

Abstract
Schizophrenia is associated with reduced numbers of spines and dendrites from layer III of the dorsolateral prefrontal cortex (dlPFC), the layer that houses the recurrent excitatory microcircuits that subserve working memory and abstract thought. Why are these synapses so vulnerable, while those in deeper or more superficial layers are little affected? This review describes the special molecular properties that govern layer III neurotransmission and neuromodulation in the primate dlPFC, and how they may render these circuits particularly vulnerable to genetic and environmental insults. These properties include a reliance on NMDAR rather than AMPAR neurotransmission, cAMP magnification of calcium signaling near the glutamatergic synapse of dendritic spines, and potassium channels opened by cAMP-PKA signaling that dynamically alter network strength, with built-in mechanisms to take dlPFC “off-line” during stress. A variety of genetic and/or environmental insults can lead to the same phenotype of weakened layer III connectivity, where mechanisms that normally strengthen connectivity are impaired, and those that normally weaken connectivity are intensified. Inflammatory mechanisms such as increased kynurenic acid and GCPII expression are especially detrimental to layer III dlPFC neurotransmission and modulation, mimicking genetic insults. The combination of genetic and inflammatory insults may cross the threshold into pathology.
Keywords: NMDAR, mGluR3, calcium, PDE4, GCPII, VIPR2
“By diverse means we arrive at the same end”
- Michel de Montaigne
Introduction
The etiology of schizophrenia remains a puzzle, with seemingly divergent mechanisms amongst the many risk factors for this complex, cognitive disorder. A large number of genetic factors confer risk, as well as environmental factors such as perinatal inflammatory events and psychological stress in adolescence. Although this landscape remains challenging, accumulating data are beginning to provide a foothold, suggesting how risk factors may weaken the recurrent excitatory circuits in the dorsolateral prefrontal cortex (dlPFC) that subserve higher cognition.
There is consistent evidence that schizophrenia involves impaired functioning of the dlPFC, with deficits in working memory and dlPFC BOLD response relating strongly to symptoms of thought disorder e.g. (1–5), including worsening with stress exposure (6). Risk genes for schizophrenia are enriched in dlPFC compared to other brain areas (7), structural imaging studies show waves of PFC gray matter loss heralding disease onset (8, 9), accompanied by elevated inflammation (9, 10), and PET markers show reduced presynaptic labeling (11). Most pertinent to the current review, post-mortem neuropathological studies show consistent reduction in the numbers of spines and dendrites in deep layer III dlPFC (12, 13), the sublayer that contains the recurrent excitatory microcircuits that subserve working memory (14). The reduction in spines shows striking laminar specificity (15), with normal levels in superficial III or deep layers V and VI (12, 16), as well as regional specificity, with relatively preserved spine numbers in the primary visual cortex (12). Thus, clues to the etiology of schizophrenia may be found in trying to understand this selective pathology, determining why spine numbers are particularly reduced in deep layer III of the dlPFC. It is possible that the distorted thinking of schizophrenia is related to this selective change in layer III, whilst more global insults to all layers of dlPFC would produce a simpler syndrome of cognitive impairment. Please note that the reduction in spine number (here termed “atrophy”) may reflect impaired spine formation and/or increased removal of existing spines, and that there are decreases in spine/synapse numbers in other cortical areas as well, e.g. in anterior cingulate cortex (17), temporal association cortex (18), and primary auditory cortex (19, 20) (although excitatory synapse numbers remain constant in auditory cortex despite loss of spines (19)). However, as little is known about the molecular regulation of these areas in primate, they will not be considered here.
The current review will discuss how a variety of genetic and environmental risk factors may lead to loss of spines on layer III dlPFC spines, based on studies of the rhesus monkey dlPFC. Layer III dlPFC spines in rhesus macaque express a concentration of proteins that are risk factors for disease, with many related to the special molecular properties needed for working memory. These properties include a reliance on NMDAR rather than AMPAR neurotransmission, cAMP magnification of calcium (Ca2+) signaling near the synapse, and potassium (K+) channels opened by cAMP-PKA signaling that dynamically alter network strength. These mechanisms render layer III spines especially vulnerable to atrophy when they are dysregulated due to genetic and/or inflammatory insults, weakening network connectivity.
Summary of dlPFC neurotransmission and neuromodulation
The dlPFC is essential to working memory –our mental sketch pad- i.e. the ability to generate, maintain and manipulate mental representations without sensory stimulation, the foundation of abstract thought and the executive functions, including cognitive control (14, 21–24). The dlPFC accomplishes these functions through widespread connections, e.g. it has reciprocal connections with the sensory association cortices, mediodorsal thalamus and hippocampus (25–27),, as well as outputs to basal ganglia, premotor cortices and pons-cerebellum to influence motor response (25–27) (28). Fuster and Goldman-Rakic found “Delay cells” in macaque dlPFC with tuned persistent firing, that represent information (e.g. a location) over a delay period of many seconds, without sensory stimulation (29, 30). (31)Goldman-Rakic showed that persistent firing arises from recurrent excitatory circuits concentrated in deep layer III of dlPFC (Fig. 1A), with information refined by lateral inhibition (14), a model affirmed by in vitro recordings (32). Thus, pyramidal cells excite each other through glutamatergic synapses on spines to keep information “in mind”.(32)
Figure 1-.
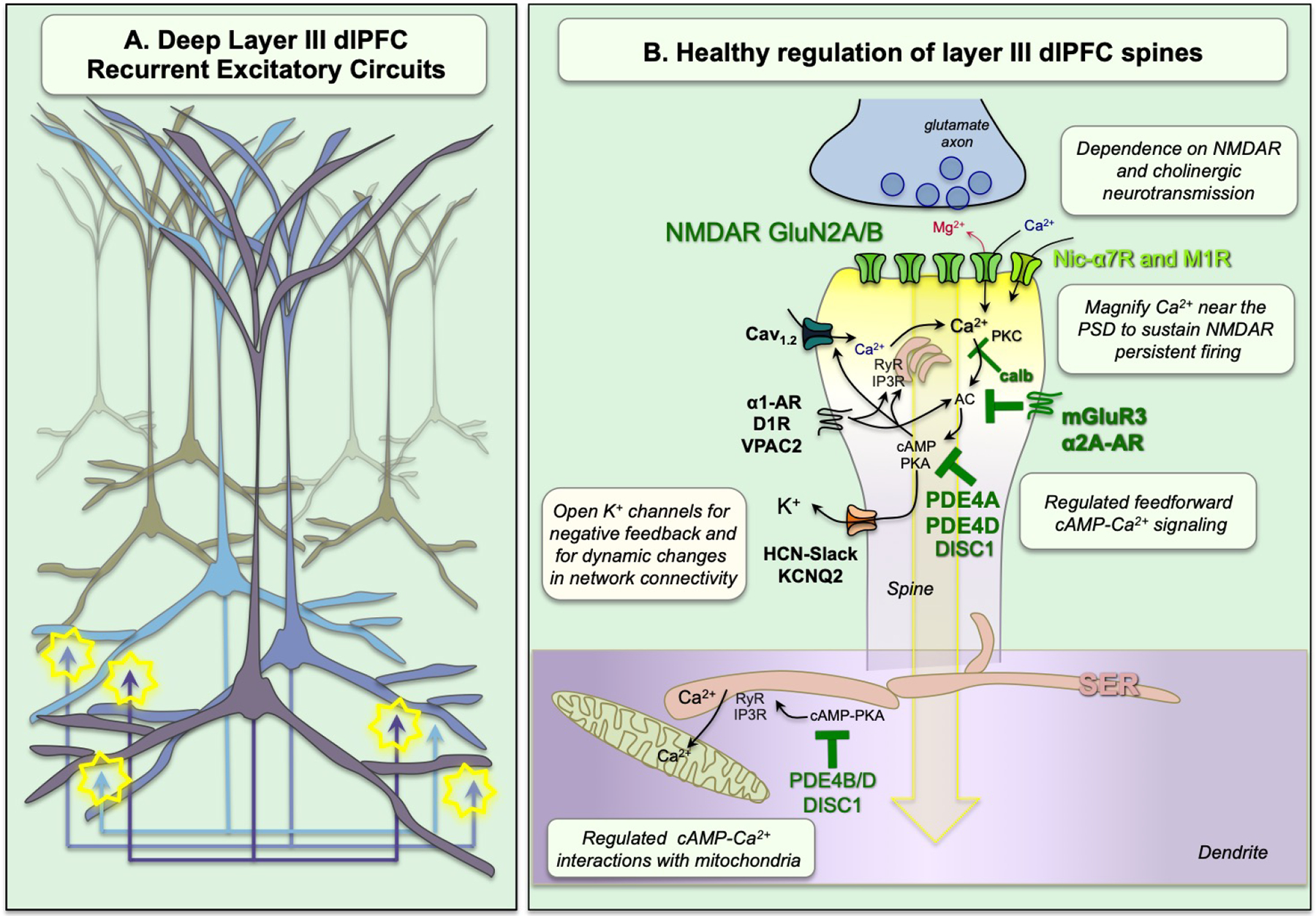
