


Abstract
RNA-binding proteins (RBPs) have been relatively overlooked in cancer research despite their contribution to virtually every cancer hallmark. Here, we use RNA interactome capture (RIC) to characterize the melanoma RBPome and uncover novel RBPs involved in melanoma progression. Comparison of RIC profiles of a non-tumoral versus a metastatic cell line revealed prevalent changes in RNA-binding capacities that were not associated with changes in RBP levels. Extensive functional validation of a selected group of 24 RBPs using five different in vitro assays unveiled unanticipated roles of RBPs in melanoma malignancy. As proof-of-principle we focused on PDIA6, an ER-lumen chaperone that displayed a novel RNA-binding activity. We show that PDIA6 is involved in metastatic progression, map its RNA-binding domain, and find that RNA binding is required for PDIA6 tumorigenic properties. These results exemplify how RIC technologies can be harnessed to uncover novel vulnerabilities of cancer cells.
INTRODUCTION
Genetic mutations of oncogenic drivers are at the base of cancer generation. However, non-genetic adaptive mechanisms are increasingly recognized to play fundamental roles in cancer cell plasticity, malignancy and resistance to therapy (1–3). Epigenetic and post-transcriptional reprogramming allow cancer cells to quickly respond to changing environments and resist a wide variety of stresses. For this reason, RNA-binding proteins (RBPs) are gaining considerable attention in the cancer field. RBPs regulate all aspects of post-transcriptional gene expression, and have been shown to modulate virtually all cancer hallmarks (4–8).
The family of RBPs is large and complex. Recent high-throughput experiments, including RNA-interactome capture (RIC) and other unbiased technologies, have greatly expanded the number of potential members of this family, which are counted in the thousands (9,10). The RBP family includes a wide spectrum of entities, from well-known RBPs containing classical RNA-binding domains (RBD) to proteins without any discernible RBD, generally containing intrinsically disordered regions that participate in RNA binding. An interesting group are enzymes that have dual roles as RBPs and may connect cellular metabolism with RNA regulation (11–13). Despite their prevalence, the number of RBPs with established roles in tumorigenesis and metastatic progression is small, and their molecular mechanisms of action are poorly understood. Here, we have searched for novel RBPs implicated in cancer progression and metastasis using melanoma as a model system.
Cutaneous melanoma is the most aggressive form of skin cancer, and a disease of increasing incidence where mortality remains high (14). Melanoma is associated with a high mutation burden due to UV-damage arising from sun exposure. Notably, the most frequent driver mutations are already present in benign nevi and are not indicative of disease outcome (15–17). Non-genomic mechanisms such as translational reprogramming have been shown to contribute to melanoma malignancy (18–20), and a handful of RBPs have been reported to play essential roles in melanoma tumorigenesis at early (CELF1, CPEB4) or advanced (DDX3X, IGF2BP1, CSDE1) stages (21–25), highlighting the importance of post-transcriptional regulation in melanoma cell plasticity. However, a global analysis of the melanoma RNA-binding proteome (RBPome) and an unbiased scrutiny of RBPs contributing to metastatic progression is missing.
In this study, we use RIC to characterize the RBPomes of two melanoma cell lines with distinct aggressiveness. We show large changes in RNA-binding capacities (hereof referred to as ‘activities’) among cell lines that are not generally accompanied by changes in RBP levels. We combine these large-scale proteomics experiments with functional validation (>1000 assays) to identify novel RBPs implicated in cancer progression and metastasis. One of these RBPs is PDIA6, a chaperone of the endoplasmic reticulum (ER) that exhibited a novel activity in RNA-binding which we associate with malignancy. These results confirm the potential of RIC to reveal novel vulnerabilities of cancerous cells, many of which could not have been uncovered by expression level profiling present in current public databases.
MATERIALS AND METHODS
Mice xenografts
Eight-week-old Swiss Nude (Crl:NU(Ico)-Foxn1nu) female mice were obtained from Charles River Laboratories and maintained at the PRBB Animal Facility under pathogen-free conditions, in ventilated racks and with a 12 h light-dark cycle.
SK-MEL-147 cells expressing both Luciferase-GFP and shRNA-RFP were FACS sorted and injected into mice. To assess primary tumor growth, 1 × 106 cells were injected subcutaneously at each flank of mice. If applicable, mice were kept under administration of 1 g/l doxycycline and 75 g/l of sucrose in the drinking water. Tumor growth was monitored every 3–4 days. Tumor volume was calculated by measuring the major and the minor diameters with a caliper (4/3 π × minor radius2 × major radius). Animals were sacrificed when ulceration or cachexia were observed, or when tumors reached a volume of 1500 mm3.
To assess metastasis, 1.5 × 106 luciferase-expressing and shRNA-expressing cells were injected via the lateral tail vein of nude mice. Luciferase signal was monitored using an IVIS-50 imaging system for 40–55 days. Ten minutes before imaging, mice were injected intraperitoneally with luciferin (150 mg/kg, Perkin Elmer 122799). At endpoint, luciferin was injected 10 min prior to sacrifice, and lungs exposed ex vivo to the imaging system. Photons were quantified using Living Image software (Caliper Life Sciences).
Cell culture
SK-MEL-147, UACC-62, 1205-LU, 451-LU, WM164, WM793, HEK293T, GP2-293 and Phoenix-Ampho cells were grown in DMEM, high glucose, pyruvate, and GlutaMax (Gibco 31966021) supplemented with 10% heat-inactivated FBS (Gibco 10270106) and 1% penicillin-streptomycin (Gibco 15070063). Mel-ST cells were grown in the same conditions except for 7% FBS. Human cell lines were maintained at 37°C with 5% CO2 and were regularly tested for mycoplasma infection. Drosophila SL2 cells were grown in Schneider medium supplemented with L-Gln (ThermoFisher), 10% heat-inactivated FBS and 1% penicillin-streptomycin at 25ºC. Melanoma SK-MEL-147, UACC-62, 451-LU, WM164, WM793 and 1205-LU cells were provided by Maria S. Soengas (CNIO), while Mel-ST cells were a gift of Corine Bertolotto (University of Nice, INSERM). All melanoma cell lines in this study were authenticated using the GenePrint® 10 System in the Genomics Unit of Centro Nacional de Investigaciones Oncológicas (CNIO).
Constructs and cloning
SMARTvector lentiviral constructs expressing a short-hairpin targeting PDIA6 (shPDIA6, ATCACTCTGAATAACTTCT, V3SH7670-225183231) or a non-targeting short-hairpin (shControl, TCACACAACATGTAAACCA, VSC11656) under doxycycline induction were obtained from Horizon Discovery. The retroviral vector pMSCV-Luc2-PGK-Neo-IRES-GFP was used to constitutively express luciferase as a reporter in mice experiments. GFP-PDIA6 constructs were cloned into the pcDNA5/FRT/TO/GFP-linker vector using Gibson cloning. Similarly, PDIA6 constructs containing 2xHA-tags immediately downstream of the signal peptide were cloned into the pRetroQ-AcGFP1-C1 vector.
Viral transduction
HEK-293T cells (for lentiviral shRNA expression), G2P-293T cells (for retroviral HA-PDIA6 or luciferase expression) or Phoenix-Ampho (for retroviral H-Ras expression) were transfected using the calcium phosphate method to produce viral particles. Briefly, 2.5 million cells were seeded in 10 cm plates the following day and transfected with a mix containing 450 μl of DNA (3.4 μg of pVSV-G, 6.3 μg of PCMV-dR8-9,1 and 9.8 μg of transfer plasmid for 293T cells; 3.5 μg of pVSV-G and 10 μg of transfer plasmids for G2P-293T cells; or 10 μg transfer plasmid for Phoenix-Ampho cells), 50 μl of 2.5M CaCl2 and 500 μl of HEPES-buffered saline solution (HBS: 140.5 mM NaCl, 50 mM HEPES, and 1.5 mM Na2HPO4, pH 7.12). Two and three days after transfection medium was collected, filtered (4.5 μm), and viral particles were concentrated in a Beckman 38.5 ml open-top tube (Beckman 326823) for 2h at 70 000g.
Reverse infections were performed with 3 × 105 SK-Mel-147 cells or 2 × 105 Mel-ST cells in 6-well plates for 24 h followed by selection with 1 μg/ml puromycin for 3 days. If applicable, expression of shRNA was induced by adding doxycycline (Sigma D9891) at a final concentration of 0.5 μg/ml for at least 5 days.
Transient depletion of RBPs
To silence RBPs, siRNA pools (siPools, siTools Biotech) were used (26). We performed reverse transfections following the company's protocol. In short, RBPs were individually depleted with 6 pmol of siPools mixed with 4 μl of Lipofectamine RNAiMax (Invitrogen 13778150) in Opti-MEM (Gibco 31985070), added to 3 × 105 cells just after seeding in 6-well plates. After 24 h, cells were split, and 72 h later, cells were trypsinized to start cell culture assays. For experiments requiring prolonged silencing, cells were re-transfected with siPools every 3 days. In this case, forward transfections were carried out with 2 pmol of siPools and 1.3 μl of RNAiMax. Depletion was assessed by RT-qPCR using primer sequences indicated in Supplementary Table S1. Knock-down was quantified relative to siControl using beta-actin mRNA levels as normalizer. Typical depletion efficiencies, as well as optimization of depletion showing significant loss of silencing after day 4 in the absence of siPool re-transfection, are shown in Supplementary Table S2.
Transient expression of PDIA6 constructs
Transient transfections to overexpress the desired PDIA6 constructs were carried out using the K4 transfection system (Biontex T080). Briefly, 2.5 × 105 SK-Mel-147 cells were seeded in 6-well plates 24 h before transfection. Thirty minutes prior to transfection, 20 μl of Multiplier were added per well. The transfection mix consisted of 0.5 μg of DNA and 1 μl of K4 in DMEM without FBS or antibiotics. Cells were recovered by centrifugation after 24 h, snap-frozen and stored at –80ºC.
Growth curves
About 1 × 104 cells per well were seeded in a 24-well plate. After 2, 4 and 6 days, cells from three wells were trypsinized, mixed with trypan blue and counted with Countess Cell Counting slides (Invitrogen C10228).
Clonogenic assays
Five hundred cells per well were seeded in 6-well plates, three wells were transfected with siControl and three with siRBP. Assays were stopped when colonies were visible, at around 7–10 days. Cells were simultaneously fixed and stained with 25% methanol, 0.5% crystal violet (Sigma) for 1 h. The number of colonies was counted with the Analyze Particles tool from ImageJ (27).
Anoikis resistance assays
One thousand cells in DMEM with 1–2% FBS were seeded in flat, ultralow attachment plates (Corning CLS3474-24EA). After 3–6 days, cell survival was quantified using CellTiter-Glo (Promega g7570). The survival of input cells was also quantified and used to normalize the data.
