

Abstract
Restless legs syndrome (RLS) is a common sensorimotor disorder, whose basic components include a sensory experience, akathisia, and a sleep-related motor sign, periodic leg movements during sleep (PLMS), both associated with an enhancement of the individual’s arousal state. The present review attempts to integrate the major clinical and experimental neurobiological findings into a heuristic pathogenetic model. The model also integrates the recent findings on RLS genetics indicating that RLS has aspects of a genetically-moderated neurodevelopmental disorder involving mainly the cortico-striatal-thalamic-cortical circuits. Brain iron deficiency (BID) remains the key initial pathobiological factor and relates to alterations of iron acquisition by the brain, also moderated by genetic factors. Experimental evidence indicates that BID leads to a hyperdopaminergic and hyperglutamatergic states that determine the dysfunction of cortico-striatal-thalamic-cortical circuits in genetically vulnerable individuals. However, the enhanced arousal mechanisms critical to RLS are better explained by functional changes of the ascending arousal systems. Recent experimental and clinical studies suggest that a BID-induced hypoadenosinergic state provides the link for a putative unified pathophysiological mechanism for sensorimotor signs of RLS and the enhanced arousal state.
Keywords: restless legs syndrome, brain iron deficiency, dopamine, glutamate, adenosine, arousal
Introduction
Restless legs syndrome (RLS), also known as Willis-Ekbom disease, is a common sensorimotor disorder with a prominent circadian pattern. According to the RLS Epidemiology, Symptoms and Treatment (REST) study, about 5% of US and European adults reported experiencing RLS symptoms at least weekly (Allen and others 2005). RLS is defined as a rest-induced, movement-responsive, mostly nocturnal, urge to move the legs. The term ‘akathisia’ is used to define the feeling of restlessness and urgent need to move. RLS can be conceptualized as an enhancement of a biological ‘drive’ whose primary purpose is to keep the individual alert, active, and moving and essentially operates as a counter to the sleep homeostatic drive. About 88% of RLS patients have an objective motor sign of repetitive periodic leg movements during sleep (PLMS) (Montplaisir and others 1997). Moderate to severe RLS also presents with enhanced arousal state (Allen and others 2010; Ferri and others 2015a). This “hyperarousal” is shown both during the night with disrupted, short sleep time of 4.0 to 5.5 hours (Saletu and others 2000) and also during the day with lack of the profound sleepiness expected given the significant sleep loss at night (Gamaldo and others 2009). This likely reflects changes at some level in neural mechanisms that govern arousal-sleep drives as well as sensory-motor function (Trenkwalder and Paulus 2010). RLS is associated with alterations in iron homeostasis, which are assumed to play a primary role in many cases and seem to determine changes in glutamatergic and dopaminergic systems (see below). As here reviewed, RLS is determined by genetically-moderated brain iron homeostasis and developmental abnormalities of cortico-striatal-thalamic-cortical circuits function. The functional changes in these brain circuits, however, fall short of explaining the enhanced arousal state. Here, we also review recent experimental and clinical studies that suggest that a brain-iron-deficiency (BID)-induced hypoadenosinergic state provides the link for a putative unified pathophysiological mechanism for both the sensorimotor signs of RLS and hyperarousal.
Neurophysiological evidence for spinal and supraspinal hyperexcitability
Qualitatively, the PLMS component of RLS resembles the spinal cord flexor reflex (dorsiflexion of ankle and flexion of the knee and hip). Patients with both primary RLS and RLS secondary to end-stage renal disease show a significantly lower threshold of the flexor reflex during sleep (Bara-Jimenez and others 2000; Aksu and Bara-Jimenez 2002). This could indicate a spinal cord hyperexcitability for RLS during sleep that might also be involved with generating PLMS. Both the spinal cord flexor reflex and the PLMS could involve the same spinal generator. Leg movements with the same qualitative pattern, duration and periodicity of PLMS have also been reported after acute spinal cord injuries (SCI), in patients with completely absent volitional activity in their legs (Salminen and others 2013; Ferri and others 2015b). The SCI-induced episodes of leg movements, however, do not a have a circadian dependence and are not associated with episodes of cortical arousal. Therefore, the spinal component of RLS, spinal hyperexcitability, most probably depends on an altered supraspinal-mediated mechanism, either a hyperfunctioning excitatory or a hypofunctioning inhibitory mechanism. The first situation seems to be the case, since cortical hyperexcitability has been well documented from transcranial magnetic stimulation (TMS) studies in RLS patients (Lanza and others 2015; Magalhaes and others 2015).
The preferential alterations in some of the TMS parameters associated with cortical hyperexcitability (such as a reduction in short-latency intracortical inhibition) favored abnormalities in sensory-motor integration by the cortico-striatal-thalamic-cortical circuits with a prominent subcortical component (Tergau and others 1999). In agreement with this interpretation, a recent functional magnetic resonance imaging (fMRI) study found particularly significant changes in regional spontaneous activity of RLS patients compared to controls in the striatum and thalamus and also significant, but less pronounced changes in frontal cortical areas (Zhuo and other 2017). A more recent MRI study has reported a significant decrease in cortical thickness in the somatosensory cortex, as well as in the region of the corpus callosum with the callosal fibers that interconnect the somatosensory cortices (Lee and others 2018). Anatomical data indicate that cortico-striatal projections from reciprocally connected cortical regions are more likely to have overlapping arborizations within the striatum (Yeterian and Van Hoesen 1978; Hoffer and Alloway 2001; Ramanathan and others 2002). In fact, motor and somatosensory cortical areas are heavily interconnected, directly, by reciprocal projections, and indirectly by convergent projections to even the same striatal neurons (Ramanathan and others 2002). In addition, interconnected motor and somatosensory cortical areas also project to the same specific thalamic areas (Hoffer and Alloway 2001), providing the framework for sensory-motor integration by tightly interconnected cortical, striatal and thalamic areas. Therefore, the akathisia and PLMS components of RLS seems to arise as a disorder of sensory-motor integration by the cortico-striatal-thalamic-cortical circuits and, more specifically, those circuits involving the motor and somatosensory cortices (Fig. 1).
Figure 1.
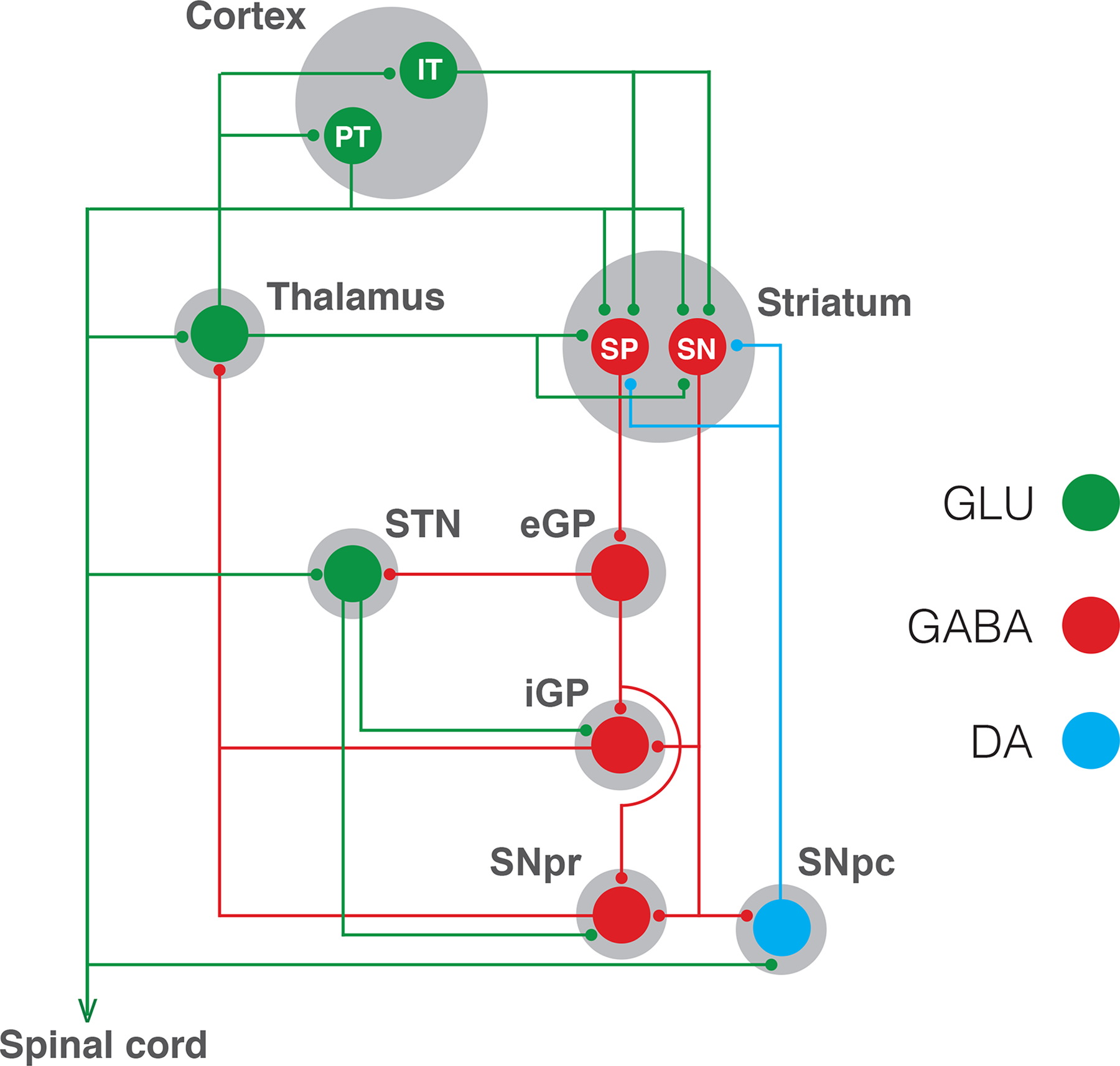
Parallel cortico-striatal-thalamic-cortical circuitry. Two types of pyramidal neurons project from the cortex to the striatum: the intratelencephalic (IT) neuron, which project ipsilaterally and bilaterally to the cortex and striatum; and the pyramidal tract (PT) neuron, which projects ipsilaterally to the striatum, but also projects to the ipsilateral thalamus, subthalamic nucleus (STN), mesencephalon (including the substantia nigra pars compacta, SNpc) and spinal cord. IT and PT neurons project to both subtypes of GABAergic striatal efferent neurons: the striatonigral (SN) neuron, which directly connect the striatum with the output structures of the basal ganglia, which are the substantia nigra pars reticulata (SNpr) and the internal segment of the globus pallidus (iGP); and the striatopallidal (SP) neuron, which connect indirectly with the output structures by a relay through the external segment of the globus pallidus (eGP) and the STN. This scheme shows only the characteristic parallel processing of the basal ganglia, but there is also a substantial level of convergent integration of cortico-striatal projections, more specifically from reciprocally connected cortical regions, such as the motor and somatosensory cortical areas. Dysfunction of cortico-striatal-thalamic-cortical circuitry involving motor and somatosensory cortical areas seem to determine the PLMS and akathia components of RLS.
