

Abstract
Recessive dystrophic epidermolysis bullosa (RDEB) is a severe skin fragility disorder caused by loss-of-function mutations in the COL7A1 gene, which encodes type VII collagen (C7), a protein that functions in skin adherence. From 36 Korean RDEB patients, we identified a total of 69 pathogenic mutations (40 variants without recurrence), including point mutations (72.5%) and insertion/deletion mutations (27.5%). For fibroblasts from two patients (Pat1 and Pat2), we applied adenine base editors (ABEs) to correct the pathogenic mutation of COL7A1 or to bypass a premature stop codon in Pat1-derived primary fibroblasts. To expand the targeting scope, we also utilized prime editors (PEs) to correct the COL7A1 mutations in Pat1- and Pat2-derived fibroblasts. Ultimately, we found that transfer of edited patient-derived skin equivalents (i.e., RDEB keratinocytes and PE-corrected RDEB fibroblasts from the RDEB patient) into the skin of immunodeficient mice led to C7 deposition and anchoring fibril formation within the dermal-epidermal junction, suggesting that base editing and prime editing could be feasible strategies for ex vivo gene editing to treat RDEB.
Keywords: recessive dystrophic epidermolysis bullosa, CRISPR, genome editing, base editing, prime editing, type VII collagen, COL7A1
Graphical abstract
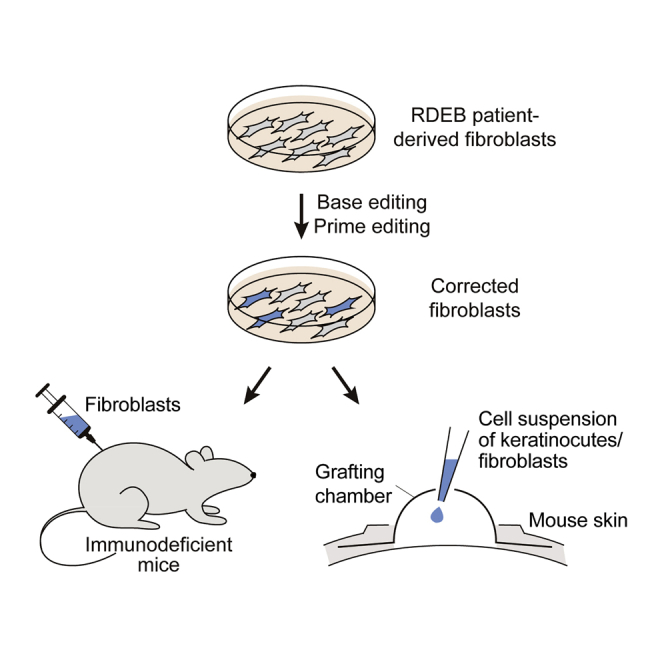
Hong et al. demonstrate adenine base editing and prime editing to correct the COL7A1 gene in recessive dystrophic epidermolysis bullosa (RDEB) patient-derived fibroblasts and keratinocytes. They ultimately transferred edited patient-derived skin equivalents into the skin of immunodeficient mice, leading to C7 deposition and anchoring fibril formation within the dermal-epidermal junction.
Introduction
Epidermolysis bullosa (EB) is a heterogeneous group of genodermatoses characterized by mucocutaneous fragility. Recessive dystrophic EB (RDEB), one of the most severe subtypes of EB, results from biallelic mutations in the COL7A1 gene, which encodes the α1 chain of type VII collagen (C7). C7 is a key component of anchoring fibrils, which create a strong attachment between the epidermis and the dermis. Loss of C7 causes extensive mucocutaneous blistering, scarring, and extracutaneous complications, leading to considerable morbidity and occasional mortality.1,2 Therefore, effective treatments are urgently needed. To date, various therapeutic strategies, including protein replacement,3 disease-modifying drugs,4,5 and allogeneic cell-based therapies using fibroblasts,6,7 mesenchymal stromal cells,8, 9, 10, 11 and bone marrow transplantation,12 have been studied, but a complete cure is not achievable with those current approaches. As a potential long-lasting therapeutic option, remarkable progress has been made in gene therapy that aims to transfer the normal COL7A1 gene into deficient cells from RDEB patients. In this strategy, it is advantageous to use autologous cells for gene transfer. Early phase clinical trials using viral vectors such as lentivirus and retrovirus to transduce COL7A1 into autologous fibroblasts and keratinocytes have resulted in C7 restoration in the treated skin of some RDEB patients for more than 1 or 2 years after treatment.13, 14, 15 Despite these promising results, such viral vector-based gene therapy has potential concerns: (1) random integration of viral vectors into the host genome that could cause tumorigenesis16, 17, 18 and frequently induce aberrant transcripts resulting in a dominant negative-like inhibition of wild-type protein19,20 and (2) different levels of constitutive expression from the virus-delivered exogenous gene regardless of the cellular environment.
To overcome these limitations, therapeutic editing of the endogenous COL7A1 gene in patients’ autologous cells via genome editing tools has been suggested for RDEB treatment.21, 22, 23, 24, 25, 26, 27, 28, 29 Conventional CRISPR nucleases rely on double-strand breaks (DSBs) in the target DNA, which are repaired by one of the cell’s repair systems, such as non-homologous end joining (NHEJ) or homology-directed repair (HDR).30,31 Previously, several groups demonstrated the feasibility of correcting the reading frame25,26 or skipping a mutant exon27, 28, 29 in mutant COL7A1 using CRISPR-coupled NHEJ repair. However, these strategies have limited value for the correction of point mutations, the most common type of mutation in RDEB. In contrast, the use of HDR enables the precise correction of point mutations, but its low editing efficacy, requirement for donor templates, and limited activity in non-dividing cells are obstacles for HDR-mediated approaches. Furthermore, recent studies have revealed that CRISPR nuclease-mediated DSBs can induce unwanted large deletions, chromosomal rearrangements,32,33 and a p53-mediated DNA damage response34,35 that results in cell death, potentially inhibiting further clinical applications.
To bypass such risks, newly developed tools that generate few DSBs, such as base editors (BEs) and prime editors (PEs), can be used.36, 37, 38, 39, 40 BEs, which include cytosine BEs (CBEs)41 and adenine BEs (ABEs),42 can convert one target nucleotide into another, C-to-T or A-to-G, by catalyzing cytosine or adenine deamination, respectively. A recent report described an ABE-mediated strategy in which two nonsense mutations in COL7A1 were corrected ex vivo in RDEB patient-derived fibroblasts and induced pluripotent stem cells, resulting in therapeutic effects such as C7 restoration.43 However, despite their therapeutic potential, BEs have limited ability to correct small insertion and deletion (indel) mutations or transversion mutations such as C-to-G/A and A-to-C/T. Alternatively, PEs can generate all types of substitutions and indels within about 40 nucleotides. A practical version of PE, PE2, which consists of a Cas9 nickase (nCas9) that contains an H840A mutation and an engineered reverse transcriptase, is recruited to the target site by a prime editing guide RNA (pegRNA).44 The pegRNA is composed of a standard single-guide RNA (sgRNA) and an extension sequence at the 3′ end that includes a primer binding site (PBS) and a reverse transcription template (RTT) that encodes the desired correction. To maximize PE efficacy, PE3 employs an additional nicking sgRNA (ngRNA) for inducing a second nick in the non-edited strand. However, prime editing has not yet been demonstrated for RDEB treatment.
In this study, we established a COL7A1 mutation database from a large cohort of South Korean patients with RDEB and analyzed the percentage of mutations that are potentially targetable by BEs and PEs. We then applied either ABE or PE3 to correct the mutations in primary fibroblasts from two patients with highly recurrent COL7A1 mutations. We further transplanted the ABE-/PE-corrected primary fibroblasts into immunodeficient mice and observed strong linear deposition of human C7 at the dermal-epidermal junction (DEJ), supporting the therapeutic potential of ex vivo ABE- or PE-mediated gene editing for treating RDEB.
Results
Establishment and analysis of a COL7A1 mutation database specific for Korean RDEB patients
Using an in silico approach, we first inspected all known RDEB-associated COL7A1 variations among the worldwide population of patients and determined which ones would be targetable with BEs and PEs. According to the COL7A1 variants database (http://www.col7a1-database.info), a total of 810 COL7A1 gene variants causing RDEB are currently registered, of which 23.6% are indel mutations and 76.4% are point mutations, including nonsense, missense, synonymous, and intron mutations (Figure 1A). Among the COL7A1 point mutations, 3.0% (i.e., A > G or T > C) and 26.3% (i.e., G > A or C > T), respectively, can theoretically be corrected by CBEs and ABEs derived from SpCas9 (Cas9 from Streptococcus pyogenes), which recognize a canonical 5′-NGG-3′ protospacer-adjacent motif (PAM). When NG-PAM-targetable BEs are used instead, 6.0% and 37.8% of mutations are covered by CBEs and ABEs, respectively.45 Moreover, when PAM-less Cas9 (i.e., 5′-NNN-3′ PAM) is used for base editing, 6.5% and 40.0% of mutations are covered by CBEs and ABEs, respectively.46 In contrast, given that PEs can correct all types of point mutations as well as indel mutations, 96.7% of the mutations can potentially be corrected by NG- or NNN-PAM-targetable PEs (Figure 1A). Similarly, we further investigated the editing scope of both BEs and PEs for correcting pathogenic COL7A1 mutations found in Korean patients suffering from RDEB. To this end, using information from the only EB referral center in South Korea, we established the largest database of COL7A1 mutations in Korean RDEB patients and identified a total of 69 pathogenic mutations (40 variants without recurrence) from a total of 36 patients. Of the 40 mutations, 72.5% were point mutations, including missense (35.0%), nonsense (25.0%), and intron (12.5%) mutations, whereas 27.5% were indel mutations (Figure 1B and Table S1). Among the point mutations, 27.5% can theoretically be corrected by NGG-PAM-targetable ABEs, and 42.5% can theoretically be corrected by NG- or NNN-PAM-targetable ABEs. When PEs were considered, we found that 97.5% of the mutations would be covered by NG- or NNN-PAM-targetable PEs (Figure 1B), consistent with the situation for the worldwide population of patients.
