

Abstract
Several preclinical studies demonstrate that antitumor efficacy of programmed cell death-1 (PD-1)/programmed death-ligand 1 (PD-L1) blockade can be improved by combination with other checkpoint inhibitors. Lymphocyte-activation gene 3 (LAG-3) is an inhibitory checkpoint receptor involved in T cell exhaustion and tumor immune escape. Here, we describe ABL501, a bispecific antibody targeting LAG-3 and PD-L1 in modulating immune cell responses against tumors. ABL501 that efficiently inhibits both LAG-3 and PD-L1 pathways enhances the activation of effector CD4+ and CD8+ T cells with a higher degree than a combination of single anti-LAG-3 and anti-PD-L1. The augmented effector T cell responses by ABL501 resulted in mitigating regulatory-T-cell-mediated immunosuppression. Mechanistically, the simultaneous binding of ABL501 to LAG-3 and PD-L1 promotes dendritic cell (DC) activation and tumor cell conjugation with T cells that subsequently mounts effective CD8+ T cell responses. ABL501 demonstrates its potent in vivo antitumor efficacy in a humanized xenograft model and with knockin mice expressing human orthologs. The immune profiling analysis of peripheral blood reveals an increased abundance of LAG-3hiPD-1hi memory CD4+ T cell subset in relapsed cholangiocarcinoma patients after gemcitabine plus cisplatin therapy, which are more responsive to ABL501. This study supports the clinical evaluation of ABL501 as a novel cancer immunotherapeutic, and a first-in-human trial has started (NCT05101109).
Keywords: cancer immunotherapy, bispecific antibody, LAG-3, PD-L1, immune checkpoint inhibitor, cholangiocarcinoma
Graphical abstract
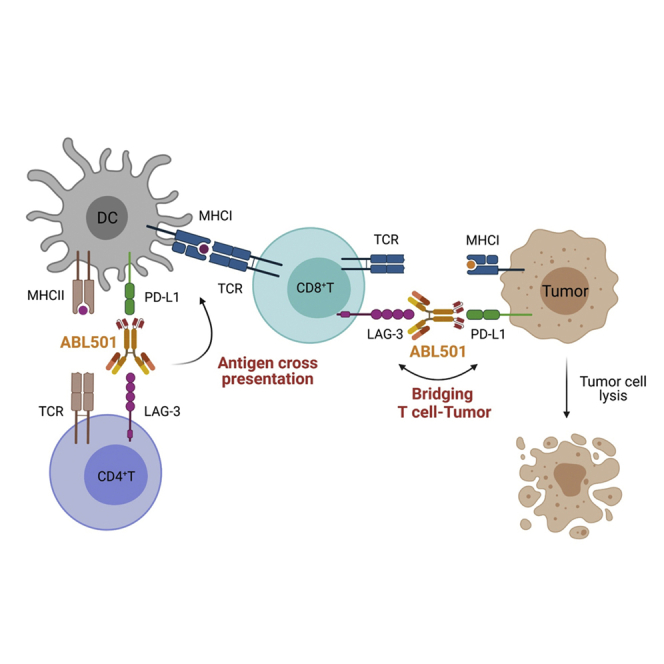
ABL501, a clinical-stage bispecific antibody, potentiates CD8+ T cell responses against tumor cells by blocking LAG-3 and PD-L1 immune-suppressive signals, promoting dendritic cell activation and bridging tumor with T cells.
Introduction
Somatic mutations in cancers can potentially generate neoantigens that are recognized and targeted by cytotoxic T cells.1 However, cancer cells evolve in various ways to evade adaptive immune-mediated killing. For example, advanced tumor cells create an immunosuppressive tumor microenvironment (TME), and tumor-infiltrating T cells progressively become dysfunctional or exhausted in the TME. Exhausted T cells are characterized by decreased proliferative capacities, altered metabolism, impaired cytokine production, and sustained high-level expression of inhibitory receptors, such as programmed cell death-1 (PD-1), T cell immunoglobulin and mucin domain-containing protein 3 (Tim-3), T cell immunoreceptor with immunoglobulin and ITIM domains (TIGIT), and lymphocyte-activation gene 3 (LAG-3).2, 3, 4 Therapeutic antibodies for blocking the interaction between PD-1 on tumor-infiltrating T cells and programmed death-ligand 1 (PD-L1) expressed on tumor cells have proven successful in the treatment of multiple cancer types. However, the proportions of patients and cancer types that can respond to anti-PD-1/PD-L1 therapies are still limited. This suggests that combination approaches may be required to generate efficient antitumor immunity and improve therapeutic outcomes.5, 6, 7 LAG-3 is a type I transmembrane protein with four extracellular immunoglobulin (Ig)-like domains. It is primarily expressed on T cells, natural killer cells, and plasmacytoid dendritic cells (DCs). LAG-3 is upregulated during activation of T cells and plays critical roles in maintaining immune tolerance. LAG-3 has structural homology with CD4 and inhibits T cell activation by outcompeting CD4 for major histocompatibility complex class II (MHC class II) binding.8, 9, 10 The studies by Maruhashi et al.11,12 showed that LAG-3 selectively recognizes a stable complex of peptides and MHC class II (pMHC-II), but not unstable pMHC-II complexes. In addition, to date, several molecules have been reported as potential ligands of LAG-3, such as LSECtin/CLEC4G, galectin-3, α-synuclein, and fibrinogen-like protein-1.13, 14, 15, 16 LAG-3 engagement on the T cell surface suppresses the proliferation, activation, and homeostasis of both CD4+ and CD8+ T cells. However, there are little data on the mechanism by which LAG-3 binding to pMHC-II modulates the activation of CD8+ T cells. Several regulatory T cells (Treg cell) populations have been shown to express a high and constitutive level of LAG-3, suggesting that its expression on Treg cells may promote immune suppression.17,18 However, the role of LAG-3 in the suppressive function of Treg cells remains unclear. Elevated LAG-3 expression on tumor-infiltrating lymphocytes (TILs) was observed in hematologic malignancies and various solid tumors.19 It has been reported that LAG-3 expression is associated with poor clinical outcomes.20, 21, 22 LAG-3 is frequently co-expressed with PD-1 in TILs, and this is positively associated with T cell exhaustion. Consistent with this finding, co-blockade of LAG-3 and PD-1 augmented the proliferation and activation of antigen-specific tumor-infiltrating CD8+ T cells. In several mouse preclinical models, combined treatment with anti-LAG-3 and anti-PD-(L)1 monoclonal antibodies (mAbs) has shown synergistic therapeutic effects compared with blocking either one alone.23, 24, 25 In addition, genetic deletion of LAG3 and PD-1 has led to increased antitumor immunity and decreased tumor growth.23,26 Several antagonist antibodies to LAG-3 are currently being evaluated in clinical trials for the treatment of solid tumors. Most recently, a LAG-3-blocking mAb (relatlimab), in combination with a PD-1 blocking mAb (nivolumab) has been approved by U.S. Food and Drug Administration (FDA) for the treatment of unresectable or metastatic melanoma. Opdualag (nivolumab and relatlimab-rmbw) showed an improved median progression-free survival compared with that in the nivolumab alone group at 10.1 (95% confidence interval [CI], 6.37–15.74) versus 4.6 (95% CI, 3.38–5.62) months, respectively, suggesting that this dual-blockade strategy is an effective treatment option.27 Along this line, there are several bispecific antibodies targeting LAG-3 and PD-(L)1, such as MGD013, FS118, and IBI323, which are currently undergoing preclinical or early clinical studies.28, 29, 30, 31 Those bispecific antibodies have exhibited stronger antitumor effect over the combination of anti-PD-(L)1 and anti-LAG-3 mAbs. However, the studies have not mechanistically elucidated the advantages of the bispecific approach over a combination of mAbs. Here, we describe the generation of ABL501, a human anti-LAG-3xPD-L1 bispecific antibody (BsAb) based on anti-LAG-3 human IgG4 mAb and single-chain variable fragments (scFv) of PD-L1. We show that ABL501 directly promoted the activation of effector T cells, which led to reduction of the suppressive effect by Treg cells. In addition, the LAG-3 binding arm of ABL501 enabled its engagement with PD-L1 on DCs in a context-dependent manner. Through this mechanism, ABL501 may enhance antigen cross-priming by DCs, resulting in heightened activation of CD8+ T cells. Furthermore, ABL501 mediated the bridging of LAG-3+ T cells and PD-L1+ tumor cells, thus augmenting the antitumor cytolytic activity of CD8+ T cells. Together, these findings indicated that bispecific antibody ABL501 may have superior antitumor potency compared with the combination of LAG-3 and PD-L1 mAbs.
Results
ABL501 enhances effector T cell responses by blocking the LAG-3 and PD-L1 pathways
ABL501, a bispecific antibody targeting human LAG-3 and PD-L1, was constructed from an anti-LAG-3 monoclonal IgG4 antibody and a scFv for PD-L1, which is fused at the C terminus of the anti-LAG-3 antibody via a (G4S)3 linker, with an amino acid substitution (S224P) in the hinge region of IgG4 to reduce the formation of half-antibodies (Figure 1A).32 Moreover, the scFv domain is composed of light- and heavy-chain variable regions connected by a (G4S)4 linker. The binding of ABL501 to either recombinant human LAG-3 or PD-L1 was measured via surface plasmon resonance (SPR) and ELISA (Figure S1). In addition, comparable binding capacity between ABL501 and anti-LAG-3 or anti-PD-L1 mAbs was observed in Jurkat cells expressing LAG-3 or SNU324 cells endogenously expressing PD-L1 (Figure 1B). As ABL501 was developed in the human IgG4 isotype, Fc-mediated effector functions, including antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC), were not elicited by ABL501 (Figure S2). We then assessed the ability of ABL501 to block the binding between PD-1 and PD-L1 or LAG-3 and MHC class II (Figures 1C and S3). A dual blocking effect by ABL501 was comparable to the combination of anti-LAG-3 and anti-PD-L1 single antibody treatment, as measured using bioassays in LAG-3 and PD-1 expressing nuclear factor κB (NF-κB)-Luc2 reporter Jurkat cells. Next, to examine the potency of ABL501 on human T cell activation under physiological conditions, we stimulated CD3+ T cells with allogeneic monocyte-derived DCs (mo-DCs) in the presence of anti-LAG-3 and/or anti-PD-L1 or ABL501. Upregulation of both LAG-3 and PD-1 was found in activated CD4+ or CD8+ T cells upon stimulation with allogeneic mo-DCs that expressed human leukocyte antigen-DR isotype (HLA-DR) and PD-L1 (Figure S4). Dual blockade of LAG-3 and PD-L1 by the combination or ABL501 treatment increased the proliferative capacity of both CD4+ and CD8+ T cells in a similar extent (Figure 1D). However, an even greater increase in interferon (IFN)-γ secretion by both CD4+ and CD8+ T cells was observed following ABL501 treatment at a low concentration of 8.35 nM than the combination of anti-LAG-3 and anti-PD-L1 that showed a synergistic effect on IFN-γ compared with PD-L1 blockade alone (Figure 1E). These results indicate that co-blockade of LAG-3 and PD-L1 by ABL501 efficiently promotes the activation of both CD4+ and CD8+ effector T cells.
