

Abstract
A promising treatment for β-hemoglobinopathies is the de-repression of γ-globin expression leading to increased fetal hemoglobin (HbF) by targeting BCL11A. Here, we aim to improve a lentivirus vector (LV) containing a single BCL11A shmiR (SS) to further increase γ-globin induction. We engineered a novel LV to express two shmiRs simultaneously targeting BCL11A and the γ-globin repressor ZNF410. Erythroid cells derived from human HSCs transduced with the double shmiR (DS) showed up to a 70% reduction of both BCL11A and ZNF410 proteins. There was a consistent and significant additional 10% increase in HbF compared to targeting BCL11A alone in erythroid cells. Erythrocytes differentiated from SCD HSCs transduced with the DS demonstrated significantly reduced in vitro sickling phenotype compared to the SS. Erythrocytes differentiated from transduced HSCs from β-thalassemia major patients demonstrated improved globin chain balance by increased γ-globin with reduced microcytosis. Reconstitution of DS-transduced cells from Berkeley SCD mice was associated with a statistically larger reduction in peripheral blood hemolysis markers compared with the SS vector. Overall, these results indicate that the DS LV targeting BCL11A and ZNF410 can enhance HbF induction for treating β-hemoglobinopathies and could be used as a model to simultaneously and efficiently target multiple gene products.
Keywords: β-hemoglobinopathies, BCL11A, ZNF410, HbF, double shmiR vector
Graphical abstract
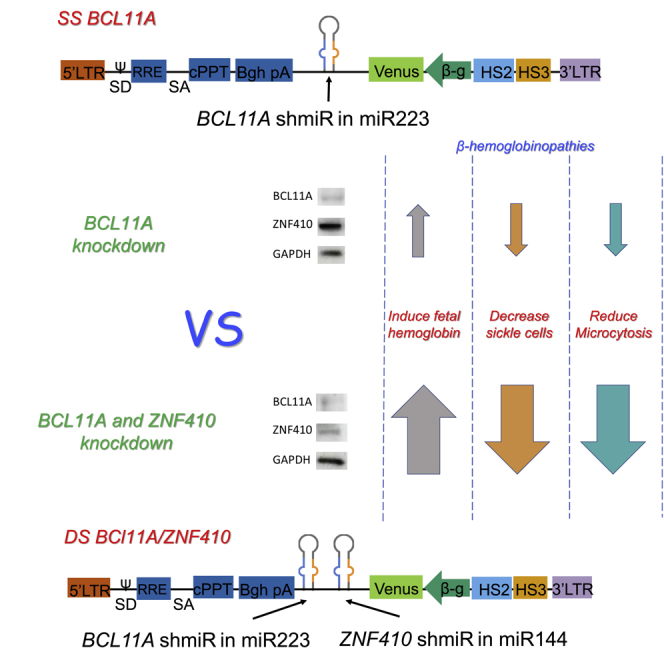
Liu et al. engineered a novel lentivirus vector to express tandem shmiRs simultaneously targeting two γ-globin repressors, BCL11A and ZNF410. Simultaneously targeting both genes significantly increases HbF induction as a potential therapy for β-hemoglobinopathies. This novel approach could be used as a model to simultaneously and efficiently target multiple gene products.
Introduction
β-Hemoglobinopathies, including sickle cell disease (SCD) and β-thalassemia (β-thal), are inherited blood disorders that have serious health effects and shorten lifespans for millions of people around the world.1, 2, 3 Both diseases are caused by mutations in the hemoglobin β gene (HBB), which affects the production or structure of adult hemoglobin (HGB). Mutant β-sickle globin (βS) is associated with intracellular HGB polymerization in the deoxygenated state, leading to sickle-shaped and stiffened red blood cells (RBCs) that block capillaries and have a short circulating half-life.4, 5, 6 This leads to lifelong morbidity, ischemic end organ damage, and a shortened lifespan in many individuals. β-Thal is caused by more than 200 different β-globin gene mutations that reduce or eliminate the production of β-globin chains and lead to ineffective erythropoiesis, intramedullary apoptosis of erythroid precursors, and chronic hemolytic anemia. Patients with β-thal major require blood transfusions and suffer from complications such as severe anemia, chronic hemolysis with medullary expansion, hepatosplenomegaly, iron overload, heart disease, and endocrine disorders.7, 8, 9
The only curative therapy available for β-hemoglobinopathies is allogeneic hematopoietic stem cell transplantation (HSCT). However, most affected individuals lack a well-matched, disease-unaffected donor.10 The current treatments for SCD in developed countries include the use of hydroxyurea (HU) to induce fetal hemoglobin (HbF), which has potent anti-polymerization properties.11,12 HU is well tolerated and effective in many patients, but some individuals do not respond with HbF elevation.12,13 The induction of HbF has been a long-term goal for the treatment of β-hemoglobinopathies, and continued production or reactivation of HbF in adults effectively reduces many of the most serious complications of the disease phenotype, especially in SCD.14, 15, 16, 17, 18
The results of genome-wide association studies (GWASs) identified BCL11A as associated with higher levels of HbF in adults.19,20 Orkin and colleagues demonstrated BCL11A as a significant repressor of γ-globin expression, and multiple lines of evidence have validated the therapeutic potential of BCL11A as a molecular target.21,22 BCL11A is a major component regulating the physiologic globin fetal to adult switch. Clinical trials aimed at downregulating BCL11A use different genetic approaches such as zinc finger nucleases,23 and CRISPR-Cas9 editing shows promise in elevating HbF and resolving disease complications.16,24,25 We have used the unique approach of a microRNA (miRNA)-adapted short hairpin RNA that targets BCL11A (shmiR BCL11A)26, 27, 28, 29 to effectively and selectively reduce expression in erythroid cells. Studies that include gene addition with lentivirus vectors (LVs) that overexpress globin in β-thalassemia patients with severe genotypes and in SCD patients show that the effectiveness of these vectors expressing a single curative gene require several integrations per genome to produce levels of the transgene that are high enough to correct the phenotype of the patients.29,30 Multiple integrations of vectors in the genome may increase the risk of insertional mutagenesis. Thus, achieving the high-efficiency gene transfer and robust γ-globin production required to correct the SCD phenotype and β-thal remains a challenge.31,32 Vectors that are more effective at lower vector copy number (VCN) to achieve therapeutic levels is an important goal of current research efforts.
The zinc-finger protein 410 (ZNF410) is a HbF repressor that was identified via transcription factor (TF) CRISPR screening. ZNF410 does not directly bind to the genes that encode γ-globin; rather, it binds and increases the transcription of the chromodomain Helicase DNA Binding Protein 4 (CHD4) gene, which encodes the nucleosome remodeling and deacetylase (NuRD) complex ATP-dependent nucleosome remodeler. NuRD itself is required for HbF repression and is in part independent of BCL11A.33,34 We hypothesized that the simultaneous reduction of both ZNF410 and BCL11A would have additive effects on HbF induction. We generated a novel shmiR targeting ZNF410 and subsequently combined a BCL11A shmiR and a ZNF410 shmiR into the same erythroid-specific LV to express these two shmiRs simultaneously. The combined double shmiR vector increases further the induction of HbF in erythrocytes compared to the BCL11A single shmiR vector currently in clinical trials. We demonstrate that the double shmiR LV is more effective in inhibiting sickling and restoring globin chain balance in erythroid cells derived from hematopoietic stem and progenitor cells (HSPCs) from SCD and β-thal patients in vitro. The double shmiR LV also more effectively attenuates the hematologic phenotypes of SCD in a murine model in vivo.
Results
Development of LVs expressing a ZNF410 shmiR
We designed three shRNAs targeting the ZNF410 mRNA into RNA Polymerase II (RNA Pol II)-driven miRNA 144-adapted shmiRs expressed in LVs from a strong spleen focus forming virus (SFFV) ubiquitous promoter (Figure S1A). These three shmiRs were further modified according to Guda et al.27 by deleting the first four bases from the guide sequence and by the addition of GCGC to the 3′ end (shmiRs modified, M defined as modified). These vectors were labeled ZNF410-shmiR-1, 1M, 2, 2M, 3, and 3M. To investigate the effect of these ZNF410 shmiR vector candidates on HbF induction, CD34+ HSPCs from healthy donors were infected with ZNF410 shmiR LVs and transduced cells were subjected to in vitro erythroid differentiation (Figure S1B). Sorted, gene-modified cells demonstrated a ∼50% reduction in ZNF410 mRNA expression (Figure S1C). There was a correlation between the reduction of ZNF410 expression and the induction of γ-globin (Figure S1D). Transduction with ZNF410 shmiR-3 yielded the highest levels of γ-globin and HbF induction in erythroid progeny (Figures S1E and S1F). The reduction of CHD4 after ZNF410 knockdown confirms the described mechanism of action, in which ZNF410 acts as an activator of CHD4, which in turn silences HbF (Figure S1G). These data demonstrate the effective knockdown of ZNF410 expression in erythroid cells using ZNF410 shmiR LVs. While all of the ZNF410 shmiRs increased γ-globin and HbF expression in CD34-derived erythroid cells, ZNF410 shmiR-3 was used in the subsequent development of a double shmiR vector. We performed RNA sequencing (RNA-seq) on ZNF410 shmiR-3 transduced CD34+ cells to measure gene expression changes (Figure S2). Based on cutoffs of fold changes >2 and adjusted p values (Padj) <0.05, 547 genes differentially expressed in ZNF410 shimiR-3 transduced CD34+ cells (84 genes were upregulated and 463 genes were downregulated). The upregulated genes related to dorsal aorta development, ventricular trabecula myocardium morphogenesis, phosphatidylglycerol acyl chain remodeling, and cardiac right ventricle morphogenesis, while downregulated genes were related to hemostasis, positive regulation of cell activation, blood coagulation, and myeloid cell differentiation.
