

Abstract
Src family kinases (SFKs) play a crucial role in the regulation of multiple cellular pathways, including mitochondrial oxidative phosphorylation (OXPHOS). Aberrant activities of one of the most predominant SFKs, c-Src, was identified as a fundamental cause for dysfunctional cell signaling and implicated in cancer development and metastasis, especially in human hepatocellular carcinoma (HCC). Recent work in our laboratory revealed that c-Src is implicated in regulation of mitochondrial energy metabolism in cancer. In this study, we investigated the effect of c-Src expression on mitochondrial energy metabolism by examining changes in the expression and activities of OXPHOS complexes in liver cancer biopsies and cell lines. An increased expression of c-Src was correlated with an impaired expression of nuclear- and mitochondrial-encoded subunits of OXPHOS complexes I and IV, respectively, in metastatic biopsies and cell lines. Additionally, we observed a similar association between high c-Src and reduced OXPHOS complex expression and activity in mouse embryonic fibroblast (MEF) cell lines. Interestingly, the inhibition of c-Src kinase activity with the SFK inhibitor PP2 and c-Src siRNA stimulated the expression of complex I and IV subunits and increased their enzymatic activities in both cancer and normal cells. Evidence provided in this study reveals that c-Src impairs the expression and function of mitochondrial OXPHOS complexes, resulting in a significant defect in mitochondrial energy metabolism, which can be a contributing factor to the development and progression of liver cancer. Furthermore, our findings strongly suggest that SFK inhibitors should be used in the treatment of HCC and other cancers with aberrant c-Src kinase activity to improve mitochondrial energy metabolism.
Keywords: Oxidative phosphorylation (OXPHOS), mitochondrial energy metabolism, human hepatocellular carcinoma, liver cancer, Src family kinases (SFK)
1. Introduction
Src family kinases (SFKs) are a major group of tyrosine (Tyr) kinases that are essential for the regulation of a large number of intracellular signaling pathways, including cell growth and proliferation, differentiation, motility, adhesion, cell death, and survival, in a variety of cell types [1, 2]. Aberrant expression of SFK members, such as c-Src, have been shown to induce malignant properties in cells, thereby establishing them as proto-oncogenes [3–5]. In fact, the increased expression and/or activity of c-Src kinase has been observed in colorectal, ovarian, lung, breast, and liver cancer [6–9], including human hepatocellular carcinoma (HCC) which is the most common type of liver cancer and one of the most malignant and metastatic cancers worldwide [10, 11].
c-Src is one of the ubiquitously expressed SFK members with a high sequence homology to Yes and Fyn and, consequently, has overlapping functions in cellular pathways [12, 13]. c-Src, as well as several other SFKs, Fyn, Fgr, Lyn, and Lck, are localized to the mitochondria and regulate mitochondrial pathways [14–17], such as pyruvate decarboxylation [18], the tricarboxylic acid (TCA) cycle [19, 20], apoptosis [21], and oxidative phosphorylation (OXPHOS) [19, 20, 22–27]. OXPHOS is supported by four electron transfer chain complexes, complex I-IV and ATP synthase, complex V. The majority of these complexes have been shown to be Tyr phosphorylated by SFKs; however, c-Src was identified as the kinase responsible for the phosphorylation of two nuclear-encoded subunits, NDUFB10 and NDUFV2, of complex I, SDHA of complex II, and the mitochondrial-encoded subunit, COII, of complex IV [23–26]. The phosphorylation of COII by c-Src kinase and the epidermal growth factor receptor (EGFR) were shown to regulate the activity of complex IV as well as cellular ATP synthesis in several normal and cancer cell lines [23]. In addition to the alteration of OXPHOS activity by SFK phosphorylation of its complexes, our laboratory recently demonstrated the implication of Fyn with mitochondrial translation which is responsible for the synthesis of the 13 core subunits of OXPHOS complexes I, III, IV, and V [12]. Although reduced mitochondrial function and c-Src activity have been implicated in promoting the Warburg effect in various cancer types [18, 28, 29], the role of c-Src in mitochondrial energy metabolism in liver cancer still remains to be established.
In this study, we investigated the role of c-Src on mitochondrial energy metabolism by studying the steady-state expression of OXPHOS complex subunits. We observed increased expression of c-Src in metastatic liver tumors and the metastatic liver cancer cell line, Hep3B, which was correlated with significant reductions in the expression and activity of OXPHOS complex I and IV subunits. Additionally, we observed the same association between high c-Src expression and impaired OXPHOS complex expression and activity in mouse embryonic fibroblast (MEF) cell lines. Interestingly, the inhibition of c-Src with the SFK inhibitor PP2 stimulated the expression of complex I and IV subunits, improved their enzymatic activities, and significantly reduced cell proliferation in both cancer and normal cell lines. The suppression of c-Src using c-Src siRNA was also shown to increase mitochondrial OXPHOS expression. Our studies strongly indicate that c-Src impairs the expression and function of OXPHOS complexes, which can be one of the underlying factors for the development and progression of liver cancer. Furthermore, our findings provide evidence that the inhibition of SFKs with PP2 can be used as possible therapeutic strategies to inhibit cell proliferation and improve mitochondrial function and energy metabolism in liver cancers
2. Materials and methods
2.1. Liver tumor samples
Liver tissue biopsies, five cancerous and four non-cancerous, were derived from patients treated at the Pennsylvania State University Health Milton S. Hershey Medical Center. Protein lysates used in Western blot analyses were obtained by the resuspension and sonication of tissues in RIPA buffer containing 1% SDS. Lysates were then separated by 12% SDS-PAGE, transferred to nitrocellulose membranes, and probed with various antibodies, as described below.
2.2. Cell culture
Human liver cancer Hep3B and HepG2 cell lines and mouse embryonic fibroblast (MEF) cells with endogenous expression of c-Src, Yes, and Fyn (WT), MEF cells with functional null mutations in both alleles of c-Src, Yes, and Fyn kinases (SYF), MEF cells with an endogenous expression of c-Src kinase and null mutations in Yes and Fyn (Src++), and SYF cells with a stable over expression of c-Src kinase (Src) were purchased from the American Type Culture Collection (ATCC). Monolayer cultures of each cell line were maintained in Dulbecco’s modified Eagles Medium (DMEM) (Hyclone, Thermo-Scientific, Waltham, MA) with 4.5 g/L glucose and adjusted to contain 10% fetal bovine serum (FBS) (Rocky Mountain Biologicals, Missoula, MT), 4 mM glutamine, 1 mM pyruvate, and 1% penicillin/streptomycin (P/S) (Corning Cellgro, Manassas, VA). The cells were grown in a humidified incubator at 37 °C with 5% CO2. Collected cells were treated with sodium orthovanadate to preserve Tyr phosphorylation [30, 31].
Transfections of siRNAs were performed using Lipofectamine transfection reagent according to the manufacturer’s protocols (Thermo Fisher Scientific). Partial knock down of human and mouse c-Src were performed using control and c-Src siRNAs obtained from Santa Cruz Biotechnology Inc. (Dallas, TX). The Hep3B, HepG2, WT, SYF, Src++, and Src cell lines were also treated with the Src family kinase inhibitor PP2 (4-amino-5-(4-chlorophenyl)-7-(dimethylethyl)pyrazolo[3,4-d]pyrimidine) (EMD Chemicals, Gibstown, NJ) [32], dissolved in dimethyl sulfoxide (DMSO), at concentrations ranging from 0 – 10 μM. Briefly, cells were seeded at 25×104 cells/mL in DMEM containing 0.2% FBS and incubated for 24 h. The media was then replaced with fresh DMEM containing 10% FBS and cells were treated with PP2 for 24 – 72 h. To measure cell proliferation, cells were grown as described above and counted using the Trypan blue exclusion assay. For reactive oxygen species (ROS) generation assays, cells were grown as described above and the amount of ROS produced by the Hep3B and HepG2 cells was measured using Amplex Red assays (Thermo Fisher Scientific) according to the manufacturer’s protocol. The results for cell proliferation and ROS generation assays are represented as the mean ± SD of at least three experiments for each group and presented as a percentage of the control cells (HepG2 cells = 100%; control cells for treatments = 100%).
2.3. Western blot analyses
Whole cell lysates obtained from liver biopsies and cell lines were lysed in RIPA buffer containing 50 mM Tris-HCl (pH 7.6), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP40, 0.1% SDS, 0.5% DOC, 1 mM PMSF, and protease and phosphatase inhibitor cocktails. Protein concentrations were determined using the bicinchoninic protein assay (BCA) (Pierce, Rockford, USA). The protein lysates (10 – 40 μg) were separated by 12% SDS-PAGE, transferred to nitrocellulose membranes (Amersham, GE Healthcare, Pittsburg, PA), stained with Ponceau S to ensure equal protein loading, blocked in a Tris-buffered saline (TBS) containing 0.05% Tween-20 (TBST), 5% (w/v) dry skim milk, and 1% BSA, and incubated with the corresponding primary antibodies overnight. Human and rodent OXPHOS complex cocktails and SDHA antibodies were purchased from Sigma Aldrigh (St. Louis, MO). Citrate synthase (CS), ACOX1, and c-Src kinase antibodies were purchased from Santa Cruz (Dallas, TX). Phosphotyrosine (PY-100), phospho-Src family (pSFK), LDHA, and PDH antibodies were purchased from Cell Signaling Technologies (Danvers, MA). The pSFK antibody detects the phosphorylation of c-Src and other family members Fyn, Lyn, Lck, Yes, and Hck at the kinase activation site Tyr416 (pTyr416) or the equivalent residue. The loading control antibody GAPDH was purchased from Fitzgerald (Acton, MA). Mitochondrial EF-Tu and TFAM antibodies were kind gifts from Dr. Linda Spremulli and Dr. Craig Cameron, respectively. The protein immunoreactivity was detected using the ECL Western blotting detection kit from Amersham (GE Healthcare, UK) and the membranes were developed per the manufacturer’s protocol. The membranes were exposed to autoradiography films and band intensities were quantified using UN-Scan-It (Silk Scientific, Inc, Orem, UT) and ImageJ [33]. The quantified values were normalized to the protein loading determined by Ponceau S staining and GAPDH antibody probing of the membranes. The expression of each protein was quantified from individual tissue and cell lysates from at least three independent experiments. The results represent the mean ± SD for each quantified sample and are expressed as a percentage of the control cells (non-cancerous liver tissues = 100%; HepG2 cells = 100%; SYF cells = 100%; control cells for treatments = 100%).
