

Abstract
Methamphetamine (METH) abuse is associated with the emergence of cognitive deficits and hypofrontality, a pathophysiological marker of many neuropsychiatric disorders that is produced by altered balance of local excitatory and inhibitory synaptic transmission. However, there is a dearth of information regarding the cellular and synaptic mechanisms underlying METH-induced cognitive deficits and associated hypofrontal states. Using PV-Cre transgenic rats that went through a METH sensitization regime or saline (SAL) followed by 7–10 days of home cage abstinence combined with cognitive tests, chemogenetic experiments, and whole-cell patch recordings on the prelimbic prefrontal cortex (PFC), we investigated the cellular and synaptic mechanisms underlying METH-induce hypofrontality. We report here that repeated METH administration in rats produces deficits in working memory and increases in inhibitory synaptic transmission onto pyramidal neurons in the PFC. The increased PFC inhibition is detected by an increase in spontaneous and evoked inhibitory postsynaptic synaptic currents (IPSCs), an increase in GABAergic presynaptic function, and a shift in the excitatory-inhibitory balance onto PFC deep-layer pyramidal neurons. We find that pharmacological blockade of D1 dopamine receptor function reduces the METH-induced augmentation of IPSCs, suggesting a critical role for D1 dopamine signaling in METH-induced hypofrontality. In addition, repeated METH administration increases the intrinsic excitability of parvalbumin-positive fast spiking interneurons (PV + FSIs), a key local interneuron population in PFC that contributes to the control of inhibitory tone. Using a cell type-specific chemogenetic approach, we show that increasing PV + FSIs activity in the PFC is necessary and sufficient to cause deficits in temporal order memory similar to those induced by METH. Conversely, reducing PV + FSIs activity in the PFC of METH-exposed rats rescues METH-induced temporal order memory deficits. Together, our findings reveal that repeated METH exposure increases PFC inhibitory tone through a D1 dopamine signaling-dependent potentiation of inhibitory synaptic transmission, and that reduction of PV + FSIs activity can rescue METH-induced cognitive deficits, suggesting a potential therapeutic approach to treating cognitive symptoms in patients suffering from METH use disorder.
Subject terms: Neuroscience, Cellular neuroscience
Introduction
Methamphetamine (METH) use disorder is a profound medical and societal problem. METH users frequently develop hypofrontality, a state of reduced frontal cortex activity associated with deficits in temporal order memory (TOM), working memory (WM), attention, and impulsivity [1–3]. METH-induced cognitive deficits can significantly reduce the quality of life and contribute to sustained drug-taking and relapse [4, 5]. In addition, METH-induced cognitive impairments are long-lasting, with chronic METH users reporting cognitive dysfunction even after 11 months of abstinence [6, 7]. However, the mechanisms underlying METH-induced hypofrontality and cognitive dysfunction during abstinence remain elusive. Along with classic substance use disorder-like behaviors, such as drug craving and seeking, rodents repeatedly exposed to METH also exhibit cognitive abnormalities during abstinence [8–10], including memory and WM deficits [9–11]. As such, repeated psychostimulant administration in rodents represents a strong, face-valid model to study the mechanisms that underlie hypofrontality and cognitive disabilities in METH use disorder.
Although inhibitory interneurons constitute only 10–20% of the neuronal population in the cortex [12], their function is critical to control the activity level of the principal glutamatergic pyramidal neurons (PNs) and to maintain the proper balance between excitatory (E) and inhibitory (I) synaptic transmission. E-I ratio alterations in the frontal cortex are thought to contribute to diverse neuropsychiatric disorders [13, 14], including substance use disorders [15–19]. The role of cortical glutamatergic excitatory neurons in cognition has been extensively studied [20–28], but we are only beginning to understand the contribution of cortical interneurons to cognitive abilities. Among the multiple GABAergic interneuron subtypes within the medial prefrontal cortex (mPFC) [12, 29–31], the parvalbumin-positive fast-spiking interneurons (PV + FSIs) exert a particularly powerful influence on PN inhibitory tone. Indeed, PV + FSIs are the most common interneuron subtype [32], they contact nearly every local PN [33], and they target PNs near the site of action potential initiation [12, 29]. PV + FSI dysfunction has been linked to numerous neuropsychiatric disorders, such as epilepsy, schizophrenia, depression, autism, and Alzheimer’s disease [34–38]. Importantly, PV + FSIs are particularly affected in patients with WM deficits [39–41]. PV + FSIs have elicited interest for their critical role in orchestrating cortical oscillations, maintaining E-I balance, and mediating a variety of cognitive behaviors, including WM [41–44]. However, while it is well known that repeated METH exposure induces cognitive dysfunction in both rodents and humans [6, 7, 9–11], its synaptic and cellular effects on PV + FSIs, and the mechanisms underlying METH-induced cognitive deficits, remain elusive.
Since METH is known to increase dopamine (DA) release in the brain, including in the mPFC [45–47], DA receptor signaling could, at least in part, regulate changes in PFC activity, and consequently elicit PFC-dependent behavioral deficits. VTA neurons project to both PFC PNs and PV + FSIs in layers I–III and V–VI [48–50], but several studies have shown that DA in the PFC preferentially acts on PV + FSIs [50–52]. In PFC layers V–VI, PV + FSIs express high levels of dopamine D1 receptors (D1R) and low levels of dopamine D2 receptors (D2R) [31]. Activation of D1R signaling on PV + FSIs increases their excitability – enhancing both evoked and spontaneous inhibitory post-synaptic currents (IPSCs) recorded in PNs [51, 53–56]. Multiple studies demonstrate that DA release and GABA interneurons influence WM via their contribution in tuning recurrent excitation in mPFC networks [53, 57–61]. In addition, WM delay cells, which are mainly PN neurons that show spiking during memory delays of WM tasks [62–64], are modulated by DA activation of D1R (but not D2R) signaling, and optimal levels of D1R stimulation are essential for WM function [53, 57, 60, 61].
In this study, we show that repeated METH administration produces hypofrontal-like states and cognitive deficits that are the consequence of a maladaptive, D1R-dependent enhancement of GABAergic transmission. Moreover, using cell type-specific chemogenetics, we demonstrate that reduction of PV + FSIs activity is sufficient to rescue the cognitive deficits produced by chronic METH administration, suggesting a circuit-specific strategy to treat cognitive symptoms in individuals suffering from METH use disorder.
Materials and methods
Animals
PV-Cre transgenic Long Evans male rats [65, 66] were obtained from the NIDA Transgenic Rat Project (Optogenetics and Transgenic Technology Core, National Institute on Drug Abuse (NIDA) [66], via the Rat Resource and Research Center (catalog #00773). They were bred and maintained on a Long Evans background at the Animal Facility at MUSC. Rats were housed under a 12 light/12 dark hour cycle in pairs, with access to food and water ad libitum. Following the NIDA Transgenic Rat Project protocol, the PV-Cre rats were genotyped via PCR using forward primer 5’GTTCTGCCGGGTCAGAAAGAATGGT3’, and reverse primer 5’CCAACCCCGAGGATAAGGGAATG3’, producing a 713 bp band.
All experiments were performed in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals. Procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the Medical University of South Carolina.
METH administration
Day 1 and day 14 of the treatment consisted of a single home cage i.p injection (0.1 ml/100 g) of METH at 1 mg/kg or saline (SAL) [67]. From day 2 to day 13, rats received home cage i.p injections (0.1 ml/100 g) of METH at 5 mg/kg or SAL. Following METH or SAL treatment, rats were given 7-10 days of home cage abstinence. One group of animals was tested for temporal order memory (TOM), followed by electrophysiological recordings (Fig. 1A), and a second group of animals was tested for working memory using a Delay Non-Match to Sample Task (DNMST; Fig. 1A), also followed by electrophysiological recordings.
Fig. 1. Experimental design and behavioral assessments.

A Schematic illustration of the experimental design. We infuse either pAAV- FLEX-tdTomato, pAAV-hsyn-DIO-hM3Dq (Gq) or pAAV-hsyn-DIO-hM4Di (Gi) DREADDS in the PFC of PV-Cre rats followed by repeated METH or SAL in home cage treatment. Cognitive tests and electrophysiological assessments are performed after 7–10 days of abstinence. B Schematic representation of the Temporal Order Memory (TOM) test. B1 Total time of exploration for each object. Saline animals spend more time with the old object [Main effect of object: SAL Object A. 70.33 ± 3.8 vs SAL Object B. 31.00 ± 3.0; Mixed effect analysis ANOVA, F(1,30) = 35.32, ****p < 0.0001., Holm–Sidak post-hoc test]. On the other hand, METH animals do not show preference for any particular object [METH Object A. 43.5 ± 3.4 vs METH Object B. 46.5 ± 5.1; Mixed effect analysis ANOVA, F(1,30) = 1.053, ns p = 0.66, Holm–Sidak post-hoc test]. B2 There was no difference in the total time of exploration between groups [SAL total time 101.3 ± 6.0; METH total time 90.5 ± 7.6; t(31) = 0.3172, ns, SAL n = 13, METH n = 20]. B3 METH induces a significant decrease in the TOM test ratio [t(31) = 5.657, ****p < 0.0001, SAL n = 13, METH n = 20]. C Schematic representation of the Delay No Match to Sample Test. The graph shows METH-induced impairment in working memory [Main effect by drug: Two-way ANOVA, F(1,115) = 11.3 **p = 0.001, SAL n = 12; METH n = 20]. The schematic illustration was created using BioRender.com.
