


Abstract
Coronaviruses generate double-stranded (ds) RNA intermediates during viral replication that can activate host immune sensors. To evade activation of the host pattern recognition receptor MDA5, coronaviruses employ Nsp15, which is a uridine-specific endoribonuclease. Nsp15 is proposed to associate with the coronavirus replication-transcription complex within double-membrane vesicles to cleave these dsRNA intermediates. How Nsp15 recognizes and processes dsRNA is poorly understood because previous structural studies of Nsp15 have been limited to small single-stranded (ss) RNA substrates. Here we present cryo-EM structures of SARS-CoV-2 Nsp15 bound to a 52nt dsRNA. We observed that the Nsp15 hexamer forms a platform for engaging dsRNA across multiple protomers. The structures, along with site-directed mutagenesis and RNA cleavage assays revealed critical insight into dsRNA recognition and processing. To process dsRNA Nsp15 utilizes a base-flipping mechanism to properly orient the uridine within the active site for cleavage. Our findings show that Nsp15 is a distinctive endoribonuclease that can cleave both ss- and dsRNA effectively.
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has infected millions worldwide and led to the unprecedented COVID-19 global health pandemic. Despite the rapid development of effective vaccines, new anti-viral treatments are urgently needed due to emerging SARS-CoV-2 variants and the prevalence of break-through infections amongst those already vaccinated. Coronaviruses are a family of large positive sense single stranded (ss) RNA viruses that utilize a multi-subunit replication-transcription complex (RTC) to replicate the viral genome and synthesize sub-genomic viral mRNA transcripts within double membrane vesicles (1,2). The RTC is composed of multiple viral non-structural proteins (Nsps), including an RNA-dependent RNA polymerase (RdRp, Nsp12), RNA helicase (Nsp13), dual exonuclease/N7-Methyltransferase (Nsp14), 2′O-Methyltransferase (Nsp16), and multiple accessory/cofactor subunits (Nsp7, Nsp8, Nsp9 and Nsp10) (2–6). The RTC is a leading target for new anti-viral therapeutics such as nucleotide analogues that specifically target the Nsp12 polymerase subunit (6–8).
During viral replication the RTC generates double-stranded (ds) RNA intermediates, which are well established activators of the innate immune defense system (2,9–11). To prevent the accumulation of long dsRNAs, which activate the pattern recognition receptor MDA5, coronaviruses employ a uridine specific endoribonuclease (EndoU, Nsp15) (12,13). EndoU like enzymes are also found across the larger nidovirus family suggesting that this endonuclease activity is critically important for single-stranded (ss) RNA viruses. However, Nsp15 is one of the most understudied non-structural proteins (5,14,15). In virus infected cells Nsp15 has been shown to cleave the polyU tail at the 5′-end of the negative strand replication intermediate as well as numerous sites within the positive strand (12,16). In vitro, Nsp15 has broad cleavage specificity towards ssRNA substrates and is largely guided to its cleavage sites by recognition of uridine (17). Structures of SARS-CoV-2 Nsp15 in complex with single and di-nucleotide substrates have revealed the molecular basis for uridine specificity and provided molecular details into the RNase A-like transesterification reaction (17–19). Recent molecular modeling suggests that beyond its role in cleaving viral RNA, Nsp15 may play a pivotal role as the central scaffold for the RTC complex (5). Nsp15 assembles into a 240 kDa hexamer and this oligomerization is required for nuclease activity (20). While the molecular basis for oligomerization is still not fully understood (19), it has been proposed that the Nsp15 hexamer functions as the central scaffold for the RTC to coordinate the RTC’s numerous enzymatic activities by serving as a platform for binding dsRNA generated during replication (5). This model is further supported by work establishing that Nsp15 co-localizes with members of the RTC (21,22).
Earlier work with SARS-CoV-1 Nsp15 revealed that the enzyme can cleave both ss- and dsRNA substrates (14). It is not known how Nsp15 can cleave dsRNA as this would require movement of a base-paired uridine into the uridine binding pocket, where it specifically interacts with a well conserved serine residue (17–19). Furthermore, outside of the EndoU active site it remains unclear how the largely electronegative surface of the Nsp15 hexamer could engage a dsRNA substrate. Central to the Nsp15 RTC scaffold model is Nsp15′s ability to engage dsRNA across multiple Nsp15 protomers, however there is no experimental to support this model of dsRNA binding (5). To address these gaps in our knowledge we solved structures of SARS-CoV-2 bound to dsRNA and determined the significance of RNA-protein interfaces by site-directed mutagenesis coupled with nuclease assays. We discovered that SARS-CoV-2 Nsp15 can process dsRNA through a unique base-flipping mechanism.
MATERIALS AND METHODS
Protein expression and purification
Wild type (WT) and mutant Nsp15 constructs were created as described previously (17,19). Nsp15 was overexpressed in E. coli C41 (DE3) competent cells in Terrific Broth with 100 mg/L ampicillin. At an optical density (600 nm) between 0.8 and 1.0, cultures were cooled prior to induction with 0.2 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). Cells were harvested after overnight expression at 16°C and stored at −80°C until use. Nsp15 purification was done as described previously (17,19). Briefly, cells were resuspended in Lysis Buffer (50 mM Tris pH 8.0, 500 mM NaCl, 5% glycerol, 5 mM β-ME, 5 mM imidazole) supplemented with cOmplete EDTA-free protease inhibitor tablets (Roche) and disrupted by sonication. The lysate was clarified at 26 915 × g for 50 min at 4°C and then incubated with TALON metal affinity resin (Clontech). His-Nsp15 was eluted from the resin with 250 mM imidazole, and buffer exchanged into Thrombin Cleavage Buffer (50 mM Tris pH 8.0, 150 mM NaCl, 5% glycerol, 2 mM β-ME, 2 mM CaCl2) for cleavage at room temperature for 4 h. The cleavage reaction was repassed over TALON resin and quenched with 1 mM phenylmethylsulfonyl fluoride (PMSF) prior to gel filtration using a Superdex-200 column equilibrated in SEC buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 5 mM MnCl2, 5 mM β-ME).
Cryo-EM sample preparation
RNA oligos (Supplementary Table 1) were annealed at 250 uM final concentration by incubating equimolar amounts for 5 min at 75°C and then cooled on the heat block for 1.5 h. Purified Nsp15 H235A was diluted in a low-salt buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 5 mM MnCl2, 5 mM β-ME) to 0.75 μM and incubated with excess RNA substrate (50 μM) for 1 h at 4°C. The purified protein was plunge frozen on homemade customized support grids made of C-flat R1.2/1.3 (Protochips) sputtered with 30 nm thick layer of gold on the grid bar side using Leica ACE-600 sputterer. Before specimen deposition, the grids were pretreated on a Tergeo plasma cleaner (Pie Scientific) in immersion mode with a power of 38 W for a period of 75 s. The Nsp15/RNA mixture (3 μl) was deposited onto the grids inside the chamber of a Leica EM-GP2 vitrification robot held at 15°C and RH of 90% humidity. The grid was back-blotted for 3 s (Whatman Grade 40 filter paper), and then quickly plunged into liquid ethane kept at 90 K. The vitrified sample on the grid was then transferred to liquid nitrogen for storage.
