

Abstract
Yes-associated protein 1 (YAP1) is a transcriptional coactivator that activates transcriptional enhanced associate domain transcription factors upon inactivation of the Hippo signaling pathway, to regulate biological processes like proliferation, survival, and differentiation. YAP1 is most prominently expressed in biliary epithelial cells (BECs) in normal adult livers and during development. In the current review, we will discuss the multiple roles of YAP1 in the development and morphogenesis of bile ducts inside and outside the liver, as well as in orchestrating the cholangiocyte repair response to biliary injury. We will review how biliary repair can occur through the process of hepatocyte-to-BEC transdifferentiation and how YAP1 is pertinent to this process. We will also discuss the liver’s capacity for metabolic reprogramming as an adaptive mechanism in extreme cholestasis, such as when intrahepatic bile ducts are absent due to YAP1 loss from hepatic progenitors. Finally, we will discuss the roles of YAP1 in the context of pediatric pathologies afflicting bile ducts, such as Alagille syndrome and biliary atresia. In conclusion, we will comprehensively discuss the spatiotemporal roles of YAP1 in biliary development and repair after biliary injury while describing key interactions with other well-known developmental pathways.
Keywords: Hippo signaling, bile duct development, cholestasis, transdifferentiation, ductular reaction
Graphical Abstract
Bile ducts that carry bile within the liver and then from the liver to the intestines can be highly susceptible to injury. Newborns can sometimes be born with defects in bile ducts ranging from their complete absence to paucity to obstruction. In adults, injury to the bile ducts can lead to impairment in bile flow and manifest as jaundice. There are very few medical treatments available, and hence, it is highly relevant to study key molecules that are expressed in bile duct cells and regulate their structure and function at baseline and help in their repair after injury. In this article, we will discuss the role of one such protein called Yes-associated protein-1, which has been shown to be important in bile duct development and in helping with various types of repair responses once bile duct gets injured.
YAP1 as a Component of Hippo Signaling Pathway
YAP1 Signaling
YAP1 is a transcriptional coactivator that works mostly through the transcriptional enhanced associate domain (TEAD) family member transcription factors to regulate genes related to cell proliferation and differentiation, often by binding to enhancer regions and collaborating with activator protein 1 (AP1).1-4 YAP1 is canonically repressed by the Hippo kinase pathway through cytoplasmic retention and degradation (Fig. 1A). A variety of signaling inputs activate the kinases, mammalian STE20-like 1 and 2 (MST1/2), which phosphorylate large tumor suppressor kinase 1 and 2 (LATS1/2), which phosphorylate YAP1 at various sites, including serine-127 (S127).5,6 Phosphorylated YAP1 is sequestered in the cytoplasm, at adherens junctions through binding α-catenin and at tight junctions through binding angiomotin.7-9 Phosphorylated YAP1 is also sequestered and degraded through interactions with 14-3-3 proteins.6,8
Fig. 1.
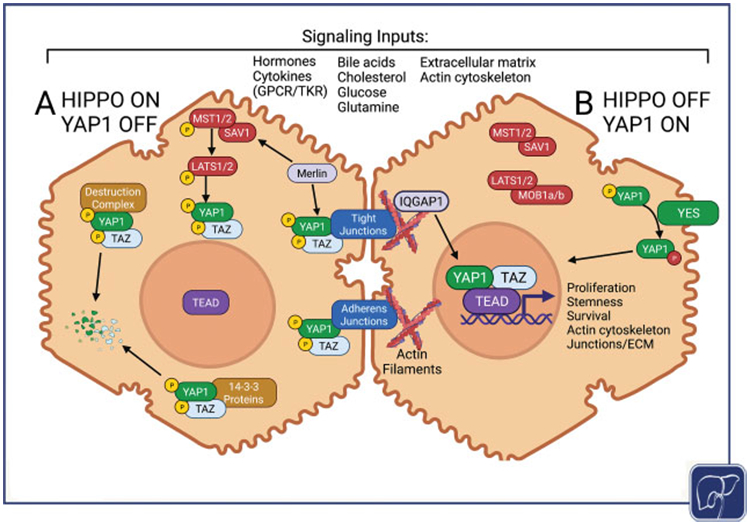
Overview of the Hippo/YAP signaling pathway. (A) When the Hippo kinases are activated YAP1 and/or TAZ are phosphorylated, which targets them for degradation by 14-3-3 proteins. YAP1 and/or TAZ are also inactivated through cytoplasmic sequestration mediated by interactions with cell junctions and other complexes. (B) When the Hippo kinase pathway activity is reduced, de-phosphorylated YAP1 and/or TAZ enters the nucleus and partners with TEAD transcription factors to regulate proliferation, stemness, survival, and mechanical properties of the cell. YAP1 can also be activated in a Hippo-independent manner through Yes kinase activity. Numerous inputs can influence the level of YAP1 activity through cell membrane receptors, nuclear receptors, or mechanosensory signaling pathways.
The decreased activity of Hippo pathway kinases allows for YAP1 activation and transport into the nucleus for the regulation of gene expression (Fig. 1B). Alternatively, YAP1 can be activated through phosphorylation at tyrosine-357 (Y357) by Yes kinase, a member of the Src kinase family; this can override S127 phosphorylation and, thus, can activate YAP1 despite maintenance of Hippo pathway activity.10,11 YAP1 activity also responds to and modulates changes in cytoskeletal organization, and YAP1 becomes activated when cells interact with stiffer extracellular matrix environments.12,13
Numerous inputs regulate Hippo pathway activity, including extracellular signals from growth factors and cytokines acting through membrane G-protein-coupled receptors and tyrosine kinase receptors, changes in actin cytoskeletal tension, and cell–cell focal adhesions and junctions.11,14,15 In particular, merlin (Nf2), a well-known tumor suppressor, recruits Hippo pathway kinases to the plasma membrane in close proximity to YAP1, thus facilitating regulatory interactions that inactivate YAP1.16 In addition, YAP1 can be regulated in a Hippo-independent manner through interactions with many key signaling pathways including Notch, Wnt/β-catenin, transforming growth factor β (TGFβ), and mammalian target of rapamycin (mTOR) pathways.17-22 Lastly, nuclear YAP1 can be prevented from binding to TEAD through interaction with the AT-rich interaction domain 1A (ARID1a)-containing SWItch/sucrose non-fermentable chromatin remodeling complex, which also responds to mechano-transduced signals.23 Thus, YAP1 integrates a variety of upstream signals allowing cells to respond actively to their environment.
Generally, YAP1 regulates gene expression to promote cell proliferation and survival, enhance metabolic activity, and alter extracellular matrix composition.12,24 Several studies have combined RNA-sequencing and ChIP-seq data to identify YAP1 transcriptional targets.1,25,26 Notably, connective tissue growth factor (CTGF, gene name CCN2) and cysteine-rich 61 (CYR61, gene name CCN1) have been recurrently identified and used experimentally as surrogate measures of YAP1 activity.27,28 However, more studies are needed to identify context-dependent transcriptional targets with functional impacts in liver pathobiology.
YAP1 is known to impact baseline liver size, regenerative response to injury, and tumorigenesis in liver.12,24 YAP1 seems to promote cell dedifferentiation in some contexts while promoting biliary differentiation in others; studies have yet to disentangle these two distinct functions in the context of liver biology and disease and identify unique YAP1 targets that mediate these processes.11,29,30 In addition, the upstream signals regulating activity and the downstream targets modulated by YAP1 remain to be further investigated in the context of biliary development and repair.
YAP1 versus TAZ: Similar but not Always Interchangeable
TAZ (transcriptional coactivator with PDZ-binding motif, also known as WWTR1, WW-domain containing transcription regulator 1) is a paralog of YAP1 which is similarly regulated by the Hippo pathway and some Hippo-independent mechanisms.31 YAP1 and TAZ together form a complex primarily with TEAD transcription factors as well as AP1, T-box 5, runt-related transcription factor 1, and small mothers against decapentaplegic 2–4 (SMADs 2–4) in different contexts, but both YAP1 and TAZ have distinct transcriptional partners such as p73 and peroxisome proliferator-activated receptor gamma (PPARγ), respectively.31 Studies have shown that YAP1 can regulate transcription of TAZ, and together they can activate expression of LATS2, forming feedback loops regulating overall Hippo pathway activity.32
Structurally, YAP1 and TAZ share most protein domains but with several key differences. First, YAP1 contains two WW-domains, while TAZ contains just one, potentially altering their ability to bind to many shared regulators such as the LATS kinases.31 Second, while both YAP1 and TAZ carry a TEAD-binding domain to form heterodimers with TEAD proteins, TAZ has the unique ability to homodimerize and, thus, form a TAZ/TEAD heterotetramer, which has the potential to bind to multiple TEAD sites nearby and result in altered transcriptional regulation.33 TAZ also lacks both a proline-rich motif (used by YAP1 to interact with pre-mRNA splicing proteins) and an SCR homology 3 domain-binding motif (used by YAP1 to interact with SRC and Yes kinases and other kinase adaptor proteins).31 Finally, TAZ contains two phosphodegron regions (compared with just one in YAP1) that can be phosphorylated by glycogen synthase kinase 3-β (GSK3), creating a binding site for β-TrCP ubiquitin-protein ligase for protein degradation, similar to β-catenin.31
Many studies in the literature have studied YAP1 and TAZ together and applied their conclusions to both proteins as a unit. Also, numerous key studies in the field have focused on double knockout models, which inactivate both YAP1 and TAZ, or models that delete upstream Hippo regulators MST1/2 or LATS1/2 and thus activate both YAP1 and TAZ but have additional downstream effects. Considering how often YAP1 and TAZ work as a complex, these studies are invaluable in understanding processes that depend on both YAP1 and TAZ. However, there is mounting evidence that YAP1 and TAZ play distinct roles in many tissue types and developmental stages which may be redundant, complementary, or completely different. For instance, whole-body knockout of YAP1 is embryonic lethal, with broad vasculogenetic defects, while whole-body knockout of TAZ results in viable offspring with focal disease in the kidney and lung.34-36 Studies in many organs show that YAP1 and TAZ regulate survival, proliferation, and stemness, but individual tissue-specific knockouts show additional subtle defects related to YAP1 or TAZ but not both, suggesting that beyond their core shared functions YAP1 and TAZ play unique tissue-specific roles that cannot be compensated by the other.31,37 For this reason, more studies are needed to dissect the individual functions of YAP1 and TAZ as well as how they regulate one another in development and disease.
In the liver, YAP1 and TAZ are both critical for liver regeneration by regulating proliferation and cell cycling, liver tumorigenesis, and regulating inflammation and fibrosis.38 However, recent studies have identified distinct roles of YAP1 and TAZ in specific liver pathologies. For instance, several studies have shown that TAZ plays a unique role in the development of non-alcoholic steatohepatitis through its effects on fatty acid and cholesterol metabolism, recruitment and activation of innate immune cells, and activation of stellate cells through Hedgehog signaling.39,40 Other studies have identified distinct roles of YAP1 and TAZ in liver cancers, with TAZ expression, in particular, serving as a poor prognostic marker in hepatocellular carcinoma even more so than YAP1.41,42 While many studies have pointed to key roles for YAP1 in biliary development and homeostasis and in regulating cell plasticity between hepatocytes and cholangiocytes, TAZ is unable to fully compensate for the absence of YAP1 in biliary development.27,43-46 Throughout this review, we will highlight the few known differences between YAP1 and TAZ in biliary development and repair and point out gaps in knowledge that remain to be investigated.
Liver Development
Hepatocyte and Biliary Differentiation during Liver Development
Here, we will discuss general principles of liver development, which have been elucidated mostly in rodent and zebrafish models. Foregut endoderm undergoes specification at E8.5 through the expression of forkhead box (Fox) A1/A2/A3 and GATA4 transcription factors. Secreted bone morphogenetic proteins (BMP) and fibroblast growth factor (FGF) ligands from the growing septum transversum mesenchyme and cardiac mesoderm promote the induction of liver progenitor cells (LPCs), hepatoblasts, from the foregut endoderm. Hepatoblasts begin to migrate into the septum transversum mesenchyme at E10.5, forming cords and proliferating to expand the growing liver bud.47 Around E13.5, hepatoblasts begin to differentiate into hepatocytes. Most hepatoblasts show the upregulation of hepatocyte nuclear factor 4 α (HNF4α) and CCAAT/enhancer-binding protein, C/EBPα, and the downregulation of HNF1β, homeobox B, among other factors as they differentiate into hepatocytes.47-49 From E13.5 onwards through the first few weeks of postnatal development, hepatoblasts gradually mature into hepatocytes and adopt the unique structural and metabolic features of the mature liver acini, with polarized canaliculi for bile transport and strictly zonated metabolic functions.