In support of the concept that RLS is an enhancement of the arousal ‘drive’ whose primary purpose is to keep the individual alert, active, and moving, are the findings of increased electroencephalographic high frequencies during the sleep onset period (Ferri and others 2014). Also, the cerebrospinal fluid content of hypocretin 1/orexin A, an important wake-promoting neuropeptide (de Lecea and others 1998), was significantly higher in RLS subjects as compared to controls (Allen and others 2002). A magnetic resonance spectroscopy (MRS) study found evidence for an increase in basal glutamate levels in the thalamus of RLS patients, which correlated significantly with the wake time during the sleep period (Allen and others 2013a). In RLS, PLMS are commonly associated with arousal from sleep (when the episodes of arousal and PLM are separated by less than 0.5 seconds; Ferri and others 2015a). Significantly, in roughly half of all cases of arousal associated with PLMS, the onset of the episode of arousal precedes the onset of the leg movement (Ferri and others 2015a). Thus, in this situation, the leg movement per se may not cause, but reflect, the episode of arousal. Furthermore, a significant correlation exists between increasing duration of the leg movements and its probability of being associated with an arousal (Ferri and others 2015a). This correlation combined with a lack of causal relation between the movements and the arousal indicates there is likely some underlying mechanism producing both of these linked CNS events.
Genetics evidence for a neurodevelopmental disorder of cortico-striatal-thalamic-cortical circuits
Family and twin studies have estimated that RLS heritability is 50–60%. Initially considered as mostly a Mendelian disease, it is now considered as a complex multifactorial disorder with both genetic and non-genetic factors contributing to the susceptibility (Schormair and Winkelmann 2011). Significant findings have been obtained in several genome-wide association studies (GWAS). GWAS is a hypothesis-free method that tests the presence of genetic association throughout the genome, whether variation in any of the approximately 20,000 human genes might contribute to disease susceptibility or other individual differences. GWAS involve analyses of several hundred to more than 1 million single nucleotide polymorphisms (SNPs) from thousands of individuals comparing those with the disease to a large sample from the general population. A first conclusion from GWAS studies in general, and RLS in particular, is that most common SNPs (by definition, with minor allele frequency greater than 1%), related to increased RLS risk confer little risk increments and explain only a small proportion of heritability in the population. Thus, heritability of RLS, like other common diseases, depends on multiple common polymorphisms (the so-called “common disease-common variant”) (Altshuler and others 2008).
A recent meta-analysis of several GWAS databases replicated previously identified six risk loci, MEIS1, BTBD9, PTPRD, MAP2K5, SKOR1 and TOX3 (Schormair and Winkelmann 2011), and 13 new risk loci, with each genetic variant increasing about 50% the risk for RLS (Schormair and others 2017). As often observed with GWAS, these associations related not to protein coding but rather regulatory regions and did not involve candidate genes thought to be possibly significant for RLS (such as those related to iron metabolism; see below). Nevertheless, the genetic findings provided new pathogenetic clues based on their relation to neurogenesis, neuronal differentiation, axonal pathfinding and synaptogenesis (Schormair and others 2017). MEIS1, PTPRD and BTBD9 have been the most studied polymorphisms.
MEIS1 polymorphism has been confirmed as the strongest genetic risk factor (Schormair and others 2017). The protein products of MEIS1 and MEIS2 belong to the TALE family of homeobox transcription factors, involved in the development of numerous organs, including the brain. In the developing telencephalon, MEIS1 and MEIS2 are particularly expressed within the ganglionic eminences (Toresson and others 2000; Spieler and others 2014), which give rise to the basal ganglia in the adult brain. Maybe not surprisingly, one of the recently added RLS risk loci has MEIS2 as a candidate gene (Schormair and others 2017). Transcripts of BTBD9, PTPRD, MAP2K5 and TOX3 are also expressed during development in the ganglionic eminences (Spieler and others 2014). In addition, the risk allele of a lead SNP in the MEIS1 cis-regulatory locus (rs12469063) has been shown to specifically reduce its enhancer activity in the murine embryonic ganglionic eminences (Spieler and others 2014) (Fig. 2). The association of RLS risk alleles with reduced MEIS1 transcription and protein expression levels in brain tissues from RLS patients (Xiong and others 2009), supports the validity of mouse models based on decreased MEIS1 expression. However, little recapitulation of the RLS phenotype was observed. Homozygous MEIS1 KO mice were not viable (Azcoitia and others 2005) and the heterozygous showed general hyperactivity with increased locomotor behavior, but without a circadian relationship or qualitative motor abnormalities matching RLS phenotype (Spieler and others 2014). Furthermore, these failed to produce the most common neurochemical abnormalities described in RLS, such as BID, hyperdopaminergic and hyperglutamatergic states (Spieler and others 2014).
Figure 2.
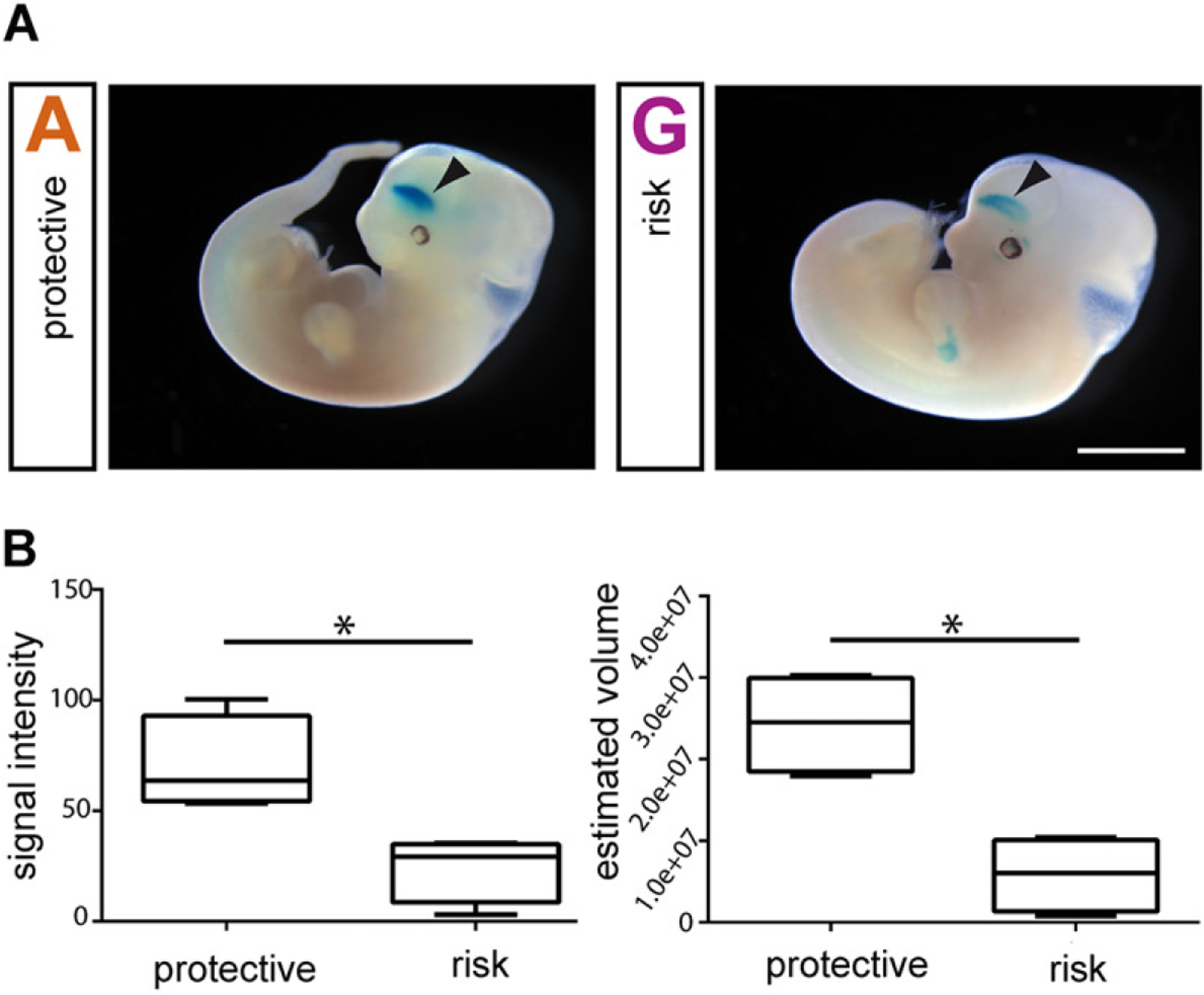
Reduction of the enhancer activity of an RLS-risk common polymorphism of the regulatory locus of the gene MEIS1 in the mouse ganglionic eminence. A. Transgenic mouse embryos after injection of the protective (left, ‘A’ for alanine) and risk allele constructs (right, ‘G’ for guanine). The blue color indicates the regions expressing the reporter gene beta-galactosidase, and the arrowheads indicate the telencephalic signal in the ganglionic eminence. B. Analysis of the signal intensity and volume demonstrated a significant reduction (*) of the enhancer activity of the risk versus the protective construct, down to 35% and 24%, respectively. Reproduced and modified with permission from Spieler and others 2014.