Figure 1.

ABL501 augments effector T cell activation by the inhibition of both LAG-3 and PD-L1 axes
(A) Schematic of LAG-3xPD-L1 BsAb, ABL501. (B) FACS analysis of ABL501 or anti-LAG-3 binding to LAG-3 and PD-1 expressing Jurkat cells (left panel) is shown (ABL501: half-maximal effective concentration [EC50] = 0.15 nM; anti-LAG-3: EC50 = 0.16 nM). FACS analysis of ABL501 or anti-PD-L1 binding to endogenously PD-L1 expressing SNU-324 cells (right panel) is shown (ABL501: EC50 = 0.87 nM; anti-PD-L1: EC50 = 0.36 nM). (C) LAG-3 and PD-1 expressing NF-κB-Luc2 reporter Jurkat cells were co-incubated with target cells expressing MHC-II and PD-L1 in the presence of the indicated antibodies at various concentrations. Blocking activity was analyzed by measuring luminance of the downstream NF-κB reporter (ABL501: EC50 = 1.78 nM, Combi: EC50 = 0.7 nM, and anti-PD-L1: EC50 = 0.53 nM). (D and E) CD4+ T or CD8+ T cells isolated from PBMCs were co-cultured with allogeneic mo-DCs in the presence of the indicated antibodies at various concentrations for 5 days. (D) Schematic illustration of mixed lymphocyte reaction (MLR) assay (left panel) is shown. FACS analysis shows CTV dilution of CD4+ and CD8+ T cells with the indicated antibodies at a final concentration of 33.4 nM. Representative FACS plots (above) and a summary plot (below) show the percentages of CTVlow CD4+ and CD8+ T cells. Each dot represents an individual human sample. (E) ELISA of IFN-γ secretion by CD4+ T cells (left; ABL501: EC50 = 7.27 nM, Combi: EC50 = 10.78 nM, anti-PD-L1: EC50 = 14.9 nM, and anti-LAG-3: EC50 = 14.07 nM) or by CD8+ T cells (right; ABL501: EC50 = 7.99 nM, Combi: EC50 = 13.61 nM, anti-PD-L1: EC50 = 18.04 nM, and anti-LAG-3: EC50 = 26.28 nM). Data were compiled from three to five independent experiments with two replications. Statistical significance was determined by one-way ANOVA with Holm-Sidak multiple comparisons. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, and ns, not significant.
ABL501 mitigates Treg-cell-mediated immunosuppression by augmenting effector T cell responses
LAG-3 has been known to be highly expressed on Treg cells, but the role of LAG-3 in modulating Treg cell responses remains controversial.33 We therefore investigated whether ABL501 contributes to weaken Treg cell function. Instead of using a Treg mixed lymphocyte reaction assay,34,35 we employed A375 cells expressing membrane-bound anti-CD3 scFv (A375-OKT3) as a stimulator to avoid disparities between effector T and Treg cells for alloantigen recognition. CD8+ or CD25−CD127+CD4+ T cells were co-cultured with A375-OKT3 cells in the presence or absence of autologous CD4+CD25+Foxp3+ Treg cells that were fluorescence-activated cell sorting (FACS) sorted (CD4+CD25+CD127−) and ex vivo expanded with interleukin-2 (IL-2) and transforming growth factor β (TGF-β) (Figures 2A and 2B). Co-culture with Treg cells induced a robust suppression of both proliferative capacity and IL-2 secretion in CD4+ or CD8+ T cells (Figures 2C and 2D). In the presence of Treg cells, the combination of anti-LAG-3 and anti-PD-L1 increased proliferation and IL-2 secretion by both effector CD4+ and CD8+ T cells, and ABL501 more efficiently relieved effector T cells from Treg-cell-mediated suppression (Figures 2E and 2F). However, when Treg cells were stimulated by A375-OKT3 cells in the absence of effector T cells, single or dual blockade of LAG-3 and/or PD-L1 largely unaltered Treg cell proliferation and phenotypic marker expressions that are known as key features of highly suppressive CD45RO+ Treg cells (Figures 2G and S5).36 Unlike Treg cells, effector T cell activation was augmented upon ABL501 treatment (Figure 2H), which suggested that co-blockade of LAG-3 and PD-L1 by ABL501 compensates the suppressive effect of Treg cells by promoting effector T cell activation rather than directly affecting Treg cell responses.
Figure 2.

ABL501 weakens Treg-mediated suppression through promoting effector T cell responses
(A–F) FACS sorted and ex vivo expanded CD25+CD127−CD4+ Treg cells were co-cultured with conventional CD4+ or CD8+ T cells isolated from the same human donor in the presence of A375 melanoma cells expressing membrane-bound anti-CD3 scFv (A375-OKT3). (A) Schematic illustration of the ex vivo human Treg cell suppression assay is shown. (B) FACS analysis of Foxp3 expression in ex vivo expanded Treg cells is shown. (C and D) FACS analysis showing CTV dilution (C) or ELISA of IL-2 secretion (D) by CD4+ T or CD8+ T cells in the presence or absence of Treg cells (effector T: Treg = 4:1) is shown. (E and F) FACS analysis shows CTV dilution (E) or ELISA of IL-2 secretion (F) by CD4+ T or CD8+ T cells with the indicated antibodies at a final concentration of 66.8 nM in the presence of Treg cells. (G) Isolated and ex vivo expanded Treg cells were co-cultured with A375-OKT3 cells in the presence of the indicated antibodies at a final concentration of 66.8 nM. Proliferation index of Treg cells was calculated by FlowJo software based on CTV dilution. (H) Isolated conventional CD4+ or CD8+ T cells were co-cultured with A375-OKT3 cells in the presence of the indicated antibodies at a final concentration of 66.8 nM. ELISA of IL-2 secretion by CD4+ T or CD8+ T cells is shown. Data were pooled from three to five independent experiments with two replications. Statistical significance was determined by unpaired t tests with two-tailed analysis in (D) or one-way ANOVA with Holm-Sidak multiple comparisons in (E)–(H). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, and ns.
ABL501 induces CD8+ T cell activation by promoting DC maturation
We then investigated the mechanism by which ABL501 promotes both CD4+ and CD8+ T cell responses. First, we assessed the effect of ABL501 on antigen-specific CD4+ memory T cell responses in cytomegalovirus (CMV)-seropositive donors upon stimulation with CMV lysate that was presented by MHC class II on DCs.37 Co-blockade of LAG-3 and PD-L1 by the combination treatment with anti-LAG-3 and anti-PD-L1 or ABL501 enhanced CMV-specific CD4+ T cell expansion compared with LAG-3 or PD-L1 blockade alone (Figure 3A). Despite the comparable expansion rate of CD4+ T cells, ABL501 treatment resulted in a higher secretion of IFN-γ, IL-2, and tumor necrosis factor alpha (TNF-α) than the co-treatment with anti-LAG-3 and anti-PD-L1 (Figures 3B and S6). Although CMV lysate primarily triggers MHC-class-II-restricted CD4+ T cell responses, CMV-specific CD8+ T cell activation can also be induced by MHC-class-I-mediated cross-presentation of CMV antigen in DCs. Indeed, we found that co-blockade of LAG-3 and PD-L1 mounted CMV-responsive CD8+ T cell populations, and this effect was more significant with ABL501 treatment (Figure 3C). ABL501 treatment not only increased the expansion rate but also the activation level, as indicated by CD25 expression frequencies in CMV-responsive CD8+ T cells, compared with anti-LAG-3 and anti-PD-L1 co-treatment (Figure 3D). This suggests that the induction of CMV-responsive CD8+ T cell expansion by ABL501 results in increased IFN-γ secretion upon CMV lysate stimulation, compared with the combination treatment with anti-LAG-3 and anti-PD-L1. It has been reported that reverse signaling via PD-L1 ligation with PD-1 inhibits DC maturation and activation,38 which can be restored by PD-L1 or PD-1 blockade.39,40 We reasoned that a strong PD-L1-blocking effect by LAG3xPD-L1 BsAb may promote DC activation. To test this hypothesis, we assessed the expression of co-stimulatory receptors, including HLA-ABC, HLA-DR, CD86, CD80, and CD40 on mo-DCs at 48 h after treatment with plate-coated or soluble anti-PD-L1. DCs incubated with plate-coated anti-PD-L1, which mimics an immobilized anti-PD-L1 by the bispecific format of ABL501, exhibited more activated phenotype with increased expression of activation markers compared with DCs treated with soluble anti-PD-L1 (Figure 3E). Furthermore, a marked increase in TNF-α, MMP-1, and IL-12 secretion was shown by DCs incubated with plate-coated anti-PD-L1, even without lipopolysaccharide (LPS) stimulation (Figure 3F). We then confirmed the strong PD-L1-blocking effect of ABL501 on DC activation by employing LAG-3-expressing Chinese hamster ovary (CHO) cells (CHO-LAG-3) as a matrix for anti-LAG-3 binding, which allowed to avoid extrinsic effects on DCs by T cell activation in response to ABL501 treatment. Both HLA-ABC and CD40 expressions were upregulated in DCs when they were co-incubated with CHO-LAG-3 cells in the presence of ABL501 (Figure 3G). Collectively, these results imply that ABL501 can promote DC maturation for antigen cross-priming and subsequent CD8+ T cell activation.
Figure 3.