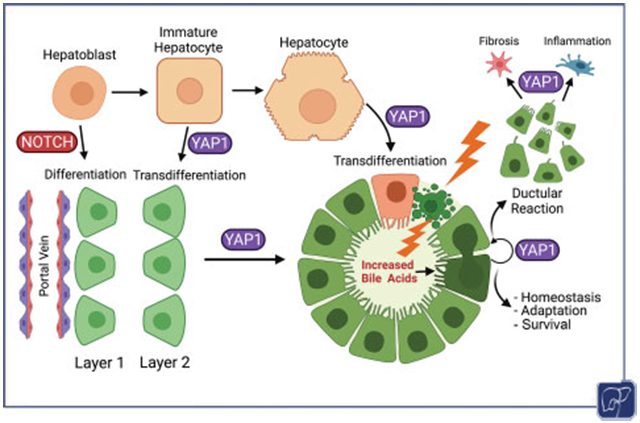
The process of intrahepatic biliary differentiation and morphogenesis is outlined in Fig. 2A. Molecular studies have identified that the process of biliary differentiation starts as early as E11.5 in mice, although most morphological studies have been able to identify primitive biliary cells around E13.5.48,49 Mesenchymal cells surrounding the immature portal veins express the Notch ligand Jagged1, which binds to the Notch2 receptor in neighboring hepatoblasts to induce the formation of the ductal plate around E13.5.50 Ductal plate cells and hepatoblasts also express TGFβ receptor II (TGFßRII), which allows them to respond to TGFβ ligands produced around and by the periportal mesenchyme.51,52 However, these ligands exert their activity only in a tightly controlled gradient extending outwards from the portal vein, regulated by a precise ratio of CCAAT/enhancer-binding proteins, C/ebpα and C/ebpβ, resulting in stimulation of a layer of hepatoblasts directly adjacent to the ductal plate.53 Notch and TGFβ signaling form part of feedback loops that contribute to a gene regulatory network by activating the biliary transcription factors, SRY-box transcription factors Sox9 and Sox4, which are essential for biliary morphogenesis, along with HNF6, Onecut2, and HNF1β, which also contribute to limiting TGFβ signaling to only two layers of portal hepatoblasts.54
Fig. 2.

Timeline of intrahepatic biliary development and potential roles of YAP1 in intrahepatic biliary differentiation and morphogenesis during development. (A) Notch activation is evident in earliest biliary cells adjacent to portal mesenchyme during early liver development from embryonic day 11 to 13 (E11–E13). These cells begin to mature as cholangiocytes, whereas a second layer of biliary cells appears from immature hepatocytes as hepatocyte-specific transcription factors like HNF4α are shut-off and biliary transcription factors like Sox-9 are tuned on, under the influence of TGFβ from portal mesenchyme. Eventually both layers of biliary cells come together to form a duct composed of maturing and polarizing cholangiocytes with appropriate surrounding laminin. (B) Potential roles of YAP1 in regulating biliary differentiation in the first and second layers of hepatoblasts which eventually form the mature bile ducts. (C) Potential roles of YAP1 in biliary cell polarization and laminin deposition during bile duct maturation.
If the ductal plate is properly induced, ductal plate cells undergo a variety of changes to mature into functional biliary epithelial cells (BECs), or cholangiocytes. Cell polarity is established very early on, as visualized by polarized expression of osteopontin1 and ezrin (a junctional protein) along with the appearance of primary cilia on the apical membrane.51 Polarization is also integral to lumen formation and apical constriction which marks the gradual morphological maturation of cholangiocytes.55,56 A combination of cytoskeletal mechanical forces and early bile acid flow from the nascent hepatocyte canaliculi contributes to the reorganization of plate cells to form ductal structures in parallel to the portal veins around E18.5.51,57 A basement membrane secreted by adjacent portal mesenchymal cells containing laminin-α1 initially supports the ductal plate, but as the cholangiocytes differentiate they secrete their own basement membrane containing laminin-α5 which wraps around the nascent duct.58 Ductal plate cells left out of the growing bile ducts continue to express Sox9 for some time but ultimately develop into periportal hepatocytes; a small population of hybrid Sox9+ hepatocytes remains into adulthood, with potential implications in liver regeneration.59,60 At the same time, the differentiating cholangiocytes and hepatocytes secrete vasculogenic factors to regulate the formation of hepatic arteries from periportal mesenchymal cells.61,62 Despite our understanding of the process of biliary morphogenesis, there remain many gaps in our knowledge of the molecular mechanisms which underlie these subtle, coordinated, and yet highly pertinent changes.
Finally, at maturity, the portal veins, bile ducts, and hepatic arteries form a parallel system of vessels known as the portal triad. Blood flows from the portal vessels through the liver sinusoids (fenestrated capillary system) toward the central veins, creating an oxygen gradient, while bile produced in hepatocytes flows in a counter-current manner toward the bile ducts.63 As hepatocytes mature, they adopt different phenotypes based on their proximity to either portal triad vessels or central veins. Periportal hepatocytes perform gluconeogenesis, cholesterol biosynthesis, and urea metabolism, whereas pericentral hepatocytes, responsive to Wnt/β-catenin signaling, perform glycolysis, bile acid biosynthesis, and glutamine synthesis, thus creating zonation of opposing metabolic processes, which is a hallmark of the mature liver.64
Development and Maturation of the Extrahepatic Biliary Tree
The extrahepatic bile ducts (EHBDs) consisting of the common bile duct, gallbladder, and cystic duct connect the intrahepatic bile ducts at the perihilar region to the pancreatic ductal system and transport bile to the intestine. The EHBDs are also closely associated with peribiliary glands (PBGs), a network of mucinous and serous acini connected with the bile duct lumina and supported by a network of connective, vascular, and nervous tissue which may act as a multipotential stem cell niche.55,65,66
Although the EHBDs are thought to arise from the hepatic bud in humans, in mice the EHBDs arise from the ventral pancreatic bud as shown by recent lineage tracing studies.67 An SRY-box transcription factor 17 (Sox17)-positive and pancreatic and duodenal homeobox 1 (Pdx1)-positive progenitor population arising from the foregut endoderm and ventral pancreatic bud give rise to the pancreas and pancreatic ductal system, the duodenum, and the EHBD network.68 Haploinsufficiency of Sox17 leads to malformation or agenesis of the gallbladder with defective contractility and function, although other parts of the EHBD network are not as drastically affected.69 Hes1 null mice showed major dysgenesis of the EHBDs along with expression of ectopic pancreatic cells, suggesting that Notch activity is involved in promoting and maintaining biliary differentiation while simultaneously blocking pancreatic acinar differentiation from common progenitors.68,70 Activation of transcription factors including hematopoietically expressed homeobox protein (Hhex), Hnf6, and Hnf1β, along with BMP and FGF signals from the adjacent mesenchyme, has also been shown to play a critical role in EHBD formation.67,71,72 Multiple mechanical signals regulated by Eph/Ephrin interactions regulate the formation of a continuous lumen from differentiating cholangiocytes.73 Finally, there is some evidence that the Wnt and Hippo pathways contribute to EHBD formation, both within the biliary cells themselves and also from nearby hepatocytes.55,67,74
However, the mechanisms regulating EHBD formation and the functions of each of these signaling pathways remain to be further elucidated. Furthermore, we still do not understand how the EHBD and intrahepatic bile duct (IHBD) networks interact during development to establish a seamless tubular network.
Role of YAP1 in Liver Development
Role of YAP1 in Intrahepatic Bile Duct Formation
At E10-12, YAP1 is present in both the nuclei and cytoplasm of hepatoblasts, but its function in this context is unknown.75 Recent analysis of a publicly available single-cell RNA-sequencing data from developing mouse livers between E10.5 and E17.5 also showed low levels of YAP1 expression in cells classified as hepatoblasts and hepatocytes, with a comparative increase in YAP1 expression in developing BECs over the course of their differentiation.46,49 Canonical YAP1 targets, Ccn1 (Cyr61) and Ccn2 (Ctgf), were also found to be minimally expressed in hepatoblasts but were comparatively much more strongly expressed in developing BECs, suggesting that YAP1 activity is restricted to the cholangiocyte population.46 Nonetheless, using FoxA3-cre, YAP1 was deleted from hepatoblasts during early murine liver development.46 This led to a complete loss of intrahepatic biliary tree in the knockout mice. Intriguingly, YAP1 loss did not impair Notch signaling in the ductal plate, and hence, YAP1 appears to not be necessary for Notch-driven initiation of biliary differentiation in the ductal plate. Interestingly, deletion of YAP1 from the early hepatoblasts did not impair hepatoblast differentiation into hepatocytes, suggesting that YAP1 is dispensable in the hepatocyte differentiation trajectory of the hepatic progenitors.46
YAP1 loss from hepatoblasts interfered with biliary morphogenesis in several ways (Fig. 2B,C). First, in the absence of YAP1, we observed persistent expression of HNF4α in hepatoblasts adjacent to the ductal plate. YAP1 seems to be necessary for the differentiation of the second layer of BECs, and the absence of YAP1 impairs the integration of this cell layer with the ductal plate, leading to failure of ductal morphogenesis and the absence of an intrahepatic biliary tree. This defect may be cell autonomous, suggesting that the two layers of developing BECs differentiate in different ways. Indeed, many developmental models of impaired bile duct morphogenesis result in what appears to be a “pause” between the formation of the first and second layers of developing BECs.58,76,77 YAP1 may play a unique role in activating a biliary program and perhaps repressing HNF4α in the second layer (Fig. 2B). It has been shown that YAP1 can regulate both HNF4α expression and its genome binding distribution in mouse hepatocyte lineage, so YAP1 may be critical for turning off a hepatoblast/hepatocyte genetic program in favor of a Notch-driven biliary program.30,43 In this case, Notch signaling may be upstream of YAP1 activation in the first layer but downstream of YAP1 signaling in the second layer (Fig. 2B). Indeed, YAP1 has been shown to upregulate Notch2 and Jagged1 gene expression, directly or indirectly via CCN1, and to activate Notch signaling in mature hepatocytes and in various liver cancers.17,18,43,78-80 Likewise, Notch signaling has been shown to activate YAP1 in various mouse models of liver cancer, particularly those with biliary phenotype, and they can engage in a positive feedback loop that promotes hepatocyte transdifferentiation into biliary-like cells.18,19 Thus, the relationship between Notch and YAP1 signaling in early biliary development remains a key area of research interest.
The observed phenotype may also result from impaired cell–cell communication from the portal mesenchyme to ductal plate cells as well as from ductal plate cells to adjacent hepatoblasts. In particular, TGFβ signaling originating from the portal mesenchyme is critical for the formation of the second layer during bile duct morphogenesis.43,52,77 Also, it was shown to drive hepatocyte-derived biliary regeneration in a mouse model of Alagille syndrome (ALGS) in which Notch signaling was impaired.81 YAP1 may be a downstream mediator of TGFβ signaling in developing hepatocytes, regulating their fate-switch to form a second layer of biliary cells and downregulating HNF4α (Fig. 2B).43 Studies have identified numerous forms of cross-talk and positive feedback between YAP1 and TGFβ signaling, including in hepatocytes, hepatic stellate cells, and various cancers, and it is known that the SMAD transcription factors can bind to YAP1 and mediate its gene regulation, although this relationship has not been clearly examined in liver development or in cholangiocytes.82-84 More conclusive studies are needed to elucidate these mechanisms.
