

Abstract
Infections impair neurological outcome and increase mortality after spinal cord injury (SCI). Emerging data show that pathogens more easily infect individuals with SCI because SCI disrupts neural and humoral control of immune cells, culminating with the development of “SCI-Induced Immune Deficiency Syndrome” (SCI-IDS). Here, we review data that implicate autonomic dysfunction and impaired neuroendocrine signaling as key determinants of SCI-IDS. Although it is widely appreciated that mature leukocyte dysfunction is a canonical feature of SCI-IDS, new data indicate that SCI impairs the development and mobilization of immune cell precursors in bone marrow. Thus, this review will also explore how the post-injury acquisition of a “bone marrow failure syndrome” may be the earliest manifestation of SCI-IDS.
Keywords: Spinal cord injury, immune deficiency, neuroendocrine, neuroplasticity, bone marrow, spleen, adrenal glands
Graphical Abstract
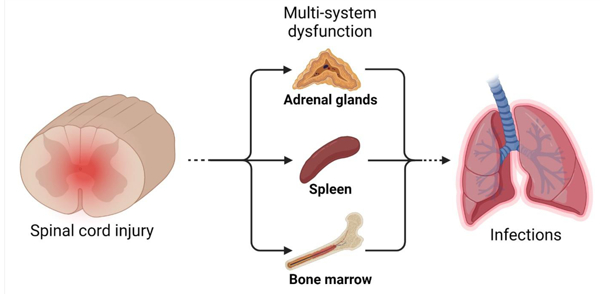
Introduction
Individuals with spinal cord injury (SCI) are at high-risk for contracting infections [1]. Because infections impair neurological outcome and increase mortality after SCI, it is important to understand why SCI increases susceptibility to infections [1–6]. Although the risk of exposure to infectious agents is increased due to complications associated with paralysis (e.g., mechanical ventilation, dysphagia, urine retention and catheterization) and extended hospitalization (e.g., nosocomial pathogens), emerging data indicate that SCI also hinders the immune system’s ability to eliminate bacteria and viruses that infect the body. This phenomenon, known as “SCI-Induced Immune Deficiency Syndrome” (SCI-IDS) [7,8], and the primary mechanisms underlying SCI-IDS, namely injury-induced impairment of both neuroendocrine signaling and the sympathetic nervous system control over lymphoid tissues, are the focus of this review.
SCI increases infection risk and worsens post-infection sequalae
The devastation of SCI goes beyond paralysis and loss of sensation. Immune function is also impaired by SCI, and this increases the risk of acquiring infections [6–9]. Indeed, infections are common after SCI and infectious complications adversely affect recovery of function and contribute to mortality [2,4,6,10]. Why then, despite significant advances in treatment for other SCI-related complications and comorbidities, has less progress been made in preventing infections [11–13]?
It is likely that infections evade early identification and empiric treatment by both health care professionals and SCI patients because common infection symptoms such as pain and fever are concealed by other, more emergent medical complications. Or, these symptoms are masked due to post-injury defects in temperature regulation and loss of sensation [14,15]. Clinical decision- making is further impaired by diagnostic ambiguity in the early identification and treatment of acquired infections, such as SCI-associated pneumonia (SCI-AP) [16]. Even after recovery and discharge from the hospital, SCI patients experience frequent medical complications that require rehospitalization [17]. Nosocomial infections can affect the lung, gastrointestinal tract and genitourinary tract through esophageal and endotracheal tubes and urinary catheters [18,19]. Dermatological complications associated with SCI (e.g., decubital ulcers) also manifest during prolonged hospitalization and because pathogens thrive in skin wounds these infections can be severe, with some cases resulting in osteomyelitis [15,20].
Still, injury to the spinal cord itself is now recognized as an independent risk factor for developing infections [5]. Thus, there is hope that new therapies may be developed that target injury-dependent mechanisms that place individuals at risk for developing infections after SCI. One such mechanism appears to be neurogenic-mediated suppression of immunity, with the magnitude and consequences of immune suppression varying as a function of spinal injury level [5,8,9,21–23]. For example, SCI occurring at or above the thoracic 6 (T6) spinal level causes greater immunological impairment with more frequent and severe infections than if a spinal injury were to occur below T6 [8,21,24]. These spinal-level dependent differences implicate a break in autonomic control over immune function as a principal component of SCI-IDS [25].
SCI-induced autonomic neuroplasticity and hyperreflexia cause immune dysfunction
The sympathetic branch of the autonomic nervous system plays an important role in regulating immune function [26,27]. Unlike the parasympathetic vagus nerve, which is a cranial (non-spinal cord derived) nerve that also regulates immunity, sympathetic preganglionic neurons (SPNs) are distributed throughout the intermediate gray matter of the thoracic and upper lumbar spinal cord. SPNs project to and control secondary noradrenergic post-ganglionic neurons in chain ganglia (i.e., pre- or paravertebral) located outside the spinal cord. Noradrenergic post-ganglionic neurons directly innervate viscera, including primary and secondary lymphoid tissues (e.g., adrenal glands, spleen, lymph nodes, and bone marrow, and gastrointestinal tract) [28–34].
Normally, bulbospinal neurons in the ventrolateral medulla and other supraspinal centers tonically inhibit SPNs, titrating sympathetic control over the viscera and lymphoid tissues. After severe, high-level SCI (above T6 spinal level), most or all supraspinal input to SPNs is abolished. Over time, these nascent autonomous spinal sympathetic circuits become “hyper-responsive”, and the net effect of spinal sympathetic reflex activity becomes pathological, creating dysfunction in most viscera including immune organs. The extent and severity of autonomic dysfunction, including suppression of immunity, is exacerbated when SCI occurs at high-spinal levels. That is because most lymphoid tissues are innervated by SPNs located at or below T6 spinal level, including the spleen (Fig 1) [33]. Thus, when SCI occurs at or above T6, more SPNs are removed from supraspinal control compared to injury at lower levels. Several reports in both animal models and humans illustrate these spinal level-dependent effects on immune function. Specifically, the incidence and severity of pneumonia, lymphoid tissue atrophy, and suppression of innate and adaptive immune function are consistently worse when SCI occurs at or above T3 when compared with SCI at or below T9 spinal level [8,9,21,35]. In humans, incomplete lesions often spare autonomic innervation whereas motor complete lesions cause >80% at- level ‘sympathetic decentralization’ [36]. Compared to motor complete injuries, incomplete lesions at or above T4 significantly reduce the risk of pneumonia, suggesting that sparing supraspinal input to SPNs critically reduces immunological impairment after high-level SCI [21].