Schematic diagram of the layer III dlPFC recurrent excitatory microcircuits subserving working memory. A. Pyramidal cells with shared properties excite each other to keep information “in mind” through glutamatergic synapses on spines. Note: There is also lateral inhibition by GABA interneurons to refine the contents of working memory; this is not shown. B. A glutamatergic NMDAR synapse on a dendritic spine in the young adult, healthy dlPFC with tightly regulated, feedforward Ca2+-cAMP-K+ channel signaling. In these microcircuits, neurotransmission depends on NMDAR with GluN2A and GluN2B subunits, with permissive activation by cholinergic Nic-α7R and M1R (M1R via closure of KCNQ5 channels, not shown). These spines also contain the molecular machinery for cAMP-PKA to magnify Ca2+ signaling needed to sustain persistent firing, including internal Ca2+ release from the SER spine apparatus, which in turn increases cAMP production, leading to feedforward cAMP-Ca2+ signaling. A variety of receptors are localized on spines that drive cAMP-Ca2+ signaling, including the DA receptor D1R, the VIP and PACAP receptor, VIPR2, and the NE receptor α1-AR. Layer III spines also express K+ channels that are opened by cAMP-PKA signaling to provide negative feedback, and for dynamic changes in network connectivity. Under healthy conditions, these intracellular signaling pathways are tightly regulated by receptors that inhibit cAMP production, PDE4s that are anchored to the SER by DISC1 and which catabolize cAMP once it is generated, and calbindin (calb) to bind cytosolic Ca2+. PDE4s are also found in dendrites near mitochondria, positioned to regulate cAMP drive on Ca2+ release from the SER into mitochondria.
Recent research shows that Delay cells have unusual neurotransmission and neuromodulation, likely related to their need to sustain dynamic, everchanging, mental representations. Delay cell neurotransmission relies heavily on glutamate stimulation of NMDAR (GluN2B and GluN2A), but not AMPAR (Fig. 1B) (33). GluN2B subunits close slowly and flux high levels of Ca2+, and reside exclusively within the post-synaptic-density (PSD) in layer III dlPFC (33). This dependence on GluN2B channels had been predicted by computational models (34). In classic circuits such as rodent hippocampus, AMPAR normally serve to depolarize the post-synaptic membrane, ejecting the Mg2+ block from the NMDAR channel, and permitting NMDAR neurotransmission. However, dlPFC Delay cells have surprisingly little reliance on AMPAR, and instead this critical permissive function is performed by acetylcholine, through actions at nicotinic-α7 receptors (Nic-α7R) (35) and/or muscarinic M1R (36), that reside within the PSD (Fig. 1B). M1R act in part by closing KCNQ5 channels (36), while Nic-α7R directly flux Na+ and Ca2+. The heavy reliance of these synapses on Nic-α7R may help explain why most patients with schizophrenia smoke cigarettes or vape nicotine.
Layer III Delay cells also have unique neuromodulation needed to sustain neuronal firing without sensory stimulation, and to dynamically alter network strength, e.g. according to arousal state (Fig. 1B). Layer III spines express the molecular machinery to magnify Ca2+ signaling near the PSD through multiple mechanisms, including 1) Ca2+ entry through NMDAR and Nic-α7R and, 2) through voltage-gated Ca2+ channels, as well as 3) internal Ca2+ release from the smooth endoplasmic reticulum (SER, called the “spine apparatus” in spines). These Ca2+ actions are all increased by cAMP-PKA signaling, and Ca2+ can also increase internal Ca2+ release, e.g. Cav1.2 channels drive Ca2+ release through ryanodine receptors (37). Cytosolic Ca2+ can in turn increase cAMP production via adenylyl cyclase, thus driving feedforward signaling (Fig. 1B). Layer III dlPFC spines express a large number of cAMP-signaling proteins, especially near the spine apparatus and the PSD (38, 39). There is also a concentration of cAMP-Ca2+ signaling near mitochondria in nearby dendrites (38, 40), coordinating energy demands.
Layer III spines also express ion channels that are opened by cAMP-PKA signaling that weaken connectivity. The open state of HCN channels (Hyperpolarization activated Cyclic Nucleotide gated cation channels) is increased by cAMP, and these channels are concentrated on layer III spines in dlPFC (38, 41), but not V1 (42). Opening HCN channels with cAMP reduces Delay cell firing, while HCN channel blockade enhances firing (41). Recent data suggest a likely partnership with Slack potassium (K+) channels (43), where cAMP opens HCN channels, and the entry of sodium opens neighboring Slack channels to have a net outflow of K+. Another key channel localized in layer III spines is the KCNQ2 K+ channel, whose open state is increased by PKA signaling (36). Opening these channels reduces, while blocking them increases, Delay cell firing (36). The capability to rapidly open and close these ion channels allows very rapid, dynamic changes in network strength, (termed Dynamic Network Connectivity) needed for a constantly changing mental sketchpad (44), including taking dlPFC “offline” during stress (see below).
In the young, healthy dlPFC, feedforward Ca2+-cAMP-K+ channel signaling is tightly regulated by mGluR3 and α2A-AR inhibition of cAMP production, and by PDE4 catabolism of cAMP once it is formed (39). Layer III dendritic spines are a focus of these proteins, e.g. with PDE4A anchored to the spine apparatus by Disrupted In Schizophrenia (DISC1; (44)). Layer III dlPFC pyramidal cells are also enriched in the Ca2+-binding protein, calbindin, which regulates Ca2+ in the cytosol (45). Loss of these regulatory proteins, e.g. due to inflammation or advancing age, leads to toxic Ca2+ dysregulation, atrophy of dendritic spines and reduced Delay cell firing (39).
Stress signaling in dlPFC weakens connectivity and takes dlPFC “offline”
Layer III dlPFC circuits have “built-in” molecular mechanisms to rapidly take dlPFC “off-line” during uncontrollable stress exposure. This has survival value under some dangerous conditions, (e.g. being cut off on the highway, where it is helpful to rapidly switch control to circuits mediating habitual/instinctive behaviors), but it is detrimental when one needs higher cognition to deal with the threat (e.g. an invisible virus). As summarized in Figure 2, high levels of catecholamine release during uncontrollable stress activate large numbers of dopamine (DA) D1R and norepinephrine α1-AR to drive high levels of Ca2+-cAMP-K+ channel signaling and disconnect dlPFC recurrent networks. For example, high levels of D1R signaling markedly reduce Delay cell firing via increased cAMP-PKA signaling, and this is prevented by HCN channel blockade (46, 47). Other Gs-coupled receptors likely contribute as well, e.g. β-AR are currently under study, and transcriptomics show very high levels of ADCYAP1 (encoding PACAP) in layer III dlPFC (48), the “master stress activating peptide” which drives cAMP signaling (49), e.g. via VPAC2 receptors (see below). High levels of α1-AR stimulation also reduce Delay cell firing via IP3-Ca2+-/PKC signaling (50, 51). Interestingly, rodent studies have shown that PKC can cause internalization and/or decoupling of α2A-AR (52, 53) and mGluR3 (54), which, if true in dlPFC as well, would reduce the inhibitory regulation of stress signaling pathways. Detrimental effects of high levels of D1R or α1-AR stimulation can also be seen at the behavioral level, where D1R or α1-AR stimulation in dlPFC impairs working memory performance, and conversely, D1R or α1-AR blockade reduces stress-induced working memory deficits (reviewed in (55, 56)). It is noteworthy that atypical antipsychotics have α1-AR blocking properties that may be beneficial in blocking stress signaling in the dlPFC (57).