Third, the formation of primary cilia was impaired in developing BECs after YAP1 loss, suggesting a defect in cell polarization in the ductal plate (Fig. 2C). Another recent study showed that impaired formation of primary cilia interfered with YAP1 activation in biliary development, which combined with our data may suggest a positive feedback loop between regulation of YAP1 activity and signaling functions associated with primary cilia in developing BECs.76 Finally, YAP1 loss resulted in the absence of laminin deposition in the basolateral side of the ductal plate (Fig. 2C). Impairment of laminin–integrin signaling has also been shown to block murine bile duct morphogenesis.58 However, whether YAP1 directly regulates the synthesis of laminins and integrins and/or whether this process depends on communication with the adjacent portal mesenchyme remains unclear.
Several other studies have also implicated YAP1 as an essential factor regulating biliary development. Deletion of YAP1 during mid-late embryonic development using Cre-recombinase driven by the Albumin promoter (Alb-Cre) results in a marked paucity of bile ducts postnatally, causing long-term cholestatic injury and failed attempts by the liver to regenerate cholangiocytes.85 On the contrary, inducing the expression of constitutively active YAP1 (S127A) in mature hepatocytes was shown to activate Notch signaling and promote the expression of biliary markers such as Sox9 and resulted in the dedifferentiation of hepatocytes into oval cells, resembling LPCs.44 Similarly, deletion of upstream regulator Nf2 causes dramatic overgrowth of bile ducts, a phenotype which is completely ablated in the absence of YAP1.85 A previous study suggested that the overgrowth of bile ducts due to Alb-Cre Nf2-deletion and subsequent YAP1 activation was ablated by Notch2 deletion, suggesting Notch activity may be downstream of YAP1 in bile duct development.79 However, Notch2 deletion in this model did not completely prevent bile duct formation, suggesting YAP1 may be regulating bile duct formation through additional signaling pathways.79
Importantly, activation of YAP1 through the Alb-Cre-mediated knockout of the upstream inhibitory kinases Lats1/2 in mid-late embryonic development resulted in abnormal overgrowth of ductular cells expressing immature biliary markers.43 Lats1/2-negative hepatoblasts differentiated much more efficiently into BECs than hepatocytes in vitro, due to YAP1-mediated direct transcriptional upregulation of Tgfβ2 and transcriptional repression of Hnf4α.43 Furthermore, YAP1 may signal upstream to activate both Notch and TGFβ signaling in both embryonic cells (in vitro) and adult liver epithelial cells, and both of these pathways play key roles in biliary development.43,44,52,81,83
Notably, deletion of both YAP1 and TAZ using the Alb-Cre model resulted in grossly similar biliary defects as Alb-Cre YAP1 single knockout.86,87 However, Lee et al examined the effect of deleting either YAP1, TAZ, or both in the context of Alb-Cre LATS1/2 deletion and found that YAP1 and TAZ each exerted some influence on the level of biliary overgrowth, but only when both were deleted did the biliary lineage disappear altogether.43 Furthermore, their histological analysis suggests some morphological differences in the biliary cell clusters after deletion of YAP1 and TAZ.43 TAZ has also been implicated in positive feedback loops with Notch and TGFβ signaling in the above-mentioned studies together with YAP1, and TAZ can also regulate cross-talk between liver epithelium and associated mesenchymal cells.19,28,38,88 However, no studies so far have looked at deletion of TAZ alone in the embryonic stages of liver development to fully differentiate the roles of YAP and TAZ in this process, which remains an open question of investigation.
Role of YAP1 in EHBD Development
Relatively less is known about the role of YAP1 in the development of EHBD. Intriguingly, one recent study showed that deletion of Hippo pathway regulator Sav1 in zebrafish resulted in abnormal or absent gallbladder development, abnormal intra- and extra-hepatic biliary morphology, and impaired bile flow through hepatocyte canaliculi.74 Loss of Sav1 led to dysplastic and proliferative phenotype in developing biliary cells causing masses resembling cholangiocarcinoma, as well as loss of cell polarity throughout the hepatobiliary epithelium. There was an elevated expression of both Yap1 and Taz, but concomitant Yap1 deletion did not rescue lethality of the Sav1 null phenotype. Re-expression of Sav1 in hepatocytes rescued the phenotype and mostly restored biliary morphogenesis and gallbladder formation, suggesting an unexpected non-cell autonomous role for Hippo/YAP1 signaling in regulating intra- and extra-hepatic biliary development through an unknown hepatocyte function. Nevertheless, Sav1 null zebrafish with hepatocyte Sav1 re-expression eventually developed cholangiocarcinoma-like gallbladder masses, suggesting that YAP1 has to be tightly controlled to regulate the homeostasis of gallbladder epithelium. Indeed, YAP1 has been found to be highly expressed in cholangiocarcinoma, both intra- and extra-hepatic, and elevated YAP1 histologic expression correlates with a worse prognosis in gallbladder cancer.89,90 Further study is needed to investigate the functions of YAP1 and TAZ in the development of the EHBDs and how this may impact associated disease.
Principles of Biliary Repair and Regeneration: A YAP1 Perspective
Overview of Cholangiocyte Response to Injury
The biliary system is sensitive to injury, and many acute and chronic liver injuries can result in damage to the bile ducts. Intriguingly, the liver reacts to such injuries and mounts diverse reparative responses, albeit with varying degrees of effectiveness. Under normal conditions, mature cholangiocytes are quiescent, secretory cells which regulate the transport and composition of bile.91 Studies in mice have distinguished two types of intrahepatic cholangiocytes by location, morphology, and function: large cholangiocytes, found in large IHBDs and EHBDs, and small cholangiocytes, found in the peripheral liver parenchyma.91,92 While large cholangiocytes respond to secretin signaling to regulate the secretion of bicarbonate and water to modulate bile fluid composition, small cholangiocytes do not normally express secretin receptor and instead rely on Ca2+-mediated signaling pathways to adjust bile fluid composition.93
All cholangiocytes are sensitive to injury and activate a variety of responses (Fig. 3A). The proliferation of cholangiocytes following to replace the demise of a damaged cholangiocyte is a common injury response. Sustained injury to bile duct cells, however, can result in the chronic proliferation of remnant cholangiocytes and their expansion to constitute what is commonly referred to as ductular reaction. The ductular reaction can be composed of orderly or more haphazard expansion of cholangiocytes of varying morphology; it occurs initially around the portal vein and can reach deeper into the liver parenchyma toward the pericentral zone depending on the type, extent, and duration of injury.94-97 The ductular reaction may arise from cholangiocytes, hepatocytes, or LPCs, such as hybrid Sox9-positive cells residing in the canals of Hering of rodents, or specific EpCAM-positive populations identified in human liver.98-100 The ductular reaction itself may be comprised of LPCs expanding in the setting of injury, and numerous markers such as EpCAM, CD24, and CD133 have been used to isolate these cells and demonstrate their bipotentiality.97,99,101-104 In addition, it has been shown that when large cholangiocytes are damaged, small cholangiocytes can expand and adopt the features of large cholangiocytes such as secretin receptor signaling to repopulate this niche, suggesting that small cholangiocytes may be more poorly differentiated within the spectrum of cholangiocyte identity.92,105 This remains a topic of intense investigation and it seems that the source, extent, and fate of a ductular reaction depend on the severity of injury and which liver cell types are primarily affected.97 The pathogenesis of the ductular reaction varies greatly based on the disease context and has been extensively reviewed elsewhere.95,106,107
Fig. 3.
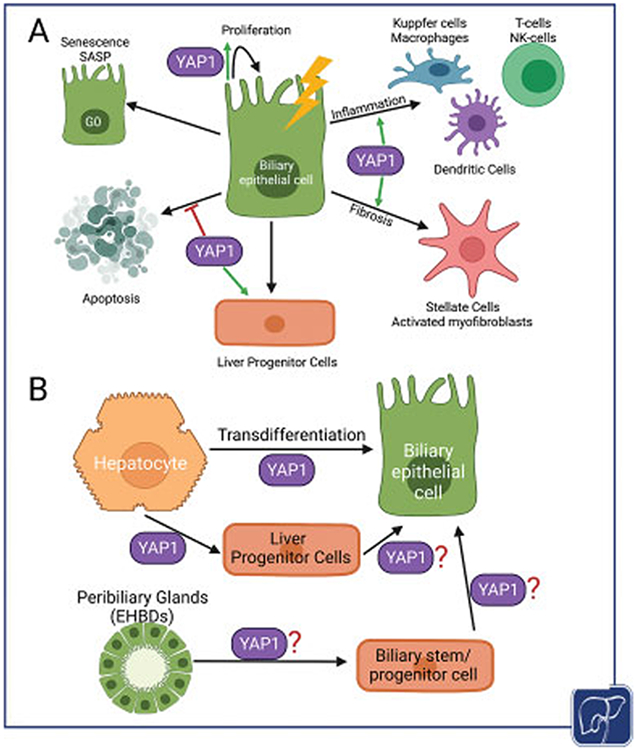
Diverse roles of YAP1 in regulating cholangiocyte and hepatocyte responses to cholestatic injury. (A) YAP1 can induce proliferation as well as promote survival of cholangiocytes during injury. Its role in activating liver progenitor compartment is also recognized. Under certain chronic and uncontrolled injury, YAP1 activation in bile duct cells could lead to ductular reaction to induce pro-inflammatory and pro-fibrogenic gene expression and in turn promote disease progression. Likewise, YAP1 might also contribute to senescence-associated secretory phenotype in bile duct cells. (B) YAP1 is known to directly induce hepatocyte to cholangiocyte transdifferentiation in adult livers. It could also lead to dedifferentiation of a mature hepatocyte to a progenitor cell which could in turn differentiate into a cholangiocyte. The role of YAP1 in liver progenitor cells arising from either peribiliary glands in the EHBDs or hepatic stem cell populations in the canal of Hering, especially in contribution to biliary repair, remains to be elucidated.
The ductular reaction has been associated with both alleviating the injury and contributing toward disease phenotype. Some studies have shown that added ductal cells contribute to collecting bile from the parenchyma and channel it out of the liver and, thus, prevent bile toxicity.96,108 Activated cholangiocytes can also secrete and respond to a variety of cytokines, chemokines, and neuroendocrine signals and are closely associated with inflammatory infiltrates of neutrophils and macrophages as well as activated myofibroblasts.94,109-111 In chronic injury, cholangiocytes may become senescent in response to DNA damage and oxidative stress, leading to cell cycle arrest and activation of a pro-inflammatory senescence-associated secretory phenotype.112,113 Senescent cholangiocytes secrete classic cytokines such as IL1, IL6, CXCL1/2, and IL8 in addition to matrix metalloproteinases and other ECM remodeling factors, leading to autocrine and paracrine signaling to neighboring Kupffercells and stellate cells which respond in kind to these injury-related stimuli.112 Chronically reactive cholangiocytes may contribute to worsening portal fibrosis and a pro-tumorigenic environment. The molecular basis of pro-inflammatory and pro-fibrogenic nature of reactive ductules is incompletely understood. Also, how the ductular reaction can be manipulated to promote favorable repair and minimize tissue damage is an area of active investigation in various laboratories. Finally, chronic severe biliary injury will often lead to duct loss through apoptosis of existing cholangiocytes and failure of proliferative regenerative mechanisms.109,110,112,114
The PBGs play an important role in the regeneration of large IHBDs and EHBDs as a source of stem cells with the potential to differentiate into hepatocytes, cholangiocytes, and pancreatic islet-like cells.65,66,99 The PBGs seem to provide a source of BEC turnover in homeostasis, although at a very slow rate, and they can be activated to expand in the setting of injury as observed in rodents and human tissue.66,99 PBGs are closely associated with loose connective tissue, the peribiliary vascular plexus, and unmyelinated nerve processes, suggesting that PBG epithelial cells can sense and respond to hormonal and neurotransmitter signals to drive the secretion of bile-modifying components and mucinous glycoproteins to create a barrier from toxic bile as well as components of mucosal immunity such as IgA and lactoferrin.66 This close relationship has been implicated in the pathogenesis of EHBD diseases such as primary sclerosing cholangitis, as increased levels of bile duct fibrosis are associated with PBG hyperplasia in patients, possibly mediated by Hedgehog signaling.115 Many more studies are needed to further characterize the similarities and differences between EHBD and IHBD responses to injury and clarify the functions of the PBGs in mediating biliary injury and repair.