PTPRD encodes the gene of the receptor type protein tyrosine phosphatase D (or PTPδ), a presynaptic cell adhesion molecule that plays an important role in synaptogenesis, particularly of excitatory synapses (Takahashi and Craig 2013). Like MEIS1, RLS-risk SNPs of the non-coding PTPRD risk locus (specially rs2381970) are associated with a reduced PTPRD expression. This led to the development of PTPRD KO mice as a putative model of RLS (Drgonova and others 2015). Homozygous PTPRD KO were viable and also hyperactive. Furthermore, they had a significant reduction in sleep time at the end of the wakefulness and beginning of the sleep periods (Drgonova and others 2015), the period with more frequent symptomatology in RLS. Nevertheless, no common RLS-associated neurochemical abnormalities were reported.
Finally, the BTBD9 KO mouse has also been developed as a putative RLS animal model (DeAndrade and others 2012), although no correlation has yet been established between any RLS-associated SNP of the non-coding BTBD9 risk locus and brain BTBD9 expression (Freeman and Rye 2013). Furthermore, a role of BTBD9 in neurodevelopment has not been established yet. Nevertheless, its putative role in protein ubiquitylation indicates a possible significant role in neurogenesis and synaptogenesis (Kawabe and Brose 2011; Freeman and Rye 2013). Similar to PTPRD KO, BTBD9 KO (homozygous) mice show hyperactivity and a disruption of the normal sleep architecture, with an increased awake time (DeAndrade and others 2012). BTBD9 KO mice also showed some neurochemical abnormalities in serum iron levels and serotonin metabolism, but no evidence for BID or dopamine metabolites in the striatum (DeAndrade and others 2012).
In summary, what we can learn from the putative genetic animal models of RLS is that a single deficiency of the products of the risk polymorphisms does not appear likely to produce the neuropathological phenotype of RLS (see below for RLS-associated neurochemical abnormalities). This should not be surprising considering, first, that RLS has a polygenic heritability with significant dependence on non-genetic factors. Second, that there is a low disease-associated specificity of the RLS risk polymorphisms. For instance, MEIS2, PTPRD and BTBD9 polymorphisms have also been significantly associated with other neuropsychiatric disorders, particularly obsessive-compulsive disorder (OCD; Nestadt and others 2011; Mattheisen and others 2015; Gazzelione and others 2016). In addition, it has been documented that comorbidity exists between RLS and OCD and attention deficit-hyperactivity disorder (Ghorayeb and others 2017), disorders characterized by basal ganglia dysfunction. Altogether, the studies of the risk loci point to a significant neurodevelopmental component in the pathogenesis of RLS, leading to an increased vulnerability to dysfunctional cortico-striatal-thalamic-cortical circuits.
Altered brain iron homeostasis as a primary pathogenetic mechanism
BID is well-recognized as a key mechanism in the development of RLS (Earley and others 2014). This association was first reported by Nordlander in 1953. Subsequently, several studies indicated a high prevalence of RLS symptoms in conditions with insufficient iron availability (Allen and Earley 2007). In fact, the prevalence of RLS in patients with iron-deficient anemia is as high as 30% (Allen and others 2013b), six times higher than the prevalence of RLS in the general population (Allen and others 2005). Nevertheless, most RLS patients do not show systemic iron deficiency, rather RLS patients have a specific iron deficiency in the brain. This CNS iron deficiency was first demonstrated by reduced ferritin (the intracellular iron storage protein) and elevated transferrin levels (the extracellular iron carrier protein) in the CSF of RLS patients who had normal serum levels for ferritin and transferrin (Earley and others 2000). This was further supported by 12 imaging studies with all but two showing decreased iron in specific regions, principally the substantia nigra (reviewed in Earley and others 2014). Postmortem studies showed alterations in the expression and function of iron management proteins in the choroid plexus and brain microvasculature of RLS patients (Connor and others 2011). BID was demonstrated in the epithelial cells of the choroid plexus by the significant reduction of intracellular iron, and ferritin, as well as the significant upregulation of transferrin receptor (Connor and others 2011; Fig. 3). The presence of RLS-associated alterations of transferrin receptors, ferritin and the transporters divalent metal protein 1 and ferroportin in the brain endothelial cells, supports that iron transport to the brain via transferrin is regulated by the endothelial cells of the blood-brain barrier and that this transport is impaired in RLS (Connor and others 2011). BID in RLS can thus result from alterations of iron acquisition by the brain, i.e. dysregulation of iron transportation by the blood-brain barrier. Different iron formulations are being introduced to treat RLS patients with or without documented iron-deficiency anemia (Allen and others 2018). The challenge is to obtain a formulation and a schedule of administration with the optimal pharmacokinetic properties that would facilitate the restoration of iron levels in the brain (Allen and others 2018).
Figure 3.
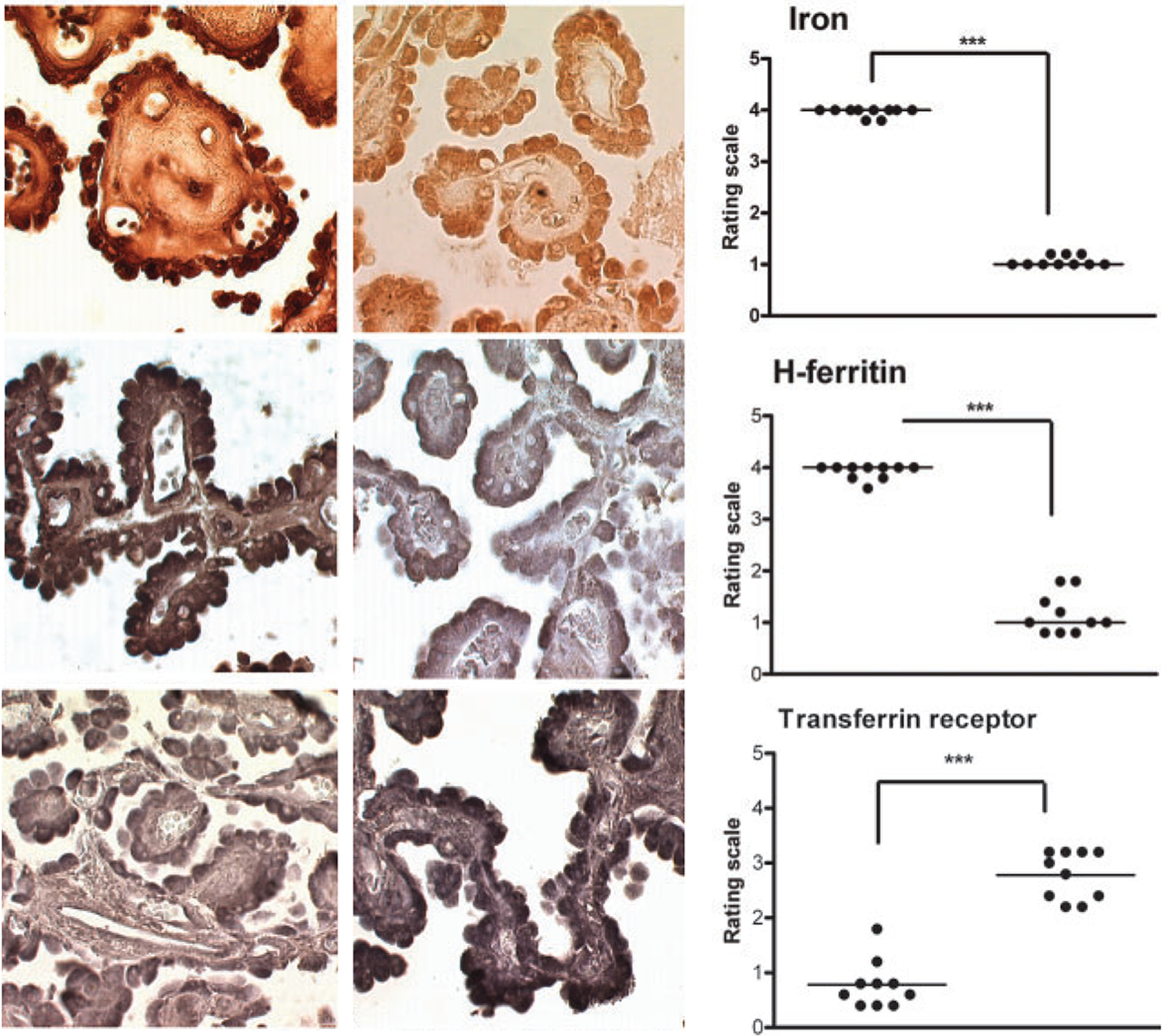
Iron deficiency in choroid plexus tissue from RLS patients. Upper, middle and lower micrographs represent iron, heavy-chain ferritin (H-ferritin) and transferrin receptor staining, respectively, from RLS (left panels) and control subjects (middle panels). Right panels represent the respective quantitative analysis, showing a very significant decrease of iron and H-ferritin densities (***) and a significant increase in transferrin receptor density (***) in RLS (left scatterplots) versus control subjects (right scatterplots). Reproduced and modified from Connor and others 2011.
BID in rodents can be induced by a severe iron-deficient diet during the post-weaning period and represents a well-accepted pathogenetic model of RLS (Connor and others 2009; Earley and others 2014). Diet-induced BID in rats has recently been shown to increase activity, reduce sleep and increase PLMS (Lai and others 2017). The BID rodent also reproduces the circadian sleep architecture of RLS, showing an increase in wakefulness at the end of the awake period (Dean ad others 2006; Allen and others 2017). But more importantly, and differently from the above-mentioned genetic RLS mouse models, the BID rodent has provided a valuable model to understand the neurochemical alterations in RLS, including alterations in the dopaminergic and glutamatergic systems, as well as putative alterations in the adenosinergic system (for reviews, see Earley and others 2014; Ferré and others 2018a).