Figure 2-.

The effects of uncontrollable stress exposure on layer III dlPFC spines. Acute exposure to an uncontrollable stressor increases catecholamine release in the PFC, driving feedforward cAMP-Ca2+-K+ channel signaling, to rapidly weaken synaptic efficacy, reduce persistent firing, and take dlPFC “offline”. Cortisol release exacerbates (or on its own, mimics) these actions, likely by blocking the extraneuronal catecholamine transporters on glia that take up catecholamines from the extrasynaptic space. With chronic stress exposure, there are additional architectural changes, with loss of spines and dendrites that correlate with cognitive deficits. Phagocytosis of spines and dendrites likely involves Ca2+ overload of mitochondria, initiating an inflammatory response. Calbindin (calb) expression is decreased by chronic stress exposure, which may further elevate cytosolic Ca2+ levels.
During stress, glucocorticoids (e.g. cortisol) are also released by the adrenal cortex and cross into brain. High levels of glucocorticoids can mimic and/or exacerbate the effects of catecholamines in PFC, e.g. impairing working memory in rats (58), and deactivating PFC in humans (59). Glucocorticoids are known to block the extraneuronal catecholamine transporters on glia that normally serve to remove catecholamines from the extracellular space (60), which would exacerbate the stress response.
With chronic stress exposure, there continues to be elevated catecholamine and glucocorticoid release, but there are additional architectural changes, with loss of layer III dendrites and spines ((61–63), reviewed in (64)). Much of this research has been done in rodent models, largely for ethical reasons. These studies show that elevated cAMP-PKA and Ca2+-PKC signaling contributes to layer III spine loss, and that spine loss correlates with working memory deficits (65, 66). In particular, dysregulated Ca2+ signaling can initiate a number of toxic actions, including Ca2+ overload of mitochondria, leading to inflammatory signals such as complement that initiate spine removal (Figs. 2–3; reviewed in (64)). In layer III dlPFC, stress may simultaneously elevate cytosolic Ca2+, and weaken synaptic efficacy through opening of nearby K+ channels. However, either of these conditions may be sufficient to initiate spine removal. It is possible that related mechanisms contribute to the loss of spines and dendrites from layer III dlPFC in schizophrenia when intracellular stress signaling pathways are dysregulated by genetic and/or environmental insults. In this regard, it is of interest that there is an increased DA innervation of macaque layer III dlPFC in adolescence (67) which can drive stress signaling (47, 56). As there are increased DA D1R in the dlPFC in the earliest stages of schizophrenia (68), magnified stress signaling in layer III may contribute to the distinctive spine loss in this layer at the onset of disease, especially if proteins that regulate the stress response are impaired by genetic and/or inflammatory insults.
Figure 3-.

Speculations regarding the sequence of events underlying spine removal. Initial events involve reduced growth factors and/or elevated, dysregulated Ca2+ signaling, followed by actin destabilization, Ca2+ overload of the mitochondria and inflammatory signaling, which engages glial removal of the spine. Complement C1q signaling is shown for illustrative purposes. Note that genetic alterations in schizophrenia may propel spine loss at multiple stages of this sequence. For more details on the mechanisms underlying spine removal, please see (64). MOAS = Mitochondria-On-A-String; ROS = reactive oxygen species.
Genetic risk factors weaken layer III dlPFC connectivity
The genetics of schizophrenia are complex, with a large number of factors of (mostly) very small effect size (7, 69). As summarized in Figure 4, many of the genetic insults that increase risk of schizophrenia would weaken the connectivity of layer III dlPFC synapses. A general pattern emerges whereby there are:
loss-of-function alterations in proteins that are necessary to strengthen connectivity, including those needed for synapse creation or structure, for NMDAR neurotransmission, for mitochondrial energy production, or for inhibitory regulation of cAMP-Ca2+-K+ channel signaling; and
gain-of-function alterations in proteins that weaken synaptic connectivity, including those that increase cAMP-Ca2+-K+ channel signaling, or those that mediate phagocytic removal of spines such as complement C4a (70).
As a thorough review of this extensive field is beyond the scope of this paper, this section will focus on those genetic alterations that directly relate to the neurotransmission and neuromodulation of layer III dlPFC circuits which may help to explain their exceptional vulnerability.
Figure 4-.

There are multiple genetic risk factors for schizophrenia that would weaken layer III dlPFC network connectivity by either reducing beneficial actions or increasing detrimental actions. The loss-of-function alterations in gene products that normally strengthen connectivity are shown in gray; the gain-of-function alterations in gene products that normally weaken connectivity are shown in red. Thus, multiple different genotypes can lead to the same phenotype of weakened layer III dlPFC connectivity.
Loss-of-function alterations in gene products that normally strengthen layer III dlPFC network connectivity-
There are multiple genes where loss-of-function genetic mutations would weaken synapses, including those involved with synaptic development and adherence (e.g. ZNF804A (71); NRXN1 (72)), actin cytoskeletal dynamics (e.g. CDC42, ARP2/3 (73, 74)) and mitochondrial energy production (e.g. (75–77)). However, these genetic insults should afflict all synapses, and thus a key question is why they would particularly afflict those in deep layer III more than others, e.g. those in layer V. The recurrent excitatory circuits in deep layer III are thought to reside on the basal dendrites of deep layer III pyramidal cells, that have remarkably high levels of dendritic branching and spine density (78–80), and thus these insults may be more evident against a background of high connectivity. It is also possible that synaptic errors could be magnified in a recurrent excitatory circuit, including interactions with additional factors that preferentially weaken connectivity in layer III dlPFC, as discussed below.
Several genetic insults linked to schizophrenia target glutamate neurotransmission in ways that may be particularly detrimental to layer III dlPFC recurrent circuits. Proteins that normally strengthen dlPFC network connectivity and are associated with loss-of-function mutations and are shown in gray in Figure 4. As discussed above, layer III dlPFC Delay cell circuits heavily rely on NMDAR with either GluN2B or GluN2A subunits (33). GRIN2A, which encodes NMDAR-GluN2A, is a replicated risk factor for schizophrenia (69, 81). Inadequate NMDAR-GluN2A neurotransmission may lead to spine pruning, as weak synapses are generally removed, e.g. in developing circuits (82). Interestingly, insults to GRIN2B are associated with even more profound intellectual disability, which may relate to the key role this receptor plays in cortical development (83).
Perhaps most distinct to layer III dlPFC would be genes that regulate feedforward, Ca2+-cAMP-K+ channel signaling in layer III spines, where loss of regulation would weaken synaptic connectivity. A key gene in this regard is GRM3, which encodes mGluR3. As mentioned above, mGluR3 are concentrated on layer III dlPFC spines (rather than axon terminals where they reside in classic circuits, e.g. rodent spinal cord (84)). In layer III dlPFC, mGluR3 inhibit cAMP-K+ channel signaling, strengthening connectivity and enhancing Delay cell firing (85). GRM3 is a consistent risk factor for schizophrenia (86), with reduced mGluR3 protein in the dlPFC of patients with schizophrenia (87). Our data suggest that loss-of-function mutations in GRM3 would be especially detrimental to layer III dlPFC connectivity (85, 88), consistent with findings that genetic alterations in mGluR3 are associated with impaired dlPFC function in schizophrenia (89) and in healthy individuals (90).
A rare, loss-of-function translocation in Disrupted in Schizophrenia (DISC1) is associated with high rates of mental disorders (91). DISC1 has multiple functions, including anchoring phosphodiesterases type 4 (PDE4s), the enzymes that catabolize cAMP (92). We have documented DISC1 anchoring PDE4A to the spine apparatus in layer III dlPFC spines (38, 93), positioned to regulate feedforward, cAMP-Ca2+-K+ channel signaling (39, 93). Thus, a loss-of-function mutation in DISC1 would result in high levels of K+ efflux and elevated cytosolic Ca2+, reducing Delay cell firing and increasing risk of dendritic atrophy. DISC1 knockdown in rat medial PFC lowered the threshold for stress-induced deficits in working memory, with no effect under nonstress conditions (94). These data suggest that phenotypic expression of reduced DISC1 may only be seen under stressful conditions when there is elevated cAMP signaling in layer III dlPFC. Similar effects in humans may help to explain the heterogeneity of the DISC1 phenotype.