A double shmiR vector targeting both ZNF410 and BCL11A efficiently knocks down expression of both genes
We hypothesized that simultaneous knockdown of BCL11A and ZNF410 using LVs could further enhance HbF induction in erythroid cells. To test this hypothesis, we transduced CD34+ cells concurrently with individual vectors targeting both genes. To generate double-transduced cells, we used the LV containing ZNF410 shmiR with a Tomato fluorescence reporter (LV-LCR-miR144 ZNF410, subsequently called SS ZNF410 (single shmiR ZNF410); Figure S3A) and a LV containing BCL11A shmiR with a Venus fluorescence reporter (LV-LCR-miR223 BCL11A, SS BCL11A (single shmiR BCL11A); Figure S3B). CD34+ cells from three different healthy donors were transduced simultaneously with both vectors, and the transduced cells were kept in culture for 18 days under conditions supporting erythroid differentiation (Figure 1A). We obtained 1.7% Venus and Tomato double-positive gene-marked cells. As seen in Figures 1B and 1C, cells transduced with both vectors (designated SS BCL11A + SS ZNF410 in Figures) demonstrated decreases in both BCL11A and ZNF410 mRNA and an incremental increase in γ-globin and HbF expression (Figures 1D and 1E), confirming an additive effect on HbF induction by simultaneously targeting both BCL11A and ZNF410.
Figure 1.

Efficient knockdown of BCL11A and ZNF410 by double shmiR vector leads to high γ-globin and HbF induction in erythroid cells differentiated in vitro from transduced gene-marked human CD34+ HSPCs
(A) Schematic of virus transduction and erythroid differentiation of human CD34+ HSPCs. (B) BCL11A and ZNF410 mRNA expression as measured by qRT-PCR with GAPDH as control on day 11 of differentiation. (C) BCL11A and ZNF410 protein expression as measured by western blot with GAPDH as a loading control on day 11 of differentiation. Densitometric quantitation is shown below each lane. (D) Induction of γ-globin mRNA as determined by qRT-PCR on day 18 of differentiation. (E) HbF of cell lysates as measured by HPLC on day 18 of differentiation. (F) Correlation of γ-globin determined by qRT-PCR versus HbF by HPLC. Black dots represent samples transduced with different shmiR vectors. The Pearson correlation coefficient (r2) is shown. (G) Differentiation status of erythroid cells after 18 days in culture using CD71 and CD235a. (H) Summary of the data shown in (G) normalized to the percentage of the CD71-CD235a+ population. (I) Enucleation of in vitro differentiated erythroid cells. (J) Summary of the data shown in (H) normalized to the percentage of the Hoechst− population. Data represent means ± SD, n = 3. ns, not significant; ∗p < 0.05.
Next, to improve the efficiency of the transduction of target cells, a LV incorporating both the BCL11A shmiR and ZNF410 shmiR cassettes, and the Venus fluorescent reporter under transcriptional control of the minimal β-globin proximal promoter linked to hypersensitive sites 2 and 3 (HS2 and HS3) of the β-globin locus control region (LCR), called LV-LCR-miR223 BCL11A-miR144 ZNF410 (double shmiR [DS] BCL11A/ZNF410) (Figure S3C), was constructed. To characterize the performance of DS BCL11A/ZNF410, we transduced mobilized human peripheral blood (mPB) CD34+ cells from three different healthy donors with the double shmiR vector (designated DS BCL11A/ZNF410 in Figures). We observed 24%, 16.9%, 15.8%, and 20.9% gene-marked cells after transduction with non-targeting (NT), SS BCL11A, SS ZNF410, and DS BCL11A/ZNF410 vectors, respectively (Figure S4), representing a 10-fold enhanced transduction efficiency of DS BCL11A/ZNF410 vector over simultaneous transduction with 2 vectors. Transduced cells were sorted for the analysis of BCL11A and ZNF410 mRNA and protein expression in erythroid cells derived from transduced CD34+ HSPCs. The use of a double shmiR led to an equivalent and simultaneous reduction in BCL11A mRNA by 80% and in ZNF410 mRNA by 70% (Figure 1B). Western blot confirmed the equivalent reduction in BCL11A and ZNF410 protein (Figure 1C) as a result of transduction with the double shmiR vector compared to single shmiR vectors targeting each gene individually.
DS BCL11A/ZNF410 effectively induces HbF expression in human erythroid cells in vitro
Consistent with a reduction in mRNA and protein of BCL11A and ZNF410, erythroid cells derived from CD34+ cells transduced with the double shmiR induced significantly more γ-globin and HbF than the induction with knockdown of either gene alone (p < 0.05 DS versus SS) and a similar level compared with cells derived after simultaneous transduction with both SS BCL11A shmiR plus SS ZNF410 shmiR vectors (p = not significant [ns]) (Figures 1D, 1E, and S5). Specifically, DS BCL11A/ZNF410 showed 49.4% HbF expression compared to 39.8% HbF induction after transduction with the SS BCL11A vector (p < 0.05 SS versus DS). γ-Globin mRNA and HbF levels were highly correlated in differentiated cells (Figure 1F), allowing for the extrapolation of HbF based on γ-globin qRT-PCR in this experimental setting. During erythroid maturation cultures, shmiR transduced HSPCs showed normal terminal erythroid maturation based on immunophenotype and enucleation frequency (Figures 1G–1J). Together, these results suggest that DS BCL11A/ZNF410 can efficiently and simultaneously knock down BCL11A and ZNF410 and further enhance HbF induction without adverse effects on erythroid differentiation and enucleation. We performed RNA-seq on SS BCL11A and DS BCL11A/ZNF410 transduced CD34+ cells to measure gene expression changes (Figure S6). Based on cutoffs of fold change >2 and Padj < 0.05, we then performed the Gene Ontology (GO) term analysis using the differentially expressed genes. Both SS BCL11A and DS BCL11A/ZNF410 demonstrated potential effects on similar pathways, except for the “positive regulation of cytokine production,” which was downregulated in SS BCL11A.
DS BCL11A/ZNF410 more effectively modify patient cells and disease cellular phenotypes than SS BCL11A shmiR vector
Next, we evaluated the potential therapeutic impact of higher HbF induction by DS BCL11A/ZNF410 after the transduction of primary HSPCs from patients with SCD and β-thal. PB CD34+ cells from three different SCD donors were used for transduction with SS BCL11A and DS BCL11A/ZNF410 vectors. NT or untransduced cells served as controls. We observed 10.5% and 11.5% gene-marked cells after transduction with SS BCL11A and DS BCL11A/ZNF410, respectively (Figure S7). Sorted gene-marked cells were differentiated into erythrocytes in vitro for further analysis. As demonstrated previously in normal cells, transduction with the double shmiR vector led to simultaneous knockdown of BCL11A and ZNF410 mRNA (Figure 2A). As seen in Figure 2A, in these cells, knockdown of BCL11A was equally effective with the double shmiR vector as with the single shmiR targeting BCL11A. Simultaneous knockdown of BCL11A and ZNF410 had no effect on erythroid cell differentiation and enucleation (Figures S8A and S8B). After transduction and erythroid cell differentiation, the mean γ-globin induction was 48.7% and 35.3% for DS BCL11A/ZNF410 and SS BCL11A, respectively (Figure 2B). Similarly, HbF induction was higher after transduction with the double shmiR vector. Compared to NT transduced erythroid cells, which showed 3.3% HbF, DS BCL11A/ZNF410 and SS BCL11A demonstrated 44.6% and 32.1% HbF, respectively (p < 0.05 DS versus SS) (Figures 2C and S9). The cell proliferation result suggested that BCL11A and ZNF410 knockdown has no effect on survival (Figure 2D). After sodium metabisulfite (MBS) treatment, we observed significantly fewer sickled cells in erythrocytes derived from cells transduced with the DS BCL11A/ZNF410 vector compared with SS BCL11A, 8.0 versus 14.9% (p < 0.01) (Figures 2E and 2F). As expected, untransduced or NT transduced erythroid cells showed robust sickling of 49.5% and 50.4%, respectively. There was a strong inverse correlation (r2 = 0.95) between HbF level and the percentage of sickled erythroid cells, again validating the anti-sickling effect of high levels of HbF (Figure 2G). Taken together, these data suggest that simultaneous knockdown of BCL11A and ZNF410 in SCD cells is feasible, further enhances HbF induction compared to knockdown of either gene alone, and the increment in HbF induction is physiologically relevant in preventing the cellular phenotype of erythrocyte sickling.
Figure 2.