Role of the dopaminergic system in the expression of primary symptoms
Altered dopaminergic neurotransmission is considered the final mechanism underlying both akathisia and PLMS in RLS. The initial basis for this assertion is the dramatic therapeutic benefit of levodopa on both clinical components of RLS. Thus, akathisia and PLMS are dopamine-response elements. Subsequent research has provided evidence for abnormalities in the dopaminergic system in RLS patients (Earley and others 2014). The dopaminergic profile in RLS patients includes abnormally high CSF levels of the dopamine metabolite 3-OMD, interpreted to indicate increased tyrosine hydrolase activity (Allen and others 2009). Single-photon emission computed tomography (SPECT) and positron emission tomography (PET) imaging studies have produces conflicting results regarding the striatal dopamine transporter (DAT) (Earley and others 2014). The differences in the findings reflect differences in what each imaging technique provides. SPECT imaging is likely to reflect the total dynamic DAT pool, while PET reflects only membrane bound DAT. The overall interpretation of the results is that, although the total cellular DAT is unchanged, the amount of the plasma membrane-bound is reduced (Earley and others 2011). On the other hand, PET and SPECT have been consistent in finding a decrease in striatal dopamine D2 receptors (D2Rs) binding (Earley and others 2014). One PET study using a difference multi-imaging technique to assess the dynamic nature of D2R binding, found changes that were most consistent with an increase in synaptic dopamine (Earley and others 2013). Autopsy studies found no difference in total striatal DAT concentration but did show a significant decrease in striatal D2R protein (Connor and others 2009). In addition, the striatal D2R protein concentration correlated with the severity of RLS symptoms (Connor and others 2009). Furthermore, there was a pronounced increase in tyrosine hydroxylase activity (increased expression of phosphorylated tyrosine hydroxylase) in the striatum and substantia nigra (Connor and others 2009). Taken in total, these results are compatible with a presynaptic hyperdopaminergic state, with increased synthesis and release of dopamine (Earley and others 2014; Ferré and others 2018a).
Importantly, BID in rodents recapitulates the same dopaminergic profile observed in RLS patients, with a decrease in striatal D2R density, DAT and an increase in expression of phosphorylated tyrosine hydroxylase as the main neurochemical findings (Connor and others 2009; Earley and others 2014). The connection between BID and alterations in the dopaminergic system could also be reproduced with genetic correlational analysis of several BXD recombinant inbred strains of mice showing differences in regional brain and liver iron content (Jones and others 2003; Jellen and others 2013). These studies provided evidence for a significant and polygenic heritability of brain iron homeostasis and its independence from peripheral iron modulation. They also showed a wide variation in susceptibility to iron loss after being fed an iron-deficient diet, both peripherally and in the brain. One of the strains, BXD 40, has been proposed as a particularly valuable rodent model of RLS, since, upon an iron-deficient diet, it shows several phenotypical signs of RLS in the presence of BID with minimal change in hemoglobin (Allen and others 2017).
The descending spinal dopaminergic system has also been suggested to be involved in the pathophysiology of RLS. This system originates in the dorsal-posterior hypothalamus and has been designated as the A11 region. The hypothetical mechanism would depend on a loss of the inhibitory influence of the A11 dopaminergic system on spinal sensory inputs (for review, see Clemens and others 2006). The rodent with a lesion of the A11 region has then been suggested as another animal model of RLS. A11-lesion animals show an increase in locomotor activity which is further accentuated by iron deprivation (Qu and others, 2007). The main tenet of the A11 spinal cord model, however, cannot be supported by neuropathological studies, which show no evidence for A11 neuronal degeneration in RLS (Earley and others 2009).
Hypersensitivity of cortico-striatal glutamatergic terminals
The clinically-based evidence for an altered glutamatergic neurotransmission in RLS comes from several independent sources. Ketamine, a non-competitive inhibitor of NMDA receptor, has been shown in a limited case study to improve RLS symptoms (Kapur and Friedman 2002). The μ-opioid receptor agonist methadone is also a non-competitive NMDA antagonist (Inturrisi 2005), and is a highly effective treatment for RLS at low, non-analgesic doses (Silver and others 2011). A study using Magnetic Resonance Spectroscopy (MRS) showed an increase in basal glutamate levels in the thalamus of RLS patients (Allen and others 2013a). Finally, medications effective in treating RLS symptoms that bind to the α2δ auxiliary subunit of voltage-gated Ca2+-channels, like gabapentin, may derive their benefits from their action on the glutamatergic system (García-Borreguero and others 2014). The α2δ subunit is preferentially localized in glutamatergic terminals through which α2δ ligands exert a presynaptic inhibition of glutamatergic transmission (Dooley and others 2007). Medications that bind to the α2δ protein are also more effective in improving sleep compared to dopamine receptor agonists (García-Borreguero and others 2014). In fact, clinical trials with careful analysis of sleep disturbances have shown that despite the marked improvement in PLMS with dopamine receptor agonists, the overall sleep efficiency was not improved (Inoue and others 2010; Oertel and others 2010). The data can therefore be interpreted as indicating two distinct, interacting, mechanisms underlying RLS symptomatology: a dopaminergic component, mostly involved in akathisia and PLMS and a glutamatergic component, which is also involved in enhanced arousal.
In keeping with the concept of BID as an underlying cause of RLS, ID in rodents leads to changes in the glutamatergic system (Shukla and others 1989; McGahan and others 2005). A quantitative trace loci analysis of more than twenty BXD recombinant inbred strains of mice with genetic differences in brain iron content found a strong correlation with the expression of the gene for the glial high-affinity glutamate transporter 1 (Glt1), which plays a main role in the uptake extracellular glutamate. (Jellen and others 2012). The experimental evidence for a BID-dependent alteration in glutamatergic transmission came from a recent optogenetic-microdialysis study in rodents with BID (Yepes and others 2017). This methodology allowed the measurement of the striatal extracellular concentration of glutamate upon local light-induced stimulation of cortico-striatal glutamatergic terminals. The method also allowed the analysis of the effect of local perfusion of different drugs directly within the same area being sampled for glutamate. The study showed that BID in rats produces hypersensitivity of cortico-striatal glutamatergic terminals (Yepes and others 2017). Furthermore, the dopamine receptor agonists pramipexole and ropinirole and the α2δ ligand gabapentin, all of them being clinically useful drugs for RLS, completely blocked the optogenetic-induced glutamate release both in BID rats and controls (Yepes and others 2017). This implied that dopamine receptors and voltage-gated Ca2+-channels localized in striatal glutamatergic terminals represent real targets for the dopamine receptor agonists and α2δ ligands in RLS (Yepes and others 2017; Fig. 4). The differential effect of several dopamine receptor antagonists in counteracting the ability of pramipexole to inhibit cortico-striatal glutamate release point to the involvement of D2R and D4R subtypes, probably forming heteromsers (Yepes and others 2017; Fig. 4). The fact that dopamine receptor agonists and α2δ ligands, which are clinically effective drugs with very different pharmacological profiles, produce the same effect in the same specific neuronal target, strongly supports that BID-induced hypersensitivity of cortico-striatal terminals represents a main pathogenetic mechanism of RLS symptoms.
Figure 4.