Figure 1.

Establishment of a COL7A1 mutation database specific for Korean RDEB patients and analysis of Korean and worldwide databases
(A) The full spectrum of pathogenic COL7A1 mutations reported to date in DEB patients. Frequencies (%) of COL7A1 alleles that can theoretically be corrected with CBE, ABE, and PE constructed with SpCas9 (NGG-PAM) or its variant with a relaxed PAM requirement (NG-PAM) are shown. (B) Mutational analysis of COL7A1 in a large cohort of Korean RDEB patients. Frequencies (%) of COL7A1 alleles that can theoretically be corrected with ABE and PE recognizing NGG- or NG-PAMs are shown. (C) The five most frequent pathogenic COL7A1 mutations in our database among the 36 Korean RDEB patients with 69 pathogenic mutations. (D) Information about the two RDEB patients enrolled in this study and a schematic representation of procollagen VII showing the locations of the COL7A1 mutations identified in these patients. (E) Immunofluorescence to visualize C7 was performed on skin samples from Pat1, Pat2, and healthy controls using polyclonal rabbit anti-COL7 antibody. The white dotted line indicates the DEJ. White arrows indicate C7 deposited at the DEJ. Representative images are shown. Scale bars, 50 μm. Epi, epidermis; Der, dermis.
In the Korean RDEB database, c.8569G > T (p.Glu2857Ter), c.2005C > T (p.Arg669Ter), and c.3631C > T (p.Gln1211Ter) were the most recurrent RDEB-causing COL7A1 mutations, representing 14.5% (10/69), 7.2% (5/69), and 7.2% (5/69) of the mutant alleles, respectively (Figure 1C). Among these, c.2005C > T in exon 15 and c.3631C > T in exon 27 affect the amino-terminal non-collagenous NC-1 domain and have been reported to induce nonsense-mediated decay of COL7A1 transcripts.47,48 In addition, these two nonsense mutations have been reported to be responsible for severe generalized RDEB (Figure 1D).48,49 Therefore, we focused on these two nonsense mutations in our cohort as targets for correction via ABEs or PEs. Two patients with severe RDEB who had compound heterozygous COL7A1 nonsense mutations were enrolled in this study; patient 1 (Pat1, hereafter) carried a c.3631C > T and c.8569G > T mutation on each allele and patient 2 (Pat2, hereafter) carried a c.2005C > T and c.8569G > T mutation on each allele, which were confirmed by Nanopore sequencing (Figures 1D and S1). Skin biopsies from both patients showed only trace staining of C7 by immunofluorescence microscopy using antibodies against the NC-1 domain, whereas skin samples from healthy donors showed clear C7 staining at the DEJ (Figure 1E).
Adenine base editing for COL7A1 gene correction in Pat1-derived fibroblasts
To correct the C > T nonsense mutations, we first used the ABE system and prepared an optimized version of ABE7.10, named ABEmax.50 We used two strategies to rescue COL7A1 gene function in Pat1-derived fibroblasts: (1) direct correction of the mutated nucleotide (i.e., c.3631C > T) using sgRNA#1 (Pat1-sg1, hereafter) and (2) readthrough of the premature stop codon (PTC) using a method called CRISPR-pass,51 which involved editing the neighboring sequences using sgRNA#2 (Pat1-sg2, hereafter) (Figure 2A). Using Pat1-sg1, correction of the pathogenic mutation at position 5 (counting from the 5′ end of the target sequence) in the protospacer sequence would occur by conversion of adenine to guanine in the template strand, resulting in T-to-C correction on the coding strand. Using Pat1-sg2, the PTC (5′-TAG-3′) caused by c.3631C > T can be converted to 5′-TGG-3′, which will be translated to tryptophan (Trp), leading to restoration of the COL7A1 reading frame. Because this amino acid change was predicted to have no deleterious effects on C7 (PROVEAN score < −2.5; PredictProtein score >50), we hypothesized that Pat1-sg2-induced PTC readthrough could contribute to C7 restoration despite this amino acid change.
Figure 2.

Correction of c.3631C > T (Q1211X) in COL7A1 using ABEs in primary RDEB fibroblasts
(A) Schematic diagram of the ABE target sequence in exon 27 of the COL7A1 gene containing a C > T nonsense mutation (c.3631C > T, p.Q1211X). The mutant sequence is shown in red. Target sequences are highlighted in bold type, and PAM sequences are underlined. A·T to G·C conversions in the ABE editing window are shown in green. (B) Heatmap visualizing A·T to G·C conversion rates analyzed by high-throughput sequencing (top) and the sequences at the target sites together with their proportions (middle and bottom). The five most common sequences in ABE-treated RDEB fibroblasts are shown, and the frequencies of normal and mutated alleles in the non-edited patient-derived fibroblasts are shown at the top. PAM sequences are underlined, and the target A·T is indicated by an arrowhead. (C and D) Conversion rates calculated by deep sequencing of genomic DNA (C) and mRNA (D) from RDEB fibroblasts treated with different sgRNAs. (E) Western blots to measure C7 abundance in cell lysates and culture supernatants using GAPDH and Ponceau as internal controls, respectively. Images are representative of three independent experiments. Protein band densities from three independent experiments are presented as bar graphs. Each density value was normalized to the loading control value and expressed relative to the value in NHDFs. Data are the mean ± SEM (n = 3); ∗∗p < 0.01, ∗∗∗p < 0.001. (F) Immunofluorescence staining to visualize the C7 protein (green) in NHDFs, non-edited RDEB fibroblasts from Pat1, and ABE-treated RDEB fibroblasts. Nuclei were stained with DAPI (blue). Shown are representative images from at least three independent cell samples. Scale bars, 50 μm. (G) Representative graphics of flow cytometry assay for C7 restoration and quantitation of the percentage of C7-positive cells (n = 2). Histograms for unstained control and secondary antibody control for each cell samples are also shown. (H) Trypsin-based cell detachment assay. Cell adhesion is represented as the percentage of cells that remain attached after the indicated period of trypsin treatment. Five independent experiments were performed. Data are the mean ± SEM; ∗p < 0.05, ∗∗p < 0.01. (I) The proliferation of NHDFs, non-edited fibroblasts from Pat1, and ABE-treated RDEB fibroblasts was evaluated by the WST-1 assay. The ratio of the absorbance at 24, 48, and 72 h to that at 0 h is shown. Five independent experiments were performed. Data are the mean ± SEM; ∗p < 0.05. Statistical analyses were performed using unpaired Student’s t test for (E), (H), and (I) (NHDF versus Pat1 group) and one-way ANOVA followed by Dunnett’s tests for (E), (H), and (I) (each group compared with Pat1).
Pat1-derived fibroblasts were then transfected with the ABEmax-encoding plasmid and each sgRNA-encoding plasmid by electroporation, maintained at least for 3 days, and harvested at 80%–90% confluency. Genomic DNA was isolated from the bulk population of cells and subjected to high-throughput sequencing for the assessment of base editing outcomes. The sequencing results showed that, through strategy (1) using Pat1-sg1, the target T (T5) was efficiently converted to C at a frequency of 30.6%, whereas bystander Ts (T7 and T9) were also edited at frequencies of 44.5% and 5.5%, respectively (Figures 2B and 2C). On the other hand, through strategy (2) using Pat1-sg2, the target A (A6) and a bystander A (A8) were converted at frequencies of 24.6% and 1.8%, respectively (Figures 2B and 2C). The frequencies of indels generated by ABE/Pat1-sg1 and ABE/Pat1-sg2 were 3.3% and 0.1%, respectively (Figure 2C). We further assessed the frequencies of COL7A1 editing outcomes at the mRNA level using complementary DNAs (cDNAs). We found that the target sequences were edited at rates that were higher than that in genomic DNA, similar to findings from previous studies (Figures 2D and S2A).43,52,53
Next, we evaluated C7 expression in ABE-treated RDEB fibroblasts from Pat1. Western blot analysis of bulk populations of such cells revealed the restoration of the full-length C7 protein, at levels that were up to 72% (Pat1-sg1) and 50% (Pat1-sg2) of the level in normal human dermal fibroblasts (NHDFs), whereas uncorrected cells showed barely detectable C7 protein (Figures 2E and S3). The amount of C7 released into the culture supernatant of the RDEB fibroblasts was also increased following ABE treatment, to up to 46% (Pat1-sg1) and 48% (Pat1-sg2) of the levels seen in the NHDF supernatant (Figures 2E and S3). Immunocytochemistry of C7 confirmed these findings and revealed increased C7 protein expression in the cytoplasm of ABE-treated RDEB fibroblasts (Figure 2F). Flow cytometric analysis revealed that 43.3% and 35.2% of the ABE/Pat1-sg1- and ABE/Pat1-sg2-treated RDEB fibroblasts expressed C7, respectively (Figure 2G). It was previously reported that RDEB fibroblasts exhibited decreased adhesion ability due to C7 deficiency, and that viral vector-mediated transduction of the full-length human COL7A1 gene restored their adhesion capacity.54,55 Thus, we further evaluated the adhesion properties of the ABE-treated RDEB fibroblasts using a trypsin-based cell detachment assay. Uncorrected RDEB fibroblasts showed poor cell adhesion, with 59%, 37%, and 19% of cells adhering at 2, 4, and 6 min after trypsin treatment, compared with 92%, 51%, and 40% of NHDFs. However, ABE/Pat1-sg2 treatment increased the percentage of adherent cells over untreated RDEB fibroblasts by 35%, 40%, and 32% at 2, 4, and 6 min. ABE/Pat1-sg1 treatment also increased the percentage of adherent cells by 42% after 6 min trypsinization compared with untreated RDEB fibroblasts (Figure 2H). We also tested the effect of COL7A1 correction on the ability of RDEB fibroblasts to proliferate using a mitochondrial activity assay (WST-1 assay). The RDEB fibroblasts showed lower rates of proliferation than did NHDFs, but ABE/Pat1-sg1-treated RDEB fibroblasts showed enhanced cell proliferation compared with uncorrected cells (Figure 2I). We found that ABE/Pat1-sg2-treated cells enhanced their adhesion strength more effectively, whereas ABE/Pat1-sg1-treated cells showed stronger enhancement of growth potential, which might imply the different effect between ABE/Pat1-sg1 (direct correction) and ABE/Pat1-sg2 strategies (CRISPR-pass). Taken together, our results indicate that both ABE-mediated strategies are relevant for gene rescue in Pat1-derived cells.