Fig 1. SCI-Induced Immune Deficiency Syndrome is caused by changes in autonomic and neuroendocrine control over the adrenal gland and lymphoid tissues.
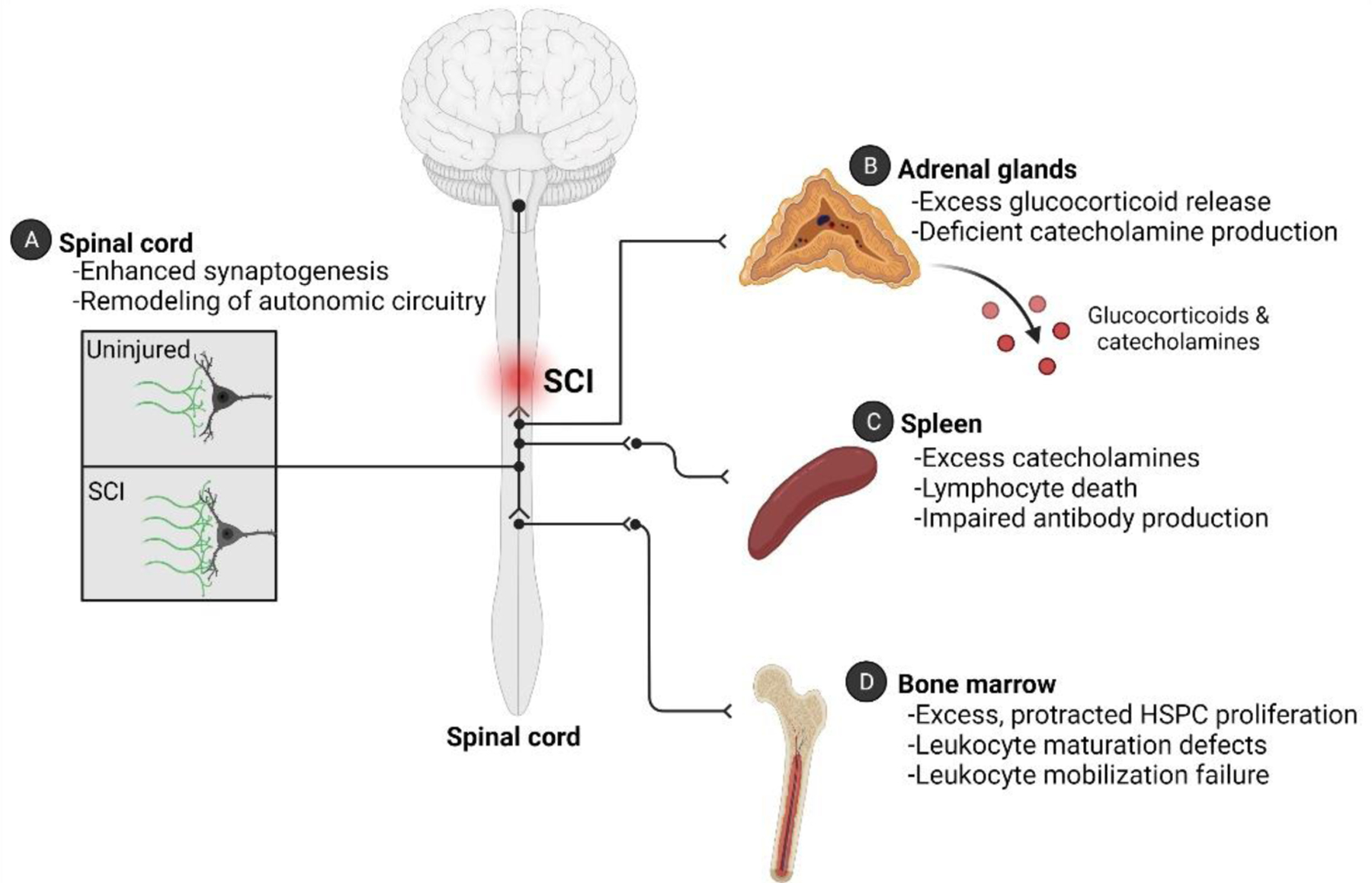
(A) Structural remodeling of intraspinal circuitry occurs below the level of injury after SCI. This “plasticity” is often referred to as being maladaptive because the new circuitry causes pathological changes in multiple organs. Experimental data have proved that these maladaptive circuits affect the spleen and adrenal glands. (B) After SCI, primary hypercortisolism develops (i.e., it is not triggered by ACTH) together with a defect in production or release of catecholamines. (C) Exaggerated spinal-splenic reflexes develop after SCI, cause post-ganglionic release and accumulation of catecholamines in the spleen. Overstimulation by glucocorticoids and catecholamines has been shown to trigger lymphocyte death and impair antibody-production after SCI. (D) SCI causes an acquired “bone marrow failure syndrome” that persists indefinitely. Defects in the bone marrow niche and in hematopoietic progenitor cells occur after SCI.
Although immunological impairment develops quickly after SCI, SCI-IDS is protracted with many indices of immune dysfunction worsening at longer survival times post-injury. Experimental data indicate that immune suppression into chronic phases is caused, in part, by structural remodeling and formation of new spinal sympathetic networks controlling the function of peripheral immune organs and the adrenal glands (Fig 1A). In rodent models of SCI, this occurs in three stages. In stage 1, supraspinal axons die back and dendritic arbors on intraspinal neurons temporarily retract causing the loss of synapses on interneurons and nearby SPNs [37–40]. Stage 2 begins at approximately two weeks post-injury and signals a period of reactive growth and sprouting, accompanied by increased expression of growth-associated proteins in axon terminals that ultimately form new synapses on SPNs and surrounding interneuronal networks [41]. Then, during stage 3, new synapse formation and axon growth continues until at least 6 weeks post-injury when supernumerary excitatory synapses occupy previously vacant synaptic sites throughout the intermediate gray matter including on SPN soma (Fig 1A) [42]. The cellular and molecular determinants, as well as the functional significance of this structural remodeling, are best documented for spinal circuitry that controls a major secondary lymphoid organ - the spleen (Fig 1A,C) [43].