ABL501 promotes DC maturation to induce CD8+ T cell activation
(A–E) CTV-stained PBMCs were stimulated with CMV lysate in the presence of the indicated antibodies at various concentrations for 5 days. (A) Schematic illustration of CMV lysate assay (above) is shown. Representative FACS plots (left panel) and a summary plot (right panel) showing the percentages of proliferating CD4+ T cells (CTVlow) with the indicated antibodies at a final concentration of 66.8 nM are shown. (B) ELISA of IFN-γ secretion by CMV-responsive T cells with the indicated antibodies at various concentrations is shown. (C) Representative FACS plots show proliferating CD8+ populations (CTVlow) in CD3+ T cells (above) or the percentages of CTVlow CD8+ T cells (gated on CD8+ T; below) with the indicated antibodies at a final concentration of 66.8 nM. (D) Representative FACS plots (left panel) and a summary plot (right panel) show the percentages of CD25 expression in CMV-specific (CTVlow) CD8+ T cells. Data were compiled from five independent experiments with two replicates. (E and F) mo-DCs were incubated with plate-coated or soluble anti-hIgG or anti-PD-L1 for 2 days. (E) Representative FACS histograms showing the expression of HLA-ABC, HLA-DR, CD86, CD80, and CD40 in mo-DCs (left panel). Summary graph shows the geometric mean fluorescence intensity (MFI) of the expression of the indicated markers in mo-DCs (right panel). (F) Multiplex cytokine assay of the secretion of the indicated cytokines in mo-DCs is shown. (G) Schematic illustration of an in vitro DC activation assay (above). mo-DCs were incubated with CHO-LAG-3 cells in the presence of the indicated antibodies at a final concentration of 66.8 nM for 2 days. Summary graph shows the geometric MFI of the expression of the indicated markers in mo-DCs (below). Data were pooled from three independent experiments with two replicates. Statistical significance was determined by one-way ANOVA with Holm-Sidak multiple comparisons. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, and ns.
ABL501 potentiates CD8+ T cell cytotoxicity by promoting conjugation with tumor cells
We next investigated whether ABL501 directly affects CD8+ T cell responses against tumor. To measure the cytotoxic activity of CD8+ T cells against tumor cells, we employed 1G4 T cell receptor (TCR)-expressing CD8+ T cells that specifically recognize NY-ESO-1 cancer testis antigen (NY-ESO-1: 157–165 epitope) expressed by cancer cells in an HLA-A∗0201-restricted manner (Figure 4A).41 Co-culture of A375 cells endogenously expressing A2-NY-ESO-1 with 1G4 TCR-CD8+ T cells led to efficient killing of A375 cells in a dose-dependent manner, compared with TCR non-transduced (NT) CD8+ T-cell-mediated A375 killing, which is probably caused by allogeneic responses of CD8+ T cells against tumor cells (Figure S7). Treatment with ABL501 resulted in an increased killing rate of A375 cells by 1G4 TCR-CD8+ T cells, compared with combination treatment with single anti-LAG-3 and anti-PD-L1 at various concentrations (Figure 4B) or at effector:target (E:T) ratio of 1:10 and 1:20 (Figure 4C). In addition, 1G4 TCR-CD8+ T cells led to higher IFN-γ levels upon ABL501 treatment, compared with co-treatment with anti-LAG-3 and anti-PD-L1 (Figure 4D). Consistent with the enhanced effector responses of 1G4 TCR-CD8+ T cells by ABL501, upregulation of co-stimulatory markers, including CD25, 4-1BB, CD226, and ICOS, was observed in ABL501-treated 1G4 TCR-CD8+ T cells at 48 h after co-culture with A375 cells (Figure 4E). We then wanted to evaluate whether ABL501 requires simultaneous binding with both LAG-3 and PD-L1 to enhance 1G4 TCR-CD8+ T cell activation. To test this, we generated HLA-DR- or PD-L1-knockout (KO) A375 cells (Figure S8). ABL501 did not show an enhanced killing effect on HLA-DR KO or PD-L1 KO A375 cells by 1G4 TCR-CD8+ T cells, whereas the combination treatment retained its PD-L1-blocking effect in HLA-DR KO A375 cells (Figure 4F), which confirm that the effect of ABL501 is dependent on simultaneous binding to LAG-3 and PD-L1. We further elaborated how ABL501 elicits a higher cytotoxic activity in 1G4 TCR-CD8+ T cells against tumor cells than the combination of anti-LAG-3 and anti-PD-L1. Because bispecific T cell engagers are known to exert antitumor immune responses by assembling immunological synapse between T cells and tumor cells,42 we hypothesized that ABL501 acts as a T cell engager by bridging between CD8+ T cells and tumor cells. To test this hypothesis, we performed a conjugation assay using A375-OKT3-PD-L1 (BFP+) and carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled CD8+ T cells expressing LAG-3 (LAG-3+CD8+ T) by pre-activation with anti-CD3 and anti-CD28 (Figure 4G). When LAG-3+CD8+ T cells were incubated with A375-OKT3-PD-L1 cells in the presence of a combination of anti-LAG-3 and anti-PD-L1, we observed no significant increase in the BFP and CFSE double-positive population, compared with incubation with the control hIgG. In contrast, ABL501 treatment increased LAG-3+CD8+ or CD4+ T cell conjugation with A375-OKT3-PD-L1 cells (Figures 4H and S9), suggesting that ABL501 acts as a bridge facilitating tumor cell engagement with T cells.
Figure 4.

ABL501 plays a bridging role between CD8+ T cells and tumors to enhance tumor cell killing
(A–E) 1G4 TCR-engineered CD8+ T (1G4 TCR-CD8+ T) cells were co-cultured with A375 cells expressing HLA-A∗0201-restricted peptide NY-ESO-1 (157-165) at variable E:T ratios for 2 days in the presence of the indicated antibodies at various concentrations. (A) Schematic illustration of 1G4 TCR-engineered CD8+ T-cell-mediated tumor cell killing is shown. (B) Percentages of NY-ESO-1-specific killing of A375 cells at E:T ratio of 1:10 in the presence of the indicated antibodies at various concentrations are shown. (C and D) Percentages of NY-ESO-1-specific killing of A375 cells (ABL501: EC50 = 13.6 nM; Combi: EC50 = 19.5 nM) (C) or ELISA of IFN-γ secretion by 1G4 TCR-CD8+ T cells (D) at the indicated E:T ratios in response to combination treatment with anti-LAG-3 and anti-PD-L1 or ABL501 at a final concentration of 66.8 nM are shown. (E) Heatmap plot shows the geometric MFI of the expression of co-stimulatory molecules by 1G4 TCR-CD8+ T cells at E:T ratio of 1:10 in the presence of the indicated antibodies at a final concentration of 66.8 nM. (F) Percentages of NY-ESO-1-specific killing of wild-type (WT), PD-L1-deficient (PD-L1 KO), or HLA-DR-deficient (HLA-DR KO) A375 cells by 1G4 TCR-CD8+ T cells at an E:T ratio of 1:10 in the presence of the indicated antibodies at a final concentration of 66.8 nM are shown. Data were pooled from three independent experiments with three replicates. (G and H) CFSE-labeled CD8+ T cells expressing LAG-3 by pre-activation with anti-CD3/CD28 antibodies were incubated with A375-OKT3 cells expressing PD-L1-BFP for 30 min in the presence of the indicated antibodies at a final concentration of 66.8 nM, followed by FACS analysis. (G) Schematic illustration of T-cell-tumor conjugation assay (above) is shown. Representative FACS plots show PD-L1-BFP expression by A375-OKT3 cells and LAG-3 expression by pre-activated CD8+ T cells (below). (H) Representative FACS plots (left panel) and a summary plot (right panel) show the percentages of BFP and CFSE double-positive populations in the presence of the indicated antibodies. Data were compiled from four independent experiments with two replicates. Statistical significance was determined by one-way ANOVA with Holm-Sidak multiple comparisons in (B)–(D), (F), and (H). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ns.
Antitumor efficacy of ABL501 in humanized mouse tumor models
Considering that the binding arms of ABL501 do not cross-react with murine LAG-3 and PD-L1 molecules, the efficacy of ABL501 in vivo was assessed in humanized mouse models. First, we utilized a humanized tumor xenograft model. This model evaluates the efficacy of therapeutics to modulate antigen-specific cytolytic activity of an infused CD8+ T cell against implanted tumors expressing specific MHC-antigen complexes. We implanted A375-PD-L1 tumors into immunodeficient non-obese diabetic (NOD)-severe combined immunodeficiency (SCID) IL-2Rγnull (NSG) mice and adoptively transferred 1G4 TCR-T cells (Figure 5A). In mice bearing A375-PD-L1 tumors, adoptive transfer of 1G4 TCR-T cells was able to control tumor growth, compared with the no T cell transfer group (Figure S10A). Treatment with the parental anti-LAG-3 alone did not suppress tumor growth compared with control IgG treatment. The parental anti-PD-L1 treatment exhibited approximately 50% tumor growth inhibition (TGI), but tumors continued to grow. Notably, treatment with ABL501 yielded significant tumor regression (Figure 5A, bottom). To explore the antitumor activity mechanisms of ABL501, we analyzed TILs following the final treatment with antibodies. Anti-PD-L1 treatment increased the percentage and number of transferred CD8+ T cells in tumors. In contrast, anti-LAG-3 antibody had no effect on CD8+ TIL persistence. The frequency and number of 1G4 TCR CD8+ TILs were significantly higher in the ABL501-treated groups than in the other groups (Figure 5B). In addition, ABL501 treatment increased the frequency and expression level of IL-2, IFN-γ, and granzyme B by 1G4 TCR CD8+ TILs, compared with treatment with either parental antibody (Figures 5C and 5D). These results indicate that ABL501 potentiates antigen-specific CD8+ T cell responses against tumor. Next, we evaluated the in vivo antitumor efficacy of ABL501 using humanized transgenic BALB/c mice expressing human LAG-3 and PD-1. Human PD-L1 knockin CT26 cells (CT26hPD−L1) were implanted into the transgenic mice, followed by intraperitoneal administration of test antibodies. ABL501 exerted potent antitumor activity over a broad dose range of 1, 3, and 10 mg/kg (TGI = 22.8%, 30.2%, and 71.9%, respectively), resulting in improved overall survival (Figure 5E). The antitumor effect of ABL501 (10 mg/kg) was further confirmed in human PD-L1 knockin MC38 cells (MC38hPD−L1) bearing humanized transgenic C57BL/6 mice expressing human LAG-3, PD-1, and PD-L1 (Figure 5F). ABL501 treatment at all doses did not produce significant toxicity, as indicated by body weight measurements in mice (Figures S10B and S10C). We also assessed the safety of ABL501 in cynomolgus monkeys that were administered ABL501 at 0, 20, 60, or 200 mg/kg twice weekly, eight times. ABL501 was well tolerated up to 200 mg/kg/dose with a safe toxicity profile in cynomolgus monkeys (Figures S11A–S11C). In addition, treatment with ABL501 did not induce the secretion of pro-inflammatory cytokines, including IL-2, IFN-γ, IL-6, and TNF-α, by peripheral blood mononuclear cells (PBMCs) (Figure S11D).