Prime editing for COL7A1 gene correction in both Pat1- and Pat2-derived fibroblasts
We next investigated the potential use of prime editing for correcting the two nonsense mutations (i.e., c.2005C > T and c.3631C > T) in the COL7A1 gene. Because PEs have a more flexible targeting scope than BEs, we sought to apply PEs for correcting both mutations. In this experiment, we used the PE3 system because of its enhanced editing efficiency compared with that of PE2. We designed pegRNAs that could correct the nonsense mutation and also induce silent mutations in the PAM sequences, because it was previously reported that such mutations in the PAM enhance the editing efficiency and reduce indel generation by inhibiting repetitive PE binding after the initial editing. We first used a pegRNA containing a 13-nt PBS and 14-nt RTT together with an ngRNA. For Pat1, pegRNA#1 (Pat1-peg1, hereafter) was designed for the correction of c.3631C > T; a T-to-C conversion would occur at position +10 (10 nt downstream from the nick site) and the ngRNA would lead to the generation of a nick 60 nt downstream of the pegRNA-induced nick (Figures 3A and S2B). For Pat2, pegRNA#2 (Pat2-peg2, hereafter) was designed for the correction of c.2005C > T; in this case, a T-to-C conversion would occur at position +12 (12 nt downstream from the nick site) and the ngRNA would direct the formation of a nick in the non-edited strand at a position 56 nt upstream of the pegRNA-induced nick site (Figures 3B and S2B). Then, PE2-, pegRNA-, and ngRNA-encoding plasmids were transfected into Pat1- or Pat2-derived primary fibroblasts via electroporation. At least 3 days after, the cells at 80%–90% confluency were harvested for assessment of the editing efficiency. Genomic DNA from the bulk population of cells was subjected to high-throughput sequencing. The results showed that the average prime editing efficiencies were 10.5% at c.3631C > T with PE3/Pat1-peg1 (Figures 3C) and 5.2% at c.2005C > T with PE3/Pat2-peg2 (Figure 3D). The average indel frequencies were 1.5% for PE3/Pat1-peg1 and 0.7% for PE3/Pat2-peg2 (Figure 3E). When we tested various pegRNAs with different PBS lengths (i.e., 11 and 15 nt), pegRNAs with a 13-nt PBS led to editing activity that was comparable to that seen with the other pegRNAs (Figure 3E).
Figure 3.

PE3-mediated correction of c.3631C > T (Q1211X) and c.2005C > T (R669X) COL7A1 in primary fibroblasts derived from two RDEB patients
(A and B) Schematic diagram of the pegRNA and ngRNA target sites in the COL7A1 gene in Pat1 (A) and Pat2 (B). Target sequences are highlighted in bold type, and PAM sequences are underlined. The pathogenic mutations, converted pathogenic mutations, and synonymous mutations for PAM disruption are shown in red, blue, and green, respectively (top). A 14-nt RTT and a 13-nt PBS were used for pegRNAs (bottom). (C and D) Heatmaps visualizing conversion rates determined by high-throughput sequencing. (E) Prime editing efficiencies and indel frequencies in the target sequence in the patient-derived fibroblasts transfected with various pegRNAs with different PBS lengths (n = 1–3). (F) Conversion rates in mRNA from PE-treated RDEB fibroblasts. (G) Western blot to measure C7 abundance in cell lysates and culture supernatants using GAPDH and Ponceau as internal controls, respectively. Images are representative of three independent experiments. Protein band densities from three independent experiments are presented as bar graphs. Each density value was normalized to the loading control value and expressed relative to the value in NHDFs. Data are the mean ± SEM (n = 3); ∗∗p < 0.01, ∗∗∗p < 0.001. (H) Immunofluorescence staining to visualize the C7 protein (green) in NHDFs, non-edited RDEB fibroblasts from Pat1, and PE3-treated RDEB fibroblasts. Nuclei were stained with DAPI (blue). Shown are representative images from three independent cell samples. Scale bars, 50 μm. (I) Representative graphics of flow cytometry assay for C7 restoration and quantitation of the percentage of C7-positive cells (n = 2). Histograms for unstained control and secondary antibody control for each cell samples are also shown. (J) Trypsin-based cell detachment assay. Cell adhesion is represented as the percentage of cells that remain attached after the indicated period of trypsin treatment. Five independent experiments were performed. Data are the mean ± SEM; ∗p < 0.05, ∗∗p < 0.01. (K) The proliferation of NHDFs, non-edited fibroblasts from Pat1, and PE3-treated RDEB fibroblasts was evaluated by the WST-1 assay. The ratio of the absorbance at 24, 48, and 72 h to that at 0 h is shown. Five independent experiments were performed. Data are the mean ± SEM; ∗p < 0.05, ∗∗p < 0.01. Statistical analyses were performed using two-tailed, unpaired Student’s t test for (G), (J), and (K) comparing NHDF versus Pat1 group and Pat1 group versus Pat1-peg1 group.
To investigate whether correction of COL7A1 by the PE3 system can restore functional C7 in RDEB fibroblasts, we selected PE-treated fibroblasts derived from Pat1 because the editing efficiency was higher than that of PE-treated fibroblasts derived from Pat2. Similar to the above experiments using ABEs, we assessed the editing efficiency in cDNAs and found that the correction frequency in cDNAs was consistently higher than that in genomic DNA (Figures 3F and S2C). Western blot analysis showed that PE-treated RDEB fibroblasts expressed increased levels of the C7 protein, to up to 55% and 37% of the level in NHDFs in cell lysates and culture supernatant, respectively (Figures 3G and S4). In addition, immunocytochemistry confirmed efficient expression of the C7 protein in PE-treated RDEB fibroblasts, whereas the unedited cells showed no antibody reactivity (Figure 3H). Flow cytometric analysis also revealed that 25.2% of PE-treated RDEB fibroblasts expressed C7 (Figure 3I). We further evaluated the adhesion properties of the PE-treated RDEB fibroblasts by the trypsin-based cell detachment assay. PE treatment increased the percentage of adherent cells over untreated RDEB fibroblasts by 35%, 46%, and 37% at 2, 4, and 6 min (Figure 3J). We also tested the effect of COL7A1 correction on the proliferation ability of RDEB fibroblasts using the WST-1 assay. RDEB fibroblasts carrying p.Gln1211Ter and p.Glu2857Ter showed lower rates of proliferation than NHDFs, but PE-treated fibroblasts showed enhanced cell proliferation compared with uncorrected cells (Figure 3K). These findings indicate that COL7A1 correction by PE can effectively rescue the impaired adhesion and proliferation properties of RDEB fibroblasts.
Deposition of human type VII collagen at the DEJ of immunodeficient mice injected with ABE-/PE-corrected RDEB fibroblasts
Next, we investigated whether ABE-/PE-corrected RDEB fibroblasts could synthesize and secrete human C7 that would then localize correctly at the DEJ in vivo after intradermal injection into immunodeficient mice. To this end, a single dose of 5 × 106 NHDFs, non-edited Pat1-derived RDEB fibroblasts, or Pat1-derived RDEB fibroblasts corrected using Pat1-sg1, Pat1-sg2, or Pat1-peg1 suspended in 150 μL of phosphate-buffered saline was intradermally injected into the back skin of immunodeficient mice (Figure 4A). Two weeks after injection, human C7 protein deposition at the DEJ was analyzed by immunofluorescence using anti-human COL7 antibody (kindly provided by Dr. Hiroaki Iwata, Hokkaido University Graduate School of Medicine). Human fibroblasts could be discriminated by a human-specific anti-vimentin antibody among the mouse skin sections in which human fibroblasts (NHDF, RDEB fibroblasts, and ABE-/PE-treated RDEB fibroblasts) were injected intradermally, indicating their survival for at least 2 weeks. Murine skin with the injection of phosphate-buffered saline alone showed no immunoreactivity for human C7 and vimentin, whereas NHDF-injected murine skin showed strong immunostaining for C7 along the DEJ and numerous vimentin-positive cells in the dermis. Human C7 was barely observed in the skin with the injection of uncorrected RDEB fibroblasts, but the skin with the injection of ABE-/PE-treated RDEB fibroblasts showed linear deposition of human C7 along the DEJ (Figure 4B).