Gain-of-function alterations in gene products that normally weaken layer III dlPFC network connectivity-
There are also multiple gain-of-function alterations in proteins that weaken layer III dlPFC connectivity by increasing Ca2+-cAMP-K+ channel signaling (Figure 4, left side). For example, a rare microduplication of the Gs-coupled receptor VIPR2 (i.e. VPAC2) increases risk of schizophrenia (95). VIPR2 are stimulated by either VIP or the “master stress peptide” PACAP, increasing cAMP signaling. Studies in patients with schizophrenia that express this microduplication show increased VIPR2 transcription and increased cAMP signaling in cultured lymphocytes (95). VIPR2 are concentrated on layer III dlPFC spines, positioned near the SER spine apparatus where they can drive feedforward cAMP-Ca2+-K+ channel signaling (96, 97). Thus, microduplications that increase VIPR2-cAMP signaling would weaken layer III dlPFC connectivity.
Genetic alterations in voltage-gated Ca2+ channels are also a replicated risk factor for schizophrenia (98). In particular, a gain-of-function mutation (99) in CACNA1C, which encodes for the alpha subunit of the L-type Ca2+ channel, Cav1.2, is consistently linked to increased risk of schizophrenia and bipolar disorder (69, 81, 100–102). This gain-of-function in Cav1.2 is associated with inefficient dlPFC function (103) and poor working memory and executive control in healthy controls and especially in patients (104, 105). In the heart, Cav1.2 is central to the “fight or flight” stress response, where its activation by noradrenergic beta-AR-PKA signaling drives internal Ca2+ release from the sarcoplasmic reticulum, increasing muscle contraction (106). Our immunoEM data find parallel localization on layer III dlPFC spines near the SER spine apparatus (96), positioned to drive Ca2+-cAMP-K+ signaling, which can weaken connectivity and increase toxic Ca2+ actions that lead to spine removal.
There also appear to be gain-of-function insults to the gene that encodes for one of the key channels that weakens synaptic connectivity in layer III spines: HCN1 (81, 107). The HCN1 risk allele is associated with impaired spatial memory (108), and physiological data are consistent with the risk allele conferring a gain-of-function (109).
All of these gain-of-function alterations would weaken the efficacy of dlPFC synapses, and increased expression of VIPR2 or Cav1.2 would additionally increase cytosolic Ca2+, which can cause Ca2+ overload of mitochondria, leading to an inflammatory response, e.g. complement activation (82) to initiate phagocytosis and the removal of spines and dendrites (Fig. 4). Alternatively, over-pruning of spines and dendrites could occur through a gain-of-function genetic alteration in complement C4a, which has the largest likelihood of being a genetic risk factor for schizophrenia (70). The C4a gain-of-function risk allele has been shown to increase phagocytosis in mouse models (70), and is associated with reduced neuropil in patients (110), consistent with excessive spine pruning contributing to the etiology of schizophrenia. The current discussion highlights how multiple genetic insults, indirectly or directly, can lead to weakening and removal of layer III dlPFC excitatory synapses on spines.
Inflammatory insults can mimic genetic insults to weaken dlPFC connectivity
Environmental insults are also major risk factors for schizophrenia, including perinatal inflammation (e.g. influenza during the second trimester, hypoxia at birth), and psychological stressors during adolescence/early adulthood are associated with symptom onset (e.g. leaving home for college) (111). It is hypothesized that perinatal inflammation may sensitize the inflammatory response and contribute to greater spine removal during adolescence. The following section describes three inflammatory responses that may be especially detrimental to layer III dlPFC connections, as summarized in Figure 5, mimicking several of the genetic insults shown in Figure 4.
Figure 5-.

Inflammation weakens layer III dlPFC network connectivity in ways that mimic genetic insults. Inflammation increases the expression and release of kynurenic acid (KYNA) and GCPII by astrocytes, and increases p38-MK2 signaling within neurons. KYNA blocks NMDAR and Nic-α7R, which would mimic loss-of-function genetic alterations to GRIN2A; GCPII reduces NAAG stimulation of mGluR3, which would mimic loss-of-function genetic alterations to GRM3; and MK2 unanchors and disinhibits PDE4s, which would mimic loss-of-function genetic alterations to DISC1. Calbindin (calb) expression may also be decreased by chronic stress exposure, which would increase cytosolic Ca2+ levels, similar to gain-of-function mutations in CACNA1C. Ca2+ overload of mitochondria can lead to complement activation, which may mimic gain-of-function genetic alterations in complement C4a. Thus, the combination of genetic and environmental insults may interact to cross the threshold into pathology.
Kynurenic acid (KYNA)
Inflammation increases tryptophan metabolism to kynurenine, which can then be further processed to KYNA, especially in astrocytes (112). Kynurenine is actively taken up into brain, so it can also originate from peripheral sources (113). Most pertinent to the current discussion, KYNA is known to block both NMDAR and Nic-α7R (114, 115), the receptors most essential to dlPFC neurotransmission. Thus, KYNA inflammatory signaling would mimic loss-of-function alterations in GRIN2A as well as reducing factors permissive for NMDAR neurotransmission in layer III dlPFC, and thus may be particularly detrimental to the circuits mediating higher cognition. Schizophrenia is associated with higher KYNA levels (116), including in the dlPFC (117, 118), and is especially evident in those with signatures of inflammation, where it is associated with reduced dlPFC volume and impaired attention regulation (118). Current treatment strategies aim to inhibit KYNA production and restore dlPFC function (119, 120).
Glutamate carboxypeptidase II (GCPII)
As shown in Figure 5, GCPII reduces mGluR3 signaling by catabolizing NAAG, the endogenous ligand for mGluR3 that is co-released with glutamate (Fig. 5; (121)). GCPII expression is increased by inflammation e.g. in the aged rat mPFC (122), and especially relevant to schizophrenia, by perinatal inflammation (123). Thus, inflammation can mimic loss-of-of function mutations in GRM3 by reducing mGluR3 signaling. As mGluR3 are concentrated on layer III spines where they enhance connectivity, loss of beneficial mGluR3 actions would be particularly deleterious to dlPFC working memory function. Elevated GCPII levels have been documented in the dlPFC of patients with schizophrenia (87), consistent with increased inflammation in this disorder. Interestingly, in healthy individuals, a gain-of-function variant in the FOLH1 gene that encodes for GCPII is associated with decreased NAAG levels, inefficient dlPFC activity, and impaired cognition (90), highlighting the importance of this signaling mechanism to human intelligence.
MAPK-activated protein kinase 2 (MAPKAPK2 or MK2)
MK2 is a downstream substrate of p38MAPK signaling, which is activated under conditions of stress and/or inflammation (124). MK2 phosphorylates the PDE4s such that they can no longer be anchored by DISC1 to the correct location (125, 126). In this way, MK2 inflammatory signaling would mimic loss of function translocation in DISC1. MK2 also prevents PKA from activating PDE4s, thus taking away negative feedback that would normally regulate stress signaling, once initiated, can “run wild”.
Psychological stressors
Although most studies of inflammation use infectious agents or hypoxia to induce an inflammatory state, it is important to remember that psychological stress also impacts many of these same signaling pathways (Fig. 2). As described above, psychological stress drives Ca2+-cAMP-K+ signaling, and chronic stress exposure leads to spines loss. There is some evidence from rodents that psychological stress exposure can reduce the expression of the Ca2+ binding protein, calbindin (127), which would further increase cytosolic Ca2+ levels, leading to inflammation and spine removal (64).
Taken together, it is evident that there are multiple ways in which inflammatory/stress signaling can weaken dlPFC network connectivity, and in several cases, increase cytosolic Ca2+ signaling, mimicking genetic insults. Thus, the interaction between environmental and genetic insults may cross the threshold into pathology, leading to atrophy of layer III dlPFC dendrites and spines.