Data collection and processing
Nsp15 images were collected using a Talos Arctica electron microscope at 200 keV with a Gatan K2 Summit detector, and a Titan Krios at 300 keV with a K3 Bioquantum detector. Beam-induced motion and drift were corrected using MotionCor2 (23) through Scipion 3 (24). CryoSPARC v2 (25) was used in all subsequent image processing. The aligned dose-weighted images were used to calculate CTF parameters using CTFFIND4 (26). For the first dataset, particles were selected using blob and template-based particle picking, downsampled by a factor of 4, extracted with a box size of 64 and subjected to an initial round of 2D classification. Full resolution particle projections from good classes were re-extracted using a box size of 256. Ab initio reconstruction was used to build an initial model, followed by several rounds of 3D classification with localized CTF refinement and per particle motion correction. No symmetry was applied.
For the combined Arctica/Krios datasets, the Topaz Extract machine learning algorithm was used to pick particles. The extracted particles were curated and segregated using a combination of 2D classifications, ab initio model reconstruction and heterogeneous refinement. The curated particles stacks from both Talos Arctica and Titan Krios were then combined. The combined particle stack that yielded the most ideal 3D map was then further refined using homogeneous, Global CTF, and Non-Uniform Refinements without symmetry applied. Maps were re-scaled to optimize RMS fit to core domain residues of reference structure PDBID 6WLC (27).
Model building
A SARS-CoV-2 Nsp15 cryo-EM structure (PDBID 7K0R) was used as a starting model and fit into the cryo-EM maps using rigid body docking in Phenix (28). A combination of rigid body and real-space refinement in Phenix as well as iterative rounds of building in COOT (29) were used to improve the fit of the models. Due to the weak RNA density the exact register of the RNA is not known. dsRNA was built manually using one possible binding configuration, supported by the higher quality density at the active site. The quality of the RNA density is poorer surrounding the termini, suggesting that the dsRNA is not fixed into position once it is no longer in contact with Nsp15. Given the weaker density of the RNA, it was restrained to model RNA parameters during refinement. Molprobity (30) was used to evaluate the model (Table 1). Figures were prepared using Chimera (31) and Chimera X (32).
Table 1.
Cryo-EM collection and processing statistics
| Data collection and processing | Nsp15/dsRNA dataset 1 (EMD-26073, PDB: 7TQV) | Nsp15/dsRNA dataset 2 (EMD-25915 PDB: 7TJ2) |
|---|---|---|
| Microscope | Talos Arctica | Titan Krios |
| Detector | Gatan K2 Summit | Gatan K3 Bioquantum |
| Nominal magnification | 45 000× | 81 000× |
| Voltage (kV) | 200 | 300 |
| Electron exposure (e–/Å2) | 54 | 60 |
| Defocus range (μm) | −0.8 to -1.8 | −1.2 to 2.2 |
| Pixel size (Å) | 0.93 | 0.53 |
| Symmetry imposed | C1 | C1 |
| Number of Micrographs | 2435 | 752 |
| Initial particle images | 1 201 304 | 254 136 |
| Final particle images | 398 516 | 97 592 |
| Refinement | Dataset 1 | Combined Datasets |
| Resolution (Å)/FSC threshold | 3.43/0.143 | 3.19/0.143 |
| B-factor used for map sharpening (Å2) | 162.7 | 171.3 |
| Map to model CC | ||
| CC (mask) | 0.69 | 0.76 |
| CC (volume) | 0.68 | 0.74 |
| CC (peaks) | 0.56 | 0.65 |
| CC (box) | 0.68 | 0.69 |
| Model composition | ||
| Non-hydrogen atoms | 17 793 | 17 084 |
| Protein residues | 2083 | 2007 |
| Nucleic acid | 66 | 62 |
| Mean B factors (Å2) | ||
| Protein | 85.95 | 58.77 |
| Nucleic acid | 277.22 | 150.81 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.002 | 0.002 |
| Bond angles (°) | 0.383 | 0.370 |
| Validation | ||
| Molprobity score | 1.52 | 1.39 |
| Clashscore | 5.00 | 3.74 |
| Poor rotamers (%) | 0.82 | 1.64 |
| Ramachandran plot | ||
| Favored (%) | 96.18 | 97.69 |
| Allowed (%) | 3.82 | 2.31 |
| Disallowed (%) | 0.00 | 0.00 |
Urea-PAGE endoribonuclease assay
DsRNA was prepared by incubating equimolar concentrations of complementary oligos at 75°C for 5 min, followed by cooling on the heat block for 1.5 h. To remove any remaining unpaired RNA, samples were purified on a 20% native polyacrylamide gel. The dsRNA band was excised, and the RNA was eluted overnight in 10 mM HEPES pH 7.5, 50 mM NaCl, 0.5 u/μl RNasin (Promega). Double fluorescently-labeled RNA substrates (5′-FI and 3′-Cy5, 500 nM) were incubated with Nsp15 (50 nM) in RNA cleavage buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 5 mM MnCl2, 5 mM DTT, 1 u/μl RNasin ribonuclease inhibitor) at room temperature for 30 min, with samples collected at 0, 1, 5, 10 and 30 min. The reaction was quenched with 2× urea loading buffer (8 M urea, 20 mM Tris pH 8.0, 1 mM EDTA). At least three independent reactions were performed with protein from at least two different purifications. Due to the expected size of cleavage products and the size of bromophenol blue, loading buffer without dye was used. To monitor the gel front, a control lane of protein only with bromophenol blue was run. A ladder composed of double and single labeled oligos of different sizes were used to help assign cleavage product identities. Cy5-labeled products run anomalously compared to FI-labeled products (33,34). The cleavage reactions were separated using 15% TBE-urea PAGE gels and visualized with a Typhoon RGB imager (Amersham) using Cy2 (λex = 488 nm, λem = 515–535 nm) and Cy5 (λex = 635 nm, λem = 655–685 nm) channels. RNA cleavage was measured by disappearance of the uncleaved RNA, and quantified using Image Studio Lite (LI-COR). Prism (Graphpad) was used to calculate significant differences using Dunnett's T3 multiple corrections test.