Figure 4.

Correct deposition of human C7 at the DEJ in immunodeficient mice after intradermal injection of ABE- or PE-treated primary RDEB fibroblasts
(A) Scheme of the experiment in which ABE-/PE-treated patient-derived fibroblasts were injected into the mouse model. (B) Immunofluorescence staining to visualize the C7 protein (row 1). NHDFs, non-edited fibroblasts from Pat1, ABE-treated RDEB fibroblasts, PE-treated RDEB fibroblasts (5 × 106 cells/150 μL of phosphate-buffered saline), or phosphate-buffered saline were intradermally injected into the back skin of immunodeficient mice. Two weeks after the injections, immunofluorescence analysis of C7 (green) was performed using a rabbit polyclonal antibody that recognizes human C7. Immunofluorescence staining using an antibody specific for human vimentin (red) revealed the presence of human fibroblasts injected into the mouse skin (row 2). Phosphate-buffered saline-injected mouse skin was used as a negative control. The mouse experiments were repeated two times, and at least three mice were included in each cell group each time. Images of a representative mouse experiment are displayed (n = 3 repeats of each cell group in two independent experiments). The white dotted line indicates the DEJ. White arrows indicate human C7 deposited at the DEJ. Scale bars, 50 μm. Epi, epidermis; Der, dermis; Fb, fibroblasts; NHDFs, normal human dermal fibroblasts.
We also investigated whether off-target DNA editing occurred in Pat1-derived RDEB fibroblasts corrected using Pat1-sg1, Pat1-sg2, or Pat1-peg1. We carefully identified potential off-target sites using Cas-OFFinder software.56 When up to three mismatched bases or up to two mismatched bases with a DNA/RNA bulge were allowed, a total of 20 potential off-target sites for Pat1-sg1, 5 sites for Pat-sg2, and 19 sites for Pat1-peg1 were identified. High-throughput sequencing revealed that off-target editing was observed in the case of an ABE/Pat1-sg1 treated cell population. (Figure S5).
Restoration of human type VII collagen deposition and anchoring fibril formation at the DEJ in skin graft generated from patient-derived skin equivalents
To evaluate the functional consequences of PE-mediated COL7A1 gene correction in RDEB fibroblasts, we generated an in vivo full-thickness human skin equivalent composed of RDEB patient-derived keratinocytes (RDEB keratinocytes) and PE-corrected RDEB fibroblasts on an immunodeficient mouse using a grafting chamber system (Figure 5A). Normal human keratinocytes and fibroblasts grafted onto the back of the immunodeficient mice reconstituted the stratified structure of human skin, which expressed the epidermal differentiation marker keratin 1. Positive immunofluorescence staining with human-specific anti-keratin 1 and anti-vimentin antibodies in the grafts and negative staining in untreated mouse skin confirmed the human origin of the graft. Immunofluorescence staining with an antibody specific for human C7 showed strong positive staining at the DEJ in the graft composed of normal human keratinocytes and fibroblasts, in contrast to the negative staining in untreated mouse skin.
Figure 5.

Restoration of C7 expression and anchoring fibril formation at the DEJ in human skin equivalents generated from PE-treated primary RDEB fibroblasts and RDEB keratinocytes
(A) Reconstruction of full-thickness human skin equivalent on an immunodeficient mouse using a grafting chamber system. Normal human keratinocytes and fibroblasts, untransduced RDEB patient-derived keratinocytes (RDEB keratinocytes) and RDEB fibroblasts, or RDEB keratinocytes and PE-corrected RDEB fibroblasts were seeded into grafting chambers implanted on the back of immunodeficient mice (n = 2). Seventeen days post-implantation, the scab fell off, and the skin graft was reconstructed. (B) Four weeks after the reconstitution of the human skin graft, frozen sections of graft samples were subjected to immunofluorescent labeling using anti-human COL7 antibody (row 1, green fluorescence). Immunofluorescence staining using antibodies specific for human vimentin and human keratin 1 revealed the human origin of the grafts (rows 2 and 3, red fluorescence). Untreated mouse skin was used as a negative control. The mouse experiments were repeated two times, and at least three mice were included in each cell group each time. Images of a representative mouse experiment are displayed (n = 3 repeats of each cell group in two independent experiments). The white dotted line indicates the DEJ. White arrows indicate human C7 deposited at the DEJ. Scale bars, 50 μm. (C) Transmission electron microscopy of graft generated using normal human keratinocytes and fibroblasts, RDEB keratinocytes and RDEB fibroblasts, or RDEB keratinocytes and PE-corrected RDEB fibroblasts. White arrowheads, anchoring fibrils. Images are representative of two independent experiments (n = 3 repeats of each cell group). Scale bars, 500 nm. Fb, fibroblasts; IF, immunofluorescence; KC, keratinocytes; TEM, transmission electron microscopy.
We also found that skin grafts generated from RDEB keratinocytes and RDEB fibroblasts showed normal skin structures with positive staining of human-specific anti-keratin 1 and anti-vimentin antibodies, but there was no detectable human C7 deposition at the DEJ. In contrast, skin grafts composed of RDEB keratinocytes and PE-corrected RDEB fibroblasts showed increased linear staining of human C7 along the DEJ, similar to that of the graft composed of normal human keratinocytes and fibroblasts, but the staining intensity of C7 was slightly lower than that in the control graft sections (Figure 5B). In addition, while anchoring fibrils were barely detected in transmission electron microscopy (TEM) of untransduced RDEB cell combinations, skin grafts composed of RDEB keratinocytes and PE-corrected RDEB fibroblasts revealed well-formed anchoring fibrils at the sublamina densa similar to those seen in the graft composed of normal human keratinocytes and fibroblasts (Figure 5C). These results suggest that PE-corrected RDEB fibroblasts restore the expression of functional human C7 that can incorporate into the basement membrane and form anchoring fibrils.
Discussion
Genome editing has emerged as a promising molecular approach for treating genetic diseases. In this study, we first established a COL7A1 mutation database containing 69 pathogenic mutations (40 variants without recurrence) from a total of 36 Korean RDEB patients. As a proof of concept, we chose two patients having two representative COL7A1 mutations (i.e., c.3631C > T [p.Gln1211Ter] and c.2005C > T [p.Arg669Ter]), which were formerly shown to cause generalized severe RDEB48,49 and have been reported to result in nonsense-mediated mRNA decay that manifests as a complete absence of C7.47,48 We first applied ABEs to correct the mutations in primary RDEB patient-derived fibroblasts through two different strategies: direct correction of target mutations to the wild-type sequence and induction of readthrough of a PTC with the CRISPR-pass method. After electroporation of the plasmid-based delivery system, ABEmax showed an editing efficacy (24.6%–30.6% in genomic DNA and 37%–58% at the mRNA level) comparable to that in previous work by Osborn et al. in which the ABEmax-induced gene correction rate was 8.2%–23.8% in genomic DNA and 17.8%–45% at the mRNA level in primary RDEB fibroblasts.43 In addition, ABEmax induced a low rate of indels (0.1%–3.3%), similar to that in the previous research (1.5%–1.9%),43 suggesting a more reliable editing approach than HDR-mediated gene correction, which resulted in higher indel rates. This point is important because of DSB-associated safety issues.
However, BEs cannot be used to correct the disease-associated mutations in more than half of RDEB patients. Furthermore, we observed that the frequency of ABE-induced bystander edits varied over a wide range, with an upper value of 44.5% (1.8%–44.5%), depending on the width of the editing window. This effect might limit further applications of BEs for RDEB treatment. As a potential alternative, we next used PEs to correct the mutations. To the best of our knowledge, this study is the first to demonstrate the feasibility of PEs for correcting pathogenic COL7A1 mutations, including a mutation that was not suitable for correction with an ABE recognizing the canonical SpCas9 PAM, to treat RDEB. In addition, we investigated whether both BEs and PEs could be applicable for RDEB patient-derived keratinocytes. For keratinocytes derived from another harboring the c.3631C > T mutation, we showed that approximately 3% and 6% were corrected by ABEs and PEs, respectively (Figure S6), indicating that gene correction of fibroblasts and/or keratinocytes could be used for RDEB treatment.
We further found that correction of these two nonsense mutations by either ABE or PE restored the synthesis and secretion of full-length C7 in RDEB fibroblasts. ABE- and PE-mediated genetic correction also rescued the poor adhesion capacity and growth potential of RDEB fibroblasts. We ultimately observed that ABE- and PE-treated RDEB fibroblasts produced functional C7 that was correctly deposited into the DEJ at the site of injection into the skin of immunodeficient mice. Moreover, we reconstituted human skin grafts of patient-derived keratinocytes and fibroblasts on the back of immunodeficient mice and showed that the gene-corrected skin equivalent composed of patient-derived keratinocytes and PE-treated RDEB fibroblasts restored C7 deposition and anchoring fibril formation at DEJ. It has been shown that C7 levels that are 35% of that in NHDFs are sufficient to provide mechanical stability of the skin in a DEB hypomorphic murine model.57 In this study, ABE- and PE-mediated correction of p.Gln1211Ter in RDEB fibroblasts respectively restored the production of full-length C7 to levels of up to 68% and 49% of that in NHDFs; furthermore, the C7 was correctly deposited along the DEJ of the immunodeficient mouse skin. Edited RDEB fibroblasts showed enhanced proliferation compared with non-edited cells, which may explain why the levels of C7 restoration are higher than would be expected from the editing frequency at the genomic DNA level.