Limitations, Future Directions and Summary
There is much that is not known about the primate cortex that limits our current hypotheses and interpretations. For example, almost nothing is known about the molecular regulation of synapses in primate cortices beyond the dlPFC and V1. Thus, it is not possible to relate spine differences in schizophrenia in other cortical areas to vulnerabilities in their underlying molecular regulation. There is also little known about the mechanisms governing gray matter thinning in the primate cortex, whether this process targets ineffective connections, and how these processes may be altered in schizophrenia. The current review is an initial attempt to begin to relate synaptic and inflammatory mechanisms under study in primate dlPFC to the striking pathology of these circuits in schizophrenia.
In summary, a unique feature of layer III dlPFC circuits is their built-in mechanisms to rapidly take them “off-line” during psychological and/or physiological stress. These include cAMP-PKA magnification of Ca2+ signaling in spines, and the concentration of cAMP-PKA opened K+ channels on spines that rapidly weaken synaptic efficacy. These properties may interact with genetic insults (e.g. gain-of-function expression of Cav1.2), and/or environmental insults (e.g. GCPII-mediated dysregulation of cAMP signaling), to cross the threshold into pathology. Flaws in neuronal signaling may also be exaggerated due to the recurrent nature of layer III microcircuits, e.g. where the reduction in neurotransmission caused by genetic insults to GRIN2A, and/or blockade of NMDAR by KYNA, would be amplified across a large number of layer III recurrent NMDAR synapses on spines. Understanding the unique needs of these circuits may help in the design of preventive treatments to protect these recently evolved circuits that are so critical to cognitive function.
Financial Disclosures-
The authors were supported by NIH grants MH108643 and AG061190 to AFTA, and MH093354 to MW. AFTA and Yale University receive royalties from the USA sales of Intuniv (extended release guanfacine) from Shire/Takeda Pharmaceuticals, but do not receive royalties from generic nor international sales. The remaining authors report no biomedical financial interests or potential conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Invited review from Bruno Averbeck and Matt Chaffee for Biological Psychiatry on schizophrenia synapses
References
- 1.Weinberger DR, Berman KF, Zec RF (1986): Physiologic dysfunction of dorsolateral prefrontal cortex in schizophrenia. I. Regional cerebral blood flow evidence. Archives General Psychiatry. 43:114–124. [DOI] [PubMed] [Google Scholar]
- 2.Docherty NM, Hawkins KA, Hoffman RE, Quinlan DM, Rakfeldt J, Sledge WH (1996): Working memory, attention, and communication disturbances in schizophrenia. J Abnormal Psychology. 105:212–219. [DOI] [PubMed] [Google Scholar]
- 3.Perlstein WM, Carter CS, Noll DC, Cohen JD (2001): Relation of prefrontal cortex dysfunction to working memory and symptoms in schizophrenia. Am J Psychiatry. 158:1105–1113. [DOI] [PubMed] [Google Scholar]
- 4.Barch DM (2005): The cognitive neuroscience of schizophrenia. Annu Rev Clin Psychol. 1:321–353. [DOI] [PubMed] [Google Scholar]
- 5.Keefe RS, Harvey PD (2012): Cognitive impairment in schizophrenia. Handb Exp Pharmacol 213:11–37. [DOI] [PubMed] [Google Scholar]
- 6.Docherty NM, Evans IM, Sledge WH, Seibyl JP, Krystal JH (1994): Affective reactivity of language in schizophrenia. J Nerv Ment Dis 182:98–102. [DOI] [PubMed] [Google Scholar]
- 7.The Schizophrenia Working Group of the Psychiatric Genomics Consortium, Ripke S, Walters JT, O’Donovan MC (2020): Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. medRxiv. DOI: 10.1101/2020.09.12.20192922. [DOI] [Google Scholar]
- 8.Cannon TD, Thompson PM, van Erp TG, Toga AW, Poutanen VP, Huttunen M, et al. (2002): Cortex mapping reveals regionally specific patterns of genetic and disease-specific gray-matter deficits in twins discordant for schizophrenia. Proc Natl Acad Sci U S A. 99:3228–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cannon TD, Chung Y, He G, Sun D, Jacobson A, van Erp TG, et al. (2014): Progressive reduction in cortical thickness as psychosis develops: A multisite longitudinal neuroimaging study of youth at elevated clinical risk. Biol Psychiatry. 77:147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Föcking M, Sabherwal S, Cates HM, Scaife C, Dicker P, Hryniewiecka M, et al. (2021): Complement pathway changes at age 12 are associated with psychotic experiences at age 18 in a longitudinal population-based study: evidence for a role of stress. Mol Psychiatry. 26:524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radhakrishnan R, Skosnik PD, Ranganathan M, Naganawa M, Toyonaga T, Finnema S, et al. (2021): In vivo evidence of lower synaptic vesicle density in schizophrenia. Mol Psychiatry. Jun 16, epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 12.Glantz LA, Lewis DA (2000): Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 57:65–73. [DOI] [PubMed] [Google Scholar]
- 13.Berdenis van Berlekom A, Muflihah CH, Snijders GJLJ, MacGillavry HD, Middeldorp J, Hol EM, et al. (2020): Synapse Pathology in Schizophrenia: A Meta-analysis of Postsynaptic Elements in Postmortem Brain Studies. Schizophr Bull. 46:374–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldman-Rakic P (1995): Cellular Basis of Working Memory. Neuron. 14:477–485. [DOI] [PubMed] [Google Scholar]
- 15.Glausier JR, Lewis DA (2013): Dendritic spine pathology in schizophrenia. Neuroscience. 251:90–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kolluri N, Sun Z, Sampson AR, Lewis DA (2005): Lamina-specific reductions in dendritic spine density in the prefrontal cortex of subjects with schizophrenia. Am J Psychiatry. 162:1200–1202. [DOI] [PubMed] [Google Scholar]
- 17.Roberts RC, Barksdale KA, Roche JK, Lahti AC (2015): Decreased synaptic and mitochondrial density in the postmortem anterior cingulate cortex in schizophrenia. Schizophr Res. 168:543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garey LJ, Ong WY, Patel TS, Kanani M, Davis A, Mortimer AM, et al. (1998): Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J Neurol Neurosurg Psychiatry. 65:446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moyer CE, Delevich KM, Fish KN, Asafu-Adjei JK, Sampson AR, Dorph-Petersen KA, et al. (2013): Intracortical excitatory and thalamocortical boutons are intact in primary auditory cortex in schizophrenia. Schizophr Res. 149:127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKinney BC, MacDonald ML, Newman JT, Shelton MA, DeGiosio RA, Kelly RM, et al. (2019): Density of small dendritic spines and microtubule-associated-protein-2 immunoreactivity in the primary auditory cortex of subjects with schizophrenia. Neuropsychopharmacology. 44:1055–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacobsen C (1936): Studies of cerebral functions in primates. Comp Psychol Monographs. 13:1–60. [Google Scholar]
- 22.Fuster J (1989): The prefrontal cortex. New York: Raven Press. [Google Scholar]
- 23.Robbins TW (1996): Dissociating executive functions of the prefrontal cortex. Phil Trans R Soc London. 351:1463–1471. [DOI] [PubMed] [Google Scholar]
- 24.Szczepanski SM, Knight RT (2014): Insights into human behavior from lesions to the prefrontal cortex. Neuron. 83:1002–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldman-Rakic PS, Selemon LD, Schwartz ML (1984): Dual pathways connecting the dorsolateral prefrontal cortex with the hippocampal formation and parahippocampal cortex in the rhesus monkey. Neuroscience. 12:719–743. [DOI] [PubMed] [Google Scholar]
- 26.Giguere M, Goldman-Rakic PS (1988): Mediodorsal nucleus: areal, laminar, and tangential distribution of afferents and efferents in the frontal lobe of rhesus monkeys. J Comp Neurol 277:195–213. [DOI] [PubMed] [Google Scholar]