Efficient knockdown of BCL11A and ZNF410 by double shmiR vector in erythrocytes differentiated in vitro from transduced gene-marked SCD patient CD34+ HSPCs
(A) BCL11A and ZNF410 mRNA expression was measured by qRT-PCR with GAPDH as control on day 11 of differentiation. Data represent mean ± SD, n = 3. (B) Induction of γ-globin mRNA was determined on day 18 of differentiation by qRT-PCR. Data represent mean ± SD, n = 3, ∗p < 0.05. (C) HbF of cell lysates was measured by HPLC on day 18 of differentiation. Data represent mean ± SD, n = 3. ∗p < 0.05. (D) Cell proliferation of shmiR transduced cells during erythroid differentiation. Data represent mean ± SD, n = 3. (E) Phase-contrast microscope image of representative sample of Hoechst 33342− sorted enucleated erythroid progeny 30 min after sodium MBS treatment from erythrocytes differentiated from nontransduced or transduced SCD CD34+ HSPCs; white arrows indicate sickle forms; scale bar, 50 μm. (F) Quantification of sickled cells from nontransduced and transced enucleated erythroid cells at 30 min after MBS treatment. Data are plotted as means, symbols indicate different SCD patients, and each data point represents an independent replicate. ∗∗p < 0.01. (G) Correlation of HbF expression assessed by HPLC versus numbers of sickled cells. Black dots represent samples transduced with different shmiR vectors or nontransduced. Correlation coefficient (r2) is shown for all data.
To further evaluate the potential translational value of simultaneous knockdown of BCL11A and ZNF410 using the double shmiR vector, we studied cells from β-thal patients. We transduced PB CD34+ HSPCs from three different patients with β0β0 (homozygous 3′ 619-bp deletion), β+β+ (IVS I-110 G > A; IVS I-110 G>A) and βEβ0 (p.Ser72fsX2; codon 26 G > A) thalassemia genotypes. Transduction efficiencies ranged from 25% to 40% with SS BCL11A and DS BCL11A/ZNF410 vectors (Figure S10). Sorted gene-marked cells were analyzed after erythroid differentiation. Transduction with DS BCL11A/ZNF410 led to the simultaneous reduction of BCL11A and ZNF410 mRNA expression in cells derived from each donor, while transduction with SS BCL11A led to knockdown of BCL11A mRNA expression alone (Figure 3A). In each donor, DS BCL11A/ZNF410 transduced erythroid cells showed significantly higher γ-globin and HbF induction compared to SS BCL11A cells (p < 0.01) (Figures 3B and 3C). We hypothesized that therapeutically relevant amelioration of globin chain imbalance, the pathophysiologic underpinning of β-thal, would result in the improvement of terminal erythroid maturation and reduced microcytosis. After shmiR vector transduction, we observed a higher frequency of erythroid cell differentiation and enucleation with both SS BCL11A and DS BCL11A/ZNF410 vector-transduced β-thal erythroid cells than control, NT transduced cells (p < 0.01) (Figures S11A and S11B). Reduced microcytosis of enucleated β-thal erythroid cells was also observed. The cell size of the NT transduced group was 74% that of healthy donors, while SS BCL11A and DS BCL11A/ZNF410 were significantly larger at 88% and 91% of healthy donors, respectively (Figure 3D), which is consistent with improved erythropoiesis. Together, these data suggested that DS BCL11A/ZNF410 vector enhances HbF induction in β-thal erythrocytes and more effectively modifies some cellular β-thal phenotypes.
Figure 3.

Efficient knockdown of BCL11A and ZNF410 by double shmiR vector in erythrocytes differentiated in vitro from β-thalassemia patient gene-marked CD34+ HSPCs
(A) BCL11A and ZNF410 mRNA expression was measured by qRT-PCR with GAPDH as control on day 11 of differentiation. (B) Induction of globin mRNA was determined on day 18 of differentiation by qRT-PCR. (C) Hemoglobin of cell lysates was measured by HPLC on day 18 of differentiation. (D) Cell size by relative forward scatter intensity of enucleated erythroid cells, normalized to healthy donor. Data represent mean ± SD; n = 3; ∗p < 0.05; ∗∗p < 0.01;.
DS BCL11A/ZNF410 gene-modified cells engraft immunodeficient mice and lead to higher levels of γ-globin induction than the SS BCL11A shmiR
We next determined whether erythroid cells derived from engrafted human transduced CD34+ cells showed target gene knockdown and HbF induction. Healthy donor CD34+ cells transduced with SS BCL11A or DS BCL11A/ZNF410 were transplanted into NOD.Cg-KitW-41J Tyr + Prkdcscid Il2rgtm1Wjl (NBSGW) immunodeficient mice. NT transduced cells and untransduced CD34+ cells served as controls. Mice were bled at weeks 4, 8, 12, and 16 to analyze the engraftment of human cells. Engraftment was calculated as a percentage of human CD45+ cells in the total human and murine CD45+ cell populations. The engraftment was ∼30% and similar among shmiR transduced and untransduced groups (Figure S12A). No significant differences were found between treatment groups in PB white blood cell (WBC), and platelet (PLT) counts, RBC or HGB and hematocrit (HCT) (Figures S12B–S12F), suggesting that the shmiR vectors have no significant effect on the hematopoietic function of transplant recipients. Lineage distribution of human erythroid cell, B cells, T cells, and myeloid cells within the PB was also similar (Figure S13).
At 16 weeks post-engraftment bone marrow (BM) cells were collected and analyzed (Figure S14). The engraftment of untransduced and shmiR transduced CD34+ HSPCs was 80% and similar between all groups (Figure 4A). Lineage distribution of erythroid cell, B cells, T cells, and myeloid cells within the BM was also similar (Figure 4B). VCN in total BM and lineage-purified cells was similar between each experimental group (Figure 4C). In the engrafted BM cells, there was no reduction in BCL11A or ZNF410 mRNA expression levels in B cells (Figure 4D), myeloid cells (Figure S15A), or CD34+ cells (Figure S15B), confirming the lack of knockdown in non-targeted lineages. Both BCL11A and ZNF410 mRNA expression was reduced in erythroid cells in mice engrafted with DS BCL11A/ZNF410 transduced CD34+ cells, while only BCL11A was reduced in mice engrafted with SS BCL11A (Figure 4E), demonstrating the erythroid specificity of these vectors. Consistent with the knockdown at the mRNA level, in human erythroid cells derived from the BM, we observed the robust induction of γ-globin, increasing from 3.5% to 22.8% with SS BCL11A and a significantly higher induction to 33.3% with DS BCL11A/ZNF410 (p < 0.05) (Figure 4F). We normalized γ-globin expression to erythroid cell VCN for each shmiR vector. As shown in Figure 4G, DS BCL11A/ZNF410 led to a higher γ-globin expression than SS BCL11A shmiR when normalized to VCN, as shown by the increased slope of the fitted line. HbF induction in erythroid cells differentiated in vitro from BM hCD34+ cells were also analyzed by high-performance liquid chromatography (HPLC) (Figure 4H). Transduction with the DS BCL11A/ZNF410 vector increased HbF to a significantly higher level than SS BCL11A (HbF = 21.6% versus 17.1%, p < 0.05). In BM erythroid cells, a robust increase in the fraction of HbF+ cells after SS BCL11A and DS BCL11A/ZNF410 transduction to an average >75% positive cells, which verifies that DS BCL11A/ZNF410 functions as a robust and pan-cellar repressor of HbF (Figure 4I). Normalized to VCN in erythroid cells, HbF expression, showed a trend toward higher HbF expression in DS BCL11A/ZNF410 transduced cells compared with BCL11A shmiR at the same VCN, as shown by the increased slope of the fitted line, but this difference failed to reach significance (Figure S16).
Figure 4.

Hematopoietic reconstitution of transduced human CD34+ HSPCs in immunodeficient NBSGW mice
Animals were euthanized 16 weeks after CD34+ HSPCs transplantation with cells derived from 3 different healthy donors, and bone marrow (BM) was collected and sorted into various subpopulations by flow cytometry. (A) Human chimerism in the BM of transplanted animals as determined by expression of hCD45. Data represent mean ± SD; each dot represents 1 animal. (B) Lineage distribution of human BM cells; data represent mean ± SD. (C) Vector copy number (VCN) determined in BM and multilineage cells; data represent mean ± SD; each dot represents 1 animal. Gene expression analysis by qRT-PCR in human cells from BM of engrafted mice. BCL11A and ZNF410 expression normalized to GAPDH in human B cells (D) or human erythroid cells (E) sorted from the BM of engrafted mice, and γ-globin expression (F) by qRT-PCR in human erythroid cells sorted from BM. Data represent mean ± SD. Each data point represents an individual mouse. ns, not significant; ∗p < 0.05; ∗∗∗p < 0.001. (G) γ-Globin shown in relation to VCN for SS BCL11A and DS BCL11A/ZNF410. (H) HbF by HPLC analysis of CD34+ cells isolated from the BM and subjected to in vitro erythroid differentiation. (I) Percentage of HbF+ cells by flow cytometry analyses in human CD235a+ erythroblasts isolated from BM. Data represent mean ± SD. Each data point represents an individual mouse, n = 10; ns, not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Genetic modification of Berkeley SCD HSCs with double shmiR vector leads to improvement of disease-associated hematological parameters
We next determined whether the double shmiR vector could ameliorate characteristic SCD disease parameters in a transplantation model using Berkeley SCD (BERK-SCD) mouse HSCs. We designed a DS BCL11A/Zfp410 for double knockdown in murine BM cells, replacing the ZNF410 shmiR with a Zfp410 shmiR. Zfp410 shmiR targeted the murine sequence and transduction led to the knock down of Zfp410 and induction of Hbb-y mRNA expression in mouse erythroid leukemia (MEL) cells (Figure S17). Lineage-negative CD45.1+ BM cells from BERK-SCD mice were transduced with DS BCL11A/Zfp410, SS BCL11A vector, NT vector, or left untransduced (as an unmanipulated disease control), and resulting transduced cells were transplanted into lethally irradiated CD45.2+ BL/6 recipient animals. An additional control group received untransduced cells isolated from CD45.1+ BoyJ (C57BL/6J, Pep Boy/J [CD45.1 congenic]) animals to quantitate the maximal correction of the phenotype. Transplantation into BoyJ mice was chosen to circumvent the increased mortality after conditioning in BERK-SCD that is required for successful engraftment. We used the CD45 isotypes to follow the engraftment of transduced cells in the irradiated wild-type (WT) recipient. In vitro VCN was determined by colony-forming unit (CFU) assay on progenitor-derived colonies from the cell product 14 days after transduction (Figure S18).