3D growth assays
One thousand cells were seeded in round, ultralow attachment plates (Corning CLS7007-24EA) and centrifuged at 1000 rpm for 10 min at 25ºC. After seeding (input control) and after 4–6 days, spheroids were visualized with bright field microscopy (5–10× magnification). The area of spheroids was quantified with ImageJ (27). CellTiter-Glo was used to quantify cell survival, which was corrected for input survival.
Invasion assays
Spheroids were first formed as in 3D growth assay. After 24 h, an invasion matrix (5 mg/ml, Trevigen 3500-096-03) was added, and cell infiltration monitored for 24h using light microscopy. The invasion index was calculated by dividing the perimeter of the spread spheroid by the calculated perimeter that the corresponding area would have if it were a perfect circle.
RNA extraction, reverse transcription and quantitative PCR
RNA was isolated either with TRIzol (Ambion 15596018) and treated with Turbo DNAse (Ambion AM1907) or Maxwell LEV simplyRNA purification kit (Promega AS1280), following the manufacturers’ protocol. Tumor samples were first homogenized on ice in homogenization buffer from the Maxwell kit using the microtube homogenizer. RNA was retrotranscribed with 50 U of Superscript II (ThermoFisher 18064014) following manufacturer's instructions. The resulting cDNA was diluted 1/4 and 1μl of this dilution was mixed with 0.5 μmol of primers and 5 μl of PowerUP SYBR Green Master Mix (Applied Biosystems 4367659). qPCR was carried out with ViiA7 Real-Time PCR system (Applied Biosystems) with primers (Sigma, IDT) detailed in Supplementary Table S1. Beta-actin was used as normalizer and relative quantifications calculated with the delta-delta CT method.
Protein extract preparation and immunoblotting
Cultured cells were lysed in RIPA buffer (150 mM NaCl, 10 mM Tris–HCl pH 7.4, 0.1% SDS, 1% DOC, 5 mM EDTA, 1% Triton X-100 and 1× EDTA-free Proteinase Inhibitor Cocktail (PIC) from Roche 11873580001) for 10 min on ice, homogenized and centrifuged at 14000g/4ºC for 10 min. For western blotting, the following commercial primary antibodies were used: anti-ß-ACTIN (Santa Cruz, sc-47778), anti-CELF1 (Santa Cruz, sc-20003), anti-PDIA6 (ThermoFisher PA3-008 or Proteintech 18233–1-AP), anti-Vinculin (Sigma V9131), anti-HA tag (Abcam ab9110), anti-GFP (Invitrogen A6455), anti-Ras (BD Biosciences 610001), anti-α-Tubulin (Sigma T9026). Anti-Stubarista and anti-CSDE1 were produced in house (24,28). For chemiluminescence detection, we used Goat Anti-Rabbit IgG and Goat Anti-Mouse IgG (Biorad, 1706515 and 1721011), and for infrared (IR) detection, IRDye 800CW and IRDye 680RD (LI-COR, 92632213 and 92668072). The Gels tool from ImageJ was used for quantification.
RNA interactome capture (RIC)
RIC was performed as published (29) using Mel-ST and SK-Mel-147 cells in biological triplicates. Briefly, cells were collected after UV-irradiation at 254 nm (0.15 J/cm2) or not and lysed in RIC lysis buffer (20 mM Tris–HCl pH 7.5, 500 mM LiCl, 0.5% LiDS, 1 mM EDTA, 5 mM DTT and 1× PIC). Lysates were homogenized by passing the sample 3 times through a syringe with a 0.4 mm diameter needle and incubated for 10 min on ice. Poly(A)+ RNA selection was achieved by three rounds of incubation of 45 ml of extract with 2 ml of Oligo-d(T)25 magnetic beads. Eluates were adjusted to a concentration of recovered RNA of 300 ng/μl per replicate.
For RNA quality control, 20 μl of each sample were digested with proteinase K (Roche 3115828001) in 10 mM Tris–HCl (pH 7.5), 150 mM NaCl, 0.2% SDS, 10 mM EDTA, 0.5 mM DTT and 5 mM CaCl2 (1 μg, 50ºC, 1 h) and RNA was isolated with the RNeasy kit (Qiagen 74106) prior analyis with a Bioanalyzer at the CRG Genomics facility.
For protein quality control, 20 μl of each sample were centrifuged twice in a 3 kDa cut-off device (Amicon UFC500396) in 10 mM Tris–HCl pH 7.5 and 50 mM NaCl (4000g, 45 min). Samples were collected in a volume of 230 μl and digested with 1 μg of RNase A (Sigma R4642) and 50 U of RNase T1 (Sigma R1003) in a buffer containing 100 mM Tris–HCl pH 7.4, 1.4 M NaCl and 5 mM DTT, in a final volume of 250 μl (37ºC, 90 min and 55ºC, 15 min). Ten μl of each sample were used for western blot analysis and silver staining. For the latter, protein complexes were separated in a 4–12% Bis–Tris Protein Gel (NuPage Novex NP0335) in MOPS buffer (1 M MOPS, 1 M Tris–HCl, 20.5 mM EDTA, 63.3 mM SDS). The gel was fixed for 30 min in methanol/acetic acid/water mix at 50:5:45 vol, incubated for 2 min in 0.002% sodium thiosulfate and with 0.1% silver nitrate for 40 min. It was developed in 0.04% formaldehyde with 2% Na2CO3 until signal appearance. Stopping the reaction with the methanol/acetic acid/water mix.
The remaining volume (240 μl) was used for protein identification. Samples were brought to 6 M urea for reduction with dithiothreitol (30 nmol, 37 ºC, 60 min) and alkylation in the dark with iodoacetamide (60 nmol, 25ºC, 30 min). Samples were further diluted to 2 M urea with 200 mM ammonium bicarbonate for digestion with endoproteinase LysC (1:10 w:w, 37ºC, o/n, Wako), and then diluted 2-fold with 200 mM ammonium bicarbonate for trypsin digestion (1:10 w:w, 37ºC, 8h, Promega). After digestion, peptide mixes were acidified with formic acid and desalted with a MicroSpin C18 column (The Nest Group, Inc.) prior to LC–MS/MS analysis.
Mass spectrometry analysis of RIC samples
RIC samples were analyzed using a LTQ-Orbitrap Velos Pro mass spectrometer coupled to an EASY-nLC 1000 (ThermoFisher). Peptides were separated by reverse phase chromatography using 90 min gradients (7–35% acetonitrile) with a C18 25-cm column.
The mass spectrometer was operated in positive ionization mode. The acquisition was performed in data-dependent acquisition (DDA) mode and full MS scans at resolution of 60 000 were used over a mass range of m/z 350–2000 with detection in the Orbitrap. In each cycle of DDA analysis, the twenty most intense ions with multiple charges above a threshold ion count of 5000 were selected for fragmentation. Fragment ion spectra were produced via collision-induced dissociation (CID) at normalized collision energy of 35% and they were acquired in the linear ion trap mass analyser. All data were acquired with Xcalibur software.
Acquired spectra were analysed using the Proteome Discoverer software suite (v1.4, ThermoFisher) and the Mascot search engine v2.5 (30). Data were searched against a SwissProt Human database plus a list of common contaminants and all the corresponding decoy entries. For peptide identification, a precursor ion mass tolerance of 7 ppm was used for MS1 level, trypsin was chosen as enzyme and up to three missed cleavages were allowed. The fragment ion mass tolerance was set to 0.5 Da for MS2 spectra. Oxidation of methionine and N-terminal protein acetylation were used as variable modifications whereas carbamidomethylation on cysteines was set as a fixed modification. False discovery rate (FDR) in peptide identification was set to a maximum of 5%.
Orthogonal organic phase separation (OOPS)
We followed OOPS (31) with some modifications. Up to 1.25 million human cells (UV-irradiated or not) were mixed with a spike-in control consisting of one million Drosophila SL2 cross-linked cells. Prior to addition of one ml TRIzol, 20 μl of 80 mM DTT (Figure 4A) or 100 μl of urea buffer (8M Urea, 1% SDS in PBS) (Figure 4C) were added to the pellet. Phase separation was achieved by adding 200 μl of chloroform (VWR) and centrifugation at 12 000g for 15 min at 4ºC. Aqueous and organic phases were removed, preserving the interphase. Two to four additional phase separations were performed. The resulting interphase was precipitated with at least 9 volumes of methanol and centrifuged at 14 000g for 10 min at RT, pellets were partially resuspended in water and digested with 2000 U of RNase T1 (ThermoFisher EN0541) and 4 μg of RNase A (Sigma) for 10 min at 4ºC. The pellet was further resuspended by adding 1% SDS and 100 mM TEAB (Honeywell 17902) followed by sonication for 6 cycles (30 s ON/OFF) in a Bioruptor Pico (Diagenode). RNases (2000 U RNase T1, 4 μg RNase A, 127 U Benzonase) were added, the mix adjusted to 1 mM MgCl2 and digested overnight at 37ºC. Samples were analyzed by western blot.
Figure 4.

PDIA6 is an RNA binding protein. (A) Assessment of RNA-binding by OOPS. Left, schematic representation of the OOPS principle, including the Drosophila spike-in control. Right, western blot showing that PDIA6 is recovered in the OOPS eluate (output) upon UV-crosslinking. Crosslinked Drosophila extracts were added to all samples to estimate sample recovery, as monitored by Western blot against Stubarista (Sta). Vinculin (VCL) was used as negative control. (B) Assessment of RNA-binding by PNK assay. PDIA6 was immunoprecipitated from 2 mg of extract from SK-Mel-147 or Mel-ST cells, and the associated RNA digested and labelled with 32P. The same membrane was first exposed to capture the 32P-RNA signal and then incubated with anti-PDIA6 antibodies for western blot (WB). The asterisk denotes a non-specific band. Quantification of 3 independent experiments is shown on the right. (C) PDIA6 binds RNA in a panel of melanoma cell lines. RNA-binding was assessed by OOPS as in (A). Only crosslinked samples are shown. Quantification of the output signal corrected for input and spike in, and normalized to SK-Mel-147 is shown on the right. Significance was assessed by Student's t-test (n = 3–4).
Polynucleotide kinase (PNK) assays
Cells were lysed in PNK lysis buffer (100 mM NaCl, 50 mM Tris–HCl pH 7.4, 0.1% SDS, 0.5% DOC, 1% NP4 and 1× PIC). Unless otherwise indicated, 150 μg of extract were digested with 2 U of Turbo DNAse (Ambion) and 10 μg of RNase A (Roche 10109142001, 37ºC, 15 min), and PDIA6 was immunoprecipitated using magnetic beads (10 μl Chromotek GFP-Trap beads for GFP-tagged proteins, 30 μl Pierce anti-HA beads for HA-tagged proteins, or 25 μl Protein A Dynabeads for the endogenous protein). Anti-HA beads were pre-blocked with 30 μg BSA for 30 min at room temperature. Dynabeads were covalently bound to 2 μg anti-PDIA6 antibody (Proteintech).