Intracranial surgeries
Adult male PV-Cre rats (270–320 g) were anesthetized with isoflurane. Viral vectors [Cre-dependent pAAV-Flex-tdTomato-AAV9; AAV-hSyn-DIO-HM3Dq-mCherry; AVV-hSyn-DIO-HM4Di-mCherry (1.5–3 µl)] were injected bilaterally into the mPFC using a stereotaxic apparatus. Injections were performed using a nanojector (Drummond Scientific Company, Broomall, PA, USA) and a glass pipette (1 µm tip diameter) using the following coordinates for prelimbic mPFC: PL-PFC [anterior-posterior (AP): +3.0–3.2 mm; medial-lateral (ML): ±0.7 mm; dorsal- ventral (DV): −3.8 mm, Paxinos and Watson 1982]. Rats were single housed for 7 days following surgery and then moved to pair housing for METH or SAL administration.
A group of PV-Cre rats (n = 14), was injected with the GqDREADDS follow by SAL treatment. A different group of PV-Cre rats (n = 8), was injected with the GiDREADDS follow by repeated METH exposure. Clozapine N-oxide (CNO) or vehicle was administered i.p, (10 mg/kg; [68]) and 30 min later behavioral test were performed.
Cognitive assessments
Temporal order memory (Fig. 1B)
Animals were habituated for 2 consecutive days to the arena (45 × 35 × 40 cm) for 5 min in the absence of objects. During the first sample trial or phase 1, a single rat was placed into the arena and allowed to explore two identical objects for 5 min. Then the rat was placed into its home cage for 30 min. Phase 2 consisted of repeating the 5 min exploration but using a different pair of identical objects, before being placed again into its home cage for 30 min. Finally, during the test, the rat was placed again into the arena with one object from each sample (i.e., Object A and B), for 5 min (Fig. 1B). The criteria for evaluating behavioral exploration of the object was defined as: sniffing the objects and physically interacting with the object. The objects were counter balanced in all the groups evaluated. The discrimination ratio was calculated as the difference in time spent by each animal exploring object A compared with object B, divided by the total time spent exploring both objects during the five minutes of the test period. The total time of exploration was recorded and considered as a control for locomotor activity. In this test, the hypothetical ratio expected in a control animal is a positive ratio, meaning that the animal spent more time exploring the “oldest” object, since in order terms, the “oldest” object is novel compared with the most recent one [68]. On the other hand, a ratio equal to or less than zero suggests a deficit in recognizing the most recent object, and means that animals spent equal time exploring the “oldest” and recent object or that they spent more time exploring the most recent object. All the phases of the TOM test (habituation, phase 1, phase 2 and test) were videotaped and the analysis and quantification were performed post hoc. All the objects used on the test were previously tested for preference by a control group of naïve rats. The rats didn’t show preference for any particular object.
Working memory (Fig. 1C)
To test reference and WM, we used a modified version of a DNMS test [69–71]. In brief, rats were food restricted (~90% of their original weight) and habituated to a Y maze for 3 days before the training phase. Training consisted of two sessions, an information run and a test run. In the information run, one arm was blocked forcing the animal into the open arm, where it received a sugar pellet. Subsequently, the blocked arm was open, and the rat was given a free choice of either arm. Rats received a sugar pellet for choosing the previously unvisited arm (correct choice), choosing the visited arm yield no reward. Left/right allocations for the information and test runs were pseudo-randomized over 10 trials/day with no more than 3 consecutive information runs to the same side. The inter-trial interval was 1 min. An entry to the arm was considered when the 4 paws of the rat and half length of the tail were on the selected arm. A correct choice was scored when the animal retrieved the sugar pellet. An incorrect choice was scored when the animal entered the previously visited arm. A no choice was scored when the animal did not enter the correct arm in a period of 2 min or entered a correct arm but did no retrieve the sugar pellet. After 3 d of training, ≥70% correct choices for 2 consecutive days were required to proceed to the WM evaluation. For the WM test, rats were assigned randomly 10 trials with delays of 90, 120, 180 and 240 s between the information run and the test run (the % of correct choices/delay was calculated for comparison).The performance in the delay component of the task was assessed for 2 consecutive days. All cognitive tests were performed during the light phase of the rats.
Brain slice preparation
Animals were deeply anesthetized using isofluorane (Minrad Inc., Orchard Park, NY, USA), rapidly decapitated, and the brain extracted. Coronal slices 300 µM thick containing prelimbic mPFC were obtained with a vibratome (Leica VT 1200 s, Leica Microsystems, Buffalo Grove, IL, USA) in ice-cod artificial cerebral spinal fluid [aCSF containing: 200 mM sucrose, 1.9 mM KCL, 1.2 mM Na2HPO4, 33 mM NaHCO3, 6 mM MgCl2, 10 mM D-glucose, and 0.4 mM ascorbic acid (pH: 7.3–7.4, 305–310 mOsm)]. Slices were incubated in a hot bath at 31 °C for 1 h before recordings, with a physiological solution containing:120 mM NaCl, 2.5 mM KCl, 1.25 mM NaH2PO4, 25 mM NaHCO3, 4 mM MgCl2, 1 mM CaCl2, 10 mM D-glucose, and 0.4 mM ascorbic acid, oxygenated 95% o2/5% CO2 (pH: 7.3–7.4, 305–310 mOsm).
Electrophysiological recordings
Whole-cell patch clamp was used to record neurons located in layers V–VI of the prelimbic mPFC identified using infrared-differential interference contrast optics and video microscopy. PV + FSIs were identified using a filter for tdTomato. Signals were recorded using an Axoclamp 2B (Molecular Devices), low-pass filtered at 3 kHz and digitalized at 10 kHz. Data acquisition was performed using Axograph-software (Axograph J. Clemens). For voltage-clamp recordings, electrodes (3–5 MΩ resistance in situ) were filled with a solution containing: 130 mM CsCl, 10 mM HEPES, 2 mM MgCl2, 0.5 mM EGTA, 2 mM Na2ATP, 0.3 mM Na-GTP, 2 mM QX-314, 10 mM phosphocreatine, (pH = 7.2–7.3, 270–280 mOsm). The membrane potential was clamped at −70 mV. Series resistance (Rs) was monitored throughout the experiment using a −5-mV pulse (50 ms duration) and recordings that exhibit changes in baseline >30% were discarded. GABAergic-mediated events were pharmacologically isolated by adding CNQX (10 µM) and AP-V (50 µM) to the bath. All recordings were performed 5–10 min after break in to allow for stabilization of the recordings. Spontaneous Inhibitory Post-Synaptic Currents (sIPSC) recordings consisted on five sweeps/10 s. Evoked inhibitory synaptic currents (eIPSCs) and paired pulse (PP) recordings consisted in 8–10 repetitions at 0.5 Hz using the minimum amount of current needed to evoke a response. In average, the stimulation currents in SAL rats were 61 ± 52.9 µA and in METH rats 76 ± 48.7 µA. The sIPSCs were analyzed using MiniAnalysis software (Synaptosoft, Fort Lee, NJ, USA). Synaptic currents were evoked using bipolar theta capillaries filled with saline solution placed in the mPFC around 300 µm from the patched neurons. Cells were recorded during baseline conditions using aCSF followed by 5–7 min of aCSF+ DA drugs. After 5–7 min of perfusion with the aCSF+ DA drugs, the perfusion solution was switched to regular aCSF. The coefficient of variation (CV) was calculated as the standard deviation of the eIPSCs amplitude/amplitude of the eIPSCs [72]. In order to normalize the CV data, we used 1/CV2. For current clamp recordings, a potassium-based internal solution was used. Parvalbumin-positive fast spiking interneurons (PV + FSI) were identified by tdTomato fluorescence and electrophysiological signature [29]. tdTomato fluorescent neurons that did not display the electrophysiological characteristics of fast spiking interneurons (brief action potentials, less than 2.0 msec duration and fast non-adapting patterns of firing) were excluded from the analysis. Current steps of 20 pA from −100 to 300 pA (1 s duration) were injected to generate I/F and input resistances curves. The duration of the action potentials (AP) was calculated from the initiation of the AP to the end of the spike. We use the 3er AP in each current step, from 120-180 pA, to measure AP duration, AHP amplitude and AHP duration. The average was then calculated for the SAL or METH groups.
