and at TSS, in association with a reduction of RNA pol II occupancy. The second mechanism is associated with increased acetylation and chromatin accessibility in the presence of SPI1 at enhancers and is independent of HDAC1.
SPI1 represses enhancers that are enriched for GATA1
The fact that the reduction of SPI1 at the chromatin was not associated with a strong chromatin closing down is consistent with a process in which SPI1 binds to active enhancers where several other TFs are present. We attempted to identify TFs located at enhancers where SPI1 repressed the H3K27ac signal (H3K27acDown) in SPI1-repressed genes and performed motif enrichment analysis. The TAL1:GATA1 and GATA1 motifs were the most significant motifs in repressed enhancers compared to enhancers of repressed genes whose H3K27ac was not restrained by SPI1 (H3K27acNotDown) and compared to activated genes (Figure 4F). Using previously published GATA1 ChIP-seq data (63) in murine erythroleukaemia (MEL) cells, we found that 63% of enhancers that were deacetylated by SPI1 (H3K27acDown) were sites also bound by GATA1 in proximity to SPI1, while this percentage was only 29% for H3K27acNotDown enhancers. Such a difference in GATA1 and SPI1 co-binding between the two types of SPI1-bound enhancers was also seen using GATA1 peaks from G1E-ER4 restored for GATA1 activity (64,65) or Ter119+ foetal liver proerythroblasts (66) (Supplementary Figure S9A), revealing a specific enrichment for GATA1 binding sites at SPI1-deacetylated enhancers.
Since deacetylation was associated with reduced chromatin accessibility in the presence of SPI1 at enhancers, we compared ATAC-seq at regions co-bound by GATA1 and SPI1 and showed that SPI1 also reduced chromatin accessibility at GATA1 binding sites (Figure 4G). However, except for Nprl3, GATA1 ChIP-qPCR experiments showed that the enrichment of GATA1 binding was similar in leukaemic cells and cells depleted of SPI1 (Supplementary Figure S9B). This result suggests that the presence of SPI1 at enhancers did not impair GATA1 occupancy at a large part of its targets.
Taken together, these results indicate that SPI1 reduces the transcription of a subset of genes by contributing to deacetylation with HDAC1 and reducing chromatin opening at active enhancers, some of which are targets for GATA1 in erythroid cells. The fact that the genes whose enhancers were not deacetylated were not enriched for GATA1 binding with SPI1 provides additional evidence for distinct modes of gene repression by SPI1.
SPI1 binding at enhancers is associated with increased H3K27me3 around the TSS of the SPI1 repressed genes
We have previously demonstrated that polycomb repressive complex 2 (PRC2), known to control the methylation of H3K27 (67), interacts physically and functionally with SPI1 and participates in the repression of Bcl2l11 gene expression in leukaemic cells from TgSpi1 mice (23). To define whether PRC2 is involved in the repressive transcriptional function of SPI1/HDAC1 in leukaemic cells, we analysed the presence of the H3K27me3 mark as a function of the SPI1 expression level. The inverse correlation of H3K27me3 intensity at the TSS with the gene expression level validated the quality of H3K27me3 ChIP-seq (Supplementary Figure S10A). Regardless of its transcriptional activity, the level of SPI1 occupancy at enhancers was inversely correlated with the level of H3K27me3 around the TSS or SPI1 (Supplementary Figure S10B). In contrast, for SPI1-repressed genes, the H3K27me3 intensity around the TSS was higher in the presence of high expression of SPI1, concordant with the fact that the RNA level was reduced by SPI1 (Figure 5A). Surprisingly, when SPI1 occupied deacetylated enhancers (repressed H3K27acDown genes), it impacted the H3K27me3 intensity around the TSS more than when it was not present (repressed SPI1 not bound genes) (Figure 5A, compare left and right panels). This stronger differential of H3K27me3 at SPI1 bound compared to not bound genes was verified when the comparison between SPI1– and SPI1+ cells was performed gene by gene (Figure 5B). This result may be due to the release of H3K27 substrate after deacetylation and/or to the activity of SPI1, since SPI1 interacts physically with PRC2 in leukaemic cells (23). To define whether SPI1 might directly act on PRC2 activity, we performed an in vitro PRC2 activity assay by measuring the incorporation of a methyl group from the S-adenosyl methionine (SAM) donor on two increasing quantities of recombinant histone H3.1 mixed with increasing molar ratios of SPI1:PRC2 (Figure 5C and Supplementary Figure S10C-E). We found that SPI1 with PRC2 stimulates trimethylation of H3 compared to PRC2 alone or PRC2 mixed with BSA (negative control). Importantly, SPI1 increased the level of H3K27me3 only in the presence of PRC2 and the quantity of the PRC2 components was not altered, consistent with specificity of action on PRC2 activity (Figure 5C and Supplementary Figure S10D). Moreover, H3K27 mono-methylation (H3K27me1), evaluated by HPLC, indicated a reduction in the mono-methylation of the H3K27 peptide with the increased amount of SPI1 (Supplementary Figure S10F), further supporting that SPI1 favours PRC2 activity.
Figure 5.

Occupancy of SPI1 at deacetylated enhancers is associated with increased H3K27me3 around the TSS of the SPI1 repressed genes. (A) Density plot profiles of H3K27me3 signals in SPI1+ (untreated) or SPI1– (54 h dox treated) cells centered on the TSS of SPI1-repressed genes with H3K27acDown SPI1-bound enhancers or not bound by SPI1. (B) Distribution of the sums of H3K27me3 signals within region ±1 kb from TSS for the same genes and groups than in (A). Means of the differential ratio of H3K27me3 signal between SPI1+ and SPI1– cells are indicated for each group by the black lane. ****P < 0.0001, Wilcoxon test. (C) PRC2 in vitro activity assays using 500 ng (light bars) or 250 ng (dark bars) of H3.1 mixed with the PRC2 complex and SPI1 or BSA (negative control) at a molar ratio of 1:1 (large circles) or 1:5 (small circles). Circles in the legend indicate the proteins or substrate added to the mix. S-Adenosyl methionine (SAM) was the PRC2 substrate. H3K27me3 was quantified by dot blot experiments (Supplementary Figure S10E) and normalized to the activity of PRC2 with SAM (fifth group of bars). Protein mixes were separated on Stain-Free polyacrylamide gradient gels detected upon activation by UV light (λ = 302 nm) as shown in the lower panel. The identity of the proteins was validated by immunoblotting (Supplementary Figure S10D).
In conclusion, these results suggest that SPI1 stimulates the tri-methylation of H3K27 to the promoters of repressed genes when it is linked to active enhancers whose acetylation is reduced. Evidence indicates that SPI1 may play an active role in this process by stimulating PRC2 activity.