Immunoprecipitation was performed for 2 h at 4ºC. Beads were then washed 5 times with wash buffer (50 mM Tris–Cl pH 7.5, 1 M NaCl, 1 mM EDTA, 1% NP40, 0.1% SDS, 0.5% DOC). When Dynabeads were used, four additional washes in urea buffer (8 M urea, 1% SDS in PBS) were included. Beads were equilibrated in PNK buffer (50 mM NaCl, 50 mM Tris–HCl pH 7.4, 10 mM MgCl2, 0.5% NP40), and incubated for 15 min at 37ºC in a reaction containing 1 mM DTT, 1× PIC, 3U of T4 PNK, and 0.3 μCi γ-32P-ATP (PerkinElmer NEG502Z250UC) under agitation at 850 rpm. Beads were washed 4 times with PNK buffer and 4 times with washing buffer. Complexes were eluted in 1× SDS buffer at 95ºC. Proteins were resolved by SDS-PAGE, transferred to nitrocellulose membranes and exposed. Autoradiography was revealed using a Typhoon TRIO Variable Mode Imager (Amersham Biosciences). Membranes were further processed by western blot.
PDI activity assays
PDI thioreductase activity was measured using the PROTEOSTAT PDI kit (Enzo Life Sciences, ENZ-51024-KP050) following the recommendations of the manufacturer. Briefly, HA-PDIA6 was immunoprecipitated from 2 mg of total protein extract obtained from shPDIA6/HA-PDIA6 over-expressing cells, using 100 μl protein A Sepharose beads coupled with anti-HA antibody. The pellet was divided into two aliquots: one was used for Western blot, and the other further divided for triplicate activity assays (10 μl beads per assay). Activity was assessed by adding the PROTEOSTAT reagent to the beads in the presence of insulin. Recombinant PDI provided in the kit was used as positive control, and 1 mM bacitracin (a PDI inhibitor) was used as negative control. Immunoprecipitation from shControl cells (expressing endogenous PDIA6) was carried as negative control.
Immunofluorescence
SK-Mel-147 cells expressing WT or del9 HA-PDIA6 seeded on glass coverslips were fixed in 4% paraformaldehyde and permeabilized with PBS–0.1% Triton. Cells were incubated with blocking solution (PBS–5% NGS-heat inactivated) for 1 h at room temperature, and incubated with anti-HA (Abcam ab9110, 1:1000) and anti-HSP47 (Enzo Life Sciences, ADI-SPA-470-F, 1:600) overnight at 4ºC. Cells were washed with PBS, incubated with secondary antibodies (AlexaFluor488 and AlexaFluor568, ThermoFisher, 1:500) for 1h at room temperature, washed and mounted with Fluoromount-G (SouthernBiotech). Images were acquired with a Leica SP5 inverted confocal microscope.
RNA sequencing
Four μg of TRIzol-extracted total RNA from SK-MEL-147 or MEL-ST cells were digested with Turbo DNase (1U, 37ºC, 10 min). Biological duplicates were processed on different days. RNA quality was assessed with the Bioanalyzer (RIN ≥ 9). PolyA + selection and library preparation for Illumina sequencing (Illumina HiSeq 2500 sequencer, 50 bp single-end reads) was performed by the CRG Genomics facility. Data were analyzed as described in computational methods.
Total proteomics
Mel-ST and SK-Mel-147 cells in triplicates (frozen vials from different passages) were lysed with RIC lysis buffer (20 mM Tris–HCl pH 7.5, 500 mM LiCl, 0.5% LiDS, 1 mM EDTA, 5 mM DTT and 1× PIC) for 10 minutes on ice, and the homogenate cleared by centrifugation at 14 000 rpm, 4ºC. One hundred micrograms of total protein were precipitated in 6 volumes of cold acetone and the resulting pellets were resuspended in 6 M urea, 20 mM ammonium bicarbonate. Ten micrograms of each sample were processed for LC–MS/MS analysis as described above for the RIC samples.
Mass spectrometry analysis of total proteome samples
Total proteome samples were analyzed using an Orbitrap Fusion Lumos mass spectrometer coupled to an EASY-nLC 1000 (ThermoFisher). Peptides were separated by reversed-phase chromatography in a C18 50-cm column (ThermoFisher) using 90 min chromatographic gradients (5–35% acetonitrile). The mass spectrometer was operated in positive ionization mode. The acquisition was performed in DDA mode and full MS scans at resolution of 120 000 were used over a mass range of m/z 350–1500 with detection in the Orbitrap mass analyzer. In each cycle of data-dependent acquisition analysis the most intense peptides determined by the ‘Top Speed’ acquisition algorithm were selected for fragmentation. Fragment ion spectra produced via high-energy collision dissociation (HCD) at normalized collision energy of 28% were acquired in the ion trap mass analyzer. All data were acquired with Xcalibur software and QCloud was used to assure instrument stability during the analyses (32,33). Acquired spectra were analysed by cell line for peptide and protein identification as described above.
Statistical analysis
Statistical analyses were performed with R (R Core Team, 2017, https://www.R-project.org/) and graphics using ggplot2 (34). Exceptionally, data from experiments in mice and PDIA activity assays were plotted using GraphPad Prism 6 software. Statistical significance between growth curves was computed with a two-sample Kolmogorov-Smirnov test. Statistical significance from luciferase signal in mice experiments were determined by two-sample Wilcoxon test. For the other assays, statistical significance was determined by unpaired, two-tailed Student's t test with unequal variance. For the functional validation, P-values were adjusted (Benjamini-Yekutieli). In bar-plots error bars represent standard deviation.
COMPUTATIONAL METHODS
RNA interactome capture
Definition of RBPs and RBDs in our data sets
After protein identification, SAINTexpress (35) was used to evaluate differences between UV-crosslinked and non-crosslinked samples. Confident RBPs were defined as proteins with BFDR ≤0.05 found in at least two replicates of the same cell line.
The RBPomes identified in our study were compared to previous human and non-human RBPome datasets (9,31,36,37). Human RIC was compared using Uniprot identifiers. For non-human RIC, InParanoid package from R (38) was used to match orthologs in mouse, fly, worm and yeast. PFAM domains of melanoma RBPomes were classified as:
Classical RNA binding domains: RRM, KH, dsRNA, Piwi, DEAD (DDX and DHX), Pumilio, CSD, ZnF-CCCH.
Non-classical RNA binding domains: any of the domains previously identified (39): Ribosomal, GTP EFTU, GTP EFTU D2, SAP, zf-CCHC, LSM, R3H, PseudoU synth, YTH, PAP assoc, MMR HSR1, WD40, Zf-C2H2, SAM, Helicase C, or any domain in RBDmap (40) that has at least three peptides supporting it.
Others.
The melanoma RBPome was compared against databases defining genes related to cancer: COSMIC (07/2017), ICGC (02/2018) and OMIM or GO terms as previously defined (12).
Differential binding and GO analysis
For analysis of differential binding of RBPs to RNA, spectral counts from each RBP were normalized by the total number of spectral counts in each run and triplicates averaged. The Log2FoldChange (L2FC) between the cell lines was calculated. Significance was assessed with the R-ibb package (41). To determine GEO terms enriched in the identified RBPs of each cell line, the ‘gseGO’ function from Cluster Profiler R package was run inputting the list of common RBPs ranked according to the L2FC. The Homo sapiens database was used with an FDR cutoff of <0.01.
RNA-Seq data analysis
RNA-Seq duplicates were analyzed with the grape pipeline (42). Briefly, reads were mapped to the GRCh38 Human Genome with STAR (43) and the transcripts quantified with RSEM (44).
Total proteome analysis
For each identified protein, the ‘Top3 Area’ was calculated, corresponding to the average peak area of the three most intense peptides. Peptide quantification data were retrieved from the ‘Precursor ion area detector’ node from Proteome Discoverer (v2.0) using 2 ppm mass tolerance for the peptide extracted ion current (XIC). For cell line comparisons, the Top3 Area was normalized by the total abundance of each sample, and a Log2 Fold change between the cell lines was computed. A q-value was calculated to assess the significance of computed differences.
Correlation between RIC and total proteome
Log2 Fold Changes previously calculated for RIC and total Proteomics were compared for the cell lines. The Pearson correlation and coefficient of regression were computed and plotted in R. Regulation at the level of protein expression or RNA binding were considered for proteins with a q-value <0.05 or a P-value <0.05, respectively. To include RBPs absent in one of the cell lines, a value of log2 = –10 was imputed for the missing values.
Correlation between RNA-Seq and total proteomics
Correlation between RNA and protein expression in each cell line was calculated. First, gene symbols corresponding to the ensemble IDs of our RNAseq dataset were retrieved with BioMart (45) to merge datasets. For each cell line, the correlation between RNA abundance and protein abundance was plotted in R. The log10 of the FPKM was used as RNA abundance measure, and the log10 of the normalized mean Top3Area was used for protein abundance measure. Spearman correlation was calculated.
Functional clustering
For all validation results, the L2FC between the siRBP and the corresponding siControl was calculated. For 2D-growth, only the last time point was considered. The data were inputted for K-means clustering with four centers on R.
DepMap analysis
The DepMap package [Gatto L, Killian T (2021). depmap: Cancer Dependency Map Data Package. R package version 1.6.0] was downloaded from bioconductor and the ‘DepMap’ and ‘ExperimentalHub’ libraries were loaded in R. The RNAi knockdown data from 21Q1 DepMap release was downloaded with the accession number EH226. The data from melanoma cells was retrieved by the grep function searching ‘SKIN’ in the cell line column and ‘melanoma’ in subtype_disease. We calculated our score by substracting 1 to the calculated log2 Fold Change (log2FC(siRBP/siControl) – 1) to obtain a score at the same range than the dependency one and correlate the two datasets. The pearson correlation coefficient was calculated in R.
RESULTS
Characterization of the melanoma RBPome
To identify RBPs binding to mRNA in melanoma cells, we used RIC (39,46). Briefly, cells were UV-crosslinked in order to covalently bind proteins to RNA, lysed with denaturing buffers, and protein-mRNA complexes captured using oligo(dT) beads (Figure 1A). Non-crosslinked samples were processed in parallel as negative controls. After extensive washing, samples were divided into two groups; one group was treated with RNases to release the RBPs and the other with proteinase-K to recover the RNA. These samples were first subjected to quality control. We performed RIC in triplicates on two cell lines with different aggressiveness: non-tumoral melanocytic Mel-ST cells, which are melanocytes that have been immortalized by SV40ER/hTERT transformation (47), and metastatic SK-Mel-147 cells, containing the NRASQ61R mutation. RNA analysis showed a dramatic loss of both tRNAs and rRNAs upon oligo(dT) selection, reflecting strong enrichment for mRNAs as expected (Figure S1A). Protein analysis using silver staining showed marked differences between eluates and inputs, and nearly no background in the non-crosslinked controls (Figure S1B). Furthermore, Western blot analysis indicated that known RBPs were enriched only in the eluates from irradiated samples, while abundant cytoskeletal proteins were absent (Figure 1B). As our RIC eluates passed these quality controls (29), we proceeded to protein identification using mass spectrometry (LC/MSMS).