Excitatory/inhibitory ratio (E-I) was recorded using a potassium-based internal solution (in mM): 120 K-gluconate,10 NaCl, 10 KCl, 0.5 EGTA, 2 Na2ATP, 0.3 Na-GTP, 10 HEPES and 10 phosphocreatine, pH 7.2–7.35 and 270–280 mOsm.. Evoked PSCs at −70 mV and 0 mV were recorded and the ratio calculated by dividing the amplitude of the AMPA response/amplitude of GABA response. Control experiments recording a 0 mV in SAL rats (n = 3) showed that application of picrotoxin at this membrane potential blocked all spontaneous activity.
All the electrophysiological recordings were performed in rats that have been either behaviorally tested after METH or SAL treatments, or in rats that have gone only through METH or SAL treatments.
Immunohistochemistry
Rats were given an overdose of ketamine/xylazine (150 mg/kg and 2.9 mg/kg respectively) diluted in 0.9% saline, then perfused transcardially with 4% paraformaldehyde (PFA). The brains were post-fixed overnight in 4% PFA, cryoprotected in 30% sucrose for 3 days and sectioned at 40 um using a cryotome. Sections were washed in PBS 1X, incubated in blocking solution (5% normal donkey serum, 1% Bovine Serum Albumin, 0.2% glycine, 0.2% lysine, 0.3% Triton X-100 diluted in PBS 1X) under shaking for 1 h at room temperature, incubated with primary antibodies diluted in blocking solution (mouse anti-parvalbumin (1:800) from Millipore #MAB1572; rabbit anti-DsRed (1:800) from LivingColors #632496) overnight at 4 °C under shaking, washed 3 times 10 min in PBS 1X under shaking at room temperature, incubated with secondary antibodies diluted in blocking solution (AlexaFluor 488 donkey anti-mouse (1:500) from Fisher #A21202; AlexaFluor 594 donkey anti-rabbit (1:500) from Fisher #A21207) for 1 h and 30 min at room temperature under shaking and protected from light, washed 3 times 10 min in PBS 1X under shaking and finally mounted in ProLong Gold Antifade Mountant (Invitrogen #P36931). Sections were imaged using a Zeiss confocal: z-stacks were made at 20X in the mPFC. All quantifications were performed using ImageJ.
Data analysis and statistics
Electrophysiological data analysis was performed using Axograph software® (J. Clemens) and MiniAnalysis® (Synaptosoft, Fort Lee, NJ USA). Electrophysiological and behavioral data were analyzed using GraphPad software. Data are presented as mean ± SEM. Normality was determined using Shapiro–Wilk test. For normal data, we used Student’s t test for comparison of two independent groups. In the case of multiple comparison, we used Two-way ANOVA following by Tukey’s post-hoc test or Two- way ANOVA Mixed effect analysis following by Holm–Sidak’s multiple comparison test.
Results
Repeated METH administration produces cognitive deficits
To examine the effects of repeated, daily METH administration and abstinence on working memory behavior in rats, we injected rats with METH or saline (SAL) for 14 consecutive days (i.p., see methods and Fig. 1A) followed by 7–10 days of home-cage abstinence. We then tested the rats in two PFC-dependent cognitive behavior tests, the temporal order memory test (TOM) and the Delayed Non-Match to Sample Test (DNMST) [68] (Fig. 1B, C). In the TOM test as expected, the saline-treated rats show a preference to interact with the non-recent object A. In contrast, METH-treated rats spent same time exploring both objects (Fig. 1B1), and there was a significant difference in the TOM ratio by METH treatment (Fig. 1B3), indicating a likely deficit in short-term spatial memory. There was no significant effect of METH treatment on total time interacting with the objects (Fig. 1B2), so METH did not appear to alter exploratory drive or motor ability. In the DNMST, a working memory task where rats must remember the most recent reward location to make the subsequent non-matching location choice, METH-treated rats show a significant decrease in the percentage of correct choices (Fig. 2C), compared to SAL controls. Together, these data demonstrate that repeated METH exposure in rats produces significant working memory deficits, similar to the cognitive symptoms in individuals suffering from METH use disorder.
Fig. 2. Repeated METH increases GABA transmission.

A Repeated METH evokes a significant increase in the amplitude of sIPSCs recorded in PNs [SAL = 15.35 ± 1.3 pA, ncells/rats = 16/8; METH = 24.24 ± 1.9 pA, ncells/rats = 28/8, t(42) = 3.18, **p = 0.0028]. B Repeated METH elicits a significant increase in the frequency of sIPSCs [SAL = 1.18 ± 0.13 Hz, ncells/rats = 15/8; METH = 2.78 ± 0.44 Hz, ncells/rats = 28/8, t(41) = 2.61, *p = 0.012]. Representative traces are shown to the right. C Repeated METH elicits a significant increase in amplitude of eIPSCs [SAL = 50.55 ± 5.7 pA, ncells/rats = 19/7; METH = 89.82 ± 12.3 pA, ncells/rats = 21/9; t(38) = 2.7, **p = 0.008]. Representative traces to the right. D Repeated METH elicits a significant paired pulse depression [SAL = 1.04 ± 0.07, ncells/rats = 20/6; METH = 0.78 ± 0.05, ncells/rats = 21/6; t (39) = 2.8, **p = 0.007]. E Repeated METH elicits a significant decrease in eIPSCs coefficient of variation [SAL = 0.38 ± 0.04, ncells/rats = 11/7; METH = 0.25 ± 0.03, ncells/rats = 15/9; t(24) = 2.25, *p = 0.0337]. F Repeated METH elicits a significant decrease in E-I ratio measured in PNs [SAL = 1.5 ± 0.2, ncells/rats = 10/7; METH = 1.0 ± 0.1, ncells/rats = 25/10; t(32) = 2.11, *p = 0.042]. Representative traces are shown to the right. All results are reported as mean (X) ± standard error (SEM). The schematic illustration was created using BioRender.com.
Repeated METH treatment increases inhibitory transmission in PFC deep layers
Working memory tasks, like TOM and DNMS, are dependent on PFC function [68, 73, 74]. To test whether repeated, daily METH administration in male adult rats produced electrophysiology changes in PFC synaptic transmission after abstinence that are similar to a hypofrontal state, we performed patch-clamp recordings of deep-layer (layer V–VI) pyramidal neurons (PNs) in acute slice preparations. Repeated METH exposure significantly increase both the amplitude (Fig. 2A) and frequency (Fig. 2B) of spontaneous inhibitory postsynaptic currents (sIPSCs) on PNs, indicating that chronic METH exposure increases PFC deep-layer GABAergic synaptic transmission. Consistent with the sIPSCs findings, METH-treated rats also show a significant increase in the amplitude of evoked IPSCs (eIPSCs) (Fig. 2C). Using paired-pulse ratio analysis (PPR, 50 ms interstimulus interval), we found that METH-treated animals show a decrease in PPR (Fig. 2D), suggesting an increase in GABA release properties of inhibitory inputs onto PNs. Chronic METH-treated rats also display a significantly reduced eIPSC coefficient of variation (CV) (Fig. 2E), consistent with an increase in GABAergic presynaptic probability of release (Pr) and/or increase in the number of synaptic contacts [72]. Indeed, plots of the CV vs % failed responses show that in SAL rats increases in CV do not correlate with changes in % of failed responses (r = −0.02, Supplementary Fig. 1), whereas in METH rats, as the CV becomes bigger, the % of failed responses increases (r = 0.35, Supplementary Fig. 1) suggesting that METH treatment is increasing Pr. Finally, we assessed whether the increased inhibition onto PNs altered the E-I ratio by recording both eEPSCs (−70mV) and eIPSCs (0 mV) from the same PNs, and we observed a significant decrease in the E-I ratio (Fig. 2F). When E-I ratio was evaluated, we did not find significant differences in the average decay of the eEPSCs (SAL rats: 19.3 ± 6.15 msec; METH rats 16.3 ± 8.9 msec) or the average decay of the eIPSCs (SAL rats: 21.0 ± 11.3; METH rats: 14.3 ± 10.8 msec). Taken together, our data indicate that repeated METH administration plus one-week of home-cage abstinence increases GABAergic synaptic transmission onto prelimbic PNs and alters E-I ratio, consistent with hypofrontal circuit function.