2.4. Mitochondrial OXPHOS complex enzymatic activity assays
Cell pellets were dissolved in a buffer containing 10 mM Tris-HCl (pH 7.0), 250 mM sucrose, and phosphatase and protease inhibitor cocktails. Briefly, for the complex I and III enzymatic activity assay, cell lysates were further diluted in a 50 mM phosphate buffer (pH 7.4) containing 1 mM potassium cyanide, 1 mg/mL BSA, and 100 μM cytochrome c. Protein concentrations were determined using the BCA protein assay and protein amounts were normalized by Ponceau S staining and GAPDH antibody probing of the membranes. The reactions were started by adding 200 μM NADH and the combined activity of complex I and III was determined spectrometrically by measuring the reduction of cytochrome c at 550 nm. The complex IV enzymatic activity protein lysates were diluted in a 50 mM phosphate buffer (pH 7.4) containing 1 mM EDTA and 100 μM reduced cytochrome c. The activity of complex IV was also measured spectrometrically by monitoring cytochrome c oxidation at 550 nm, as previously described [34]. The rate of cytochrome c reduction and oxidation were calculated by dividing the change in absorbance at 550 nm between two linear points by the difference in time using the formula: Rate = (Abs 1 – Abs 2) / (Time 1 – Time 2). Each sample was run in triplicate for each experiment. The data are expressed as the mean ± SD of at least three experiments and are presented as a percentage of the control (HepG2 cells = 100%; SYF cells = 100%; control for treated cells = 100%).
2.5. Statistical analysis
Statistical analyses were performed using GraphPad Prism 6.07. Statistically significant differences between non-cancerous/cancerous liver biopsies, HepG2/Hep3B cell lines, MEF (WT/SYF//Src++/Src) cell lines, and control/treated cells were detected using unpaired Student’s t-tests (2-tailed), *P < 0.05. The non-cancerous tissues, HepG2, SYF, and control cells for treatments were set to 100% during statistical analyses. All data are expressed as mean ± SD, unless otherwise described.
3. Results and Discussion
3.1. c-Src is overexpressed in metastatic liver cancers
Aberrant expression and activity of c-Src was reported in patients with HCC and was positively correlated with tumor stage and metastasis. Thus, c-Src is believed to play an important role in the malignant transformation of hepatocytes [10, 11]. Although c-Src has been located in the mitochondria and established as one of the major regulators of mitochondrial energy metabolism and OXPHOS in various cell lines [14, 15] and has been identified as a promoter of the aggressiveness of breast cancer cells by reduced mitochondrial activity [9], the effects of c-Src on the expression of OXPHOS complex subunits in liver cancer have yet to be determined. Therefore, a better understanding of c-Src signaling and mitochondrial energy metabolism in liver cancer is essential to improve disease prognosis and treatments. To investigate c-Src kinase in liver cancer, we examined and compared the expression of c-Src in two groups of liver biopsies, cancerous and non-cancerous (patient tissue characteristics described in Table 1), by Western blot analyses. The expression of c-Src was detected in all liver tissues with a significantly higher expression of approximately 61% in the cancerous tissues compared to the non-cancerous tissues (Fig. 1). We also assessed the activation of c-Src by observing its phosphorylation at Tyr416, which is located in the activation loop of the kinase domain and is responsible for the enzymatic activity of c-Src when phosphorylated [2, 35]. The phospho-Src family (pSFK) antibody detects the endogenous c-Src phosphorylation at Tyr416, in addition to the phosphorylation of other SFK members, Lyn, Fyn, Lck, Yes, and Hck, at equivalent activation sites. An increase in SFK phosphorylation was also detected in liver cancer biopsies with an increased phosphorylation in cancerous tissues, especially those correlated with a higher c-Src expression (Fig. 1). The SFK phosphorylation was increased in the non-cancerous tissue from patient 7; however, this increase in pSFK may be due to the detection of another active SFK member. Interestingly, the tissues with the highest expression of c-Src and pSFK were metastatic adenocarcinomas (Table 1) from patients 2, 3, and 5 (Fig. 1), implying that increased c-Src expression and activity may contribute to the metastatic state of the tumors.
Table 1.
Characterization of liver tumor and non-cancerous tissues.
| Patient | Age | Tissue type | Metastasis | Disease state |
|---|---|---|---|---|
|
| ||||
| 1 | 18 | T | + | Adenocarcinoma |
| 2 | 20 | T | + | Adenocarcinoma |
| 3 | 28 | T | + | Adenocarcinoma |
| 4 | 31 | T | − | Adenoma |
| 5 | 43 | T | + | Adenocarcinoma |
| 6 | 58 | N | − | Benign cyst |
| 7 | 36 | N | − | Hyperplasia |
| 8 | 69 | N | − | Bilary cystadenoma |
| 9 | 73 | N | − | Macrovesicular steatosis |
T: primary tumor
N: normal tissue
Figure 1.
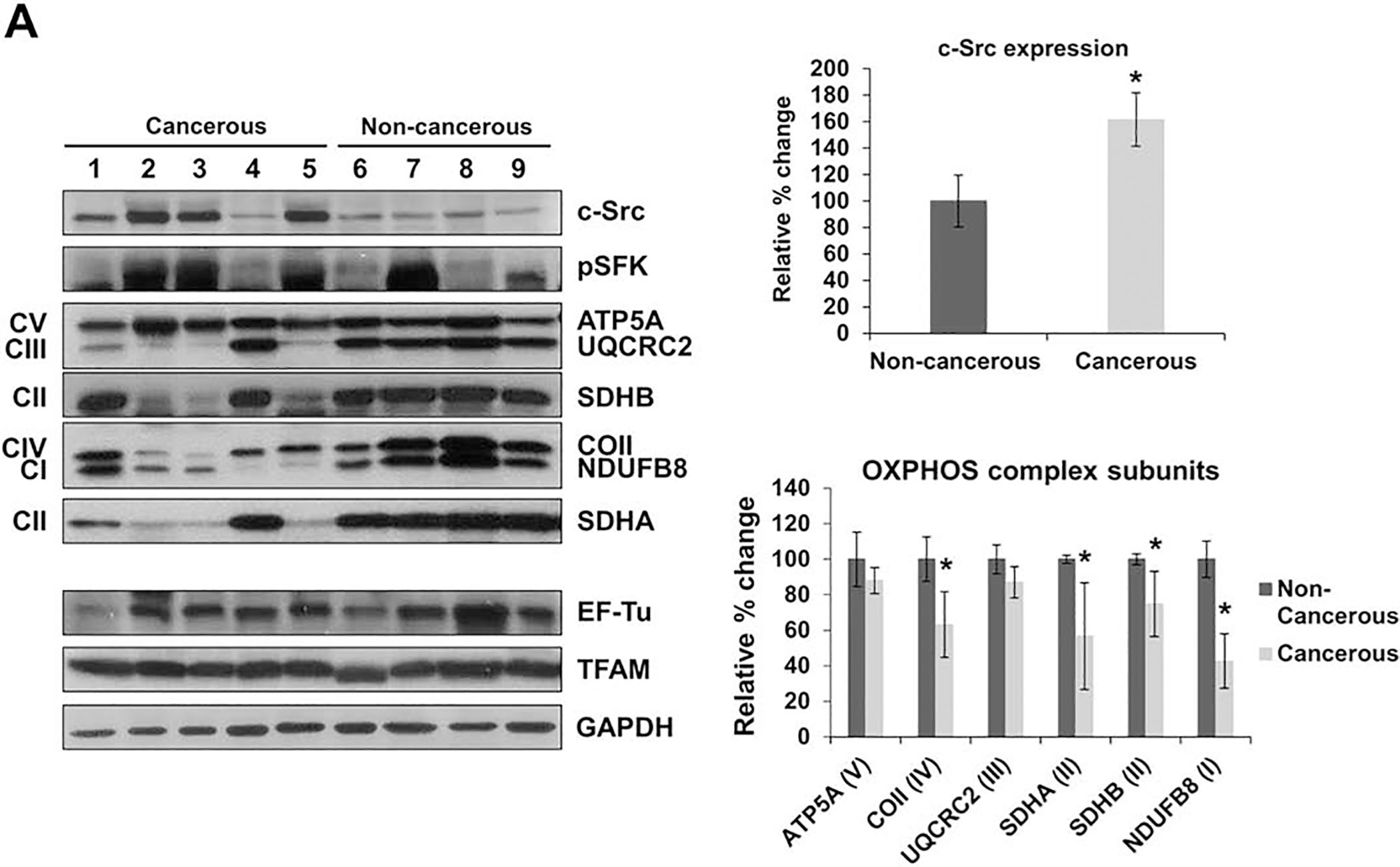
c-Src expression is increased in metastatic liver cancer tissues. The expression of OXPHOS subunits, including NDUFB8 (complex I), SDHA and SDHB (complex II), UQCRC2 (complex III), COII (complex IV), and ATP5A (complex V) were detected by Western blot analyses. The expression of c-Src kinase, mitochondrial transcription factor A (TFAM), and mitochondrial elongation factor Tu (EF-Tu) were also detected. Approximately 30 μg of whole cell lysates from each tissue was separated by 12% SDS-PAGE, and equal protein loading was determined by GAPDH antibody and Ponceau S staining of the membranes. The bar graphs represent the average quantitative analyses from densitometry of the expression of c-Src (top right panel) and the OXPHOS complex subunits (bottom right panel) of the two groups. Significant differences were identified in c-Src (P=0.0488), COII (P=0.0028), SDHA (P=0.0253), SDHB (P=0.0001), and NDUFB8 (P=0.0001). The data is represented as the mean ± SD of at least three experiments for each tissue group and are presented as a percentage of the non-cancerous tissues (non-cancerous tissues = 100%) (see Table 1 for details). Significant differences were observed between the non-cancerous and cancerous tissues via unpaired Student’s t-tests (2-tailed), *P < 0.05.