RESULTS
Structure of SARS-CoV-2 Nsp15 bound to dsRNA
To experimentally determine how SARS CoV-2 Nsp15 interacts with dsRNA, we determined a cryo-EM structure of the Nsp15 hexamer bound to a 52-nt blunt termini dsRNA (35) (Figure 1, Supplementary Table S1, Table 1 and Supplementary Figure S1). To prevent RNA cleavage, we used the H235A Nsp15 catalytic deficient mutant for structural studies (19). We incubated Nsp15 with an excess of annealed dsRNA for 1 h prior to vitrification and could visualize dsRNA extending from the Nsp15 hexamer in the 2D classes (Figure 1A). Following 3D classification and refinement the Nsp15 dsRNA complex converged on a single asymmetric class with one dsRNA bound to the Nsp15 hexamer (Supplementary Figure S1). The overall resolution of the reconstruction goes to 3.4 Å, and there is clear density for dsRNA engaged by the Nsp15 hexamer however, the local resolution is lower for the dsRNA (Figure 1B, Supplementary Figure S2). This variable local resolution led to high B-factors in our dsRNA model outside of the active site (Supplementary Figure S3). The dsRNA substrate we used contains multiple uridine nucleotides in each strand, leading to heterogeneity in the register of the dsRNA (Figure 1B). Despite the nucleotide heterogeneity we were able to dock in a model of dsRNA containing 35 nucleotides in each strand and spanning a length of ∼100 Å (Supplementary Figure S4). The exact register of the dsRNA is not known. The local resolution of the EndoU domains that do not engage the dsRNA is also lower supporting our earlier observation that substrate is required to fix the EndoU domain into position (19) (Figure 1B, Supplementary Figure S2). To improve the resolution of the dsRNA surrounding the engaged EndoU active site we collected an additional dataset to increase the number of particles, resulting in a second reconstruction with an overall resolution to 3.2 Å and improved resolution in the engaged EndoU active site (Supplementary Figure S5 and Table 1). The improved density led to better model B-factors for the RNA, though they are still higher outside of the active site (Supplementary Figure S6). Overall, the reconstructions are very similar to one another. The first reconstruction in which we can see more overall density for the dsRNA was used for global analysis and docking of the dsRNA, while the second reconstruction was used for a specific analysis of the engaged endoU active site.
Figure 1.

DsRNA binds Nsp15 through interactions with two platforms in addition to the EndoU active site. (A) Selected 2D classifications of Nsp15 bound to dsRNA. DsRNA, indicated by arrows is visible extending away from the complex. Some classes suggest binding of more than one dsRNA substrate; however these were not the predominant views. (B) Side view of the complex in ribbon (left), EM density (middle), and local resolution (right) views.
Nsp15 engages dsRNA through multiple interfaces
In both cryo-EM reconstructions the Nsp15 hexamer asymmetrically engages one dsRNA substrate. Nsp15 forms a hexamer of back-to-back trimers. The dsRNA interacts with three of the six Nsp15 protomers; two from the top trimer (P1 and P3), and one from the bottom trimer (P6, Figure 1B). Furthermore, across the three interacting protomers the dsRNA ultimately interacts with residues spanning all three domains that comprise an Nsp15 protomer (N-terminal, Middle, and catalytic EndoU domain; Figure 2, Supplementary Figure S7). We identified 4 key RNA-protein interfaces within the structure (Figure 2A, Supplementary Figure S7). The N-terminal domain (NTD) of Nsp15, including residues Q19 and Q20, interacts with the dsRNA through P1 in the top trimer (P1-B). Two different interfaces stem from the middle domain (MD): P1 in the top trimer (P1-A), and P6 in the bottom trimer, both of which predominantly interact with the backbone through the major groove. Key residues in these interfaces include K65, K111 and K150. Finally, the EndoU domain of P3 in the top trimer interacts with the dsRNA through interactions with both the minor and major groove. The minor groove interaction is mediated by EndoU residues such as H243 and Q245, while the major groove interaction includes residues W333, Y343 and E340. The structure revealed that Nsp15 interacts with both strands of the dsRNA, and except for the active site uridine, all of the protein-RNA interactions are RNA backbone mediated. This supports earlier work demonstrating that Nsp15 has broad specificity towards uridine containing substrates (16,17).
Figure 2.
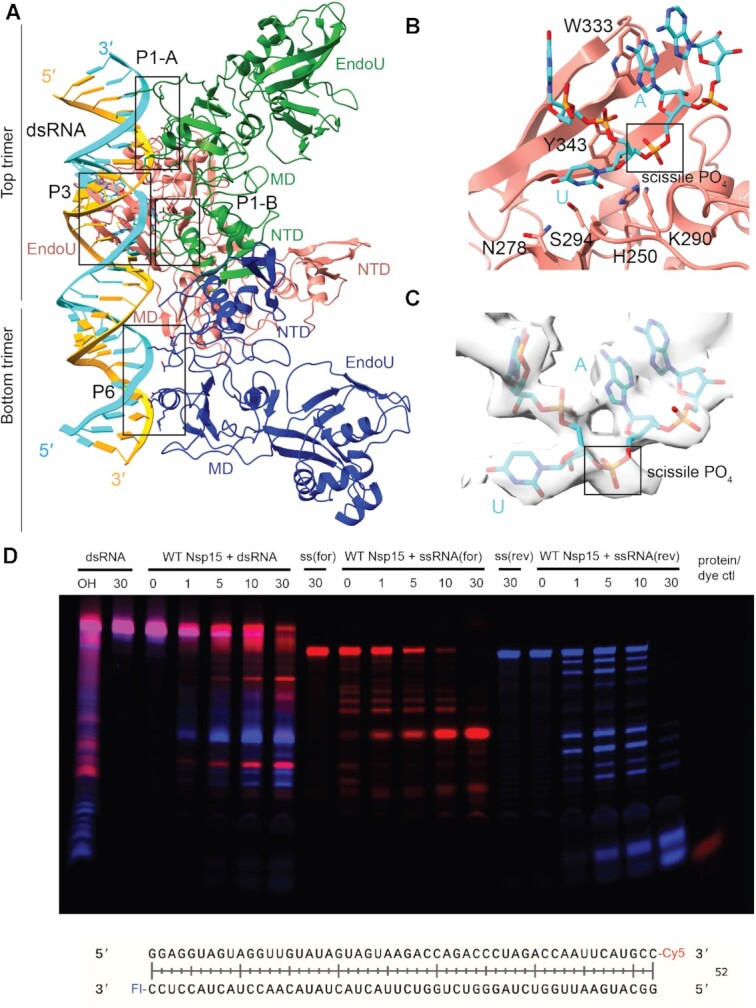
Nsp15 can cleave both ss- and dsRNA. (A) DsRNA interacts with three Nsp15 protomers, across both trimers. P1 and P6 form platforms that support RNA cleavage by P3. (B) Close-up of the active site (P3). Critical residues are shown in stick format. Uridine flips out to interact with S294 and N278, which provides optimal positioning for the catalytic triad. The 3′ base is stabilized by W333. (C) Cryo-EM density for the RNA engaged in the active site. (D) Time-course cleavage reaction for the dsRNA as well as each strand alone. Nsp15 cleaves ssRNA more quickly than dsRNA, and prefers different positions depending on that context. OH: alkaline hydrolysis of dsRNA. ssRNA(for) is the Cy5 labeled strand (red); ssRNA(rev) is the FI-labeled strand (blue).