Engraftment as determined by flow cytometric enumeration of CD45.1+ donor cells; RBC counts and HGB/HCT concentrations were measured on PB samples acquired 4, 8, 12, and 16 weeks post-transplantation. PB engraftment was >95% at week 16 (Figure 5A) and similar in all recipient groups receiving BERK-SCD HSCs, suggesting that transduction did not alter the fitness of transplanted cells. To analyze the SCD phenotype, RBC, HGB/HCT, reticulocyte level, and the percentages of sickled erythrocytes were measured. Engraftment of untransduced BERK-SCD HSCs was associated with significantly lower RBC, HGB, and HCT levels and increased reticulocytes and sickled cells, indicative of severe hemolytic anemia, supporting the validity of the experimental model.
Figure 5.

Correction of in vivo hematologic parameters in Berkeley SCD mouse model
Mice were bled at weeks 4, 8, 12, and 16 and peripheral blood (PB) was analyzed. (A) PB engraftment in transplanted mice. (B) Red blood cell (RBC) count, (C) percentage of hematocrit (HCT), (D) hemoglobin (HGB) level, and (E) reticulocyte counts. (F) The percentage of sickled RBCs in PB after MBS treatment was quantitated. (G) Percentage of total β-like globin as γ-globin mRNA expression of erythroid cells in PB determined by qRT-PCR. (H) Representative fluorescence-activated cell sorting (FACS) plots of PB cells stained for the erythroid differentiation markers CD71 and Ter119 at week 16. (I) Summary of the percentages of CD71+ Ter119+ high erythroid precursor cell population. Data represent mean ± SD. Symbols indicate mice transplanted with different shmiR vectors or nontransduced cells; each data point represents an individual mouse. ns, not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Mice transplanted with BERK-SCD HSCs transduced with the NT vector showed similar results compared with the untransduced group, while SS BCL11A and DS BCL11A/Zfp410 transduced groups showed significant improvements in all blood parameters. We observed that the animals engrafted with cells transduced with the DS BCL11A/Zfp410 vector demonstrated significant improvement in hematologic parameters compared with animals engrafted with cells transduced with the SS BCL11A shmiR vector. After engraftment with DS BCL11A/Zfp410 transduced cells, the RBC count at week 16 was 9.4 × 106 cell/μL compared with 8.4 × 106 cell/μL for SS BCL11A (Figure 5B); the HCT percentage was 45% in DS BCL11A/Zfp410 compared with 42% in the SS BCL11A transduced group (p < 0.05) (Figure 5C), and the HGB expression level was 12.8 g/dL of DS BCL11A/Zfp410 compared with 11.4 g/dL of SS BCL11A (p < 0.05) (Figure 5D). Reticulocyte counts (Figure 5E) were decreased from 35% in the NT group to 8% in the DS BCL11A/Zfp410 transduced group, which was significantly lower than the SS BCL11A transduced group at 12% (p < 0.05), indicative of reduced erythropoietic stress because of reduced hemolysis and improved RBC survival.
The number of irreversibly sickled RBCs after sodium MBS treatment of collected PB was next tested. PB from animals transplanted with the DS BCL11A/Zfp410 vector transduced cells demonstrated significantly fewer sickled cells compared with the SS BCL11A group. PB taken from mice transplanted with untransduced or NT shmiR transduced HSPCs at week 16 displayed 49.5% and 50.4% irreversibly sickled RBC, respectively, while mice transplanted with SS BCL11A or DS BCL11A/Zfp410 transduced HSPCs displayed a decrease in the percentage of sickled cells in PB after treatment with MBS with 14.8%, and 10.0%, respectively (p < 0.05) (Figure 5F). Consistent with decreasing rates of hemolysis and lower reticulocyte and sickled cells counts, the average level of γ-globin was 1.4%, 13.4%, and 20.1% for the NT group, SS BCL11A, and DS BCL11A/Zfp410, respectively (Figure 5G). We also observed a striking reduction in circulating erythrocyte precursors between treatment groups, with a significant difference between the SS BCL11A and DS BCL11A/Zfp410 groups. The frequency of CD71+ Ter119+ erythroid precursors in the PB at week 16 (Figures 5H and 5I), was reduced from 35% (untransduced and NT groups) to 8.8% in the SS BCL11A group and 6.2% in DS BCL11A/Zfp410 group. These data show that the transplantation of DS BCL11A/Zfp410 transduced SCD HSPCs leads to the durable production of RBCs that are resistant to sickling both in vivo and in vitro.
After sacrifice, additional studies were performed on the spleen and BM. BM engraftment was >97% (Figure 6A) and similar in all of the recipient groups receiving BERK-SCD HSCs. The average BM VCN was similar among all of the experimental arms (Figure 6B) and matched the average VCN of in vitro cultured cells before transplantation. To determine whether shmiR vectors retained the ability to achieve therapeutic levels of expression in BM cells, we measured γ-globin by qRT-PCR. The DS BCL11A/Zfp410-treated group showed significantly higher γ-globin expression than the SS BCL11A-treated group (p < 0.05). The average levels of γ-globin were 1.7%, 18.5%, and 25.4% for the NT group, SS BCL11A, and DS BCL11A/Zfp410, respectively (Figure 6C). To compare the difference in expression per vector genome between SS BCL11A and DS BCL11A/Zfp410, we normalized γ-globin expression to BM VCN for different shmiR vectors. As seen in Figure S19, erythrocytes derived from DS BCL11A/Zfp410 transduced cells demonstrated a trend toward higher normalized γ-globin expression compared with SS BCL11A, as shown by the increased slope of the fitted line, but this difference failed to reach significance. We observed a strong negative correlation between the reduction of sickled RBCs in PB after MBS treatment and induction of γ-globin in BM erythroid cells (Figure 6D). In BM erythroid cells, we saw a robust increase in the fraction of HbF+ cells after SS BCL11A and DS BCL11A/Zfp410 transduction, which verifies a near pan-cellular HbF induction by DS BCL11A/Zfp410 (Figure 6E).
Figure 6.

HbF expression in vivo in BM cells from Berkeley SCD mouse model
Whole BM was taken from each mouse at week 16 and analyzed. (A) BM engraftment in transplanted mice. (B) VCN in BM was determined. (C) Percentages of γ-globin mRNA expression of erythroid cells in BM determined by qRT-PCR. (D) Correlation of γ-globin expression assessed by qRT-PCR in erythroid cells of BM versus sickled cells in PB after MBS treatment; correlation coefficient (r2) is shown for all of the data. (E) Percentage of HbF+ cells by flow cytometry analyses in human CD235a+ erythroblasts isolated from BM. (F) Spleen weights at week 16. Data represent mean ± SD. Each data point represents an individual mouse. ns, not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
The mitigation of the sickling phenotype was also associated with reduced spleen size, with the DS BCL11A/Zfp410-treated group showing significantly less spleen mass than the SS BCL11A-treated group (p < 0.05). The average spleen mass of the SS BCL11A vector and the DS BCL11A/Zfp410 vector group decreased to 0.25 ± 0.06 and 0.18 ± 0.04 g, respectively, compared to 0.53 ± 0.06 g in the NT group, while mice that received healthy cells showed spleen weights of 0.09 ± 0.02 g (Figure 6F). Thus, the persistence of DS BCL11/Zfp410 transduced cells leads to a more robust rescue of all of the SCD cellular phenotypes examined.
Discussion
The downregulation of BCL11A in erythroid cells leads to the sustained reactivation of γ-globin, the production of HbF resulting in reduced polymerization of sickle-containing HGB, and significant mitigation of the hematologic effects of SCD in a murine model.21,22 We previously reported a lentiviral vector that mediates knockdown of BCL11A via a shmiR expressed selectively in erythroid cells.26,27 This design adds a Drosha processing site to the shRNA construct and has been shown to greatly increase knockdown efficiency. In particular, the hairpin stem of a shmiR can comprise 21 nt of double-stranded RNA (dsRNA) and a 15-nt loop from a human miRNA. Adding the miRNA loop and flanking sequences on either or both sides of the hairpin results in a >10-fold increase in Drosha and Dicer processing of the expressed hairpins when compared with conventional shRNA designs without miRNA scaffold.35 Increased Drosha and Dicer processing translates into greater siRNA/miRNA production and greater potency for expressed hairpins. Subsequent clinical studies have used this vector design to validate BCL11A as a therapeutic target in SCD in humans.15,28 Additional clinical studies under way36, 37, 38 add back copies of a mutated form of adult β-globin, which is resistant to sickling. However, to be maximally effective, these gene transfer strategies require the sustained engraftment of a large number of transduced HSCs10 to effect polyclonal reconstitution. The approach of adding back additional globin gene sequences also appears to require a higher number of vector insertions per cell to effect maximal transgene expression. High VCNs may increase the genotoxic risks, an outcome of particular relevance in SCD patients who may have an increased risk of developing hematological malignancies as compared to the general population.39 While promising approaches aimed at improving LV-derived gene expression have been investigated,40 reaching therapeutic gene expression with a low VCN is still challenging.