Subsequently, the steady-state expression of OXPHOS complex subunits was also examined in the two tissue groups using an antibody cocktail that detects the nuclear-encoded subunits of complex I (NDUFB8), II (SDHB), III (UQCRC2), and V (ATP5A) and the mitochondrial-encoded subunit of complex IV (COII) (Fig. 1), along with an antibody for complex II (SDHA). The lysates were also probed with GAPDH antibody as a protein loading control. Compared to the non-cancerous biopsies, the expression of complex III and V subunits (UQCRC2 and ATP5A, respectively) were reduced in the cancerous tissues, while the subunits of complexes I (NDUFB8) and II (SDHA and SDHB) were significantly decreased by roughly 55%, 45%, and 25%, respectively, as shown in Fig. 1. The expression of the mitochondrial-encoded complex IV subunit (COII) was also substantially decreased by approximately 35% in the cancerous liver tissues (Fig. 1). Surprisingly, the lowest expression of OXPHOS complexes was also identified in the metastatic tissues from patients 2, 3, and 5 (Fig. 1), potentially implicating impaired OXPHOS complex expression in cancer metastasis. The altered expression of both nuclear- and mitochondrial-encoded OXPHOS subunits implies that there may be a possible defect in protein synthesis. Due to these findings, we investigated two factors essential to the synthesis of mitochondrial proteins. The expression of both mitochondrial transcription factor A (TFAM) and mitochondrial elongation factor Tu (EF-Tu) was comparable between the two tissue types, signifying that the synthesis of nuclear- and mitochondrial-encoded OXPHOS subunits may be regulated by c-Src kinase. These results strongly suggest that increased c-Src expression is correlated with reduced OXPHOS subunit expression and may play a role in liver cancer.
To further examine the impaired OXPHOS complex expression observed in the cancerous liver tissues above, we analyzed the expression of several crucial metabolic enzymes that control OXPHOS by regulating pathways such as the citric acid cycle (CAC), fatty acid beta oxidation, and pyruvate metabolism, including peroxisomal acyl-coenzyme A oxidase 1 (ACOX1), citrate synthase (CS), pyruvate dehydrogenase (PDH), as well as mitochondrial markers heat shock protein 60 (HSP60) and voltage-dependent anion channel (VDAC)/porin. The expression of ACOX1, the first enzyme of the fatty acid beta oxidation pathway, is significantly decreased in the cancerous tissues from patients 2, 3, and 5, indicating that fatty acid beta oxidation is reduced in these patients which possibly implies lower energy production through oxidative phosphorylation, as observed by reduced expression of OXPHOS complexes (Fig. S1). Next, we also examined the expression of citrate synthase, one of the major checkpoints for the CAC. CS expression was comparable between the cancerous and non-cancerous liver tissues (Fig. S1) suggesting that the CAC activity is relatively equal between the two tissue types. Due to these findings, we further analyzed other metabolic pathways and enzymes that converge onto the CAC, such as PDH. The expression of PDH, which is a critical enzyme involved in the regulation of pyruvate metabolism by catalyzing the conversion of pyruvate to acetyl-CoA and connecting glycolysis to the CAC and mitochondrial oxidative metabolism. The expression of PDH was higher in the cancerous tissues of patients 2, 3, and 5 (Fig. S1), implying that pyruvate metabolism is high in liver cancer tissues. Interestingly, c-Src expression, which has been shown to phosphorylate, inactivate PDH, and cause a restraint on the pyruvate flux into mitochondrial metabolism [18], was increased in these cancerous liver tissues (Fig. 1). Therefore, although the expression of PDH was increased, the presence of high c-Src expression may inhibit the activity of PDH, causing these metastatic tissues to rely more on glycolysis than mitochondrial oxidative metabolism, the Warburg effect. Combined, our results suggest that c-Src may be contributing to the metabolic reprogramming of cancer tissues and inducing cancer metastasis by impairing mitochondrial oxidative phosphorylation.
To ensure the reduction in the expression of OXPHOS complexes was not a result of decreased mitochondrial mass, we assessed the expression of two mitochondrial markers, VDAC/porin and HSP60. The expression of VDAC/porin, found in the mitochondrial outer membrane, was comparable between cancerous and non-cancerous tissues, implying comparable amounts of mitochondria between each liver tissue analyzed (Fig. S1). The expression of HSP60, a mitochondrial chaperone protein that is responsible for the folding and assembly of newly imported proteins in the mitochondria, was slightly reduced (Fig. S1). Additionally, minor differences were observed in the expression of mitochondrial matrix protein, CS, between the cancerous and non-cancerous liver tissues, demonstrating that mitochondrial content was almost equal levels in both tissue types. These findings suggest that the decrease in OXPHOS complex expression was largely a result of the expression and activation of c-Src kinase.
3.2. c-Src is expressed in metastatic liver cancer cell line
The findings from the liver biopsies led us to further explore the relationship between c-Src and OXPHOS complex expression by studying their expressions in whole cell lysates from two of the most common liver cancer cell lines, Hep3B and HepG2, which are isolated from patients with primary HCC and hepatoblastoma (HB), respectively [36–38]. HepG2 cells express proteins that are characteristic of normal hepatocytes, while proteins expressed in Hep3B cells are involved in the induction of the epithelial mesenchymal transition (EMT) [38]. Due to the increased expression of c-Src in the metastatic liver biopsies, we first assessed its expression in both cell lines. c-Src expression was clearly increased in Hep3B cells compared to almost no expression in HepG2 cells (Fig. 2A). Surprisingly, we found relatively similar SFK phosphorylation between Hep3B and HepG2 cell lines (Fig. 2A), indicating other SFK members are active in the two cell lines. Since a handful of SFKs have been located in the mitochondria [14, 15], we analyzed the expression of some of these kinases. The expression of Lyn was comparable between the two cell lines (data not shown), while the expression of Lck and Fyn (data not shown) were increased in HepG2 cells, as previously described [12]. Therefore, the expression of Lyn, Lck, Fyn, c-Src, and other SFKs contribute to the pSFK signal detected in Hep3B and HepG2 cells. Yet, our results clearly show c-Src expression is only found in the metastatic Hep3B cell line.
Figure 2.
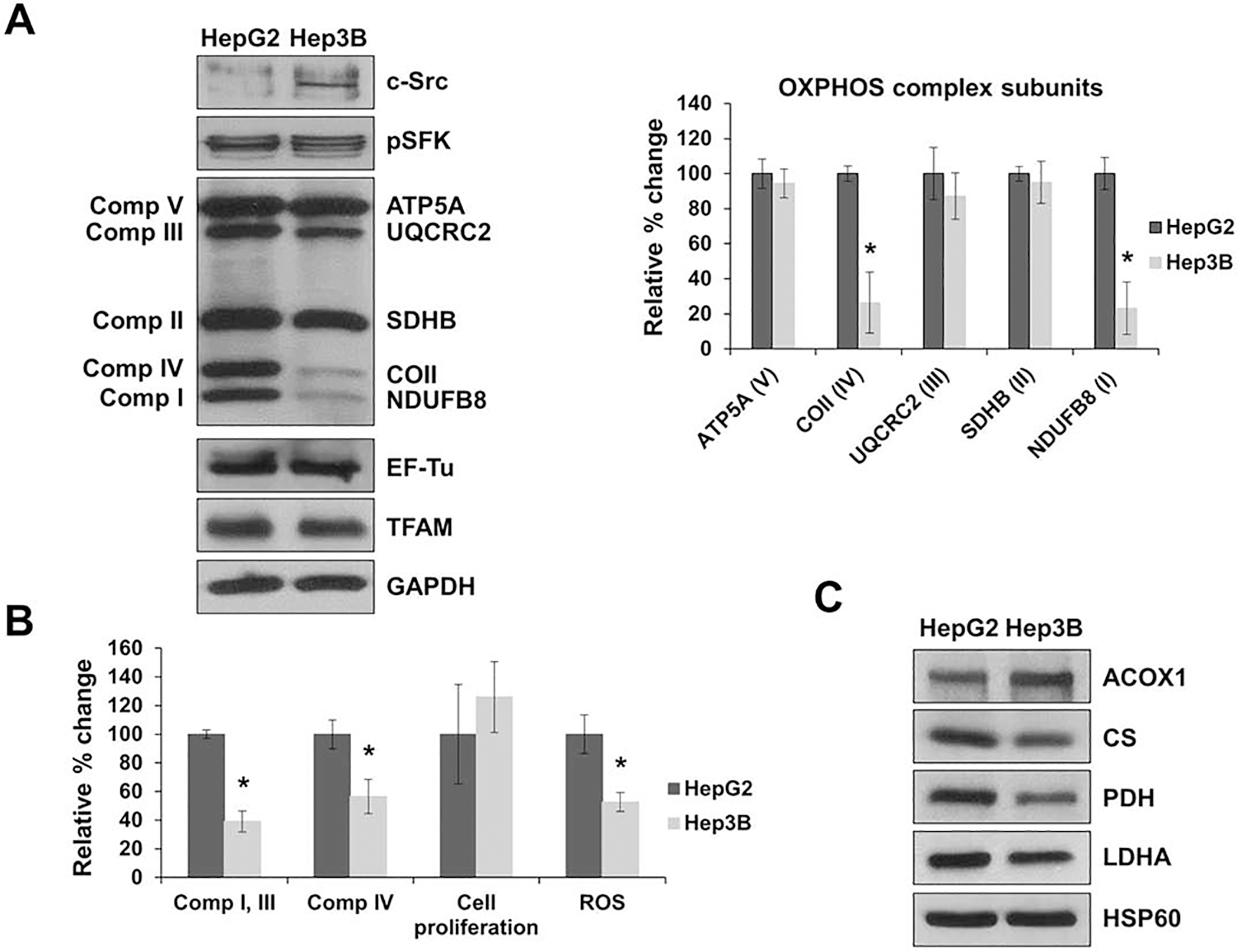
c-Src is overexpressed in liver cancer cell lines. A) The expression of OXPHOS complex subunits, c-Src kinase, and mitochondrial proteins TFAM and EF-Tu were detected by Western blot analyses in Hep3B and HepG2 cell lines. Additionally, the phosphorylation of SFK members at the activation site, Tyr416 (pSFK), was also observed. Equal protein concentration was determined by GAPDH antibody expression and Ponceau S staining of the membranes. Quantification of the average expression of OXPHOS complex subunits between the two cell lines is shown in the bar graph (right panel). The results represent the mean ± SD of at least three independent experiments. Significant difference was observed between HepG2 and Hep3B cells in OXPHOS subunits COII (P = 0.0104) and NDUFB8 (P = 0.0001). The data is presented as a percentage of HepG2 protein expression (HepG2 cells = 100%). B) HepG2 and Hep3 cells were grown for 48 h and proliferation was measured by the Trypan blue exclusion assay (cell proliferation). Combined complex I and III activity was determined by measuring the rate of cytochrome c reduction at 550 nm using equal amounts (10 μg) of whole cell lysates obtained from HepG2 and Hep3B cells. Complex IV enzymatic activity was determined by measuring the rate of cytochrome c oxidation spectrometrically at 550 nm using equal amounts of HepG2 and Hep3B whole cell lysates. ROS generation was assessed with the Amplex Red assay kit using equal concentrations of HepG2 and Hep3B cell lysates. Significant differences were observed between Hep3B and HepG2 cells in complex I and III activity (P = 0.0001), complex IV activity (P = 0.0139), cell proliferation (P = 0.013), and ROS generation (P = 0.0002). C) The expression of metabolic enzymes peroxisomal acyl-coenzyme A oxidase 1 (ACOX1), citrate synthase (CS), pyruvate dehydrogenase (PDH), lactate dehydrogenase A (LDHA), and heat shock protein 60 (HSP60) were detected by Western blot analyses of HepG2 and Hep3B cell lines. See Fig. 1 legend for details. Values are presented as mean ± SD for at least three experiments and presented as a percentage of HepG2 (HepG2 = 100%). Significant differences were observed between Hep3B and HepG2 cells via unpaired student’s t-test (2-tailed) with Welch’s correction, *P < 0.05.