Figure 5.

ABL501 treatment exerts antitumor effects in mouse tumor models
(A–D) 1G4 T cells expressing an NY-ESO-specific TCR were adoptively transferred into NSG mice bearing A375-PD-L1 tumors. At 1 day following T cell transfer, the mice were intraperitoneally treated with hIgG4 (10 mg/kg), anti-LAG3 (10 mg/kg), anti-PD-L1 (10 mg/kg), or ABL501 (14 mg/kg, molar equivalent amount) on days 11, 13, 17, and 20 (five mice/group). Tumor samples were collected on day 21 and processed for single-cell suspensions and then subjected to FACS analysis. (A) Experimental scheme of an in vivo xenograft mouse tumor model (above). Mice were monitored for tumor growth, and means ± SEM of n = 5 mice/group are shown (below). (B) Summary plots showing the percentages (left panel) and numbers (right panel) of 1G4 TCR-CD8+ T cells in tumors per treatment group. (C) Representative FACS plot (left panel) and summary plot (right panel) show the percentages of IL-2, IFN-γ, or granzyme B by 1G4 TCR-CD8+ T cells in tumors per treatment group. (D) Heatmap plot shows the geometric MFI of the expression of the indicated cytokines by tumor infiltrating 1G4 TCR-CD8+ T cells. (E) CT26 cells with knockin human PD-L1 were subcutaneously injected into human LAG-3/PD-1 knockin BALB/c mice (n = 6–8/group). Mice were treated with the indicated concentrations of ABL501 three times at 7-day intervals. Tumor growth curves of individual mice (right panel) are shown. (F) MC38 cells with knockin human PD-L1 were subcutaneously injected into human LAG-3/PD-1/PD-L1 knockin C57BL/6 mice (n = 5/group). Mice were treated with ABL501 (10 mg/kg) or isotype control (8.5 mg/kg) four times at 3-day intervals. Tumor growth curves of individual mice (right panel) are shown. Significant differences between groups were determined by two-way ANOVA Tukey’s multiple comparison test. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.001, ∗∗∗∗p < 0.001, and ns.
LAG-3hiPD-1hi memory CD4+ T cell signatures contribute to predicting the response to ABL501 in cholangiocarcinoma patients
Immune checkpoint inhibitor therapies are commonly used in the second-line setting in a variety of advanced solid malignancies after progression on standard-of-care treatment. To identify characteristics of peripheral blood T cells that relate to LAG-3xPD-L1 BsAb responsiveness, we next analyzed the expression of immune cell markers in the peripheral blood of cholangiocarcinoma (CCA) patients after therapy with gemcitabine plus cisplatin (GemCis), a standard regimen for advanced CCA (Figure S12).43,44 High-dimensional flow cytometry and self-organizing map (FlowSOM) algorithm were used to cluster immune cells based on the expression of immune cell markers, including CD3, CD4, CD8, Foxp3, CD19, CD56, CD14, and CD11c, and also to compare the abundance of each metacluster between relapsed and non-relapsed patients after GemCis therapy (Figures 6A and 6B). Frequencies of cells in cluster 4, which represents non-Treg CD4+ T cells (Figures 6B and 6C), were increased in relapsed patients compared with those in non-relapsed patients, suggesting a role of non-Treg CD4+ T cells in clinical responses. The abundance of non-Treg CD4+ T cell populations in relapsed patients was also shown by cluster identification, characterization, and regression (CITRUS) algorithm that allows the discovery of statistically significant signatures by unsupervised hierarchical clustering (Figure S13).45 We then further assess the phenotypic characteristics of both CD4+ and CD8+ T cells in CCA patients using CITRUS analysis (Figure 6D). Among four clusters (red dots) that were identified by CITRUS analysis, two (84953 and 84978) had a phenotype of CD62L−CD45RO− effector CD4+ T cells with a lower expression of LAG-3, 4-1BB, PD-1, Tim-3, and TIGIT, and the abundance of these clusters was increased in non-relapsed patients after GemCis therapy (Figures 6E and 6F). In contrast, the other two clusters (84981 and 84993) that represented CD62LhiCD45ROhi/int memory CD4+ T cells were more abundant in relapsed patients than in non-relapsed patients. CD4+ T cells in these clusters presented an activated phenotype with high expression levels of LAG-3, 4-1BB, PD-1, Tim-3, and TIGIT, showing an opposite expression profile from clusters 84993 and 84978. Similarly, these phenotypic differences of CD4+ T cells between relapsed and non-relapsed patients after GemCis therapy were visualized by t-distribution stochastic neighbor embedding (viSNE) analysis (Figure 6G). We next questioned which CD4+ T cell signatures were more responsive to ABL501 treatment. CD3+ T cells from patients were co-cultured with A375-OKT3 cells in the presence of anti-LAG-3, anti-PD-L1, or ABL501. CD3+ T cells in relapsed patients who had a greater abundance of CD62Lhi memory CD4+ T cells with high expression levels of LAG-3 and PD-1 were more responsive to ABL501 treatment than CD3+ T cells in non-relapsed patients (relapse: 3.08-fold; non-relapse: 1.85-fold) (Figure 6H). Similar results were observed for PBMC stimulation with a peptide pool (CEF+NY-ESO-1+MART1+) that induces antigen-specific CD8+ T cell responses. ABL501 efficiently augmented IFN-γ secretion by antigen-specific CD8+ T cells in relapsed patients compared with that in non-relapsed patients (relapse: 2.52-fold; non-relapse: 1.64-fold) (Figure 6I), indicating that the abundance of LAG-3hiPD-1hi memory CD4+ T cell subset has potential predictive value for ABL501 therapy.
Figure 6.

Abundance of LAG-3hiPD-1hi memory CD4+ T cells in peripheral blood of CCA patients was correlated with response to ABL501
(A–C) FlowSOM analysis of immune cell subsets in the peripheral blood of relapsed (n = 10) or non-relapsed (n = 9) CCA patients. (A) viSNE plot shows immune cell subset clusters to identify FlowSOM clustering. (B) Bar graph shows the frequencies of each cluster in two groups (relapse versus non-relapse). (C) Heatmap shows the MFI of each cluster. (D–F) CITRUS analysis of T cell signatures in the peripheral blood of relapsed (n = 9) or non-relapsed (n = 8) CCA patients is shown. (D) CITRUS plot shows clusters from CCA patients in two groups (relapse versus non-relapse). Red dots represent clusters that showed a different abundance with a statistical significance between groups. (E) Bar graphs show the relevance of abundance for the selected clusters (red dots) between groups. (F) Histogram plots show each marker expression by the selected clusters (red) over background (light blue). (G) Representative viSNE plots of CD4+ T cells in the peripheral blood of relapsed or non-relapsed CCA patients overlaid with the expression of naive, memory, co-inhibitory, or co-stimulatory markers. (H) Fold increase of IFN-γ secretion by CD3+ T cells from relapsed (n = 12) or non-relapsed (n = 9) CCA patients upon co-culture with A375-OKT3 in the presence of the indicated antibodies at a final concentration of 66.8 nM is shown. (I) Fold increase of IFN-γ secretion by peptide pool-treated peripheral blood CD8+ T cells obtained from relapsed (n = 11) or non-relapsed (n = 9) CCA patients in the presence of the indicated antibodies at a final concentration of 66.8 nM is shown. Statistical significance was determined by paired t tests with two-tailed analysis in (B) or one-way ANOVA with Holm-Sidak multiple comparisons in (H) and (I). ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.001, ∗∗∗∗p < 0.001, and ns.
Discussion
Therapeutic strategies for restoring T cell activity via co-blocking of LAG-3 and PD-1/PD-L1 with mAbs have proven to be beneficial in preclinical tumor models23,24 and humans.46, 47, 48 Especially with the FDA approval of Opdualag (nivolumab and relatlimab-rmbw),27 new approaches targeting LAG-3 and PD-(L)1 with BsAbs, which are expected to allow more selective binding of antibodies and a simultaneous blockade of both LAG-3 and PD-(L)1, have been actively developed in the preclinical and early clinical stages.28, 29, 30, 31 However, despite the apparent benefits of BsAb, the functional and mechanistic advantages of dual blocking of LAG-3 and PD-(L)1 by BsAb over a combination of anti-LAG-3 and anti-PD-(L)1 mAbs are still unclear.
Our study revealed that ABL501, a BsAb targeting LAG-3 and PD-L1, promoted antigen cross-presentation by DCs and subsequently induced CD8+ T cell activation to a higher extent than a combination treatment with anti-LAG-3 and anti-PD-L1 mAbs. This result highlights the benefit of co-blocking LAG-3 and PD-L1 with the BsAb. As it has been shown that PD-1/PD-L1 pathway blockade enhances DC maturation and activation,38, 39, 40 the observed effect of ABL501 in DCs might come from the anti-PD-L1 part of the BsAb, which is expected to provide either a stronger blocking effect or more stable binding to PD-L1 on DCs than anti-PD-L1 mAbs. Indeed, we observed enhanced mo-DC activation with the plate-coated anti-PD-L1, but not with the soluble anti-PD-L1 treatment.
Although PD-L1 blockade directly affected DC activation, co-blockade of LAG-3 and PD-L1 mounted a higher level of CD8+ T cell responses upon CMV lysate stimulation compared with LAG-3 or PD-L1 blockade alone, which implies that LAG-3 blockade can contribute to PD-L1 blockade-mediated DC activation. One possible explanation is that LAG-3 blockade can augment CMV-specific CD4+ T cell responses, accompanied by an increased secretion of IFN-γ, IL-2, and TNF-α, supporting DC activation and leading to the subsequent cross-priming of CD8+ T cells.49, 50, 51 A recent study suggested that treatment with LAG-3xPD-L1 BsAb increased soluble LAG-3 (sLAG-3) levels by promoting the shedding of LAG-3 from the cell surface, although it is unclear how LAG-3xPD-L1 BsAb induced the proteolytic cleavage of surface LAG-3.29 Because a recombinant sLAG-3Ig protein has been reported to stimulate the maturation of DCs or macrophages by competitively binding to MHC class II against surface LAG-3,52, 53, 54, 55 increased sLAG-3 by LAG-3xPD-L1 BsAb was suggested as an indicator of T cell or DC activation.29 However, unlike recombinant sLAG-3Ig protein, the role of sLAG-3 or its binding to MHC class II is unknown,56,57 warranting further investigation of its biological relevance.