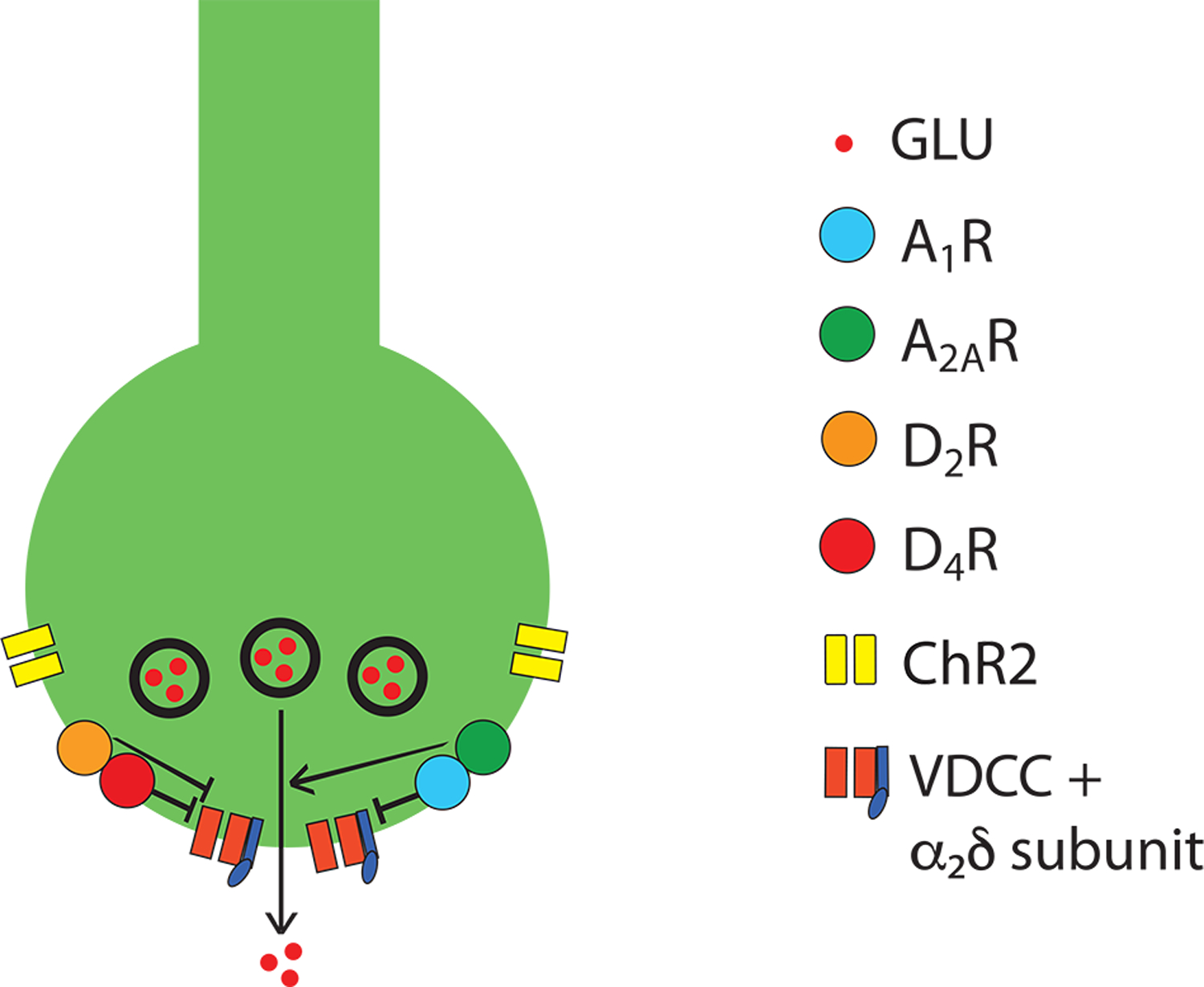
Schematic representation of a cortico-striatal glutamatergic terminal and its modulatory dopamine and adenosine receptors. Dopamine and adenosine modulate cortico-striatal glutamate (GLU) release by acting on A1R-A2AR and D2R-D4R heteromers. The A1R-A2AR heteromer acts as an adenosine concentration-dependent switch, by which a low adenosine concentration activates preferentially A1R, which produces inhibition of glutamate release, and a high adenosine concentration also activates A2AR, which shuts down A1R signaling and promotes and A2AR-mediated stimulation of glutamate release. The D2R-D4R heteromer provides a dopamine concentration-dependent stepwise inhibitory mechanism of glutamate release, that depends on the higher affinity of dopamine for the D4R and on a D4R-mediated-increase of D2R signaling. The BID-dependent increase in the excitability of the glutamatergic terminal to release glutamate seems to depend, predominantly, on functional downregulation of A1R, which can be counteracted by D2R or D4R agonists and α2δ ligands (see text). The function of voltage-dependent calcium channels (VDCC), which activation promotes vesicular fusion and neurotransmitter release, is regulated by Gi-coupled receptors, including A1R, D2R and D4R, as well as by accessory α2δ subunits, the targets of gabapentin-like compounds. Reproduced and modified from Yepes and others 2017.
The question becomes: is hypersensitivity of cortico-striatal glutamatergic terminals a primary determinant of the dysfunction of the cortico-striatal-thalamic-cortical circuits in RLS. There are two types of cortico-striatal glutamatergic neurons, defined by their long-range projections (see Fig. 1): the intratelencephalic (IT) neuron, which projects ipsi- or bilaterally (via the external capsule and corpus callosum) within the telencephalon, to the cortex and striatum; and the pyramidal tract (PT) neuron, which projects ipsilaterally to both the brainstem (including dopaminergic mesencephalic cells) and the spinal cord (via the internal capsule, cerebral peduncle and pyramidal tract), and also branches to the ipsilateral cortex, striatum, thalamus and subthalamic nucleus (Shepherd 2013). Importantly, the PT neuron is multiprojectional (Shepherd 2013). Therefore, it becomes tempting to speculate that BID-induced hypersensitivity occurring in the cortico-striatal terminals of PT neurons also occur in its other glutamatergic terminals, in the mesencephalon, thalamus and the spinal cord, which could lead to the presynaptic hyperdopaminergic state, the thalamic increase in glutamate levels and the spinal hyperexcitability (see above and Fig. 1). An additional glutamate-dependent component of the BID-induced hyperdopaminergic state could be related to the ability of striatal glutamate to locally control dopamine release. Thus, we recently demonstrated that optogenetically-induced glutamate release from cortico-striatal terminals locally induces striatal dopamine release (Quiroz and others 2016a).
Putative pivotal role of adenosine
As mentioned earlier, clinically there appears to be at least two distinct, but yet interrelated, elements within the underlying biology of RLS: a hyperdopaminergic state (mainly involved in akathisia and PLMS) and a hyperglutamatergic state (also involved in enhanced arousal). Recent studies have provided a third mechanism that may link BID to the hyperdopaminergic and hyperglutamatergic states: a hypoadenosinergic state (Ferré and others 2018a). Firstly, adenosine exerts a brake in the function of the ascending dopaminergic system, mainly by means of specific interactions between subtypes of adenosine and dopamine receptors (Ferré and others 1997). There are two main subtypes of adenosine receptors in the brain tonically inhibited by endogenous adenosine, A1 and A2A receptors (A1R and A2AR). These establish specific intermolecular interactions with dopamine D1 receptors (D1R) and D2R, respectively. Recent studies indicate that these A1R-D1R and A2AR-D2R heteromers form part of macromolecular complexes that include G proteins and the effector adenylyl cyclase (Ferré and others 2018b). These complexes allow an elaborated fine-tuned inhibitory modulation of dopamine signals by adenosine and they are highly expressed in the striatum. They are segregated in the two most populated types of striatal neurons, with A1R-D1R and A2AR-D2R being respectively localized in the striatonigral and striatopallidal neurons (SN and SP in Fig. 1; for a more detailed description of these adenosine receptor-dopamine receptor heteromers, see Ferré and others 2018a and 2018b). Secondly, adenosine acts as a universal presynaptic inhibitor of glutamate transmission. This function is mediated by A1R receptors localized in basically all glutamatergic terminals, that when activated inhibit glutamate release (Dunwiddie and Masino 2001). Finally, adenosine is a main modulator of homeostatic sleep and it mediates the sleepiness induced by prolonged wakefulness.
The sleep function of adenosine depends on the ability of adenosine to directly inhibit the function of the multiple interconnected ascending arousal systems, by acting both on their cells of origin and also their broadly targeted cortex (Ferré 2010; Brown and others 2012) (Fig. 5). Those arousal systems include: the ascending reticular activating system, which originates in the pontomesencephalic tegmentum (including the laterodorsal tegmental nucleus, pedunculopontine nucleus and locus coeruleus), uses glutamate, acetylcholine and noradrenaline as neurotransmitters, and relays in the midline-intralaminar and reticular nucleus of the thalamus or directly broadcasts to the cortex; the corticopetal basal forebrain systems (including the nucleus basalis of Meynert), which uses acetylcholine, glutamate and GABA; and the ascending hypothalamic histamine (which originates in the tuberomammillary nucleus) and hypocretin/orexin ascending arousal systems (Ferré 2010; Brown and others 2012). A1R are particularly involved in the adenosine-mediated regulation of these ascending arousal systems. Infusion of A1R agonists in the basal forebrain pontomesencephalic tegmentum, lateral hypothalamus or prefrontal cortex increases sleep, whereas infusion of A1R antagonists in the same areas increases waking (reviewed in Brown and others 2012). The work by Haydon and others has established vesicular release of ATP by astrocytes as one of the main cellular mechanisms that provides the A1R-dependent adenosine-mediated control of homeostatic sleep. Extracellular metabolization of ATP then leads to accumulation of adenosine in the extracellular levels of adenosine (reviewed in Blustein and Haydon 2013). However, although adenosine A1R directly control the effects of all the ascending arousal systems, to date adenosine levels have only been shown to increase with prolonged wakefulness in the basal forebrain and neocortex (Brown and others 2012). Nevertheless, an additional adenosine-controlled system to be considered is the prefrontal corticofugal neurons that innervate the cells of origin of the pontomesencephalic tegmentum (Van Dort and others 2009) (Fig. 5). Through this connection, adenosine and A1R within the prefrontal cortex comprise part of a descending system that inhibits wakefulness by inhibiting the function of the interconnected ascending arousal systems (Van Dort and others 2009).
Figure 5.