PRC2 synergizes with HDAC1 to mediate SPI1 transcriptional repressive activity that enables differentiation blockage and leukaemic cell survival
Next, we investigated whether HDAC1 and PRC2 cooperate in mediating transcriptional repression by SPI1. To this end, leukaemic cells were cultured in the presence of a pharmacological inhibitor of EZH1/2, UNC1999 and the HDAC1 inhibitor entinostat. As expected, HDAC1 inhibition by entinostat increased H3K27 acetylation, and EZH1/2 inhibition by UNC1999 abolished the trimethylation of H3K27 (Figure 6A). Notably, the combination of the two inhibitors synergistically exacerbated acetylation of H3K27. We analysed the transcriptional consequences of the pharmacological inhibition of HDAC1 and/or PRC2 on SPI1 repressed genes involved in erythroid differentiation, whose SPI1 induces deacetylation of their enhancers (Alas2, St3gal6, Sox6 and Nprl3). As reference, Figure 6B shows the re-activation of these SPI1 repressed genes after SPI1 knockdown measured by RT-qPCR. Treatment with UNC1999 showed few effects on gene expression and treatment with entinostat increased transcription of some of these genes (Figure 6C). The combined treatment of HDAC1/3 and PRC2 inhibitors induced a strong and synergistic increase in the expression of the four genes repressed by SPI1 (Figure 6C), indicating that optimal repression requires both HDAC1 and PRC2. Moreover, the transcriptional activation due to the combination of HDAC1 and PRC2 inhibitors in leukemic cells were mitigated in the presence of low SPI1 expression, demonstrating that the repressive activity of SPI1 is mediated by HDAC1 and PRC2 (Figure 6D).
Figure 6.

PRC2 functionally cooperates with HDAC1 to maintain gene repression, erythroid differentiation blockage and erythroid blast survival. (A) Leukaemic cells were treated with Entinostat (150 nM) or UNC1999 (10 μM), or both, for 48 h. Whole-cell extracts were analysed by immunoblotting with antibodies against H3K27ac, H3K27me3 and H3 (loading control). (B) Expression of SPI1 repressed genes was quantified by RT-qPCR in untreated leukaemic cells (SPI1+, red) or dox treated (SPI1–, blue) for 48 h. Data represent the mean ± SEM of expression relative to untreated cells normalized to PolR2a mRNA (three independent experiments). (C) Expression of SPI1 repressed genes was quantified by RT-qPCR in leukaemic cells treated with DMSO (control, black), Entinostat 150 nM (green), UNC1999 10 μM (blue) or both inhibitors (purple) for 48 h. Data represent the mean ± SEM of expression relative to DMSO-treated cells normalized to PolR2a mRNA (three independent experiments). (D) Schematic diagram of the treatment of cells whose expression of SPI1 repressed genes was quantified by RT-qPCR at the end of the 96 h of treatment. Data represent the mean ± SEM of expression relative to DMSO of SPI1+ or SPI1–cells and normalized to PolR2a mRNA (three independent experiments). (E) Upper panel: Leukaemic cells were treated or not with dox (SPI1– or SPI1+ cells) concomitantly with DMSO or Entinostat 150 nM or UNC1999 10 μM or the combination of both inhibitors for 5 days in cells. Expression of CD71 and the level of forward scattering (FSC-A) analyzed by flow cytometry is shown. Three distinct population were defined: CFU-E/Pro-E (FSC-AhighCD71high, blue), early differentiation (FSC-AlowCD71high/medium, green), late differentiation (FSC-AlowCD71low, red) as shown in Supplementary Figure S11B. Means ± SEM of the percentage of each population for three independent experiments are indicated. Lower panel: Benzidine staining of representative colonies grown in methylcellulose with or without dox (SPI1– or SPI1+ cells) and with DMSO or Entinostat + UNC1999 for 5 days. (F) Differentiation state described in (E) are presented as the ratio of the percentage of early and late differentiated cells relative to CFU-E/Pro-E. Mean of the ratios were compared by a Wilcoxon test. *P < 0.05, ns = not significant. (G) Cells were treated for 48 h with different concentrations of Entinostat with or without UNC1999. The synergy score calculated by the ZIP method on the number of live cells or percentage of dead cells is represented by a 3D surface heatmap. Green, significant synergy; red, significant antagonism; white, additivity. (H) Cells were treated with DMSO (control), Entinostat 150 nM, UNC1999 12.5 μM or the combination of both inhibitors for the indicated hours. Mean ± SEM of the number of live cells and percentage of dead cells measured by DAPI exclusion assay (Three independent experiments). (I) Cells were treated (SPI1–) or not (SPI1+) with dox for the indicated hours. Mean ± SEM of the number of live cells and percentage of dead cells measured by DAPI exclusion assay (Three independent experiments).
Since a large portion of SPI1-repressed genes that are bound by SPI1 encode proteins involved in erythroid differentiation (Figure 1D), we investigated whether the two inhibitors could restore erythroid differentiation. The combination of UNC1999 and entinostat in the presence of SPI1 did not induce the appearance of differentiated erythroid cells, as evidenced by the absence of change in FSC-A and CD71 or benzidine staining of haemoglobin (Figure 6E and F). This suggests that other functions of SPI1 independent of HDAC1 and PRC2 might be involved in blocking differentiation, such as repression of additional genes (Figure 2D) or SPI1-activating functions. Consequently, to investigate the consequences of HDAC1 and PRC2 inhibition on erythroid differentiation, we co-treated the erythroleukemic cells with UNC1999 and entinostat simultaneously with dox to decrease SPI1 expression. Under these conditions, reactivation of SPI1-repressed genes was enhanced by treatment with the inhibitors (SPI1–/Ent + UNC) compared to conditions where only SPI1 expression was decreased (SPI1–/DMSO) (Supplementary Figure S11A). Consequently, these culture conditions allowed rapid reactivation of SPI1-repressed genes in addition to other consequences due to decreased SPI1 expression. We examined the fraction of differentiated cells by FACS or by benzidine staining cells after 5 days of treatment (Figure 6E, F and Supplementary Figure S11B). Figure 6F presents the ratio of % early and late differentiated cells over the CFU-E/ProE progenitors presented in the Figure 6E. While individual treatment with HDAC1 or PRC2 inhibitors had small and no significant effect on differentiation in the absence of SPI1, combined inhibition of HDAC1 with PRC2 strongly increased progression to terminal erythroid differentiation. This result indicates a synergistic effect of HDAC1 and PRC2 inhibition on the recovery of erythroid differentiation. Altogether, our data provide evidence that HDAC1 and PRC2 cooperate to the erythroid differentiation blockage. The finding that their inhibition alone is not sufficient to rescue the differentiation potential suggests an additional role for SPI1 in erythroid blockage independent of HDAC1 and PRC2.
We next investigated whether both inhibitors may be used to inhibit proliferation and/or survival of the erythroleukemic cells. Erythroleukemic cells were cultured in the presence of increasing doses of entinostat combined with increasing doses of UNC1999, and the percentage of dead and alive cells was quantified by flow cytometry using DAPI staining. Calculation of a synergy score per combination of doses demonstrated the synergistic effect of the combination of the two inhibitors on stopping proliferation and killing leukaemic cells (Figure 6G). The cytotoxic and cytostatic effects of the combination of the two drugs were illustrated by the increase in the percentage of dead cells and the inhibition of cell proliferation over time (Figure 6H), resembling to those triggered by the reduction of SPI1 expression alone (Figure 6I). The synergistic cytotoxic and cytostatic effects of HDAC1 and PRC2 inhibitors were also validated for erythroleukemic cells derived from two others sick TgSpi1 mice, #722 and #683 mice (Supplementary Figure S12A-B). Importantly, the cellular response of PRC2 and HDAC1 inhibitors varied extensively in three non-erythroid cancer cell lines derived from B Lymphoma (A20), chronic myeloid leukemia (K562) and AML (KG1) and no synergistic effects of the combination of the two inhibitors was observed (Supplementary Figure S12C).
Overall, these experiments indicate a synergy between PRC2 and HDAC1 complexes in mediating the transcriptional repressive activity of SPI1, the combined inhibition of which results in synergistic adverse consequences on the proliferation and survival of leukaemic cells.