- 27.Selemon LD, Goldman-Rakic PS (1988): Common cortical and subcortical targets of the dorsolateral prefrontal and posterior parietal cortices in the rhesus monkey: evidence for a distributed neural network subserving spatially guided behavior. J Neurosci. 8:4049–4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldman-Rakic PS (1987): Circuitry of the primate prefrontal cortex and the regulation of behavior by representational memory. In: Plum F, editor. Handbook of Physiology, The Nervous System, Higher Functions of the Brain. Bethesda: American Physiological Society, pp 373–417. [Google Scholar]
- 29.Fuster J, Alexander G (1971): Neuron activity related to short-term memory. Science. 173:652–654. [DOI] [PubMed] [Google Scholar]
- 30.Funahashi S, Bruce CJ, Goldman-Rakic PS (1989): Mnemonic coding of visual space in the monkey’s dorsolateral prefrontal cortex. J Neurophysiol 61:331–349. [DOI] [PubMed] [Google Scholar]
- 31.Li D, Constantinidis C, Murray JD (2021): Trial-to-trial variability of spiking delay activity in prefrontal cortex constrains burst-coding models of working memory. J Neurosci. 41:8928–8945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.González-Burgos G, Barrionuevo G, Lewis DA (2000): Horizontal synaptic connections in monkey prefrontal cortex: an in vitro electrophysiological study. Cereb Cortex. 10:82–92. [DOI] [PubMed] [Google Scholar]
- 33.Wang M, Yang Y, Wang CJ, Gamo NJ, Jin LE, Mazer JA, et al. (2013): NMDA receptors subserve working memory persistent neuronal firing In dorsolateral prefrontal cortex. Neuron. 77:736–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang XJ (1999): Synaptic basis of cortical persistent activity: the importance of NMDA receptors to working memory. J Neurosci. 19:9587–9603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Y, Paspalas CD, Jin LE, Picciotto MR, Arnsten AFT, Wang M (2013): Nicotinic α7 receptors enhance NMDA cognitive circuits in dorsolateral prefrontal cortex. Proc Nat Acad Sci USA. 110:12078–12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galvin VC, Yang S-T, Paspalas CD, Yang Y, Jin LE, Datta D, et al. (2020): Muscarinic M1 receptors modulate working memory performance and activity via KCNQ potassium channels in primate prefrontal cortex. Neuron. 106:649–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vierra NC, Kirmiz M, van der List D, Santana LF, Trimmer JS (2019): Kv2.1 mediates spatial and functional coupling of L-type calcium channels and ryanodine receptors in mammalian neurons. Elife. 8:e49953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paspalas CD, Wang M, Arnsten AFT (2013): Constellation of HCN Channels and cAMP regulating proteins in dendritic spines of the primate prefrontal cortex — Potential substrate for working memory deficits in schizophrenia. Cereb Cortex. 23:1643–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arnsten AFT, Datta D, Wang M (2021): The Genie in the Bottle- Magnified calcium signaling in dorsolateral prefrontal cortex. Molecular Psychiatry. 26:3684–3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Datta D, Enwright JF, Arion D, Paspalas CD, Morozov YM, Lewis DA, et al. (2020): Mapping phosphodiesterase 4D (PDE4D) in macaque dorsolateral prefrontal cortex: Postsynaptic compartmentalization in higher-order layer III pyramidal cell circuits. Frontiers in Neuroanatomy. 14:578483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang M, Ramos B, Paspalas C, Shu Y, Simen A, Duque A, et al. (2007): Alpha2A-adrenoceptor stimulation strengthens working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 129:397–410. [DOI] [PubMed] [Google Scholar]
- 42.Yang ST, Wang M, Paspalas CP, Crimins JL, Altman MT, Mazer JA, et al. (2018): Core differences in synaptic signaling between primary visual and dorsolateral prefrontal cortex. Cereb Cortex. 28:1458–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.El-Hassar L, Datta D, Chatterjee M, Arnsten AFT, Kaczmarek LK (2019): Interaction Between HCN and Slack Channels Regulates mPFC pyramidal cell excitability and Working Memory function. Neurosci Abstracts. 462.05. [DOI] [PubMed] [Google Scholar]
- 44.Arnsten AFT, Wang M, Paspalas CD (2012): Neuromodulation of thought: Flexibilities and vulnerabilities in prefrontal cortical network synapses. Neuron. 76:223–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Datta D, Leslie Sc-fa, Wang M, Yang S-T, Morozov Y, Mentone S, et al. (2021): Age-related calcium dysregulation linked with tau pathology and impaired cognition in non-human primates. Alzheimer’s & Dementia. 17:920–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vijayraghavan S, Wang M, Birnbaum SG, Bruce CJ, Williams GV, Arnsten AFT (2007): Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nature Neuroscience. 10:376–384. [DOI] [PubMed] [Google Scholar]
- 47.Gamo NJ, Lur G, Higley MJ, Wang M, Paspalas CD, Vijayraghavan S, et al. (2015): Stress impairs prefrontal cortical function via D1 dopamine receptor interactions with HCN channels. Biol Psychiatry. 78:860–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.González-Burgos G, Miyamae T, Krimer Y, Gulchina Y, Pafundo DE, Krimer O, et al. (2019): Distinct Properties of Layer 3 Pyramidal Neurons from Prefrontal and Parietal Areas of the Monkey Neocortex. J Neurosci. 39:7277–7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mustafa T (2013): Pituitary adenylate cyclase-activating polypeptide (PACAP): a master regulator in central and peripheral stress responses. Adv Pharmacol. 68:445–457. [DOI] [PubMed] [Google Scholar]
- 50.Birnbaum SB, Yuan P, Wang M, Vijayraghavan S, Bloom A, Davis D, et al. (2004): Protein kinase C overactivity impairs prefrontal cortical regulation of working memory. Science. 306:882–884. [DOI] [PubMed] [Google Scholar]
- 51.Datta D, Yang ST, Galvin VC, Solder J, Luo F, Morozov YM, et al. (2019): Noradrenergic α1-Adrenoceptor Actions in the Primate Dorsolateral Prefrontal Cortex. J Neurosci. 39:2722–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liang M, Eason MG, Jewell-Motz EA, Williams MA, Theiss CT, Dorn GWn, et al. (1998): Phosphorylation and functional desensitization of the alpha2A-adrenergic receptor by protein kinase C. Mol Pharmacol. 54:44–49. [DOI] [PubMed] [Google Scholar]
- 53.Zhu Q, Qi LJ, Shi A, Abou-Samra A, Deth RC (2004): Protein kinase C regulates alpha(2A/D)-adrenoceptor constitutive activity. Pharmacology. 71:80–90. [DOI] [PubMed] [Google Scholar]
- 54.Macek TA, Schaffhauser H, Conn PJ (1998): Protein kinase C and A3 adenosine receptor activation inhibit presynaptic metabotropic glutamate receptor (mGluR) function and uncouple mGluRs from GTP-binding proteins. J Neurosci. 18:6138–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arnsten AFT (2009): Stress signaling pathways that impair prefrontal cortex structure and function. Nature Reviews Neuroscience 10:410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arnsten AF (2015): Stress weakens prefrontal networks: molecular insults to higher cognition. Nat Neurosci. 18:1376–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baldessarini RJ, Huston-Lyons D, Campbell A, Marsh E, Cohen BM (1992): Do central antiadrenergic actions contribute to the atypical properties of clozapine? Br J Psychiatry. 160 S17:12–16. [PubMed] [Google Scholar]
- 58.Barsegyan A, Mackenzie SM, Kurose BD, McGaugh JL, Roozendaal B (2010): Glucocorticoids in the prefrontal cortex enhance memory consolidation and impair working memory by a common neural mechanism. Proc Natl Acad Sci U S A. 107:16655–16660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Stegeren AH, Roozendaal B, Kindt M, Wolf OT, Joëls M (2010): Interacting noradrenergic and corticosteroid systems shift human brain activation patterns during encoding. Neurobiol Learn Mem. 93:56–65. [DOI] [PubMed] [Google Scholar]