Collectively, our data demonstrate that both ABEs and PEs enable efficient correction of pathogenic COL7A1 mutations with higher ratios of the desired edit per indel than HDR in patient-derived primary cells, restore C7 expression to levels known to rescue the phenotype of the DEB murine model, and induce the formation of functional C7 that incorporates into the DEJ of immunodeficient mice. By using a non-viral delivery method, electroporation, we minimize safety concerns for therapeutic translation of these editing technologies to treat RDEB. Furthermore, transient expression of genome editing tools via ribonucleoprotein (RNP) complex or mRNA and/or the use of high-fidelity Cas proteins will further mitigate the off-target effects.37,58 Despite the higher editing efficacy of ABEs over PEs for correcting COL7A1 nonsense mutations, PEs would be a more plausible tool considering that PEs exhibit precise correction without bystander edit, the ability to target almost all pathogenic mutations, and negligible genome-wide off-target editing effects.59 In the near future, we expect that BEs or PEs will be used for treating RDEB patients via the transplantation of ex vivo gene-corrected autologous cells.
Materials and methods
Analysis of targetable disease mutations in COL7A1
RDEB-associated variants were collected from the COL7A1 variants database (http://www.col7a1-database.info). The information on reference sequence and CDS position of each variant were obtained from the National Center for Biotechnology Information (NCBI) website (NG_007065). The number of possible BE-targetable variants was calculated when the mutations were located within editing activity windows, third to ninth positions counting from the 5’ end of the target sequence. The number of possible PE-mediated gene corrections was counted when distances between the mutations and Cas9-mediated nick sites were 12 bp or less. The analysis program was developed using Python3.
Nanopore long-read sequencing for genotyping of RDEB patients
RNA was extracted from the patient-derived fibroblasts using NucleoSpin RNA Plus (Macherey-Nagel), and cDNA was synthesized using ReverTra Ace qPCR RT master mix (TOYOBO, FSK-101). Then, the target region was amplified using KOD-Multi & Epi (TOYOBO, KME-101). Nanopore libraries were prepared with 100 fmol for Flongle flow cells (Oxford Nanopore Technologies, FLO-FLG001) using the Ligation sequencing kit (Oxford Nanopore Technologies, SQK-LSK110) and were sequenced on a MinION sequencer (Oxford Nanopore Technologies, Mk1B) for 24 h. Fastq files were obtained using guppy-basecaller (guppy 6.0.6 version) with default option from the fast5 files that are result files of nanopore sequencing. Fastq files were aligned to COL7A1 cDNA sequence (GenBank: NM_000094) using guppy-aligner with the default option. To divide into two the index data in one nanopore sequencing result, the front 20-bp pair from the beginning of aligning in the reads was extracted, and the index of read was classified using the k-mer scoring system for the extracted 20-bp sequence. After the aligned results were divided by their index, the divided files were classified into two files by substitution at c.3631 in the Pat1 sample and by substitution at c.2005 in the Pat2 sample using the Python (3.8.8) code. The Bam file was read using pysam (0.18.0). The Python codes were uploaded to Github (https://github.com/Gue-ho/RDEB_Nanopore).
Establishment of a COL7A1 mutation database specific for Korean RDEB patients
RDEB patients from all regions of South Korea were referred to Gangnam Severance Hospital, Seoul, Korea, for molecular diagnosis. The results, which include information about 38 patients from 35 unrelated families, make up the largest Korean database for RDEB. RDEB was diagnosed based on clinical features, immunofluorescence antigen mapping, and next-generation sequencing (NGS) and/or Sanger sequencing of COL7A1. All participants or their legal guardians gave their written informed consent, and this study was approved by the Institutional Review Board (IRB) at Gangnam Severance Hospital in accordance with the principles of the Declaration of Helsinki. Genomic DNA was extracted from peripheral blood lymphocytes from patients. DNA from five families was analyzed by NGS, and DNA from 30 families was analyzed by traditional Sanger sequencing. All 118 COL7A1 exons and exon-intron borders were amplified by polymerase chain reaction (PCR) and the products were subsequently sequenced. For all mutations other than nonsense mutations, 100 control alleles were studied to rule out the possibility that the putative disease-associated mutation might be a frequent polymorphism.
Study approval and human subjects
Three patients with RDEB carrying compound heterozygous COL7A1 mutations (c.3631C > T, p.Gln1211Ter, exon 27, and c.8569G > T, p.Glu2857Ter, exon 116, in patient 1; c.2005C > T, p.Arg669Ter, exon 15, and c.8569G > T, p.Glu2857Ter, exon 116, in patient 2; c.3631C > T, p.Gln1211Ter, exon27, and c.3717_3721delTACTC, p.Thr1239ThrfsTer118, exon 27, in patient 3) were enrolled in this study, approved by the Gangnam Severance Hospital IRB (no. 3-2021-0485). Declaration of Helsinki protocols were followed, and both subjects gave written informed consent for the donation of skin cells.
Isolation and culture of primary cells
Skin samples from the RDEB patients and healthy controls, which were obtained by 3-mm punch biopsies, were dissected into 10 pieces with sharp scalpels. For skin explant culture, the pieces were placed in and attached to the well of a 100-mm dish and maintained at 37°C with 5% CO2 in Dulbecco’s modified Eagle medium (DMEM), supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). After 1 week, the medium was changed to Keratinocyte-SFM medium (Thermo Fisher Scientific) supplemented with 1% P/S for keratinocyte culture and DMEM supplemented with 10% FBS and 1% P/S for fibroblasts. Primary human keratinocytes and fibroblasts were cryopreserved at the second passage and stored at −80°C until use.
Sanger sequencing
Genomic DNA was extracted from patient blood samples using an Exgene Blood SV mini kit (GeneAll, Seoul, South Korea). The COL7A1 gene was amplified by PCR using targeted primers (Table S2).
Immunofluorescence for C7 in human skin
Frozen skin tissues from a normal individual and the patients were sectioned at 6 μm and stained with a polyclonal rabbit anti-COL7 antibody (anti-FNIII7-FNIII8 antibody, kindly provided by Dr. Hiroaki Iwata, Department of Dermatology, Hokkaido University Graduate School of Medicine, Sapporo, Japan)25,60 at a 1:500 dilution. Alexa Fluor 488-conjugated goat anti-rabbit IgG (Thermo Fisher Scientific) was used as secondary antibody. Sections were stained with 4,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific). Images were captured using an LSM 980 confocal microscope (Carl Zeiss, Oberkochen, Germany).
Western blots
Total proteins from primary fibroblasts were isolated using RIPA buffer (Cell Signaling Technology, Danvers, MA) supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF). The fibroblast culture supernatants were mixed with acetone and centrifuged at 4,000 rpm for 20 min, after which the resulting pellets were washed with phosphate-buffered saline. Total proteins from these supernatant-derived pellets were isolated using RIPA buffer (Cell Signaling Technology, Danvers, MA) supplemented with 1 mM PMSF. After protein isolation, equal amounts of proteins from each group were loaded onto Nupage Novex Bis-Tris gels (Thermo Fisher Scientific), and electrophoresis was performed using an X-cell SureLock mini-cell (Thermo Fisher Scientific). After electrophoresis, proteins were transferred onto polyvinylidene difluoride membranes, which were then incubated with rabbit anti-COL7 polyclonal antibody (reference 234192, Calbiochem) that was diluted in Tris-buffered saline (TBS) containing 0.05% Tween 20 (TBS-T), at a dilution of 1:3,000. Blots were washed with 0.05% TBS-T and then incubated with horseradish peroxidase-conjugated anti-mouse and anti-rabbit secondary antibodies (Thermo Fisher Scientific) in 0.05% TBS-T at a dilution of 1:2,000. Blots were developed using ECL PLUS reagent (Pierce, Rockford, IL). The densities of the resulting protein bands were analyzed using ImageJ densitometry software (National Institutes of Health, Bethesda, MD).
Immunocytochemistry
For immunocytochemistry, fibroblasts were cultured in chamber slides (LabTek; Thermo Fisher Scientific), fixed with 4% paraformaldehyde for 10 min, blocked with 0.5% bovine serum albumin for 30 min, and then incubated with a polyclonal rabbit anti-COL7 antibody (anti-FNIII7-FNIII8 antibody; 200-fold dilution) overnight at 4°C. After being washed, the cells were incubated with Alexa Fluor 488-conjugated goat anti-rabbit IgG (Thermo Fisher Scientific) as the secondary antibody at a 1:2,000 dilution and DAPI (Thermo Fisher Scientific). Images were captured using an LSM 780 confocal microscope (Carl Zeiss, Oberkochen, Germany).
Flow cytometry
To investigate the proportion of C7-expressing cells quantitatively in the ABE- and PE-corrected RDEB fibroblasts, flow cytometry was performed. Cells (1 × 106) were fixed and permeabilized using an eBioscience Foxp3/transcription factor staining buffer set (Thermo Fisher Scientific) according to the manufacturer’s instructions. After being washed with permeabilization buffer, the pelleted cells were resuspended in permeabilization buffer with C7 antibody (LH7.2; Santa Cruz Biotechnologies) at a 1:25 dilution and incubated at 4°C overnight. After being washed with permeabilization buffer, the cells were pelleted and resuspended in permeabilization buffer with Alexa 488 goat anti-mouse secondary antibody (Invitrogen) at a 1:200 dilution, and incubated at 4°C for 1 h. Unstained cells and cells incubated only with the secondary antibody were used as negative controls. Flow cytometry was performed with an LSRFortessa X-20 flow cytometer (BD Biosciences) and analyzed using FlowJo software (BD Biosciences) (n = 3).