DISCUSSION
SPI1 is a major haematopoietic TF with pioneering function whose positive activity in transcription has been widely described in normal haematopoiesis. Recently, accumulating evidence has also indicated a major activity of SPI1 in repressive transcriptional function in normal processes of differentiation. Distinct mechanisms according to haematopoietic lineages with direct or indirect binding to regulatory regions of repressed genes have been reported (12,14–16). In this study, we characterized a new mechanism involving the synergistic combination of the two epigenetic repressors, PRC2 and HDAC1, used by SPI1 to repress gene expression, encoding proteins that play a major role in its leukemic functions of blocking apoptosis and differentiation. This function is associated with SPI1 occupancy to regulatory intergenic or intragenic regions and involves deacetylation at both bound enhancers and distant TSSs by HDAC1. PRC2 reinforces the inhibition of gene expression, and its activity at promoters is actively favoured by SPI1 when bound at distant regulatory regions (Figure 7).
Figure 7.
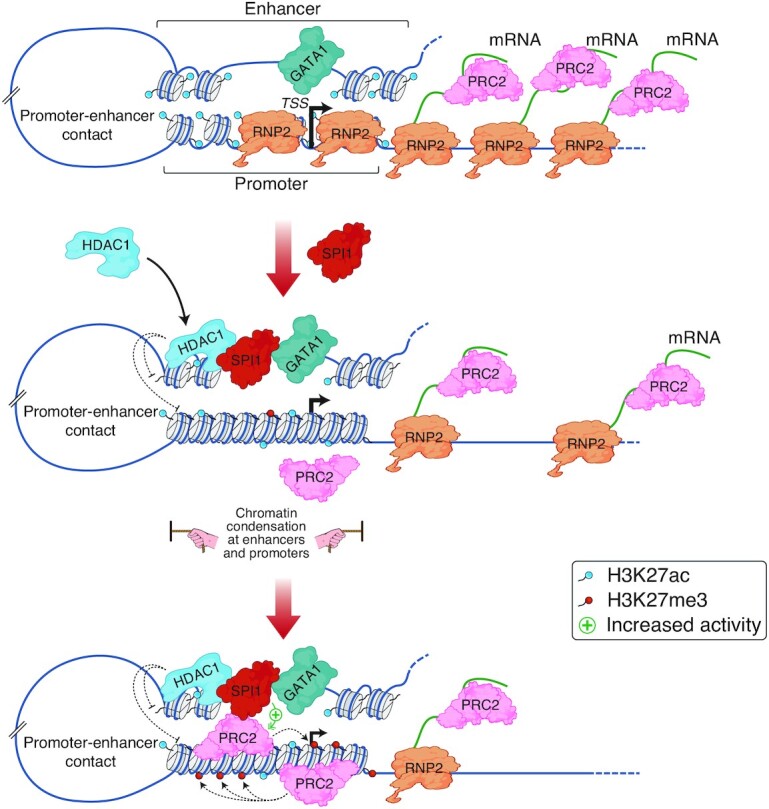
Model of SPI1-mediated gene repression in murine erythroleukaemia. Working hypothesis representing one of the mechanisms of gene repression by SPI1 based on the data described in this manuscript. During normal erythroid differentiation, GATA1 binds active enhancers and induces erythroid gene expression; SPI1 is barely expressed in CFU-E. When SPI1 is highly expressed, it binds to SPI1 binding sites at enhancers and, by interacting with HDAC1, triggers HDAC1-mediated deacetylation of enhancers and associated gene promoters. Consequently, chromatin is slightly condensed at enhancers and closed down around the TSS, the total RNA pol II (RNP2) quantity is reduced at the TSS and, the RNA transcription is decreased. Reduction of transcribed RNA molecules is associated with the venue of PRC2 to the chromatin and PRC2-mediated H3K27 methylation activity is potentially emphasized by SPI1. Thereby, the PRC2 complex reinforces HDAC1 transcriptional inhibition.
SPI1 is a determining lineage factor whose expression level controls the engagement of progenitors towards lineage specification. In myeloid and B-lymphoid cells, SPI1 activates lineage-specific genes due to lineage-specific coactivators at enhancers (6,7,68). Instead, activation of common housekeeping genes is associated with an enrichment of SPI1 occupancy at promoters without requiring lineage-specific coactivators but with a higher affinity of binding to chromatin compared to enhancers (6,7,68). In the erythroleukaemic context, SPI1 activates genes mainly by multiple promoter occupancies independent of lineage determining cofactors (36). In contrast, in this study, we found that gene repression by SPI1, including repression of genes controlling the erythroid differentiation process (69), is mainly associated with an enrichment of SPI1 at active enhancers instead of promoters.
RIME experiments allowed identification of HDAC1 at chromatin with SPI1. Interactions of SPI1 with HDAC1 and HDAC2 in myeloid progenitors and AML1-ETO AML have been previously described (52,70). Recently, SPI1 has been described to restrain neutrophil activation by antagonizing the AP-1 transcription factor JUNB through its interaction with HDAC1 (14). In this study, we show that this interaction mediates SPI1 repressive activity and participates in the transformation activity of SPI1 in erythroleukaemia, emphasizing the major function of the SPI1 and HDAC1 interaction in different lineages. In the erythroleukaemic cells, SPI1 interacts with the HDAC1 belonging to the SIN3A complex, an interaction previously shown to be mediated by SIN3A (71). Additionally, we observed that the CHD4 member of the NuRD complex co-immunoprecipitates with SPI1, indicating that SPI1 may interfere with several HDAC1 complexes.
Our work distinguishes at least two modes of transcriptional repression intrinsic in SPI1 occupancy to active intergenic or gene body enhancers.
For one subgroup of genes, including erythroid differentiation function encoding genes, we described a decrease in acetylated H3K27 by SPI1 taking place locally at SPI1-bound enhancers and at distal TSS not bound by SPI1 in association with weak but significant decreased chromatin accessibility. We showed that (i) HDAC1 was detected with SPI1 at enhancers, (ii) HDAC1 inhibits acetylation of SPI1-bound enhancers and TSS, (iii) HDAC1 represses the expression of SPI1-repressed genes and (iv) HDAC1 inhibition decreases SPI1 activity in repressing transcription. Together, these data provide evidence that HDAC1 mediates SPI1 repressive transcriptional activity by creating a deacetylated and, at least in a part of genes, a nonpermissive chromatin structure. More research is necessary to define whether, as with immune-related genes in neutrophils (14), SPI1 recruits or stabilizes HDAC1 or improves its activity in erythroleukaemic cells.
Since SPI1 also interacts with the histone acetyltransferases (HAT) CBP/P300, questions have been raised regarding the role played by SPI1 in the equilibrium between HATs and HDACs. In particular, it would be interesting to define whether SPI1’s partner transcription factors on chromatin might determine the nature of SPI1’s interactivity, by changing the interactions with co-repressors or co-activators, as it has been shown in the AML containing the RUNX1:ETO fusion (52,70).
Deacetylated enhancers of this subgroup of genes are specifically enriched with GATA1 binding at the TAL1:GATA1 composite motif, associated to the reduction in chromatin accessibility at these sites. Previous reports proposed that SPI1, by binding to GATA1, impedes histone or GATA1 acetylation, which is required for the stabilization of GATA1 binding at the chromatin (72,73). This occurs at promoters of GATA1 targets, by directly inhibiting CBP acetyltransferase activity (30) or by establishing a H3K9me3 context through pRB recruitment (28,29). Our results, which indicate the presence of GATA1 at enhancers whose histone acetylation is controlled by SPI1, are consistent with these propositions.