Overall, our structures are in good agreement with the predicted position of dsRNA binding interfaces derived from the computational model of Nsp15 proposed in the RTC Nsp15 based scaffold (Supplementary Figure S8) (5). However, unlike the model which proposes multiple copies of RNA bound to the Nsp15 hexamer, the majority of our cryo-EM 2D classes revealed only one RNA bound per hexamer (Figure 1). Previously determined cryo-EM structures of Nsp15 determined in the presence and absence of small RNA substrates have revealed both symmetric and asymmetric states, thus we assume it is possible for Nsp15 to engage more than one dsRNA substrate at a time (17,19). We hypothesize that the Nsp15 scaffold is a very transient part of the RTC as this platform positions the dsRNA within the EndoU active site. This Nsp15-scaffolded RTC may only form during certain phases of replication (i.e. discontinuous transcription). Alternatively, the assembly of the RTC complex with Nsp15 in the center could lock Nsp15 into an inactive conformation incapable of cleaving dsRNA.
Nsp15 base flipping mechanism
While the register for the RNA is ambiguous in our structure, we observed density for a nucleotide engaged in the active site, which we modeled as a uridine. The engaged uridine flips out from the dsRNA helix and is positioned in the active site as previously observed in UMP, and di-nucleotide bound structures (Supplementary Figure S9) (17–19). As a result of the flipping of the engaged uridine the base 3′ of the uridine, which we modeled as an A, is destabilized from the helix. In our structure this base engages with W333, which likely stabilizes that base to support cleavage of the phosphate backbone 3′ of the engaged uridine (Figure 2B). Collectively both the flipping of the uridine and the engagement the 3′ base with W333 re-position the scissile phosphate within the Nsp15 active site composed of the catalytic residues H235, H250 and K290. Aside from the flipped base, we do not have sufficient density to determine if the global dsRNA structure is otherwise perturbed. Many nucleic acid processing enzymes utilize a base-flipping mechanism to position substrates within their active sites, however most of these enzymes do not flip bases from normal, A-form dsRNA (36–41). The base-flipping mechanism in Nsp15 is reminiscent to the mechanism used by adenosine deaminases acting on RNA (ADARs), which convert adenosine to inosine in dsRNA. Like Nsp15, ADARs use a base flipping mechanism to position the reactive adenosine within the editing active site. Thus, Nsp15 appears to join a small group of RNA enzymes, including ADARS, known to flip a base from a normal duplex RNA substrate (36). It remains to be seen if Nsp15 actively flips out the engaged uridine or if it stabilizes the flipped-out base through a passive process.
Nsp15 cleaves ss- and dsRNA
Following structure determination, we confirmed that SARS-CoV-2 Nsp15 cleaves dsRNA substrates. Previous studies with SARS-CoV-1 and human coronavirus 299E (HCov-229E) Nsp15 using a 22-nt RNA substrate containing a GUU sequence suggested that Nsp15 cleaves dsRNA more efficiently than ssRNA (14). To confirm that SARS-CoV2 Nsp15 can cleave dsRNA we carried out a time course using the 52-nt dsRNA from the cryo-EM structure with labels on the 3′ ends. We observed that WT Nsp15 can cleave the duplex 52-nt substrate as well as the individual strands at numerous positions; this was not observed with catalytically inactive Nsp15 H235A (Figure 2D; Supplementary Figure S10). We observed that the accumulation of cleavage products is different suggesting that the cleavage specificity for ssRNA vs dsRNA varies. A similar observation of altered specificity was previously made with SARS-CoV-1 and HCov-299E Nsp15 (14). We hypothesize that this could be a result of the base flipping mechanism. Uridines followed by strong base pairs may not be as accessible to the EndoU active site. This is supported by RNA sequencing data from mouse hepatitis virus (MHV) infected cells which showed a strong preference for adenines 3′ of Nsp15 cleavage sites detected in the positive strand (16). Similarly, ADARs have a nearest neighbor preference for specific base-pairs before and after the reactive adenine that flips into the active site (36,42).
To further probe Nsp15′s ability to cleave dsRNA we measured cleavage of an alternative dsRNA substrate. Our structure suggests that dsRNA of approximately 35-nts is sufficient to interact across the Nsp15 hexamer, so we expanded on the nucleocapsid protein transcriptional regulatory sequence (TRS-N) previously used to characterize ssRNA cleavage preferences (Supplementary Table S1, (17)). TRS sequences are found upstream in many of the viral subgenomic RNAs, are important for facilitating discontinuous transcription and are known to form dsRNA intermediates (2,43). To compare nuclease activity, a 60 min time course reaction with 35-nt ss- and dsRNA was performed and the disappearance of the uncleaved RNA band quantified (Figure 3). Similar to our results with the 52-nt dsRNA substrate, Nsp15 can cleave both the ss- and dsRNA TRS and we observe that the ssRNA is cleaved faster than the dsRNA under the same reaction conditions. We hypothesize that ssRNA is cleaved faster because it is more accessible to the Nsp15 EndoU active site. We also observe different cleavage patterns for the ss- and dsTRS (Figure 3A) further suggesting that Nsp15′s substrate specificity is altered for ss and dsRNA. Some of the prominent dsRNA cleavage products occur near the termini of the dsRNA for the TRS-N substrate. We cannot rule out the possibility that cleavage products near the termini do not need to undergo base-flipping and rather are made accessible to the EndoU active site through a breathing mechanism.
Figure 3.