Figure 1.

Characterization of the melanoma RBPome. (A) Schematic representation of the RNA interactome capture (RIC) procedure. (B) RBP enrichment assessed by western blot. (C) Number of total and significant (BFDR ≤ 0.05) proteins recovered in RIC eluates. Most significant proteins were also recovered with a BFDR ≤0.01 (dashed line). Significance was calculated using SAINTexpress by comparing crosslinked versus non-crosslinked conditions. The overlap of significant RBPs in both cell lines is shown on the right. (D) Comparison of the melanoma RBPome (excluding histones and ribosomal proteins) with previously published RBPomes. (E) Analysis of RNA binding domains (RBDs) present in the melanoma RBPome. Domain word clouds indicate the frequency of protein domains in the data sets.
Comparison of the spectral counts for each cell line showed a good correlation among triplicates (R2 ≥ 0.74, Supplementary Figure S1C). We used SAINTexpress to determine true RNA interactors (35), considered as proteins detected in at least two replicates from the same cell line with a Bayesian False Discovery Rate (BFDR) ≤0.05. We excluded common contaminants detected in proteomics studies featured in the CRAPome, including proteins from the keratin, tubulin and actin families (48), yielding 438 RBPs reliably detected in the non-tumoral cell line and 529 RBPs in the metastatic cell line. Most RBPs were shared, consistent with the notion that RBPs perform housekeeping functions (Figure 1C). We further excluded ribosomal proteins due to the minor rRNA contamination of the eluates, and histones because they have been reported as common RIC contaminants (49,50). Altogether, we considered a final list of 477 RBPs as components of the melanoma RBPome (Supplementary Table S3). Comparison of this list with RBPs identified in previous high-throughput experiments indicated that most melanoma RBPs have been previously detected in other human RBPomes or had orthologs in RBPomes from other species (Figure 1D and Supplementary Table S4). Four RBPs, although not present in RBPome studies, had been characterized as RBPs in the literature, while two are newly reported in this study (Figure 1D).
In line with previous studies, 57% of our data set include proteins with classical and/or non-classical RBDs (see methods for definition) (39). Among these, the RNA Recognition Motif (RRM) and the helicase-C motif predominate (Figure 1E). Almost half of the melanoma RBPome (43%) lack recognizable RBDs. These proteins contain a diversity of other domains, some of which are present in translation factors (OB_NTP_BIND, MIF4G, MA3, GFP_EFTU_D3, HA2) or regulators of RNA metabolism (Brix, DUF4217, G-patch). Overall, the melanoma RBPome is in good agreement with RBPomes of other human cells.
RNA interactome capture uncovers novel dependencies of metastatic cells
To identify RBPs with potential roles in melanoma progression, we compared the RBPomes of the two cell lines. Interestingly, a number of RBPs were significantly retrieved in only one cell line, suggesting dedicated roles (Figure 1C, Supplementary Table S3). Significant differences in RIC recovery were also detected for those RBPs that were common in both cell lines (Figure 2A). Notably, proteins recovered with higher efficiency in the non-tumoral cell line were related to RNP granules, while those recovered with higher efficiency in the metastatic cell line were enriched in GO terms related to ribosome biogenesis (Supplementary Figure S2A). To understand if changes in RIC recovery reflected changes at the protein or RNA levels, we performed transcriptomics (n = 2) and total proteome (n = 3) analyses of the two cell lines. Strikingly, although RBP mRNA levels showed a good correlation with protein abundance (Supplementary Figure S2B), changes in mRNA or protein levels did not match the observed variations in RIC recovery (Figure 2B and Supplementary Figure S2C). Indeed, regression analysis to assess whether total proteomics data were predictive of RIC data yielded a poor correlation (R2 = 0.17), revealing prominent changes in RNA-binding activity across cell lines. These results imply that the capacity to bind RNA is specifically regulated in the different cell lines, and that changes observed in RIC can provide unique information on RNA-binding dependencies of cancerous cells.
Figure 2.

RIC uncovers novel dependencies of cancer cells. (A) Volcano plot showing differential RBP recovery in RIC eluates of non-tumoral and metastatic cells. Some RBPs are indicated. (B) Scatter plot showing the correlation between changes at the level of RIC and changes at the level of total proteomics for RBPs identified in both cell lines. Proteins with a significant Log2FC (P< 0.05) only in RIC (blue), levels (red) or both (yellow) are highlighted. Pearson correlation and the coefficient of determination are shown in the lower-right corner of the graphs. Some RBPs are indicated. (C) RIC identifies RBP dependencies of metastatic SK-Mel-147 cells. Boxplots show the outcomes of RBP depletions within the decreased binder (green) or increased binder (orange) groups in clonogenicity, anoikis resistance, invasion, 3D growth (spheroid size and ATP) and 2D-growth assays. Each dot represents the averaged results of siRBP normalized to siControl (red line) for one RBP (n = 6–9). Significance between groups was assessed by Student's t-test. (D) Unbiased K-means clustering of results from functional validation. Clustering did not take into account the efficiency of depletion of each RBP. The colored sidebar on the left indicates the original classification of RBPs based on RIC. (E) Comparison of our functional validation with RNAi data for melanoma cells in DepMap. Each dot represents one RBP.
To evaluate if changes in RNA-binding activities are informative of the capacity of RBPs to influence the tumorigenic potential of cells, we selected proteins for functional validation. We first divided proteins into increased binders and decreased binders, by considering RBPs with the highest difference in RNA-binding activity between cell lines. Increased binders show a higher activity in the metastatic cell line (unique in SK-Mel-147, or with Log2FC > 1 and P-value < 0.05) while decreased binders show higher activity in the non-tumoral cell line (unique in Mel-ST, or with Log2FC < –1 and P-value < 0.05). The 216 RBPs meeting these conditions were further filtered using public cancer databases including COSMIC, OMIM, IntOGen and GeneOntology (51–54) (Supplementary Table S5, Supplementary Figure S2D). We favored proteins either absent from cancer databases or without a described RNA-binding activity linked to cancer progression. Among these, we randomly selected a final group of 24 RBPs for functional validation (Supplementary Table S5, Supplementary Figure S2D). This group contains a variety of molecular functions: 13 RBPs (BICC1, DDX3Y, DDX60L, EIF2A, ERAL1, FTSJ3, GNL2, MTPAP, PABPC5, PARP12, POPLRMT, SPEN, ZCCHC8) are involved in a wide range of RNA-related roles including transcription, ribosome biogenesis, translation, RNA degradation and mitochondrial RNA metabolism, while 11 (AHNAK, CKAP2, EZR, FAM120C, FLNC, GTPBP4, KIF1C, MAGED1, PDIA6, SPATS2L, TKT) have no described roles in RNA regulation and include enzymes, cytoskeleton-binding proteins and proteins with unknown function (Supplementary Table S5). One candidate, DDX3Y, was used as a negative control as this RBP is not expressed in SK-Mel-147 cells, while an additional protein known to promote melanoma metastasis, CSDE1, was included as positive control (24).
Functional validation consisted of individual depletion of each RBP from metastatic SK-Mel-147 cells followed by five different in vitro assays that are informative of the tumorigenic potential of cells (Supplementary Figure S2E). These include: (i) growth in monolayer (2D-growth) as an indicator of proliferation; (ii) clonogenicity as an indicator of the capacity to grow in isolation, a common stress that cancer cells need to overcome when colonizing new niches; (iii) growth in three dimensions (3D-growth), which better resembles growth in the tissue environment; (iv) resistance to anoikis, a form of programmed cell death triggered by lack of cell anchorage, which is important for cancer cell dissemination and metastasis; and (v) invasion, performed by evaluating the capacity of spheroids to invade a matrigel mimicking the extracellular environment. To silence the expression of the RBPs, we transfected siRNA pools (siPools) targeting each RBP individually, obtaining efficient depletion (Supplementary Table S2). siPools are a mix of 30 siRNAs hybridizing along the length of an mRNA, used at a low concentration that results in more specific and efficient depletion compared to single siRNAs (26). Detailed results for each of these assays and RBPs are found in Supplementary Figure S3. We found a variety of behaviors, from proteins whose depletion causes a dramatic loss of cell survival affecting all measured parameters (e.g. FTSJ3, GNL2, GTPBP4) to, interestingly, proteins whose depletion does not affect growth per se but influence several other parameters (e.g. TKT, PDIA6, EIF2A, etc). Considering proteins belonging to the same group collectively, metastatic SK-Mel-147 cells were strongly dependent on increased binders, while they showed lesser or no dependence on decreased binders, suggesting a correlation between RNA-binding activity and tumor promoting potential (Figure 2C). Among all assays, clonogenicity, anoikis resistance, 2D-growth and survival in 3D (ATP levels) showed the highest differences between the two groups of proteins. The invasion assay did not yield significant differences between groups, perhaps because of the intrinsic ability of melanocytes to migrate and invade (55). Importantly, unbiased k-means clustering of the functional validation results was broadly concordant with our classification based on RIC, that is, most decreased binders clustered together and separately from most increased binders (Figure 2D). These results indicate that RNA binding activity assessed by RIC informs on the tumorigenic potential of RBPs.
We next tested whether the role of these RBPs in melanoma progression could have been inferred by evaluation of public cancer databases. Comparison of our functional data with the Dependency Map (DepMap) portal (56), an integrated database that includes large-scale RNAi screens in cancer cells, revealed a reasonable correlation with 2D-growth and clonogenicity, as expected from the fact that the DepMap readout is primarily based on cell proliferation (Figure 2E, top). However, the correlation with other assays such as anoikis resistance and 3D-growth was poor (Figure 2E, bottom), indicating that these assays contribute novel information that may increase the power to distinguish true RBPs involved in melanoma malignancy.
Inspection of the TCGA database using cBioportal (57,58) for RNA expression differences in primary vs metastatic melanoma samples showed that 57% of the RBPs scored a significant difference, most of them in a direction that was inconsistent with our expectations (Supplementary Figure S4A). For instance, only 4 of the 12 RBPs in the increased binder group, shown to promote tumorigenic traits, showed increased mRNA levels in metastatic samples (Supplementary Figure S4A). In addition, analysis of patient outcome based on mRNA levels for the selected RBPs indicated that only 4 of the 24 RBPs (GTPBP4, PARP12, EIF2A and ZCCHC8) could predict disease-free survival in patients after primary tumor resection, whereas only 3 of the 24 (DDX60L, FAM120C and PARP12) predicted overall survival in patients with metastasis (Supplementary Figure S4B, C and Supplementary Table S6). While these data highlight some RBPs with potential clinical value, they also indicate that RNA levels alone currently present in public databases are not sufficiently informative, and that comparative RIC provides new and valuable information to discriminate RBPs with roles in tumorigenesis.