By deleting a SPI1 binding consensus sequence at the Alas2 enhancer occupied by SPI1, with GATA1 and HDAC1 at very close locations, we brought evidence that SPI1 requires direct binding to chromatin to repress genes in erythroid cells. Previous reports showed that SPI1 requires GATA1 to occupy chromatin at GATA1-responsive reporter sequence belonging to the α-globin promoter that did not contain SPI1 binding motif (28,29). Thus, SPI1 appears to repress gene through diverse and complex mechanisms in relation with GATA1.
Here, we showed that, HDAC1, through the SPI1 interaction, is one of the mediators of histone deacetylation and it may also participate in GATA1 deacetylation and oppose to its transcriptional activity (74). However, in agreement with a previous report (29), GATA1 was still able to bind to most of its target enhancers, excluding that SPI1 opposes to GATA1 activity by impeding GATA1 chromatin occupancy as a general mechanism.
HDAC1 activity in losing accessibility of enhancers pre-bound by SPI1 has been reported in the myelogenous leukemia line K562, but in contrast to the erythroid leukemic cells, SPI1 was not instructive in the HDACi transcriptional changes of these genes (75).
In addition to controlling chromatin accessibility, histone acetylation also affects the binding of bromodomain and extraterminal motif proteins (BETs), such as BRD2, BRD3 and BRD4 (bromodomain-containing protein), at chromatin (76,77), which stimulate transcription by promoting RNA pol II elongation and stability (78,79). Our results demonstrate that at the TSS, deacetylation of enhancers by the SPI1/HDAC1 axis is associated with a reduction of RNA pol II at TSS in this fraction of repressed genes and is compatible with a decrease in BRD chromatin occupancy consequent to deacetylation. The decreased enhancer-promoter interactions in SPI1 repressed genes may also be due in part to the observed deacetylation of H3K27 at enhancers and promoters. Indeed, recent data indicate that H3K27ac may also change gene expression by interfering with interaction frequency between enhancer and promoters (62).
Our findings also identified a second interesting group of repressed genes whose distal regulatory regions are bound by SPI1 and, in contrast to the first group, exhibits more acetylation and increased chromatin accessibility with SPI1. These epigenetic modifications are opposite to deacetylating HDAC1 activity and suggest that SPI1 represses those genes independently of HDAC1. Notably, searches for TF motif enrichment indicate that SPI1 binding occurs mainly at class I ETS-specific binding sites and not in association with GATA1 binding. Further studies are needed to understand what mechanisms are responsible for the reduction in transcription of these genes.
It should be noted that for upregulated genes, SPI1 binding at enhancers was strongly associated with chromatin opening up of these regions, which is in keeping with its role as a pioneer TF in the positive regulation of genes. This gene upregulation takes place without impacting chromatin accessibility at the TSS while strongly increasing RNA pol II loading. The absence of a correlation between chromatin accessibility at the TSS and transcriptional activation was also observed for inflammatory SPI1-activated genes in neutrophils, in which TSS ATAC signal intensity was inversely correlated with transcription (14), or in macrophages in which nucleosomes were not evicted at promoters in contrast to enhancers (80).
In addition to HDAC1, our work reveals a role of PRC2 as a complementary process to mediate the activity of SPI1 in repressing transcription. Recent data indicate that the polycomb complex, which controls the methylation of H3K27, maintains the repressed state of a gene (32,33) but is not the initiator of repression (67). Indeed, a model of recruitment of polycomb proposes that a decrease in newly synthesized RNA during gene repression allows the transfer of PRC2 from RNA to chromatin with consequent amplification of methylation at histone H3K27 (81–83). In this situation, PRC2 acts in the context of a weakly transcribed gene to maintain repression by favouring trimethylation through the TSS position (84). Consistent with this model, we found that H3K27me3 increased at promoters of SPI1-repressed genes bound or not bound by SPI1. Moreover, we determined that the fraction of genes that were repressed through the SPI1/HDAC1 axis displayed an even higher level of H3K27me3 marks at the TSS than the SPI1-repressed genes that were not occupied by SPI1. SPI1 and PRC2 interact physically (23,85), and SPI1 is able to favour PRC2 activity, at least in vitro. We proposed that PRC2 activity may be triggered as a result of the decreased RNA expression level due to HDAC1 and that the presence of SPI1 at chromatin may favour PRC2 activity, further maintaining low expression of RNA (model described in Figure 7). Consequently, PRC2 reinforces the repression of the transcriptional level of genes. Interestingly, this action is not only additive to the repressive activity of HDAC1, but HDAC1 and PRC2 activities synergistically enhance gene repression. Even though PRC2 and HDAC1 participate synergistically in SPI1-erythroid differentiation blockage, the combination of PRC2 and HDAC1 inhibitors was not sufficient to reverse this blockage. Indeed, our results suggest that SPI1-activated genes and/or HDAC1/PRC2 independent repressed genes also play a role in erythroid blockage. One of these SPI1-activated genes may be Cbfa2t3 (encoding ETO2) that was shown to be an oncogenic factor involved in erythroleukaemia (25) and that is a SPI1 target gene.
Alterations in PRC2 complex components appear to be relatively rare events in AML (86,87), and when identified, they are associated with loss of function. However, PRC2 activity has also been shown to be required for RUNX1-ETO9a or MLL-AF9 leukaemic cell growth, revealing a dual role for PRC2 in AML and its interest as a therapeutic target for certain types of leukaemia (88–90). Here, we described a specific sensitivity of combined HDAC1/3 and PRC2 inhibitors to kill TgSpi1 murine erythroleukaemic cells. HDACs inhibitors are efficient in killing tumor cells including human cell lines and established mouse models of AEL (91). It would be interesting to determine whether the combination of HDACs and PRC2 inhibitors results in a specific and higher sensitivity on primary human AEL cells.
Similar to PRC2, SPI1 plays a dual role in AML. Indeed, a functional decrease in activity participates in leukaemic development, and minimal residual activity of SPI1 is required for cell survival and to maintain stemness of leukaemic cells (92,93). Based on this feature, Antony-Debré et al. provided proof of concept that SPI1 inhibition has potential as a therapeutic strategy for the treatment of AML by inducing leukaemic cell death (94). Consequently, understanding the molecular pathways by which SPI1 controls its function, including transcriptional repression, may improve the range of possible therapeutic combinations to inhibit SPI1 activity.
DATA AVAILABILITY
Data of ChIP-seq, CUT&Tag, RNA-seq, ATAC-seq and 4C-seq are available under accession number GSE172088. Publicly available raw data used for SPI1 in TgSpi1 erythroid cells and GATA1 in MEL cells ChIP-seq were found with the accession number GSE33611 (GEO) and ERR007441-44 (ENA), respectively. SIN3A (CistromeDB #46488) and EP300 (CistromeDB #46468) signal files (.bw) data were obtained from Cistrome DB (95) analysis of ChIP-seq performed in MEL cells (56). GATA1 ChIP-seq peak files (.bed) were obtained from ES-G1ER cells restored for GATA1 activity (64,65) (CistromeDB #502 and CistromeDB #56614–16) or from foetal liver E14.5 Ter119+ proerythroblasts (CistromeDB #3270–71) (66) publicly available on CistromeDB.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank F. Moreau-Gachelin, F. Rosselli and E. Elvira-Matelot for helpful scientific discussions and critical reading of the manuscript; N. Droin (Gustave Roussy genomic platform, France); C. Thibault-Carpentier, S. Legras and B. Jost for sequencing and analysis (GenomEast platform, France Genomique consortium); Y. Lecluse, P. Rameau and C. Catelain (Imaging and Cytometry Platform, Gustave Roussy, France) for flow cytometry. We thank the technical platform ‘BioProfiler’ (BFA, CNRS UMR 8251, Université de Paris) for provision of UFLC facilities.