The mitochondrial localization of c-Src has been shown to increase [26, 39] or decrease [18, 23] the OXPHOS activity and expression in various cell lines. Due to our findings in the liver biopsies and the expression of c-Src in the Hep3B liver cancer cell line only, we investigated the relationship between c-Src and changes in mitochondrial energy metabolism by observing the expression of OXPHOS subunits. Similar to what was found in the metastatic cancerous liver tissues from patients 2, 3, and 5 (Fig. 1), the subunits of complexes II, III, and V were decreased in Hep3B cells compared to HepG2 cells (Fig. 2A), with significant reductions in the expression of the nuclear-encoded subunit of complex I and the mitochondrial-encoded subunit of complex IV (NDUFB8 and COII, respectively) in Hep3B cells (Fig. 2A). Even with a lower expression of mitochondrial-encoded COII, the expression of EF-Tu and TFAM were comparable between the two cell lines (Fig. 2A). These results imply that c-Src may regulate the expression of both nuclear- and mitochondrial-encoded OXPHOS subunits in liver cancer tissues and cell lines.
Next, we determined the effects of the reduced expression of NDUFB8 and COII on mitochondrial oxidative phosphorylation and energy metabolism by performing complex I and III and complex IV enzymatic activity assays in both cell lines. As shown in Fig. 2B, a significant decrease of approximately 60% was observed in the complex I and III activity and a reduction of about 50% was found in the complex IV activity in Hep3B cells compared to HepG2 cells. The impaired complexes I and III and complex IV activities in Hep3B cells can be attributed to the reduction of the expression of nuclear-encoded NDUFB8 and mitochondrial-encoded COII, which are the two core subunits of complexes I and IV, respectively. The reduced COII expression and activity are in agreement with previous studies that demonstrate reduced complex IV activity due to the phosphorylation of COII by c-Src and EGFR [23, 40]. Since ROS are major byproducts of OXPHOS, we examined the ROS production in Hep3B and HepG2 cell lines. ROS generation was significantly reduced in Hep3B cells, which is possibly a result of the decreased OXPHOS expression and activity found in Hep3B cells. Although Hep3B cells had significantly lower OXPHOS function, cell proliferation increased compared to HepG2 cells.
To further study the decrease in OXPHOS activity and subunit expression in Hep3B cells, we analyzed the expression of several metabolic enzymes that control OXPHOS. The expression of ACOX1 was increased in Hep3B cells compared to HepG2 cells, demonstrating increased peroxisomal fatty acid oxidation in these cells (Fig. 2C). The expression of CS, PDH, and lactate dehydrogenase A (LDHA) were decreased in Hep3B cells implying that the reduction in OXPHOS may be a result of impaired CAC and pyruvate metabolism and further suggesting that Hep3B cells may rely more on anaerobic glycolysis for energy metabolism. In fact, both PDH and LDHA have known to be phosphorylated by c-Src induce a more metastatic phenotype in various cancers [18, 41, 42], similar to our observation in Hep3B cells. Reductions in the expression of these metabolic enzymes could be caused by the diminished OXPHOS subunit expression and/or activity in Hep3B cell lines. Although the expression of mitochondrial marker, HSP60, is comparable between Hep3B and HepG2 cells indicating similar mitochondrial presence in the two cell lines, mitochondrial OXPHOS is regulated by c-Src kinase expression and activity. Along with an aberrant activity of c-Src, decreased expression of OXPHOS complexes I and IV and reduced mitochondrial function have been correlated with cancer metastasis and the induction of EMT [43, 44]. The expression of several EMT factors analyzed were increased in Hep3B compared to HepG2 cell lines (data not shown), confirming the metastatic state of Hep3B cells, as previously described [37, 38]. Furthermore, our findings suggest that increased c-Src expression may impair OXPHOS expression and activity which could contribute to the metastatic phenotype of the Hep3B cell line.
3.3. Inhibition of c-Src stimulates the expression of OXPHOS subunits
Our data provide strong evidence that c-Src expression is associated with impaired OXPHOS complex expression and activity in liver cancer cells. Since aberrant expression of c-Src was shown in metastatic liver cancer tissues and cell lines (Figs. 1 and 2) and has been correlated with cancer metastasis, recurrence, adverse prognosis, and resistance to therapies [8, 11, 45], we investigated c-Src kinase as a potential target for liver cancer therapies by treating Hep3B and HepG2 cell lines with the selective SFK inhibitor PP2 at concentrations ranging from 0 – 10 μM for 72 h and investigated the steady-state expression of OXPHOS complex subunits. PP2 is a membrane-permeable inhibitor that blocks the ATP binding site of c-Src and prevents activation without altering its expression [17, 32].
We first determined the effect of PP2 treatments on c-Src kinase in Hep3B cells and observed a substantial decrease of approximately 40% in phosphorylation at Tyr416 (pSFK) using the pSFK and the phospho-Tyr antibody, along with a reduction in the overall Tyr phosphorylation at PP2 concentrations ranging from 0 – 5 μM (Fig. 3A). Although SFK phosphorylation was lower, minor differences in the expression of c-Src were observed in the presence of PP2 (Fig. 3A). Interestingly, the suppression of c-Src kinase stimulated the expression of nuclear-encoded NDUFB8 and mitochondrial-encoded COII in a concentration-dependent manner, with a significant increase of approximately 50% in their expression at 5 μM PP2, as shown in Fig. 3A. However, minor changes were found in most of the nuclear-encoded OXPHOS complex subunits, SDHB, UQCRC2, and ATP5A in the presence of PP2 (Fig. 3A). Despite the higher expression of COII and NDUFB8, the expression of EF-Tu and TFAM were comparable between the treated and control cells (Fig. 3A), demonstrating that the increased expression of OXPHOS subunits may be a result of c-Src inhibition and decreased Tyr phosphorylation. To determine if the increases in the expression of NDUFB8 and COII altered the function of complexes I and IV, respectively, we performed complex I and III and complex IV enzymatic activity assays. Significant increases were measured in the complex I and III activity of Hep3B cells treated with 2.5 and 5 μM PP2 (Fig. 3B). Similar increases were found in the complex IV activity of PP2 treated Hep3B cells (Fig. 3B). Additionally, the proliferation of Hep3B cells was decreased by approximately 65% and 80% at 2.5 and 5 μM PP2, respectively (Fig. 3B). The results of these studies demonstrated that the inhibition of c-Src kinase by PP2 stimulated OXPHOS complex expression which may be therapeutically beneficial to increase mitochondrial energy metabolism and possibly contribute to the decrease in cell proliferation.
Figure 3.

Inhibition of c-Src increases OXPHOS expression in the Hep3B cell line. A) Hep3B cells were treated with 0 – 5 μM Src family kinase (SFK) inhibitor PP2 for 72 h before collection. Changes in the expression of OXPHOS complex subunits, c-Src, pSFK, Tyr phosphorylation (pTyr), EF-Tu, and TFAM were measured by Western blot analyses with respect to GAPDH antibody probing and Ponceau S staining of the membranes as equal protein loading controls. The phosphorylation of SFK members (pSFK) was detected by the pTyr antibody, indicated by an arrow, as well as the pSFK antibody that detects phosphorylation at Tyr416. The quantitative analyses of the expression of OXPHOS subunits are represented as mean ± SD of at least three experiments for each group (bar graph, right panel). Significant differences were observed in the expression of COII (P = 0.0091) and NDUFB8 (P = 0.024) at 5 μM PP2. B) Cell proliferation of PP2 treated Hep3B cells were measured with the Trypan blue exclusion assays after 72 h treatment. Statistical difference was shown between the control and 2.5 μM (P = 0.001) and 5 μM (P = 0.0001) PP2. Complex I and III and complex IV activities were measured by determining the rates of cytochrome c reduction and oxidation, respectively, at 550 nm. Significant differences were observed in the complex I and III activity at 2.5 μM (P = 0.0122) and 5 μM (P = 0.0006) PP2 and complex IV activity at 2.5 μM (P = 0.0002) and 5 μM (P = 0.0001) PP2. C) Western blot analyses were used to identify changes in Hep3B cells transfected with with c-Src siRNA (c-Src) relative to control siRNA (cont). The quantitative analyses of OXPHOS expression are represented as the mean ± SD of at least three experiments for each group (bar graph, right panel). Significant differences were observed in the expression of COII (P = 0.0397) and NDUFB8 (P = 0.0022) between the control and c-Src siRNA transfected Hep3B cells. Data represents the mean ± SD of at least three independent experiments. The results are presented as a percentage of the control cells (cont = 100%). Statistical difference was observed between the control and treatment groups via unpaired Student’s t-test (2-tailed), *P < 0.05.