Dopamine D1 receptors are required for METH-induced changes in PRL inhibitory synaptic transmission
METH produces a strong increase in dopamine release [46, 47, 75] and dopamine receptor signaling [76], and D1 dopamine receptors (D1R) are expressed in PNs and PV + FSIs in the PFC [56, 77, 78]. To test whether D1R signaling influences the chronic METH-induced increase in GABAergic synaptic transmission onto PFC PNs, we prepared PFC acute slices from saline- or METH-treated rats and assessed the effects of the selective D1R antagonist SCH 23390 (10 µM). While the D1R antagonist had no significant effects on sIPSCs, eIPSCs, or PPR on PNs from saline-control treated rats (Supplementary Fig. 2 B1-4), it significantly decreased the sIPSCs amplitude (Fig. 3A), induced a statistical trend for reduced sIPSCs frequency (Fig. 3A), and significantly decreased the eIPSCs amplitude (Fig. 3B) in slices from METH-treated rats. Interestingly, SCH 23390 did not alter the METH-induced changes in PPR (Fig. 3C), which suggests that the METH-induced increases in s/eIPSCs are largely independent of the changes in presynaptic short-term plasticity. Finally, to test whether activation of D1Rs is sufficient to enhance IPSCs onto deep-layer PNs, we also treated slices from saline-treated rats with the D1R agonist SKF 81297 (5 µM). However, acute pharmacological activation of D1R signaling did not alter sIPSCs, eIPSCs, or PPR (Supplementary Fig. 2 A1-4), suggesting that activation of D1R it is necessary, but not sufficient, for METH-induced changes in GABAergic synaptic transmission.
Fig. 3. Bath application of the D1 antagonist SCH 23390 reduces repeated METH-mediated increases in inhibitory activity in PNs recorded in mPFC.
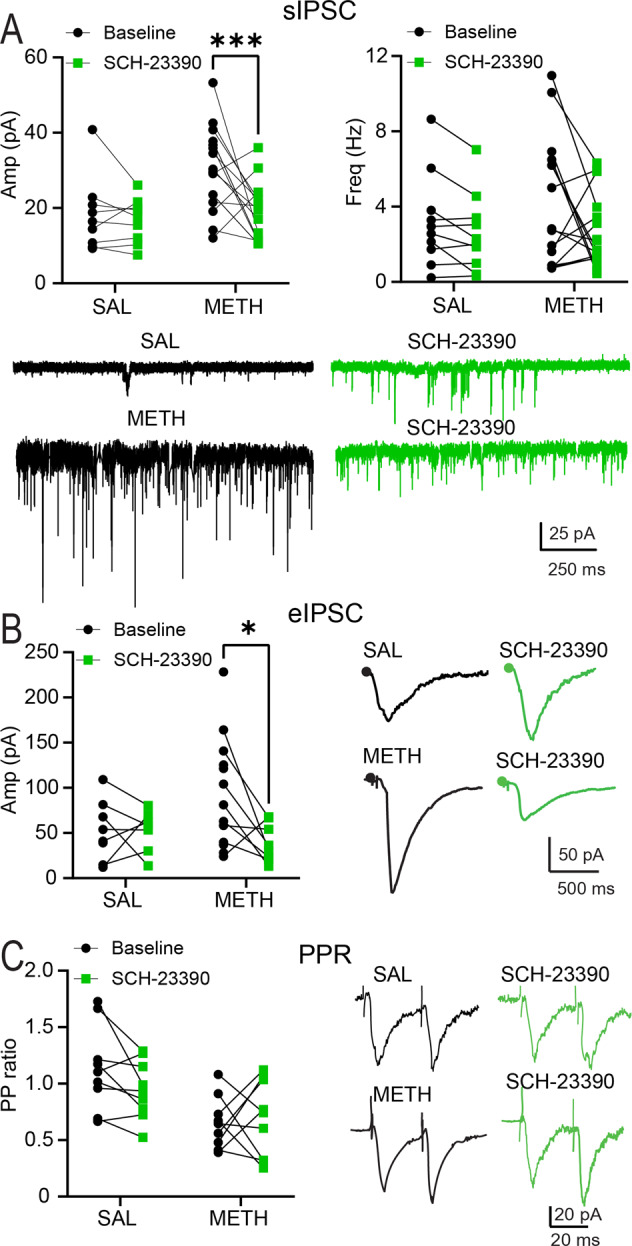
A Bath application of the selective D1R antagonist SCH 23390 (5 µM) produces significant decreases in sIPSCs amplitude in rats treated with repeated METH [Main effect by SCH application: METH = 24.3 ± 2.5 pA; SCH = 19.0 ± 1.6 pA, Mixed effect analysis ANOVA, F(1,23) = 4.559, ***p = 0.0009, Sidak’s post-hoc test, ncells/rats = 18/7] Left Panel. Bath application of SCH 23390 produces a trend for decreased frequency of sIPSCs recorded in rats treated with repeated METH [METH = 3.3 ± 0.5 Hz; SCH = 1.9 ± 0.2 Hz; Mixed effect analysis ANOVA, F (1, 23) = 0.7995, p = 0.0550, ncells/rats = 18/7], Right Panel. B Bath application of SCH 23390 elicits a significant decrease in the amplitude of eIPSCs [Main effect of SCH application: METH = 90.2 ± 20.7 pA; SCH = 46.8 ± 10.5 pA, Mixed effect analysis ANOVA, F (1, 13) = 3.718, *p = 0.023; Sidak’s post-hoc test, ncells/rats = 13/6]. C Bath application of SCH 23390 does not alter the PP of PNs recorded in rats treated with repeated METH or SAL [No effect in drug and no effect after SCH application, Mixed effect analysis ANOVA, F(1,16) = 1.763, p = 0.2029]. Representative traces treated with repeated METH or SAL before and after bath perfusion of SCH 23390. All results are reported as mean (X) ± standard error (SEM).
Repeated METH administration alters the intrinsic excitability of PV-FSIs
Since chronic METH administration increased inhibitory synaptic transmission onto PFC PNs, we tested whether chronic METH altered PV + FSIs function, a major contributor to PFC inhibitory transmission. To this end, we injected a Cre-dependent tdTomato-expressing virus into the PFC of PV-Cre rats (Fig. 4A), and after 3 weeks, we found that 94% of the tdTomato-positive cells were parvalbumin-positive (Fig. 4B). Using whole-cell current clamp configuration, we recorded PV-FSIs from chronic saline (n = 21) or METH-treated animals (n = 13). From the cells recorded in SAL rats we excluded 4 tdTomato-labeled neurons and from the METH recorded rats we excluded 3 tdTomato-labeled neurons because did not exhibit the canonical characteristics of FSIs as reported by Kawaguchi and Kubota [29] and McCormick et al. [79]. Compared to saline-treated rats, chronic METH treatment significantly increased the frequency of action potentials elicited by a range of injected currents (Fig. 4C), indicating that METH produces an increase in PFC PV-FSIs intrinsic excitability. There were no significant differences in the duration of action potentials (SAL: 2.0 ± 0.8 msec, METH: 1.9 ± 0.9 msec); duration of AHP (SAL: 3.9 ± 0.72 msec METH: 5.8 ± 1.6 msec), amplitude of AHP (SAL: 25.2 ± 6.8 mV; METH: 25.2 ± 9.1 mV) or input resistance of PV + FSIs between SAL and METH groups (SAL: 279 ± 203.4 MΩ; METH: 207.4 ± 120.4 MΩ).
Fig. 4. Repeated METH alters the firing frequency of interneurons in the mPFC.

A Schematic representation of intracranial injection of pAVV-FLEX-tdTomato in the mPFC to label parvalbumin-positive interneurons (PV + FSIs). B Colocalization of PV (green) and pAVV-FLEX-tdTomato (red) show selectivity for labeling PV + FSIs (yellow). C Rats treated with repeated METH exhibit a significant increase in PV + FSIs intrinsic excitability [Main effect of drug treatment, Two-way ANOVA, F(1,400) = 77.37, ****p < 0.0001, SAL ncells/rats = 17/8; METH ncells/rats = 10/7]. Representative traces are shown to the right. The schematic illustration was created with BioRender.com.
Chemogenetic reduction of PFC PV + FSI activity rescues METH-induced cognitive deficits
To test whether an increase in PV + FSIs function is sufficient to mimic METH-induced deficits in temporal order memory, we injected the PFC of saline-treated PV-Cre rats with a Cre-dependent excitatory DREADD (AAV-hSyn-DIO-HM3Dq-mCherry). After 3 weeks of virus expression, we observed strong co-localization of GqDREADD/mCherry with endogenous parvalbumin (Fig. 5A), confirming the selective expression of DIO-HM3Dq in PV + FSIs. The Gq DREADD-expressing rats were injected with either vehicle or CNO (10 mg/kg; i.p.) to activate PFC PV + FSIs 30 min before testing in the temporal order memory task. Compared to animals that received vehicle, the CNO-treated rats showed a significant decrease in the TOM ratio (Fig. 5C), indicating that increasing PFC PV + FSIs activity is sufficient to reduce temporal order memory function. To determine whether the increased PFC PV + FSIs activity is necessary for the chronic METH-induced temporal order memory deficits, we injected chronic METH-treated PV-Cre rats with a Cre-dependent inhibitory DREADD virus (AAV-hSyn-DIO-HM4Di-mCherry) in the prelimbic cortex. We confirmed that 87% of the GiDREADD/mCherry-expressing cells colocalized with PFC parvalbumin (Fig. 5B). As expected, vehicle control-treated rats after repeated METH administration displayed significant deficits in the TOM ratio (Fig. 5C); whereas CNO-treated rats after repeated METH displayed a TOM ratio similar to the saline-treated rats and significantly different than the METH + vehicle group (Fig. 5C). Together, these findings suggest that increasing PFC PV + FSIs activity is both necessary and sufficient for chronic METH-induced deficits in temporal order memory performance.