Notes
Present address: Lélia Polit, CNRS UMR7238, Sorbonne Université, F-75006 Paris, France.
Present address: Emilie Evanno, SGS Life Science Services, F-86000 Poitiers, France.
Present address: Isabelle Gallais, Université Rennes, Inserm UMR_S1085, EHESP, Institut de Recherche en Santé, Environnement et Travail, F-35000 Rennes, France.
Present address: Jérémy Berthelet, CNRS UMR7216, Université Paris Cité, F-75013 Paris, France.
Contributor Information
Sebastian Gregoricchio, Inserm U1170, Université Paris-Saclay, Gustave Roussy Cancer Campus, F-94800 Villejuif, France; Equipe Labellisée Ligue Nationale Contre le Cancer, France; Division of Oncogenomics, Oncode Institute, The Netherlands Cancer Institute, Amsterdam, The Netherlands.
Lélia Polit, CNRS UMR8104, Inserm U1016, Université Paris Cité, Cochin Institute, F-75014 Paris, France.
Michela Esposito, Inserm U1170, Université Paris-Saclay, Gustave Roussy Cancer Campus, F-94800 Villejuif, France; Equipe Labellisée Ligue Nationale Contre le Cancer, France.
Jérémy Berthelet, Université Paris Cité, CNRS UMR8251, F-75013 Paris, France.
Laure Delestré, Inserm U1170, Université Paris-Saclay, Gustave Roussy Cancer Campus, F-94800 Villejuif, France; Equipe Labellisée Ligue Nationale Contre le Cancer, France.
Emilie Evanno, Curie Institute, Inserm U830, F-75005Paris, France.
M’Boyba Diop, Inserm U1170, Université Paris-Saclay, Gustave Roussy Cancer Campus, F-94800 Villejuif, France; Equipe Labellisée Ligue Nationale Contre le Cancer, France.
Isabelle Gallais, Curie Institute, Inserm U830, F-75005Paris, France.
Hanna Aleth, Institute of Molecular Tumor Biology, University of Münster, Münster, Germany.
Mathilde Poplineau, CNRS UMR7258, Inserm U1068, Université Aix Marseille, Paoli-Calmettes Institute, CRCM, F-13009 Marseille, France; Equipe Labellisée Ligue Nationale Contre le Cancer, France.
Wilbert Zwart, Division of Oncogenomics, Oncode Institute, The Netherlands Cancer Institute, Amsterdam, The Netherlands.
Frank Rosenbauer, Institute of Molecular Tumor Biology, University of Münster, Münster, Germany.
Fernando Rodrigues-Lima, Université Paris Cité, CNRS UMR8251, F-75013 Paris, France.
Estelle Duprez, CNRS UMR7258, Inserm U1068, Université Aix Marseille, Paoli-Calmettes Institute, CRCM, F-13009 Marseille, France; Equipe Labellisée Ligue Nationale Contre le Cancer, France.
Valentina Boeva, CNRS UMR8104, Inserm U1016, Université Paris Cité, Cochin Institute, F-75014 Paris, France; Department of Computer Science and Department of Biology, ETH Zurich, 8092 Zurich, Switzerland.
Christel Guillouf, Inserm U1170, Université Paris-Saclay, Gustave Roussy Cancer Campus, F-94800 Villejuif, France; Equipe Labellisée Ligue Nationale Contre le Cancer, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Inserm (to C.G., V.B.); CNRS (to V.B., C.G.); Ligue Nationale contre le Cancer (to C.G.); l’Institut National du Cancer [20141PLBIO-06-1 to C.G., M.E., M.P. and E.D.]; Société Française d’Hématologie (to S.G.); Gustave Roussy Genomic Core Facilities [TA2019]; Institut Thématique Multi-Organisme-Cancer de l’Alliance Nationale pour les Sciences de la Vie et de la Santé (AVIESAN) [C13104L to C.G., L.D. and E.D.]; ATIP-Avenir (to V.B.); ARC Foundation [ARC-RAC16002KSA-R15093KS to V.B.]; ‘Who Am I?’ Laboratory of Excellence [ANR-11-LABX-0071], funded by the French Government through its Investissement d′Avenir program, operated by the French National Research Agency [ANR-11-IDEX-0005-02 to V.B.]; Ministère de l’Enseignement Supérieur (to S.G.); Région Ile-de-France [ARDoC, 17012698 to L.P.]; Université Paris Cité (to F.R.L.); Excellence Initiative of Aix-Marseille University – A∗MIDEX, a French ‘Investissements d’Avenir’ program (to E.D.); Fondation de France [2017-00076284 to M.P.] Funding for open access charge: Inserm.
Conflict of interest statement. None declared.
REFERENCES
- 1. Lara-Astiaso D., Weiner A., Lorenzo-Vivas E., Zaretsky I., Jaitin D.A., David E., Keren-Shaul H., Mildner A., Winter D., Jung S.et al.. Immunogenetics. Chromatin state dynamics during blood formation. Science. 2014; 345:943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang J., Liu X., Li D., Shao Z., Cao H., Zhang Y., Trompouki E., Bowman T.V., Zon L.I., Yuan G.C.et al.. Dynamic control of enhancer repertoires drives lineage and stage-specific transcription during hematopoiesis. Dev. Cell. 2016; 36:9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kamath M.B., Houston I.B., Janovski A.J., Zhu X., Gowrisankar S., Jegga A.G., DeKoter R.P.. Dose-dependent repression of T-cell and natural killer cell genes by PU.1 enforces myeloid and B-cell identity. Leukemia. 2008; 22:1214–1225. [DOI] [PubMed] [Google Scholar]
- 4. McKercher S.R., Torbett B.E., Anderson K.L., Henkel G.W., Vestal D.J., Baribault H., Klemsz M., Feeney A.J., Wu G.E., Paige C.J.et al.. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. 1996; 15:5647–5658. [PMC free article] [PubMed] [Google Scholar]
- 5. Scott E.W., Simon M.C., Anastasi J., Singh H.. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994; 265:1573–1577. [DOI] [PubMed] [Google Scholar]
- 6. Ghisletti S., Barozzi I., Mietton F., Polletti S., De Santa F., Venturini E., Gregory L., Lonie L., Chew A., Wei C.L.et al.. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010; 32:317–328. [DOI] [PubMed] [Google Scholar]
- 7. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fernandez Garcia M., Moore C.D., Schulz K.N., Alberto O., Donague G., Harrison M.M., Zhu H., Zaret K.S. Structural features of transcription factors associating with nucleosome binding. Mol. Cell. 2019; 75:921–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Minderjahn J., Schmidt A., Fuchs A., Schill R., Raithel J., Babina M., Schmidl C., Gebhard C., Schmidhofer S., Mendes K.et al.. Mechanisms governing the pioneering and redistribution capabilities of the non-classical pioneer PU.1. Nat. Commun. 2020; 11:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barozzi I., Simonatto M., Bonifacio S., Yang L., Rohs R., Ghisletti S., Natoli G.. Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers. Mol. Cell. 2014; 54:844–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McAndrew M.J., Gjidoda A., Tagore M., Miksanek T., Floer M.. Chromatin remodeler recruitment during macrophage differentiation facilitates transcription factor binding to enhancers in mature cells. J. Biol. Chem. 2016; 291:18058–18071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de la Rica L., Rodriguez-Ubreva J., Garcia M., Islam A.B., Urquiza J.M., Hernando H., Christensen J., Helin K., Gomez-Vaquero C., Ballestar E.. PU.1 target genes undergo Tet2-coupled demethylation and DNMT3b-mediated methylation in monocyte-to-osteoclast differentiation. Genome Biol. 2013; 14:R99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lio C.W., Zhang J., Gonzalez-Avalos E., Hogan P.G., Chang X., Rao A.. Tet2 and Tet3 cooperate with B-lineage transcription factors to regulate DNA modification and chromatin accessibility. Elife. 2016; 5:e18290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fischer J., Walter C., Tonges A., Aleth H., Jordao M.J.C., Leddin M., Groning V., Erdmann T., Lenz G., Roth J.et al.. Safeguard function of PU.1 shapes the inflammatory epigenome of neutrophils. Nat. Immunol. 2019; 20:546–558. [DOI] [PubMed] [Google Scholar]