PDIA6 promotes melanoma metastasis
To showcase the value of our results, we selected PDIA6 (also known as ERp5 or P5) for further evaluation. PDIA6 is a thioreductase mainly located in the ER lumen, where it modulates the unfolded protein response (59–61). PDIA6 has also been found in the plasma membrane, where it facilitates antigen shedding and immune evasion (62,63). We selected PDIA6 because: (i) it belongs to the increased binder group, containing RBPs for which melanoma cells show the strongest dependency (Figures 2D, S2C); (ii) it is a novel RBP that, although detected in other RIC studies, its binding to RNA has not been validated and there is no mechanistic understanding of this novel function.
An interesting observation from our validation is that, while PDIA6 depletion does not affect 2D-growth, it impacts phenotypes relevant for metastatic colonization. Supporting this notion, PDIA6 has been implicated in breast cancer metastasis (64) and treatment-resistance of lung adenocarcinoma (65). To test the relevance of PDIA6 in malignancy of melanoma in vivo, we performed xenograft experiments using cells with inducible PDIA6 depletion.
We first confirmed that, similar to the results obtained with transient depletion with siPools, stable shRNA-mediated depletion of PDIA6 from SK-Mel-147 cells reduced clonogenicity, anoikis resistance and 3D-growth without affecting proliferation (Figure 3A). Furthermore, strong effects of PDIA6 depletion on clonogenicity with minor effects on cell viability were observed for melanoma cells of a different genetic background (UACC62 and 1205-LU cells, which contain the BRAFV600E mutation) (Figure 3B). In contrast, despite the fact that non-tumoral Mel-ST cells express comparable levels of PDIA6 (Supplementary Figure S5A), depletion of PDIA6 from this cell line had weak effects on supported traits (Supplementary Figure S5B, C). These include 2D-growth, clonogenicity and to a lesser extent 3D-growth, while anoikis resistance is not supported by this cell line consistent with its less aggressive behaviour (Supplementary Figure S5B). These results suggest that metastatic cells have acquired a new dependency on PDIA6.
Figure 3.

PDIA6 promotes melanoma metastasis. (A) Outcomes of PDIA6 depletion from metastatic SK-Mel-147 cells using either siPools or shRNAs. Typical depletion efficiency at the start of cellular assays is indicated at the top. siRNA data for PDIA6 was reproduced from Supplementary Figure S3. Significance was assessed by the Kolmogorov–Smirnov test (2D-growth) or Student's t-test (clonogenicity, anoikis resistance and 3D-growth). Data were normalized to depletion controls. (B) Outcomes of PDIA6 depletion in viability (left) and clonogenicity (right) of UACC-62 and 1205-LU cells. PDIA6 was depleted with shRNAs and the values normalized to shControl. Significance was assessed by Student's t-test (n = 3–4). (C) Effect of PDIA6 depletion on primary tumor growth. PDIA6 was depleted using a doxycycline-inducible shRNA and its levels were assessed at the beginning (western blot, left) and end (RT-qPCR, right) of the experiment. One million cells were injected per mouse flank (n = 10) and tumor volume was monitored until animals had to be sacrificed (bottom). (D) Effect of PDIA6 depletion on metastasis. Mice were injected in the tail vein with 1.5 million metastatic SK-Mel-147 cells labeled with luciferase, either expressing inducible shPDIA6 or shControl, and luciferase signal followed using an IVIS Spectrum. Quantification of the signal is shown at the bottom. The box limits indicate the 25th and 75th quartiles and the upper whisker extends from the minimum to the maximum values. The level of depletion attained before injection is shown at the top. Wilcoxon t-test was used to assess significance (n = 5).
We next performed xenograft experiments using subcutaneous or tail-vein injections in nude mice in order to assess primary tumor growth or metastasis, respectively. Depletion of PDIA6 showed at most a delay on primary tumor growth (Figure 3C), but a dramatic impairment in the capacity of melanoma cells to metastasize to the lung (Figure 3D). These data indicate that PDIA6 is relevant for melanoma metastasis.
PDIA6 is an RNA-binding protein
To strengthen the observed RNA binding activity of PDIA6, we first employed Orthogonal Organic Phase Separation (OOPS) (31). This method is based on the property of RNA-RBP complexes to remain at the interphase of an aqueous-organic phase partition. We applied OOPS to melanoma SK-Mel-147 cells subjected or not to UV-crosslinking, and added crosslinked Drosophila extracts to all human samples as a spike-in control in order to estimate recovery. As expected, PDIA6 was found in the interphase (output) only under crosslinked conditions, while Vinculin, an abundant cytoplasmic protein, was not recovered (Figure 4A). We further employed polynucleotide kinase (PNK) assays. These involve immunoprecipitation of PDIA6 from UV-irradiated cells, digestion with RNase and detection of associated RNA fragments by radioactive labeling with PNK. A radioactive signal migrating at the molecular mass of PDIA6 in SDS-PAGE was detected in SK-Mel-147 cells, which was absent in Mel-ST cells confirming that PDIA6 RNA-binding activity is higher in the melanoma cell line (Figure 4B). Finally, we evaluated PDIA6 RNA-binding activity in a panel of aggressive melanoma cell lines, showing comparable high RNA-binding activity of PDIA6 (Figure 4C). Altogether, these results show that PDIA6 is a bona fide RBP whose RNA-binding activity increases in metastatic cells.
To assess the relationship between PDIA6 RNA-binding and oncogenic activation, we over-expressed H-RasV12 in Mel-ST generating Mel-STR cells (Supplementary Figure S5D, left panel). It has been reported that, contrary to Mel-ST, Mel-STR cells are able to form colonies in suspension and tumors in vivo, indicating increased malignancy (47). We found that Mel-STR cells were more resistant to anoikis, while their ability to grow in 2D and 3D was similar to that of Mel-ST (Supplementary Figure S5D). Interestingly, depletion of PDIA6 from Mel-STR did not affect these traits, nor did oncogenic transformation increase PDIA6 RNA-binding activity (Supplementary Figure S5D, E). These results strengthen the notion that dependency on PDIA6 is associated to its RNA-binding activity, and suggest that this dependency may be acquired at a step posterior to oncogene activation.
Identification of the PDIA6 RNA-binding domain
PDIA6 contains two thioredoxin domains and is flanked by signals that drive its localization to the ER lumen, a signal peptide at the N-terminus and a KDEL ER-retention/retrieval signal at the C-terminus (Figure 5A). As an atypical RBP, PDIA6 does not harbor any recognizable RBD. To identify regions needed for RNA binding, we first decided to split PDIA6 into three fragments of similar size preserving the thioredoxin domains, which have been previously shown to exhibit RNA binding capacity (66). These fragments were fused to the C-terminus of GFP, expressed in SK-Mel-147 cells, and tested for RNA binding using PNK assays. While fragment 1, containing the first thioredoxin domain, showed no detectable RNA-binding activity, fragment 2, containing the second thioredoxin domain, bound to RNA weakly and fragment 3 retained most of
Figure 5.

PDIA6 binds RNA through its C-terminal thioredoxin-like fold and disordered region. (A) Domain organization of PDIA6. The signal peptide and the KDEL retention/retrieval signal are indicated. Peptides retrieved from pCLAP and OOPS databases, as well as disordered tracks, are shown. Dashed lines mark the limits of the fragments chosen for RNA binding analysis. TRX, thioredoxin domain. (B) PDIA6 binds RNA via the C-terminal domain. PDIA6 fragments were fused to the C-terminus of GFP, transfected into melanoma SK-Mel-147 cells and tested in PNK assays. Cells expressing GFP alone were used as control. Western blot (top) and 32P signal (bottom) from a representative example are shown. Dashed squares indicate the regions of focus. Quantification of the 32P signal corrected for the amount of immunoprecipitated protein is shown at the bottom. Significance was assessed by Student's t-test (n = 3). (C) Structural model of the C-terminal domain of PDIA6 (I281-L440). Regions 4 and 5 are indicated in orange and regions 8 and 9 in yellow. (D) Scanning mutagenesis of fragment 3. Nine regions (1–9) were serially deleted, as indicated by alternating black and red colors (top). Representative results from PNK assays of UV-irradiated cells (Western blot and 32P signal) are shown. Dashed squares indicate the regions of focus. Quantification of the 32P signal corrected for the amount of immunoprecipitated protein is shown at the bottom. Significance was assessed by Student's t-test (n = 2–3). (E) The C-terminal disordered region of PDIA6 is essential for RNA binding. Regions 8 or 9 were removed from full length, HA-tagged PDIA6 (left) and tested in PNK assays (middle). Quantification of the 32P signal corrected for immunoprecipitated protein is shown (right). Significance was assessed by Student's t-test (n = 3).
the RNA-binding activity (Figure 5B).
Previous high-throughput studies, including pCLAP and OOPS, have provided a wealth of potential RNA-binding peptides present in RBPs (31,67). To further narrow down the PDIA6 RNA-binding region, we examined the RNA-binding peptides assigned to PDIA6 in these proteome-wide efforts (Figure 5A). Deletion of several of these peptides showed no effect on RNA binding (Supplementary Figure S6A and data not shown). Furthermore, we searched for stretches enriched in positively charged or aromatic residues suggestive of RNA-binding activity, several of which were found in fragment 3. Only deletion of one of them (365-RKMKF-369) resulted in a modest reduction of the 32P signal (Supplementary Figure S6A), suggesting that other regions in fragment 3 hold most of the RNA-binding activity.
The C-terminal domain of PDIA6 is thought to fold into a thioredoxin-like domain that lacks catalytic activity because of the absence of the CGHC active site (68). Modeling of this domain using iTasser (69) indeed revealed a thioredoxin-like fold followed by a disordered region (Figure 5C). To identify the RNA-binding domain, we performed scanning mutagenesis on fragment 3 by serially deleting individual alpha-helices, beta-strands or disordered regions based on the structural model (Figure 5D, top). The different mutants were expressed in SK-Mel-147 cells and tested in PNK assays. The results indicated that individual deletion of several regions decreased the RNA-binding signal, one of them (region 5) containing the KMKF residues (Figure 5D). Importantly, major effects were observed after deletion of the C-terminal 31 amino acids of PDIA6 (regions 8 and 9, Figure 5D), predicted to be highly disordered (Figure 5C, yellow).
To confirm the impact of the C-terminal amino acids of PDIA6 on RNA binding, we individually deleted regions 8 and 9 from full-length PDIA6. In order to more accurately mimic the endogenous protein, we tagged the constructs with two HA-tags inserted after the signal peptide, and preserved the KDEL ER-retention/retrieval signal (Figure 5E, left). Strikingly, we observed a complete loss of RNA binding after deletion of region 9, with a strong but lesser effect of deletion of region 8 (Figure 5E, right). We conclude that the disordered C-terminal amino acids of PDIA6 are required for RNA binding.