Injecting GFP-tagged pseudorabies virus (PRV), a trans-synaptic retrograde neuronal tracer, into the spleen of mice with a high (T3) or mid-thoracic (T9) SCI, revealed marked changes in the anatomical boundaries and relative connectivity of spinal-splenic neuronal networks [43]. In the uninjured spinal cord, PRV–GFP+ cells exist only ipsilateral to the intrasplenic injection and labeled cells are restricted to the lateral gray matter at the T4–9 spinal levels. This labeling pattern is unchanged after T9 SCI. However, after T3 SCI, PRV-GFP labeling is redistributed with more than twice as many labeled neurons found in intermediate and medial gray matter throughout thoracic, lumbar, and sacral spinal cord [43]. This newly formed and expanded spinal-splenic circuit contains large numbers of excitatory glutamatergic (VGlut2+) interneurons that form new synaptic contacts with and influence the activity of SPNs.
Precisely how SCI induces neuroplastic changes in VGlut2+ neurons in autonomic circuitry is unclear, although cytokines and growth factors released at the injury site have been implicated as key mediators of this process [44–47]. More recently, it was shown that the neuronal voltage-gated calcium channel subunit α2δ−1 enhances synaptogenesis within spinal autonomic circuitry after SCI [42,48–50]. α2δ−1 is the cognate receptor for gabapentin [51], an FDA-approved drug commonly used to treat neuropathic pain. When injected into mice with high-level SCI beginning at 1 dpi (prior to stage 1 remodeling), gabapentin blocked the onset of multi-segmental excitatory synaptogenesis and remodeling of the neuronal networks that are responsible for cardiovascular and immune dysfunction [42].
Even before aberrant remodeling of intraspinal autonomic circuitry begins, SCI disrupts autonomic and neuroendocrine mechanisms that control immune cell survival and function (Fig 1). Specifically, because SCI removes tonic inhibition by supraspinal neurons, normal visceral or somatic stimuli (e.g., bowel/bladder filling) trigger exaggerated spinal autonomic reflexes which increases the release of norepinephrine from post-ganglionic adrenergic neurons into lymphoid tissues and other end organs (Fig 1). In SCI mice, excess norepinephrine in immune organs (e.g., spleen) triggers apoptosis in immune cells with suppression of antibody production within the first two weeks post-injury [8]. Transection of the splenic nerve or pharmacological blockade of β2 adrenergic receptors (β2ARs) prevents acute leukopenia, prevents splenic atrophy, and restores antibody production, confirming that aberrant noradrenergic signaling in lymphoid tissues contributes to early immune dysfunction after SCI (Fig 1C) [21,52].
The physical trauma/stress of SCI also activates neuroendocrine signaling (e.g., hypothalamic-pituitary-adrenal axis; HPA axis), marked by an abrupt but transient elevation of circulating norepinephrine and adrenal steroids that exert profound immunomodulatory effects [8,35,52,53]. Indeed, in the context of SCI, the adrenal glands are often overlooked as potent and systemic regulators of immunity.
Adrenal gland dysfunction after SCI
The adrenal glands are located at the top of each kidney and are comprised of two distinct anatomical regions- the cortex and medulla. The adrenal cortices, derived from embryonic mesoderm, produce steroid hormones including glucocorticoids, while the adrenal medullae are derived from the neuroectoderm (neural crest) and function as sympathetic post-ganglionic neurons that synthesize and release catecholamines (e.g., norepinephrine, dopamine, and epinephrine) [54,55]. Although usually considered anatomically and functionally independent, anatomical specializations may support local signaling networks between the adrenal medullae and cortices. Specifically, adrenal cortical cells extend filopodia to nearby chromaffin cells and medullary chromaffin cell islands are dispersed throughout the adrenal cortex [55,56].
The adrenal glands are essential organs that maintain body homeostasis. Diseases like Addison’s disease or Cushing’s disease that either impair or cause excess adrenal hormone production, respectively, profoundly disrupt metabolism, blood pressure and immune function [57]. The adrenal glands also play a critical role in regulating the body’s response to stress. During stress, endocrine and neural signaling pathways converge in the adrenal glands to stimulate release of catecholamines and glucocorticoids. Endocrine signaling, mediated via the HPA axis, is initiated when the hypothalamus, sensing stressors, secretes corticotrophin releasing hormone which in turn triggers the secretion of adrenocorticotrophin hormone (ACTH) from the anterior pituitary. ACTH ultimately elicits the synthesis and release of glucocorticoids from the adrenal cortices (Fig 1B) [58]. Neural control over adrenal hormone production is also regulated by the autonomic nervous system. The adrenal capsule, all three cortical layers, and the medullae, are richly innervated by both parasympathetic and sympathetic nerve fibers, although the sympathoadrenal axis is likely to be most affected after SCI [30,59–64].
Sympathetic nervous system regulation of adrenal output requires integration between supraspinal, intraspinal, and adrenal networks (Fig 1). Pre-sympathetic supraspinal projections from the paraventricular nucleus of the hypothalamus and brainstem (e.g., rostral ventrolateral and medial medulla, raphe nuclei) project to thoracic spinal cord gray matter where they regulate the activity of SPNs that innervate the adrenal glands (Fig 1B) [30,59–64]. Sympathetic innervation of the adrenal cortices controls the diurnal sensitivity of cortical cells to ACTH, which in turn regulates circadian production and release of glucocorticoids [65]. At the same time, sympathetic input to the adrenal medullae controls the production and release of catecholamines (Fig 1B) [65]. After SCI, the sympathoadrenal axis is compromised and stress-mediated activation of endocrine signaling is defunct [35,66].
Indeed, a break in diurnal rhythms becomes evident within hours post-SCI and is associated with progressive accumulation of glucocorticoids in the circulation over several weeks, months and even years post-injury [5,8,35,67,68]. Post-SCI hypercortisolism occurs without increased plasma ACTH, suggesting an adrenal-autonomous (i.e., primary hypercortisolism) or neurogenic mechanism (e.g., via the sympathoadrenal axis) is causing aberrant glucocorticoid release (Fig 1B) [21,35,69]. Just as SCI causes VGlut2+ spinal interneurons to become structurally and functionally integrated with neurons that comprise the spinal-splenic sympathetic network (Fig 1C), creating de novo circuitry that subserves exaggerated spinal autonomic reflexes, recent data indicate that an amplified spinal-adrenal circuit also forms after SCI (Fig 1B) [70]. Although unproven, it is possible that autonomic nerve terminals within the adrenal gland undergo plasticity and remodeling, just as they do in diseased cardiac muscle [71] or in secondary lymphoid tissues in response to immune challenges [72].