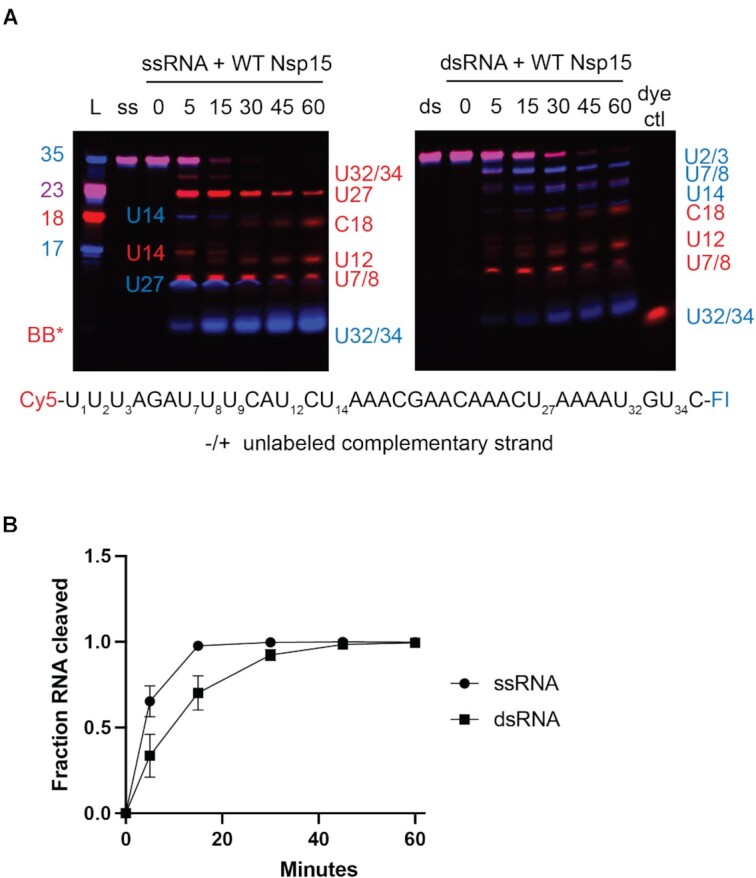
Comparison of WT Nsp15 activity on ss- and ds-RNA. (A) Time course reactions with WT Nsp15 were performed on double-labeled ssRNA ± the unlabeled complementary strand. Cleavage products are labeled to the right of the gel, with the color corresponding to which product (Cy5 or FI) it is. (B) RNA cleaved was quantified by disappearance of the uncleaved RNA band, normalized to the 0 time point for that reaction. The average and standard deviation for at least three independent reactions are graphed.
W333 mediates dsRNA cleavage
To test the importance of dsRNA-interacting residues identified by our structure, we made point mutants for several residues spanning the three domains of Nsp15 (Figure 4-5, Supplementary Table S2, and Supplementary Figure S11–S13). We compared the activity of these mutants on both 35-nt ss- and dsRNA using gel-based cleavage assays (Figure 5, Supplementary Figures S11, S12). Most of the single point mutants exhibited little to no defects in ssRNA and small defects in dsRNA processing, likely because of the large number of residues that support dsRNA engagement. Given that the majority of the single point mutants did not have a large impact on dsRNA cleavage, we reasoned multiple mutations to the interaction surface might be necessary. We created an Nsp15 mutant with eight point mutations across the interacting regions on P1, P3 and P6 (Figure 4A) and showed that dsRNA cleavage was selectively impaired (Figure 4B), demonstrating the importance of these regions for engaging dsRNA. One single point mutant, E340A gave rise to an increase in cleavage activity (Supplementary Figure S12). Given that RNA is negatively charged, the positioning of E340 in the active site may be important for regulating nuclease activity. We also identified a critical active site mutation that is important for dsRNA cleavage. Our structure suggests that W333 plays a crucial role in stabilizing the dsRNA, with a flipped-out base by interacting with the base 3′ of the cleavage site. While W333A results in a 20% reduction of ssRNA cleavage at 60 min, the defect in dsRNA is much more severe, resulting in 75% less cleavage at 60 min (Figure 5). This observation is further supported by our earlier structure of Nsp15 bound to ssRNA in the pre-cleavage state, in which we could not observe strong density for the base 3′ of the cleavage site (17). Collectively this suggests that W333 is a critical regulator for stabilizing the base 3′ of the cleavage site in dsRNA but is not essential for positioning the same base within a flexible ssRNA substrate (Figure 6).
Figure 4.
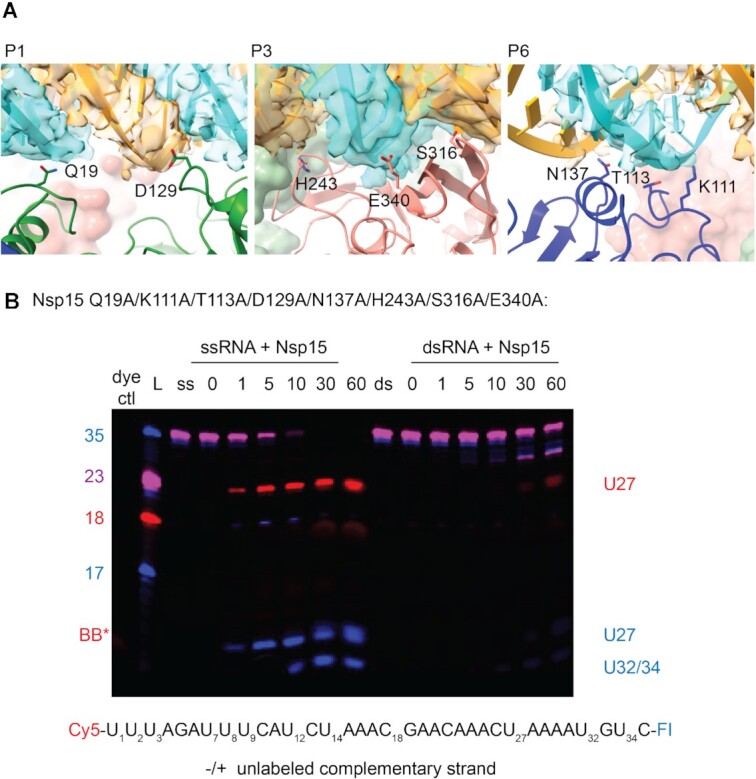
Combined dsRNA surface mutant disrupts dsRNA cleavage. (A) Panels depicting the mutated residues at the P1, P3, and P6 interfaces (left to right). RNA cryo-EM density is shown as a transparent surface, with the modeled RNA shown as ribbons and slabs. (B) Nsp15 with mutations in residues near the dsRNA binding interface (Q19A, K111A, T113A, D129A, N137A, H243A, S136A and E340A) was tested for cleavage activity with ss- and dsRNA. A representative time-course cleavage gel from three technical replicates is shown.
Figure 5.

EndoU domain mutations in Nsp15 affect dsRNA cleavage. (A) Zoomed in depiction of the active site mutants, focused on W333. (B) Time course reactions for Nsp15 active site mutants were performed with double-labeled ssRNA ± the unlabeled complementary strand. A representative time course is shown for Nsp15 W333A. Cleavage products are labeled to the right of the gel, with the color corresponding to which product (Cy5 or FI) it is. (C, D) RNA cleaved was quantified by disappearance of the uncleaved RNA band, normalized to the 0 time point for that reaction. The average and standard deviation for at least three independent reactions are graphed.