Role of YAP1 in the Cholangiocyte Response to Injury
YAP1 has been implicated in many aspects of the cholangiocyte response to injury, partly because it plays an important role in biliary homeostasis and response to environmental cues. YAP1 has been shown to be active in a subset of mature cholangiocytes during homeostasis and responds dynamically to increased levels of bile acids in mice, subsequently regulating transcription and activating downstream pathways like mitogen-associated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) signaling.27 This process has been shown to depend on apical sodium-dependent bile acid transporter (ASBT)-mediated apical transport of bile acids into the BECs, as well as IQ motif containing GTPase Activating Protein 1 (IQGAP1)-mediated regulation of YAP1 nuclear localization in response to bile acid signaling.27,116 Loss of YAP1 in mature BECs results in cell death and activation of inflammation even under homeostatic conditions in a bile acid-dependent manner, while YAP1 helps prevent BEC cell death in the presence of pathologically elevated bile acids, such as the 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC)-diet or cholic acid feeding.27 Thus, YAP1 is critical for BEC survival and adaptation to changing levels of bile acids in homeostasis and disease settings.
YAP1 has also been shown to be essential for BEC proliferation and ductular reaction formation in varied injury models such as DDC-diet, bile duct ligation, choline-deficient ethionine-supplemented diet, and carbon tetrachloride injury.117-120 Ablation of YAP1 in BECs, hepatocytes, or both causes dramatically reduced ductular reaction, with consequently worsened cholestatic liver injury. YAP1 is critical in regulating the ductular reaction originating from both BECs and hepatocytes in cell-autonomous ways, but YAP1 in hepatocytes may also indirectly regulate BEC proliferation and ductular reaction expansion.117-120 This process may depend on mTOR activation and increased Survivin expression mediating pro-proliferative and anti-apoptotic signals.118,120 In addition, YAP1-target CCN1 (CYR61) signals in an autocrine manner in BECs to drive BEC proliferation and ductular reaction through integrin binding and subsequent activation of NF-κB and Jagged/Notch signaling.80 Importantly, studies have not determined what role YAP1 may play in the activation and expansion of various stem cell populations in the liver, which remains to be investigated. The aforementioned studies use genetic models, which alter and/or label the majority of cholangiocytes and/or hepatocytes without targeting specific stem cell markers or fully characterizing the stem-cell properties of the ductular reaction. However, the profound impact of YAP1 loss on ductular reaction in these diverse injury models suggests that YAP1 is required for the ductular reaction to occur regardless of its source.
YAP1 activation in BECs also contributes to inflammation and fibrosis by regulating the production of cytokines such as CYR61 and CTGF from BECs and hepatocytes. CYR61 can act as a macrophage chemoattractant and also promote the activation of hepatic stellate cells, leading to increased fibrosis.28,80,121 CTGF from BECs can act in an autocrine manner through integrin binding to promote TGFβ signaling and collagen deposition associated with the ductular reaction, and deletion of CTGF reduces markers of fibrosis in the setting of cholestatic injury such as the DDC-diet model.119 While TAZ has been shown to regulate similar gene targets as YAP1, so far we do not know what distinct role TAZ may play in BECs in homeostasis and regeneration.
Overview of Hepatocyte-Driven Biliary Repair via Transdifferentiation
Although once a controversial idea, numerous studies in rodents have demonstrated using lineage-tracing experiments that hepatocytes have significant plasticity and are capable of transdifferentiating into cholangiocytes to promote repair and regeneration, especially in the setting of chronic biliary injury (Fig. 3B).122 Various types of injury models targeting the murine biliary system have been used to stimulate this response, such as the DDC-diet model, bile duct ligation, and 4,4’-diaminodiphenylmethane biliary toxin. These studies have implicated pathways including Notch, Wnt-β-catenin, Hippo/YAP1, and TGFβ.27,44,81,123-126 While many studies suggest that all hepatocytes have this potential, some studies have identified specific sub-populations of hepatocytes that may have greater repopulation capacity, such as telomerase-expressing hepatocytes or a subset of Sox9-expressing portal hepatocytes, which may have different or enhanced cholangiocyte transdifferentiation capabilities.60,127 Also, despite the clear evidence of hepatocyte-derived cholangiocytes, questions remain about whether such response is durable especially when the injury abrogates and also whether these transdifferentiatied cells may be predisposed to neoplastic transformation in the long-term either spontaneously or following an additional insult.128
Recently, this regenerative response was convincingly observed in an animal model of ALGS with liver-specific developmental ablation of Notch signaling and HNF6.81 Intriguingly, despite the total failure of intrahepatic bile duct formation, many of these mice recovered and survived long term due to hepatocyte-derived de novo generation of bile ducts forming a three-dimensional, functional network.81 This study demonstrated a role of TGFβ-signaling through TGFßRII for transdifferentiation and regeneration to occur. Phenotypic recovery over time has also been observed in some (but not all) murine models of ALGS.129,130 Similar observations have been reported in a subset of Alagille patients making hepatocyte-derived biliary regeneration a likely therapeutic modality for improved biliary function in humans.131
Role of YAP1 in Hepatocyte-Driven Biliary Repair via Transdifferentiation
Several studies have implicated YAP1 in hepatocyte transdifferentiation into biliary-type cells (Fig. 3B). Notably, Yimlamai et al showed that conditional doxycycline-induced expression of YAP1-S127A (a mutant constitutionally active form of YAP1) in hepatocytes led them to express biliary markers such as pan-cytokeratin, SOX9, and HNF1β and change their morphology from large cuboidal epithelia to small, progenitor-like cells with increased nuclear-to-cytoplasmic ratio.44 Using lineage tracing, they rule out activation of a pre-existing progenitor cell type and demonstrate that YAP1 activation caused almost 75% of individual, mature hepatocytes to dedifferentiate and adopt a ductal or progenitor-like state in a cell-autonomous manner. Transcriptional analysis showed that YAP1-activated hepatocytes underwent reprogramming with activation of Notch, TGFβ, and epidermal growth factor receptor signaling and downregulation of HNF4α expression. In particular, YAP1 was shown to regulate transcription of Notch2 and Sox9 and blocking Notch signaling through simultaneous deletion of RBPJ significantly abrogated hepatocyte transdifferation. Importantly, when YAP1-S127A expression was shut off, approximately 20% of transdifferentiated cells began to re-express HNF4α and return to a hepatocyte-like morphology, suggesting that continuous YAP1 activation may be necessary to maintain ductal differentiation.
Other studies have examined the role of YAP1 in mediating hepatocyte transdifferentiation in the context of biliary injury. For example, both Pepe-Mooney et al and Planas-Paz et al showed that biliary injury caused by DDC-diet treatment in mice could induce expression of YAP1 targets in hepatocytes and led to the formation of hepatocyte-derived duct-like structures, and this response was significantly abrogated when YAP1 was specifically deleted from hepatocytes.27,118 Another study demonstrated that YAP1-mediated hepatocyte transdifferentiation and ductular reaction in the setting of DDC-diet injury was dependent on Arid1a-mediated chromatin remodeling, which opened the chromatin of thousands of genes implicated in hepatocyte reprogramming and created a permissive environment for gene expression modulation by YAP1 and other factors.132 Classic pathways implicated in hepatocyte transdifferentiation as well as biliary development were impacted by Arid1a-mediated chromatin remodeling, including targets of Hippo/YAP1, TGFβ, MAPK, and WNT signaling. An indispensable role of YAP1 in the process of hepatocyte-to-biliary transdifferentiation was also indicated on our recently characterized mouse model of Alagille-like syndrome due to YAP1 deletion from hepatoblasts by FoxA3-Cre.46 Although other studies have shown that YAP1 activation drives the expression of hepatoblast markers and biliary markers in hepatocytes and promotes the formation of hepatocyte-derived hepatoblastoma or cholangiocarcinoma,18,44,133,134 our study provides evidence that without YAP1 the liver cell identity shift cannot occur.46
Hepatocyte Adaptation to Cholestasis
One of the major functions of the hepatocytes is the production of bile which is channeled through biliary canaliculi, intrahepatic bile ducts, and, eventually, through extrahepatic biliary tree to the small intestine. However, the imperfect flow of the bile can lead to stagnation and bile accumulation, a feature referred to as cholestasis. Although cholestasis can be caused by a wide variety of pathologies, the end result is an increase in hepatic bile acids (as well as bilirubin, toxins, and heavy metals also excreted in bile) due to impaired bile processing and flow out from the liver through the biliary system. Since bile acids are in fact detergents essential for carrying lipids and for solubilizing lipids for absorption, these can themselves lead to severe cell injury and death both directly and indirectly by binding to cell death receptors and inducing oxidative damage.135,136 Bile acids also deplete cell membranes of cholesterol, resulting in altered lipid raft-associated signaling, and the subsequent elevated cholesterol synthesis results in hyper-cholesteremia and feedback effects on lipid metabolism.137,138
There is a complex system of feedback regulation by which hepatocytes respond to elevated bile acid levels, especially through interactions among bile acids and various nuclear receptors (Fig. 4). One central regular is the farnesoid X receptor (FXR) together with the small heterodimer partner (SHP), which responds to elevated bile acid levels in hepatocytes by downregulating transcription of Cyp7a1, a key rate-limiting enzyme in bile acid synthesis.136 In addition, FXR/SHP, pregnane X receptor, and vitamin D receptor among others form heterodimers with the retinoid X receptor α to coordinate the expression of bile acid transporters (altering influx and efflux of bile acids from hepatocytes into the canaliculi or the serum to reduce intracellular accumulation) and phase I/II metabolizing enzymes (which conjugate bile acids to reduce their toxicity and increase hydrophilicity).136,139 The PPARα, a key regulator of lipid and glucose metabolism, regulates genes promoting bile acid conjugation and phospholipid secretion into the bile, overall decreasing bile toxicity.136,139,140 Moreover, many of these receptors also regulate the secretion of inflammatory and pro-fibrotic cytokines as well as regulating the balance of pro- and anti-apoptotic signals.136,139,141 This complex signaling network lies at the heart of the liver’s adaptive response to cholestasis and is the subject of an intense investigation to develop therapeutics that amplify protective responses while reducing pro-apoptotic, pro-inflammatory, and pro-fibrotic signals.
Fig. 4.

Overview of hepatocyte adaptation to cholestatic injury. Hepatocytes can adapt to increased cholestatic injury seen due to enhanced bile acids in the liver. Several key nuclear receptors sense an increased bile acid pool and begin inhibiting de novo synthesis in an attempt to decrease total bile acid content. They also reduce bile acid hydrophobicity through increased conjugation, increase phospholipid secretion into bile to decrease toxicity, reduce apical transport of bile acids if there is obstruction or limited biliary canalicular or ductal flow, and enhance basolateral efflux of bile acids into sinusoidal blood to promote their exit from the liver. Bile acids can also directly or indirectly result in YAP1 activation through changing the mechanical tension in the bile canalicular wall and altering Hippo pathway activity. Hepatocyte can, thus, undergo metabolic reprogramming of bile acids and at the same time decrease its general synthetic and metabolic functions while it turns on proliferative program to maintain mass, as was seen in an extreme case of complete intrahepatic biliary tree absence due to hepatoblast-specific YAP1 deletion in mice.