Ultimately, loss of adrenal gland homeostasis after SCI has profound effects on immune function. Excess or sustained elevation of glucocorticoids can impair inflammatory signaling in immune cells by blocking key transcription factors (e.g., Activating Protein-1 and Nuclear Factor kappa beta). Glucocorticoids also enhance the effects of the sympathetic nervous system; glucocorticoids increase the expression and binding affinity of β2ARs and prevent downregulation of these receptors. In leukocytes, glucocorticoid-induced amplification of β2AR signaling causes mitochondrial dysfunction and programmed cell death [73]. In SCI mice, blocking norepinephrine and glucocorticoid signaling in leukocytes improves leukocyte survival and restores immune function, providing hope that similar interventions could be used to prevent the onset of SCI-IDS in humans, or mitigate the immune suppressive effects of aberrant spinal autonomic reflexes in chronic SCI [25,52].
Hematopoietic dysfunction after spinal cord injury
Aberrant autonomic and neuroendocrine signaling after SCI might also impair immune function directly at the source of all immune cells, i.e., the bone marrow [74]. All blood cells, including innate and adaptive immune cells, are born in the bone marrow from hematopoietic stem and progenitor cells (HSPCs) during the process of hematopoiesis. At the top of the hematopoietic pyramid are long-term hematopoietic stem cells (LT-HSCs). LT-HSCs are multi-potent stem cells that differentiate into lineage-committed hematopoietic progenitor cells (HPCs) or self-renew without differentiating. HPCs mature into distinct functional immune cells. Despite the well-documented effects of SCI on mature immune cells in secondary lymphoid tissue (e.g., spleen, lymph nodes), less is known about how SCI affects HSPCs in bone marrow, although data from two independent studies indicate that in humans, SCI may permanently disrupt hematopoiesis [75,76].
A recent study in mice revealed that SCI causes an acquired “bone marrow failure syndrome” (BMFS) marked by excess proliferation of bone marrow lineage- (Lin-)Sca1+cKit+ (LSK) HSPCs within 3 days post-SCI (Fig 1D, Fig 2) [77]. Rather than be released as a normal part of hematopoiesis, these highly proliferative HSPCs remained sequestered indefinitely in bone marrow after SCI, even after challenging the host with a potent inflammatory stimulus [77]. The aberrant sequestration phenomenon, caused in part by an increase in chemokine-dependent signaling (CXCR4-CXCL12) in LSKs (Fig 2), could be overcome by injecting SCI mice with AMD3100, an FDA-approved drug and CXCR4 antagonist used to mobilize HSPCs and reduce infection risk during chemotherapy [77]. Bone marrow failure after SCI can again be attributed to injury-dependent autonomic and neuroendocrine dysfunction.
Fig 2. SCI induces bone marrow failure.
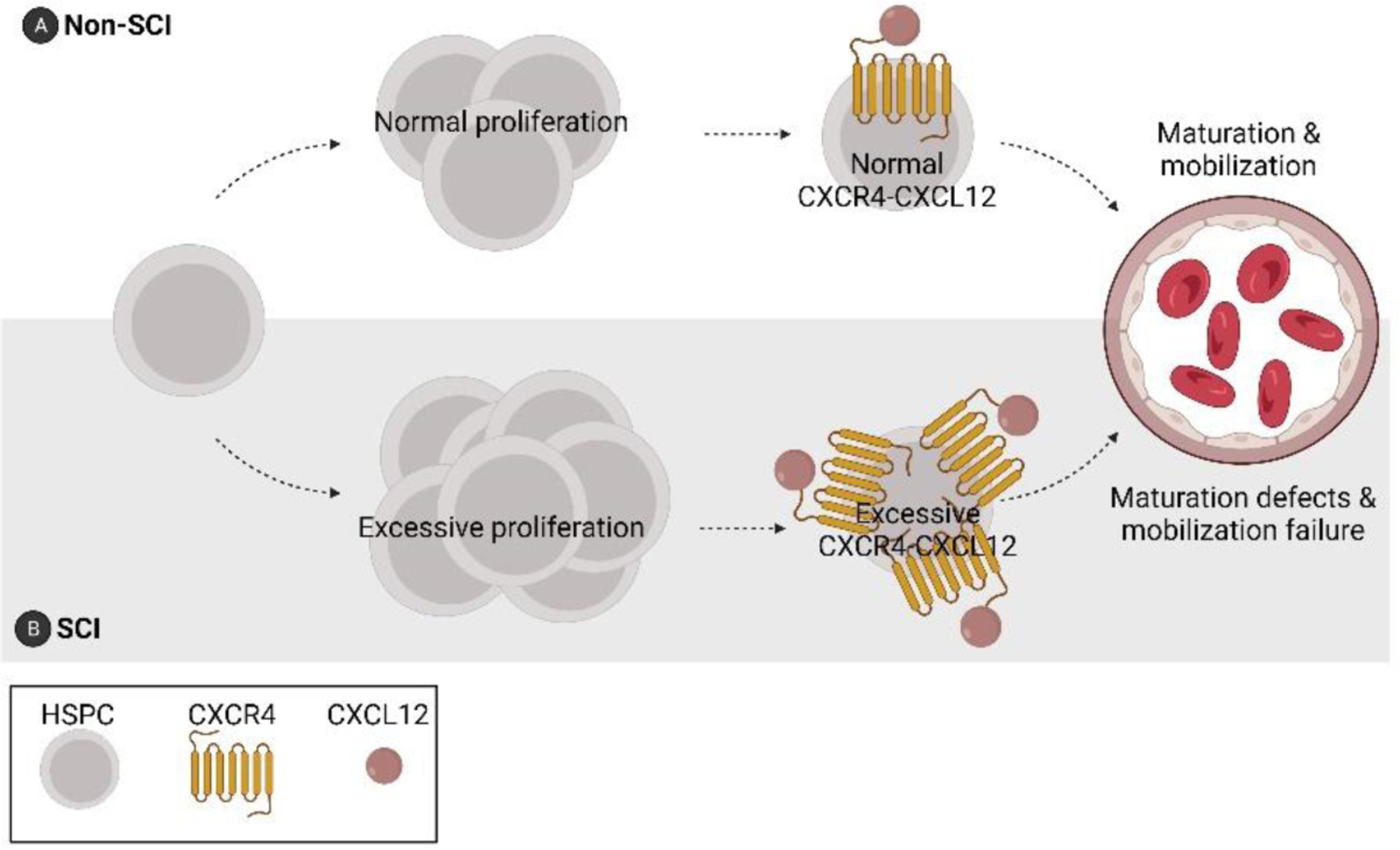
As early as 3 days post-injury, aberrant proliferation and sequestration of HSPCs occurs in the bone marrow. This phenomenon is associated with enhanced CXCR4-CXCL12 signaling and a reduction in lymphocyte maturation [77].