The induction of HbF has been a long-term goal for the treatment of β-hemoglobinopathies; its expression can be effective in reducing many of the most serious complications of the disease phenotype. Targeting BCL11A to reverse the physiological fetal-to-adult globin switch to increase HbF and concurrently reduce HbS has clear advantages. HbS content and the percentage of HbF are the two main modulators of clinical severity. HbF has potent anti-sickling characteristics, and a level of HbF of approximately one-third of the total cellular content of HGB would likely prevent HbS polymerization, while the concurrent reduction in intracellular HbS further attenuates the tendency for polymer formation.41, 42, 43, 44, 45 The induction of high levels of HbF distributed broadly in red cells is the most promising approach to the pharmacologic treatment of sickle cell anemia because it targets the proximal pathophysiologic trigger of disease.46 In our previously published work,26,28 the use of a lentivirus shmiR vector targeting BCL11A led to a significant induction of HbF at VCN <1, a clear advantage of this approach. However, due to the complex biology of γ-globin repression,47 the maximal induction of HbF may not be obtained in all cells using this approach.
Two studies have previously identified ZNF410 as a HbF repressor that specifically activates the expression of CHD4. The specificity of ZNF410 is conveyed by two highly evolutionarily conserved clusters of ZNF410 binding sites near the CHD4 gene, with no counterparts elsewhere in the genome.33,34 In this study, we developed erythroid-specific double shmiR-expressing vectors that simultaneously knock down two genes, BCL11A and ZNF410, and consistently enhance HbF induction an additional ∼10% compared to knockdown of BCL11A alone. One advantage of this approach is the efficiency of the transduction of rare and difficult-to-transduce HSCs compared to simultaneous transduction with two different vectors. Thus, as expected, we show in xenograft experiments using human CD34+ cells that the transduction efficiency of DS BCL11A/ZNF410 on CD34+ and progeny B cells, myeloid cells, and erythroid cells was similar to that of SS BCL11A. In addition, a positive correlation was observed between HbF induction and VCNs in the erythroid cells of DS BCL11A/ZNF410-transplanted animals, and the ratios of HbF induction per VCN were higher in DS BCL11A/ZNF410 transplanted mice compared to SS BCL11A transplanted animals. The enhanced induction of HbF expression per red cell may therefore lead to a more effective induction of total HbF while maintaining the safety of lower VCN per cell in humans, as demonstrated here in the SCD mouse model and in the xenograft models using human cells.
The vector reported here uniquely combines two shmiR in the same vector to induce HbF. HbF expression can be induced by knockdown of not only BCL11A and ZNF410 but also other regulatory genes, such as LRF48 and POGZ,49 so additional combinatorial approaches are possible. We found the DS did not alter significantly GO-defined pathways compared with SS, except for one pathway. The biological relevance of this finding is unclear and is under investigation. This strategy may also be combined with other strategies to achieve the maximal mitigation of sickle HGB polymerization. For instance, Uchida et al.32 combined BCL11A knockdown and thEpoR co-expression for improved HSC-targeted gene therapy for HGB disorders in humans. The co-expression of a BCL11A shmiR and a therapeutic β-globin gene in one vector to enhance functional HGB production has recently been described.29 In addition, recent genome editing technologies allow for the development of other HbF induction methods, such as targeting the erythroid-specific BCL11A enhancer,50 a BCL11A binding site in γ-globin promoters similar to the Greek variant of the hereditary persistence of HbF (HPFH),51 and inducing a large deletion in the δ-globin gene mimicking Sicilian HPFH.52 Ramadier et al. developed therapeutic approaches combining LV-based gene addition and CRISPR-Cas9 strategies aimed to increase the incorporation of an anti-sickling globin (AS3) and induce the expression of HbF.53
Many of these strategies appear effective, but which is safest or most effective in humans is still unknown. However, the DS shmiR vector approach demonstrated here offers several potential advantages. First, lentiviral shmiR for HbF induction leaves HBBS alleles intact, which largely avoids DSBs generated by nucleases such as Cas9 that lead to uncontrolled mixtures of indels at the target site as well as the potential for large deletions, translocations, chromosomal loss, chromothripsis, and activation of the p53 DNA damage response.54, 55, 56 Second, since the DS BCL11A/ZNF410 uses a physiologic switch, this approach concurrently and proportionately reduces the concentration of sickle HGB in RBCs, the primary determinant of pathogenic HGB polymerization, more effectively than targeting BCL11A alone. Although other approaches, including a single BCL11A shmiR vector with an anti-sickling HBB transgene vector, can decrease the fraction of βS in erythroid progeny by 30%–70%,26,57 we achieved even greater βS percentage reduction in erythroid populations by DS BCL11A/ZNF410 shmiR without changing red cell differentiation kinetics. Double shmiR-transduced patient-derived CD34+ cells thus provide a promising basis for the autologous treatment of SCD and β-thal. This strategy can also be combined with other induction strategies to achieve sustained HbF induction or express an anti-sickling HBB transgene.
In summary, we developed erythroid-specific lentiviral vectors encoding a double shmiR targeting two repressors of γ-globin, allowing for an enhanced HbF induction at a similar VCN compared to targeting only BCL11A. The double shmiR vector can effectively knock down target genes in cells at the same time with high transduction efficiency, which can be used as a model to target multiple gene products simultaneously and efficiently. We report the functional characterization of a novel and efficient LV expressing double shmiR in clinically relevant cells from SCD and β-thal patients as part of a program of work aimed at the clinical translation of an effective LV-based gene therapy approach for these diseases. Finally, multi-shmiR LVs can have a wider range of potential applications in this field since multiple shmiRs could be exploited as an approach in other diseases with complex pathophysiology affecting multiple pathways.
Materials and methods
Construction of shRNAmiR constructs
The generation of mir223 BCL11A vectors has been described previously.27 For mir144 ZNF410 vectors to introduce desired sequence substitution, we used sets of reverse-oriented primers with the replaced sequence to PCR amplify the mir223 BCL11A plasmid backbone by the Q5 Site-Directed Mutagenesis Kit (New England Biolabs, Ipswich, MA, USA) and then phosphorylated and ligated the resultant linearized plasmids. The LV-LCR-mir223 BCL11A-mir144 ZNF410 vector was created by using the NEBuilder HiFi DNA Assembly kit (New England Biolabs) to fuse together PCR-amplified mir144 ZNF410 shmiR fragments to mir223 BCL11A linearized plasmids that digested with Mul1 (New England Biolabs). mi223 NT vectors with non-target shmiR cassettes were used as controls. All of the sequences of the shmiRs are shown in Figure S20.
Virus production and titration
LV supernatants were produced by adding 10 μg LV, 5 μg gag-pol, 2.5 μg rev, and 2.5 μg of vesicular stomatitis virus G (VSVG) packaging plasmids into HEK 293T cells grown in 10-cm plates. Plasmids were mixed with 1 mL DMEM (Cytiva, Marlborough, MA, USA) and 60 μL of 1 mg/mL linear PEI (Polysciences, Warrington, PA, USA), incubated for 15–20 min at room temperature, and added to the culture dish. The medium was changed 14 h later and virus supernatants were collected 48 and 72 h post-transfection, filtered through a 0.45-μm membrane (Corning, Corning, NY, USA), and then concentrated by ultracentrifugation at 23,000 rpm for 2 h in a Beckmann XL-90 centrifuge with SW-28 swinging buckets.
Infectious titers were determined on MEL cells by applying serial dilutions of vector supernatant followed by erythroid differentiation for 4 days in RPMI (Cytiva) supplemented with 1.25% DMSO (Sigma-Aldrich, St. Louis, MO, USA), and 5% fetal calf serum (Summerlin Scientific, Hampton, NH, USA).
Cell culture
293T and MEL cells were maintained in DMEM (Cytiva) or RPMI medium (Cytiva) supplemented with 10% fetal calf serum (Summerlin Scientific), 1% penicillin-streptomycin (Thermo Fisher, Waltham, MA, USA), respectively.
Transduction of human CD34+ cells
Human CD34+ HSPCs from the mPB of anonymized healthy donors were obtained from the Fred Hutchinson Cancer Research Center, Seattle, Washington. SCD patient and β-thal patient CD34+ HSPCs were isolated from unmobilized PB following Boston Children’s Hospital institutional review board (IRB) approval and informed patient consent. The CD34+ HSPCs were enriched using the Miltenyi CD34 Microbead kit (Miltenyi Biotec, Auburn, CA, USA). CD34+ cells were prestimulated for 44–48 h at 1×106 cells/mL in Stem Cell Growth Media (CellGenix, Portsmouth, NH, USA) supplemented with stem cell factor (SCF), FMS-like tyrosinekinase 3 ligand (FLT3L), and thrombopoietin (TPO), all from Peprotech (Rocky Hill, NJ, USA). Cells were then enumerated and transduced with the virus at an MOI as indicated for 24 h before downstream processing.