- 15. Hosokawa H., Ungerbäck J., Wang X., Matsumoto M., Nakayama K.I., Cohen S.M., Tanaka T., Rothenberg E.V.. Transcription factor PU.1 represses and activates gene expression in early T cells by redirecting partner transcription factor binding. Immunity. 2018; 48:1119–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ungerbäck J., Hosokawa H., Wang X., Strid T., Williams B.A., Sigvardsson M., Rothenberg E.V.. Pioneering, chromatin remodeling, and epigenetic constraint in early T-cell gene regulation by SPI1 (PU.1). Genome Res. 2018; 28:1508–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moreau-Gachelin F., Wendling F., Molina T., Denis N., Titeux M., Grimber G., Briand P., Vainchenker W., Tavitian A.. Spi-1/PU.1 transgenic mice develop multistep erythroleukemias. Mol. Cell. Biol. 1996; 16:2453–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Seki M., Kimura S., Isobe T., Yoshida K., Ueno H., Nakajima-Takagi Y., Wang C., Lin L., Kon A., Suzuki H.et al.. Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat. Genet. 2017; 49:1274–1281. [DOI] [PubMed] [Google Scholar]
- 19. Roos-Weil D., Decaudin C., Armand M., Della-Valle V., Diop M.K., Ghamlouch H., Ropars V., Herate C., Lara D., Durot E.et al.. A recurrent activating missense mutation in waldenstrom macroglobulinemia affects the DNA binding of the ETS transcription factor SPI1 and enhances proliferation. Cancer Discov. 2019; 9:796–811. [DOI] [PubMed] [Google Scholar]
- 20. Rosenbauer F., Owens B.M., Yu L., Tumang J.R., Steidl U., Kutok J.L., Clayton L.K., Wagner K., Scheller M., Iwasaki H.et al.. Lymphoid cell growth and transformation are suppressed by a key regulatory element of the gene encoding PU.1. Nat. Genet. 2006; 38:27–37. [DOI] [PubMed] [Google Scholar]
- 21. Back J., Dierich A., Bronn C., Kastner P., Chan S.. PU.1 determines the self-renewal capacity of erythroid progenitor cells. Blood. 2004; 103:3615–3623. [DOI] [PubMed] [Google Scholar]
- 22. Nutt S.L., Metcalf D., D’Amico A., Polli M., Wu L.. Dynamic regulation of PU.1 expression in multipotent hematopoietic progenitors. J. Exp. Med. 2005; 201:221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ridinger-Saison M., Evanno E., Gallais I., Rimmele P., Selimoglu-Buet D., Sapharikas E., Moreau-Gachelin F., Guillouf C.. Epigenetic silencing of Bim transcription by Spi-1/PU.1 promotes apoptosis resistance in leukaemia. Cell Death Differ. 2013; 20:1268–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rimmele P., Kosmider O., Mayeux P., Moreau-Gachelin F., Guillouf C.. Spi-1/PU.1 participates in erythroleukemogenesis by inhibiting apoptosis in cooperation with Epo signaling and by blocking erythroid differentiation. Blood. 2007; 109:3007–3014. [DOI] [PubMed] [Google Scholar]
- 25. Fagnan A., Bagger F.O., Piqué-Borràs M.R., Ignacimouttou C., Caulier A., Lopez C.K., Robert E., Uzan B., Gelsi-Boyer V., Aid Z.et al.. Human erythroleukemia genetics and transcriptomes identify master transcription factors as functional disease drivers. Blood. 2020; 136:698–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rekhtman N., Radparvar F., Evans T., Skoultchi A.I.. Direct interaction of hematopoietic transcription factors PU.1 and GATA-1: functional antagonism in erythroid cells. Genes Dev. 1999; 13:1398–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang P., Behre G., Pan J., Iwama A., Wara-Aswapati N., Radomska H.S., Auron P.E., Tenen D.G., Sun Z.. Negative cross-talk between hematopoietic regulators: GATA proteins repress PU.1. Proc. Nat. Acad. Sci. U.S.A. 1999; 96:8705–8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rekhtman N., Choe K.S., Matushansky I., Murray S., Stopka T., Skoultchi A.I.. PU.1 and pRB interact and cooperate to repress GATA-1 and block erythroid differentiation. Mol. Cell. Biol. 2003; 23:7460–7474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stopka T., Amanatullah D.F., Papetti M., Skoultchi A.I.. PU.1 inhibits the erythroid program by binding to GATA-1 on DNA and creating a repressive chromatin structure. EMBO J. 2005; 24:3712–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hong W., Kim A.Y., Ky S., Rakowski C., Seo S.B., Chakravarti D., Atchison M., Blobel G.A.. Inhibition of CBP-mediated protein acetylation by the Ets family oncoprotein PU.1. Mol. Cell. Biol. 2002; 22:3729–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wontakal S.N., Guo X., Will B., Shi M., Raha D., Mahajan M.C., Weissman S., Snyder M., Steidl U., Zheng D.et al.. A large gene network in immature erythroid cells is controlled by the myeloid and B cell transcriptional regulator PU.1. PLos Genet. 2011; 7:e1001392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R.S., Zhang Y.. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002; 298:1039–1043. [DOI] [PubMed] [Google Scholar]
- 33. Kuzmichev A., Nishioka K., Erdjument-Bromage H., Tempst P., Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the enhancer of Zeste protein. Genes Dev. 2002; 16:2893–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Labun K., Montague T.G., Krause M., Torres Cleuren Y.N., Tjeldnes H., Valen E.. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019; 47:W171–W174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Concordet J.P., Haeussler M.. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018; 46:W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ridinger-Saison M., Boeva V., Rimmele P., Kulakovskiy I., Gallais I., Levavasseur B., Paccard C., Legoix-Ne P., Morle F., Nicolas A.et al.. Spi-1/PU.1 activates transcription through clustered DNA occupancy in erythroleukemia. Nucleic Acids Res. 2012; 40:8927–8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yadav B., Wennerberg K., Aittokallio T., Tang J.. Searching for drug synergy in complex dose-response landscapes using an interaction potency model. Comput. Struct. Biotechnol. J. 2015; 13:504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. He L., Kulesskiy E., Saarela J., Turunen L., Wennerberg K., Aittokallio T., Tang J.. Methods for High-throughput drug combination screening and synergy scoring. Methods Mol. Biol. 2018; 1711:351–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Emiliani S., Fischle W., Van Lint C., Al-Abed Y., Verdin E.. Characterization of a human RPD3 ortholog, HDAC3. Proc. Nat. Acad. Sci. U.S.A. 1998; 95:2795–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van den Boom V., Maat H., Geugien M., Rodríguez López A., Sotoca A.M., Jaques J., Brouwers-Vos A.Z., Fusetti F., Groen R.W., Yuan H.et al.. Non-canonical PRC1.1 targets active genes independent of H3K27me3 and is essential for leukemogenesis. Cell Rep. 2016; 14:332–346. [DOI] [PubMed] [Google Scholar]