Normally, the release of HSPCs from the bone marrow is cyclical, waxing and waning throughout day and night. This circadian control is regulated by the sympathetic nervous system [78,79]. Norepinephrine, released cyclically in bone marrow by sympathetic nerve terminals, downregulates CXCL12 production by bone marrow stromal cells [80–82]. As CXCL12 gradients decrease, CXCR4+ HSPCs and mature leukocytes are released from the bone marrow microenvironment into the circulation. After SCI, circadian rhythmicity is impaired and this could disrupt hematopoiesis and HSPC egress indefinitely. Indeed, injecting endotoxin, a potent mobilizer of HSPCs, failed to mobilize HSPCs or mature lymphocytes from the bone marrow of chronic SCI mice [77].
Cell-intrinsic defects may also develop in HSPCs and mature leukocytes after SCI. In support of this hypothesis, bone marrow cells isolated from mice three days after SCI were ineffective in engrafting the lethally irradiated bone marrow of recipient mice, signaling a defect in the self-renewal capacity of LT-HSCs [77]. It is unclear why SCI impairs the long-term function of these cells or how this occurs so rapidly (within 3 days) after SCI. DNA damage to rapidly proliferating HSPCs is unlikely since γH2AX, a marker of DNA damage, was unchanged in chronic SCI mice [77]. However, both catecholamines and glucocorticoids, released because of aberrant sympathetic or neuroendocrine signaling, could cause epigenetic modifications in HSPCs, imprinting them with new functional identities [83,84]. Future research is needed to determine if epigenetic mechanisms contribute to SCI-induced bone marrow failure and the extent to which hematopoietic defects are responsible for acute or chronic immune dysfunction.
Conclusion
Infections are common after SCI and propagate morbidity and mortality. Understanding how to prevent infections or treat them more effectively is essential for improving health care and quality of life for individuals with SCI. In this review, we describe how SCI adversely affects autonomic and neuroendocrine control over lymphoid tissues, adrenal glands, and bone marrow, leading to the onset and persistence of SCI-IDS. Although the consequences of SCI-IDS are described in the literature as SCI-induced changes in mature leukocytes in blood or secondary lymphoid tissues, new data indicate that SCI impairs the development and mobilization of leukocyte precursors from their primary site of production in bone marrow. A better understanding of the predominant mechanism(s) underlying SCI-IDS is likely to yield new interventions for treating or preventing infections after SCI.
Highlights.
Acquired infections are the leading cause of death after SCI.
Infections are caused by abnormal autonomic and neuroendocrine control over the adrenal glands and lymphoid tissues.
Intraspinal remodeling of sympathetic circuitry contributes to immune deficiency.
SCI induces bone marrow dysfunction with early deficits in hematopoiesis.
Modulation of sympathetic-neuroendocrine signaling may prevent infections after SCI.
Funding:
This work was supported in part by NINDS-NIH grants R01NS099532 (PGP), R01NS083942 (PGP) and R35NS111582 (PGP) and R01NS118200 (JMS), the National Institute of Disability, Independent Living and Rehabilitation Research (NIDILRR Grant 90SI5020 to JMS), the Ray W. Poppleton Endowment (PGP), the European Union (EU Era Net – Neuron Program, SILENCE Grant 01EW170A to JMS), and the William E. Hunt and Charlotte M. Curtis endowment (JMS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: The authors declare no competing financial interests.
References
- 1.Sekhon LH, Fehlings MG: Epidemiology, Demographics, and Pathophysiology of Acute Spinal Cord Injury. Spine 2001, 26:S2–S12. [DOI] [PubMed] [Google Scholar]
- 2.Failli V, Kopp MA, Gericke C, Martus P, Klingbeil S, Chen Y, Devivo MJ, Dirnagl U, Brommer B, Schwab JM: Functional neurological recovery after spinal cord injury is impaired in patients with infections. Brain 2012, 135:3238–3250. [DOI] [PubMed] [Google Scholar]
- 3.DeVivo MJ, Stover SL, Black KJ: Prognostic factors for 12-year survival after spinal cord injury. Archives of Physical Medicine and Rehabilitation 1992, 73:156–162. [PubMed] [Google Scholar]
- 4.DeVivo MJ, Kartus PL, Stover SL, Rutt RD, Fine PR: Cause of Death for Patients With Spinal Cord Injuries. Archives of Internal Medicine 1989, 149:1761–1766. [PubMed] [Google Scholar]
- 5.Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U: Central nervous system injury-induced immune deficiency syndrome. Nature Reviews Neuroscience 2005, 6:775–786. [DOI] [PubMed] [Google Scholar]
- 6.Kopp MA, Watzlawick R, Martus P, Failli V, Finkenstaedt FW, Chen Y, DeVivo MJ, Dirnagl U, Schwab JM: Long-term functional outcome in patients with acquired infections after acute spinal cord injury. Neurology 2017, 88:892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riegger T, Conrad S, Liu K, Schluesener HJ, Adibzahdeh M, Schwab JM: Spinal cord injury-induced immune depression syndrome (SCI-IDS). European Journal of Neuroscience 2007, 25:1743–1747. [DOI] [PubMed] [Google Scholar]
- 8.Lucin KM, Sanders VM, Jones TB, Malarkey WB, Popovich PG: Impaired antibody synthesis after spinal cord injury is level dependent and is due to sympathetic nervous system dysregulation. Experimental Neurology 2007, 207:75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kopp MA, Druschel C, Meisel C, Liebscher T, Prilipp E, Watzlawick R, Cinelli P, Niedeggen A, Schaser K-D, Wanner GA, et al. : The SCIentinel study-prospective multicenter study to define the spinal cord injury-induced immune depression syndrome (SCI-IDS)-study protocol and interim feasibility data. BMC Neurology 2013, 13:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nyquist RH: Mortality in Spinal Cord Injuries. California Medicine 1965, 103:417–419. [PMC free article] [PubMed] [Google Scholar]