Intradermal injection of RDEB fibroblasts into immunodeficient mice
All animal experiments were approved by the Animal Care Committee of Yonsei University College of Medicine. Male athymic nude mice (nu/nu) (Central Lab Animal, Inc., Seoul, Korea) were maintained under specific-pathogen-free conditions with water, food, and supportive nutrition ad libitum. Three fibroblast populations (NHDFs, RDEB fibroblasts, and ABE- and PE-treated RDEB fibroblasts) were expanded to obtain the required number of cells for intradermal injections. Then, cells were harvested using trypsin and ethylenediaminetetraacetic acid (EDTA) (Life Technologies), after which they were washed gently three times with phosphate-buffered saline. Five million of each fibroblast type were resuspended in 150 μL of phosphate-buffered saline and were intradermally injected with a 24G needle in a 1-cm2 area. A single 150-μL volume was delivered via two injections of 75 μL. Each mouse was subjected to three or four different intradermal injections in the back and at least three different mice per cell group were analyzed. Two weeks after injection, mouse skin samples were obtained for immunofluorescence staining for human C7.
Generation of the human skin equivalent on an immunodeficient mouse
We generated the human skin equivalent on an immunodeficient mouse by seeding human keratinocytes and fibroblasts in a grafting chamber that was surgically implanted on the back of immunodeficient mice, as previously described.61,62 Briefly, grafting chambers (12-mm inner diameter, 20-mm outer diameter, and 10 mm tall) with two manually punched holes were implanted onto the muscle fascia of an immunodeficient mouse. Primary cultured fibroblasts and keratinocytes from healthy volunteers and RDEB patients were used to reconstitute human skin. Three grafting chambers were implanted into each mouse for (1) control graft (normal human keratinocytes and fibroblasts), (2) patient graft (keratinocytes and fibroblasts from an RDEB patient), and (3) PE-corrected patient graft (keratinocytes from an RDEB patient and PE-corrected RDEB fibroblasts) (n = 3 mice/cell group). After implantation of grafting chambers, a mixture of 5 × 106 fibroblasts and 5 × 106 keratinocytes was seeded in the grafting chamber in a total volume of 400 μL in DMEM. Seven days after implantation of the cells, the grafting chambers were removed and the wound was left to heal. Around 10 days after the removal of the grafting chambers, the scab fell off. Then, transplanted mice with the human engrafted skin equivalent were followed for 4 weeks to evaluate the human C7 deposition and anchoring fibril formation.
Immunofluorescence staining of mouse skin
For immunofluorescence detection of human C7 in injected mouse skin and human skin equivalent that was grafted onto immunodeficient mice, frozen skin tissues were sectioned at 6 μm and stained with polyclonal rabbit anti-COL7 antibody (anti-FNIII7-FNIII8 antibody, kindly provided by Dr. Hiroaki Iwata, Department of Dermatology, Hokkaido University Graduate School of Medicine, Sapporo, Japan)25,60 at a dilution of 1:500, polyclonal rabbit anti-KRT1 antibody (BioLegend) at a dilution of 1:1,000, and monoclonal (SP20) rabbit anti-vimentin antibody (Abcam) at a dilution of 1:500 at 4°C overnight. Alexa Fluor 488-conjugated goat anti-rabbit IgG (Thermo Fisher Scientific) was used as the secondary antibody. Sections were stained with DAPI (Thermo Fisher Scientific). Images were captured using an LSM 780 confocal microscope (Carl Zeiss, Oberkochen, Germany).
Transmission electron microscopy
For detection of anchoring fibrils in human skin equivalent that was grafted onto immunodeficient mice, the skin tissue sections were fixed in Karnovsky’s fixative and examined under a transmission electron microscope (H 7600, Hitachi, Japan).
Cell detachment assay
Fibroblasts were seeded at a density of 6 × 104 cells/well in a 96-well plate and cultured for 24 h. After being washed with 1× phosphate-buffered saline, confluent layers of fibroblasts were treated with 0.05% trypsin/EDTA for 6, 4, 2, 1, and 0 min, followed by washing once with 10% FBS/DMEM to inactivate the trypsin and then twice with phosphate-buffered saline. The adherent cells were stained with 0.5% crystal violet (Sigma Aldrich) for 30 min and lysed with 1% sodium dodecyl sulfate (Sigma Aldrich). The percentage of adherent cells was determined by measuring the absorbance at 590 nm using a spectrophotometer.
Proliferation assay
NHDFs, unedited RDEB fibroblasts, and corrected RDEB fibroblasts were seeded at a concentration of 5 × 103 cells/well into microplates (tissue culture grade, 96 wells, flat bottom) in 100 μL of 10% FBS/DMEM culture medium per well. At 24, 48, or 72 h after incubation at 37°C with 5% CO2, cellular proliferation was evaluated using a WST-1 assay (05015944001, Roche, Basel, Switzerland). Briefly, the cells were incubated with the WST-1 reagent for 4 h, and absorbance at 450 and 650 nm (reference wavelength) was detected using a microplate reader (iMark, Bio-Rad).
Construction of sgRNA- and pegRNA-expressing plasmids
The target sequences were selected using Cas-designer (http://www.rgenome.net/cas-designer/).63 To construct sgRNA- and ngRNA-expressing plasmids, complementary oligos representing target sequences were annealed and cloned into pRG2 (Addgene, no. 104174). To construct pegRNA-expressing plasmids, complementary oligos representing target sequences, the sgRNA scaffold, and 3′ extensions were annealed and cloned into pU6-pegRNA-GG-acceptor (Addgene, no. 132777). The oligos are listed in Table S3.
Transfection
Patient-derived fibroblasts (150,000) were transfected using a Neon transfection system (Thermo Fisher) with the following parameters: voltage, 1,650; width, 10 ms; number, 3. For base editing, the fibroblasts were transfected with 900 ng of ABEmax-encoding plasmid (Addgene, no. 112095) and 250 ng of sgRNA-encoding plasmid. For prime editing, the fibroblasts were transfected with 900 ng of PE2-encoding plasmid (Addgene, no. 132775), 300 ng of pegRNA-encoding plasmid, and 83 ng of ngRNA-encoding plasmid. One million keratinocytes were transfected using an AMAXA Human Keratinocyte Nucleofector kit (Lonza Cologne, Germany) according to the manufacturer’s instructions. For base editing, the keratinocytes were transfected with 4.5 μg of ABEmax-encoding plasmid and 1.25 μg of sgRNA-encoding plasmid. For prime editing, the keratinocytes transfected with 4.5 μg of PE2-encoding plasmid, 1.5 μg of pegRNA-encoding plasmid, and 415 ng of ngRNA-encoding plasmid.
Cell lysis and high-throughput sequencing
Cell pellets were resuspended in proteinase K extraction buffer (40 mM Tris-HCl [pH 8.0] [Sigma], 1% Tween 20 [Sigma], 0.2 mM EDTA [Sigma], 10 mg of proteinase K, 0.2% Nonidet P-40 [VWR Life Science]) and then incubated at 60°C for 15 min and 98°C for 5 min. Proteinase K extraction solution (1–3 μL) containing genomic DNA was amplified for high-throughput sequencing. ABE and PE target sites were amplified using SUN-PCR blend (Sun Genetics). The PCR products were purified using Expin PCR SV mini (GeneAll) and sequenced using a MiniSeq sequencing system (Illumina). The results were analyzed using Cas-Analyzer (http://www.rgenome.net/cas-analyzer/), BE-Analyzer (http://www.rgenome.net/be-analyzer/), and PE-Analyzer (http://www.rgenome.net/pe-analyzer/).64, 65, 66 The primers are listed in Table S2.
Data and code availability
High-throughput sequencing data have been deposited in the NCBI Sequence Read Archive database (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession no. PRJNA739484.
The authors declare that all unreported custom Python code used in this study is available from the corresponding author upon reasonable request.
Acknowledgments
This research was supported by grants from the National Research Foundation of Korea (NRF), 2021R1A2C3012908 and 2021M3A9H3015389 to S.B. and 2021R1A2C2013170 to S.E.L.
Author contributions
S.B. and S.E.L. conceived this project; S.-E.K., S.-A.H., and A.-y.L. performed and analyzed the experiments; G.-H.H. performed bioinformatics analyses; J.H.K., H.I., and S.-C.K. gave critical comments; S.-A.H., S.B., and S.E.L. wrote the manuscript with the approval of all other authors.
Declaration of interests
S.-A.H., S.-E.K., S.B., and S.E.L. are filing a patent application based on this work.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.06.005.
Contributor Information
Sangsu Bae, Email: sbae7@snu.ac.kr.
Sang Eun Lee, Email: jennifer823@yuhs.ac.