In addition to Hep3B cells, we treated the HepG2 cell line with PP2 at concentrations ranging from 0 – 10 μM for 72 h. Minor differences were found in the expression of OXPHOS complex subunits in HepG2 treated cells compared to the control cells (Fig. S2). Yet, we showed a clear reduction in the proliferation of HepG2 cells, which was associated with reduced SFK phosphorylation at increasing concentrations of PP2 (data not shown). Although PP2 treatments reduced cell proliferation in both Hep3B and HepG2 cell lines by suppressing phosphorylation at Tyr416, the increase in OXPHOS complex subunits COII and NDUFB8 were only observed in Hep3B cells. These findings strongly imply that c-Src impairs OXPHOS subunit expression, which can be alleviated when c-Src activity is suppressed.
As shown in Fig. 3C, siRNA mediated knock down of c-Src kinase was reduced c-Src expression and activity relative to the cells transfected with control siRNA in Hep3B cells. The expression of NDUFB8 and COII of complexes I and IV, respectively, were slightly increased by approximately 30% in cells transfected with c-Src siRNA (Fig. 3C). The expression of SDHB and UQCRC2, of complexes II and III, respectively, were also increased; however, the expression of ATP5A of complex V remained unchanged (Fig. 3C). Minor differences were also observed in the expression of TFAM and EF-Tu (data not shown), implying that the increase found in the expression of OXPHOS complexes may be a result of the suppression of c-Src. The increase in the expression of NDUFB8 and COII in Hep3B cells transfected with c-Src siRNA is similar to what was observed in PP2 treated Hep3B cells (Fig. 3A). These findings provide further evidence that c-Src inhibition stimulates OXPHOS expression and increases mitochondrial energy metabolism. Therefore, c-Src should be closely evaluated as a therapeutic target for carcinomas with elevated c-Src expression and activity.
3.4. c-Src expression impairs the expression of OXPHOS complexes
Above, we clearly showed increased c-Src expression was associated with impaired OXPHOS subunit expression and complexes I and III and IV activities, in metastatic liver cancer tissues and cell lines. To further examine the functional significance of c-Src on mitochondrial energy metabolism, we investigated changes in the expression of OXPHOS complexes in more homogenous cellular systems. For this purpose, we used the mouse embryonic fibroblast (MEF) cell line (WT cells), MEF cells with functional null mutations in both alleles of c-Src, Yes, and Fyn (SYF cells), MEF cells with an endogenous expression of wild-type c-Src and null mutations in Yes and Fyn (Src++ cells), and SYF cells with a stable over expression of wild-type c-Src kinase (Src cells). Detection of c-Src expression by Western blot analyses in all four cell lines revealed varying expression of c-Src in WT, Src++ and Src cells (Fig. 4A) compared to its complete knock-down in SYF cells. The pSFK level was significantly higher in Src++ and Src cells compared to SYF cells with the highest phosphorylation in WT cells (Fig. 4A). The phosphorylation of Tyr416 in WT cells detected by pSFK antibody could be a result of the activation of c-Src as well as Yes, Fyn, and other SFKs expressed in WT cells. The levels of pSFK detection in WT, Src++, and Src cell lines can also be correlated to level of Tyr phosphorylation in these cell lines (Fig. S3) confirming both c-Src expression and activity in Src++, Src, and WT cells compared to SYF cells.
Figure 4.
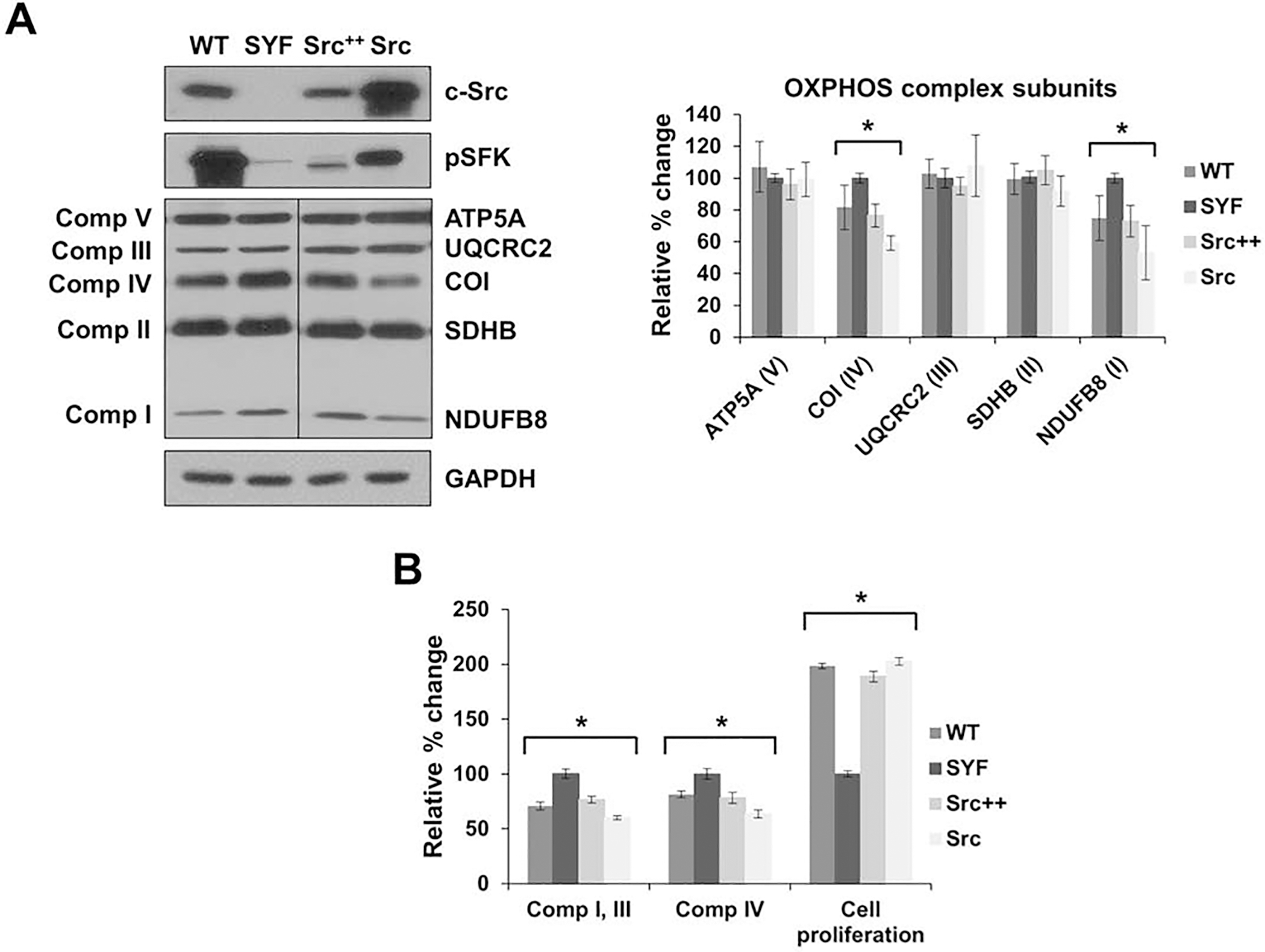
c-Src expression reduces OXPHOS expression in fibroblast cells. A) Mouse embryonic fibroblast (MEF) cells with functional null mutations in both alleles of three SFK members, c-Src, Yes, and Fyn (SYF), MEF cells with the endogenous expression of c-Src and null mutations in Yes and Fyn (Src++), SYF cells with the overexpression of c-Src (Src), and wild-type embryonic fibroblast cells (WT) were cultured. Expression of OXPHOS complex subunits, including NDUFB8 (complex I), SDHB (complex II), UQCRC2 (complex III), COI (complex IV), and ATP5A (complex V) were detected by Western blot analyses of WT, SYF, Src++, and Src cell lines. The expression of c-Src and pSFK were also detected with respect to GAPDH and ponceau S staining as a control for equal protein loading. Quantification of the average expression of OXPHOS complex subunits between SYF and WT, Src++, and Src cells is shown in the bar graph (right panel). Significant differences were observed in the expression of COI between SYF and WT (P = 0.0426), Src++ (P = 0.0017), and Src (P = 0.0001) and in the expression of NDUFB8 between SYF and WT (P = 0.0163), Src++ (P = 0.0033), and Src (P = 0.0016). B) WT, SYF, Src++, and Src cells were grown for 24 h and proliferation was measured by the Trypan blue exclusion assay (cell proliferation). Significant differences were observed in the cell proliferation between SYF and WT (P = 0.0007), Src++ (P = 0.0018), and Src (P = 0.0014). The complex I and III and complex IV enzymatic activities were determined by measuring the reduction and oxidation rate, respectively, of cytochrome c using equal amounts of whole cell lysates obtained from WT, SYF, Src++, and Src cells. Significant differences were observed in the complex I and III activity between SYF and WT (P = 0.0071), Src++ (P = 0.0001), and Src (P = 0.0002) and complex IV activity (P = 0.0001). Results are expressed as mean ± SD for at least three experiments and are presented as a percentage of SYF cells (SYF = 100%). Significant differences were observed between SYF, WT, Src++, and Src cells via unpaired student’s t-tests (2-tailed) to measure statistical significance, *P < 0.05. See Fig. 2 legend for details.
We also assessed the steady-state expression of OXPHOS subunits in these cell lines using a rodent OXPHOS antibody cocktail that detects subunits from nuclear-encoded complex I (NDUFB8), II (SDHB), III (UQCRC2), V (ATP5A), and the mitochondrial-encoded complex IV (COI) (Fig. 4A). Significant reductions were observed in the expression of the nuclear-encoded complex I (NDUFB8) and mitochondrial-encoded complex IV (COI) subunits with increasing c-Src expression in WT, Src++, and Src cells compared to the SYF cells (Fig. 4A). On the contrary, the expression complexes II, III, and V subunits were comparable among all the cell lines (Fig. 4A). These results strongly suggest that c-Src expression impairs the expression of complexes I and IV subunits in mouse embryonic fibroblast cells.