Fig. 5. Chemogenetic manipulation of PV + FSIs.
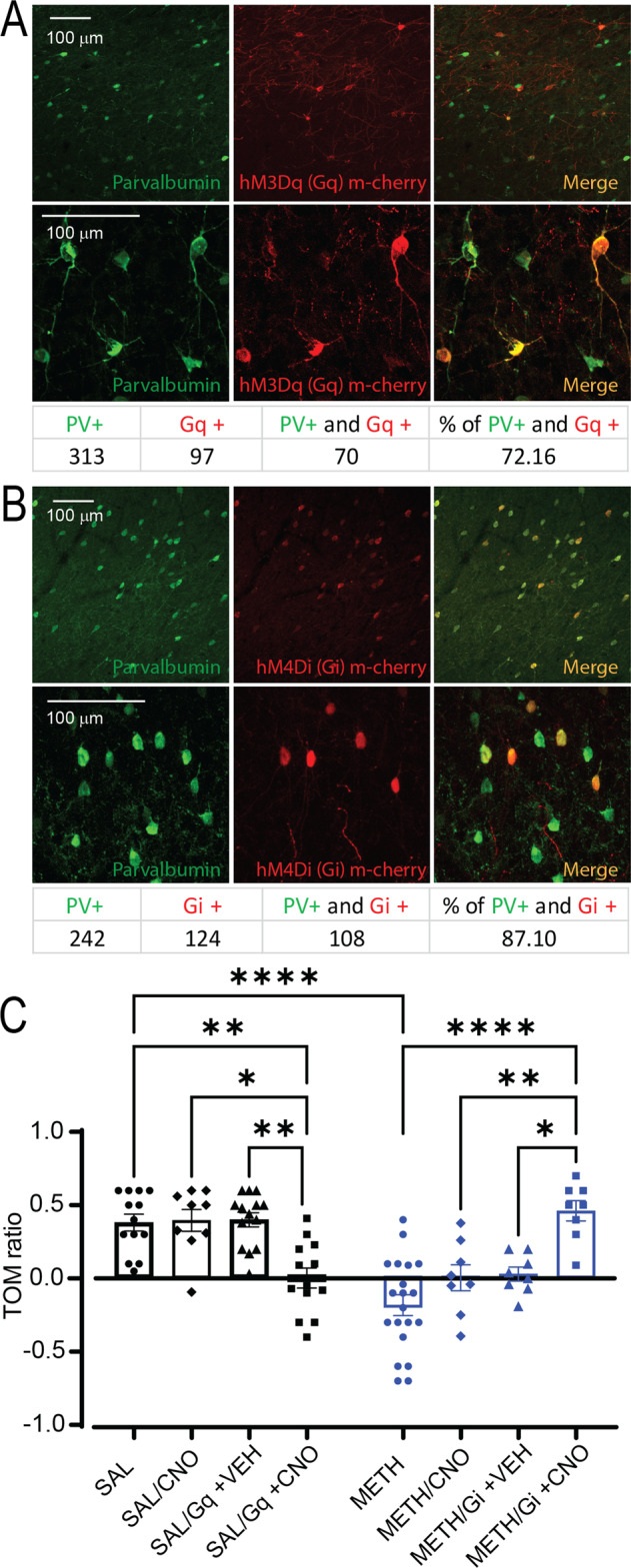
A Representative pictures of the colocalization of PV + cells with hM3Dq m-cherry virus (61.05%) and hM4Di m-cherry virus (82.48%). B Temporal order memory test (TOM) following activation of DREADDs in the mPFC. Activation of PV + FSIs in SAL rats using Gq-DREADD replicates the deficits in TOM elicited by repeated METH. On the other hand, inactivation of PV + FSIs using Gi-DREADD in rats treated with repeated METH ameliorates the deficits in TOM elicited by repeated psychostimulant administration [Main effect by CNO administration and drug treatment: Two-way ANOVA F (7,66) = 11.04, **p < 0.01, ***p < 0.001, ****p < 0.0001, n = 8–19, Tukey’s post-hoc test]. Temporal order memory ratio was calculated as the difference in time spent by each animal exploring object A compared with object B, divided by the total time spent exploring both objects.
Discussion
We report here that repeated METH administration followed by 7–10 days of abstinence elicits significant reductions in WM function and long-lasting increases in PFC GABAergic transmission. D1R signaling was required for the METH-induced increase in PFC GABAergic synaptic transmission. In addition, repeated METH increase PFC PV + FSIs excitability, and chemogenetic reduction of PFC PV + FSIs activity rescue the METH-induced temporal order memory deficits, suggesting that a maladaptive increase in PV + FSIs activity, and possibly other PFC interneurons, underlies the METH-induced hypofrontality and cognitive dysfunction.
In our study, we noted multiple changes in GABAergic synaptic transmission and cell function following repeated METH administration, including: 1) an increase in the amplitude and frequency of sIPSCs onto deep-layer PNs in the PFC, 2) an increase in the amplitude of eIPSCs, 3) a decrease in E-I ratio, 4) the presence of inhibitory paired-pulse depression, 5) a decrease in eIPSCs CV, and 6) an increase in PV + FSIs intrinsic excitability. The paired-pulse depression and decrease in eIPSC CV are consistent with an increase in GABAergic presynaptic function [72, 80], although a change in CV could also reflect a change in GABAergic synapse number, which could also contribute to the observed changes in IPSCs. Future studies will be important to assess that possibility, and to investigate the relative contributions of various GABAergic interneurons to the noted changes. Nonetheless, our studies suggest that enhanced PV + FSIs activity and excitability could underlie, at least in part, the METH-induced electrophysiology and behavioral effects.
Studies have shown that in layer V of the PFC, only PV neurons show increases in FOS expression following an amphetamine regimen [81] and chronic cocaine elicits an increase in PV + FSIs excitability [82]. Similarly, Shi and colleagues [83] showed that in mice, repeated cocaine administration increases mIPSCs and our previous results showed that a repeated cocaine regime increases the D1R-mediated amplitude of eIPSCs [84]. However, to the best of our knowledge, this is the first report describing the effects of repeated METH on the activity of PV + FSIs in PFC.
Our results show that the selective D1R antagonist SCH 23390 significantly decreases METH-enhanced amplitude and frequency of sIPSCs in chronic METH-treated rats, but without affecting synaptic measures in SAL-treated rats. Furthermore, the D1R antagonist significantly decrease the amplitude of eIPSCs and induce a trend for an increase in E-I ratio, suggesting that blockade of D1R decreases GABAergic synaptic transmission. However, increases in amplitude of eIPSCs could also suggest an increase in GABAR and/or changes in GABAR affinity, and the lack of effects of the D1 antagonist in paired-pulse ratio might suggest that the METH-induced increases in eIPSCs and sIPSCs are not cause by changes in presynaptic probability of release, but rather by increases in GABAergic cell firing, PN GABAergic synapse strength, and/or the number or sub-cellular location of GABAergic synapses on PNs. Also, the lack of effect of the D1R antagonis on the inhibitory synaptic activity in SAL-treated rats, compared with the significant decreases in METH-treated rats, might suggest that repeated drug exposure produces D1R signaling sensitization and/or increases in DAergic tone that are nonexistent in the SAL animals. Future studies investigating D1 signaling in PNs and PV + FSIs will be important to test this possibility. However, it is worth noting that increased D1R signaling is not sufficient to explain our observations of increased GABAergic function, since treatment of SAL-treated rat slices with the selective D1R agonist, SKF 81297, did not mimic the effects of repeated METH administration, suggesting that other cellular events are needed to produce the changes in inhibitory synaptic transmission in METH-treated animals.
Cognitive deficits produced by METH have been extensively reported [4, 5, 11, 85–88], and our results confirm that repeated METH elicits temporal order memory and working memory deficits. More importantly, our results are the first to demonstrate that activation of Gq-linked receptors in PFC PV + FSIs of SAL-treated rats mimics the cognitive deficits produces by repeated METH, while activation of Gi-linked DREADDs in PFC PV + FSIs of repeated METH rats ameliorates the cognitive deficits. These results suggest that maladaptive changes in PFC PV + FSI activity are necessary and sufficient for METH-induced cognitive deficits, likely by shifting the PFC E-I balance. It is important to note that not all of the Tdtomato-labeled neurons exhibit electrophysiological characteristics of FSI (~7% did not exhibit canonical electrophysiological signature), thus it is possible that neurons other than FSIs are involved in the effects elicited by the DREADDS manipulations. Indeed, Wearne and colleagues (2017) have shown that METH sensitization increases the transcriptional expression of markers for calbindin, calretinin, somatostatin, cholecyctokinin and VIP, besides PV, in the PFC.