Bile acids can regulate YAP1 activity in murine hepatocytes in various manners (Fig. 4). A recent study showed that MST1/2, core kinases of the Hippo pathway, act downstream of FGF15/19-FGFR4 to regulate the activity of SHP in a feedback loop between the liver and the intestine which regulates bile acid synthesis.142 In the absence of MST1/2, SHP was destabilized, resulting in increased bile acid synthesis and an increased bile acid pool, causing direct and indirect activation of YAP1.142 In addition, bile acid exposure alters hepatocyte cell adhesion and actin cytoskeleton structure, which can impact the distribution of scaffolding proteins like IQGAP1 which can transport YAP1 into the nucleus.116 Another study showed that the quantity of bile acids being transported through hepatocyte canaliculi influences the size and tension of the actin cytoskeletal network attached to the canalicular junctions such that an increase in bile acid flow can increase the tension felt by the actin fibers resulting in increased YAP1 translocation to the nucleus.143 Bile acid-dependent YAP1 activation in hepatocytes can also impact fibrosis and inflammation through many of the same mechanisms described above for cholangiocytes. For example, a recent study showed that taurocholate could induce the expression of CTGF in hepatocytes in a YAP1-ERK-dependent manner.144 YAP1 activation also leads to hepatocyte proliferation and regeneration, which has been reviewed elsewhere.11,15,145
Besides managing bile acid toxicity directly, hepatocytes in the setting of cholestasis undergo a global transcriptional and functional reprogramming in response to injury (Fig. 4). This has been described in several studies of the MDR2 knockout mouse model, which mimics human progressive familial intrahepatic cholestasis (PFIC) by impairing phosphatidylcholine secretion into the bile canaliculi, resulting in chronic obstructive cholestasis.146 In the first few months of injury, MDR2 KO livers show an increase in oncogenic pathways, pro-survival and pro-proliferative pathways, DNA-damage response pathways, and oxidative stress response.146 While these pathways contributed to survival in the short term, the long-term activation of oncogenic pathways resulted in the development of hepatocellular carcinoma in MDR2 KO mice over 1 year of age.146-150 Interestingly, it has been shown that bile acids can signal through the scaffold protein IQGAP1 to directly activate YAP1, which contributed to carcinoma formation in a model of severe cholestasis due to loss of FXR/SHP.116 These models demonstrate the double-edged sword of liver adaptations to injury.
Adaptive transcriptional reprogramming was also observed following YAP1 deletion in hepatoblasts by FoxA3-Cre, after which YAP1 KO mice were able to survive long term despite the severity of disease due to a complete absence of IHBDs.46 Similar to MDR2 KO mice, YAP1 KO livers activated pathways promoting hepatocyte proliferation, regeneration, and survival. Interestingly, however, YAP1 KO hepatocytes completely reversed the direction of bile acid transport to overcome the lack of plumbing for bile excretion, while also altering bile acid metabolism to favor a more hydrophilic, less toxic profile (Fig. 4).46 While this led to elevated levels of bile acids and bilirubin in the blood, it also prevented these toxic components from accumulating in the hepatic parenchyma, thereby reducing hepatocellular injury. The exact regulatory mechanisms responsible for this adaptation remain to be elucidated. Persistently elevated serum total and conjugated bilirubin levels in young childrenwith ALGS are associated with more severe liver disease and decreased likelihood of spontaneous improvement over time, similar to that seen in the FoxA3-Cre YAP1 KO model.151 The same adaptive changes in bilirubin and bile acid transport observed in YAP1 KO mice may be occurring in patients with severe disease and may indicate maximal hepatocyte adaptation in the context of failed biliary regeneration. Thus, YAP1 activation (or inactivation) in hepatocytes may be an important disease modifier in patients with ALGS and other biliary disorders requiring further studies. Overall, the surpris-ing capacity of the liver to survive and adapt may be harnessed therapeutically to better understand how to support patients with chronic liver injury.
Clinical Significance of YAP1 in Diseases with Developmental Loss of Bile Ducts
There are a variety of pediatric diseases affecting the biliary tree, caused by both genetic and environmental factors. Some diseases like PFIC involve not just cholangiocyte but hepatocyte dysfunction in biliary metabolism and transport. Not much is known on the role of YAP1 in PFIC, and more studies are needed to determine whether YAP1 impacts the role of hepatocytes in both causing and repairing cholestatic injury. In this section, we will focus on the potential roles of YAP1 in the pathogenesis of ALGS and biliary atresia (BA), two major pediatric cholestatic diseases caused by genetic developmental defects and/or injury to the developing biliary system.
Alagille Syndrome
ALGS is an autosomal dominant disorder arising from mutations in the JAGGED1 (> 90%) or NOTCH2 genes which causes multi-system malformations including impaired formation of bile ducts in embryonic development.131 Children also exhibit congenital cardiovascular abnormalities, vascular anomalies, renal disease, and skeletal abnormalities among others.131 In addition, children with ALGS exhibit marked growth deficiencies, associated with decreased caloric intake and chronic fat malabsorption due to decreased bile secretion into the gut, although there may be additional factors.152 According to the National Organization for Rare Disorders, the incidence of ALGS is estimated at 1 in 30,000 to 1 in 45,000 births.
One longitudinal study found that 89% of children with ALGS have cholestasis, ranging from mild to severe, and 75% exhibit bile duct paucity histologically.153 According to a recent prospective study, only approximately 24% of children with bile duct paucity reach adulthood without a liver transplant, indicating the serious need for alternative therapies.154 Interestingly, the penetrance of these mutations varies widely leading to variability in the extent of cholestasis and disease presentation, even among family members with the same mutation.131 We lack a full understanding of the disease modifiers and relevant biomarkers that can help stratify or distinguish these patients during a critical treatment window.131,155,156 Children with mild cholestasis, as measured by levels of serum bilirubin and alkaline phosphatase among others, often show improvement and resolution of disease over the first few years of life.131,155,156 In contrast, persistently elevated serum total and conjugated bilirubin levels in young children with ALGS are associated with more severe liver disease and decreased likelihood of spontaneous improvement over time.151
The disease phenotype has not been correlated with the location or type of mutation in the JAGGED1 gene, suggesting that additional genetic or environmental modifiers greatly affect disease presentation and time course.157,158 Studies in mice have shown that inactivating glycosyltransferases, such as Rumi which directly modify JAG1 and NOTCH2 proteins, further worsens the course of disease.130,159 In addition, a genome-wide association study identified a single-nucleotide polymorphism in the THROMBOSPONDIN2 gene which was associated with more severe disease.160 Further studies are needed to identify mechanisms of action of these modifiers and determine whether they can be targeted clinically.
As described above, multiple mouse models have shown that deletion of YAP1 or YAP1/TAZ in early or late liver development leads to bile duct paucity ranging from mild to severe, resembling the clinical phenotype of ALGS.46,85,86 Whole-body knockout of YAP1 is embryonic lethal in mice due to defects in yolk sac vasculogenesis, suggesting that complete inactivation of YAP1 may be incompatible with survival and thus would not be found in children.34 However, altered YAP1 regulation and/or decreased YAP1 function whether by genetic mutation in the YAP1 gene or associated signaling partners could compound the effect of decreased Notch signaling resulting in worsened overall phenotype. Likewise, specific studies need to assess YAP1 activity status in ALGS-like pathologies without known mutations in Jagged/Notch pathway or in cases of idiopathic bile duct paucity.
In addition, hepatocyte-derived regeneration of the biliary system may be an important component of the compensatory adaptation of livers in the setting of ALGS. While atopic expression of biliary markers such as CK7 in human hepato cytes is a common feature of most liver diseases, one study observed a dramatic accumulation of bipotential cells expressing both biliary and hepatocyte markers in the portal area of ALGS livers but noted that these cells did not express transcription factors classically regulated by Notch signaling, hypothesizing that they were arrested and unable to completely transdifferentiate into BECs.161 Given the observational nature of most studies using human tissue, it is difficult to determine the origin and fate of these bipotential-appearing cells. Nevertheless, since YAP1 is a necessary driver of hepatocyte transdifferentiation to BECs, it seems a reasonable candidate for modulation to better understand and stimulate a functional regenerative response in patients.
Biliary Atresia
BA is a severe disease of infancy occurring in approximately 1 of every 15,000 U.S. births, characterized by severe inflammation and fibrosis of the extrahepatic biliary tree resulting in obstruction of bile flow and consequently acholic stools, jaundice, and hyperbilirubinemia.162 While the extrahepatic biliary tree is often obliterated by the time of diagnosis, the intrahepatic biliary tree exhibits extensive proliferation and expansion associated with immune infiltration and activation of stellate cells, consistent with a ductular reaction attempting to increase biliary flow out of the liver.163,164 Early diagnosis is the key for surgeons to attempt improvement of bile drainage via Kasai hepatoportoenterostomy, but even after surgical intervention many patients still have poor bile flow and progressive fibrosis leading to end-stage cirrhosis, and most patients with BA eventually require liver transplantation.162,165 Studies have uncovered multiple factors contributing to the pathogenesis of BA, including exposure in utero to environmental toxins or viruses, developmental abnormalities in bile duct morphogenesis, and dysregulated immune responses. Recent research has also identified multiple classes of BA based on incidence and histopathological features: (1) “developmental” BA with early onset and potential syndromic associations, (2) perinatal non-syndromic BA, (3) cystic BA, and (4) CMV-associated BA.162,166 Studies are ongoing to further validate these classifications and target underlying mechanisms of disease, including reducing fibrogenic inflammation and improving biliary drainage.
Multiple studies have found significantly elevated cytoplasmic and nuclear expression of YAP1 in proliferative BECs in patients with BA as compared with BECs in non-BA neonatal cholestasis (including a few cases of ALGS, PFIC, and idiopathic bile duct paucity) as well as pediatric and adult control BECs.163,164,167 In fact, nuclear YAP1 staining in BECs was highly sensitive and specific for a diagnosis of BA as opposed to other causes of neonatal cholestasis.163,164 Increased YAP1 expression in BECs was also correlated with elevated fibrosis scores in patients with BA.167 Zheng et al also found that out of several known YAP1 targets, CTGF and ANKRD1 were significantly upregulated in BA livers and specifically localized in BECs.167 A Hippo/YAP-related gene set was also found to be upregulated in BA liver samples as compared with non-BA cholestatic disease and control samples.168 Consistent with many murine studies as well as in vitro experiments using BA-like organoids,169 YAP1 may be contributing to BEC proliferation in the ductular reaction, which is a classical feature of BA, as well as the activation of fibrogenic pathways in both BECs and activated stellate cells. Thus, YAP1 may be playing an important role in worsening the chronic, reactive component of BA disease. Persistent, progressive fibrosis after Kasai hepatoportoenterostomy is a major factor leading to mortality and/or liver transplantation in children with BA, for which there is no targeted treatment.165,170
In addition, YAP1 may be involved in embryonic defects leading to ductal plate malformations, which have been reportedly observed in approximately 10 to 50% of BA cases.171-173 Indeed, several studies have shown that at least some patients with BA exhibit a hilar and perihilar ductal structure more closely resembling immature 11 to 15 weeks fetal ducts which have not coalesced into a continuous network, rather than postnatal ductal systems with distinct large branching ducts.174,175 These malformations contribute to poor bile flow despite Kasai hepatoportoenterostomy and are associated with worsened survival, shorter time to liver transplantation, and longer periods of uncontrolled jaundice with an increased need for steroid treatment.171,172 A few genetic studies among BA patients have begun to identify single nucleotide polymorphisms in genes such as JAG1 (Jagged1), ITGB2 (integrin subunit beta 2), VEGF (vascular endothelial growth factor), and GPC1 (Glypican 1) among others, pointing to possible roles of Notch signaling, cell polarization, and epithelial-mesenchymal cell communication in BA pathogenesis, all pathways which YAP1 has been shown to modulate as described above.162 In addition to known infectious, toxic, and inflammatory components of BA pathogenesis, underlying defects in embryogenesis play a significant role in dictating the severity and progression of the disease and remain to be fully investigated.
Concluding Remarks
YAP1 signaling adds to the resilience of liver, especially during chronic cholestatic injuries. YAP1 plays key roles both in cholangiocytes and in hepatocytes to allow for repair through the regulation of key target genes in these cells. YAP1 plays a key role in bile duct development independent of the Notch signaling pathway by directing the differentiation of immature hepatocytes to a second layer of bile ducts during prenatal murine hepatic development. YAP1 continues to play important role in bile duct homeostasis throughout development and adulthood. Its role in both biliary proliferation and promoting cholangiocyte survival during insults is unquestionable. However, prolonged YAP1 activation in cholangiocytes could result in chronic ductular reaction with increased inflammation and fibrosis and, hence, also contribute to disease pathogenesis. YAP1 in hepatocytes can also play an important role in repair. While its role in regulating hepatocyte proliferation normally is of lesser relevance, its ectopic expression in hepatocytes plays a profound role in switching their cell fate to a cholangiocyte. This reprogramming allows hepatocytes to transdifferentiate into cholangiocytes during chronic injury to the ducts helping with their repair and maintaining bile flow.