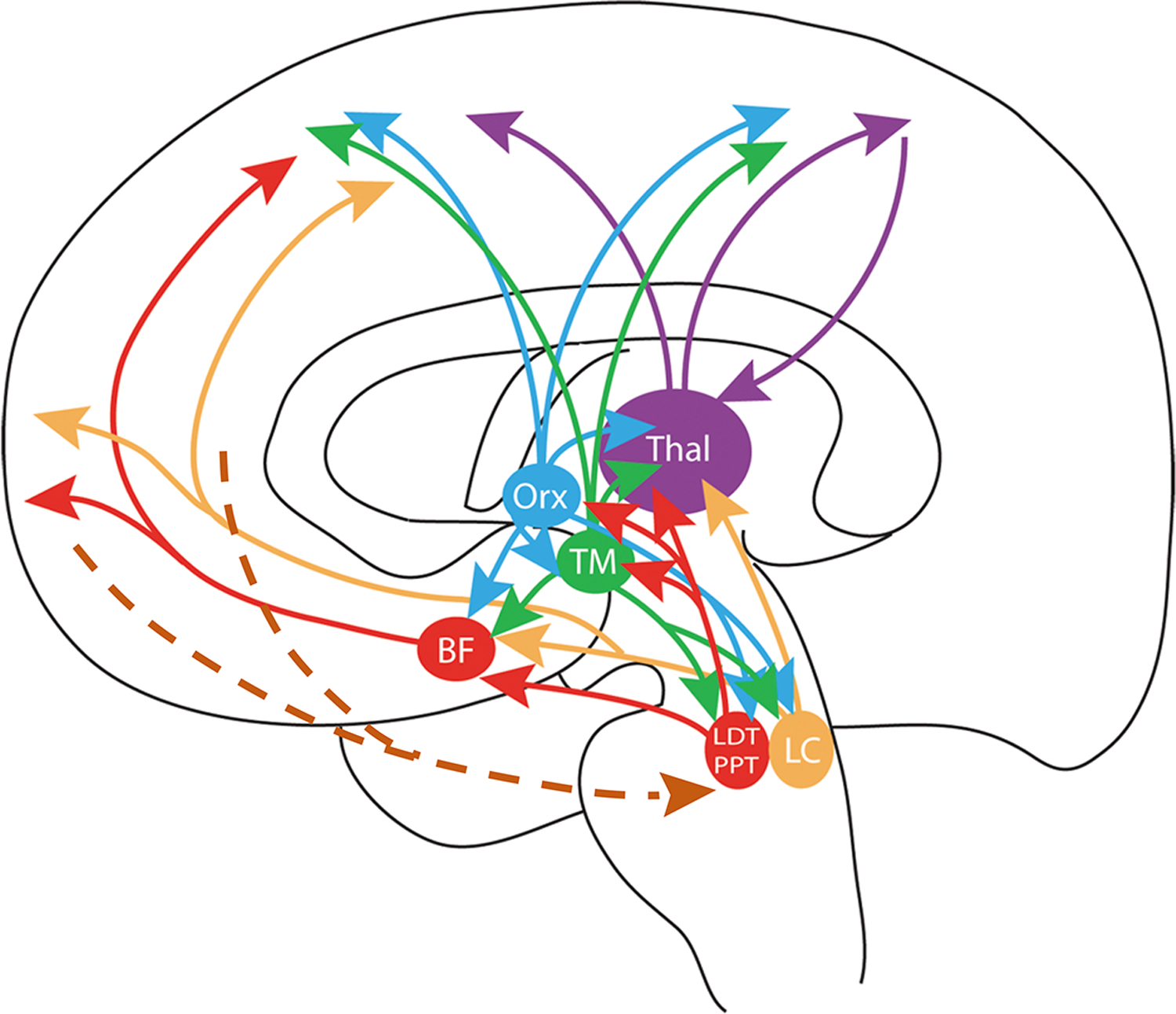
Scheme of the highly interconnected multiple ascending arousal systems, which are directly or indirectly involved in the homeostatic sleep function of adenosine. This homeostatic function depends mostly on the accumulation of extracellular adenosine in basal forebrain and cortex. BF: area of origin of the corticopetal basal forebrain system; LC: locus coeruleus; LDT and PPT: laterodorsal and pedunculopontine nuclei; TM: tuberomammillary nucleus; Orx: area of origin of the hypocretin/orexin system; Thal: thalamus. The brown broken lines indicate the existence of descending prefrontal corticofugal neurons that influence the pontomesencephalic tegmentum (see text). Reproduced and modified from Ferré 2010
BID in rodents was initially shown to produce a significant upregulation of striatal A2AR, which could provide an explanation for the increased sensitivity of cortico-striatal terminals (reviewed in Ferré and others 2018a). Thus, both A1R and A2AR are localized in cortico-striatal terminals, where they form A1R-A2AR heteromers that operate as an adenosine concentration-dependent switch, by which low and high concentrations inhibit and facilitate glutamate release, respectively (Ciruela and others 2006; Fig. 4). Nevertheless, recent experiments in BID rodents indicated that A1R was more sensitive than A2AR to BID. Thus, a less severe iron deficient diet (still associated with BID, as demonstrated by significant upregulation of transferrin receptors) was associated with a significant downregulation of A1R in the striatum and cortex (Quiroz and others 2016b). BID-induced A1R downregulation would determine a hypoadenosinergic state, which could play a significant role in the hyperdopaminergic and hyperglutamatergic states involved in the PLMS and the hyperarousal state of RLS (Ferré and others 2018a). In fact, A1R has been suggested to be a marker of the homeostatic sleep response, of the need for recovery from lack of sleep (reviewed in Ferré and others 2018a). Recent preliminary findings support this hypothesis. Dipyridamole, a blocker of the adenosine transporters ENT1 and ENT2, should restore the BID decreased extracellular levels of adenosine in the CNS and thereby provide effective treatment for RLS. Dipyridamole showed in a recent open-label study a pronounced therapeutic effect on dysesthesias, PLMS and sleep disturbances in RLS patients (García-Borreguero and others 2018). This, however, needs to be confirmed in further clinical trials.
Conclusion and future directions
The pathogenesis of RLS presented in this paper can be summarized in the scheme shown in Fig. 6. Genetic and environmental factors lead to BID, which is the main initial pathogenetic mechanism leading to RLS. Experimental and clinical evidence indicate that restoration of normal brain iron levels should lead to reversal of the RLS phenotype (reviewed in Earley and others 2014). A BID-induced hypoadenosinergic state seems to be the next pathogenetic link, which leads to the hyperglutamatergic and hyperdopaminergic states. This leads to the dysfunction of the cortico-striatal-thalamic-cortical circuits producing PLMS and akathisia. Genetic factors that operate during the development of the CNS determine the vulnerability of these circuits to acquire BID-induced PLMS and akathisia. Hyperarousal would also be determined by the hypoadenosinergic state. The hypoadenosinergic state would disinhibit the ascending arousal systems, including the glutamatergic component of the ascending reticular activating system, as part of the hyperglutamatergic state (Fig. 6).
Figure 6.
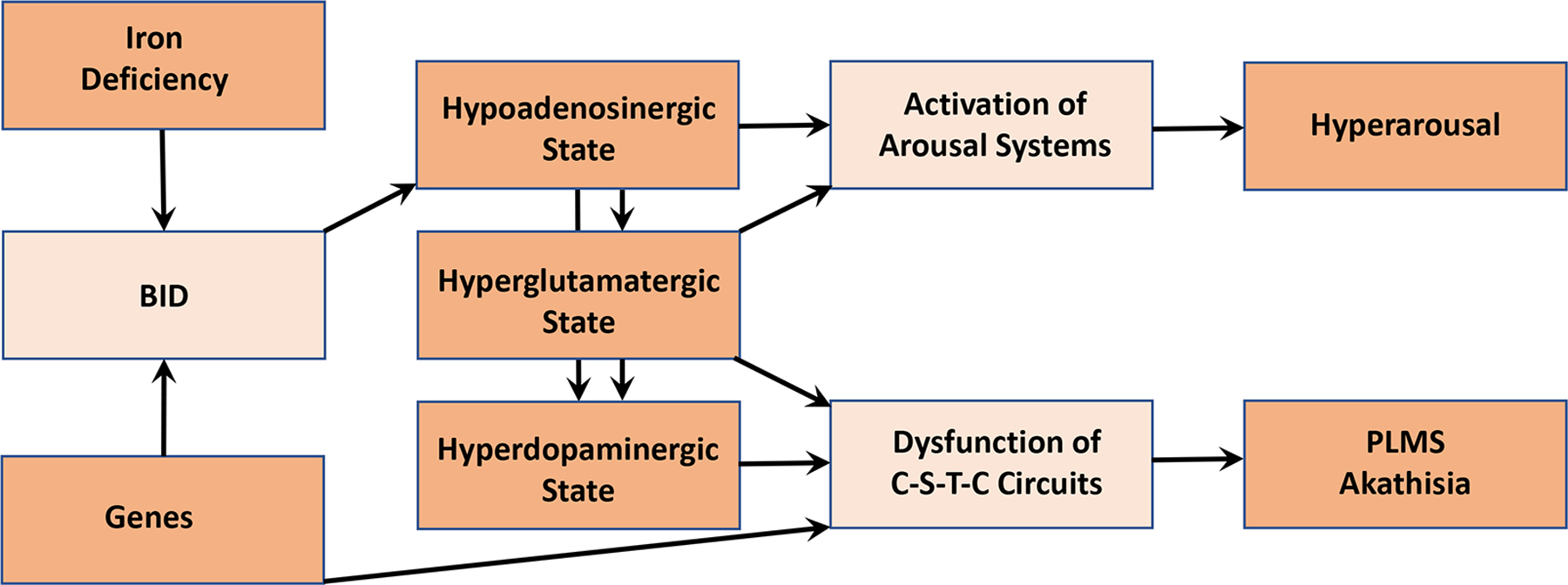
Integrative scheme of the pathogenetic mechanisms involved in the PLMS, akathisia and arousal components of RLS (see text: Conclusion and future directions).
At this moment, we are in particular need for alternative treatments for RLS. The initially very efficacious dopaminergic agonists with continued use produce side effect eventually leading to discontinuation of the drug (García-Borreguero and Williams 2011). The α2δ ligands and opioids (low doses) provide efficacious alternative treatments but also have their limitations and side effects. The mechanism of action responsible for the beneficial effects of opioids is at this moment very unclear (Trenkwakser and others 2017). In fact, it cannot be discarded that at least part of the beneficial effects of any of the medications used to treat RLS are related to their ability to directly decrease the spinal hyperexcitability (Clemens and others 2006; Kumru and others 2015). However, efforts for developing future treatments should specially be devoted to the elucidation and targeting of the initial pathogenetic mechanisms, that lead to BID and to BID-induced changes in several specific neurotransmitter systems (Earley and others 2014; Ferré and others 2018a).
Acknowledgements
We thank Prof. Juliane Winkelmann for allowing the reproduction of her work (Fig. 2).