To determine the effect of reduced NDUFB8 and COI expressions, we performed mitochondrial complex I and III and complex IV activity assays in WT, SYF, Src++, and Src cells. The activity of complex I and III was significantly decreased with an increasing expression of c-Src found in WT, Src++, and Src cells compared to SYF cells (Fig. 4B). Similarly, the complex IV activity was also decreased in WT, Src++, and Src cells compared to the SYF cell line (Fig. 4B). Therefore, the impaired complex I and III and complex IV activities can be attributed to the reduced expression of the core subunits, NDUFB8 and COI, and associated with an increase in c-Src expression in these cells.
c-Src kinase has also shown to control cell proliferation, migration, and adhesion; consequently, we examined the cell proliferation of fibroblast cells with and without c-Src expression. In cells expressing c-Src, cell proliferation was significantly higher compared to SYF cells (Fig. 4B). Increased cell proliferation and c-Src activity are correlated with high glycolytic metabolism by inhibiting PDH activity and OXPHOS, as previously described [18]. Therefore, the increased c-Src expression could also cause inhibition of PDH activity, resulting in diminished OXPHOS as observed in MEF cell lines (Fig. 4). These findings are in agreement with our Hep3B cell line data shown above and further imply that c-Src alters the enzymatic activity of OXPHOS complexes, possibly by changing the expression of their subunits. Together, our results obtained with liver biopsies and cell lines suggest that c-Src expression regulates mitochondrial function in both normal and cancerous cells.
3.5. c-Src inhibition with PP2 stimulates the expression of OXPHOS subunits
Our data provide strong evidence that c-Src expression is associated with impaired OXPHOS subunit expression and activity in normal and cancerous cells. To investigate if the inhibition of c-Src kinase activity could improve OXPHOS subunit expression and function, we first treated MEF cell lines expressing the highest c-Src kinase, Src cells, with PP2 for 24 – 48 h. As expected PP2 treatments decreased overall Tyr phosphorylation and the phosphorylation of c-Src at Tyr416 without influencing the c-Src expression (Fig. 5A). Interestingly, the steady-state expression of nuclear-encoded NDUFB8 and mitochondrial-encoded COI subunits of complexes I and IV, respectively, were significantly increased with PP2 treatments compared to the control cells, while minor changes were observed in the expression of the remainder of the OXPHOS subunits detected by the rodent antibody cocktail (Fig. 5A). Despite the higher expression in COI and NDUFB8, the expression of EF-Tu and TFAM (data not shown) were comparable, suggesting that the increase in the expression of OXPHOS subunits may be a result of the inhibition of c-Src kinase activity and the associated changes in Tyr phosphorylation.
Figure 5.
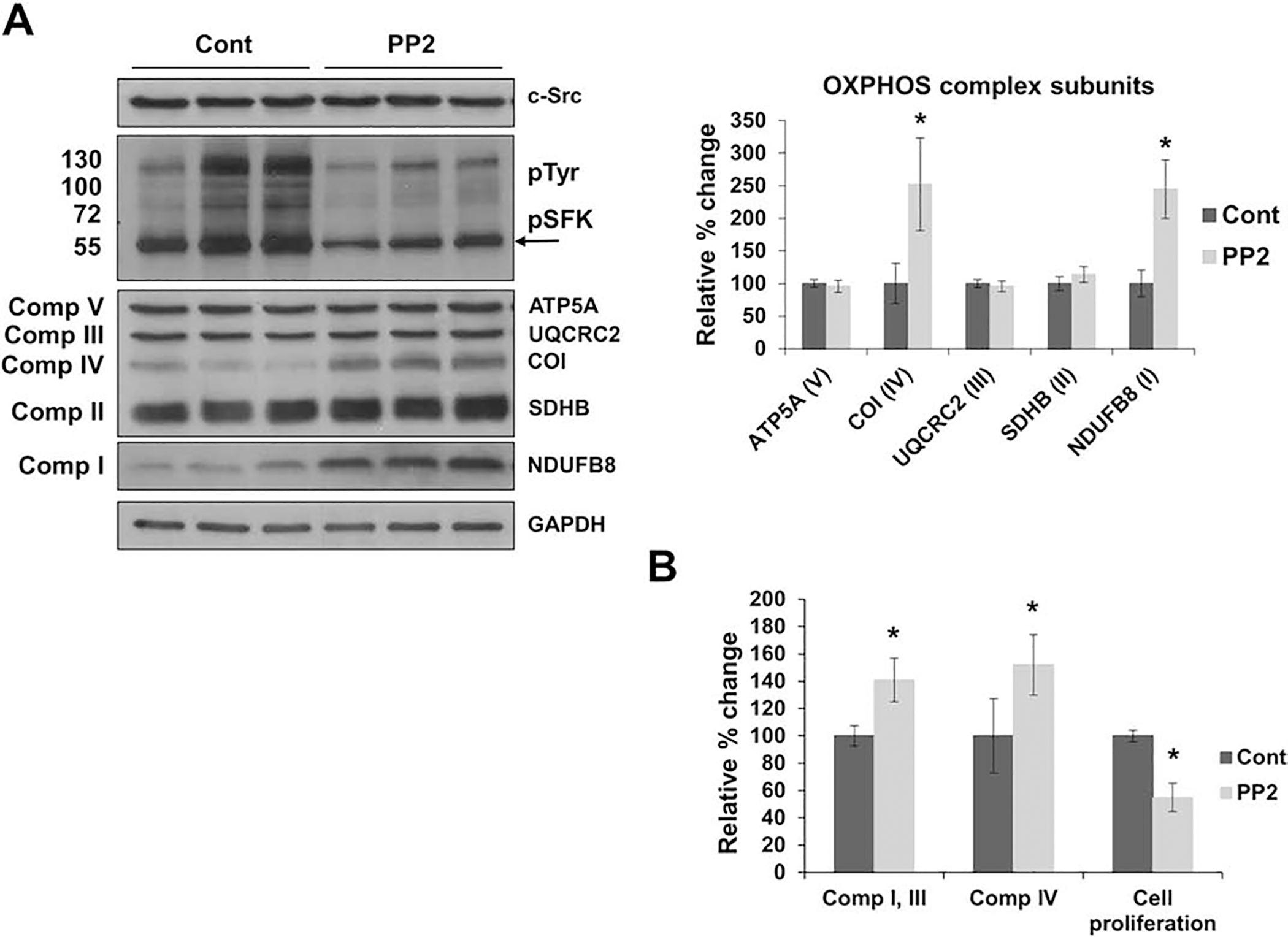
Inhibition of c-Src stimulates mitochondrial OXPHOS expression in Src cells. A) Src cells were treated with DMSO (cont) or 5 μM Src family kinase (SFK) inhibitor PP2 (PP2) for 24 h before collection. Changes in OXPHOS complex subunits, c-Src, and EF-Tu expression were measured by Western blot analyses with respect to GAPDH as a loading control. The phosphorylation of SFK members (pSFK), around 60 kDa, detected by the pTyr antibody, is indicated by an arrow. Quantitation of mitochondrial protein expression of OXPHOS complex subunits are represented as the mean ± SD of at least three experiments (right panel) and expressed as a percentage of the control cells (cont = 100%). Significant difference was observed between PP2 treated Src cells in mitochondrial-encoded COI and nuclear-encoded NDUFB8 subunits via an unpaired student’s t-test (2-tailed), *P-value < 0.05. B) Cell proliferation, complex I and III activity, and complex IV enzymatic activity of PP2 treated Src cells were measured after 24 h treatments. Significant differences were observed between the cont and PP2 treated Src cells in the cell proliferation (P = 0.0003), complex I and III activity (P = 0.0001), and complex IV activity (P = 0.0001). Results are presented as the mean ± SD of at least three experiments and expressed as a percentage of the control cells (cont = 100%). The statistical significance was analyzed by unpaired student’s t-tests (2-tailed), *P < 0.05. See Fig. 4 legend for details.
Subsequently, significant increases of roughly 40% and 50% were observed in the enzymatic activities of complexes I and III and complex IV in PP2 treated Src cells, respectively, which may be a result of the stimulated expression of the NDUFB8 and COI subunits (Fig. 5B). In fact, the OXPHOS subunit expression and activity found in PP2 treated Src cells was comparable to the subunit expression SYF cell lines (Figs. 4 and 5). The morphology of PP2-treated Src cells became similar to that of SYF cells (data not shown). Clearly, cell proliferation was also reduced by approximately 50% in PP2 treated Src cells compared to the control cells (Fig. 5B), further demonstrating the role of c-Src on cell proliferation and morphology.
In addition to the treatment of Src cells with PP2, we also treated the WT, Src++, and SYF cell lines with PP2. Similar to what was observed in Src cells, we detected an increase in the expression of NDUFB8 and COI of complexes I and IV, respectively, in PP2 treated Src++ cells (Fig. S4) and WT cells (data not shown). The increases in the expression of NDUFB8 and COI were correlated with a decrease in c-Src activity, determined by the changes in the pSFKs at Tyr416 and total Tyr phosphorylation. Additionally, increasing concentrations of PP2 were also correlated with significant decreases in cell proliferation in Src++ (Fig. S4) and WT (data not shown) cell lines. To ensure the increases in the expression of OXPHOS complexes I and IV were a result of reduced c-Src activity, we treated SYF cell lines with PP2 for 48 h and observed the effects on OXPHOS complexes. Although the cell proliferation was diminished in SYF cells treated with PP2, the OXPHOS subunit expressions were comparable between the control and PP2 treated SYF cells (Fig. S5). Combined, these results demonstrate the negative effects of c-Src on mitochondrial OXPHOS and energy metabolism, cell proliferation, and cell morphology which can be alleviated in the presence of an SFK inhibitor, such as PP2.
3.6. c-Src knock down increases OXPHOS subunit expression
Above, we demonstrated that the c-Src expression was correlated with a decrease in OXPHOS subunit expression and activity in cells treated with PP2. To further investigate c-Src inhibition, we transfected WT, Src++, and Src cell lines with mouse specific c-Src siRNA. The c-Src expression was significantly reduced in WT, Src++, and Src cell lines (Figs. 6A, 6B, and 6C, respectively) transfected with c-Src siRNA relative to cells treated with control siRNA. A significant increase was observed in the expression of NDUFB8 and COI of complexes I and IV, respectively, in all three cell lines transfected with c-Src siRNA (Fig. 6). The remainder of the OXPHOS subunits, were comparable between the cells transfected with the control and c-Src siRNAs. Clearly, the increases in OXPHOS subunit expression was a result of the c-Src knock down in these MEF cell lines expressing c-Src to varying degree. Therefore, our findings strongly suggest that c-Src regulates mitochondrial energy metabolism by inhibiting the expression and activity of OXPHOS complexes, specifically complexes I and IV. Furthermore, our results demonstrate that therapies targeting c-Src kinase alleviate the inhibition on mitochondrial oxidative phosphorylation and could be beneficial for the treatment of cancers with elevated c-Src expression and activity.