Several general and specific questions remain in the field of YAP1 and TAZ function in the liver. YAP1 seems to promote cell dedifferentiation in some contexts while promoting biliary differentiation in others. The molecular basis of these two distinct functions along with their specific genetic targets needs to be addressed. How exactly YAP1 acts to commit cells to a biliary lineage during hepatic development also needs further mechanistic elucidation, whether it is turning off the hepatocyte genetic program in favor of a Notch-driven biliary program or acting as a downstream effector of TGFβ signaling to induce hepatocyte fate-switch to form the second layer of biliary cells, or regulating the effects of extracellular matrix on BEC differentiation state. Likewise, we do not know the specific downstream targets that YAP1 directly regulates to induce and sustain biliary fate in development or repair. In addition, no studies have looked individually at TAZ to identify its unique role in liver development. How much TAZ can compensate for the absence of YAP1 is an open question that will be important to address their redundant and unique roles in the liver, particularly when considering how to design pharmacological targets to modulate YAP1 and/or TAZ activity. How YAP1 and/or TAZ may regulate EHBD formation remains to be further elucidated, along with a better understanding of how the EHBD and IHBD networks interact during development to eventually establish a seamless tubular network. Most importantly, further studies are needed to determine if perturbations in YAP1 can contribute to the pathogenesis of ALGS and BA as well as as-of-yet uncharacterized biliary defects in pediatric and even adult patients. There is a significant need for new treatment strategies to promote biliary repair and regeneration in pediatric cholestatic disease, and it may be useful to learn how to activate the pro-regenerative properties of YAP1 signaling while blocking excess inflammation and fibrosis to achieve functional biliary repair rather than maintaining chronic disease.
Funding
Funding was provided by 2T32EB001026-16A1 (S.P.M and L.M.); 1F30DK121393 to L.M., and 1R01DK62277, 1R01DK100287, 1R01DK116993, R01CA204586, 1R01CA251155 and Endowed Chair for Experimental Pathology to S.P.M., and by National Institutes of Health grant 1P30DK120531–01 to Pittsburgh Liver Research Center (S.P.M.).
Footnotes
Conflict of Interest
None declared.
References
- 1.Zanconato F, Forcato M, Battilana G, et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol 2015;17(09):1218–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galli GG, Carrara M, Yuan W-C, et al. YAP drives growth by controlling transcriptional pause release from dynamic enhancers. Mol Cell 2015;60(02):328–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stein C, Bardet AF, Roma G, et al. YAP1 exerts its transcriptional control via TEAD-mediated activation of enhancers. PLoS Genet 2015;11(08):e1005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao B, Ye X, Yu J, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 2008;22(14):1962–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou D, Conrad C, Xia F, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 2009;16(05):425–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao B, Li L, Lei Q, Guan KL. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev 2010;24(09):862–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moleirinho S, Hoxha S, Mandati V, et al. Regulation of localization and function of the transcriptional co-activator YAP by angiomotin. eLife 2017;3(06):e23966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robinson BS, Moberg KH. Cell-cell junctions: α-catenin and E-cadherin help fence in Yap1. Curr Biol 2011;21(21):R890–R892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev 2010;24(01):72–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sugihara T, Werneburg NW, Hernandez MC, et al. YAP tyrosine phosphorylation and nuclear localization in cholangiocarcinoma cells are regulated by LCK and independent of LATS activity. Mol Cancer Res 2018;16(10):1556–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patel SH, Camargo FD, Yimlamai D. Hippo Signaling in the liver regulates organ size, cell fate, and carcinogenesis. Gastroenterology 2017;152(03):533–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Panciera T, Azzolin L, Cordenonsi M, Piccolo S. Mechanobiology of YAP and TAZ in physiology and disease. Nat Rev Mol Cell Biol 2017;18(12):758–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gumbiner BM, Kim N-G. The Hippo-YAP signaling pathway and contact inhibition of growth. J Cell Sci 2014;127(Pt 4):709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heng BC, Zhang X, Aubel D, et al. An overview of signaling pathways regulating YAP/TAZ activity. Cell Mol Life Sci 2021;78(02):497–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moya IM, Halder G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat Rev Mol Cell Biol 2019;20(04):211–226 [DOI] [PubMed] [Google Scholar]
- 16.Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell 2013;154(06):1342–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang S, Wang J, Wang H, et al. Hippo cascade controls lineage commitment of liver tumors in mice and humans. Am J Pathol 2018;188(04):995–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Dong M, Xu Z, et al. Notch2 controls hepatocyte-derived cholangiocarcinoma formation in mice. Oncogene 2018;37(24):3229–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim W, Khan SK, Gvozdenovic-Jeremic J, et al. Hippo signaling interactions with Wnt/β-catenin and Notch signaling repress liver tumorigenesis. J Clin Invest 2017;127(01):137–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim W, Khan SK, Yang Y. Interacting network of Hippo, Wnt/β-catenin and Notch signaling represses liver tumor formation. BMB Rep 2017;50(01):1–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kriz V, Korinek V. Wnt, RSPO and hippo signalling in the intestine and intestinal stem cells. Genes (Basel) 2018;9(01):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taniguchi K, Moroishi T, de Jong PR, et al. YAP-IL-6ST autoregulatory loop activated on APC loss controls colonic tumorigenesis. Proc Natl Acad Sci U S A 2017;114(07):1643–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang L, Azzolin L, Di Biagio D, et al. The SWI/SNF complex is a mechanoregulated inhibitor of YAP and TAZ. Nature 2018;563(7730):265–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ibar C, Irvine KD. Integration of Hippo-YAP signaling with metabolism. Dev Cell 2020;54(02):256–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fitamant J, Kottakis F, Benhamouche S, et al. YAP inhibition restores hepatocyte differentiation in advanced HCC, leading to tumor regression. Cell Rep 2015;10(10):1692–1707. Doi: 10.1016/j.celrep.2015.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan WC, Pepe-Mooney B, Galli GG, et al. NUAK2 is a critical YAP target in liver cancer. Nat Commun 2018;9(01):4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pepe-Mooney BJ, Dill MT, Alemany A, et al. Single-cell analysis of the liver epithelium reveals dynamic heterogeneity and an essential role for YAP in homeostasis and regeneration. Cell Stem Cell 2019;25(01):23–38.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mooring M, Fowl BH, Lum SZC, et al. Hepatocyte stress increases expression of Yes-associated protein and transcriptional coactivator with PDZ-binding motif in hepatocytes to promote parenchymal inflammation and fibrosis. Hepatology 2020;71(05):1813–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen Q, Anders RA, Alpini G, Bai H. Yes-associated protein in the liver: regulation of hepatic development, repair, cell fate determination and tumorigenesis. Dig Liver Dis 2015;47(10):826–835 [DOI] [PubMed] [Google Scholar]
- 30.Alder O, Cullum R, Lee S, et al. Hippo signaling influences HNF4A and FOXA2 enhancer switching during hepatocyte differentiation. Cell Rep 2014;9(01):261–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reggiani F, Gobbi G, Ciarrocchi A, Sancisi V. YAP and TAZ are not identical twins. Trends Biochem Sci 2021;46(02):154–168 [DOI] [PubMed] [Google Scholar]
- 32.Moroishi T, Park HW, Qin B, et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev 2015;29(12):1271–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hau JC, Erdmann D, Mesrouze Y, et al. The TEAD4-YAP/TAZ protein-protein interaction: expected similarities and unexpected differences. ChemBioChem 2013;14(10):1218–1225 [DOI] [PubMed] [Google Scholar]
- 34.Morin-Kensicki EM, Boone BN, Howell M, et al. Defects in yolk sac vasculogenesis, chorioallantoic fusion, and embryonic axis elongation in mice with targeted disruption of Yap65. Mol Cell Biol 2006;26(01):77–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Makita R, Uchijima Y, Nishiyama K, et al. Multiple renal cysts, urinary concentration defects, and pulmonary emphysematous changes in mice lacking TAZ. Am J Physiol Renal Physiol 2008;294(03):F542–F553 [DOI] [PubMed] [Google Scholar]
- 36.Hossain Z, Ali SM, Ko HL, et al. Glomerulocystic kidney disease in mice with a targeted inactivation of Wwtr1. Proc Natl Acad Sci U S A 2007;104(05):1631–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heng BC, Zhang X, Aubel D, et al. Role of YAP/TAZ in cell lineage fate determination and related signaling pathways. Front Cell Dev Biol 2020;8:735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Driskill JH, Pan D. The hippo pathway in liver homeostasis and pathophysiology. Annu Rev Pathol 2021;16:299–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, Zheng Z, Caviglia JM, et al. Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab 2016;24(06):848–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hagenbeek TJ, Webster JD, Kljavin NM, et al. The Hippo pathway effector TAZ induces TEAD-dependent liver inflammation and tumors. Sci Signal 2018;11(547):11. [DOI] [PubMed] [Google Scholar]
- 41.Wang H, Wang J, Zhang S, et al. Distinct and overlapping roles of Hippo Effectors YAP and TAZ during human and mouse hepatocarcinogenesis. Cell Mol Gastroenterol Hepatol 2021;11(04):1095–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Haele M, Moya IM, Karaman R, et al. YAP and TAZ heterogeneity in primary liver cancer: an analysis of its prognostic and diagnostic role. Int J Mol Sci 2019;20(03):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee D-H, Park JO, Kim T-S, et al. LATS-YAP/TAZ controls lineage specification by regulating TGFβ signaling and Hnf4α expression during liver development. Nat Commun 2016;7:11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yimlamai D, Christodoulou C, Galli GG, et al. Hippo pathway activity influences liver cell fate. Cell 2014;157(06):1324–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dong J, Feldmann G, Huang J, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007;130(06):1120–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Molina LM, Zhu J, Li Q, et al. Compensatory hepatic adaptation accompanies permanent absence of intrahepatic biliary network due to YAP1 loss in liver progenitors. Cell Rep 2021;36(01):109310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gordillo M, Evans T, Gouon-Evans V. Orchestrating liver development. Development 2015;142(12):2094–2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Su X, Shi Y, Zou X, et al. Single-cell RNA-Seq analysis reveals dynamic trajectories during mouse liver development. BMC Genomics 2017;18(01):946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang L, Wang W-H, Qiu W-L, Guo Z, Bi E, Xu CR. A single-cell transcriptomic analysis reveals precise pathways and regulatory mechanisms underlying hepatoblast differentiation. Hepatology 2017;66(05):1387–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tanimizu N, Miyajima A. Notch signaling controls hepatoblast differentiation by altering the expression of liver-enriched transcription factors. J Cell Sci 2004;117(Pt 15):3165–3174 [DOI] [PubMed] [Google Scholar]
- 51.Ober EA, Lemaigre FP. Development of the liver: insights into organ and tissue morphogenesis. J Hepatol 2018;68(05):1049–1062 [DOI] [PubMed] [Google Scholar]
- 52.Clotman F,Jacquemin P, Plumb-Rudewiez N, et al. Control of liver cell fate decision by a gradient of TGF beta signaling modulated by Onecut transcription factors. Genes Dev 2005;19(16):1849–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takayama K, Kawabata K, Nagamoto Y, et al. CCAAT/enhancer binding protein-mediated regulation of TGFβ receptor 2 expression determines the hepatoblast fate decision. Development 2014;141(01):91–100 [DOI] [PubMed] [Google Scholar]