Gamma oscillations are key components of cognitive processes [89, 90] and involve the reciprocal interaction between PV + FSIs and principal pyramidal cells [91]. The predominant mechanism underlying this type of rhythm is the phasic excitation of interneurons following spike generation in PNs [92]. Gamma oscillations are thought to help to organize ensembles of synchronously firing neurons in various brain regions, and may play a key role in top-down mechanisms controlling cortical processing, such as attention [93], working memory [94] and decision-making [95]. Augmented firing of PV + FSIs could affect gamma oscillations and perturb normal information processing in PFC and associated cognitive processes.
Our experiments were performed using non-contingent METH administration, because we used METH as a tool to produce cognitive deficits related with hypofrontality. Therefore, we do not intend to imply that, in our experiments, hypofrontality is an indicator of loss of control over drug taking. Future experiments using METH self-administration can investigate this issue.
Finally, we cannot exclude the possibility that the increases in GABAergic transmission reported in here are the homeostatic response to a METH-mediated increase in glutamatergic transmission. In summary, our results show that repeated METH administration alters PV + FSIs excitability, GABAergic synaptic transmission in the mPFC, and E-I ratio onto deep-layer mPFC PNs. Moreover, we show that D1R signaling is required for some of the METH-induced changes in PFC GABAergic synaptic transmission, and that modulation of PV + FSIs activity is necessary and sufficient for METH-induced deficits in PFC-dependent working memory.
Supplementary information
Acknowledgements
We thank Dr. Rachel Penrod-Martin for the advice with statistical analysis. We thank NIDA for providing Methamphetamine. The authors report no biomedical financial interests or potential conflicts of interest.
Author contributions
Substantial contribution to the conception and design of the work: AL, MAR, CC and AA; acquisition of the data: MAR and ET; analysis and interpretation of data: AL and MAR; drafting the work or revising it critically for important intellectual content: AL, MAR, CC and AA. All of the authors approved the final version to be published and they agreed to be accountable for all aspects of the work.
Funding
This work was supported by funding from NIDA R21 DA045889 and R01 DA054589 (AL), R01 DA032708 and P50 DA046373 (CWC), and NIDA/ORWH U54 DA016511 in partnership with MUSC (MAR).
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41386-022-01371-9.
References
- 1.Kim SJ, Lyoo IK, Hwang J, Sung YH, Lee HY, Lee DS, et al. Frontal glucose hypometabolism in abstinent methamphetamine users. Neuropsychopharmacology. 2005;30:1383–91. doi: 10.1038/sj.npp.1300699. [DOI] [PubMed] [Google Scholar]
- 2.Kim YT, Lee SW, Kwon DH, Seo JH, Ahn BC, Lee J. Dose-dependent frontal hypometabolism on FDG-PET in methamphetamine abusers. J Psychiatr Res. 2009;43:1166–70. doi: 10.1016/j.jpsychires.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 3.Volkow ND, Chang L, Wang G-J, Fowler JS, Ding Y-S, Sedler M, et al. Low level of brain dopamine d 2 receptors in methamphetamine abusers: association with metabolism in the orbitofrontal cortex. Am J Psychiatry. 2001;158:2015–21. [DOI] [PubMed]
- 4.Peters J, Scofield MD, Reichel CM. Chemogenetic activation of the perirhinal cortex reverses methamphetamine-induced memory deficits and reduces relapse. Learn Mem. 2018;25:410–5. [DOI] [PMC free article] [PubMed]
- 5.Bernheim A, See RE, Reichel CM. Chronic methamphetamine self-administration disrupts cortical control of cognition. Neurosci Biobehav Rev. 2016;69:36–48. doi: 10.1016/j.neubiorev.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Homer BD, Solomon TM, Moeller RW, Mascia A, DeRaleau L, Halkitis PN. Methamphetamine abuse and impairment of social functioning: a review of the underlying neurophysiological causes and behavioral implications. Psychol Bull. 2008. 10.1037/0033-2909.134.2.301. [DOI] [PubMed]
- 7.Salo R, Ursu S, Buonocore MH, Leamon MH, Carter C. Impaired prefrontal cortical function and disrupted adaptive cognitive control in methamphetamine abusers: a functional magnetic resonance imaging study. Biol Psychiatry. 2009;65:706–9. doi: 10.1016/j.biopsych.2008.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dalley JW, Theobald DEH, Berry D, Milstein JA, Lääne K, Everitt BJ, et al. Cognitive sequelae of intravenous amphetamine self-administration in rats: Evidence for selective effects on attentional performance. Neuropsychopharmacology. 2005;30:525–37. doi: 10.1038/sj.npp.1300590. [DOI] [PubMed] [Google Scholar]
- 9.Mizoguchi H, Ibi D, Takase F, Nagai T, Kamei H, Toth E, et al. Nicotine ameliorates impairment of working memory in methamphetamine-treated rats. Behav Brain Res. 2011;220:159–63. doi: 10.1016/j.bbr.2011.01.036. [DOI] [PubMed] [Google Scholar]
- 10.Nagai T, Takuma K, Dohniwa M, Ibi D, Mizoguchi H, Kamei H, et al. Repeated methamphetamine treatment impairs spatial working memory in rats: Reversal by clozapine but not haloperidol. Psychopharmacology. 2007;194:21–32. doi: 10.1007/s00213-007-0820-1. [DOI] [PubMed] [Google Scholar]
- 11.Shoblock JR, Maisonneuve IM, Glick SD. Differences between d-methamphetamine and d-amphetamine in rats: Working memory, tolerance, and extinction. Psychopharmacology. 2003. 10.1007/s00213-003-1522-y. [DOI] [PubMed]
- 12.Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807. doi: 10.1038/nrn1519. [DOI] [PubMed] [Google Scholar]
- 13.Foss-Feig JH, Adkinson BD, Ji JL, Yang G, Srihari VH, McPartland JC, et al. Searching for cross-diagnostic convergence: neural mechanisms governing excitation and inhibition balance in schizophrenia and autism spectrum disorders. Biol Psychiatry. 2017;81:848–61. doi: 10.1016/j.biopsych.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopatina OL, Malinovskaya NA, Komleva YK, Gorina YV, Shuvaev AN, Olovyannikova RY, et al. Excitation/inhibition imbalance and impaired neurogenesis in neurodevelopmental and neurodegenerative disorders. Rev Neurosci. 2019. 10.1515/revneuro-2019-0014. [DOI] [PubMed]
- 15.Goldstein RZ, Volkow ND. Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex NIH public access. Am J Psychiatry. 2002;159:1642–52. [DOI] [PMC free article] [PubMed]
- 16.Goldstein RZ, Volkow ND. Dysfunction of the prefrontal cortex in addiction: neuroimaging findings and clinical implications. Nat Rev Neurosci. 2011;12:652–69. doi: 10.1038/nrn3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Volkow N, Fowler J, Wolf A, Hitzemann R, Dewey S, Bendriem B, et al. Changes in brain glucose metabolism in cocaine dependence and withdrawal. Amrican J Psychiatry. 1991;148:621–6. doi: 10.1176/ajp.148.5.621. [DOI] [PubMed] [Google Scholar]
- 18.Volkow ND, Fowler JS, Wang GJ, Telang F, Logan J, Jayne M, et al. Cognitive control of drug craving inhibits brain reward regions in cocaine abusers. Neuroimage. 2010;49:2536–43. doi: 10.1016/j.neuroimage.2009.10.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bolla KI, Eldreth DA, London ED, Kiehl KA, Mouratidis M, Contoreggi C, et al. Orbitofrontal cortex dysfunction in abstinent cocaine abusers performing a decision-making task. Neuroimage. 2003;19:1085–94. [DOI] [PMC free article] [PubMed]
- 20.Goldman-Rakic PS. Development of cortical circuitry and cognitive function. Child Dev. 1987;58:601–22. [PubMed]
- 21.Miller EK, Cohen JD. An integrative theory of prefrontal cortex function. Annu Rev Neurosci. 2001;24:167–202. [DOI] [PubMed]
- 22.Levy R, Goldman-Rakic PS. Segregation of working memory functions within the dorsolateral prefrontal cortex. Exp Brain Res. 2000;133:23–32. doi: 10.1007/s002210000397. [DOI] [PubMed] [Google Scholar]
- 23.Wilson FA, Ó Scalaidhe SP, Goldman-Rakic PS. Dissociation of object and spatial processing domains in primate prefrontal cortex. Science. 1993;25:1955–8. doi: 10.1126/science.8316836. [DOI] [PubMed] [Google Scholar]
- 24.Zarahn E, Aguirre GK, Esposito MD. Temporal isolation of the neural correlates of spatial mnemonic processing with fMRI. Brain Res Cogn Brain Res. 1999;7:255–68. [DOI] [PubMed]