Figure 6.

Model comparing ss- and dsRNA cleavage by Nsp15. Nsp15 cleaves ssRNA mainly through engagement of the U in the active site, with limited binding by upstream or downstream nucleotides. ssRNA is readily accessible to the Nsp15 active site. Nsp15 cleaves dsRNA by multiple interaction sites, spanning three protomers and both trimers that compose the hexamer. The U base must flip out of the base-paired helix to be cleaved; distortion in the RNA is stabilized through π-stacking with W333. Figure made in Biorender.
DISCUSSION
Overall, our structures and biochemical assays provide molecular insight into the mechanism of dsRNA processing by SARS-CoV-2 Nsp15. We observed that the Nsp15 hexamer forms a platform across multiple protomers to engage dsRNA. Three of the six protomers of Nsp15 form an electropositive surface for dsRNA of ∼100 Å in length. There are numerous ways that ribonucleases and other RNA binding proteins interact with RNA but many of these interactions are mediated by small RNA binding domains (44). For example, the small structurally conserved dsRNA Binding Domain (dsRBD) is utilized by many RNA processing enzymes either on its own or in combination with other domains to mediate dsRNA binding (45). This domain has been well-studied in RNase III family members, such as dicer and drosha, where it has been shown to specifically recognize features within the minor groove of A-form RNA (46). ADARs also contain multiple dsRBDs, outside of the catalytic deaminase domain, that contribute to RNA binding and are required for editing of some dsRNA substrates (47). In contrast Nsp15 does not use a single domain or even a single protomer to bind RNA, rather it uses all three of its domains across multiple protomers to engage dsRNA. This unique RNA interface provides the molecular basis for Nsp15′s dependence on oligomerization. Our study reveals that the oligomerization of Nsp15 is necessary to form the platform for RNA binding with contributions from three of the six protomers.
Studies with Nsp15 from other coronaviruses, as well as Nsp11, the EndoU containing Nsp in arteriviruses, have demonstrated oligomerization is necessary for endoribonuclease activity although the type of oligomer (dimer, hexamer) varies across viruses (27,48). We compared our Nsp15/dsRNA structure with previous structural work that assessed the role of oligomerization and activity of nidovirus EndoU enzymes (Supplementary Figure S14). Structures of MERS-CoV and SARS-CoV-1 Nsp15 superpose very well with SARS-CoV-2 Nsp15 (27), suggesting that the dsRNA platform is conserved across CoVs. In contrast to Nsp15, Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Nsp11 forms a dimer, not a hexamer. We modeled dsRNA onto the PRRSV Nsp11 dimer by superposition of the EndoU domains and observed that the dsRNA fits in the groove formed between Nsp11 protomers. The π-stacking tryptophan is conserved and positioned to stabilize the dsRNA when the uridine is flipped out. This observation suggests that the base-flipping mechanism is conserved across nidovirus EndoU enzymes while the dsRNA platform differs.
Beyond facilitating dsRNA binding and cleavage, the Nsp15 RNA platform may further support the viral RTC complex to coordinate replication and transcription. Molecular modeling suggests that Nsp15 could be the central scaffold for the entire RTC complex (5). Nsp15 has been proposed to be the central platform that coordinates the formation of an RTC superstructure including 6 copies of Nsp11, Nsp12, Nsp13, Nsp14, Nsp15 and Nsp16. This superstructure provides a putative mechanism for the movement of the newly transcribed RNA into the capping and nuclease active sites within the RTC (5). While experimental work is needed to confirm the formation of the RTC superstructure our structures reveal that the extensive dsRNA binding interface within the Nsp15 hexamer could support the RTC. However, we hypothesize that the RTC superstructure forms very transiently otherwise the Nsp15 EndoU active sites would likely degrade a significant portion of the viral RNA.
Our work further establishes how Nsp15 cleaves dsRNA, broadening the repertoire of Nsp15 cleavage activity against a variety of RNA substrates (Figure 6). From earlier RNA bound structures of Nsp15 it was unclear how dsRNA would be able to access the EndoU active site. Flexible ssRNA substrates are positioned into the active site through uridine recognition in the uridine binding pocket. Our structure and mutagenesis revealed that Nsp15 can support dsRNA cleavage through a base flipping mechanism that positions the engaged uridine within the uridine binding pocket (Figure 6). Nsp15 also utilizes a conserved tryptophan residue to position the base 3′ of the uridine. The combined action of flipping the uridine and stabilizing the 3′ base moves the phosphodiester bond following the engaged uridine into the Nsp15 active site, where it undergoes an RNase A-like transesterification reaction. Through this unique mechanism Nsp15 can process both ss- and dsRNA substrates. The ability to cleave structurally diverse RNA substrates under the same conditions is rare in endoribonucleases, and points towards the evolution of Nsp15′s activity to be able to process both ss- and ds-viral RNA to regulate the overall level in infected cells. Bovine viral diarrhea virus, a pestivirus, also encodes for an endoribonuclease that can process ss- and ds-viral RNAs, further emphasizing the importance of viral enzymes to be multi-functional given their small genomes (49).
Finally, Nsp15 is a promising anti-viral target, and this work reveals multiple RNA interfaces that could be targeted for structural based drug design. Nsp15 antagonizes the host immune system by limiting the accumulation of dsRNA intermediates that form during viral replication (12,13,15,50,51). Inactivating Nsp15 in porcine epidemic diarrhea virus has been shown to elicit a strong immune response in infected animals and reduced viral shedding and mortality (52,53). Our work establishes that Nsp15 contains four previously unknown RNA binding interfaces that support dsRNA engagement, and further emphasizes that blocking Nsp15 oligomerization could have therapeutic benefit.
DATA AVAILABILITY
The cryo-EM structures have been deposited in the PDB and EMDB with the following accession codes: EMD-25915, PDB: 7TJ2, EMD-26073, PDB: 7TQV. All other data presented in this manuscript are available in the main text or the supplementary materials.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank the NIEHS Mass Spectrometry Research and Support Group, especially Jason Williams, for protein identification analyses. We would like to thank Drs Traci Hall and Marcos Morgan for their critical review of this manuscript. We would like to thank all members of the NIEHS Molecular Microscopy Consortium for their help with data collection. We would like to thank Dr Rick Huang and Allison Zeher for help with cryo-EM data collection. This work utilized the NCI/NICE Cryo-EM Facility.
Notes
Present address: Lucas B. Dillard. Program in Molecular Biophysics, Johns Hopkins University, 3400 N Charles St. Baltimore, MD 21218, USA.