- 54.Gérard C, Tys J, Lemaigre FP. Gene regulatory networks in differentiation and direct reprogramming of hepatic cells. Semin Cell Dev Biol 2017;66:43–50 [DOI] [PubMed] [Google Scholar]
- 55.Lemaigre FP. Development of the intrahepatic and extrahepatic biliary tract: a framework for understanding congenital diseases. Annu Rev Pathol 2020;15:1–22 [DOI] [PubMed] [Google Scholar]
- 56.Benhamouche-Trouillet S, O’Loughlin E, Liu C-H, et al. Proliferation-independent role of NF2 (merlin) in limiting biliary morphogenesis. Development 2018;145(09):145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanimizu N, Kaneko K, Itoh T, et al. Intrahepatic bile ducts are developed through formation of homogeneous continuous luminal network and its dynamic rearrangement in mice. Hepatology 2016;64(01):175–188 [DOI] [PubMed] [Google Scholar]
- 58.Tanimizu N, Kikkawa Y, Mitaka T, Miyajima A. α1- and α5-containing laminins regulate the development of bile ducts via β1 integrin signals. J Biol Chem 2012;287(34):28586–28597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carpentier R, Suñer RE, van Hul N, et al. Embryonic ductal plate cells give rise to cholangiocytes, periportal hepatocytes, and adult liver progenitor cells. Gastroenterology 2011;141:1432–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Font-Burgada J, Shalapour S, Ramaswamy S, et al. Hybrid periportal hepatocytes regenerate the injured liver without giving rise to cancer. Cell 2015;162(04):766–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Walter TJ, Cast AE, Huppert KA, Huppert SS. Epithelial VEGF signaling is required in the mouse liver for proper sinusoid endothelial cell identity and hepatocyte zonation in vivo. Am J Physiol Gastrointest Liver Physiol 2014;306(10):G849–G862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fabris L, Cadamuro M, Libbrecht L, et al. Epithelial expression of angiogenic growth factors modulate arterial vasculogenesis in human liver development. Hepatology 2008;47(02):719–728 [DOI] [PubMed] [Google Scholar]
- 63.Kietzmann T Metabolic zonation of the liver: the oxygen gradient revisited. Redox Biol 2017;11:622–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Russell JO, Monga SP. Wntβ-catenin signaling in liver development, homeostasis, and pathobiology. Annu Rev Pathol 2018;13:351–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cardinale V, Wang Y, Carpino G, et al. Multipotent stem/progenitor cells in human biliary tree give rise to hepatocytes, cholangiocytes, and pancreatic islets. Hepatology 2011;54(06):2159–2172 [DOI] [PubMed] [Google Scholar]
- 66.de Jong IEM, van Leeuwen OB, Lisman T, Gouw ASH, Porte RJ. Repopulating the biliary tree from the peribiliary glands. Biochim Biophys Acta Mol Basis Dis 2018;1864(4 Pt B):1524–1531 [DOI] [PubMed] [Google Scholar]
- 67.Villasenor A, Stainier DYR. On the development of the hepatopancreatic ductal system. Semin Cell Dev Biol 2017;66:69–80 [DOI] [PubMed] [Google Scholar]
- 68.Spence JR, Lange AW, Lin SC, et al. Sox17 regulates organ lineage segregation of ventral foregut progenitor cells. Dev Cell 2009;17(01):62–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Uemura M, Higashi M, Pattarapanawan M, et al. Gallbladder wall abnormality in biliary atresia of mouse Sox17 +/− neonates and human infants. Dis Model Mech 2020;13(04):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sumazaki R, Shiojiri N, Isoyama S, et al. Conversion of biliary system to pancreatic tissue in Hes1-deficient mice. Nat Genet 2004;36(01):83–87 [DOI] [PubMed] [Google Scholar]
- 71.Villasenor A, Gauvrit S, Collins MM, Maischein HM, Stainier DYR. Hhex regulates the specification and growth of the hepatopancreatic ductal system. Dev Biol 2020;458(02):228–236 [DOI] [PubMed] [Google Scholar]
- 72.Dong PD, Munson CA, Norton W, et al. Fgf10 regulates hepatopancreatic ductal system patterning and differentiation. Nat Genet 2007;39(03):397–402 [DOI] [PubMed] [Google Scholar]
- 73.Thestrup MI, Caviglia S, Cayuso J, et al. A morphogenetic EphB/EphrinB code controls hepatopancreatic duct formation. Nat Commun 2019;10(01):5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brandt ZJ, Echert AE, Bostrom JR, North PN, Link BA. Core Hippo pathway components act as a brake on Yap and Taz in the development and maintenance of the biliary network. Development 2020;147(12):147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wei W, Lotto J, Hoodless PA. Expression patterns of Yes-associated protein 1 in the developing mouse liver. Gene Expr Patterns 2018;29:10–17 [DOI] [PubMed] [Google Scholar]
- 76.Airik M, Schüler M, McCourt B, et al. Loss of Anks6 leads to YAP deficiency and liver abnormalities. Hum Mol Genet 2020;29(18):3064–3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Antoniou A, Raynaud P, Cordi S, et al. Intrahepatic bile ducts develop according to a new mode of tubulogenesis regulated by the transcription factor SOX9. Gastroenterology 2009;136(07):2325–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tschaharganeh DF, Chen X, Latzko P, et al. Yes-associated protein up-regulates Jagged-1 and activates the Notch pathway in human hepatocellular carcinoma. Gastroenterology 2013;144:1530–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu N, Nguyen Q, Wan Y, et al. The Hippo signaling functions through the Notch signaling to regulate intrahepatic bile duct development in mammals. Lab Invest 2017;97(07):843–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim KH, Chen CC, Alpini G, Lau LF. CCN1 induces hepatic ductular reaction through integrin αvβ5-mediated activation of NF-κB. J Clin Invest 2015;125(05):1886–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schaub JR, Huppert KA, Kurial SNT, et al. De novo formation of the biliary system by TGFβ-mediated hepatocyte transdifferentiation. Nature 2018;557(7704):247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Noguchi S, Saito A, Nagase T. YAP/TAZ signaling as a molecular link between fibrosis and cancer. Int J Mol Sci 2018;19(11):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oh S-H, Swiderska-Syn M, Jewell ML, Premont RT, Diehl AM. Liver regeneration requires Yap1-TGFβ-dependent epithelial-mesenchymal transition in hepatocytes. J Hepatol 2018;69(02):359–367. Doi: 10.1016/j.jhep.2018.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Grannas K, Arngården L, Lönn P, et al. Crosstalk between Hippo and TGFβ: subcellular localization of YAP/TAZ/Smad Complexes. J Mol Biol 2015;427(21):3407–3415 [DOI] [PubMed] [Google Scholar]
- 85.Zhang N, Bai H, David KK, et al. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell 2010;19(01):27–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Verboven E, Moya IM, Sansores-Garcia L, et al. Regeneration defects in Yap and Taz mutant mouse livers are caused by bile duct disruption and cholestasis. Gastroenterology 2021;160(03):847–862 [DOI] [PubMed] [Google Scholar]
- 87.Lu L, Finegold MJ, Johnson RL. Hippo pathway coactivators Yap and Taz are required to coordinate mammalian liver regeneration. Exp Mol Med 2018;50(01):e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nishio M, Sugimachi K, Goto H, et al. Dysregulated YAP1 /TAZ and TGF-β signaling mediate hepatocarcinogenesis in Mob1a/1b-deficient mice. Proc Natl Acad Sci U S A 2016;113(01):E71–E80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.García P, Rosa L, Vargas S, et al. Hippo-YAP1 is a prognosis marker and potentially targetable pathway in advanced gallbladder cancer. Cancers (Basel) 2020;12(04):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pei T, Li Y, Wang J, et al. YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget 2015;6(19):17206–17220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Banales JM, Huebert RC, Karlsen T, Strazzabosco M, LaRusso NF, Gores GJ. Cholangiocyte pathobiology. Nat Rev Gastroenterol Hepatol 2019;16(05):269–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wu N, Baiocchi L, Zhou T, et al. Functional role of the secretin/secretin receptor signaling during cholestatic liver injury. Hepatology 2020;72(06):2219–2227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Maroni L, Haibo B, Ray D, et al. Functional and structural features of cholangiocytes in health and disease. Cell Mol Gastroenterol Hepatol 2015;1(04):368–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sato K, Meng F, Giang T, Glaser S, Alpini G. Mechanisms of cholangiocyte responses to injury. Biochim Biophys Acta Mol Basis Dis 2018;1864(4 Pt B):1262–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sato K, Marzioni M, Meng F, Francis H, Glaser S, Alpini G. Ductular reaction in liver diseases: pathological mechanisms and translational significances. Hepatology 2019;69(01):420–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kamimoto K, Nakano Y, Kaneko K, Miyajima A, Itoh T. Multidimensional imaging of liver injury repair in mice reveals fundamental role of the ductular reaction. Commun Biol 2020;3(01):289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ko S, Russell JO, Molina LM, Monga SP. Liver progenitors and adult cell plasticity in hepatic injury and repair: knowns and unknowns. Annu Rev Pathol 2020;15:23–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Safarikia S, Carpino G, Overi D, et al. Distinct EpCAM-positive stem cell niches are engaged in chronic and neoplastic liver diseases. Front Med (Lausanne) 2020;7:479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Carpino G, Renzi A, Franchitto A, et al. Stem/progenitor cell niches involved in hepatic and biliary regeneration. Stem Cells Int 2016;2016:3658013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kordes C, Häussinger D. Hepatic stem cell niches. J Clin Invest 2013;123(05):1874–1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Raven A, Lu W-Y, Man TY, et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature 2017;547(7663):350–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lu W-Y, Bird TG, Boulter L, et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat Cell Biol 2015;17(08):971–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Boulter L, Lu W-Y, Forbes SJ. Differentiation of progenitors in the liver: a matter of local choice. J Clin Invest 2013;123(05):1867–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Huch M, Gehart H, van Boxtel R, et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015;160(1–2):299–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.LeSage GD, Glaser SS, Marucci L, et al. Acute carbon tetrachloride feeding induces damage of large but not small cholangiocytes from BDL rat liver. Am J Physiol 1999;276(05):G1289–G1301 [DOI] [PubMed] [Google Scholar]
- 106.Sun T, Annunziato S, Tchorz JS. Hepatic ductular reaction: a double-edged sword. Aging (Albany NY) 2019;11(21):9223–9224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wilson DB, Rudnick DA. Invasive ductular reaction: form and function. Am J Pathol 2019;189(08):1501–1504 [DOI] [PubMed] [Google Scholar]
- 108.Clerbaux LA, Manco R, Van Hul N, et al. Invasive ductular reaction operates hepatobiliary junctions upon hepatocellular injury in rodents and humans. Am J Pathol 2019;189(08):1569–1581 [DOI] [PubMed] [Google Scholar]
- 109.Yokoda RT, Rodriguez EA. Review: pathogenesis of cholestatic liver diseases. World J Hepatol 2020;12(08):423–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lazaridis KN, LaRusso NF. The cholangiopathies. Mayo Clin Proc 2015;90(06):791–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Alvaro D, Mancino MG, Glaser S, et al. Proliferating cholangiocytes: a neuroendocrine compartment in the diseased liver. Gastroenterology 2007;132(01 ):415–431 [DOI] [PubMed] [Google Scholar]
- 112.Meng L, Quezada M, Levine P, et al. Functional role of cellular senescence in biliary injury. Am J Pathol 2015;185(03):602–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tabibian JH, O’Hara SP, Splinter PL, Trussoni CE, LaRusso NF. Cholangiocyte senescence by way of N-ras activation is a characteristic of primary sclerosing cholangitis. Hepatology 2014;59(06):2263–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xia X, Demorrow S, Francis H, et al. Cholangiocyte injury and ductopenic syndromes. Semin Liver Dis 2007;27(04):401–412 [DOI] [PubMed] [Google Scholar]
- 115.Carpino G, Cardinale V, Renzi A, et al. Activation of biliary tree stem cells within peribiliary glands in primary sclerosing cholangitis. J Hepatol 2015;63(05):1220–1228 [DOI] [PubMed] [Google Scholar]