Cross-presenting conventional DC1s (cDC1s) that efficiently mount CD8+ T cell responses against exogenous antigens, such as tumor antigens, have been highlighted as a target of cancer vaccines.58, 59, 60 Recent approaches to deliver cancer vaccines specifically into cDC1s have shown promising results in preclinical settings, with an efficient induction of antigen-specific effector or memory CD8+ T cells and tumor regression.61, 62, 63 In this regard, the novel mechanism of ABL501 in regulating cross-presenting DCs not only suggests a non-redundant benefit of our bispecific antibody approach but also provides a rationale for therapeutic strategies, including combination with cross-presenting, DC-targeting cancer vaccines.
Aside from this DC-mediated mechanism through which ABL501 promotes CD8+ T cell activation, we also found that ABL501 can directly enhance the cytotoxic activity of CD8+ T cells against tumor cells by promoting CD8+ T cell conjugation with tumor cells. A recent study also showed a bridging function of LAG-3xPD-L1 BsAb in LAG-3- or PD-L1-overexpressing cell lines without TCR engagement,64 but it insufficiently addressed the bridging role of LAG-3xPD-L1 BsAb in a situation where T cells are engaged with tumor cells through pMHC-I-TCR binding. We employed NY-ESO-1-specific, TCR-engineered (1G4 TCR) CD8+ T cells to address the mechanistic and functional roles of ABL501 against NY-ESO-1-expressing tumor cells under a more physiological condition. ABL501 treatment resulted in nearly complete tumor regression in A375-PD-L1-bearing NSG mice subjected to adoptive transfer of 1G4 TCR-CD8+ T cells, which argues that ABL501 can sufficiently support the antitumor responses of CD8+ T cells without the intervention of CD4+ T cells. Another advantage of this model is that it provides a unique opportunity to study the biology of human tumor antigen-specific CD8+ TILs, which would allow better prediction of patients’ responses to T-cell-targeting reagents, including ABL501.65 Regardless of these obvious benefits, this model cannot fully recapitulate human tumor microenvironment due to the NSG mouse background. We exploited human LAG-3, PD-1, and PD-L1 knockin (KI) syngeneic mouse models to compensate for the limitation of the xenograft model. ABL501 showed an ability to elicit efficient antitumor responses by immune cells in the intact tumor microenvironment, which further supports its potential as cancer immunotherapeutics. However, we cannot rule out the possibility that the reconstitution of human receptors affects endogenous immune responses in KI mice. Therefore, further investigation about the modulation of TILs by ABL501 and its translational value is warranted.
Identification and characterization of the major immune cell populations responding to immune checkpoint inhibitor (ICI) therapies are crucial for their clinical success, emphasizing the importance of immune-monitoring studies in cancer patients. Compared with CD8+ T cell subsets that have been intensively investigated in the context of predicting clinical responses to ICI therapies,66,67 the characteristics and predictive value of CD4+ T cell subsets, other than Treg cells, are less well known, despite the importance of CD4+ T cell support for the antitumor immune responses of CD8+ T cells.68 Recent studies have reported the correlation between CD4+ T cell immunity and clinical responses to PD-1/PD-L1 blockade in cancer patients.69, 70, 71 In particular, one study used the immune monitoring of peripheral blood from patients with non-small cell lung cancer to show that the frequency of CD62Llo CD4+ T cells is a predictive biomarker.70
Our immune-monitoring studies also revealed that the decreased abundance of CD62LloCD45ROlo effector CD4+ T cell populations in peripheral blood was correlated to disease recurrence in patients with CCA after GemCis therapy. Conversely, LAG-3hiPD-1hiCD62Lhi memory CD4+ T cells exhibiting an activated phenotype with high expression of both co-stimulatory and inhibitory markers were more abundant in relapsed CCA patients, which implicates the slower kinetics of GemCis-therapy-induced CD4+ T cell activation or repopulation. A correlation between the LAG-3+ immunotype in peripheral blood and poor clinical outcomes was also found in patients with melanoma or urothelial carcinoma after anti-PD-1 or anti-CTLA-4 therapies.64 This CD4+ T cell phenotype in relapsed CCA patients turned out to be beneficial for eliciting T cell responses upon ABL501 treatment. In relapsed patients, ABL501-mediated T cell activation might involve not only the CD62L+ memory characteristics but also the increased LAG-3 expression on CD4+ T cells. Although the expression level of immune checkpoint receptors on T cells is not always positively correlated with the clinical response rate, as shown in PD-1 expression and anti-PD-1 blockade,72 it was reported in a small cohort study that melanoma patients with higher LAG-3 expression on TILs were more responsive to a combination of relatlimab (BMS-986016) plus nivolumab.46 Our study therefore proposes new treatment strategies for LAG-3xPD-L1 BsAb therapy in combination with chemotherapies and offers novel criteria for patient stratification.
In summary, our study illustrates the divergent ability of LAG-3xPD-L1 bispecific antibody to enhance the potency of T cell activation and highlights how this bispecific antibody can exert the immune modulation not achievable by a combination of mAbs. The favorable toxicity profile and potent antitumor activity of ABL501 support its clinical evaluation in patients with cancer.
Materials and methods
Cell lines
A375, Jurkat, and HCC1954 cells were purchased from American Type Culture Collection (CRL-1619, TIB-152, and CRL-2338). SNU-324 cells were obtained from Korean Cell Line Bank (KCLB) (00324). CHO-S cells were purchased from Invitrogen. All cells were maintained as recommended by the suppliers.
Stable cell line generation
A375 cell lines stably expressing firefly luciferase and/or human PD-L1 were generated (A375-Fluc, A375-PD-L1, and A375-Fluc-PD-L1) using a Sleeping Beauty transposon system.73 Full-length human PD-L1 gene was cloned into a bicistronic BFP expression vector, pSBbi-BP (Addgene plasmid no. 60512). For the generation of T cell stimulator cells, A375-Fluc and A375-Fluc-PD-L1 cells were infected with a lentivirus encoding mouse anti-human CD3 (OKT3) scFv fused to CD28 transmembrane domain (A375-Fluc-OKT3 and A375-Fluc-PD-L1-OKT3). OKT3 scFv expression on cell surface was verified by FACS using an anti-mouse IgG antibody. PD-L1 or HLA-DR in A375-Fluc cells was knocked out via CRISPR-Cas9. The guide sequences (PD-L1: 5′-TCTTACCACTCAGGACTTGA-3′, HLA-DR: 5′-GAGTACTGGAACAGCCAGA-3′, designed by the CRIPSPR Gold online tool) were cloned into pX330-mCherry vector (Addgene plasmid no. 98750). A375-Fluc cells were transfected with single guide RNA (sgRNA)-expressing plasmids, and the resulting transfected cells were single cell subcloned.74
Mixed lymphocyte reaction (MLR) assay
Human CD3+ T cells were separated form PBMCs using an EasySep Human T cell Isolation Kit (STEMCELL Technologies). CD3+ T cells were stained with CellTrace Violet (CTV) (Invitrogen). The proliferation of T cells was evaluated by measuring CTV dilution by flow cytometry. To assess allospecific T cell activation, CD14+ monocytes were separated from allo-PBMCs using MojoSort Human CD14 Nanobeads (BioLegend) and cultured in complete RPMI 1640 medium with granulocyte-macrophage colony-stimulating factor (GM-CSF) (80 ng/mL) and IL-4 (80 ng/mL) for 5 days. Allogenic immature DCs were co-cultured with CTV-stained CD3+ T cells for 5 days at a ratio of 15:1 (T:DC).
1G4 TCR-T cell assay
A lentiviral vector encoding a 1G4 TCR that recognizes the HLA-A∗0201 restricted NY-ESO-1 peptide (NY-ESO-1:157-165) was generated in the pHR lentiviral backbone (Addgene plasmid no. 79125). Lentivirus was generated by transient transfection of 293FT cells with the packaging plasmids psPAX2 (Addgene plasmid no. 12260) and pVSVg (Addgene plasmid no. 8454). Human primary CD8+ T cells were isolated from PBMCs and stimulated with Dynabeads Human T-Activator CD3/CD28 (Invitrogen) in the presence of IL-2 (100 ng/mL; Peprotech). A lentivirus encoding a 1G4 TCR was transduced into CD8+ T cells 3 days after the stimulation by spinoculation in the presence of polybrene (8 μg/mL). The expression of 1G4 TCR was assessed by flow cytometric analysis using an anti-human Vβ13.1 TCR chain antibody (BioLegend).
CMV lysate assay
To analyze recall responses of memory T cells in an antigen-specific manner, PBMCs were treated with 100 ng/mL of CMV lysates (Microbix Biosystems) for 5 days. T cell responses were evaluated by IFN-γ ELISA and CTV dilution assays.
Human Treg cell suppression assay
CD3+CD4+CD25+CD127− Treg cells were isolated from PBMCs and stimulated with Dynabeads Human T-Activator CD3/CD28 (Invitrogen) at a cell-to-bead ratio of 4:1 in complete RPMI 1640 medium with IL-2 (100 ng/mL; Peprotech) and TGF-β (20 ng/mL; Peprotech). To avoid allogenic immune responses, PBMCs from the same donor with Treg cells were used for isolating CD4+ T cells and CD8+ T cells. CD4+ and CD8+ T cells were separated using Mojosort Human CD4 Nanobeads (BioLegend) and EasySep Human CD8+ T cell Enrichment Kit (STEMCELL Technologies), respectively. T cells were stained with CTV. A375-PD-L1-OKT3-Fluc cells were treated mitomycin C for 20 min at 37°C and then co-cultured with CTV-stained T cells at a T:A375 cell ratio of 5:1. Next, the isolated immunosuppressive autogenic Treg cells were stained with CFSE (Invitrogen) and added into the A375-T cell co-culture. After 5 days of incubation, the immunosuppressive activity of Treg cells was measured from the proliferation of CTV-stained CD4+ and CD8+ T cells via quantification of CTV dilution. The culture medium was collected and analyzed with ELISA for IL-2 (BioLegend).
Cell conjugation assay
CFSE-stained human CD3+ T cells were stimulated with Dynabeads Human T-Activator CD3/CD28 (Invitrogen) for 48 h, and surface LAG3 expression on T cells was verified by flow cytometry. LAG3+ T cells were incubated with A375-Fluc-PD-L1 cells (BFP-positive) at a T to A375 ratio of 1:1 for 1 h at 37°C. The conjugation ratio was calculated as the portion of CFSE/BFP double-positive events.