Funding
SF is supported by the intramural funds of the National Institute on Drug Abuse
Footnotes
Declaration of Conflicting Interests
The authors declare no conflict of interests
References
- Aksu M, Bara-Jimenez W. 2002. State dependent excitability changes of spinal flexor reflex in patients with restless legs syndrome secondary to chronic renal failure. Sleep Med 3:427–30. [DOI] [PubMed] [Google Scholar]
- Allen RP, Mignot E, Ripley B, Nishino S, Earley CJ. 2002. Increased CSF hypocretin-1 (orexin-A) in restless legs syndrome. Neurology 59:639–41. [DOI] [PubMed] [Google Scholar]
- Allen RP, Walters AS, Montplaisir J, Hening W, Myers A, Bell TJ, and others. 2005. Restless legs syndrome prevalence and impact: REST general population study. Arch Intern Med 165:1286–92. [DOI] [PubMed] [Google Scholar]
- Allen RP, Earley CJ. 2007. The role of iron in restless legs syndrome. Mov Disord 18:S440–8. [DOI] [PubMed] [Google Scholar]
- Allen RP, Connor JR, Hyland K, Earley CJ. 2009. Abnormally increased CSF 3-Ortho-methyldopa (3-OMD) in untreated restless legs syndrome (RLS) patients indicates more severe disease and possibly abnormally increased dopamine synthesis. Sleep Med 10:123–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RP, Stillman P, Myers AJ. 2010. Physician-diagnosed restless legs syndrome in a large sample of primary medical care patients in western Europe: Prevalence and characteristics. Sleep Med 11:31–7. [DOI] [PubMed] [Google Scholar]
- Allen RP, Barker PB, Horská A, Earley CJ. 2013a. Thalamic glutamate/glutamine in restless legs syndrome: increased and related to disturbed sleep. Neurology 80:2028–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RP, Auerbach S, Bahrain H, Auerbach M, Earley CJ. 2013b. The prevalence and impact of restless legs syndrome on patients with iron deficiency anemia. Am J Hematol 88:261–4. [DOI] [PubMed] [Google Scholar]
- Allen RP, Donelson NC, Jones BC, Li Y, Manconi M, Rye DB, and others. 2017. Animal models of RLS phenotypes. Sleep Med 31:23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RP, Picchietti DL, Auerbach M, Cho YW, Connor JR, Earley CJ, and others. 2018. International Restless Legs Syndrome Study Group (IRLSSG). Evidence-based and consensus clinical practice guidelines for the iron treatment of restless legs syndrome/Willis-Ekbom disease in adults and children: an IRLSSG task force report. Sleep Med 41:27–44. [DOI] [PubMed] [Google Scholar]
- Altshuler D, Daly MJ, Lander ES. 2008. Genetic mapping in human disease. Science 322:881–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azcoitia V, Aracil M, Martínez-A C, Torres M. 2005. The homeodomain protein Meis1 is essential for definitive hematopoiesis and vascular patterning in the mouse embryo. Dev Biol 280:307–20. [DOI] [PubMed] [Google Scholar]
- Bara-Jimenez W, Aksu M, Graham B, Sato S, Hallett M. 2000. Periodic limb movements in sleep: state-dependent excitability of the spinal flexor reflex. Neurology 54:1609–16. [DOI] [PubMed] [Google Scholar]
- Blutstein T, Haydon PG. 2013. The Importance of astrocyte-derived purines in the modulation of sleep. Glia 61:129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Basheer R, McKenna JT, Strecker RE, McCarley RW. 2012. Control of sleep and wakefulness. Physiol Rev 92:1087–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciruela F, Casadó V, Rodrigues RJ, Luján R, Burgueño J, Canals M, and others. 2006. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J Neurosci 26:2080–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens S, Rye D, Hochman S. 2006. Restless legs syndrome: revisiting the dopamine hypothesis from the spinal cord perspective. Neurology 67:125–30. [DOI] [PubMed] [Google Scholar]
- Connor JR, Wang XS, Allen RP, Beard JL, Wiesinger JA, Felt BT, and others. 2009. Altered dopaminergic profile in the putamen and substantia nigra in restless leg syndrome. Brain 132:2403–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JR, Ponnuru P, Wang XS, Patton SM, Allen RP, Earley CJ. 2011. Profile of altered brain iron acquisition in restless legs syndrome. Brain 134:959–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean T Jr, Allen RP, O’Donnell CP, Earley CJ. 2006. The effects of dietary iron deprivation on murine circadian sleep architecture. Sleep Med 7:634–40. [DOI] [PubMed] [Google Scholar]
- DeAndrade MP, Johnson RL Jr, Unger EL, Zhang L, van Groen T, Gamble KL, and others. 2012. Motor restlessness, sleep disturbances, thermal sensory alterations and elevated serum iron levels in Btbd9 mutant mice. Hum Mol Genet 21:3984–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS 2nd , Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG 1998. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA 95:322–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley DJ, Taylor CP, Donevan S, Feltner D. 2007. Ca2+ channel alpha2delta ligands: novel modulators of neurotransmission. Trends Pharmacol Sci 28:75–82. [DOI] [PubMed] [Google Scholar]
- Drgonova J, Walther D, Wang KJ, Hartstein GL, Lochte B, Troncoso J, and others. 2015. Mouse model for PTPRD associations with WED/RLS and addiction: reduced expression alters locomotion, sleep behaviors and cocaine-conditioned place preference. Mol Med 21:717–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA. 2001. The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci 4:31–55. [DOI] [PubMed] [Google Scholar]
- Earley CJ, Connor JR, Beard JL, Malecki EA, Epstein DK, Allen RP. 2000. Abnormalities in CSF concentrations of ferritin and transferrin in restless legs syndrome. Neurology 54:1698–700. [DOI] [PubMed] [Google Scholar]
- Earley CJ, Allen RP, Connor JR, Ferrucci L, Troncoso J. 2009. The dopaminergic neurons of the A11 system in RLS autopsy brains appear normal. Sleep Med 10:1155–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley CJ, Kuwabara H, Wong DF, Gamaldo C, Salas R, Brasic J, Ravert HT, Dannals RF, Allen RP. 2011. The dopamine transporter is decreased in the striatum of subjects with restless legs syndrome. Sleep 34:41–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley CJ, Kuwabara H, Wong DF, Gamaldo C, Salas RE, Brašić JR, Ravert HT, Dannals RF, Allen RP. 2013. Increased synaptic dopamine in the putamen in restless legs syndrome. Sleep 36:51–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley CJ, Connor J, Garcia-Borreguero D, Jenner P, Winkelman J, Zee PC, and others. 2014. Altered brain iron homeostasis and dopaminergic function in Restless Legs Syndrome (Willis-Ekbom Disease). Sleep Med 15:1288–301. [DOI] [PubMed] [Google Scholar]
- Ferré S, Fredholm BB, Morelli M, Popoli P, Fuxe K. 1997. Adenosine-dopamine receptor-receptor interactions as an integrative mechanism in the basal ganglia. Trends Neurosci 20:482–7. [DOI] [PubMed] [Google Scholar]
- Ferré S. 2010. Role of the central ascending neurotransmitter systems in the psychostimulant effects of caffeine. J Alzheimers Dis 20 Suppl 1:S35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Quiroz C, Guitart X, Rea W, Seyedian A, Moreno E, and others. 2018a. Pivotal Role of Adenosine Neurotransmission in Restless Legs Syndrome. Front Neurosci 11:722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Bonaventura J, Zhu W, Hatcher-Solis C, Taura J, Quiroz C, and others. 2018b. Essential control of the function of the striatopallidal neuron by pre-coupled complexes of adenosine A(2A)-dopamine D(2) receptor heterotetramers and adenylyl cyclase. Front Pharmacol 9:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri R, Cosentino FI, Manconi M, Rundo F, Bruni O, Zucconi M. 2014. Increased electroencephalographic high frequencies during the sleep onset period in patients with restless legs syndrome. Sleep 37:1375–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri R, Rundo F, Zucconi M, Manconi M, Bruni O, Ferini-Strambi L, and others. 2015a. An Evidence-based Analysis of the Association between Periodic Leg Movements during Sleep and Arousals in Restless Legs Syndrome. Sleep 38:919–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri R, Proserpio P, Rundo F, Lanza A, Sambusida K, Redaelli T, and others. 2015b. Neurophysiological correlates of sleep leg movements in acute spinal cord injury. Clin Neurophysiol 126:333–8. [DOI] [PubMed] [Google Scholar]
- Freeman AA, Rye DB. 2013. The molecular basis of restless legs syndrome. Curr Opin Neurobiol 23:895–900. [DOI] [PubMed] [Google Scholar]
- Gamaldo C, Benbrook AR, Allen RP, Oguntimein O, Earley CJ. 2009. Evaluating daytime alertness in individuals with Restless Legs Syndrome (RLS) compared to sleep restricted controls. Sleep Med 10:134–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Borreguero D, Williams AM. 2011. Dopaminergic augmentation of restless legs syndrome: the scope of the problem. Sleep Med 12:425–6 [DOI] [PubMed] [Google Scholar]
- Garcia-Borreguero D, Patrick J, DuBrava S, Becker PM, Lankford A, Chen C, and others. 2014. Pregabalin versus pramipexole: effects on sleep disturbance in restless legs syndrome. Sleep 37:635–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Borreguero D, Guitart X, Garcia Malo C, Cano-Pumarega I, Granizo JJ, Ferré S. 2018. Treatment of restless legs syndrome/Willis-Ekbom disease with the non-selective ENT1/ENT2 inhibitor dipyridamole: testing the adenosine hypothesis. Sleep Med 45:94–7. [DOI] [PubMed] [Google Scholar]
- Gazzellone MJ, Zarrei M, Burton CL, Walker S, Uddin M, Shaheen SM, and others. 2016. Uncovering obsessive-compulsive disorder risk genes in a pediatric cohort by high-resolution analysis of copy number variation. J Neurodev Disord 8:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghorayeb I, Gamas A, Mazurie Z, Mayo W. 2017.Attention-deficit hyperactivity and obsessive-compulsive symptoms in adult patients with primary restless legs syndrome: Different phenotypes of the same disease? Behav Sleep Med 30:1–8. [DOI] [PubMed] [Google Scholar]