The disordered C-terminal RNA-binding region mediates the tumorigenic properties of PDIA6
To evaluate the possible link between the oncogenic properties of PDIA6 and its novel RNA-binding activity, we established a PDIA6 depletion/reconstitution system and tested the impact of RNA-binding mutants in anoikis assays. SK-Mel-147 cells expressing control or PDIA6 shRNAs were infected with sh-resistant wild type PDIA6 or the RNA-binding defective del9 mutant, or with GFP as control (Figure 6A). As expected, anoikis resistance was reduced upon PDIA6 knock-down, and while this effect was restored by expression of sh-resistant PDIA6, expression at similar levels of the PDIA6 del9 mutant failed to rescue the phenotype (Figure 6B). Both WT and del9 PDIA6 showed a strong co-localization with the ER marker HSP47 (Supplementary Figure S6B). Furthermore, both proteins showed similar thioreductase activities, indicating that the effects on anoikis resistance were not indirectly due to changes in the enzymatic activity of PDIA6 (Figure 6C). These results strongly argue that the RNA-binding activity associated with the C-terminal region of PDIA6 is required to sustain the tumor-promoting properties of the protein.
Figure 6.
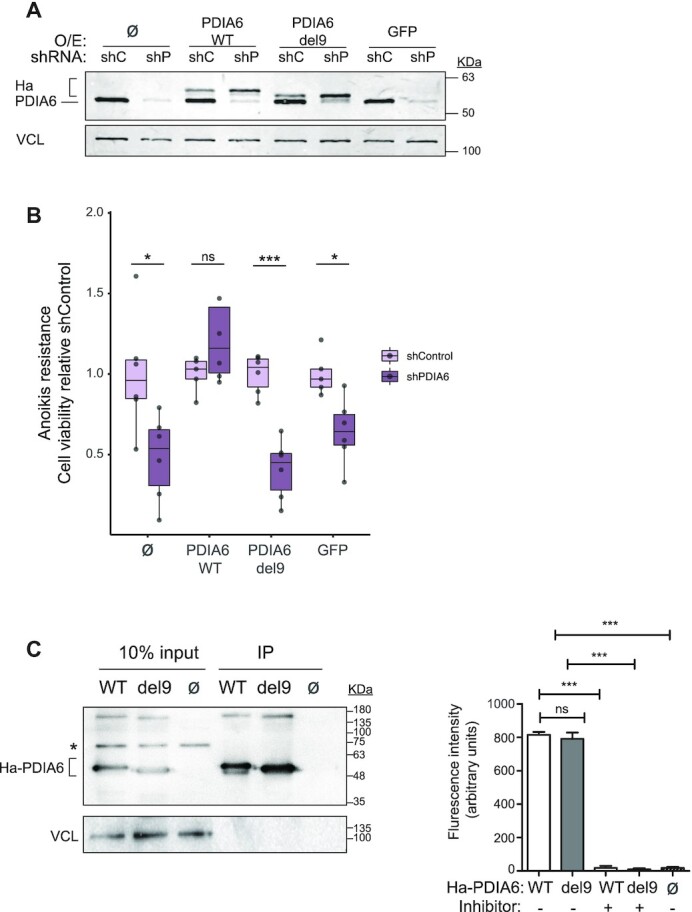
RNA-binding by PDIA6 mediates its tumorigenic properties. (A) PDIA6 expression. SK-Mel-147 cells expressing shControl (shC) or shPDIA6 (shP) were infected with sh-resistant WT or del9 HA-tagged PDIA6 constructs. Non-infected (Ø) and GFP-infected cells were carried as controls. Endogenous and exogenous (Ha) PDIA6 are indicated. (B) Cells in (A) were tested in anoikis resistance assays. Quantification is shown normalized to shControl. Significance was assessed by Student's t-test (n = 6). (C) Deletion of the C-terminal RNA-binding domain of PDIA6 does not affect its enzymatic activity. HA-PDIA6 was immunoprecipitated from shP/WT and shP/del9 cells, and the thioredoxin activity in the pellet measured. Non-infected cells (shC/ Ø) were used as control. Left, Western blot showing the efficiency of immunoprecipitation. The asterisk denotes a non-specific band. Right, PDI thioredoxin activity assay. The PDI inhibitor bacitracin was used in some reactions as control. Bars represent the average of at least two independent biological replicates with 1–3 technical replicates. Significance was assessed by Student's t-test.
DISCUSSION
Comparative RIC technologies have been previously used to understand the dynamic RBPome under a variety of conditions and organisms, either combined with label-free mass spectrometry, as in our study, or with SILAC or TMT labeling (31,36,50,70–76). To our knowledge, however, it has not been used to reveal RBPs important for metastatic progression. Here, based on RNA-binding patterns, we identify RBPs that contribute to tumor progression. The function of most validated RBPs on melanoma progression was not obvious from current literature or database mining. Data currently available in cancer databases (e.g. TCGA) typically rely on mutation frequency or RNA levels, and a few contain information on protein levels (e.g. CPTAC (77)). Yet, neither RNA nor protein levels bear correlation with changes in RNA-binding activity between cell lines. Several scenarios can be envisaged to explain these activity changes, including differential RBP isoform expression, post-translational modification, changes in expression of protein partners or mRNA targets, or dynamic target RNA modification (e.g. m6A). Indeed, a recent report showed a similar discordance between protein abundance and RNA-binding activity during SARS-CoV2 infection, and a substantial correlation between RNA-binding activity and post-translational modifications (78). Regardless of the specific mechanism operating for each melanoma RBP, our data show that comparative RIC is a powerful technology to identify vulnerabilities of cancer cells that might be missed in bulk transcriptomic or proteomic studies.
Little is known about the implications in cancer of several of the proteins selected in this study, including PABPC5, FTSJ3, MTPAP and ZCCHC8. In addition, although roles for some of the remaining factors in non-melanoma tumors have been reported, their functions in melanoma remain poorly understood. Interestingly, among them ERAL1 and POLRMT have been found to interact with the melanoma pro-metastatic factor p62 (21), suggesting connections with functional relevance in this cancer type.
Among increased binders, we found several enzymes that had not been previously related to RNA regulation (EZR, PDIA6, TKT, GTPBP4). Enzymes with RNA binding capacity are known since long in the literature (e.g. PKR, ACO1), but their numbers have dramatically increased with recent RNA interactome studies, revealing an intricate emergent connection between RNA-mediated regulation, cellular metabolism and a variety of cellular processes (11,13,79).
PDIA6 is a chaperone of the ER lumen, also present at the cell surface where it aids in antigen processing (68). As an enzyme, it is one of the less processive members of the PDIA family, displaying only a few dedicated substrates (80). Two of them are the UPR effectors IRE1α and PERK, that engage in covalent interactions with PDIA6 resulting in dampening of the UPR response and increased cell survival (60). Whether the RNA-binding activity of PDIA6 intersects with these processes remains to be determined. Although the ER lumen is not a frequent site for RNA location, recent bio-ID experiments indicate the presence of uncharacterized RNA species in this cellular compartment, raising the interesting possibility of a connection between PDIA6 RNA binding and the UPR response (81). Alternatively, PDIA6 may bind RNA at the cell surface. However, we have not been able to detect PDIA6 at the surface of melanoma cells using immunofluorescence or cell fractionation approaches (data not shown). Determining which cellular processes are influenced by PDIA6 RNA-binding and understanding the underlying molecular mechanisms are exciting avenues for future research. Regardless, we show here that PDIA6 RNA-binding is important to confer resistance to anoikis, a process essential for cellular metastasis, highlighting PDIA6 as a novel RNA-binding protein whose activity might be targeted to prevent metastatic spread.
RNA-binding by PDIA6 is mediated by residues in the thioredoxin-like domain and, critically, the C-terminal disordered region. In line with their relevance, these residues are highly conserved across evolution (Supplementary Figure S7). The involvement of the C-terminal region was unexpected because it is highly acidic and, although negatively charged amino acids can interact with nitrogenated bases (82), acidic regions usually engage into repulsive interactions with the negative RNA backbone. A possibility to reconcile this observation is that the C-terminal disordered region allows for interaction with a factor that licenses PDIA6 to bind RNA. Further biochemical characterization combined with structural analysis should help resolve these questions and aid in the design of compounds targeting PDIA6 to fight metastasis.
DATA AVAILABILITY
Transcriptomics data generated in this study are available through Gene Expression Omnibus (GEO) under accession number GSE174821. The mass spectrometry data have been deposited in the PRIDE repository with the dataset identifier PXD029927 (83).
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge the CRG/UPF Proteomics Unit and the CRG Genomics Facility for protein and RNA sequencing. We thank Eneko Villanueva for advice on OOPS, Marisol Soengas and Corine Bertolotto for cell lines, and Juan Valcárcel for carefully reading this manuscript.
Notes
Present address: Santiago Guerrero. Escuela de Medicina, Facultad de Ciencias Médicas de la Salud y de la Vida, Universidad Internacional del Ecuador, 170113 Quito, Ecuador.
Present address: Alberto Indacochea, Department of Medical Oncology, IDIBAPS, Hospital Clinic, 08036 Barcelona, Spain.
Present address: Alberto Indacochea, Medical Oncology Department, Fundació Privada Hospital Asil de Granollers, 08402 Barcelona, Spain.
Present address: Alfredo Castello, MRC-University of Glasgow Centre for Virus Research, University of Glasgow, Glasgow G61 1QH, UK.
Contributor Information
Neus Mestre-Farràs, Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, 08003 Barcelona, Spain.
Santiago Guerrero, Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, 08003 Barcelona, Spain.
Nadine Bley, Institute of Molecular Medicine, Section for Molecular Cell Biology, Faculty of Medicine, Martin Luther University Halle-Wittenberg, 06120 Halle, Germany.
Ezequiel Rivero, Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, 08003 Barcelona, Spain.
Olga Coll, Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, 08003 Barcelona, Spain.
Eva Borràs, Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, 08003 Barcelona, Spain; Department of Health and Experimental Sciences, Universitat Pompeu Fabra (UPF), 08003 Barcelona, Spain.
Eduard Sabidó, Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, 08003 Barcelona, Spain; Department of Health and Experimental Sciences, Universitat Pompeu Fabra (UPF), 08003 Barcelona, Spain.
Alberto Indacochea, Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, 08003 Barcelona, Spain.
Carlos Casillas-Serra, Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, 08003 Barcelona, Spain.
Aino I Järvelin, Department of Biochemistry, University of Oxford, South Parks Road, Oxford OX1 3QU, UK.
Baldomero Oliva, Department of Health and Experimental Sciences, Universitat Pompeu Fabra (UPF), 08003 Barcelona, Spain.
Alfredo Castello, Department of Biochemistry, University of Oxford, South Parks Road, Oxford OX1 3QU, UK.
Stefan Hüttelmaier, Institute of Molecular Medicine, Section for Molecular Cell Biology, Faculty of Medicine, Martin Luther University Halle-Wittenberg, 06120 Halle, Germany.