Antibodies and flow cytometry
Mouse and human single cells were stained with fluorochrome-conjugated antibodies for 20 min at 4°C after blocking with anti-mouse CD16/32 (BioLegend) and human TruStain FcX (BioLegend) in FACS buffer (phosphate-buffered saline [PBS] containing 1% BSA and 0.1% sodium azide). For intracellular staining, cells were fixed and permeabilized using a Cytofix/Cytoperm Kit (BD Biosciences) or Foxp3/Transcription Factor Staining Buffer Set (Invitrogen). The following fluorochrome-conjugated anti-human antibodies were used: anti-CD3 (HIT3a), anti-CD4 (OKT4), anti-CD8 (SK1), anti-CD11c (3.9), anti-CD14 (63D3), anti-CD19 (HIB19), anti-CD25 (BC96), anti-CD45RO (UCHL1), anti-CD56 (5.1H11), anti-CD62L (DREG-56), anti-CD69 (FN50), anti-CD80 (2D10), anti-CD86 (IT2.2), anti-CD96 (6F9), anti-4-1BB (4B4-1), anti-Lag3 (11C3C65), anti-CD226 (11A8), anti-PD-1 (EH12.2H7), anti-Tim3 (F38-2E2), anti-TIGIT (MBSA43), anti-HLA-DR (BB7.2), anti-HLA-ABC (W6/32), anti-Foxp3 (236A-E7), and anti-mouse-TCRβ (H57-597).
Luminex multiplex cytokine assay
To measure multi-cytokine production, human monocyte-derived DCs were stimulated with plate-bound parental anti-PD-L1 (10 μg/mL) or soluble parental anti-PD-L1 (10 μg/mL) for 48 h. Supernatants were collected, and cytokines were detected using Luminex assay kit (R&D systems) according to the manufacturer’s instructions (R&D Systems).
Human samples and study approval
Blood samples were obtained from healthy donors and CCA patients who received adjuvant gemcitabine plus cisplatin (GemCis) after curative resection under the clinical trial (ClinicalTrials.gov identifier NCT03079427) approved by Asan Medical Center Institutional Review Board (2021-0677).
Mouse studies
Murine experiments were approved and performed in accordance with the guidelines of the Institutional Animal Care and Use Committees of the Asan Medical Center (201904103) or GemPharmatech (GPTAP20200713-4 and GPTAP20201230-2).
Xenogeneic tumor model
A375-PD-L1 tumor cells were implanted in the right flank of immune-deficient NSG mice (JA Bio, Suwon, Korea). After tumors were established (mean volume of 150 mm3), the mice were randomly assigned into five groups. One group received a single intravenous tail-vein injection of PBS, and four groups were administered 1G4 TCR-expressing human CD8+ T cells (2 × 106 cells). At 1 day following T cell transfer, mice were treated intraperitoneally every 3 days 4 times with human IgG (10 mg/kg), anti-PD-L1 (10 mg/kg), anti-LAG3 (10 mg/kg), or a molar equivalent amount of ABL501 (14 mg/kg). Tumor growth and body weight were measured every 3 days.
Syngeneic tumor model
Syngeneic tumor studies were carried out in BALB/c mice expressing human LAG-3 and PD-1 (BALB/c-hPD1/hLAG3, T004622; GemPharmatech) and C57BL/6 mice expressing human LAG-3, PD-1, and PD-L1 (C57BL/6-hPD1/hPDL1/hLAG3, T006876; GemPharmatech). hPD1/hLAG3 humanized mice (seven mice/group) were implanted with 2 × 106 CT26-hPD-L1(Tg)-mPD-L1(KO) colon cancer cells (GemPharmatech) by subcutaneous injection on the right flank. When the average tumor size reached 87 mm3, antibodies were administered via intraperitoneal injection of hIgG1 (7.5 mg/kg), hPD-L1 (7.5 mg/kg), or ABL501(1, 3 or 10 mg/kg) three times at 7-day intervals. hLAG-3/hPD-1/hPD-L1 humanized mice (five mice/group) were implanted with 2 × 106 MC38-hPD-L1(Tg)-mPD-L1(KO) colon cancer cells (GemPharmatech) by subcutaneous injection on the right flank. ABL501 (10 mg/kg) or human IgG1 isotype control (8.5 mg/kg) was subsequently administered by intraperitoneal injection four times every 3 days. Tumor growth was monitored over time via caliper measurements. Mice were euthanized when the tumor size reached 2,500 mm3.
Cytobank analysis
viSNE, FlowSOM, and CITRUS analyses were performed using Cytobank software (Beckman Coulter). For viSNE analysis, equal number of cells (1,000 cells) from each flow cytometry standard (FCS) file were used, and resulting viSNE maps were fed into FlowSOM analysis.75 For each cell subset, a new self-organizing map was generated using hierarchical consensus clustering on the tSNE axes. For CITRUS analysis, association models of significance analysis of microarrays and nearest shrunken centroid were selected as statistical methods. Abundance mode was selected to quantify and characterize individual clusters in samples. The minimal cluster size was 3%, and the cross-validation fold was 5.45
Good Laboratory Practices toxicity study in cynomolgus monkeys
The cynomolgus monkey study was conducted in accordance with guidelines of the Institutional Animal Care and Use Committee (IACUC) (SZ20200416-monkeys). Cynomolgus monkeys (five/sex/group) were administered a slow intravenous injection of ABL501 at 0, 20, 60, or 200 mg/kg twice weekly, eight times (days 1, 4, 8, 11, 15, 18, 22, and 25). Necropsy was performed on day 29 for the main groups (three/sex/group), followed by a recovery period for 56 days (two/sex/group). Safety and toxicity were assessed based on standard parameters. The study was conducted in accordance with the Safety and Quality Assurance guidelines in the Guideline for Experiments Document of WuXiAppTec.
Statistical analysis
Statistical analyses were performed using the appropriate statistical comparison, including the two-tailed paired or unpaired Student’s t test, one-way ANOVA with Holm-Sidak multiple comparisons, or two-way ANOVA with Tukey’s multiple comparisons. p < 0.05 were considered statistically significant (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001).
Acknowledgments
We thank the core facilities of Genetically Engineered Animal Core and flow cytometry at the Convergence Medicine Research Center (CREDIT), Asan Medical Center. This work was supported by an intramural grant of KIST, by the National Research Foundation of Korea grants (NRF-2021R1A2C1003551 to H.J., NRF-2020M3A9G7103935 to H.J., and NRF-2021R1A2C2006647 to Y.P.), and by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health & Welfare of South Korea (HI20C0117 to J. Jung).
Author contributions
Conception and design, E.S., E.P., Jonghwa Won, H.J., J. Jung, and Y.P.; development of methodology, E.S., M.K., H.J., J. Jung, and Y.P.; acquisition of data, Ju-young Won, Y.J., E.C., U.J., J. Jeon, Y.K., H.A., D.C., S.C., Y.H., H.P., H.L., Y.-G.S., and K.P.; technical and material support, S.J.O., S.L., K.K., C.Y., and H.K.S.; analysis and interpretation of data, E.S., E.P., H.K., H.P., H.L., Jonghwa Won, H.J., J. Jung, and Y.P.; writing the manuscript, E.S., H.J., J. Jung, and Y.P.; study supervision, H.J., J. Jung, and Y.P.
Declaration of interests
E.S., E.P., H.K., U.J., J. Jeon, Y.K., Y.H., H.P., H.L., Y.-G.S., K.P., Jonghwa Won, and J. Jung are employees of ABL Bio Inc. The other authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.05.003.
Contributor Information
Hyung-seung Jin, Email: hsjin@amc.seoul.kr.
Jaeho Jung, Email: jaeho.jung@ablbio.com.
Yoon Park, Email: ypark@kist.re.kr.