In vitro erythroid differentiation of CD34+ cells
The erythroid differentiation protocol we used is based on a three-phase protocol adapted from Giarratana et al.58 The cells were cultured in erythroid differentiation medium (EDM) consisting of Iscove modified Dulbecco’s medium (Cellgro, Manassas, VA, USA) supplemented with 1% l-glutamine (Thermo Fisher), and 1% penicillin-streptomycin (Thermo Fisher), 330 μg/mL holo-human transferrin (Sigma-Aldrich), 10 μg/mL recombinant human insulin (Sigma-Aldrich), 2 IU/mL heparin (Sigma-Aldrich), 5% human solvent detergent pooled plasma AB (Rhode Island Blood Center, Providence, RI, USA), and 3 IU/mL erythropoietin (Amgen, Thousand Oaks, CA, USA). During the first phase of expansion (days 0–7), CD34+ cells were cultured in EDM in the presence of 10−6 mol/L hydrocortisone (Sigma-Aldrich), 100 ng/mL SCF (Peprotech), 5 ng/mL IL-3 (R&D Systems, Minneapolis, MN, USA), as EDM-1. In the second phase (days 7–11), the cells were resuspended in EDM supplemented with SCF, as EDM-2. For the third phase (days 11–18), the cells were cultured in EDM without additional supplements, as EDM-3.
Western blot analysis
On day 11 of erythroid differentiation, differentiated CD34+ cells were lysed in lysis buffer (RIPA) with phosphatase inhibitors (Santa Cruz Biotechnology, Dallas, TX, USA). Total protein extracts were suspended in 2× Laemmli (Bio-Rad, Hercules, CA, USA) sample buffer, boiled, and loaded onto a 10% sodium dodecyl sulfate (SDS)-–polyacrylamide gel, then transferred to a polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA). To block nonspecific binding sites, the membranes were treated for 1 h with TBST (mixture of Tris-buffered saline and Tween 20) containing 5% milk. Membranes were next incubated overnight at 4°C with mouse anti-BCL11A antibody (Abcam, Cambridge, UK) or rabbit anti-ZNF410 polyclonal antibody (Proteintech, Rosemont, IL, USA). After washing, the membranes were incubated for 1 h with horseradish peroxidase (HRP)-linked anti-rabbit or anti-mouse immunoglobulin G (IgG) secondary antibody (Cell Signaling, Danvers, MA, USA). The expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) in the cells was also measured as a control.
RNA extraction and qRT-PCR
Total RNA was extracted using an RNeasy Micro Kit (QIAGEN, Venlo, the Netherlands). Reverse transcription of mRNA used the SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA, USA) with oligo(dT) primers. qRT-PCR was performed using the SYBR Green PCR master mix (Applied Biosystems, Foster City, CA, USA) as a detection system.
RNA-seq
On day 11 of erythroid differentiation, total RNA of differentiated CD34+ cells was extracted using an RNeasy Micro Kit (QIAGEN). Purified RNA was treated with DNase I. mRNA libraries were prepared and sequenced by the Molecular Biology Core Facilities at the Dana-Farber Cancer Institute. The adapter trimming of raw fastq files was performed by fastp.59 Then, the reads were mapped and quantified using default parameters with Salmon.60 The differentially expressed genes were identified by the DEseq2.61 The threshold of differentially expressed genes was Padj <0.05 by the Benjamini-Hochberg method and fold change >2. The GO analysis was conducted using clusterProfiler.62
VCN assay
Genomic DNA was extracted using the QIAGEN DNeasy protocol. VCN was assessed by qRT-PCR, performed with the use of TaqMan Fast Advanced Master Mix (Applied Biosystems). VCN was calculated by using primers and probes HIV-1 PSI (forward 5′-CAGGACTCGGCTTGCTGAAG-3′, reverse 5′-TCCCCCGCTTAATACTGACG-3′, probe FAM-5′-CGCACGGCAAGAGGCGAGG-3′) as a target and the human glycosyltransferase Like Domain Containing 1 gene (GTDC1) as an internal reference standard (forward 5′-GAAGTTCAGGTTAATTAGCTGCTG-3′, reverse 5′-TGGCACCTTAACATTTGGTTCTG-3′, probe VIC-5′-ACGAACTTCTTGGAGTTGTTTGCT-3′). Standard curves were obtained by serial dilutions of a plasmid containing one copy of PSI and GTDC1 sequences. The number of PSI and GTDC1 copies in test samples was extrapolated from the standard curves.
HGB analysis by HPLC
After 18 days of differentiation, 1 million erythroid cells were lysed by using Hemolysate reagent (Helena Laboratories, Beaumont, TX, USA), and then incubated on ice for 15–20 min and vortexed every 5 min. Hemolysates were prepared by centrifugation at 15,000 rpm for 5 min and analyzed with the use of the D-10 Hemoglobin Analyzer (Bio-Rad) to identify the HGB variants HbF and HbA and determine their levels.
In vitro sickling assay
At the completion of erythroid differentiation, enucleated RBCs were sorted, with the use of Hoechst 33342 (5 mg/mL; Invitrogen), and subjected to an in vitro sickling assay. Sickling was induced by adding 500μL of freshly prepared 2% sodium MBS (Sigma-Aldrich) solution prepared in PBS into enucleated cells resuspended with 500μL EDM-3 in a 24-well plate, followed by incubation at 37°C for 30 min. Live cell images were acquired using a Nikon Eclipse Ti inverted microscope (Nikon, Tokyo, Japan). More than 500 cells with irregular structure, protruding spikes, or sickle shape were counted as sickling cells.
Flow cytometry for enucleation and cell size analysis
For the enucleation analysis, cells were stained with 2μg/mL of the cell-permeable DNA dye Hoechst 33342 (Invitrogen) for 20 min at 37°C. The Hoechst 33,342-negative cells were further gated for cell size analysis with the Forward Scatter A parameter. The median value of forward scatter intensity normalized by data from healthy donors collected at the same time was used to characterize the cell size.
Human CD34+ HSPC transplant and flow cytometry analysis
All of the animal experiments were approved by the Boston Children’s Hospital Institutional Animal Care and Use Committee. NBSGW mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA;stock 026622). Non-irradiated NBSGW female mice (4–6 weeks of age) were infused by retro-orbital injection with 1×106 virus transduced CD34+ HSPCs (without sort, resuspended in 150μL Dulbecco’s phosphate-buffered saline [DPBS]) derived from healthy donors. PB samples were collected at weeks 4, 8, 12, and 16 to measure engraftment by flow cytometry (hCD45/mCD45) and determine RBC indices. At week 16, mice were euthanized, and BM was isolated for human xenograft analysis. A portion of the BM cells was performed to erythroid differentiation in vitro. For flow cytometric analyses of BM, the following antibodies were used: hCD45, mCD45, and fixable viability dye eFluor 780 (Thermo Fisher), and hCD235a, hCD33, hCD19, hCD34, and hCD3 (BioLegend, San Diego, CA, USA).
In vivo experiment in SCD mouse model
Lineage-negative mouse BM cells were isolated by flushing femurs, tibias, and iliac crests of 6- to 8-week-old CD45.1 BoyJ (B6.SJL-Ptprca Pepcb/BoyJ) or CD45.1 Berkeley SCD mice (BERK-SCD, JAX stock #003342) followed by lineage depletion using the Mouse Lineage Cell Depletion Kit (Miltenyi Biotec). Lin cells were pre-stimulated at 1×106 cells/mL in Stem Cell Growth Media (CellGenix) supplemented with mouse SCF (mSCF) (100 ng/mL), hTPO (100 ng/mL), mouse interleukin-3 (mIL-3) (20 ng/mL), and hFlt3-L (100 ng/mL), all from Peprotech. Following a 24-h pre-stimulation, cells were transduced at a density of 1 × 106 cells/mL at an MOI of 40, and transduced cells (without sorting) were transplanted by retro-orbital injection into lethally irradiated (7 + 4 Gy, split dose) CD45.2 recipients 3 days after isolation. A portion of the transduced cells was used to seed a methylcellulose-based CFU assay to determine the VCN in the cell products. PB samples were collected at weeks 4, 8, 12, and 16 to measure engraftment by flow cytometry (CD45.2/CD45.1), determine RBC indices, and quantitate sickled cells. At week 16, mice were euthanized, and BM cells were used to measure engraftment by flow cytometry (CD45.2/CD45.1), VCN, and mRNA expression. For flow cytometric analyses of BM, the following antibodies were used: CD45.1, CD45.2, CD11b, and CD3 (BioLegend), B220, CD71, and Ter119 (BD Pharmingen, Woburn, MA), and fixable viability dye eFluor 780 (Thermo Fisher).
Statistical analysis
All of the data are reported as mean ± SD unless otherwise stated. Statistical analyses were performed using GraphPad Prism version 8.0 (GraphPad Software, San Diego, CA, USA). The statistical significance between two averages was established using the unpaired t test. When the statistical significance between three or more averages were evaluated, a one-way ANOVA was applied, followed by multiple paired comparisons for normally distributed data (Tukey test). All of the statistical tests were two-tailed; statistical significance differences are indicated with asterisks (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005), and N.S. denotes p > 0.05.
Acknowledgments
We thank Kurt Whittemore and Kayla E. Wright for technical assistance and Teresa Ortiz for administrative assistance. The funding sources included the Bill and Melinda Gates Foundation (INV-021791). D.E.B. was supported by NHLBI (OT2HL154984, P01HL053749), St. Jude Children’s Research Hospital Collaborative Research Consortium, and the Burroughs Wellcome Fund.
Author contributions
B.L. performed the experimental work; B.L., C.B., and D.A.W. designed the experiments; Y.Z. analyzed the RNA-seq; J.P.M. and D.E.B. provided the patient samples; H.X., D.S.V., C.H., and M.M. provided technical assistance and advice. B.L. and D.A.W. wrote and edited the paper.
Declaration of interests
D.A.W. has received research funding from bluebird bio for research in hemoglobinopathies. Boston Children’s Hospital has licensed certain intellectual property (IP) relevant to hemoglobinopathies to bluebird bio.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.05.002.