- 11.van Dyke M, Greer S, Odom E, Schieb L, Vaughan A, Kramer M, Casper M: Heart Disease Death Rates Among Blacks and Whites Aged ≥35 Years — United States, 1968–2015. Morbidity and Mortality Weekly Report Surveillance Summaries 2018, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strauss DJ, DeVivo MJ, Paculdo DR, Shavelle RM: Trends in Life Expectancy After Spinal Cord Injury. Archives of Physical Medicine and Rehabilitation 2006, 87:1079–1085. [DOI] [PubMed] [Google Scholar]
- 13.Shavelle RM, Devivo MJ, Brooks JC, Strauss DJ, Paculdo DR: Improvements in long-term survival after spinal cord injury? Archives of Physical Medicine and Rehabilitation 2015, 96:645–651. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Arguello LY, O’Horo JC, Farrell A, Blakney R, Sohail MR, Evans CT, Safdar N: Infections in the spinal cord-injured population: A systematic review. Spinal Cord 2017, 55:526–534. [DOI] [PubMed] [Google Scholar]
- 15.Burns SP, Weaver FM, Parada JP, Evans CT, Chang H, Hampton RY, Kapur V: Management of community-acquired pneumonia in persons with spinal cord injury. Spinal Cord 2004, 42:450–458. [DOI] [PubMed] [Google Scholar]
- 16.Druschel C, Saidy RRO, Grittner U, Nowak CP, Meisel A, Schaser K-D, Niedeggen A, Liebscher T, Kopp MA, Schwab JM: Clinical decision-making on spinal cord injury-associated pneumonia: a nationwide survey in Germany. Spinal Cord 2020, 58:873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cardenas DD, Hoffman JM, Kirshblum S, McKinley W: Etiology and incidence of rehospitalization after traumatic spinal cord injury: A multicenter analysis. Archives of Physical Medicine and Rehabilitation 2004, 85:1757–1763. [DOI] [PubMed] [Google Scholar]
- 18.Skelton-Dudley F, Doan J, Suda K, Holmes SA, Evans C, Trautner B: Spinal cord injury creates unique challenges in diagnosis and management of catheter-associated urinary tract infection. Topics in Spinal Cord Injury Rehabilitation 2019, 25:331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goetz LL, Howard M, Cipher D, Revankar SG: Occurrence of candiduria in a population of chronically catheterized patients with spinal cord injury. Spinal Cord 2010, 48:51–54. [DOI] [PubMed] [Google Scholar]
- 20.Evans CT, LaVela SL, Weaver FM, Priebe M, Sandford P, Niemiec P, Miskevics S, Parada JP: Epidemiology of Hospital-Acquired Infections in Veterans With Spinal Cord Injury and Disorder. Infection Control and Hospital Epidemiology 2008, 29:234–242. [DOI] [PubMed] [Google Scholar]
- 21.Brommer B, Engel O, Kopp MA, Watzlawick R, Müller S, Prüss H, Chen Y, DeVivo MJ, Finkenstaedt FW, Dirnagl U, et al. : Spinal cord injury-induced immune deficiency syndrome enhances infection susceptibility dependent on lesion level. Brain 2016, 139:692–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao Y, DiPiro N, Krause JS: Health factors and spinal cord injury: a prospective study of risk of cause-specific mortality. Spinal Cord 2019, 57:594–602. [DOI] [PubMed] [Google Scholar]
- 23.Savic G, Devivo MJ, Frankel HL, Jamous MA, Soni BM, Charlifue S: Causes of death after traumatic spinal cord injury — a 70-year British study. Spinal Cord 2017, 55:891–897. [DOI] [PubMed] [Google Scholar]
- 24.Jung W-C, Levesque J-P, Ruitenberg MJ: It takes nerve to fight back: The significance of neural innervation of the bone marrow and spleen for immune function. Seminars in Cell & Developmental Biology 2017, 61:60–70. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Guan Z, Reader B, Shawler T, Mandrekar-Colucci S, Huang K, Wei Z, Bratasz A, Wells J, Powell ND, et al. : Autonomic dysreflexia causes chronic immune suppression after spinal cord injury. Journal of Neuroscience 2013, 33:12970–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES: The Sympathetic Nerve — An Integrative Interface between Two Supersystems : The Brain and the immune system 2000, 52:595–638. [PubMed] [Google Scholar]
- 27.Bellinger DL, Millar BA, Perez S, Carter J, Wood C, ThyagaRajan S, Molinaro C, Lubahn C, Lorton D: Sympathetic modulation of immunity: Relevance to disease. Cellular Immunology 2008, 252:27–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kleitman N, Holzwarth MA: Catecholaminergic innervation of the rat adrenal cortex. Cell and Tissue Research 1985, 241:139–147. [DOI] [PubMed] [Google Scholar]
- 29.Mignini F, Streccioni V, Amenta F: Autonomic innervation of immune organs and neuroimmune modulation. Autonomic and Autacoid Pharmacology 2003, 23:1–25. [DOI] [PubMed] [Google Scholar]
- 30.Toth IE, Vizi ES, Hinson JP, Vinson GP: Innervation of the Adrenal Cortex, Its Physiological Relevance, With Primary Focus on the Noradrenergic Transmission. Microsc Res Tech 1997, 36:534–545. [DOI] [PubMed] [Google Scholar]
- 31.Dénes Á, Boldogkoi Z, Uhereczky G, Hornyák Á, Rusvai M, Palkovits M, Kovács KJ: Central autonomic control of the bone marrow: Multisynaptic tract tracing by recombinant pseudorabies virus. Neuroscience 2005, 134:947–963. [DOI] [PubMed] [Google Scholar]
- 32.Tang Y, Shankar R, Gamelli R, Jones S: Dynamic norepinephrine alterations in bone marrow: evidence of functional innervation 1999. [DOI] [PubMed] [Google Scholar]
- 33.Noble BT, Brennan FH, Popovich PG: The spleen as a neuroimmune interface after spinal cord injury. Journal of Neuroimmunology 2018, 321:1–11. [DOI] [PubMed] [Google Scholar]
- 34.Hu D, Nicholls PK, Claus M, Wu Y, Shi Z, Greene WK, Ma B: Immunofluorescence characterization of innervation and nerve-immune cell interactions in mouse lymph nodes. European Journal of Histochemistry 2019, 63:3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prüss H, Tedeschi A, Thiriot A, Lynch L, Loughhead SM, Stutte S, Mazo IB, Kopp MA, Brommer B, Blex C, et al. : Spinal cord injury-induced immunodeficiency is mediated by a sympathetic-neuroendocrine adrenal reflex. Nature Neuroscience 2017, 20:1549–1559. [DOI] [PubMed] [Google Scholar]