Supplemental information
References
- 1.Bardhan A., Bruckner-Tuderman L., Chapple I.L.C., Fine J.D., Harper N., Has C., Magin T.M., Marinkovich M.P., Marshall J.F., McGrath J.A., et al. Epidermolysis bullosa. Nat. Rev. Dis. Prim. 2020;6:78. doi: 10.1038/s41572-020-0210-0. [DOI] [PubMed] [Google Scholar]
- 2.Has C., Bauer J.W., Bodemer C., Bolling M.C., Bruckner-Tuderman L., Diem A., Fine J.D., Heagerty A., Hovnanian A., Marinkovich M.P., et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 2020;183:614–627. doi: 10.1111/bjd.18921. [DOI] [PubMed] [Google Scholar]
- 3.Wang X., Ghasri P., Amir M., Hwang B., Hou Y., Khilili M., Lin A., Keene D., Uitto J., Woodley D.T., Chen M. Topical application of recombinant type VII collagen incorporates into the dermal-epidermal junction and promotes wound closure. Mol. Ther. 2013;21:1335–1344. doi: 10.1038/mt.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nyström A., Thriene K., Mittapalli V., Kern J.S., Kiritsi D., Dengjel J., Bruckner-Tuderman L. Losartan ameliorates dystrophic epidermolysis bullosa and uncovers new disease mechanisms. EMBO Mol. Med. 2015;7:1211–1228. doi: 10.15252/emmm.201505061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tamai K., Yamazaki T., Chino T., Ishii M., Otsuru S., Kikuchi Y., Iinuma S., Saga K., Nimura K., Shimbo T., et al. PDGFRα-positive cells in bone marrow are mobilized by high mobility group box 1 (HMGB1) to regenerate injured epithelia. Proc. Natl. Acad. Sci. USA. 2011;108:6609–6614. doi: 10.1073/pnas.1016753108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wong T., Gammon L., Liu L., Mellerio J.E., Dopping-Hepenstal P.J., Pacy J., Elia G., Jeffery R., Leigh I.M., Navsaria H., McGrath J.A. Potential of fibroblast cell therapy for recessive dystrophic epidermolysis bullosa. J. Invest. Dermatol. 2008;128:2179–2189. doi: 10.1038/jid.2008.78. [DOI] [PubMed] [Google Scholar]
- 7.Petrof G., Martinez-Queipo M., Mellerio J.E., Kemp P., McGrath J.A. Fibroblast cell therapy enhances initial healing in recessive dystrophic epidermolysis bullosa wounds: results of a randomized, vehicle-controlled trial. Br. J. Dermatol. 2013;169:1025–1033. doi: 10.1111/bjd.12599. [DOI] [PubMed] [Google Scholar]
- 8.Conget P., Rodriguez F., Kramer S., Allers C., Simon V., Palisson F., Gonzalez S., Yubero M.J. Replenishment of type VII collagen and re-epithelialization of chronically ulcerated skin after intradermal administration of allogeneic mesenchymal stromal cells in two patients with recessive dystrophic epidermolysis bullosa. Cytotherapy. 2010;12:429–431. doi: 10.3109/14653241003587637. [DOI] [PubMed] [Google Scholar]
- 9.Petrof G., Lwin S.M., Martinez-Queipo M., Abdul-Wahab A., Tso S., Mellerio J.E., Slaper-Cortenbach I., Boelens J.J., Tolar J., Veys P., et al. Potential of systemic allogeneic mesenchymal stromal cell therapy for children with recessive dystrophic epidermolysis bullosa. J. Invest. Dermatol. 2015;135:2319–2321. doi: 10.1038/jid.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rashidghamat E., Kadiyirire T., Ayis S., Petrof G., Liu L., Pullabhatla V., Ainali C., Guy A., Aristodemou S., McMillan J.R., et al. Phase I/II open-label trial of intravenous allogeneic mesenchymal stromal cell therapy in adults with recessive dystrophic epidermolysis bullosa. J. Am. Acad. Dermatol. 2020;83:447–454. doi: 10.1016/j.jaad.2019.11.038. [DOI] [PubMed] [Google Scholar]
- 11.Lee S.E., Lee S.J., Kim S.E., Kim K., Cho B., Roh K., Kim S.C. Intravenous allogeneic umbilical cord blood-derived mesenchymal stem cell therapy in recessive dystrophic epidermolysis bullosa patients. JCI Insight. 2021;6:143606. doi: 10.1172/jci.insight.143606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wagner J.E., Ishida-Yamamoto A., McGrath J.A., Hordinsky M., Keene D.R., Woodley D.T., Chen M., Riddle M.J., Osborn M.J., Lund T., et al. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N. Engl. J. Med. 2010;363:629–639. doi: 10.1056/NEJMoa0910501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siprashvili Z., Nguyen N.T., Gorell E.S., Loutit K., Khuu P., Furukawa L.K., Lorenz H.P., Leung T.H., Keene D.R., Rieger K.E., et al. Safety and wound outcomes following genetically corrected autologous epidermal grafts in patients with recessive dystrophic epidermolysis bullosa. JAMA. 2016;316:1808. doi: 10.1001/jama.2016.15588. [DOI] [PubMed] [Google Scholar]
- 14.Eichstadt S., Barriga M., Ponakala A., Teng C., Nguyen N.T., Siprashvili Z., Nazaroff J., Gorell E.S., Chiou A.S., Taylor L., et al. Phase 1/2a clinical trial of gene-corrected autologous cell therapy for recessive dystrophic epidermolysis bullosa. JCI Insight. 2019;4:130554. doi: 10.1172/jci.insight.130554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lwin S.M., Syed F., Di W.L., Kadiyirire T., Liu L., Guy A., Petrova A., Abdul-Wahab A., Reid F., Phillips R., et al. Safety and early efficacy outcomes for lentiviral fibroblast gene therapy in recessive dystrophic epidermolysis bullosa. JCI Insight. 2019;4:126243. doi: 10.1172/jci.insight.126243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hacein-Bey-Abina S., von Kalle C., Schmidt M., Le Deist F., Wulffraat N., McIntyre E., Radford I., Villeval J.L., Fraser C.C., Cavazzana-Calvo M., Fischer A. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 17.Hacein-Bey-Abina S., Von Kalle C., Schmidt M., McCormack M.P., Wulffraat N., Leboulch P., Lim A., Osborne C.S., Pawliuk R., Morillon E., et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 18.Montini E., Cesana D., Schmidt M., Sanvito F., Bartholomae C.C., Ranzani M., Benedicenti F., Sergi L.S., Ambrosi A., Ponzoni M., et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Invest. 2009;119:964–975. doi: 10.1172/JCI37630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moiani A., Paleari Y., Sartori D., Mezzadra R., Miccio A., Cattoglio C., Cocchiarella F., Lidonnici M.R., Ferrari G., Mavilio F. Lentiviral vector integration in the human genome induces alternative splicing and generates aberrant transcripts. J. Clin. Invest. 2012;122:1653–1666. doi: 10.1172/JCI61852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tucker T.A., Fortenberry J.A., Zsembery A., Schwiebert L.M., Schwiebert E.M. The ΔF508-CFTR mutation inhibits wild-type CFTR processing and function when co-expressed in human airway epithelia and in mouse nasal mucosa. BMC Physiol. 2012;12:12. doi: 10.1186/1472-6793-12-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hainzl S., Peking P., Kocher T., Murauer E.M., Larcher F., Del Rio M., Duarte B., Steiner M., Klausegger A., Bauer J.W., et al. COL7A1 editing via CRISPR/Cas9 in recessive dystrophic epidermolysis bullosa. Mol. Ther. 2017;25:2573–2584. doi: 10.1016/j.ymthe.2017.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Izmiryan A., Ganier C., Bovolenta M., Schmitt A., Mavilio F., Hovnanian A. Ex vivo COL7A1 correction for recessive dystrophic epidermolysis bullosa using CRISPR/Cas9 and homology-directed repair. Mol. Ther. Nucleic Acids. 2018;12:554–567. doi: 10.1016/j.omtn.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacków J., Guo Z., Hansen C., Abaci H.E., Doucet Y.S., Shin J.U., Hayashi R., DeLorenzo D., Kabata Y., Shinkuma S., et al. CRISPR/Cas9-based targeted genome editing for correction of recessive dystrophic epidermolysis bullosa using iPS cells. Proc. Natl. Acad. Sci. USA. 2019;116:26846–26852. doi: 10.1073/pnas.1907081116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonafont J., Mencía A., Chacón-Solano E., Srifa W., Vaidyanathan S., Romano R., Garcia M., Hervás-Salcedo R., Ugalde L., Duarte B., et al. Correction of recessive dystrophic epidermolysis bullosa by homology-directed repair-mediated genome editing. Mol. Ther. 2021;29:2008–2018. doi: 10.1016/j.ymthe.2021.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takashima S., Shinkuma S., Fujita Y., Nomura T., Ujiie H., Natsuga K., Iwata H., Nakamura H., Vorobyev A., Abe R., Shimizu H. Efficient gene reframing therapy for recessive dystrophic epidermolysis bullosa with CRISPR/Cas9. J. Invest. Dermatol. 2019;139:1711–1721.e4. doi: 10.1016/j.jid.2019.02.015. [DOI] [PubMed] [Google Scholar]
- 26.Kocher T., March O.P., Bischof J., Liemberger B., Hainzl S., Klausegger A., Hoog A., Strunk D., Bauer J.W., Koller U. Predictable CRISPR/Cas9-Mediated COL7A1 reframing for dystrophic epidermolysis bullosa. J. Invest. Dermatol. 2020;140:1985–1993.e5. doi: 10.1016/j.jid.2020.02.012. [DOI] [PubMed] [Google Scholar]