Supplemental information
References
- 1.Cavazzana M., Antoniani C., Miccio A. Gene therapy for beta-hemoglobinopathies. Mol. Ther. 2017;25:1142–1154. doi: 10.1016/j.ymthe.2017.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghiaccio V., Chappell M., Rivella S., Breda L. Gene therapy for beta-hemoglobinopathies: milestones, New therapies and challenges. Mol. Diagn. Ther. 2019;23:173–186. doi: 10.1007/s40291-019-00383-4. [DOI] [PubMed] [Google Scholar]
- 3.Al-Saif A.M. Gene therapy of hematological disorders: current challenges. Gene Ther. 2019;26:296–307. doi: 10.1038/s41434-019-0093-4. [DOI] [PubMed] [Google Scholar]
- 4.Demirci S., Uchida N., Tisdale J.F. Gene therapy for sickle cell disease: an update. Cytotherapy. 2018;20:899–910. doi: 10.1016/j.jcyt.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoban M.D., Orkin S.H., Bauer D.E. Genetic treatment of a molecular disorder: gene therapy approaches to sickle cell disease. Blood. 2016;127:839–848. doi: 10.1182/blood-2015-09-618587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aygun B., Odame I. A global perspective on sickle cell disease. Pediatr. Blood Cancer. 2012;59:386–390. doi: 10.1002/pbc.24175. [DOI] [PubMed] [Google Scholar]
- 7.Asadov C., Alimirzoeva Z., Mammadova T., Aliyeva G., Gafarova S., Mammadov J. β-Thalassemia intermedia: a comprehensive overview and novel approaches. Int. J. Hematol. 2018;108:5–21. doi: 10.1007/s12185-018-2411-9. [DOI] [PubMed] [Google Scholar]
- 8.Taher A.T., Musallam K.M., Cappellini M.D. β-Thalassemias. N. Engl. J. Med. 2021;384:727–743. doi: 10.1056/nejmra2021838. [DOI] [PubMed] [Google Scholar]
- 9.Thompson A.A., Walters M.C., Kwiatkowski J., Rasko J.E.J., Ribeil J.A., Hongeng S., Magrin E., Schiller G.J., Payen E., Semeraro M., et al. Gene therapy in patients with transfusion-dependent beta-thalassemia. N. Engl. J. Med. 2018;378:1479–1493. doi: 10.1056/nejmoa1705342. [DOI] [PubMed] [Google Scholar]
- 10.Magrin E., Miccio A., Cavazzana M. Lentiviral and genome-editing strategies for the treatment of beta-hemoglobinopathies. Blood. 2019;134:1203–1213. doi: 10.1182/blood.2019000949. [DOI] [PubMed] [Google Scholar]
- 11.Charache S., Terrin M.L., Moore R.D., Dover G.J., Barton F.B., Eckert S.V., McMahon R.P., Bonds D.R. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N. Engl. J. Med. 1995;332:1317–1322. doi: 10.1056/NEJM199505183322001. [DOI] [PubMed] [Google Scholar]
- 12.Rodgers G.P., Dover G.J., Noguchi C.T., Schechter A.N., Nienhuis A.W. Hematologic responses of patients with sickle cell disease to treatment with hydroxyurea. N. Engl. J. Med. 1990;322:1037–1045. doi: 10.1056/nejm199004123221504. [DOI] [PubMed] [Google Scholar]
- 13.Wong T.E., Brandow A.M., Lim W., Lottenberg R. Update on the use of hydroxyurea therapy in sickle cell disease. Blood. 2014;124:3850–3857. doi: 10.1182/blood-2014-08-435768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castro O., Brambilla D.J., Thorington B., Reindorf C.A., Scott R.B., Gillette P., Vera J.C., Levy P.S. The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood. 1994;84:643–649. doi: 10.1182/blood.v84.2.643.bloodjournal842643. [DOI] [PubMed] [Google Scholar]
- 15.Esrick E.B., Lehmann L.E., Biffi A., Achebe M., Brendel C., Ciuculescu M.F., Daley H., MacKinnon B., Morris E., Federico A., et al. Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. N. Engl. J. Med. 2021;384:205–215. doi: 10.1056/nejmoa2029392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frangoul H., Altshuler D., Cappellini M.D., Chen Y.S., Domm J., Eustace B.K., Foell J., de la Fuente J., Grupp S., Handgretinger R., et al. CRISPR-Cas9 gene editing for sickle cell disease and beta-thalassemia. N. Engl. J. Med. 2021;384:252–260. doi: 10.1056/nejmoa2031054. [DOI] [PubMed] [Google Scholar]
- 17.Platt O.S., Brambilla D.J., Rosse W.F., Milner P.F., Castro O., Steinberg M.H., Klug P.P. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N. Engl. J. Med. 1994;330:1639–1644. doi: 10.1056/nejm199406093302303. [DOI] [PubMed] [Google Scholar]
- 18.Platt O.S., Thorington B.D., Brambilla D.J., Milner P.F., Rosse W.F., Vichinsky E., Kinney T.R. Pain in sickle cell disease. Rates and risk factors. N. Engl. J. Med. 1991;325:11–16. doi: 10.1056/NEJM199107043250103. [DOI] [PubMed] [Google Scholar]
- 19.Menzel S., Garner C., Gut I., Matsuda F., Yamaguchi M., Heath S., Foglio M., Zelenika D., Boland A., Rooks H., et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat. Genet. 2007;39:1197–1199. doi: 10.1038/ng2108. [DOI] [PubMed] [Google Scholar]
- 20.Uda M., Galanello R., Sanna S., Lettre G., Sankaran V.G., Chen W., Usala G., Busonero F., Maschio A., Albai G., et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc. Natl. Acad. Sci. U S A. 2008;105:1620–1625. doi: 10.1073/pnas.0711566105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sankaran V.G., Menne T.F., Xu J., Akie T.E., Lettre G., Van Handel B., Mikkola H.K., Hirschhorn J.N., Cantor A.B., Orkin S.H. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;112:487. doi: 10.1182/blood.v112.11.487.487. [DOI] [PubMed] [Google Scholar]
- 22.Xu J., Peng C., Sankaran V.G., Shao Z., Esrick E.B., Chong B.G., Ippolito G.C., Fujiwara Y., Ebert B.L., Tucker P.W., et al. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science. 2011;334:993–996. doi: 10.1126/science.1211053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith A.R., Schiller G.J., Vercellotti G.M., Kwiatkowski J.L., Krishnamurti L., Esrick E.B., Williams D.A., Miller W.P., Woolfson A., Walters M.C. Preliminary results of a phase 1/2 clinical study of zinc finger nuclease-mediated editing of BCL11A in autologous hematopoietic stem cells for transfusion-dependent beta thalassemia. Blood. 2019;134:3544. doi: 10.1182/blood-2019-125743. [DOI] [Google Scholar]
- 24.Demirci S., Zeng J., Wu Y., Uchida N., Shen A.H., Pellin D., Gamer J., Yapundich M., Drysdale C., Bonanno J., et al. BCL11A enhancer-edited hematopoietic stem cells persist in rhesus monkeys without toxicity. J. Clin. Invest. 2020;130:6677–6687. doi: 10.1172/jci140189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salinas Cisneros G., Thein S.L. Recent advances in the treatment of sickle cell disease. Front. Physiol. 2020;11:435. doi: 10.3389/fphys.2020.00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brendel C., Guda S., Renella R., Bauer D.E., Canver M.C., Kim Y.J., Heeney M.M., Klatt D., Fogel J., Milsom M.D., et al. Lineage-specific BCL11A knockdown circumvents toxicities and reverses sickle phenotype. J. Clin. Invest. 2016;126:3868–3878. doi: 10.1172/jci87885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guda S., Brendel C., Renella R., Du P., Bauer D.E., Canver M.C., Grenier J.K., Grimson A.W., Kamran S.C., Thornton J., et al. miRNA-embedded shRNAs for lineage-specific BCL11A knockdown and hemoglobin F induction. Mol. Ther. 2015;23:1465–1474. doi: 10.1038/mt.2015.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brendel C., Negre O., Rothe M., Guda S., Parsons G., Harris C., McGuinness M., Abriss D., Tsytsykova A., Klatt D., et al. Preclinical evaluation of a novel lentiviral vector driving lineage-specific BCL11A knockdown for sickle cell gene therapy. Mol. Ther. Methods Clin. Dev. 2020;17:589–600. doi: 10.1016/j.omtm.2020.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pires Lourenco S., Jarocha D., Ghiaccio V., Guerra A., Abdulmalik O., La P., Zezulin A., Smith-Whitley K., Kwiatkowski J.L., Guzikowski V., et al. Inclusion of a short hairpin RNA targeting BCL11A into a β-globin expressing vector allows concurrent synthesis of curative adult and fetal hemoglobin. Haematologica. 2021;106:2740–2745. doi: 10.3324/haematol.2020.276634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weber L., Poletti V., Magrin E., Antoniani C., Martin S., Bayard C., Sadek H., Felix T., Meneghini V., Antoniou M.N., et al. An optimized lentiviral vector efficiently corrects the human sickle cell disease phenotype. Mol. Ther. Methods Clin. Dev. 2018;10:268–280. doi: 10.1016/j.omtm.2018.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fitzhugh C.D., Cordes S., Taylor T., Coles W., Roskom K., Link M., Hsieh M.M., Tisdale J.F. At least 20% donor myeloid chimerism is necessary to reverse the sickle phenotype after allogeneic HSCT. Blood. 2017;130:1946–1948. doi: 10.1182/blood-2017-03-772392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uchida N., Ferrara F., Drysdale C.M., Yapundich M., Gamer J., Nassehi T., DiNicola J., Shibata Y., Wielgosz M., Kim Y.S., et al. Sustained fetal hemoglobin induction in vivo is achieved by BCL11A interference and coexpressed truncated erythropoietin receptor. Sci. Transl. Med. 2021;13:eabb0411. doi: 10.1126/scitranslmed.abb0411. [DOI] [PubMed] [Google Scholar]