- 41. Lawrence A.M., Besir H.U.. Staining of proteins in gels with Coomassie G-250 without organic solvent and acetic acid. J. Visual. Exp. 2009; 30:1350–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ashoor H., Hérault A., Kamoun A., Radvanyi F., Bajic V.B., Barillot E., Boeva V.. HMCan: a method for detecting chromatin modifications in cancer samples using ChIP-seq data. Bioinformatics. 2013; 29:2979–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Polit L., Kerdivel G., Gregoricchio S., Esposito M., Guillouf C., Boeva V.. CHIPIN: ChIP-seq inter-sample normalization based on signal invariance across transcriptionally constant genes. BMC Bioinf. 2021; 22:407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Corces M.R., Trevino A.E., Hamilton E.G., Greenside P.G., Sinnott-Armstrong N.A., Vesuna S., Satpathy A.T., Rubin A.J., Montine K.S., Wu B.et al.. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods. 2017; 14:959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tanimura N., Miller E., Igarashi K., Yang D., Burstyn J.N., Dewey C.N., Bresnick E.H.. Mechanism governing heme synthesis reveals a GATA factor/heme circuit that controls differentiation. EMBO Rep. 2016; 17:249–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mavrothalassitis G., Ghysdael J.. Proteins of the ETS family with transcriptional repressor activity. Oncogene. 2000; 19:6524–6532. [DOI] [PubMed] [Google Scholar]
- 49. Papachristou E.K., Kishore K., Holding A.N., Harvey K., Roumeliotis T.I., Chilamakuri C.S.R., Omarjee S., Chia K.M., Swarbrick A., Lim E.et al.. A quantitative mass spectrometry-based approach to monitor the dynamics of endogenous chromatin-associated protein complexes. Nat. Commun. 2018; 9:2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Delva L., Gallais I., Guillouf C., Denis N., Orvain C., Moreau-Gachelin F.. Multiple functional domains of the oncoproteins Spi-1/PU.1 and TLS are involved in their opposite splicing effects in erythroleukemic cells. Oncogene. 2004; 23:4389–4399. [DOI] [PubMed] [Google Scholar]
- 51. Hallier M., Lerga A., Barnache S., Tavitian A., Moreau-Gachelin F.. The transcription factor Spi-1/PU.1 interacts with the potential splicing factor TLS. J. Biol. Chem. 1998; 273:4838–4842. [DOI] [PubMed] [Google Scholar]
- 52. Gu X., Hu Z., Ebrahem Q., Crabb J.S., Mahfouz R.Z., Radivoyevitch T., Crabb J.W., Saunthararajah Y.. Runx1 regulation of Pu.1 corepressor/coactivator exchange identifies specific molecular targets for leukemia differentiation therapy. J. Biol. Chem. 2014; 289:14881–14895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Delcuve G.P., Khan D.H., Davie J.R.. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin. Epigenetics. 2012; 4:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang T., Jian W., Luo Y., Fu X., Noguchi C., Bungert J., Huang S., Qiu Y.. Acetylation of histone deacetylase 1 regulates NuRD corepressor complex activity. J. Biol. Chem. 2012; 287:40279–40291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang Z., Zang C., Rosenfeld J.A., Schones D.E., Barski A., Cuddapah S., Cui K., Roh T.Y., Peng W., Zhang M.Q.et al.. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008; 40:897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T., Sandstrom R., Ma Z., Davis C., Pope B.D.et al.. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014; 515:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bauer W.R., Hayes J.J., White J.H., Wolffe A.P.. Nucleosome structural changes due to acetylation. J. Mol. Biol. 1994; 236:685–690. [DOI] [PubMed] [Google Scholar]
- 58. Filippakopoulos P., Picaud S., Mangos M., Keates T., Lambert J.P., Barsyte-Lovejoy D., Felletar I., Volkmer R., Müller S., Pawson T.et al.. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012; 149:214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kanno T., Kanno Y., LeRoy G., Campos E., Sun H.W., Brooks S.R., Vahedi G., Heightman T.D., Garcia B.A., Reinberg D.et al.. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat. Struct. Mol. Biol. 2014; 21:1047–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kanno T., Kanno Y., Siegel R.M., Jang M.K., Lenardo M.J., Ozato K.. Selective recognition of acetylated histones by bromodomain proteins visualized in living cells. Mol. Cell. 2004; 13:33–43. [DOI] [PubMed] [Google Scholar]
- 61. Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K.. High-resolution profiling of histone methylations in the human genome. Cell. 2007; 129:823–837. [DOI] [PubMed] [Google Scholar]
- 62. Sungalee S., Liu Y., Lambuta R.A., Katanayeva N., Donaldson Collier M., Tavernari D., Roulland S., Ciriello G., Oricchio E.. Histone acetylation dynamics modulates chromatin conformation and allele-specific interactions at oncogenic loci. Nat. Genet. 2021; 53:650–662. [DOI] [PubMed] [Google Scholar]
- 63. Soler E., Andrieu-Soler C., de Boer E., Bryne J.C., Thongjuea S., Stadhouders R., Palstra R.J., Stevens M., Kockx C., van Ijcken W.et al.. The genome-wide dynamics of the binding of Ldb1 complexes during erythroid differentiation. Genes Dev. 2010; 24:277–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cheng Y., Wu W., Kumar S.A., Yu D., Deng W., Tripic T., King D.C., Chen K.B., Zhang Y., Drautz D.et al.. Erythroid GATA1 function revealed by genome-wide analysis of transcription factor occupancy, histone modifications, and mRNA expression. Genome Res. 2009; 19:2172–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Han G.C., Vinayachandran V., Bataille A.R., Park B., Chan-Salis K.Y., Keller C.A., Long M., Mahony S., Hardison R.C., Pugh B.F.. Genome-wide organization of GATA1 and TAL1 determined at high resolution. Mol. Cell. Biol. 2016; 36:157–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wu W., Cheng Y., Keller C.A., Ernst J., Kumar S.A., Mishra T., Morrissey C., Dorman C.M., Chen K.B., Drautz D.et al.. Dynamics of the epigenetic landscape during erythroid differentiation after GATA1 restoration. Genome Res. 2011; 21:1659–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Holoch D., Margueron R.. Mechanisms regulating PRC2 recruitment and enzymatic activity. Trends Biochem. Sci. 2017; 42:531–542. [DOI] [PubMed] [Google Scholar]
- 68. Pham T.H., Minderjahn J., Schmidl C., Hoffmeister H., Schmidhofer S., Chen W., Längst G., Benner C., Rehli M.. Mechanisms of in vivo binding site selection of the hematopoietic master transcription factor PU.1. Nucleic Acids Res. 2013; 41:6391–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wontakal S.N., Guo X., Smith C., Maccarthy T., Bresnick E.H., Bergman A., Snyder M.P., Weissman S.M., Zheng D., Skoultchi A.I.. A core erythroid transcriptional network is repressed by a master regulator of myelo-lymphoid differentiation. Proc. Nat. Acad. Sci. U.S.A. 2012; 109:3832–3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hu Z., Gu X., Baraoidan K., Ibanez V., Sharma A., Kadkol S., Munker R., Ackerman S., Nucifora G., Saunthararajah Y.. RUNX1 regulates corepressor interactions of PU.1. Blood. 2011; 117:6498–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kihara-Negishi F., Yamamoto H., Suzuki M., Yamada T., Sakurai T., Tamura T., Oikawa T.. In vivo complex formation of PU.1 with HDAC1 associated with PU.1-mediated transcriptional repression. Oncogene. 2001; 20:6039–6047. [DOI] [PubMed] [Google Scholar]