- 116.Anakk S, Bhosale M, Schmidt VA, Johnson RL, Finegold MJ, Moore DD. Bile acids activate YAP to promote liver carcinogenesis. Cell Rep 2013;5(04):1060–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jin L, Huang H, Ni J, et al. Shh-Yap signaling controls hepatic ductular reactions in CCl4 -induced liver injury. Environ Toxicol 2021;36(02):194–203 [DOI] [PubMed] [Google Scholar]
- 118.Planas-Paz L, Sun T, Pikiolek M, et al. YAP, but Not RSPO-LGR4/5, signaling in biliary epithelial cells promotes a ductular reaction in response to liver injury. Cell Stem Cell 2019;25(01):39–53.e10 [DOI] [PubMed] [Google Scholar]
- 119.Pi L, Robinson PM, Jorgensen M, et al. Connective tissue growth factor and integrin αvβ6: a new pair of regulators critical for ductular reaction and biliary fibrosis in mice. Hepatology 2015;61(02):678–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bai H, Zhang N, Xu Y, et al. Yes-associated protein regulates the hepatic response after bile duct ligation. Hepatology 2012;56(03):1097–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li ZQ, Wu WR, Zhao C, et al. CCN1/Cyr61 enhances the function of hepatic stellate cells in promoting the progression of hepatocellular carcinoma. Int J Mol Med 2018;41(03):1518–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nejak-Bowen K If it looks like a duct and acts like a duct: on the role of reprogrammed hepatocytes in cholangiopathies. Gene Expr 2020;20(01):19–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Michalopoulos GK, Barua L, Bowen WC. Transdifferentiation of rat hepatocytes into biliary cells after bile duct ligation and toxic biliary injury. Hepatology 2005;41(03):535–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sekiya S, Suzuki A. Hepatocytes, rather than cholangiocytes, can be the major source of primitive ductules in the chronically injured mouse liver. Am J Pathol 2014;184(05):1468–1478 [DOI] [PubMed] [Google Scholar]
- 125.Yanger K, Zong Y, Maggs LR, et al. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev 2013;27(07):719–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Okabe H, Yang J, Sylakowski K, et al. Wnt signaling regulates hepatobiliary repair following cholestatic liver injury in mice. Hepatology 2016;64(05):1652–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lin S, Nascimento EM, Gajera CR, et al. Distributed hepatocytes expressing telomerase repopulate the liver in homeostasis and injury. Nature 2018;556(7700):244–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tarlow BD, Pelz C, Naugler WE, et al. Bipotential adult liver progenitors are derived from chronically injured mature hepatocytes. Cell Stem Cell 2014;15(05):605–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Andersson ER, Chivukula IV, Hankeova S, et al. Mouse model of alagille syndrome and mechanisms of Jagged1 missense mutations. Gastroenterology 2018;154(04):1080–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Thakurdas SM,Lopez MF,Kakuda S, et al. Jagged1 heterozygosity in mice results in a congenital cholangiopathy which is reversed by concomitant deletion of one copy of Poglut1 (Rumi). Hepatology 2016;63(02):550–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mitchell E, Gilbert M, Loomes KM. Alagille Syndrome. Clin Liver Dis 2018;22(04):625–641 [DOI] [PubMed] [Google Scholar]
- 132.Li W, Yang L, He Q et al. A homeostatic arid1a-dependent permissive chromatin state licenses hepatocyte responsiveness to liver-injury-associated YAP signaling. Cell Stem Cell 2019;25(01):54–68.e5 [DOI] [PubMed] [Google Scholar]
- 133.Li X, Tao J, Cigliano A, et al. Co-activation of PIK3CA and Yap promotes development of hepatocellular and cholangiocellular tumors in mouse and human liver. Oncotarget 2015;6(12):10102–10115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tao J, Calvisi DF, Ranganathan S, et al. Activation of β-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology 2014;147(03):690–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Claudel T, Zollner G, Wagner M, Trauner M. Role of nuclear receptors for bile acid metabolism, bile secretion, cholestasis, and gallstone disease. Biochim Biophys Acta 2011;1812(08):867–878 [DOI] [PubMed] [Google Scholar]
- 136.Wagner M, Zollner G, Trauner M. Nuclear receptor regulation of the adaptive response of bile acid transporters in cholestasis. Semin Liver Dis 2010;30(02):160–177 [DOI] [PubMed] [Google Scholar]
- 137.Nuño-Lámbarri N, Barbero-Becerra VJ, Uribe M, Chávez-Tapia NC. Elevated cholesterol levels have a poor prognosis in a cholestasis scenario. J Biochem Mol Toxicol 2017;31(01):1–6 [DOI] [PubMed] [Google Scholar]
- 138.Chisholm JW, Nation P, Dolphin PJ, Agellon LB. High plasma cholesterol in drug-induced cholestasis is associated with enhanced hepatic cholesterol synthesis. Am J Physiol 1999;276(05):G1165–G1173 [DOI] [PubMed] [Google Scholar]
- 139.Chiang JYL. Bile acid metabolism and signaling in liver disease and therapy. Liver Res 2017;1(01):3–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zollner G, Trauner M. Nuclear receptors as therapeutic targets in cholestatic liver diseases. BrJ Pharmacol 2009;156(01):7–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kosters A, Karpen SJ. The role of inflammation in cholestasis: clinical and basic aspects. Semin Liver Dis 2010;30(02):186–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ji S, Liu Q, Zhang S, et al. FGF15 activates hippo signaling to suppress bile acid metabolism and liver tumorigenesis. Dev Cell 2019;48(04):460–474.e9 [DOI] [PubMed] [Google Scholar]
- 143.Meyer K, Morales-Navarrete H, Seifert S, et al. Bile canaliculi remodeling activates YAP via the actin cytoskeleton during liver regeneration. Mol Syst Biol 2020;16(02):e8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yu B, Jin GN, Ma M, et al. Taurocholate induces connective tissue growth factor expression in hepatocytes through ERK-YAP signaling. Cell Physiol Biochem 2018;50(05):1711–1725 [DOI] [PubMed] [Google Scholar]
- 145.Preziosi ME, Monga SP. Update on the mechanisms of liver regeneration. Semin Liver Dis 2017;37(02):141–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Katzenellenbogen M, Pappo O, Barash H, et al. Multiple adaptive mechanisms to chronic liver disease revealed at early stages of liver carcinogenesis in the Mdr2-knockout mice. Cancer Res 2006;66(08):4001–4010 [DOI] [PubMed] [Google Scholar]
- 147.Lanton T, Shriki A, Nechemia-Arbely Y, et al. Interleukin 6-dependent genomic instability heralds accelerated carcinogenesis following liver regeneration on a background of chronic hepatitis. Hepatology 2017;65(05):1600–1611 [DOI] [PubMed] [Google Scholar]
- 148.Barash H R Gross E, Edrei Y, et al. Accelerated carcinogenesis following liver regeneration is associated with chronic inflammation-induced double-strand DNA breaks. Proc Natl Acad Sci U S A 2010;107(05):2207–2212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Katzenellenbogen M, Mizrahi L, Pappo O, et al. Molecular mechanisms of liver carcinogenesis in the mdr2-knockout mice. Mol CancerRes 2007;5(11):1159–1170 [DOI] [PubMed] [Google Scholar]
- 150.Katzenellenbogen M, Mizrahi L, Pappo O, et al. Molecular mechanisms of the chemopreventive effect on hepatocellular carcinoma development in Mdr2 knockout mice. Mol Cancer Ther 2007;6(04):1283–1291 [DOI] [PubMed] [Google Scholar]
- 151.Kamath BM, Munoz PS, Bab N, et al. A longitudinal study to identify laboratory predictors of liver disease outcome in Alagille syndrome. J Pediatr Gastroenterol Nutr 2010;50(05):526–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sokol RJ, Stall C. Anthropometric evaluation of children with chronic liver disease. Am J Clin Nutr 1990;52(02):203–208 [DOI] [PubMed] [Google Scholar]
- 153.Subramaniam P, Knisely A, Portmann B, et al. Diagnosis of Alagille syndrome-25 years of experience at King’s College Hospital. J Pediatr Gastroenterol Nutr 2011;52(01):84–89 [DOI] [PubMed] [Google Scholar]
- 154.Kamath BM, Ye W, Goodrich NP, et al. Childhood Liver Disease Research Network (ChiLDReN) Outcomes of childhood cholestasis in Alagille syndrome: results of a multicenter observational study. Hepatol Commun 2020;4(03):387–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology 1999;29(03):822–829 [DOI] [PubMed] [Google Scholar]
- 156.Lykavieris P, Hadchouel M, Chardot C, Bernard O. Outcome of liver disease in children with Alagille syndrome: a study of 163 patients. Gut 2001;49(03):431–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Spinner NB, Colliton RP, Crosnier C, Krantz ID, Hadchouel M, Meunier-Rotival M. Jagged1 mutations in alagille syndrome. Hum Mutat 2001;17(01):18–33 [DOI] [PubMed] [Google Scholar]
- 158.Crosnier C, Driancourt C, Raynaud N, et al. Mutations in JAGGED1 gene are predominantly sporadic in Alagille syndrome. Gastroenterology 1999;116(05):1141–1148 [DOI] [PubMed] [Google Scholar]
- 159.Ryan MJ, Bales C, Nelson A, et al. Bile duct proliferation in Jag1/fringe heterozygous mice identifies candidate modifiers of the Alagille syndrome hepatic phenotype. Hepatology 2008;48(06):1989–1997 [DOI] [PubMed] [Google Scholar]
- 160.Tsai EA, Gilbert MA, Grochowski CM, et al. THBS2 is a candidate modifier of liver disease severity in Alagille syndrome. Cell Mol Gastroenterol Hepatol 2016;2(05):663–675.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Fabris L, Cadamuro M, Guido M, et al. Analysis of liver repair mechanisms in Alagille syndrome and biliary atresia reveals a role for notch signaling. Am J Pathol 2007;171(02):641–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Asai A, Miethke A, Bezerra JA. Pathogenesis of biliary atresia: defining biology to understand clinical phenotypes. Nat Rev Gastroenterol Hepatol 2015;12(06):342–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Abo-Zeinah SS, Behairy B, Hussein MH, et al. Histopathological expression of Yes-associated protein in neonatal cholestasis. Clin Res Hepatol Gastroenterol 2020;44(02):189–194 [DOI] [PubMed] [Google Scholar]
- 164.Gurda GT, Zhu Q, Bai H, Pan D, Schwarz KB, Anders RA. The use of Yes-associated protein expression in the diagnosis of persistent neonatal cholestatic liver disease. Hum Pathol 2014;45(05):1057–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Shen WJ, Chen G, Wang M, Zheng S. Liver fibrosis in biliary atresia. World J Pediatr 2019;15(02):117–123 [DOI] [PubMed] [Google Scholar]
- 166.Ortiz-Perez A, Donnelly B, Temple H, Tiao G, Bansal R, Mohanty SK. Innate immunity and pathogenesis of biliary atresia. Front Immunol 2020;11:329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zheng C, Luo J, Yang Y, Dong R, Yu FX, Zheng S. YAP activation and implications in patients and a mouse model of biliary atresia. Front Pediatr 2021;8:618226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tharehalli U, Svinarenko M, Kraus JM, et al. YAP activation drives liver regeneration after cholestatic damage induced by Rbpj deletion. Int J Mol Sci 2018;19(12):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Tian L, Ye Z, Kafka K, et al. Biliary atresia relevant human induced pluripotent stem cells recapitulate key disease features in a dish. J Pediatr Gastroenterol Nutr 2019;68(01):56–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gunadi S, Sirait DN, Budiarti LR, et al. Histopathological findings for prediction of liver cirrhosis and survival in biliary atresia patients after Kasai procedure. Diagn Pathol 2020;15(01):79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Safwan M, Ramachandran P, Vij M, Shanmugam N, Rela M. Impact of ductal plate malformation on survival with native liver in children with biliary atresia. Pediatr Surg Int 2015;31(09):837–843 [DOI] [PubMed] [Google Scholar]
- 172.Shimadera S, Iwai N, Deguchi E, et al. Significance of ductal plate malformation in the postoperative clinical course of biliary atresia. J Pediatr Surg 2008;43(02):304–307 [DOI] [PubMed] [Google Scholar]
- 173.Pacheco MC, Campbell KM, Bove KE. Ductal plate malformation-like arrays in early explants after a Kasai procedure are independent of splenic malformation complex (heterotaxy). Pediatr Dev Pathol 2009;12(05):355–360 [DOI] [PubMed] [Google Scholar]
- 174.Vijayan V, El Tan C. Computer-generated three-dimensional morphology of the hepatic hilar bile ducts in biliary atresia. J Pediatr Surg 2000;35(08):1230–1235 [DOI] [PubMed] [Google Scholar]
- 175.Tan CE, Chan VS, Yong RY, et al. Distortion in TGF beta 1 peptide immunolocalization in biliary atresia: comparison with the normal pattern in the developing human intrahepatic bile duct system. Pathol Int 1995;45(11):815–824 [DOI] [PubMed] [Google Scholar]