- 27.Mencía Á., Chamorro C., Bonafont J., Duarte B., Holguin A., Illera N., Llames S.G., Escámez M.J., Hausser I., Del Río M., et al. Deletion of a pathogenic mutation-containing exon of COL7A1 allows clonal gene editing correction of RDEB patient epidermal stem cells. Mol. Ther. Nucleic Acids. 2018;11:68–78. doi: 10.1016/j.omtn.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chamorro C., Mencía A., Almarza D., Duarte B., Büning H., Sallach J., Hausser I., Del Río M., Larcher F., Murillas R. Gene editing for the efficient correction of a recurrent COL7A1 mutation in recessive dystrophic epidermolysis bullosa keratinocytes. Mol. Ther. Nucleic Acids. 2016;5:e307. doi: 10.1038/mtna.2016.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bonafont J., Mencía Á., García M., Torres R., Rodríguez S., Carretero M., Chacón-Solano E., Modamio-Høybjør S., Marinas L., León C., et al. Clinically relevant correction of recessive dystrophic epidermolysis bullosa by dual sgRNA CRISPR/Cas9-Mediated gene editing. Mol. Ther. 2019;27:986–998. doi: 10.1016/j.ymthe.2019.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaj T., Gersbach C.A., Barbas C.F., 3rd ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jang H.K., Song B., Hwang G.H., Bae S. Current trends in gene recovery mediated by the CRISPR-Cas system. Exp. Mol. Med. 2020;52:1016–1027. doi: 10.1038/s12276-020-0466-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin H.Y., Wang C., Lee H.K., Yoo K.H., Zeng X., Kuhns T., Yang C.M., Mohr T., Liu C., Hennighausen L. CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat. Commun. 2017;8:15464. doi: 10.1038/ncomms15464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018;24:927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 35.Ihry R.J., Worringer K.A., Salick M.R., Frias E., Ho D., Theriault K., Kommineni S., Chen J., Sondey M., Ye C., et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018;24:939–946. doi: 10.1038/s41591-018-0050-6. [DOI] [PubMed] [Google Scholar]
- 36.Anzalone A.V., Koblan L.W., Liu D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020;38:824–844. doi: 10.1038/s41587-020-0561-9. [DOI] [PubMed] [Google Scholar]
- 37.Jeong Y.K., Song B., Bae S. Current status and challenges of DNA base editing tools. Mol. Ther. 2020;28:1938–1952. doi: 10.1016/j.ymthe.2020.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu P., Liang S.Q., Zheng C., Mintzer E., Zhao Y.G., Ponnienselvan K., Mir A., Sontheimer E.J., Gao G., Flotte T.R., et al. Improved prime editors enable pathogenic allele correction and cancer modelling in adult mice. Nat. Commun. 2021;12:2121. doi: 10.1038/s41467-021-22295-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ryu S.M., Koo T., Kim K., Lim K., Baek G., Kim S.T., Kim H.S., Kim D.E., Lee H., Chung E., Kim J.S. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol. 2018;36:536–539. doi: 10.1038/nbt.4148. [DOI] [PubMed] [Google Scholar]
- 40.Kim Y., Hong S.A., Yu J., Eom J., Jang K., Yoon S., Hong D.H., Seo D., Lee S.N., Woo J.S., et al. Adenine base editing and prime editing of chemically derived hepatic progenitors rescue genetic liver disease. Cell Stem Cell. 2021;28:1614–1624.e5. doi: 10.1016/j.stem.2021.04.010. [DOI] [PubMed] [Google Scholar]
- 41.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A∗T to G∗C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Osborn M.J., Newby G.A., McElroy A.N., Knipping F., Nielsen S.C., Riddle M.J., Xia L., Chen W., Eide C.R., Webber B.R., et al. Base editor correction of COL7A1 in recessive dystrophic epidermolysis bullosa patient-derived fibroblasts and iPSCs. J. Invest. Dermatol. 2020;140:338–347.e5. doi: 10.1016/j.jid.2019.07.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nishimasu H., Shi X., Ishiguro S., Gao L., Hirano S., Okazaki S., Noda T., Abudayyeh O.O., Gootenberg J.S., Mori H., et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science. 2018;361:1259–1262. doi: 10.1126/science.aas9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walton R.T., Christie K.A., Whittaker M.N., Kleinstiver B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science. 2020;368:290–296. doi: 10.1126/science.aba8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yonei N., Ohtani T., Furukawa F. Recessive dystrophic epidermolysis bullosa: case of non-Hallopeau-Siemens variant with premature termination codons in both alleles. J. Dermatol. 2006;33:802–805. doi: 10.1111/j.1346-8138.2006.00182.x. [DOI] [PubMed] [Google Scholar]
- 48.Murai T., Tamai K., Nakano H., Hanada K., Hashimoto I., Sawamura D.J.H.M.J. Novel COL7A1 mutations of hallopeau-siemens type recessive dystrophic epidermolysis bullosa. Hirosaki Med. 2007;59:15. [Google Scholar]
- 49.Dang N., Murrell D.F. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp. Dermatol. 2008;17:553–568. doi: 10.1111/j.1600-0625.2008.00723.x. [DOI] [PubMed] [Google Scholar]
- 50.Koblan L.W., Doman J.L., Wilson C., Levy J.M., Tay T., Newby G.A., Maianti J.P., Raguram A., Liu D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018;36:843–846. doi: 10.1038/nbt.4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee C., Hyun Jo D., Hwang G.H., Yu J., Kim J.H., Park S.E., Kim J.S., Kim J.H., Bae S. CRISPR-pass: gene rescue of nonsense mutations using adenine base editors. Mol. Ther. 2019;27:1364–1371. doi: 10.1016/j.ymthe.2019.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Villiger L., Grisch-Chan H.M., Lindsay H., Ringnalda F., Pogliano C.B., Allegri G., Fingerhut R., Häberle J., Matos J., Robinson M.D., et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 2018;24:1519–1525. doi: 10.1038/s41591-018-0209-1. [DOI] [PubMed] [Google Scholar]
- 53.Jo D.H., Jang H.-K., Cho C.S., Han J.H., Ryu G., Jung Y., Bae S., Kim J.H. Therapeutic adenine base editing corrects nonsense mutation and improves visual function in a mouse model of Leber congenital amaurosis. bioRxiv. 2021 doi: 10.1101/2021.01.07.425822. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jacków J., Titeux M., Portier S., Charbonnier S., Ganier C., Gaucher S., Hovnanian A. Gene-corrected fibroblast therapy for recessive dystrophic epidermolysis bullosa using a self-inactivating COL7A1 retroviral vector. J. Invest. Dermatol. 2016;136:1346–1354. doi: 10.1016/j.jid.2016.02.811. [DOI] [PubMed] [Google Scholar]
- 55.Chen M., Kasahara N., Keene D.R., Chan L., Hoeffler W.K., Finlay D., Barcova M., Cannon P.M., Mazurek C., Woodley D.T. Restoration of type VII collagen expression and function in dystrophic epidermolysis bullosa. Nat. Genet. 2002;32:670–675. doi: 10.1038/ng1041. [DOI] [PubMed] [Google Scholar]
- 56.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kern J.S., Loeckermann S., Fritsch A., Hausser I., Roth W., Magin T.M., Mack C., Müller M.L., Paul O., Ruther P., Bruckner-Tuderman L. Mechanisms of fibroblast cell therapy for dystrophic epidermolysis bullosa: high stability of collagen VII favors long-term skin integrity. Mol. Ther. 2009;17:1605–1615. doi: 10.1038/mt.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jang H.K., Jo D.H., Lee S.N., Cho C.S., Jeong Y.K., Jung Y., Yu J., Kim J.H., Woo J.S., Bae S. High-purity production and precise editing of DNA base editing ribonucleoproteins. Sci. Adv. 2021;7:eabg2661. doi: 10.1126/sciadv.abg2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Habib O., Habib G., Hwang G.H., Bae S. Comprehensive analysis of prime editing outcomes in human embryonic stem cells. Nucleic Acids Res. 2022;50:1187–1197. doi: 10.1093/nar/gkab1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vorobyev A., Ujiie H., Recke A., Buijsrogge J.J.A., Jonkman M.F., Pas H.H., Iwata H., Hashimoto T., Kim S.C., Hoon Kim J., et al. Autoantibodies to multiple epitopes on the non-collagenous-1 domain of type VII collagen induce blisters. J. Invest. Dermatol. 2015;135:1565–1573. doi: 10.1038/jid.2015.51. [DOI] [PubMed] [Google Scholar]
- 61.Wang C.K., Nelson C.F., Brinkman A.M., Miller A.C., Hoeffler W.K. Spontaneous cell sorting of fibroblasts and keratinocytes creates an organotypic human skin equivalent. J. Invest. Dermatol. 2000;114:674–680. doi: 10.1046/j.1523-1747.2000.00938.x. [DOI] [PubMed] [Google Scholar]
- 62.Bremer J., Bornert O., Nyström A., Gostynski A., Jonkman M.F., Aartsma-Rus A., van den Akker P.C., Pasmooij A.M. Antisense oligonucleotide-mediated exon skipping as a systemic therapeutic approach for recessive dystrophic epidermolysis bullosa. Mol. Ther. Nucleic Acids. 2016;5:e379. doi: 10.1038/mtna.2016.87. [DOI] [PubMed] [Google Scholar]
- 63.Park J., Bae S., Kim J.S. Cas-Designer: a web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics. 2015;31:4014–4016. doi: 10.1093/bioinformatics/btv537. [DOI] [PubMed] [Google Scholar]
- 64.Park J., Lim K., Kim J.S., Bae S. Cas-analyzer: an online tool for assessing genome editing results using NGS data. Bioinformatics. 2017;33:286–288. doi: 10.1093/bioinformatics/btw561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hwang G.H., Park J., Lim K., Kim S., Yu J., Yu E., Kim S.T., Eils R., Kim J.S., Bae S. Web-based design and analysis tools for CRISPR base editing. BMC Bioinf. 2018;19:542. doi: 10.1186/s12859-018-2585-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hwang G.H., Jeong Y.K., Habib O., Hong S.A., Lim K., Kim J.S., Bae S. PE-Designer and PE-Analyzer: web-based design and analysis tools for CRISPR prime editing. Nucleic Acids Res. 2021;49:W499–W504. doi: 10.1093/nar/gkab319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
High-throughput sequencing data have been deposited in the NCBI Sequence Read Archive database (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession no. PRJNA739484.
The authors declare that all unreported custom Python code used in this study is available from the corresponding author upon reasonable request.