Contributor Information
Meredith N Frazier, Signal Transduction Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, Department of Health and Human Services, 111 T. W. Alexander Drive, Research Triangle Park, NC 27709, USA.
Isha M Wilson, Signal Transduction Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, Department of Health and Human Services, 111 T. W. Alexander Drive, Research Triangle Park, NC 27709, USA.
Juno M Krahn, Genome Integrity and Structural Biology Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, Department of Health and Human Services, 111 T. W. Alexander Drive, Research Triangle Park, NC 27709, USA.
Kevin John Butay, Genome Integrity and Structural Biology Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, Department of Health and Human Services, 111 T. W. Alexander Drive, Research Triangle Park, NC 27709, USA.
Lucas B Dillard, Genome Integrity and Structural Biology Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, Department of Health and Human Services, 111 T. W. Alexander Drive, Research Triangle Park, NC 27709, USA.
Mario J Borgnia, Genome Integrity and Structural Biology Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, Department of Health and Human Services, 111 T. W. Alexander Drive, Research Triangle Park, NC 27709, USA.
Robin E Stanley, Signal Transduction Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, Department of Health and Human Services, 111 T. W. Alexander Drive, Research Triangle Park, NC 27709, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
US National Institutes of Health Intramural Research Program; US National Institute of Environmental Health Sciences [NIEHS, NIEHS/NIH ZIA ES103247(RES)]; NIEHS/NIH ZIC ES103326 (MJB)); NIH Intramural Targeted Anti-COVID-19 (ITAC) Program funded by the National Institute of Allergy and Infectious Diseases [NIAID, NIAID/NIH 1ZIAES103340 (RES)]. Funding for open access charge: NIEHS.
Conflict of interest statement. None declared.
REFERENCES
- 1. Hartenian E., Nandakumar D., Lari A., Ly M., Tucker J.M., Glaunsinger B.A.. The molecular virology of coronaviruses. J. Biol. Chem. 2020; 295:12910–12934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. V’Kovski P., Kratzel A., Steiner S., Stalder H., Thiel V.. Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021; 19:155–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yan L., Yang Y., Li M., Zhang Y., Zheng L., Ge J., Huang Y.C., Liu Z., Wang T., Gao S.et al.. Coupling of N7-methyltransferase and 3′-5′ exoribonuclease with SARS-CoV-2 polymerase reveals mechanisms for capping and proofreading. Cell. 2021; 184:3474–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yan L., Ge J., Zheng L., Zhang Y., Gao Y., Wang T., Huang Y., Yang Y., Gao S., Li M.et al.. Cryo-EM structure of an extended SARS-CoV-2 replication and transcription complex reveals an intermediate state in cap synthesis. Cell. 2021; 184:184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Perry J.K., Appleby T.C., Bilello J.P., Feng J.Y., Schmitz U., Campbell E.A.. An atomistic model of the coronavirus replication-transcription complex as a hexamer assembled around nsp15. J. Biol. Chem. 2021; 297:101218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Malone B., Urakova N., Snijder E.J., Campbell E.A.. Structures and functions of coronavirus replication-transcription complexes and their relevance for SARS-CoV-2 drug design. Nat. Rev. Mol. Cell Biol. 2022; 23:21–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kokic G., Hillen H.S., Tegunov D., Dienemann C., Seitz F., Schmitzova J., Farnung L., Siewert A., Hobartner C., Cramer P.. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 2021; 12:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wahl A., Gralinski L.E., Johnson C.E., Yao W., Kovarova M., Dinnon K.H. 3rd, Liu H., Madden V.J., Krzystek H.M., De C.et al.. SARS-CoV-2 infection is effectively treated and prevented by EIDD-2801. Nature. 2021; 591:451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hur S. Double-Stranded RNA sensors and modulators in innate immunity. Annu. Rev. Immunol. 2019; 37:349–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weber F., Wagner V., Rasmussen S.B., Hartmann R., Paludan S.R.. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006; 80:5059–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen Y.G., Hur S.. Cellular origins of dsRNA, their recognition and consequences. Nat. Rev. Mol. Cell Biol. 2021; 23:286–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hackbart M., Deng X., Baker S.C.. Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:8094–8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Deng X., Hackbart M., Mettelman R.C., O’Brien A., Mielech A.M., Yi G., Kao C.C., Baker S.C.. Coronavirus nonstructural protein 15 mediates evasion of dsRNA sensors and limits apoptosis in macrophages. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E4251–E4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ivanov K.A., Hertzig T., Rozanov M., Bayer S., Thiel V., Gorbalenya A.E., Ziebuhr J.. Major genetic marker of nidoviruses encodes a replicative endoribonuclease. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:12694–12699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Deng X., Baker S.C.. An “Old” protein with a new story: coronavirus endoribonuclease is important for evading host antiviral defenses. Virology. 2018; 517:157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ancar R., Li Y., Kindler E., Cooper D.A., Ransom M., Thiel V., Weiss S.R., Hesselberth J.R., Barton D.J.. Physiologic RNA targets and refined sequence specificity of coronavirus endoU. RNA. 2020; 26:1976–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frazier M.N., Dillard L.B., Krahn J.M., Perera L., Williams J.G., Wilson I.M., Stewart Z.D., Pillon M.C., Deterding L.J., Borgnia M.J.et al.. Characterization of SARS2 nsp15 nuclease activity reveals it's mad about U. Nucleic Acids Res. 2021; 49:10136–10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim Y., Wower J., Maltseva N., Chang C., Jedrzejczak R., Wilamowski M., Kang S., Nicolaescu V., Randall G., Michalska K.et al.. Tipiracil binds to uridine site and inhibits nsp15 endoribonuclease NendoU from SARS-CoV-2. Commun. Biol. 2021; 4:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pillon M.C., Frazier M.N., Dillard L.B., Williams J.G., Kocaman S., Krahn J.M., Perera L., Hayne C.K., Gordon J., Stewart Z.D.et al.. Cryo-EM structures of the SARS-CoV-2 endoribonuclease nsp15 reveal insight into nuclease specificity and dynamics. Nat. Commun. 2021; 12:636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guarino L.A., Bhardwaj K., Dong W., Sun J., Holzenburg A., Kao C.. Mutational analysis of the SARS virus nsp15 endoribonuclease: identification of residues affecting hexamer formation. J. Mol. Biol. 2005; 353:1106–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Athmer J., Fehr A.R., Grunewald M., Smith E.C., Denison M.R., Perlman S.. In situ tagged nsp15 reveals interactions with coronavirus replication/transcription complex-associated proteins. Mbio. 2017; 8:e02320-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu W., Pei G., Liu H., Ju X., Wang J., Ding Q., Li P.. Compartmentalization-aided interaction screening reveals extensive high-order complexes within the SARS-CoV-2 proteome. Cell Rep. 2021; 36:109482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zheng S.Q., Palovcak E., Armache J.P., Verba K.A., Cheng Y., Agard D.A.. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods. 2017; 14:331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de la Rosa-Trevin J.M., Quintana A., Del Cano L., Zaldivar A., Foche I., Gutierrez J., Gomez-Blanco J., Burguet-Castell J., Cuenca-Alba J., Abrishami V.et al.. Scipion: a software framework toward integration, reproducibility and validation in 3D electron microscopy. J. Struct. Biol. 2016; 195:93–99. [DOI] [PubMed] [Google Scholar]