Fátima Gebauer, Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, 08003 Barcelona, Spain; Department of Health and Experimental Sciences, Universitat Pompeu Fabra (UPF), 08003 Barcelona, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
N.M. was supported by an FPI fellowship from the Ministry of Economy and Competitiveness; F.G. was supported by national grants BFU2015-68741-P from MINECO, PGC2018-099697-B-I00 from MICINN, ‘la Caixa’ Foundation [100010434] under the agreement LCF/PR/HR17/52150016, Marató-TV3 Foundation [20131430]; Catalan Agency for Research and Universities [2017SGR534]; CRG/UPF Proteomics Unit is part of the Spanish Infrastructure for Omics Technologies (ICTS OmicsTech) and it is member of ProteoRed PRB3 consortium which is supported by grant PT17/0019 of the PE I + D + i 2013–2016 from the Instituto de Salud Carlos III (ISCIII), ERDF, and ‘Secretaria d’Universitats i Recerca del Departament d’Economia i Coneixement de la Generalitat de Catalunya’ [2017SGR595]. A.C. is funded by the MRC Career Development Award (MR/L019434/1) and the ERC Consolidator 101001634 vRNP-capture. We acknowledge the support of the Spanish Ministry of Science and Innovation through the Centro de Excelencia Severo Ochoa (CEX2020-001049-S, MCIN/AEI /10.13039/501100011033), and the Generalitat de Catalunya through the CERCA Programme. Funding for open access charge: MINECO [PGC2018-099697-B-I00].
Conflict of interest statement. None declared.
REFERENCES
- 1. Marine J.-.C., Dawson S.-.J., Dawson M.A.. Non-genetic mechanisms of therapeutic resistance in cancer. Nat. Rev. Cancer. 2020; 20:743–756. [DOI] [PubMed] [Google Scholar]
- 2. Salgia R., Kulkarni P.. The genetic/non-genetic duality of drug ‘resistance’ in cancer. Trends Cancer. 2018; 4:110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Flavahan W.A., Gaskell E., Bernstein B.E.. Epigenetic plasticity and the hallmarks of cancer. Science. 2017; 357:eaal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pereira B., Billaud M., Almeida R.. RNA-Binding proteins in cancer: old players and new actors. Trends Cancer. 2017; 3:506–528. [DOI] [PubMed] [Google Scholar]
- 5. Mino T., Takeuchi O.. Post-transcriptional regulation of immune responses by RNA binding proteins. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2018; 94:248–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. García-Cárdenas J.M., Guerrero S., López-Cortés A., Armendáriz-Castillo I., Guevara-Ramírez P., Pérez-Villa A., Yumiceba V., Zambrano A.K., Leone P.E., Paz-y-Miño C.. Post-transcriptional regulation of colorectal cancer: a focus on RNA-binding proteins. Front. Mol. Biosci. 2019; 6:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Masuda K., Kuwano Y.. Diverse roles of RNA-binding proteins in cancer traits and their implications in gastrointestinal cancers. Wiley Interdiscip. Rev. RNA. 2019; 10:e1520. [DOI] [PubMed] [Google Scholar]
- 8. Wurth L., Gebauer F.. RNA-binding proteins, multifaceted translational regulators in cancer. Biochim. Biophys. Acta. 2015; 1849:881–886. [DOI] [PubMed] [Google Scholar]
- 9. Hentze M.W., Castello A., Schwarzl T., Preiss T.. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018; 19:327–341. [DOI] [PubMed] [Google Scholar]
- 10. Gebauer F., Schwarzl T., Valcárcel J., Hentze MW.. RNA-binding proteins in human genetic disease. Nat. Rev. Genet. 2021; 22:185–198. [DOI] [PubMed] [Google Scholar]
- 11. Castello A., Hentze M.W., Preiss T.. Metabolic enzymes enjoying new partnerships as RNA-Binding proteins. Trends Endocrinol. Metab. 2015; 26:746–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moore S., Järvelin A.I., Davis I., Bond G.L., Castello A.. Expanding horizons: new roles for non-canonical RNA-binding proteins in cancer. Curr. Opin. Genet. Dev. 2018; 48:112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Albihlal W., Gerber A.. Unconventional RNA-binding proteins: an uncharted zone in RNA biology. FEBS Lett. 2018; 592:2917–2931. [DOI] [PubMed] [Google Scholar]
- 14. Schadendorf D., Akkooi A.C.J., Berking C., Griewank K.G., Gutzmer R., Hauschild A., Stang A., Roesch A., Ugurel S.. Melanoma. Lancet. 2018; 392:971–984. [DOI] [PubMed] [Google Scholar]
- 15. Merlino G., Herlyn M., Fisher D.E., Bastian B.C., Flaherty K.T., Davies M.A., Wargo J.A., Curiel-Lewandrowski C., Weber M.J., Leachman S.A.et al.. The state of melanoma: challenges and opportunities. Pigment Cell Melanoma Res. 2016; 29:404–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shain A.H., Bastian BC.. From melanocytes to melanomas. Nat. Rev. Cancer. 2016; 16:345–358. [DOI] [PubMed] [Google Scholar]
- 17. Damsky W.E., Bosenberg M.. Melanocytic nevi and melanoma: unraveling a complex relationship. Oncogene. 2017; 36:5771–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Falletta P., Sanchez-del-Campo L., Chauhan J., Effern M., Kenyon A., Kershaw C.J., Siddaway R., Lisle R., Freter R., Daniels M.J.et al.. Translation reprogramming is an evolutionarily conserved driver of phenotypic plasticity and therapeutic resistance in melanoma. Genes Dev. 2017; 31:18–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cerezo-Wallis D., Contreras-Alcalde M., Troulé K., Catena X., Mucientes C., Calvo T.G., Cañón E., Tejedo C., Pennacchi P.C., Hogan S.et al.. Midkine rewires the melanoma microenvironment toward a tolerogenic and immune-resistant state. Nat. Med. 2020; 26:1865–1877. [DOI] [PubMed] [Google Scholar]
- 20. Rapino F., Delaunay S., Rambow F., Zhou Z., Tharun L., de Tullio P., Sin O., Shostak K., Schmitz S., Piepers J.et al.. Codon-specific translation reprogramming promotes resistance to targeted therapy. Nature. 2018; 558:605–609. [DOI] [PubMed] [Google Scholar]
- 21. Karras P., Riveiro-Falkenbach E., Cañón E., Tejedo C., Calvo T.G., Martínez-Herranz R., Alonso-Curbelo D., Cifdaloz M., Perez-Guijarro E., Gómez-López G.et al.. p62/SQSTM1 fuels melanoma progression by opposing mRNA decay of a selective set of Pro-metastatic factors. Cancer Cell. 2019; 35:46–63. [DOI] [PubMed] [Google Scholar]
- 22. Phung B., Cieśla M., Sanna A., Guzzi N., Beneventi G., Cao Thi Ngoc P., Lauss M., Cabrita R., Cordero E., Bosch A.et al.. The X-linked DDX3X RNA helicase dictates translation reprogramming and metastasis in melanoma. Cell Rep. 2019; 27:3573–3586. [DOI] [PubMed] [Google Scholar]
- 23. Cifdaloz M., Osterloh L., Graña O., Riveiro-Falkenbach E., Ximénez-Embún P., Muñoz J., Tejedo C., Calvo T.G., Karras P., Olmeda D.et al.. Systems analysis identifies melanoma-enriched pro-oncogenic networks controlled by the RNA binding protein CELF1. Nat. Commun. 2017; 8:2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wurth L., Papasaikas P., Olmeda D., Bley N., Calvo G.T., Guerrero S., Cerezo-Wallis D., Martinez-Useros J., García-Fernández M., Hüttelmaier S.et al.. UNR/CSDE1 drives a Post-transcriptional program to promote melanoma invasion and metastasis. Cancer Cell. 2016; 30:694–707. [DOI] [PubMed] [Google Scholar]
- 25. Pérez-Guijarro E., Karras P., Cifdaloz M., Martínez-Herranz R., Cañón E., Graña O., Horcajada-Reales C., Alonso-Curbelo D., Calvo T.G., Gómez-López G.et al.. Lineage-specific roles of the cytoplasmic polyadenylation factor CPEB4 in the regulation of melanoma drivers. Nat. Commun. 2016; 7:13418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hannus M., Beitzinger M., Engelmann J.C., Weickert M.-.T., Spang R., Hannus S., Meister G.. siPools: highly complex but accurately defined siRNA pools eliminate off-target effects. Nucleic Acids Res. 2014; 42:8049–8061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schneider C.A., Rasband W.S., Eliceiri K.W.. NIH image to imagej: 25 years of image analysis. Nat. Methods. 2012; 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Coll O., Guitart T., Villalba A., Papin C., Simonelig M., Gebauer F.. Dicer-2 promotes mRNA activation through cytoplasmic polyadenylation. RNA. 2018; 24:529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Castello A., Horos R., Strein C., Fischer B., Eichelbaum K., Steinmetz L.M., Krijgsveld J., Hentze M.W.. System-wide identification of RNA-binding proteins by interactome capture. Nat. Protoc. 2013; 8:491–500. [DOI] [PubMed] [Google Scholar]
- 30. Perkins D.N., Pappin D.J., Creasy D.M., Cottrell J.S.. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999; 20:3551–3567. [DOI] [PubMed] [Google Scholar]
- 31. Queiroz R.M.L., Smith T., Villanueva E., Marti-Solano M., Monti M., Pizzinga M., Mirea D.-.M., Ramakrishna M., Harvey R.F., Dezi V.et al.. Comprehensive identification of RNA–protein interactions in any organism using orthogonal organic phase separation (OOPS). Nat. Biotechnol. 2019; 37:169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chiva C., Olivella R., Borràs E., Espadas G., Pastor O., Solé A., Sabidó E.. QCloud: a cloud-based quality control system for mass spectrometry-based proteomics laboratories. PLoS One. 2018; 13:e0189209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Olivella R., Chiva C., Serret M., Mancera D., Cozzuto L., Hermoso A., Borràs E., Espadas G., Morales J., Pastor O.et al.. QCloud2: an improved Cloud-based quality-control system for Mass-Spectrometry-based proteomics laboratories. J. Proteome Res. 2021; 20:2010–2013. [DOI] [PubMed] [Google Scholar]
- 34. Wickham H. Ggplot2: elegant graphics for data analysis. 2016; NY: Springer-Verlag. [Google Scholar]
- 35. Teo G., Liu G., Zhang J., Nesvizhskii A.I., Gingras A.-.C., Choi H.. SAINTexpress: improvements and additional features in significance analysis of interactome software. J. Proteomics. 2014; 100:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Trendel J., Schwarzl T., Horos R., Prakash A., Bateman A., Hentze M.W., Krijgsveld J.. The human RNA-Binding proteome and its dynamics during translational arrest. Cell. 2019; 176:391–403. [DOI] [PubMed] [Google Scholar]
- 37. Urdaneta E.C., Vieira-Vieira C.H., Hick T., Wessels H.-.H., Figini D., Moschall R., Medenbach J., Ohler U., Granneman D., Selbach M.et al.. Purification of cross-linked RNA-protein complexes by phenol-toluol extraction. Nat. Commun. 2019; 10:990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. O’Brien K.P., Remm M., Sonnhammer E.L.L.. Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res. 2005; 33:D476–D480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Castello A., Fischer B., Eichelbaum K., Horos R., Beckmann B.M., Strein C., Davey N.E., Humphreys D.T., Preiss T., Steinmetz L.M.et al.. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012; 149:1393–1406. [DOI] [PubMed] [Google Scholar]