- 25.Funahashi S, Bruce CJ, Goldman-Rakic PS. Dorsolateral prefrontal lesions and oculomotor delayed-response performance: evidence for mnemonic ‘Scotomas’. J Neurosci. 1993;13:1479–97. [DOI] [PMC free article] [PubMed]
- 26.Procyk E, Goldman-Rakic PS. Modulation of dorsolateral prefrontal delay activity during self-organized behavior. J Neurosci. 2006;26:11313–23. doi: 10.1523/JNEUROSCI.2157-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joaquin F. Unit activity in prefrontal cortex during delayed-response performance: neuronal correlates of transient memory. J Neurosci. 1973;36:61–78. [DOI] [PubMed]
- 28.Bauer RH, Fuster JM. Delayed-matching and delayed-response deficit from cooling dorsolateral prefrontal cortex in monkeys. J Comp Physiol Psychol. 1976;90:293–302. doi: 10.1037/h0087996. [DOI] [PubMed] [Google Scholar]
- 29.Kawaguchi Y, Kubota Y. GABAergic cell subtypes and theirsynaptic connections in rat frontal cortex. Cereb Cortex. 1997;7:476–86. doi: 10.1093/cercor/7.6.476. [DOI] [PubMed] [Google Scholar]
- 30.Gabbott PLA, Dickie BGM, Vaid RR, Headlam AJN, Bacon SJ. Local-circuit neurones in the medial prefrontal cortex (Areas 25, 32 and 24b) in the rat: morphology and quantitative distribution. J Comp Neurol. 1997;377:465–99. [DOI] [PubMed]
- 31.Markram H, Muller E, Ramaswamy S, Reimann MW, Abdellah M, Sanchez CA, et al. Reconstruction and simulation of neocortical microcircuitry. Cell. 2015;163:456–92. doi: 10.1016/j.cell.2015.09.029. [DOI] [PubMed] [Google Scholar]
- 32.Ferguson BR, Gao WJ. Thalamic control of cognition and social behavior via regulation of gamma-aminobutyric acidergic signaling and excitation/inhibition balance in the medial prefrontal cortex. Biol Psychiatry. 2018;83:657–69. doi: 10.1016/j.biopsych.2017.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Packer AM, Yuste R. Dense, unspecific connectivity of neocortical parvalbumin-positive interneurons: a canonical microcircuit for inhibition? J Neurosci. 2011;31:13260–71. doi: 10.1523/JNEUROSCI.3131-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rossignol E. Genetics and function of neocortical GABAergic interneurons in neurodevelopmental disorders. Neural Plast. 2011;2011:649325. [DOI] [PMC free article] [PubMed]
- 35.Dienel SJ, Lewis DA. Alterations in cortical interneurons and cognitive function in schizophrenia. Neurobiol Dis. 2019;131:104208. [DOI] [PMC free article] [PubMed]
- 36.Duman RS, Sanacora G, Krystal JH. Altered connectivity in depression: GABA and glutamate neurotransmitter deficits and reversal by novel treatments. Neuron. 2019;102:75–90. doi: 10.1016/j.neuron.2019.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rapanelli M, Frick LR, Xu M, Groman SM, Jindachomthong K, Tamamaki N, et al. Targeted interneuron depletion in the dorsal striatum produces autism-like behavioral abnormalities in male but not female mice. Biol Psychiatry. 2017;82:194–203. doi: 10.1016/j.biopsych.2017.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Palop JJ, Mucke L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat Rev Neurosci. 2016;17:777–92. doi: 10.1038/nrn.2016.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lewis DA. Cortical circuit dysfunction and cognitive deficits in schizophrenia - implications for preemptive interventions. Eur J Neurosci. 2012;35:1871–8. doi: 10.1111/j.1460-9568.2012.08156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rao SG, Williams GV, Goldman-Rakic PS. Destruction and creation of spatial tuning by disinhibition: GABA a blockade of prefrontal cortical neurons engaged by working memory. J Neurosci. 2000;20:485–94. [DOI] [PMC free article] [PubMed]
- 41.Ruden JB, Dugan LL, Konradi C. Parvalbumin interneuron vulnerability and brain disorders. Neuropsychopharmacology. 2021;46:279–87. doi: 10.1038/s41386-020-0778-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Isaacson JS, Scanziani M. How inhibition shapes cortical activity. Neuron. 2011;72:231–43. doi: 10.1016/j.neuron.2011.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marín O. Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci. 2012;13:107–20. doi: 10.1038/nrn3155. [DOI] [PubMed] [Google Scholar]
- 44.Uhlhaas PJ, Singer W. Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci. 2010;11:100–13. doi: 10.1038/nrn2774. [DOI] [PubMed] [Google Scholar]
- 45.Haddar M, Uno K, Azuma K, Muramatsu S ichi, Nitta A. Inhibitory effects of Shati/Nat8l overexpression in the medial prefrontal cortex on methamphetamine-induced conditioned place preference in mice. Addict Biol. 2020;25:e12749. [DOI] [PMC free article] [PubMed]
- 46.Parsegian A, See RE. Dysregulation of dopamine and glutamate release in the prefrontal cortex and nucleus accumbens following methamphetamine self-administration and during reinstatement in rats. Neuropsychopharmacology. 2014;39:811–22. doi: 10.1038/npp.2013.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stephans SE, Yamamoto BK. Effect of repeated methamphetamine administrations on dopamine and glutamate efflux in rat prefrontal cortex. Brain Res. 1995;700:99–106. [DOI] [PubMed]
- 48.Buchta WC, Mahler SV, Harlan B, Aston-Jones GS, Riegel AC. Dopamine terminals from the ventral tegmental area gate intrinsic inhibition in the prefrontal cortex. Physiol Rep. 2017;5:e13198. [DOI] [PMC free article] [PubMed]
- 49.Popescu AT, Zhou MR, Poo MM. Phasic dopamine release in the medial prefrontal cortex enhances stimulus discrimination. Proc Natl Acad Sci USA. 2016;113:E3169–E3176. doi: 10.1073/pnas.1606098113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhong P, Qin L, Yan Z. Dopamine differentially regulates response dynamics of prefrontal cortical principal neurons and interneurons to optogenetic stimulation of inputs from ventral tegmental area. Cereb Cortex. 2020;30:4402–9. doi: 10.1093/cercor/bhaa027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gorelova N, Seamans JK, Yang CR. Mechanisms of dopamine activation of fast-spiking interneurons that exert inhibition in rat prefrontal cortex. J Neurophysiol. 2002;88:3150–66. doi: 10.1152/jn.00335.2002. [DOI] [PubMed] [Google Scholar]
- 52.Zhou F-M, Hablitz JJ. Dopamine modulation of membrane and synaptic properties of interneurons in rat cerebral cortex. J Neurophysiol. 1999;81:967–76. [DOI] [PubMed]
- 53.Gao W-J, Wang Y, Goldman-Rakic PS. Dopamine modulation of perisomatic and peridendritic inhibition in prefrontal cortex. J Neurosci. 2003;23:1622–30. [DOI] [PMC free article] [PubMed]
- 54.Le Moine C, Gaspar P. Subpopulations of cortical GABAergic interneurons differ by their expression of D1 and D2 dopamine receptor subtypes. Mol Brain Res. 1998;58:231–6. doi: 10.1016/S0169-328X(98)00118-1. [DOI] [PubMed] [Google Scholar]
- 55.Durstewitz D, Seamans JK, Sejnowski TJ, Se TJ. Dopamine-mediated stabilization of delay-period activity in a network model of prefrontal cortex. J Neurophysiol. 2000;83:1733–50. [DOI] [PubMed]
- 56.Santana N, Mengod G, Artigas F. Quantitative analysis of the expression of dopamine D1 and D2 receptors in pyramidal and GABAergic neurons of the rat prefrontal cortex. Cereb Cortex. 2009;19:849–60. doi: 10.1093/cercor/bhn134. [DOI] [PubMed] [Google Scholar]
- 57.Sawaguchi T, Goldman-Rakic P. D1 dopamine receptors in prefrontal cortex: involvement in working memory. Science. 1991;22:947–50. doi: 10.1126/science.1825731. [DOI] [PubMed] [Google Scholar]
- 58.Senkowski D, Gallinat J. Dysfunctional prefrontal gamma-band oscillations reflect working memory and other cognitive deficits in schizophrenia. Biol Psychiatry. 2015;77:1010–9. doi: 10.1016/j.biopsych.2015.02.034. [DOI] [PubMed] [Google Scholar]
- 59.Goldman-Rakic P. Working memory dysfunction in schizophrenia. J Neuropsychiatry Clin Neurosci. 1994;6:348–57. doi: 10.1176/jnp.6.4.348. [DOI] [PubMed] [Google Scholar]
- 60.Arnsten A, Cai J, Murphy B, Goldman-Rakic P. Dopamine D1 receptor mechanisms in the cognitive performance of young adult and aged monkeys. Psychopharmacology. 1994;116:143–51. doi: 10.1007/BF02245056. [DOI] [PubMed] [Google Scholar]
- 61.Arnsten A. Catecholamine regulation of the prefrontal cortex. Psychopharmacology. 1997;11:151–62. doi: 10.1177/026988119701100208. [DOI] [PubMed] [Google Scholar]