- 36.Previnaire JG, Soler JM, el Masri W, Denys P: Assessment of the sympathetic level of lesion in patients with spinal cord injury. Spinal Cord 2009, 47:122–127. [DOI] [PubMed] [Google Scholar]
- 37.Llewellyn-Smith IJ, Weaver LC, Keast JR: Effects of spinal cord injury on synaptic inputs to sympathetic preganglionic neurons. Progress in Brain Research 2006, 152:11–26. [DOI] [PubMed] [Google Scholar]
- 38.Llewellyn-Smith IJ, Weaver LC: Changes in synaptic inputs to sympathetic preganglionic neurons after spinal cord injury. Journal of Comparative Neurology 2001, 435:226–240. [DOI] [PubMed] [Google Scholar]
- 39.Llewellyn-Smith IJ, Cassam AK, Krenz NR, Krassioukov A v, Weaver LC: Glutamate- and Gaba-Immunoreactive Synapses On Sympathetic Preganglionic Neurons Caudal To A Spinal Cord Transection In Rats. Neuroscience 1997, 80:1225–35. [DOI] [PubMed] [Google Scholar]
- 40.Krassioukov A v, Weaver LC: Reflex and Morphological Changes in Spinal Preganglionic Neurons After Cord Injury in Rats. Clinical and Experimental Hypertension 1995, 17:361–373. [DOI] [PubMed] [Google Scholar]
- 41.Weaver LC, Cassam AK, Krassioukov A v, Llewellyn-Smith IJ: Changes in immunoreactivity for growth associated protein-43 suggest reorganization of synapses on spinal sympathetic neurons after cord transection. Neuroscience 1997, 81:535–551. [DOI] [PubMed] [Google Scholar]
- 42.Brennan FH, Noble BT, Wang Y, Guan Z, Davis H, Mo X, Harris C, Eroglu C, Ferguson AR, Popovich PG: Acute post-injury blockade of α2δ−1 calcium channel subunits prevents pathological autonomic plasticity after spinal cord injury. Cell Reports 2021, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ueno M, Ueno-Nakamura Y, Niehaus J, Popovich PG, Yoshida Y: Silencing spinal interneurons inhibits immune suppressive autonomic reflexes caused by spinal cord injury. Nature Neuroscience 2016, 19:784–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mironets E, Fischer R, Bracchi-Ricard V, Saltos TM, Truglio TS, O’Reilly ML, Swanson KA, Bethea JR, Tom VJ: Attenuating neurogenic sympathetic hyperreflexia robustly improves antibacterial immunity after chronic spinal cord injury. Journal of Neuroscience 2020, 40:478–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cameron AA, Smith GM, Randall DC, Brown DR, Rabchevsky AG: Genetic manipulation of intraspinal plasticity after spinal cord injury alters the severity of autonomic dysreflexia. Journal of Neuroscience 2006, 26:2923–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krenz NR, Meakin SO, Krassioukov A v, Weaver LC, Canada NA: Neutralizing intraspinal nerve growth factor blocks autonomic dysreflexia caused by spinal cord injury. Journal of Neuroscience 1999, 19:7405–7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mironets E, Osei-Owusu P, Bracchi-Ricard V, Fischer X, Owens EA, Ricard J, Wu D, Saltos T, Collyer E, Hou S, et al. : Soluble TNF signaling within the spinal cord contributes to the development of autonomic dysreflexia and ensuing vascular and immune dysfunction after spinal cord injury. Journal of Neuroscience 2018, 38:4146– 4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davies A, Hendrich J, van Minh AT, Wratten J, Douglas L, Dolphin AC: Functional biology of the α2δ subunits of voltage-gated calcium channels. Trends in Pharmacological Sciences 2007, 28:220–228. [DOI] [PubMed] [Google Scholar]
- 49.Risher WC, Kim N, Koh S, Choi JE, Mitev P, Spence EF, Pilaz LJ, Wang D, Feng G, Silver DL, et al. : Thrombospondin receptor α2δ−1 promotes synaptogenesis and spinogenesis via postsynaptic Rac1. Journal of Cell Biology 2018, 217:3747–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eroglu Ç, Allen NJ, Susman MW, O’Rourke NA, Park CY, Özkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, et al. : Gabapentin Receptor α2δ−1 Is a Neuronal Thrombospondin Receptor Responsible for Excitatory CNS Synaptogenesis. Cell 2009, 139:380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Field MJ, Cox PJ, Stott E, Melrose H, Offord J, Su TZ, Bramwell S, Corradini L, England S, Winks J, et al. : Identification of the α2-δ−1 subunit of voltage-calcium calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin. Proceedings of the National Academy of Sciences of the United States of America 2006, 103:17537–17542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lucin KM, Sanders VM, Popovich PG: Stress hormones collaborate to induce lymphocyte apoptosis after high level spinal cord injury. Journal of Neurochemistry 2009, 110:1409–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tibbs PA, Young B, Ziegler MG, Mcallister RG: Studies of experimental cervical spinal cord transection. Journal of Neurosurgery 1979, 50:629–632. [DOI] [PubMed] [Google Scholar]
- 54.Kim JH, Choi MH: Embryonic development and adult regeneration of the adrenal gland. Endocrinology and Metabolism 2021, 35:765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bechmann N, Berger I, Bornstein SR, Steenblock C: Adrenal medulla development and medullary-cortical interactions. Molecular and Cellular Endocrinology 2021, 528:111258. [DOI] [PubMed] [Google Scholar]
- 56.Bornstein SR, Ehrhart-Bornstein M, Scherbaum WA, Pfeiffer EF, Holst JJ: Effects of splanchnic nerve stimulation on the adrenal cortex may be mediated by chromaffin cells in a paracrine manner. Endocrinology 1990, 127:900–906. [DOI] [PubMed] [Google Scholar]
- 57.Hasenmajer V, Sbardella E, Sciarra F, Minnetti M, Isidori AM, Venneri MA: The Immune System in Cushing’s Syndrome. Trends in Endocrinology and Metabolism 2020, 31:655–669. [DOI] [PubMed] [Google Scholar]
- 58.Berger I, Werdermann M, Bornstein SR, Steenblock C: The adrenal gland in stress – Adaptation on a cellular level. Journal of Steroid Biochemistry and Molecular Biology 2019, 190:198–206. [DOI] [PubMed] [Google Scholar]
- 59.Li Q, Johansson H, Grimelius L: Innervation of human adrenal gland and adrenal cortical lesions. Virchows Arch 1999, 435:580–589. [DOI] [PubMed] [Google Scholar]