- 25. Punjani A., Rubinstein J.L., Fleet D.J., Brubaker M.A.. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods. 2017; 14:290–296. [DOI] [PubMed] [Google Scholar]
- 26. Rohou A., Grigorieff N.. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015; 192:216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim Y., Jedrzejczak R., Maltseva N.I., Wilamowski M., Endres M., Godzik A., Michalska K., Joachimiak A.. Crystal structure of nsp15 endoribonuclease NendoU from SARS-CoV-2. Protein Sci. 2020; 29:1596–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W.et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta. Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of coot. Acta. Crystallogr. D Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Williams C.J., Headd J.J., Moriarty N.W., Prisant M.G., Videau L.L., Deis L.N., Verma V., Keedy D.A., Hintze B.J., Chen V.B.et al.. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 2018; 27:293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 32. Goddard T.D., Huang C.C., Meng E.C., Pettersen E.F., Couch G.S., Morris J.H., Ferrin T.E.. UCSF chimerax: meeting modern challenges in visualization and analysis. Protein Sci. 2018; 27:14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Killelea T., Saint-Pierre C., Ralec C., Gasparutto D., Henneke G.. Anomalous electrophoretic migration of short oligodeoxynucleotides labelled with 5′-terminal Cy5 dyes. Electrophoresis. 2014; 35:1938–1946. [DOI] [PubMed] [Google Scholar]
- 34. Niewoehner O., Jinek M.. Structural basis for the endoribonuclease activity of the type III-A CRISPR-associated protein csm6. RNA. 2016; 22:318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sinha N.K., Iwasa J., Shen P.S., Bass B.L.. Dicer uses distinct modules for recognizing dsRNA termini. Science. 2018; 359:329–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matthews M.M., Thomas J.M., Zheng Y., Tran K., Phelps K.J., Scott A.I., Havel J., Fisher A.J., Beal P.A.. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat. Struct. Mol. Biol. 2016; 23:426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Roberts R.J., Cheng X.. Base flipping. Annu. Rev. Biochem. 1998; 67:181–198. [DOI] [PubMed] [Google Scholar]
- 38. Slupphaug G., Mol C.D., Kavli B., Arvai A.S., Krokan H.E., Tainer J.A.. A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature. 1996; 384:87–92. [DOI] [PubMed] [Google Scholar]
- 39. Hoang C., Ferre-D’Amare A.R.. Cocrystal structure of a tRNA psi55 pseudouridine synthase: nucleotide flipping by an RNA-modifying enzyme. Cell. 2001; 107:929–939. [DOI] [PubMed] [Google Scholar]
- 40. Stephan N.C., Ries A.B., Boehringer D., Ban N.. Structural basis of successive adenosine modifications by the conserved ribosomal methyltransferase ksgA. Nucleic Acids Res. 2021; 49:6389–6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Klimasauskas S., Kumar S., Roberts R.J., Cheng X.. HhaI methyltransferase flips its target base out of the DNA helix. Cell. 1994; 76:357–369. [DOI] [PubMed] [Google Scholar]
- 42. Kuttan A., Bass B.L.. Mechanistic insights into editing-site specificity of ADARs. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E3295–E3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. D'Souza A.R., Buckingham A.B., Salasc F., Ingemarsdotter C.K., Iaconis G., Jarvis I., Groom H.C.T., Kenyon J.C., Lever A.M.L.. Duplex formation between the template and the nascent strand in the transcription-regulating sequences is associated with the site of template switching in SARS - CoV-2. RNA Biol. 2021; 18:148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Corley M., Burns M.C., Yeo G.W.. How RNA-Binding proteins interact with RNA: molecules and mechanisms. Mol. Cell. 2020; 78:9–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tian B., Bevilacqua P.C., Diegelman-Parente A., Mathews M.B.. The double-stranded-RNA-binding motif: interference and much more. Nat. Rev. Mol. Cell Biol. 2004; 5:1013–1023. [DOI] [PubMed] [Google Scholar]
- 46. Abou Elela S., Ji X.. Structure and function of rnt1p: an alternative to RNAi for targeted RNA degradation. Wiley Interdiscip. Rev. RNA. 2019; 10:e1521. [DOI] [PubMed] [Google Scholar]
- 47. Thomas J.M., Beal P.A.. How do ADARs bind RNA? New protein-RNA structures illuminate substrate recognition by the RNA editing ADARs. Bioessays. 2017; 39: 10.1002/bies.201600187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang M., Li X., Deng Z., Chen Z., Liu Y., Gao Y., Wu W., Chen Z.. Structural biology of the arterivirus nsp11 endoribonucleases. J. Virol. 2017; 91:e01309-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lussi C., Sauter K.S., Schweizer M.. Homodimerisation-independent cleavage of dsRNA by a pestiviral nicking endoribonuclease. Sci. Rep. 2018; 8:8226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Comar C.E., Otter C.J., Pfannenstiel J., Doerger E., Renner D.M., Tan L.H., Perlman S., Cohen N.A., Fehr A.R., Weiss S.R.. MERS-CoV endoribonuclease and accessory proteins jointly evade host innate immunity during infection of lung and nasal epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 2022; 119:e2123208119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Volk A., Hackbart M., Deng X., Cruz-Pulido Y., O’Brien A., Baker S.C.. Coronavirus endoribonuclease and deubiquitinating interferon antagonists differentially modulate the host response during replication in macrophages. J. Virol. 2020; 94:e00178-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Deng X., Buckley A.C., Pillatzki A., Lager K.M., Faaberg K.S., Baker S.C.. Inactivating three interferon antagonists attenuates pathogenesis of an enteric coronavirus. J. Virol. 2020; 94:e00565-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Deng X., van Geelen A., Buckley A.C., O’Brien A., Pillatzki A., Lager K.M., Faaberg K.S., Baker S.C.. Coronavirus endoribonuclease activity in porcine epidemic diarrhea virus suppresses type i and type III interferon responses. J. Virol. 2019; 93: 10.1128/JVI.02000-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM structures have been deposited in the PDB and EMDB with the following accession codes: EMD-25915, PDB: 7TJ2, EMD-26073, PDB: 7TQV. All other data presented in this manuscript are available in the main text or the supplementary materials.