Figure 6.
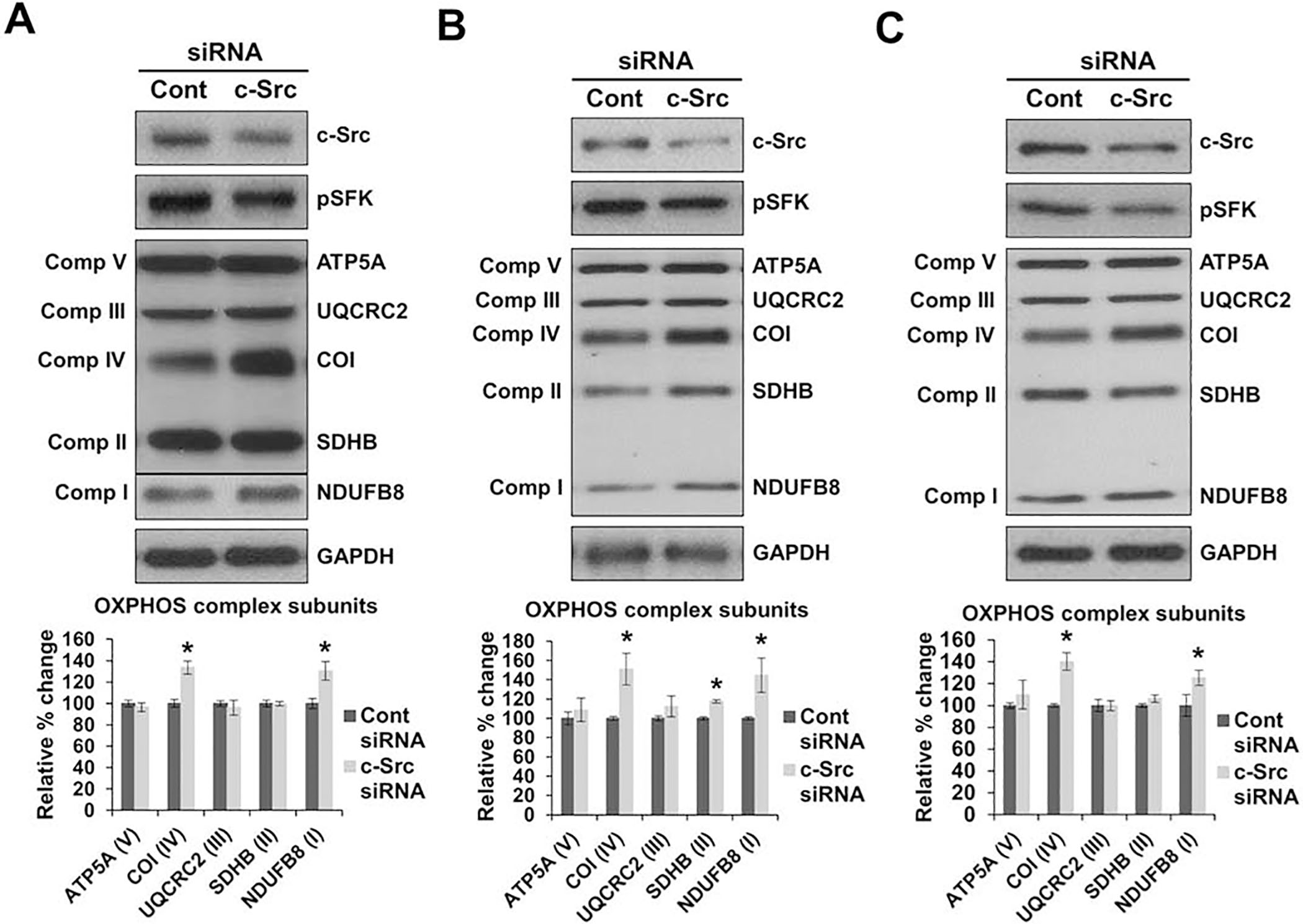
c-Src knock down increases the expression of OXPHOS complexes. A) WT, Src++ (B), and Src (C) cells were transfected with c-Src specific siRNA (c-Src) and control siRNA (cont). The expression of OXPHOS complex subunits were detected by Western blot analyses with respect to GAPDH antibody probing and Ponceau S membrane staining as loading controls. Quantitation of the expression of OXPHOS complex subunits are represented as the mean ± SD of at least three experiments (right panel) and expressed as a percentage of the control siRNA cells (cont = 100%). Significant difference was observed between c-Src and control siRNA transfected cells in the expression of NDUFB8 and COI in WT cells (P = 0.0001 and P = 0.0029, respectively), Src++ cells (P = 0.0020 and P = 0.0056, respectively), and Src cells (P = 0.0004 and P = 0.0356, respectively). Statistical differences were analyzed between the cont and c-Src siRNA transfected cells with the unpaired student’s t-tests (2-tailed), *P < 0.05.
4. Conclusions
Increased c-Src expression and activity are recognized as major underlying factors in the development of various diseases, including cancer, and is also suggested to contribute to increased metastatic, recurrence, and resistance rates in HCC [10, 11]. Although many systemic agents have been studied, to date, Sorafenib is the only approved drug for the systemic treatment of HCC, which has been shown to only extend the life of patients by 3–4 months [46–48]. Interestingly, c-Src is also located in the mitochondria and shown to alter mitochondrial energy metabolism via regulating OXPHOS complexes [23–26]. In some cases, c-Src has also been shown to induce the Warburg effect by inhibiting mitochondrial energy metabolism [18, 29]. Therefore, due to the various functions of c-Src in cell signaling, a better understanding of its role in mitochondrial energy metabolism in cancer and normal cells is essential.
In this study, we examined the effects of aberrant c-Src expression on mitochondrial energy metabolism by determining the relative changes in the expression of OXPHOS subunits in human liver cancer biopsies and cell lines. We clearly demonstrated that increased expression of c-Src was associated with an impaired expression of OXPHOS subunits in metastatic liver cancer biopsies and the Hep3B cell line. More importantly, we showed diminished OXPHOS subunit expression and activity in mouse embryonic fibroblast cells expressing c-Src kinase alone. These results demonstrated an inhibitory effect of c-Src on mitochondrial function in both health and disease.
Both c-Src kinase and mitochondrial energy metabolism have become important targets for cancer therapy. We investigated the effects of c-Src inhibition on mitochondrial energy metabolism by treating cells with the SFK inhibitor PP2 as well as transfecting cells with c-Src siRNA. A significant increase in the expression of complexes I and IV and their corresponding activities was shown in the metastatic Hep3B cell line in addition to c-Src expressing mouse fibroblast, WT, Src++, and Src, cell lines treated with PP2 and c-Src siRNA. Interestingly, the inhibition of c-Src was also shown to reduce cell proliferation in Hep3B and c-Src expressing MEF cells, suggesting that c-Src regulates cell proliferation by modulating OXPHOS. Our results provide a better understanding of the relationship between c-Src and OXPHOS expression in normal and cancerous cells. Additionally, our results indicate that c-Src inhibition should be utilized in anti-cancer therapies to reduce cell proliferation and improve mitochondrial function.
Supplementary Material
Figure S1. Metabolic pathways are altered between cancerous and non-cancerous liver tissues. The expression of peroxisomal acyl-coenzyme A oxidase (ACOX1), citrate synthase (CS), pyruvate dehydrogenase (PDH), heat shock protein 60 (HSP60), and voltage-dependent anion channel (VDAC)/porin were detected by Western blot analyses. See Fig. 1 legend for details.
{kind=link}
Figure S2. PP2 treatments of HepG2 cells reduce cell proliferation without altering OXPHOS expression. HepG2 cells were treated with 0 – 10 μM PP2 for 72 h before collection. Expression of OXPHOS complex subunits, including NDUF8 (complex I), SDHA and SDHB (complex II), UQCRC2 (complex III), COII (complex IV), and ATP5A (complex V) were detected by Western blot analyses. Approximately 30 μg of protein lysates from HepG2 cells treated with PP2 were separated by 12% SDS-PAGE and equal protein loading was assessed by Ponceau S staining and GAPDH antibody probing of the membranes.
{kind=link}
Figure S3. Increased tyrosine phosphorylation is correlated with increased c-Src. The overall tyrosine phosphorylation and the expression of citrate synthase (CS) and pyruvate dehydrogenase (PDH) were detected by Western blot analyses in WT, SYF, Src++, and Src cells. See Fig. 4 legend for details.
{kind=link}
Figure S4. Inhibition of c-Src with PP2 stimulates OXPHOS expression in Src++ cells. A) Src++ cells were treated with 0 – 5 μM PP2 for 24 h before collection. The expression of OXPHOS complex subunits were detected by Western blot analyses with respect to GAPDH as a loading control. The quantitation of the protein expression of OXPHOS subunits are represented as the mean ± SD of at least three experiments (right panel) and are expressed as a percentage of the control cells (cont = 100%). Significant difference was observed between PP2 treated Src++ cells in mitochondrial-encoded COI (P = 0.0084) and nuclear-encoded NDUFB8 (P = 0.0022) subunits. B) Cell proliferation was measured between the control and PP2 treated Src++ cells and significant difference was observed (P = 0.0003). Results are presented as the mean ± SD of at least three experiments and expressed as a percentage of the control cells (cont = 100%). The statistical significance was analyzed by unpaired student’s t-tests (2-tailed), *P < 0.05. See Fig. 5 legend for details.
{kind=link}
Figure S5. PP2 treatments of SYF cells reduce cell proliferation without changing the expression of OXPHOS complexes. SYF cells were treated with 0 – 10 μM PP2 for 48 h before collection. Expression of OXPHOS complex subunits were detected by Western blot analyses with respect to Ponceau S staining of the membrane. See Fig. 5 legend for details.