- 60.Grundemann D, Schechinger B, Rappold GA, Schomig E (1998): Molecular identification of the cortisone-sensitive extraneuronal catecholamine transporter. Nature Neuroscience. 1:349–351. [DOI] [PubMed] [Google Scholar]
- 61.Liston C, Miller MM, Goldwater DS, Radley JJ, Rocher AB, Hof PR, et al. (2006): Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J Neurosci. 26:7870–7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Radley JJ, Rocher AB, Miller M, Janssen WG, Liston C, Hof PR, et al. (2006): Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb Cortex. 16:313–320. [DOI] [PubMed] [Google Scholar]
- 63.Ota KT, Liu RJ, Voleti B, Maldonado-Aviles JG, Duric V, Iwata M, et al. (2014): REDD1 is essential for stress-induced synaptic loss and depressive behavior. Nat Med. 20:531–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Woo E, Sansing LH, Arnsten AFT, Datta D (2021): Chronic Stress Weakens Connectivity in the Prefrontal Cortex: Architectural and Molecular Changes. Chronic Stress. 5:24705470211029254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hains AB, Vu MA, Maciejewski PK, van Dyck CH, Gottron M, Arnsten AF (2009): Inhibition of protein kinase C signaling protects prefrontal cortex dendritic spines and cognition from the effects of chronic stress. Proc Natl Acad Sci U S A. 106:17957–17962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hains AB, Yabe Y, Arnsten AFT (2015): Chronic stimulation of alpha-2A-adrenoceptors with guanfacine protects rodent prefrontal cortex dendritic spines and cognition from the effects of chronic stress. Neurobiology of Stress. 2:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rosenberg DR, Lewis DA (1994): Changes in the dopaminergic innervation of monkey prefrontal cortex during late postnatal development: A tyosine hydroxylase immunohistochemical study. Biol Psychiat. 36:272–277. [DOI] [PubMed] [Google Scholar]
- 68.Abi-Dargham A, Xu X, Thompson JL, Gil R, Kegeles LS, Urban NB, et al. (2012): Increased prefrontal cortical D1 receptors in drug naive patients with schizophrenia: a PET study with [11C]NNC112. J Psychopharmacol. 26:794–805. [DOI] [PubMed] [Google Scholar]
- 69.Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014): Biological insights from 108 schizophrenia-associated genetic loci. Nature. 511:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. (2016): Schizophrenia risk from complex variation of complement component 4. Nature. 530:177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou Y, Dong F, Lanz TA, Reinhart V, Li M, Liu L, et al. (2018): Interactome analysis reveals ZNF804A, a schizophrenia risk gene, as a novel component of protein translational machinery critical for embryonic neurodevelopment. Mol Psychiatry. 23:952–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kirov G, Rujescu D, Ingason A, Collier DA, O’Donovan MC, Owen MJ (2009): Neurexin 1 (NRXN1) deletions in schizophrenia. Schizophr Bull. 35:851–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Datta D, Arion D, Corradi JP, Lewis DA (2015): Altered expression of CDC42 signaling pathway components in cortical layer 3 pyramidal cells in schizophrenia. Biol Psychiatry. 78:775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Datta D, Arion D, Roman KM, Volk DW, Lewis DA (2017): Altered Expression of ARP2/3 Complex Signaling Pathway Genes in Prefrontal Layer 3 Pyramidal Cells in Schizophrenia. Am J Psychiatry. 174:163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hjelm BE, Rollins B, Mamdani F, Lauterborn JC, Kirov G, Lynch G, et al. (2015): Evidence of Mitochondrial Dysfunction within the Complex Genetic Etiology of Schizophrenia. Mol Neuropsychiatry. 1:201–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Arion D, Corradi JP, Tang S, Datta D, Boothe F, He A, et al. (2015): Distinctive transcriptome alterations of prefrontal pyramidal neurons in schizophrenia and schizoaffective disorder. Mol Psychiatry. 20:1397–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schulmann A, Ryu E, Goncalves V, Rollins B, Christiansen M, Frye MA, et al. (2019): Novel Complex Interactions between Mitochondrial and Nuclear DNA in Schizophrenia and Bipolar Disorder. Mol Neuropsychiatry. 5:13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Elston GN (2006): Specialization of the Neocortical Pyramidal Cell during Primate Evolution. In: Kaas JH, Striedter GF, Bullock TH, Preuss TM, Rubenstein J, Krubutzer LA, editors. Evolution of Nervous Systems: Oxford: Academic Press, pp 191–242. [Google Scholar]
- 79.Elston GN, Benavides-Piccione R, Elston A, Zietsch B, Defelipe J, Manger P, et al. (2006): Specializations of the granular prefrontal cortex of primates: implications for cognitive processing. Anat Rec A Discov Mol Cell Evol Biol. 288:26–35. [DOI] [PubMed] [Google Scholar]
- 80.Elston GN, Benavides-Piccione R, Elston A, Manger PR, Defelipe J (2011): Pyramidal cells in prefrontal cortex of primates: marked differences in neuronal structure among species. Front Neuroanat. 5:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lencz T, Malhotra A (2015): Targeting the schizophrenia genome: a fast track strategy from GWAS to clinic. Mol Psychiatry. 20:820–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stephan AH, Barres BA, Stevens B (2012): The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci 35:369–389. [DOI] [PubMed] [Google Scholar]
- 83.Myers SJ, Yuan H, Kang JQ, Tan FCK, Traynelis SF, Low CM (2019): Distinct roles of GRIN2A and GRIN2B variants in neurological conditions. F1000Res. 8:F1000 Faculty Rev-1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Di Prisco S, Merega E, Bonfiglio T, Olivero G, Cervetto C, Grilli M, et al. (2016): Presynaptic, release-regulating mGlu2 -preferring and mGlu3 -preferring autoreceptors in CNS: pharmacological profiles and functional roles in demyelinating disease. Br J Pharmacol 173:1465–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jin LE, Wang M, Galvin VC, Lightbourne TC, Conn PJ, Arnsten AFT, et al. (2018): mGluR2 vs. mGluR3 in Primate Prefrontal Cortex: Postsynaptic mGluR3 Strengthen Cognitive Networks. Cerebral Cortex. 28:974–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Saini SM, Mancuso SG, Mostaid MS, Liu C, Pantelis C, Everall IP, et al. (2017): Meta-analysis supports GWAS-implicated link between GRM3 and schizophrenia risk. Transl Psychiatry. 7:e1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ghose S, Gleason KA, Potts BW, Lewis-Amezcua K, Tamminga CA (2009): Differential expression of metabotropic glutamate receptors 2 and 3 in schizophrenia: a mechanism for antipsychotic drug action? Am J Psychiatry. 166:812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Arnsten AFT, Wang M (2020): The Evolutionary Expansion of mGluR3-NAAG-GCPII Signaling: Relevance to Human Intelligence and Cognitive Disorders. Am J Psychiatry. 177:1103–1106. [DOI] [PubMed] [Google Scholar]
- 89.Egan MF, Straub RE, Goldberg TE, Yakub I, Callicott JH, Hariri AR, et al. (2004): Variation in GRM3 affects cognition, prefrontal glutamate, and risk for schizophrenia. Proc Natl Acad Sci U S A. 101:12604–12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zink C, Barker P, Sawa A, Weinberger D, Wang A, Quillian H, et al. (2020): Missense Mutation in FOLH1 is Associated with Decreased NAAG Levels and Impaired Working Memory Circuitry and Cognition. Am J Psychiatry. 177:1129–1139. [DOI] [PubMed] [Google Scholar]
- 91.Millar JK, Wilson-Annan JC, Anderson SL, Christie S, Taylor MS, Semple CA, et al. (2000): Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 9:1415–1423. [DOI] [PubMed] [Google Scholar]
- 92.Millar JK, Pickard BS, Mackie S, James RS, Christie S, Buchanan SR, et al. (2005): DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science. 310:1187–1191. [DOI] [PubMed] [Google Scholar]
- 93.Arnsten AF, Jin LE (2012): Guanfacine for the treatment of cognitive disorders: a century of discoveries at Yale. Yale J Biol Med.45–58. [PMC free article] [PubMed] [Google Scholar]