Supplemental information
References
- 1.Dunn G.P., Old L.J., Schreiber R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004;22:329–360. doi: 10.1146/annurev.immunol.22.012703.104803. [DOI] [PubMed] [Google Scholar]
- 2.Callahan M.K., Postow M.A., Wolchok J.D. Targeting T cell Co-receptors for cancer therapy. Immunity. 2016;44:1069–1078. doi: 10.1016/j.immuni.2016.04.023. [DOI] [PubMed] [Google Scholar]
- 3.McLane L.M., Abdel-Hakeem M.S., Wherry E.J. CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol. 2019;37:457–495. doi: 10.1146/annurev-immunol-041015-055318. [DOI] [PubMed] [Google Scholar]
- 4.Shen R.L., Postow M.A., Adamow M., Arora A., Hannum M., Maher C., Wong P., Curran M.A., Hollmann T.J., Jia L.W., et al. LAG-3 expression on peripheral blood cells identifies patients with poorer outcomes after immune checkpoint blockade. Sci. Transl. Med. 2021;13:eabf5107. doi: 10.1126/scitranslmed.abf5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Topalian S.L., Hodi F.S., Brahmer J.R., Gettinger S.N., Smith D.C., McDermott D.F., Powderly J.D., Sosman J.A., Atkins M.B., Leming P.D., et al. Five-year survival and correlates among patients with advanced melanoma, renal cell carcinoma, or non-small cell lung cancer treated with nivolumab. Jama Oncol. 2019;5:1411–1420. doi: 10.1001/jamaoncol.2019.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garon E.B., Rizvi N.A., Hui R.N., Leighl N., Balmanoukian A.S., Eder J.P., Patnaik A., Aggarwal C., Gubens M., Horn L., et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015;372:2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 7.Zou W.P., Wolchok J.D., Chen L.P. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016;8:328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Triebel F., Jitsukawa S., Baixeras E., Romanroman S., Genevee C., Viegaspequignot E., Hercend T. Lag-3, a novel lymphocyte-activation gene closely related to Cd4. J. Exp. Med. 1990;171:1393–1405. doi: 10.1084/jem.171.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Triebel F. LAG-3: a regulator of T-cell and DC responses and its use in therapeutic vaccination. Trends Immunol. 2003;24:619–622. doi: 10.1016/j.it.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 10.Maruhashi T., Sugiura D., Okazaki I.M., Okazaki T. LAG-3: from molecular functions to clinical applications. J. Immunother. Cancer. 2020;8:e001014. doi: 10.1136/jitc-2020-001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maruhashi T., Okazaki I.M., Sugiura D., Takahashi S., Maeda T.K., Shimizu K., Okazaki T. LAG-3 inhibits the activation of CD4(+) T cells that recognize stable pMHCII through its conformation-dependent recognition of pMHCII. Nat. Immunol. 2018;19:1415–1426. doi: 10.1038/s41590-018-0217-9. [DOI] [PubMed] [Google Scholar]
- 12.Maruhashi T., Sugiura D., Okazaki I.M., Shimizu K., Maeda T.K., Ikubo J., Yoshikawa H., Maenaka K., Ishimaru N., Kosako H., et al. Binding of LAG-3 to stable peptide-MHC class II limits T cell function and suppresses autoimmunity and anti-cancer immunity. Immunity. 2022;55:912–924.e8. doi: 10.1016/j.immuni.2022.03.013. [DOI] [PubMed] [Google Scholar]
- 13.Xu F., Liu J., Liu D., Liu B.A., Wang M., Hu Z.Y., Du X.M., Tang L., He F.C. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. 2014;74:3418–3428. doi: 10.1158/0008-5472.Can-13-2690. [DOI] [PubMed] [Google Scholar]
- 14.Kouo T., Huang L.Q., Pucsek A.B., Cao M.W., Solt S., Armstrong T., Jaffee E. Galectin-3 shapes antitumor immune responses by suppressing CD8(+) T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol. Res. 2015;3:412–423. doi: 10.1158/2326-6066.Cir-14-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mao X., Ou M., Karuppagounder S., Kam T.I., Yin X., Xiong Y., Ge P., Umanah G., Brahmachari S., Shin J., et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science. 2017;353:aah3374. doi: 10.1126/science.aah3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J., Sanmamed M.F., Datar I., Su T.T., Ji L., Sun J.W., Chen L., Chen Y.S., Zhu G.F., Yin W.W., et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell. 2019;176:334–347.e12. doi: 10.1016/j.cell.2018.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang C.T., Workman C.J., Flies D., Pan X., Marson A.L., Zhou G., Hipkiss E.L., Ravi S., Kowalski J., Levitsky H.I., et al. Role of LAG-3 in regulatory T cells. Immunity. 2004;21:503–513. doi: 10.1016/j.immuni.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Q.X., Chikina M., Szymczak-Workman A.L., Horne W., Kolls J.K., Vignali K.M., Normolle D., Bettini M., Workman C.J., Vignali D.A.A. LAG3 limits regulatory T cell proliferation and function in autoimmune diabetes. Sci. Immunol. 2017;2:eaah4569. doi: 10.1126/sciimmunol.aah4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsuzaki J., Gnjatic S., Mhawech-Fauceglia P., Beck A., Miller A., Tsuji T., Eppolito C., Qian F., Lele S., Shrikant P., et al. Tumor-infiltrating NY-ESO-1-specific CD8(+) T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. U S A. 2010;107:7875–7880. doi: 10.1073/pnas.1003345107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y.X., Dong T.Y., Xuan Q.J., Zhao H., Qin L., Zhang Q.Y. Lymphocyte-activation gene-3 expression and prognostic value in neoadjuvant-treated triple-negative breast cancer. J. Breast Cancer. 2018;21:124–133. doi: 10.4048/jbc.2018.21.2.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shapiro M., Herishanu Y., Katz Ben-Zion, Dezorella N., Sun C., Kay S., Polliack A., Avivi I., Wiestner A., Perry C. Lymphocyte activation gene 3: a novel therapeutic target in chronic lymphocytic leukemia. Haematologica. 2017;102:874–882. doi: 10.3324/haematol.2016.148965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yi Q., Zhixin F., Yuanxiang G., Wei X., Bushu X., Jingjing Z., Huoying C., Xinke Z., Musheng Z., Yao L., Xing Z. LAG-3 expression on tumor-infiltrating T cells in soft tissue sarcoma correlates with poor survival. Cancer Biol. Med. 2019;16:331. doi: 10.20892/j.issn.2095-3941.2018.0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woo S.R., Turnis M.E., Goldberg M.V., Bankoti J., Selby M., Nirschl C.J., Bettini M.L., Gravano D.M., Vogel P., Liu C.L., et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72:917–927. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang R.Y., Eppolito C., Lele S., Shrikant P., Matsuzaki J., Odunsi K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget. 2015;6:27359–27377. doi: 10.18632/oncotarget.4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waugh K.A., Leach S.M., Moore B.L., Bruno T.C., Buhrman J.D., Slansky J.E. Molecular profile of tumor-specific CD8(+) T cell hypofunction in a transplantable murine cancer model. J. Immunol. 2016;197:1477–1488. doi: 10.4049/jimmunol.1600589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goding S.R., Wilson K.A., Xie Y., Harris K.M., Baxi A., Akpinarli A., Fulton A., Tamada K., Strome S.E., Antony P.A. Restoring immune function of tumor-specific CD4(+) T cells during recurrence of melanoma. J. Immunol. 2013;190:4899–4909. doi: 10.4049/jimmunol.1300271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tawbi H.A., Schadendorf D., Lipson E.J., Ascierto P.A., Matamala L., Castillo Gutierrez E., Rutkowski P., Gogas H.J., Lao C.D., De Menezes J.J., et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N. Engl. J. Med. 2022;386:24–34. doi: 10.1056/NEJMoa2109970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J., Asch A.S., Hamad N., Weickhardt A., Tomaszewska-Kiecana M., Dlugosz-Danecka M., Pylypenko H., Bahadur B., Ulahannan S., Koucheki J., et al. A phase 1, open-label study of MGD013, a bispecific DART® molecule binding PD-1 and LAG-3 in patients with relapsed or refractory diffuse large B-cell lymphoma. Blood. 2020;136:21–22. doi: 10.1182/blood-2020-139868. [DOI] [Google Scholar]
- 29.Kraman M., Faroudi M., Allen N.L., Kmiecik K., Gliddon D., Seal C., Koers A., Wydro M.M., Batey S., Winnewisser J., et al. FS118, a bispecific antibody targeting LAG-3 and PD-L1, enhances T-cell activation resulting in potent antitumor activity. Clin. Cancer Res. 2020;26:3333–3344. doi: 10.1158/1078-0432.Ccr-19-3548. [DOI] [PubMed] [Google Scholar]
- 30.Yap T., Wong D., Hu-Lieskovan S., Papadopoulos K., Morrow M., Grabowska U., Gliddon D., Holz J.B., LoRusso P. 395 A first-in-human study of FS118, a tetravalent bispecific antibody targeting LAG-3 and PD-L1, in patients with advanced cancer and resistance to PD-(L)1 therapy. J. Immunother. Cancer. 2020;8:A240. doi: 10.1136/jitc-2020-SITC2020.0395. [DOI] [PubMed] [Google Scholar]
- 31.Jiang H.P., Ni H.Q., Zhang P., Guo X.L., Wu M., Shen H.R., Wang J., Wu W.W., Wu Z.H., Ding J.Z., et al. PD-L1/LAG-3 bispecific antibody enhances tumor-specific immunity. Oncoimmunology. 2021;10:1943180. doi: 10.1080/2162402x.2021.1943180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Angal S., King D.J., Bodmer M.W., Turner A., Lawson A.D., Roberts G., Pedley B., Adair J.R. A single amino acid substitution abolishes the heterogeneity of chimeric mouse/human (IgG4) antibody. Mol. Immunol. 1993;30:105–108. doi: 10.1016/0161-5890(93)90432-b. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Q., Chikina M., Szymczak-Workman A.L., Horne W., Kolls J.K., Vignali K.M., Normolle D., Bettini M., Workman C.J., Vignali D.A.A. LAG3 limits regulatory T cell proliferation and function in autoimmune diabetes. Sci. Immunol. 2017;2:eaah4569. doi: 10.1126/sciimmunol.aah4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Godfrey W.R., Ge Y.G., Spoden D.J., Levine B.L., June C.H., Blazar B.R., Porter S.B. In vitro-expanded human CD4(+)CD25(+) T-regulatory cells can markedly inhibit allogeneic dendritic cell-stimulated MLR cultures. Blood. 2004;104:453–461. doi: 10.1182/blood-2004-01-0151. [DOI] [PubMed] [Google Scholar]
- 35.Levitsky J., Miller J., Leventhal J., Huang X., Flaa C., Wang E., Tambur A., Burt R.K., Gallon L., Mathew J.M. The human "Treg MLR": immune monitoring for FOXP3+ T regulatory cell generation. Transplantation. 2009;88:1303–1311. doi: 10.1097/TP.0b013e3181bbee98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Booth N.J., McQuaid A.J., Sobande T., Kissane S., Agius E., Jackson S.E., Salmon M., Falciani F., Yong K., Rustin M.H., et al. Different proliferative potential and migratory characteristics of human CD4+ regulatory T cells that express either CD45RA or CD45RO. J. Immunol. 2010;184:4317–4326. doi: 10.4049/jimmunol.0903781. [DOI] [PubMed] [Google Scholar]