- 62.Fuster JM, Alexander GE. Neuron activity related to short-term memory. Science. 1971;173:652–4. doi: 10.1126/science.173.3997.652. [DOI] [PubMed] [Google Scholar]
- 63.Goldman-Rakic PS. Cellular basis of working memory. Neuron. 1995;14:477–85. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- 64.Kamigaki T, Dan Y. Delay activity of specific prefrontal interneuron subtypes modulates memory-guided behavior. Nat Neurosci. 2017;20:854–63. doi: 10.1038/nn.4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yau JOY, Chaichim C, Power JM, McNally GP. The roles of basolateral amygdala parvalbumin neurons in fear learning. J Neurosci. 2021;41:9223–34. doi: 10.1523/JNEUROSCI.2461-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wright AM, Zapata A, Hoffman AF, Necarsulmer JC, Coke LM, Svarcbahs R, et al. Effects of withdrawal from cocaine self-administration on rat orbitofrontal cortex parvalbumin neurons expressing cre recombinase: sex-dependent changes in neuronal function and unaltered serotonin signaling. ENeuro. 2021;8:1–19. doi: 10.1523/ENEURO.0017-21.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wearne TA, Parker LM, Franklin JL, Goodchild AK, Cornish JL. GABAergic mRNA expression is differentially expressed across the prelimbic and orbitofrontal cortices of rats sensitized to methamphetamine: relevance to psychosis. Neuropharmacology. 2016;111:107–18. doi: 10.1016/j.neuropharm.2016.08.038. [DOI] [PubMed] [Google Scholar]
- 68.Barker GRI, Bird F, Alexander V, Warburton EC. Recognition memory for objects, place, and temporal order: a disconnection analysis of the role of the medial prefrontal cortex and perirhinal cortex. J Neurosci. 2007;27:2948–57. doi: 10.1523/JNEUROSCI.5289-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Winters BD, Saksida LM, Bussey TJ. Object recognition memory: neurobiological mechanisms of encoding, consolidation and retrieval. Neurosci Biobehav Rev. 2008;32(5):1055–70. [DOI] [PubMed]
- 70.Aultman JM, Moghaddam B. Distinct contributions of glutamate and dopamine receptors to temporal aspects of rodent working memory using a clinically relevant task. Psychopharmacology. 2001;153:353–64. doi: 10.1007/s002130000590. [DOI] [PubMed] [Google Scholar]
- 71.Aggleton JP, Hunt PR, Shaw C. The effects of mammillary body and combined amygdalar-fornix lesions on tests of delayed non-matching-to-sample in the rat. Behav Brain Res. 1990;40:145–57. [DOI] [PubMed]
- 72.Brock JA, Thomazeau A, Watanabe A, Li SSY, Sjöström PJ. A practical guide to using CV analysis for determining the locus of synaptic plasticity. Front Synaptic Neurosci. 2020;12:11. [DOI] [PMC free article] [PubMed]
- 73.Hannesson DK, Howland JG, Phillips AG. Interaction between perirhinal and medial prefrontal cortex is required for temporal order but not recognition memory for objects in rats. J Neurosci. 2004;24:4596–604. doi: 10.1523/JNEUROSCI.5517-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Clapcote SJ, Lipina TV, Millar JK, Mackie S, Christie S, Ogawa F, et al. Behavioral phenotypes of disc1 missense mutations in mice. Neuron. 2007;54:387–402. doi: 10.1016/j.neuron.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 75.Nishijima K, Kashiwa A, Hashimoto A, Iwama H, Umino A, Nishikawa T. Differential effects of phencyclidine and methamphetamine on dopamine metabolism in rat frontal cortex and striatum as revealed by in vivo dialysis. Synapse. 1996;22:304–12. doi: 10.1002/(SICI)1098-2396(199604)22:4<304::AID-SYN2>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 76.Wang GJ, Wiers CE, Shumay E, Tomasi D, Yuan K, Wong CT, et al. Expectation effects on brain dopamine responses to methylphenidate in cocaine use disorder. Transl Psychiatry. 2019;9:93. [DOI] [PMC free article] [PubMed]
- 77.Anastasiades PG, Boada C, Carter AG. Cell-type-specific D1 dopamine receptor modulation of projection neurons and interneurons in the prefrontal cortex. Cereb Cortex. 2019;29:3224–42. doi: 10.1093/cercor/bhy299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Santana N, Artigas F. Laminar and cellular distribution of monoamine receptors in rat medial prefrontal cortex. Front Neuroanat. 2017;11:87. [DOI] [PMC free article] [PubMed]
- 79.Mccormick DA, Prince DA. Two types of muscarinic response to acetylcholine in mammalian cortical neurons (clngulate/M current/cholinergic/pirenzepine). Proc Natl Acad Sci USA. 1985;82:6344–8. [DOI] [PMC free article] [PubMed]
- 80.Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. [DOI] [PubMed]
- 81.Morshedi MM, Meredith GE. Differential laminar effects of amphetamine on prefrontal parvalbumin interneurons. Neuroscience. 2007;149:617–24. doi: 10.1016/j.neuroscience.2007.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Campanac E, Hoffman DA. Repeated cocaine exposure increases fast-spiking interneuron excitability in the rat medial prefrontal cortex. J Neurophysiol. 2013;109:2781–92. doi: 10.1152/jn.00596.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shi P, Nie J, Liu H, Li Y, Lu X, Shen X, et al. Adolescent cocaine exposure enhances the GABAergic transmission in the prelimbic cortex of adult mice. FASEB J. 2019;33:8614–22. doi: 10.1096/fj.201802192RR. [DOI] [PubMed] [Google Scholar]
- 84.Kroener S, Lavin A. Altered dopamine modulation of inhibition in the prefrontal cortex of cocaine-sensitized rats. Neuropsychopharmacology. 2010;35:2292–304. doi: 10.1038/npp.2010.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kuczenski R, Segal DS, Melega WP, Lacan G, McCunney SJ. Human methamphetamine pharmacokinetics simulated in the rat: behavioral and neurochemical effects of a 72-h binge. Neuropsychopharmacology. 2009;34:2430–41. doi: 10.1038/npp.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Scofield MD, Trantham-Davidson H, Schwendt M, Leong KC, Peters J, See RE, et al. Failure to recognize novelty after extended methamphetamine self-administration results from loss of long-term depression in the perirhinal cortex. Neuropsychopharmacology. 2015;40:2526–35. doi: 10.1038/npp.2015.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Reichel CM, Ramsey LA, Schwendt M, McGinty JF, See RE. Methamphetamine-induced changes in the object recognition memory circuit. Neuropharmacology. 2012;62:1119–26. doi: 10.1016/j.neuropharm.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Parsegian A, Glen WB, Lavin A, See RE. Methamphetamine self-administration produces attentional set-shifting deficits and alters prefrontal cortical neurophysiology in rats. Biol Psychiatry. 2011;69:253–9. doi: 10.1016/j.biopsych.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Buzsáki G, Draguhn A. Neuronal oscillations in cortical networks. Science. 2004;304:1926–9. [DOI] [PubMed]
- 90.Fries P. Neuronal gamma-band synchronization as a fundamental process in cortical computation. Annu Rev Neurosci. 2009;32:209–24. doi: 10.1146/annurev.neuro.051508.135603. [DOI] [PubMed] [Google Scholar]
- 91.Whittington MA, Traub RD, Faulkner HJ, Stanford IM, Jefferys JGR. Recurrent excitatory postsynaptic potentials induced by synchronized fast cortical oscillations (neuronal network40 Hzsynaptic potentiation). Proc Natl Acad Sci USA. 1997;94:12198–203. [DOI] [PMC free article] [PubMed]
- 92.Whittington MA, Traub RD, Kopell N, Ermentrout B, Buhl EH. Inhibition-based rhythms: experimental and mathematical observations on network dynamics. Int J Psychophysiol. 2000;38:315–36. [DOI] [PubMed]
- 93.Fries P, Reynolds J, Rorie A, Desimone R. Modulation of oscillatory neuronal synchronization by selective visual attention. Science. 2001;291:1560–3. doi: 10.1126/science.1055465. [DOI] [PubMed] [Google Scholar]
- 94.Korotkova T, Fuchs EC, Ponomarenko A, von Engelhardt J, Monyer H. NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron. 2010;68:557–69. doi: 10.1016/j.neuron.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 95.Van Wingerden M, Vinck M, Lankelma JV, Pennartz CMA. Learning-associated gamma-band phase-locking of action-outcome selective neurons in orbitofrontal cortex. J Neurosci. 2010;30:10025–38. doi: 10.1523/JNEUROSCI.0222-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.