- 72. Lamonica J.M., Vakoc C.R., Blobel G.A.. Acetylation of GATA-1 is required for chromatin occupancy. Blood. 2006; 108:3736–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Boyes J., Byfield P., Nakatani Y., Ogryzko V.. Regulation of activity of the transcription factor GATA-1 by acetylation. Nature. 1998; 396:594–598. [DOI] [PubMed] [Google Scholar]
- 74. Yan B., Yang J., Kim M.Y., Luo H., Cesari N., Yang T., Strouboulis J., Zhang J., Hardison R., Huang S.et al.. HDAC1 is required for GATA-1 transcription activity, global chromatin occupancy and hematopoiesis. Nucleic Acids Res. 2021; 49:9783–9798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Frank C.L., Manandhar D., Gordân R., Crawford G.E.. HDAC inhibitors cause site-specific chromatin remodeling at PU.1-bound enhancers in K562 cells. Epigenetics Chromatin. 2016; 9:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dey A., Chitsaz F., Abbasi A., Misteli T., Ozato K.. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Nat. Acad. Sci. U.S.A. 2003; 100:8758–8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dhalluin C., Carlson J.E., Zeng L., He C., Aggarwal A.K., Zhou M.M.. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999; 399:491–496. [DOI] [PubMed] [Google Scholar]
- 78. Jang M.K., Mochizuki K., Zhou M., Jeong H.S., Brady J.N., Ozato K.. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell. 2005; 19:523–534. [DOI] [PubMed] [Google Scholar]
- 79. Yang Z., Yik J.H., Chen R., He N., Jang M.K., Ozato K., Zhou Q.. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell. 2005; 19:535–545. [DOI] [PubMed] [Google Scholar]
- 80. Gjidoda A., Tagore M., McAndrew M.J., Woods A., Floer M.. Nucleosomes are stably evicted from enhancers but not promoters upon induction of certain pro-inflammatory genes in mouse macrophages. PLoS One. 2014; 9:e93971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Beltran M., Yates C.M., Skalska L., Dawson M., Reis F.P., Viiri K., Fisher C.L., Sibley C.R., Foster B.M., Bartke T.et al.. The interaction of PRC2 with RNA or chromatin is mutually antagonistic. Genome Res. 2016; 26:896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Davidovich C., Zheng L., Goodrich K.J., Cech T.R.. Promiscuous RNA binding by Polycomb repressive complex 2. Nat. Struct. Mol. Biol. 2013; 20:1250–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kaneko S., Son J., Shen S.S., Reinberg D., Bonasio R.. PRC2 binds active promoters and contacts nascent RNAs in embryonic stem cells. Nat. Struct. Mol. Biol. 2013; 20:1258–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Young M.D., Willson T.A., Wakefield M.J., Trounson E., Hilton D.J., Blewitt M.E., Oshlack A., Majewski I.J.. ChIP-seq analysis reveals distinct H3K27me3 profiles that correlate with transcriptional activity. Nucleic Acids Res. 2011; 39:7415–7427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cheng J.X., Chen L., Li Y., Cloe A., Yue M., Wei J., Watanabe K.A., Shammo J.M., Anastasi J., Shen Q.J.et al.. RNA cytosine methylation and methyltransferases mediate chromatin organization and 5-azacytidine response and resistance in leukaemia. Nat. Commun. 2018; 9:1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Larsson C.A., Cote G., Quintas-Cardama A.. The changing mutational landscape of acute myeloid leukemia and myelodysplastic syndrome. Mol. Cancer Res. 2013; 11:815–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ley T.J., Miller C., Ding L., Raphael B.J., Mungall A.J., Robertson A., Hoadley K., Triche T.J. Jr, Laird P.W., Baty J.Det al.. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013; 368:2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Basheer F., Giotopoulos G., Meduri E., Yun H., Mazan M., Sasca D., Gallipoli P., Marando L., Gozdecka M., Asby R.et al.. Contrasting requirements during disease evolution identify EZH2 as a therapeutic target in AML. J. Exp. Med. 2019; 216:966–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Neff T., Sinha A.U., Kluk M.J., Zhu N., Khattab M.H., Stein L., Xie H., Orkin S.H., Armstrong S.A.. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc. Nat. Acad. Sci. U.S.A. 2012; 109:5028–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tanaka S., Miyagi S., Sashida G., Chiba T., Yuan J., Mochizuki-Kashio M., Suzuki Y., Sugano S., Nakaseko C., Yokote K.et al.. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood. 2012; 120:1107–1117. [DOI] [PubMed] [Google Scholar]
- 91. Iacobucci I., Qu C., Varotto E., Janke L.J., Yang X., Seth A., Shelat A., Friske J.D., Fukano R., Yu J.et al.. Modeling and targeting of erythroleukemia by hematopoietic genome editing. Blood. 2021; 137:1628–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Aikawa Y., Katsumoto T., Zhang P., Shima H., Shino M., Terui K., Ito E., Ohno H., Stanley E.R., Singh H.et al.. PU.1-mediated upregulation of CSF1R is crucial for leukemia stem cell potential induced by MOZ-TIF2. Nat. Med. 2010; 16:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhou J., Wu J., Li B., Liu D., Yu J., Yan X., Zheng S., Wang J., Zhang L., Zhang L.et al.. PU.1 is essential for MLL leukemia partially via crosstalk with the MEIS/HOX pathway. Leukemia. 2014; 28:1436–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Antony-Debre I., Steidl U.. Functionally relevant RNA helicase mutations in familial and sporadic myeloid malignancies. Cancer Cell. 2015; 27:609–611. [DOI] [PubMed] [Google Scholar]
- 95. Mei S., Qin Q., Wu Q., Sun H., Zheng R., Zang C., Zhu M., Wu J., Shi X., Taing L.et al.. Cistrome data browser: a data portal for ChIP-Seq and chromatin accessibility data in human and mouse. Nucleic Acids Res. 2017; 45:D658–d662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data of ChIP-seq, CUT&Tag, RNA-seq, ATAC-seq and 4C-seq are available under accession number GSE172088. Publicly available raw data used for SPI1 in TgSpi1 erythroid cells and GATA1 in MEL cells ChIP-seq were found with the accession number GSE33611 (GEO) and ERR007441-44 (ENA), respectively. SIN3A (CistromeDB #46488) and EP300 (CistromeDB #46468) signal files (.bw) data were obtained from Cistrome DB (95) analysis of ChIP-seq performed in MEL cells (56). GATA1 ChIP-seq peak files (.bed) were obtained from ES-G1ER cells restored for GATA1 activity (64,65) (CistromeDB #502 and CistromeDB #56614–16) or from foetal liver E14.5 Ter119+ proerythroblasts (CistromeDB #3270–71) (66) publicly available on CistromeDB.