- 94.Gamo NJ, Duque A, Paspalas CD, Kata A, Fine R, Boven L, et al. (2013): Role of Disrupted in Schizophrenia 1 (DISC1) in stress-induced prefrontal cognitive dysfunction. Translational Psychiatry. 3:e328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vacic V, McCarthy S, Malhotra D, Murray F, Chou HH, Peoples A, et al. (2011): Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature. 47:499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Datta D, Mentone SA, Morozov Y, Arnsten A (2021): Subcellular Localization of Schizophrenia Risk Genes Encoding Cav1. 2 (CACNA1C) and VIPR2 in Rhesus Macaque Dorsolateral Prefrontal Cortex. Biological Psychiatry. 89:S308. [Google Scholar]
- 97.Datta D, Mentone SA, Morozov YM, Arnsten AFT (2021): VPAC2 receptors (VIPR2), a risk factor for schizophrenia, are concentrated on dendritic spines in layer III of the dorsolateral prefrontal cortex. Schizophrenia. in submission. [Google Scholar]
- 98.Andrade A, Brennecke A, Mallat S, Brown J, Gomez-Rivadeneira J, Czepiel N, et al. (2019): Genetic Associations between Voltage-Gated Calcium Channels and Psychiatric Disorders. Int J Mol Sci. 20:3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yoshimizu T, Pan JQ, Mungenast AE, Madison JM, Su S, Ketterman J, et al. (2015): Functional implications of a psychiatric risk variant within CACNA1C in induced human neurons. Mol Psychiatry. 20:162–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bhat S, Dao DT, Terrillion CE, Arad M, Smith RJ, Soldatov NM, et al. (2012): CACNA1C (Cav1.2) in the pathophysiology of psychiatric disease. Prog Neurobiol. 99:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ripke S, O’Dushlaine C, Chambert K, Moran JL, Kähler AK, al. e (2013): Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 45:1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gordovez FJA, McMahon FJ (2020): The genetics of bipolar disorder. Mol Psychiatry. 25:544–559. [DOI] [PubMed] [Google Scholar]
- 103.Zink CF, Giegerich M, Prettyman GE, Carta KE, van Ginkel M, O’Rourke MP, et al. (2020): Nimodipine improves cortical efficiency during working memory in healthy subjects. Transl Psychiatry.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Thimm M, Kircher T, Kellermann T, Markov V, Krach S, Jansen A, et al. (2011): Effects of a CACNA1C genotype on attention networks in healthy individuals. Psychol Med. 41:1551–1561. [DOI] [PubMed] [Google Scholar]
- 105.Cosgrove D, Mothersill O, Kendall K, Konte B, Harold D, Giegling I, et al. (2017): Cognitive Characterization of Schizophrenia Risk Variants Involved in Synaptic Transmission: Evidence of CACNA1C’s Role in Working Memory. Neuropsychopharmacology. 42:2612–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Catterall WA (2015): Regulation of Cardiac Calcium Channels in the Fight-or-Flight Response. Curr Mol Pharmacol. 8:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pardiñas AF, Holmans PA, Pocklington AJ, Escott-Price V, Ripke S, Carrera N, et al. (2018): Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet 50:381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Greenwood TA, Lazzeroni LC, Maihofer AX, Swerdlow NR, Calkins ME, Freedman R, et al. (2019): Genome-wide Association of Endophenotypes for Schizophrenia From the Consortium on the Genetics of Schizophrenia (COGS) Study. JAMA Psychiatry. 76:1274–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Refisch A, Chung HY, Komatsuzaki S, Schumann A, Mühleisen TW, Nöthen MM, et al. (2020): A common variation in HCN1 is associated with heart rate variability in schizophrenia,. Schizophrenia Research. 229:73–79. [DOI] [PubMed] [Google Scholar]
- 110.Prasad KM, Chowdari KV, D’Aiuto LA, Iyengar S, Stanley JA, Nimgaonkar VL (2018): Neuropil contraction in relation to Complement C4 gene copy numbers in independent cohorts of adolescent-onset and young adult-onset schizophrenia patients-a pilot study. Transl Psychiatry. 8:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Breier A, Wolkowitz O, Pickar D (1991): Stress and schizophrenia: Advances in neuropsychiatry and psychopharmacology. In: Tamminga C, Schult S, editors. Schizophrenia Research. New York: Raven Press, Ltd. [Google Scholar]
- 112.Parrott JM, O’Connor JC (2015): Kynurenine 3-Monooxygenase: An Influential Mediator of Neuropathology. Front Psychiatry. 6:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gál EM, Sherman AD (1980): L-kynurenine: its synthesis and possible regulatory function in brain. Neurochem Res 5:223–239. [DOI] [PubMed] [Google Scholar]
- 114.Pullan LM, Cler JA (1989): Schild plot analysis of glycine and kynurenic acid at the N-methyl-D-aspartate excitatory amino acid receptor. Brain Res 497:59–63. [DOI] [PubMed] [Google Scholar]
- 115.Albuquerque EX, Schwarcz R (2013): Kynurenic acid as an Antagonist of α7 Nicotinic Acetylcholine Receptors in the Brain: Facts and Challenges. Biochem Pharmacol 85:1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Plitman E, Iwata Y, Caravaggio F, Nakajima S, Chung JK, Gerretsen P, et al. (2017): Kynurenic Acid in Schizophrenia: A Systematic Review and Meta-analysis Schizophrenia Bulletin. 43:764–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sathyasaikumar KV, Stachowski EK, Wonodi I, Roberts RC, Rassoulpour A, McMahon RP, et al. (2011): Impaired kynurenine pathway metabolism in the prefrontal cortex of individuals with schizophrenia. Schizophr Bull. 37:1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kindler J, Lim CK, Weickert CS, Boerrigter D, Galletly C, Liu D, et al. (2020): Dysregulation of kynurenine metabolism is related to proinflammatory cytokines, attention, and prefrontal cortex volume in schizophrenia. Mol Psychiatry. 25:2860–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schwarcz R, Pellicciari R (2002): Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. J Pharmacol Exp Ther. 303:1–10. [DOI] [PubMed] [Google Scholar]
- 120.Blanco-Ayala T, Sathyasaikumar KV, Uys JD, Pérez-de-la-Cruz V, Pidugu LS, Schwarcz R (2020): N-Acetylcysteine Inhibits Kynurenine Aminotransferase II. Neuroscience. 444:160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vornov JJ, Hollinger KR, Jackson PF, Wozniak KM, Farah MH, Majer P, et al. (2016): Still NAAG’ing After All These Years: The Continuing Pursuit of GCPII Inhibitors. Adv Pharmacol. 76:215–255. [DOI] [PubMed] [Google Scholar]
- 122.Datta D, Leslie SN, Woo E, Amancharla N, Elmansy A, Lepe M, et al. (2021): Glutamate Carboxypeptidase II in aging rat prefrontal cortex impairs working memory performance. Frontiers in Aging Neuroscience. 13:760270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang Z, Bassam B, Thomas AG, Williams M, Liu J, Nance E, et al. (2016): Maternal inflammation leads to impaired glutamate homeostasis and up-regulation of glutamate carboxypeptidase II in activated microglia in the fetal/newborn rabbit brain. Neurobiology of Disease 94:116–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Duraisamy S, Bajpai M, Bughani U, Dastidar SG, Ray A, Chopra P (2008): MK2: a novel molecular target for anti-inflammatory therapy. Expert Opin Ther Targets. 12:921–936. [DOI] [PubMed] [Google Scholar]
- 125.MacKenzie KF, Wallace DA, Hill EV, Anthony DF, Henderson DJ, Houslay DM, et al. (2011): Phosphorylation of cAMP-specific PDE4A5 (phosphodiesterase-4A5) by MK2 (MAPKAPK2) attenuates its activation through protein kinase A phosphorylation. Biochem J 435:755–769. [DOI] [PubMed] [Google Scholar]
- 126.Houslay KF, Christian F, MacLeod R, Adams DR, Houslay MD, Baillie GS (2017): Identification of a multifunctional docking site on the catalytic unit of phosphodiesterase-4 (PDE4) that is utilised by multiple interaction partners. Biochem J 474:597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Li JT, Xie XM, Yu JY, Sun YX, Liao XM, Wang XX, et al. (2017): Suppressed Calbindin Levels in Hippocampal Excitatory Neurons Mediate Stress-Induced Memory Loss. Cell Rep. 21:891–900. [DOI] [PubMed] [Google Scholar]