- 60.Jansen ASP, Nguyen X van, Karpitskiy, Vladimir Mettenleiter TC, Loewy AD: Central Command Neurons of the Sympathetic Nervous Sytem: Basis of the Fight-or-Flight Response. Science 1995, 270:644–646. [DOI] [PubMed] [Google Scholar]
- 61.Schramm LP, Adair JR, Stribling JM, Gray LP: Preganglionic innervation of the adrenal gland of the rat: A study using horseradish peroxidase. Experimental Neurology 1975, 49:540–553. [DOI] [PubMed] [Google Scholar]
- 62.Parker TL, Kesse WK, Mohamed AA, Afework M: The innervation of the mammalian adrenal gland. J Anat 1993, 183:265–276. [PMC free article] [PubMed] [Google Scholar]
- 63.Edwards A v., Jones CT: Autonomic control of adrenal function. Journal of anatomy 1993, 183 ( Pt 2:291–307. [PMC free article] [PubMed] [Google Scholar]
- 64.Engeland WC: Functional Innervation of the Adrenal Cortex by the Splanchnic Nerve. Hormone and Metabolic Research 1998, 30:311–314. [DOI] [PubMed] [Google Scholar]
- 65.Ulrich-Lai YM, Arnhold MM, Engeland WC: Adrenal splanchnic innervation contributes to the diurnal rhythm of plasma corticosterone in rats by modulating adrenal sensitivity to ACTH. American Journal of Physiology - Regulatory Integrative and Comparative Physiology 2006, 290:1128–1135. [DOI] [PubMed] [Google Scholar]
- 66.Leman S, Sequeira H: Activation of adrenal preganglionic neurons during autonomic dysreflexia in the chronic spinal cord-injured rat. Autonomic Neuroscience: Basic and Clinical 2002, 98:94–98. [DOI] [PubMed] [Google Scholar]
- 67.Gaudet AD, Fonken LK, Ayala MT, Bateman EM, Schleicher WE, Smith EJ, D’angelo HM, Maier SF, Watkins LR: Spinal cord injury in rats disrupts the circadian system. eNeuro 2018, 5:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nitsche B, Perschak H, Curt A, Dietz V: Loss of circadian blood pressure variability in complete tetraplegia. Journal of Human Hypertension 1996, 10:311–317. [PubMed] [Google Scholar]
- 69.Cruse JM, Lewis RE, Bishop GR, Kliesch WF, Gaitan E: Neuroendocrine-immune interactions associated with loss and restoration of immune system function in spinal cord injury and stroke patients. Immunologic Research 1992, 11:104–116. [DOI] [PubMed] [Google Scholar]
- 70.Noble BT, Brennan FH, Wang Y, Guan Z, Mo X, Schwab JM, Popovich PG: Thoracic VGlut2+ spinal interneurons regulate structural and functional plasticity of sympathetic networks after high-level spinal cord injury 2022, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ng J, Villuendas R, Cokic I, Schliamser JE, Gordon D, Koduri H, Benefield B, Simon J, Murthy SNP, Lomasney JW, et al. : Autonomic remodeling in the left atrium and pulmonary veins in heart failure. Circulation: Arrhythmia and Electrophysiology 2011, 4:388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bottasso E: Toward the Existence of a Sympathetic Neuroplasticity Adaptive Mechanism Influencing the Immune Response . A Hypothetical View — Part I. Frontiers in Endocrinology 2019, 10:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sionov RV, Cohen O, Kfir S, Zilberman Y, Yefenof E: Role of mitochondrial glucocorticoid receptor in glucocorticoid-induced apoptosis. Journal of Experimental Medicine 2006, 203:189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, Frenette PS: Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 2006, 124:407–421. [DOI] [PubMed] [Google Scholar]
- 75.Chernykh ER, Shevela EY, Leplina OY, Tikhonova MA, Ostanin AA, Kulagin AD, Pronkina N v, Muradov ZM, Stupak V v, Kozlov VA: Characteristics of Bone Marrow Cells under Conditions of Impaired Innervation in Patients with Spinal Trauma. Cell Technologies in Biology and Medicine 2006, 2:117–120. [DOI] [PubMed] [Google Scholar]
- 76.Iversen PO, Hjeltnes N, Holm B, Flatebo T, Strom-Gundersen I, Ronning W, Stanghelle J, Benestad HB: Depressed immunity and impaired proliferation of hematopoietic progenitor cells in patients with complete spinal cord injury. Blood 2000, 96:2081– 2083. [PubMed] [Google Scholar]
- 77.Carpenter RS, Marbourg JM, Brennan FH, Mifflin KA, Hall JCE, Jiang RR, Mo XM, Karunasiri M, Burke MH, Dorrance AM, et al. : Spinal cord injury causes chronic bone marrow failure. Nature Communications 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mendez-Ferrer S, Chow A, Merad M, Frenette PS: Circadian rhythms influence hematopoietic stem cells. Curr Opin Hematol 2009, doi: 10.1097/MOH.0b013e32832bd0f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Méndez-Ferrer S, Lucas D, Battista M, Frenette PS: Haematopoietic stem cell release is regulated by circadian oscillations. Nature 2008, 452:442–447. [DOI] [PubMed] [Google Scholar]
- 80.Sugiyama T, Kohara H, Noda M, Nagasawa T: Maintenance of the Hematopoietic Stem Cell Pool by CXCL12-CXCR4 Chemokine Signaling in Bone Marrow Stromal Cell Niches. Immunity 2006, 25:977–988. [DOI] [PubMed] [Google Scholar]
- 81.Pinho S, Frenette PS: Haematopoietic stem cell activity and interactions with the niche. Nature Reviews Molecular Cell Biology 2019, 20:303–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Matsushita Y, Chu AKY, Ono W, Welch JD, Ono N: Intercellular Interactions of an Adipogenic CXCL12-Expressing Stromal Cell Subset in Murine Bone Marrow. Journal of Bone and Mineral Research 2021, doi: 10.1002/jbmr.4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Guo B, Huang X, Cooper S, Broxmeyer HE, Author NM: Glucocorticoid hormone-induced chromatin remodeling enhances human hematopoietic stem cell homing and engraftment. Nature Medicine 2017, 23:424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Maity S, Jarome TJ, Blair J, Lubin FD, Nguyen P v.: Noradrenaline goes nuclear: Epigenetic modifications during long-lasting synaptic potentiation triggered by activation of β-adrenergic receptors. Journal of Physiology 2016, 594:863–881. [DOI] [PMC free article] [PubMed] [Google Scholar]