- Hoffer ZS, Alloway KD. 2001. Organization of corticostriatal projections from the vibrissal representations in the primary motor and somatosensory cortical areas of rodents. J Comp Neurol 439:87–103. [DOI] [PubMed] [Google Scholar]
- Inoue Y, Hirata K, Kuroda K, Fujita M, Shimizu T, Emura N, Uchimura N, Kagimura T, Sha K, Nozawa T. 2010. Efficacy and safety of pramipexole in Japanese patients with primary restless legs syndrome: A polysomnographic randomized, double-blind, placebo-controlled study. Sleep Med 11:11–6. [DOI] [PubMed] [Google Scholar]
- Inturrisi CE. 2005. Pharmacology of methadone and its isomers. Minerva Anestesiol 71:435–7. [PubMed] [Google Scholar]
- Jellen LC, Unger EL, Lu L, Williams RW, Rousseau S, Wang X, Earley CJ, Allen RP, Miles MF, Jones BC. 2012. Systems genetic analysis of the effects of iron deficiency in mouse brain. Neurogenetics 13:147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellen LC, Lu L, Wang X, Unger EL, Earley CJ, Allen RP, and others. 2013. Iron deficiency alters expression of dopamine-related genes in the ventral midbrain in mice. Neuroscience 252:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BC, Reed CL, Hitzemann R, Wiesinger JA, McCarthy KA, Buwen JP, and others. 2003. Quantitative genetic analysis of ventral midbrain and liver iron in BXD recombinant inbred mice. Nutr Neurosci 6:369–77. [DOI] [PubMed] [Google Scholar]
- Kapur N, Friedman R. 2002. Oral ketamine: a promising treatment for restless legs syndrome. Anesth Analg 94:1558–9. [DOI] [PubMed] [Google Scholar]
- Kawabe H, Brose N. 2011. The role of ubiquitylation in nerve cell development. Nat Rev Neurosci 12:251–68. [DOI] [PubMed] [Google Scholar]
- Kumru H, Vidal J, Benito J, Barrio M, Portell E, Valles M, and others. 2015. Restless leg syndrome in patients with spinal cord injury. Parkinsonism Relat Disord 21:1461–4. [DOI] [PubMed] [Google Scholar]
- Lanza G, Cantone M, Lanuzza B, Pennisi M, Bella R, Pennisi G, and others. 2015. Distinctive patterns of cortical excitability to transcranial magnetic stimulation in obstructive sleep apnea syndrome, restless legs syndrome, insomnia, and sleep deprivation. Sleep Med Rev 19:39–50. [DOI] [PubMed] [Google Scholar]
- Lai YY, Cheng YH, Hsieh KC, Nguyen D, Chew KT, Ramanathan L, Siegel JM. 2017. Motor hyperactivity of the iron-deficient rat - an animal model of restless legs syndrome. Mov Disord 32:1687–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BY, Kim J, Connor JR, Podskalny GD, Ryu Y, Yang QX. 2018. Involvement of the central somatosensory system in restless legs syndrome: A neuroimaging study Neurology doi: 10.1212/WNL.0000000000005562. [DOI] [PubMed] [Google Scholar]
- Magalhães SC, Kaelin-Lang A, Sterr A, do Prado GF, Eckeli AL, Conforto AB. 2015. Transcranial magnetic stimulation for evaluation of motor cortical excitability in restless legs syndrome/Willis-Ekbom disease. Sleep Med 16:1265–73. [DOI] [PubMed] [Google Scholar]
- McGahan MC, Harned J, Mukunnemkeril M, Goralska M, Fleisher L, Ferrell JB. 2005. Iron alters glutamate secretion by regulating cytosolic aconitase activity. Am J Physiol Cell Physiol 288:C1117–24. [DOI] [PubMed] [Google Scholar]
- Montplaisir J, Boucher S, Poirier G, Lavigne G, Lapierre O, Lesperance P. 1997. Clinical, polysomnographic, and genetic characteristics of restless legs syndrome: a study of 133 patients diagnosed with new standard criteria. Mov Disord 12:61–5. [DOI] [PubMed] [Google Scholar]
- Mattheisen M, Samuels JF, Wang Y, Greenberg BD, Fyer AJ, McCracken JT, and others. 2015. Genome-wide association study in obsessive-compulsive disorder: results from the OCGAS. Mol Psychiatry 20:337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestadt G, Wang Y, Grados MA, Riddle MA, Greenberg BD, Knowles JA, and others. 2011. Homeobox genes in obsessive-compulsive disorder. Am J Med Genet B Neuropsychiatr Genet 159B:53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordlander NB. 1953. Therapy in restless legs. Acta Med Scand 145:453–7. [PubMed] [Google Scholar]
- Oertel WH, Benes H, Garcia-Borreguero D, Högl B, Poewe W, Montagna P, Ferini-Strambi L, Sixel-Döring F, Trenkwalder C, Partinen M, Saletu B, Polo O, Fichtner A, Schollmayer E, Kohnen R, Cassel W, Penzel T, Stiasny-Kolster K. 2010. Rotigotine transdermal patch in moderate to severe idiopathic restless legs syndrome: a randomized, placebo-controlled polysomnographic study. Sleep Med 11:848–56. [DOI] [PubMed] [Google Scholar]
- Qu S, Le W, Zhang X, Xie W, Zhang A, Ondo WG. 2007. Locomotion is increased in A11-lesioned mice with iron deprivation: a possible animal model for restless legs syndrome. J Neuropathol Exp Neurol 66:383–8. [DOI] [PubMed] [Google Scholar]
- Quiroz C, Orrú M, Rea W, Ciudad-Roberts A, Yepes G, Britt JP, Ferré S. 2016a. Local Control of Extracellular Dopamine Levels in the Medial Nucleus Accumbens by a Glutamatergic Projection from the Infralimbic Cortex. J Neurosci 36:851–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroz C, Gulyani S, Ruiqian W, Bonaventura J, Cutler R, Pearson V, and others. 2016b. Adenosine receptors as markers of brain iron deficiency: Implications for Restless Legs Syndrome. Neuropharmacology 111:160–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan S, Hanley JJ, Deniau JM, Bolam JP. 2002. Synaptic convergence of motor and somatosensory cortical afferents onto GABAergic interneurons in the rat striatum. J Neurosci 22:8158–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saletu B, Gruber G, Saletu M, Brandstatter N, Hauer C, Prause W and others. 2000. Sleep laboratory studies in restless legs syndrome patients as compared with normals and acute effects of ropinirole. 1. Findings on objective and subjective sleep and awakening quality. Neuropsychobiology 41:181–9. [DOI] [PubMed] [Google Scholar]
- Salminen AV, Manconi M, Rimpilä V, Luoto TM, Koskinen E, Ferri R, and others. 2013. Disconnection between periodic leg movements and cortical arousals in spinal cord injury. J Clin Sleep Med 9:1207–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla A, Agarwal KN, Shukla GS. 1989. Latent iron deficiency alters gamma-aminobutyric acid and glutamate metabolism in rat brain. Experientia 45:343–5. [DOI] [PubMed] [Google Scholar]
- Silver N, Allen RP, Senerth J, Earley CJ. 2011. A 10-year, longitudinal assessment of dopamine agonists and methadone in the treatment of restless legs syndrome. Sleep Medicine 12:440–4. [DOI] [PubMed] [Google Scholar]
- Spieler D, Kaffe M, Knauf F, Bessa J, Tena JJ, Giesert F, and others. 2014. Restless legs syndrome-associated intronic common variant in Meis1 alters enhancer function in the developing telencephalon. Genome Res 24:592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schormair B, Winkelmann J. 2011. Genetics of restless legs syndrome: Mendelian, complex, and everything in between. Sleep Med Clin 6:203–15. [Google Scholar]
- Schormair B, Zhao C, Bell S, Tilch E, Salminen AV, Pütz B, and others. 2017. Identification of novel risk loci for restless legs syndrome in genome-wide association studies in individuals of European ancestry: a meta-analysis. Lancet Neurol 16:898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GM. 2013. Corticostriatal connectivity and its role in disease. Nat Rev Neurosci 14:278–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Craig AM. 2013. Protein tyrosine phosphatases PTPδ, PTPσ, and LAR: presynaptic hubs for synapse organization. Trends Neurosci 36:522–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tergau F, Wischer S, Paulus W. 1999. Motor system excitability in patients with restless legs syndrome. Neurology 52:1060–3. [DOI] [PubMed] [Google Scholar]
- Toresson H, Parmar M, Campbell K. 2000. Expression of Meis and Pbx genes and their protein products in the developing telencephalon: implications for regional differentiation. Mech Dev 2000. 94:183–7. [DOI] [PubMed] [Google Scholar]
- Trenkwalder C, Paulus W. 2010. Restless legs syndrome: pathophysiology, clinical presentation and management. Nat Rev Neurol 6:337–46. [DOI] [PubMed] [Google Scholar]
- Trenkwalder C, Zieglgänsberger W, Ahmedzai SH, Högl B. 2017. Pain, opioids, and sleep: implications for restless legs syndrome treatment. Sleep Med 31:78–85. [DOI] [PubMed] [Google Scholar]
- Van Dort CJ, Baghdoyan HA, Lydic R. 2009. Adenosine A(1) and A(2A) receptors in mouse prefrontal cortex modulate acetylcholine release and behavioral arousal. J Neurosci 29(3):871–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong L, Catoire H, Dion P, Gaspar C, Lafrenière RG, Girard SL, and others. 2009. MEIS1 intronic risk haplotype associated with restless legs syndrome affects its mRNA and protein expression levels. Hum Mol Genet 18:1065–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yepes G, Guitart X, Rea W, Newman AH, Allen RP, Earley CJ, and others. 2017. Targeting hypersensitive corticostriatal terminals in restless legs syndrome. Ann Neurol 82:951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeterian EH, Van Hoesen GW. 1978. Cortico-striate projections in the rhesus monkey: the organization of certain cortico-caudate connections. Brain Res 139:43–63. [DOI] [PubMed] [Google Scholar]
- Zhuo Y, Wu Y, Xu Y, Lu L, Li T, Wang X, and others. 2017. Combined resting state functional magnetic resonance imaging and diffusion tensor imaging study in patients with idiopathic restless legs syndrome. Sleep Med 38:96–103. [DOI] [PubMed] [Google Scholar]