- 40. Castello A., Horos R., Strein C., Fischer B., Eichelbaum K., Steinmetz L.M., Krijgsveld J., Hentze M.W.. Comprehensive identification of RNA-Binding proteins by RNA interactome capture. Methods Mol. Biol. 2016; 1358:131–139. [DOI] [PubMed] [Google Scholar]
- 41. Pham T.V., Piersma S.R., Warmoes M., Jimenez C.R.. On the beta-binomial model for analysis of spectral count data in label-free tandem mass spectrometry-based proteomics. Bioinformatics. 2010; 26:363–369. [DOI] [PubMed] [Google Scholar]
- 42. Knowles D.G., Röder M., Merkel A., Guigó R.. Grape RNA-Seq analysis pipeline environment. Bioinformatics. 2013; 29:614–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dobin A., Gingeras T.R.. Mapping RNA-seq reads with STAR. Curr. Protoc. Bioinformatics. 2015; 51:11.14.1–11.14.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li B., Dewey C.N.. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011; 12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Durinck S., Moreau Y., Kasprzyk A., Davis S., de Moor B., Brazma A., Huber W.. BioMart and bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005; 21:3439–3440. [DOI] [PubMed] [Google Scholar]
- 46. Baltz A.G., Munschauer M., Schwanhäusser B., Vasile A., Murakawa Y., Schueler M., Youngs N., Penfold-Brown D., Drew K., Milek M.et al.. The mRNA-Bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell. 2012; 46:674–690. [DOI] [PubMed] [Google Scholar]
- 47. Gupta P.B., Kuperwasser C., Brunet J.-.P., Ramaswamy S., Kuo W.-.L., Gray J.W., Naber S.P., Weinberg R.A.. The melanocyte differentiation program predisposes to metastasis following neoplastic transformation. Nat. Genet. 2005; 37:1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mellacheruvu D., Wright Z., Couzens A.L., Lambert J.-.P., St-Denis N.A., Li T., Hauri S., Sardiu M.E., Low T.Y., Halim V.A.et al.. The CRAPome: a contaminant repository for affinity purification–mass spectrometry data. Nat. Methods. 2013; 10:730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Conrad T., Albrecht A.-.S., de Melo Costa V.R., Sauer S., Meierhofer D., Ørom U.A.. Serial interactome capture of the human cell nucleus. Nat. Commun. 2016; 7:11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Perez-Perri J.I., Rogell B., Schwarzl T., Stein F., Zhou Y., Rettel M., Brosig A., Hentze M.W.. Discovery of RNA-binding proteins and characterization of their dynamic responses by enhanced RNA interactome capture. Nat. Commun. 2018; 9:4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E.et al.. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019; 47:D941–D947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hamosh A., Scott A.F., Amberger J., Valle D., Mckusick VA.. Online mendelian inheritance in man (OMIM). Hum. Mutat. 2000; 15:57–61. [DOI] [PubMed] [Google Scholar]
- 53. Harris M.A., Clark J., Ireland A., Lomax J., Ashburner M., Foulger R., Eilbeck K., Lewis S., Marshall B., Mungall C.et al.. The gene ontology (GO) database and informatics resource. Nucleic Acids Res. 2004; 32:D258–D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Martínez-Jiménez F., Muiños F., Sentís I., Deu-Pons J., Reyes-Salazar I., Arnedo-Pac C., Mularoni L., Pich O., Bonet J., Kranas H.et al.. A compendium of mutational cancer driver genes. Nat. Rev. Cancer. 2020; 20:555–572. [DOI] [PubMed] [Google Scholar]
- 55. Haass N.K., Herlyn M.. Normal human melanocyte homeostasis as a paradigm for understanding melanoma. J. Investig. Dermatol. Symp. Proc. 2005; 10:153–163. [DOI] [PubMed] [Google Scholar]
- 56. Tsherniak A., Vazquez F., Montgomery P.G., Weir B.A., Kryukov G., Cowley G.S., Gill S., Harrington W.F., Pantel S., Krill-Burger J.M.et al.. Defining a cancer dependency map. Cell. 2017; 170:564–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cerami1 E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E.et al.. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E.et al.. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013; 6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Eletto D., Eletto D., Boyle S., Argon Y.. PDIA6 regulates insulin secretion by selectively inhibiting the RIDD activity of IRE1. FASEB J. 2016; 30:653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Eletto D., Eletto D., Dersh D., Gidalevitz T., Argon Y.. Protein disulfide isomerase A6 controls the decay of IRE1α signaling via disulfide-dependent association. Mol. Cell. 2014; 53:562–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Groenendyk J., Peng Z., Dudek E., Fan X., Mizianty M.J., Dufey E., Urra H., Sepulveda D., Rojas-Rivera D., Lim Y.et al.. Interplay between the oxidoreductase PDIA6 and microRNA-322 controls the response to disrupted endoplasmic reticulum calcium homeostasis. Sci. Signal. 2014; 7:ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kaiser B.K., Yim D., Chow I.-.T., Gonzalez S., Dai Z., Mann H.H., Strong R.K., Groh V., Spies T.. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature. 2007; 447:482–486. [DOI] [PubMed] [Google Scholar]
- 63. Poggi A., Zocchi M.R.. How to exploit stress-related immunity against hodgkin's lymphoma: targeting ERp5 and ADAM sheddases. Oncoimmunology. 2013; 2:e27089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gumireddy K., Sun F., Klein-Szanto A.J., Gibbins J.M., Gimotty P.A., Saunders A.J., Schultz P.G., Huang Q.. In vivo selection for metastasis promoting genes in the mouse. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:6696–6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tufo G., Jones A.W.E., Wang Z., Hamelin J., Tajeddine N., Esposti D.D., Martel C., Boursier C., Gallerne C., Migdalet C.et al.. The protein disulfide isomerases PDIA4 and PDIA6 mediate resistance to cisplatin-induced cell death in lung adenocarcinoma. Cell Death Differ. 2014; 21:685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Castello A., Fischer B., Frese C.K., Horos R., Alleaume A.M., Foehr S., Curk T., Krijgsveld J., Hentze M.W.. Comprehensive identification of RNA-Binding domains in human cells. Mol. Cell. 2016; 63:696–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mullari M., Lyon D., Jensen L.J., Nielsen M.L.. Specifying RNA-binding regions in proteins by peptide cross-linking and affinity purification. J. Proteome Res. 2017; 16:2762–2772. [DOI] [PubMed] [Google Scholar]
- 68. Jordan P.A., Gibbins J.M.. Extracellular disulfide exchange and the regulation of cellular function. Antioxid. Redox. Signal. 2006; 8:312–324. [DOI] [PubMed] [Google Scholar]
- 69. Yang J., Zhang Y.. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 2015; 43:W174–W181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Matia-González A.M., Laing E.E., Gerber A.P.. Conserved mRNA-binding proteomes in eukaryotic organisms. Nat. Struct. Mol. Biol. 2015; 22:1027–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sysoev V.O., Fischer B., Frese C.K., Gupta I., Krijgsveld J., Hentze M.W., Castello A., Ephrussi A.. Global changes of the RNA-bound proteome during the maternal-to-zygotic transition in drosophila. Nat. Commun. 2016; 7:12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Milek M., Landthaler M.. Systematic detection of poly(a) + RNA-Interacting proteins and their differential binding. Methods Mol. Biol. 2018; 1649:405–417. [DOI] [PubMed] [Google Scholar]
- 73. Liepelt A., Naarmann-De Vries I.S., Simons N., Eichelbaum K., Föhr S., Archer S.K., Castello A., Usadel B., Krijgsveld J., Preiss T.et al.. Identification of RNA-binding proteins in macrophages by interactome capture. Mol. Cell. Proteomics. 2016; 15:2699–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Despic V., Dejung M., Gu M., Krishnan J., Zhang J., Herzel L., Straube K., Gerstein M.B., Butter F., Neugebauer KM.. Dynamic RNA–protein interactions underlie the zebrafish maternal-to-zygotic transition. Genome Res. 2017; 27:1184–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Garcia-Moreno M., Noerenberg M., Ni S., Järvelin A.I., González-Almela E., Lenz C.E., Bach-Pages M., Cox V., Avolio R., Davis T.et al.. System-wide profiling of RNA-Binding proteins uncovers key regulators of virus infection. Mol. Cell. 2019; 74:196–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Esmaillie R., Ignarski M., Bohl K., Krüger T., Ahmad D., Seufert L., Schermer B., Benzing T., Müller R.-.U., Fabretti F.. Activation of hypoxia-inducible factor signaling modulates the RNA protein interactome in caenorhabditis elegans. iScience. 2019; 22:466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rudnick P.A., Markey S.P., Roth J., Mirokhin Y., Yan X., Tchekhovskoi D.V., Edwards N.J., Thangudu R.R., Ketchum K.A., Kinsinger C.R.et al.. A description of the clinical proteomic tumor analysis consortium (CPTAC) common data analysis pipeline. J. Proteome Res. 2016; 15:1023–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kamel W., Noerenberg M., Cerikan B., Chen H., Järvelin A.I., Kammoun M., Lee J.Y., Shuai N., Garcia-Moreno M., Andrejeva A.et al.. Global analysis of protein-RNA interactions in SARS-CoV-2-infected cells reveals key regulators of infection. Mol. Cell. 2021; 81:2851–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Beckmann B.M., Castello A., Medenbach J.. The expanding universe of ribonucleoproteins: of novel RNA-binding proteins and unconventional interactions. Pflügers Arch. 2016; 468:1029–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Schulman S., Bendapudi P., Sharda A., Chen V., Bellido-Martin L., Jasuja R., Furie B.C., Flaumenhaft R., Furie B.. Extracellular thiol isomerases and their role in thrombus formation. Antioxid. Redox. Signal. 2016; 24:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fazal F.M., Han S., Parker K.R., Kaewsapsak P., Xu J., Boettiger A.N., Chang H.Y., Ting A.Y.. Atlas of subcellular RNA localization revealed by APEX-Seq. Cell. 2019; 178:473–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kondo J., Westhof E.. Classification of pseudo pairs between nucleotide bases and amino acids by analysis of nucleotide-protein complexes. Nucleic Acids Res. 2011; 39:8628–8637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Vizcaíno J.A., Csordas A., del-Toro N., Dianes J.A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T.et al.. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016; 44:D447–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Transcriptomics data generated in this study are available through Gene Expression Omnibus (GEO) under accession number GSE174821. The mass spectrometry data have been deposited in the PRIDE repository with the dataset identifier PXD029927 (83).