- 33.Vinjamur D.S., Yao Q., Cole M.A., McGuckin C., Ren C., Zeng J., Hossain M., Luk K., Wolfe S.A., Pinello L., et al. ZNF410 represses fetal globin by singular control of CHD4. Nat. Genet. 2021;53:719–728. doi: 10.1038/s41588-021-00843-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lan X., Ren R., Feng R., Ly L.C., Lan Y., Zhang Z., Aboreden N., Qin K., Horton J.R., Grevet J.D., et al. ZNF410 uniquely activates the NuRD component CHD4 to silence fetal hemoglobin expression. Mol. Cell. 2021;81:239–254.e8. doi: 10.1016/j.molcel.2020.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boden D., Pusch O., Silbermann R., Lee F., Tucker L., Ramratnam B. Enhanced gene silencing of HIV-1 specific siRNA using microRNA designed hairpins. Nucleic Acids Res. 2004;32:1154–1158. doi: 10.1093/nar/gkh278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanter J., Walters M.C., Hsieh M.M., Krishnamurti L., Kwiatkowski J., Kamble R.T., von Kalle C., Kuypers F.A., Cavazzana M., Leboulch P., et al. Interim results from a phase 1/2 clinical study of lentiglobin gene therapy for severe sickle cell disease. Blood. 2016;128:1176. doi: 10.1182/blood.v128.22.1176.1176. [DOI] [Google Scholar]
- 37.Poletti V., Urbinati F., Charrier S., Corre G., Hollis R.P., Campo Fernandez B., Martin S., Rothe M., Schambach A., Kohn D.B., et al. Pre-clinical development of a lentiviral vector expressing the anti-sickling βAS3 globin for gene therapy for sickle cell disease. Mol. Ther. Methods Clin. Dev. 2018;11:167–179. doi: 10.1016/j.omtm.2018.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Urbinati F., Wherley J., Geiger S., Fernandez B.C., Kaufman M.L., Cooper A., Romero Z., Marchioni F., Reeves L., Read E., et al. Preclinical studies for a phase 1 clinical trial of autologous hematopoietic stem cell gene therapy for sickle cell disease. Cytotherapy. 2017;19:1096–1112. doi: 10.1016/j.jcyt.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 39.Brunson A., Keegan T.H.M., Bang H., Mahajan A., Paulukonis S., Wun T. Increased risk of leukemia among sickle cell disease patients in California. Blood. 2017;130:1597–1599. doi: 10.1182/blood-2017-05-783233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morgan R.A., Unti M.J., Aleshe B., Brown D., Osborne K.S., Koziol C., Ayoub P.G., Smith O.B., O'Brien R., Tam C., et al. Improved titer and gene transfer by lentiviral vectors using novel, small beta-globin locus control region elements. Mol. Ther. 2020;28:328–340. doi: 10.1016/j.ymthe.2019.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maier-Redelsperger M., Noguchi C.T., de Montalembert M., Rodgers G.P., Schechter A.N., Gourbil A., Blanchard D., Jais J.P., Ducrocq R., Peltier J.Y. Variation in fetal hemoglobin parameters and predicted hemoglobin S polymerization in sickle cell children in the first two years of life: Parisian Prospective Study on Sickle Cell Disease. Blood. 1994;84:3182–3188. doi: 10.1182/blood.v84.9.3182.bloodjournal8493182. [DOI] [PubMed] [Google Scholar]
- 42.Poillon W.N., Kim B.C., Rodgers G.P., Noguchi C.T., Schechter A.N. Sparing effect of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S at physiologic ligand saturations. Proc. Natl. Acad. Sci. USA. 1993;90:5039–5043. doi: 10.1073/pnas.90.11.5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brittenham G.M., Schechter A.N., Noguchi C.T. Hemoglobin S polymerization: primary determinant of the hemolytic and clinical severity of the sickling syndromes. Blood. 1985;65:183–189. doi: 10.1182/blood.v65.1.183.bloodjournal651183. [DOI] [PubMed] [Google Scholar]
- 44.Hebert N E.E., Armant M., Brendel C., Ciuculescu M.F., Audureau E., Higgins J.M., Williams D.A., Bartolucci P. Effects of BCL11A shmir-induced post-transcriptional silencing on distributions of HbF in single-RBCs and reticulocytes. Blood. 2021;138:967. (ASH Annual Meeting Abstracts) [Google Scholar]
- 45.De Souza DC E.E., Hebert N., Di Caprio G., Ciuculescu M.F., Morris E., Armant M., Gonҫalves B.P., Schonbrun E., Brendel C., Wood D.K., et al. Effects of BCL11A shmiR-induced post-transcriptional silencing on hemoglobin polymer inhibition in single red blood cells at physiologic oxygen tension. Blood. 2021;138:964. (ASH Annual Meeting Abstracts) [Google Scholar]
- 46.Steinberg M.H., Chui D.H.K., Dover G.J., Sebastiani P., Alsultan A. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood. 2014;123:481–485. doi: 10.1182/blood-2013-09-528067. [DOI] [PubMed] [Google Scholar]
- 47.Khandros E., Huang P., Peslak S.A., Sharma M., Abdulmalik O., Giardine B.M., Zhang Z., Keller C.A., Hardison R.C., Blobel G.A. Understanding heterogeneity of fetal hemoglobin induction through comparative analysis of F and A erythroblasts. Blood. 2020;135:1957–1968. doi: 10.1182/blood.2020005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masuda T., Wang X., Maeda M., Canver M.C., Sher F., Funnell A.P.W., Fisher C., Suciu M., Martyn G.E., Norton L.J., et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science. 2016;351:285–289. doi: 10.1126/science.aad3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gudmundsdottir B., Gudmundsson K.O., Klarmann K.D., Singh S.K., Sun L., Singh S., Du Y., Coppola V., Stockwin L., Nguyen N., et al. POGZ is required for silencing mouse embryonic beta-like hemoglobin and human fetal hemoglobin expression. Cell Rep. 2018;23:3236–3248. doi: 10.1016/j.celrep.2018.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bauer D.E., Kamran S.C., Lessard S., Xu J., Fujiwara Y., Lin C., Shao Z., Canver M.C., Smith E.C., Pinello L., et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342:253–257. doi: 10.1126/science.1242088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Metais J.Y., Doerfler P.A., Mayuranathan T., Bauer D.E., Fowler S.C., Hsieh M.M., Katta V., Keriwala S., Lazzarotto C.R., Luk K., et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv. 2019;3:3379–3392. doi: 10.1182/bloodadvances.2019000820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ye L., Wang J., Tan Y., Beyer A.I., Xie F., Muench M.O., Kan Y.W. Genome editing using CRISPR-Cas9 to create the HPFH genotype in HSPCs: an approach for treating sickle cell disease and beta-thalassemia. Proc. Natl. Acad. Sci. U S A. 2016;113:10661–10665. doi: 10.1073/pnas.1612075113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramadier S., Chalumeau A., Felix T., Othman N., Aknoun S., Casini A., Maule G., Masson C., De Cian A., Frati G., et al. Combination of lentiviral and genome editing technologies for the treatment of sickle cell disease. Mol. Ther. 2021;30:145–163. doi: 10.1016/j.ymthe.2021.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zuccaro M.V., Xu J., Mitchell C., Marin D., Zimmerman R., Rana B., Weinstein E., King R.T., Palmerola K.L., Smith M.E., et al. Allele-specific chromosome removal after Cas9 cleavage in human embryos. Cell. 2020;183:1650–1664.e15. doi: 10.1016/j.cell.2020.10.025. [DOI] [PubMed] [Google Scholar]
- 55.Song Y., Liu Z., Zhang Y., Chen M., Sui T., Lai L., Li Z. Large-fragment deletions induced by Cas9 cleavage while not in the BEs system. Mol. Ther. Nucleic Acids. 2020;21:523–526. doi: 10.1016/j.omtn.2020.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Enache O.M., Rendo V., Abdusamad M., Lam D., Davison D., Pal S., Currimjee N., Hess J., Pantel S., Nag A., et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat. Genet. 2020;52:662–668. doi: 10.1038/s41588-020-0623-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Demirci S., Gudmundsdottir B., Li Q., Haro-Mora J.J., Nassehi T., Drysdale C., Yapundich M., Gamer J., Seifuddin F., Tisdale J.F., et al. βT87Q-Globin gene therapy reduces sickle hemoglobin production, allowing for Ex Vivo anti-sickling activity in human erythroid cells. Mol. Ther. Methods Clin. Dev. 2020;17:912–921. doi: 10.1016/j.omtm.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Giarratana M.C., Rouard H., Dumont A., Kiger L., Safeukui I., Le Pennec P.Y., Francois S., Trugnan G., Peyrard T., Marie T., et al. Proof of principle for transfusion of in vitro-generated red blood cells. Blood. 2011;118:5071–5079. doi: 10.1182/blood-2011-06-362038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation (N Y) 2021;2:100141. doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.