- 37.Sylwester A.W., Mitchell B.L., Edgar J.B., Taormina C., Pelte C., Ruchti F., Sleath P.R., Grabstein K.H., Hosken N.A., Kern F., et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005;202:673–685. doi: 10.1084/jem.20050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuipers H., Muskens F., Willart M., Hijdra D., van Assema F.B., Coyle A.J., Hoogsteden H.C., Lambrecht B.N. Contribution of the PD-1 ligands/PD-1 signaling pathway to dendritic cell-mediated CD4+ T cell activation. Eur. J. Immunol. 2006;36:2472–2482. doi: 10.1002/eji.200635978. [DOI] [PubMed] [Google Scholar]
- 39.Park S.J., Namkoong H., Doh J., Choi J.C., Yang B.G., Park Y., Chul Sung Y. Negative role of inducible PD-1 on survival of activated dendritic cells. J. Leukoc. Biol. 2014;95:621–629. doi: 10.1189/jlb.0813443. [DOI] [PubMed] [Google Scholar]
- 40.Versteven M., Van den Bergh J.M.J., Marcq E., Smits E.L.J., Van Tendeloo V.F.I., Hobo W., Lion E. Dendritic cells and programmed death-1 blockade: a joint venture to combat cancer. Front. Immunol. 2018;9:394. doi: 10.3389/fimmu.2018.00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robbins P.F., Li Y.F., El-Gamil M., Zhao Y., Wargo J.A., Zheng Z., Xu H., Morgan R.A., Feldman S.A., Johnson L.A., et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J. Immunol. 2008;180:6116–6131. doi: 10.4049/jimmunol.180.9.6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tian Z., Liu M., Zhang Y., Wang X. Bispecific T cell engagers: an emerging therapy for management of hematologic malignancies. J. Hematol. Oncol. 2021;14:75. doi: 10.1186/s13045-021-01084-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valle J., Wasan H., Palmer D.H., Cunningham D., Anthoney A., Maraveyas A., Madhusudan S., Iveson T., Hughes S., Pereira S.P., et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010;362:1273–1281. doi: 10.1056/NEJMoa0908721. [DOI] [PubMed] [Google Scholar]
- 44.Park J.O., Oh D.Y., Hsu C., Chen J.S., Chen L.T., Orlando M., Kim J.S., Lim H.Y. Gemcitabine plus cisplatin for advanced biliary tract cancer: a systematic Review. Cancer Res. Treat. 2015;47:343–361. doi: 10.4143/crt.2014.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bruggner R.V., Bodenmiller B., Dill D.L., Tibshirani R.J., Nolan G.P. Automated identification of stratifying signatures in cellular subpopulations. Proc. Natl. Acad. Sci. U S A. 2014;111:E2770–E2777. doi: 10.1073/pnas.1408792111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ascierto P.A., Melero I., Bhatia S., Bono P., Sanborn R.E., Lipson E.J., Callahan M.K., Gajewski T., Gomez-Roca C.A., Hodi F.S., et al. Initial efficacy of anti-lymphocyte activation gene-3 (anti-LAG-3; BMS-986016) in combination with nivolumab (nivo) in pts with melanoma (MEL) previously treated with anti-PD-1/PD-L1 therapy. J. Clin. Oncol. 2017;35:9520. doi: 10.1200/JCO.2017.35.15_suppl.9520. [DOI] [Google Scholar]
- 47.Sordo-Bahamonde C., Lorenzo-Herrero S., Gonzalez-Rodriguez A.P., Payer A.R., Gonzalez-Garcia E., Lopez-Soto A., Gonzalez S. LAG-3 blockade with relatlimab (BMS-986016) restores anti-leukemic responses in chronic lymphocytic leukemia. Cancers. 2021;13:2112. doi: 10.3390/cancers13092112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heinhuis K.M., Ros W., Kok M., Steeghs N., Beijnen J.H., Schellens J.H.M. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann. Oncol. 2019;30:219–235. doi: 10.1093/annonc/mdy551. [DOI] [PubMed] [Google Scholar]
- 49.Trevejo J.M., Marino M.W., Philpott N., Josien R., Richards E.C., Elkon K.B., Falck-Pedersen E. TNF-alpha -dependent maturation of local dendritic cells is critical for activating the adaptive immune response to virus infection. Proc. Natl. Acad. Sci. U S A. 2001;98:12162–12167. doi: 10.1073/pnas.211423598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raeber M.E., Rosalia R.A., Schmid D., Karakus U., Boyman O. Interleukin-2 signals converge in a lymphoid-dendritic cell pathway that promotes anticancer immunity. Sci. Transl. Med. 2020;12:eaba5464. doi: 10.1126/scitranslmed.aba5464. [DOI] [PubMed] [Google Scholar]
- 51.Frasca L., Nasso M., Spensieri F., Fedele G., Palazzo R., Malavasi F., Ausiello C.M. IFN-gamma arms human dendritic cells to perform multiple effector functions. J. Immunol. 2008;180:1471–1481. doi: 10.4049/jimmunol.180.3.1471. [DOI] [PubMed] [Google Scholar]
- 52.Casati C., Camisaschi C., Rini F., Arienti F., Rivoltini L., Triebel F., Parmiani G., Castelli C. Soluble human LAG-3 molecule amplifies the in vitro generation of type 1 tumor-specific immunity. Cancer Res. 2006;66:4450–4460. doi: 10.1158/0008-5472.CAN-05-2728. [DOI] [PubMed] [Google Scholar]
- 53.Avice M.N., Sarfati M., Triebel F., Delespesse G., Demeure C.E. Lymphocyte activation gene-3, a MHC class II ligand expressed on activated T cells, stimulates TNF-alpha and IL-12 production by monocytes and dendritic cells. J. Immunol. 1999;162:2748–2753. [PubMed] [Google Scholar]
- 54.Andreae S., Piras F., Burdin N., Triebel F. Maturation and activation of dendritic cells induced by lymphocyte activation gene-3 (CD223) J. Immunol. 2002;168:3874–3880. doi: 10.4049/jimmunol.168.8.3874. [DOI] [PubMed] [Google Scholar]
- 55.Andreae S., Buisson S., Triebel F. MHC class II signal transduction in human dendritic cells induced by a natural ligand, the LAG-3 protein (CD223) Blood. 2003;102:2130–2137. doi: 10.1182/blood-2003-01-0273. [DOI] [PubMed] [Google Scholar]
- 56.Graydon C.G., Mohideen S., Fowke K.R. LAG3's enigmatic mechanism of action. Front. Immunol. 2021;11:615317. doi: 10.3389/fimmu.2020.615317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li N., Wang Y., Forbes K., Vignali K.M., Heale B.S., Saftig P., Hartmann D., Black R.A., Rossi J.J., Blobel C.P., et al. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J. 2007;26:494–504. doi: 10.1038/sj.emboj.7601520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matsuo K., Yoshie O., Kitahata K., Kamei M., Hara Y., Nakayama T. Recent progress in dendritic cell-based cancer immunotherapy. Cancers. 2021;13:2495. doi: 10.3390/cancers13102495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.den Haan J.M., Lehar S.M., Bevan M.J. CD8(+) but not CD8(-) dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 2000;192:1685–1696. doi: 10.1084/jem.192.12.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pooley J.L., Heath W.R., Shortman K. Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8- dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J. Immunol. 2001;166:5327–5330. doi: 10.4049/jimmunol.166.9.5327. [DOI] [PubMed] [Google Scholar]
- 61.Mizumoto Y., Hemmi H., Katsuda M., Miyazawa M., Kitahata Y., Miyamoto A., Nakamori M., Ojima T., Matsuda K., Nakamura M., et al. Anticancer effects of chemokine-directed antigen delivery to a cross-presenting dendritic cell subset with immune checkpoint blockade. Br. J. Cancer. 2020;122:1185–1193. doi: 10.1038/s41416-020-0757-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alexandre Y.O., Ghilas S., Sanchez C., Le Bon A., Crozat K., Dalod M. XCR1+ dendritic cells promote memory CD8+ T cell recall upon secondary infections with Listeria monocytogenes or certain viruses. J. Exp. Med. 2016;213:75–92. doi: 10.1084/jem.20142350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Matsuo K., Kitahata K., Kawabata F., Kamei M., Hara Y., Takamura S., Oiso N., Kawada A., Yoshie O., Nakayama T. A highly active form of XCL1/lymphotactin functions as an effective adjuvant to recruit cross-presenting dendritic cells for induction of effector and memory CD8(+) T cells. Front. Immunol. 2018;9:2775. doi: 10.3389/fimmu.2018.02775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jiang H., Ni H., Zhang P., Guo X., Wu M., Shen H., Wang J., Wu W., Wu Z., Ding J., et al. PD-L1/LAG-3 bispecific antibody enhances tumor-specific immunity. Oncoimmunology. 2021;10:1943180. doi: 10.1080/2162402X.2021.1943180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moon E.K., Ranganathan R., Eruslanov E., Kim S., Newick K., O'Brien S., Lo A., Liu X., Zhao Y., Albelda S.M. Blockade of programmed death 1 augments the ability of human T cells engineered to target NY-ESO-1 to control tumor growth after adoptive transfer. Clin. Cancer Res. 2016;22:436–447. doi: 10.1158/1078-0432.CCR-15-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dolina J.S., Van Braeckel-Budimir N., Thomas G.D., Salek-Ardakani S. CD8(+) T cell exhaustion in cancer. Front. Immunol. 2021;12:715234. doi: 10.3389/fimmu.2021.715234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ghoneim H.E., Zamora A.E., Thomas P.G., Youngblood B.A. Cell-intrinsic barriers of T cell-based immunotherapy. Trends Mol. Med. 2016;22:1000–1011. doi: 10.1016/j.molmed.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Borst J., Ahrends T., Babala N., Melief C.J.M., Kastenmuller W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018;18:635–647. doi: 10.1038/s41577-018-0044-0. [DOI] [PubMed] [Google Scholar]
- 69.Oh D.Y., Kwek S.S., Raju S.S., Li T., McCarthy E., Chow E., Aran D., Ilano A., Pai C.C.S., Rancan C., et al. Intratumoral CD4(+) T cells mediate anti-tumor cytotoxicity in human bladder cancer. Cell. 2020;181:1612–1625.e13. doi: 10.1016/j.cell.2020.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kagamu H., Kitano S., Yamaguchi O., Yoshimura K., Horimoto K., Kitazawa M., Fukui K., Shiono A., Mouri A., Nishihara F., et al. CD4(+) T-cell immunity in the peripheral blood correlates with response to anti-PD-1 therapy. Cancer Immunol. Res. 2020;8:334–344. doi: 10.1158/2326-6066.CIR-19-0574. [DOI] [PubMed] [Google Scholar]
- 71.Takeuchi Y., Tanemura A., Tada Y., Katayama I., Kumanogoh A., Nishikawa H. Clinical response to PD-1 blockade correlates with a sub-fraction of peripheral central memory CD4+ T cells in patients with malignant melanoma. Int. Immunol. 2018;30:13–22. doi: 10.1093/intimm/dxx073. [DOI] [PubMed] [Google Scholar]
- 72.Sun J.Y., Zhang D., Wu S., Xu M., Zhou X., Lu X.J., Ji J. Resistance to PD-1/PD-L1 blockade cancer immunotherapy: mechanisms, predictive factors, and future perspectives. Biomark. Res. 2020;8:35. doi: 10.1186/s40364-020-00212-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jin H.S., Ko M., Choi D.S., Kim J.H., Lee D.H., Kang S.H., Kim I., Lee H.J., Choi E.K., Kim K.P., et al. CD226(hi)CD8(+) T cells are a prerequisite for anti-TIGIT immunotherapy. Cancer Immunol. Res. 2020;8:912–925. doi: 10.1158/2326-6066.CIR-19-0877. [DOI] [PubMed] [Google Scholar]
- 74.Lee D.H., Kang S.H., Choi D.S., Ko M., Choi E., Ahn H., Min H., Oh S.J., Lee M.S., Park Y., Jin H.S. Genome wide CRISPR screening reveals a role for sialylation in the tumorigenesis and chemoresistance of acute myeloid leukemia cells. Cancer Lett. 2021;510:37–47. doi: 10.1016/j.canlet.2021.04.006. [DOI] [PubMed] [Google Scholar]
- 75.Quintelier K., Couckuyt A., Emmaneel A., Aerts J., Saeys Y., Van Gassen S. Analyzing high-dimensional cytometry data using FlowSOM. Nat. Protoc. 2021;16:3775–3801. doi: 10.1038/s41596-021-00550-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.