{kind=link}
Highlights.
Mitochondrial oxidative phosphorylation is impaired in metastatic liver cancer
Expression and activity of complexes I and IV are reduced in liver cancer
c-Src kinase is associated with decreased oxidative phosphorylation
c-Src inhibition stimulates oxidative phosphorylation expression and activity
Oxidative phosphorylation and energy metabolism are regulated by c-Src kinase
Acknowledgements
The authors would like to thank Tissue Procurement Center at the Pennsylvania State University Health Milton S. Hershey Medical Center for providing liver biopsies. We also gratefully acknowledge Biomedical Sciences Department at Joan C. Edwards School of Medicine, Marshall University for its support.
Funding
This work was partly supported by National Institutes of Health [GM071034 to ECK] and NASA WV Space Grant Consortium issued by NASA Goddard Space Flight Center [NNG05GF80H].
Footnotes
Conflict of interest
The authors declare they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Parsons SJ and Parsons JT, Src family kinases, key regulators of signal transduction. Oncogene, 2004. 23(48): p. 7906–9. [DOI] [PubMed] [Google Scholar]
- 2.Thomas SM and Brugge JS, Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol, 1997. 13: p. 513–609. [DOI] [PubMed] [Google Scholar]
- 3.Blume-Jensen P and Hunter T, Oncogenic kinase signalling. Nature, 2001. 411(6835): p. 355–65. [DOI] [PubMed] [Google Scholar]
- 4.Irby RB and Yeatman TJ, Increased Src activity disrupts cadherin/catenin-mediated homotypic adhesion in human colon cancer and transformed rodent cells. Cancer Res, 2002. 62(9): p. 2669–74. [PubMed] [Google Scholar]
- 5.Kim MP, et al. , Src family kinases as mediators of endothelial permeability: effects on inflammation and metastasis. Cell Tissue Res, 2009. 335(1): p. 249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Irby RB and Yeatman TJ, Role of Src expression and activation in human cancer. Oncogene, 2000. 19(49): p. 5636–42. [DOI] [PubMed] [Google Scholar]
- 7.Rosen N, et al. , Analysis of pp60c-src protein kinase activity in human tumor cell lines and tissues. J Biol Chem, 1986. 261(29): p. 13754–9. [PubMed] [Google Scholar]
- 8.Yeatman TJ, A renaissance for SRC. Nat Rev Cancer, 2004. 4(6): p. 470–80. [DOI] [PubMed] [Google Scholar]
- 9.Djeungoue-Petga MA, et al. , Intramitochondrial Src kinase links mitochondrial dysfunctions and aggressiveness of breast cancer cells. Cell Death Dis, 2019. 10(12): p. 940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masaki T, et al. , pp60c-src activation in hepatocellular carcinoma of humans and LEC rats. Hepatology, 1998. 27(5): p. 1257–64. [DOI] [PubMed] [Google Scholar]
- 11.Zhao R, et al. , Elevated Src expression associated with hepatocellular carcinoma metastasis in northern Chinese patients. Oncol Lett, 2015. 10(5): p. 3026–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koc EC, Miller-Lee JL, and Koc H, Fyn kinase regulates translation in mammalian mitochondria. Biochim Biophys Acta, 2016. 1861(3): p. 533–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas SM, Soriano P, and Imamoto A, Specific and redundant roles of Src and Fyn in organizing the cytoskeleton. Nature, 1995. 376(6537): p. 267–71. [DOI] [PubMed] [Google Scholar]
- 14.Salvi M, et al. , Characterization and location of Src-dependent tyrosine phosphorylation in rat brain mitochondria. Biochim Biophys Acta, 2002. 1589(2): p. 181–95. [DOI] [PubMed] [Google Scholar]
- 15.Tibaldi E, et al. , Src-Tyrosine kinases are major agents in mitochondrial tyrosine phosphorylation. J Cell Biochem, 2008. 104(3): p. 840–9. [DOI] [PubMed] [Google Scholar]
- 16.Vahedi S, et al. , Lymphocyte-specific protein tyrosine kinase (Lck) interacts with CR6-interacting factor 1 (CRIF1) in mitochondria to repress oxidative phosphorylation. BMC Cancer, 2015. 15: p. 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hebert-Chatelain E, Src kinases are important regulators of mitochondrial functions. Int J Biochem Cell Biol, 2013. 45(1): p. 90–8. [DOI] [PubMed] [Google Scholar]
- 18.Jin Y, et al. , Src drives the Warburg effect and therapy resistance by inactivating pyruvate dehydrogenase through tyrosine-289 phosphorylation. Oncotarget, 2016. 7(18): p. 25113–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewandrowski U, et al. , Identification of new tyrosine phosphorylated proteins in rat brain mitochondria. FEBS Lett, 2008. 582(7): p. 1104–10. [DOI] [PubMed] [Google Scholar]
- 20.Salvi M, et al. , Identification of the flavoprotein of succinate dehydrogenase and aconitase as in vitro mitochondrial substrates of Fgr tyrosine kinase. FEBS Lett, 2007. 581(29): p. 5579–85. [DOI] [PubMed] [Google Scholar]
- 21.Lopez J, et al. , Src tyrosine kinase inhibits apoptosis through the Erk1/2- dependent degradation of the death accelerator Bik. Cell Death Differ, 2012. 19(9): p. 1459–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Augereau O, et al. , Identification of tyrosine-phosphorylated proteins of the mitochondrial oxidative phosphorylation machinery. Cell Mol Life Sci, 2005. 62(13): p. 1478–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demory ML, et al. , Epidermal growth factor receptor translocation to the mitochondria: regulation and effect. J Biol Chem, 2009. 284(52): p. 36592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hebert-Chatelain E, et al. , Preservation of NADH ubiquinone-oxidoreductase activity by Src kinase-mediated phosphorylation of NDUFB10. Biochim Biophys Acta, 2012. 1817(5): p. 718–25. [DOI] [PubMed] [Google Scholar]
- 25.Miyazaki T, et al. , Regulation of cytochrome c oxidase activity by c-Src in osteoclasts. J Cell Biol, 2003. 160(5): p. 709–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogura M, et al. , Mitochondrial c-Src regulates cell survival through phosphorylation of respiratory chain components. Biochem J, 2012. 447(2): p. 281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Acin-Perez R, et al. , ROS-triggered phosphorylation of complex II by Fgr kinase regulates cellular adaptation to fuel use. Cell Metab, 2014. 19(6): p. 1020–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wallace DC, Mitochondria and cancer: Warburg addressed. Cold Spring Harb Symp Quant Biol, 2005. 70: p. 363–74. [DOI] [PubMed] [Google Scholar]
- 29.Wallace DC, Mitochondria and cancer. Nat Rev Cancer, 2012. 12(10): p. 685–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hebert Chatelain E, et al. , Functional impact of PTP1B-mediated Src regulation on oxidative phosphorylation in rat brain mitochondria. Cell Mol Life Sci, 2011. 68(15): p. 2603–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishikawa Y, et al. , Suppressive effect of orthovanadate on hepatic stellate cell activation and liver fibrosis in rats. Am J Pathol, 2009. 174(3): p. 881–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanke JH, et al. , Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem, 1996. 271(2): p. 695–701. [DOI] [PubMed] [Google Scholar]
- 33.Schneider CA, Rasband WS, and Eliceiri KW, NIH Image to ImageJ: 25 years of image analysis. Nat Methods, 2012. 9(7): p. 671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Birch-Machin MA and Turnbull DM, Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol, 2001. 65: p. 97–117. [DOI] [PubMed] [Google Scholar]
- 35.Hunter T, A tail of two src’s: mutatis mutandis. Cell, 1987. 49(1): p. 1–4. [DOI] [PubMed] [Google Scholar]
- 36.Lopez-Terrada D, et al. , Hep G2 is a hepatoblastoma-derived cell line. Hum Pathol, 2009. 40(10): p. 1512–5. [DOI] [PubMed] [Google Scholar]
- 37.Qiu GH, et al. , Distinctive pharmacological differences between liver cancer cell lines HepG2 and Hep3B. Cytotechnology, 2015. 67(1): p. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Slany A, et al. , Cell characterization by proteome profiling applied to primary hepatocytes and hepatocyte cell lines Hep-G2 and Hep-3B. J Proteome Res, 2010. 9(1): p. 6–21. [DOI] [PubMed] [Google Scholar]
- 39.Arachiche A, et al. , Localization of PTP-1B, SHP-2, and Src exclusively in rat brain mitochondria and functional consequences. J Biol Chem, 2008. 283(36): p. 24406–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boerner JL, et al. , Phosphorylation of Y845 on the epidermal growth factor receptor mediates binding to the mitochondrial protein cytochrome c oxidase subunit II. Mol Cell Biol, 2004. 24(16): p. 7059–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jin L, et al. , Phosphorylation-mediated activation of LDHA promotes cancer cell invasion and tumour metastasis. Oncogene, 2017. 36(27): p. 3797–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, et al. , c-Src phosphorylation and activation of hexokinase promotes tumorigenesis and metastasis. Nat Commun, 2017. 8: p. 13732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gaude E and Frezza C, Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat Commun, 2016. 7: p. 13041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Porporato PE, et al. , A mitochondrial switch promotes tumor metastasis. Cell Rep, 2014. 8(3): p. 754–66. [DOI] [PubMed] [Google Scholar]
- 45.Zhang S, et al. , Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med, 2011. 17(4): p. 461–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Daher S, et al. , Current and Future Treatment of Hepatocellular Carcinoma: An Updated Comprehensive Review. J Clin Transl Hepatol, 2018. 6(1): p. 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Llovet JM, et al. , Sorafenib in advanced hepatocellular carcinoma. N Engl J Med, 2008. 359(4): p. 378–90. [DOI] [PubMed] [Google Scholar]
- 48.Wilhelm SM, et al. , Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther, 2008. 7(10): p. 3129–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Metabolic pathways are altered between cancerous and non-cancerous liver tissues. The expression of peroxisomal acyl-coenzyme A oxidase (ACOX1), citrate synthase (CS), pyruvate dehydrogenase (PDH), heat shock protein 60 (HSP60), and voltage-dependent anion channel (VDAC)/porin were detected by Western blot analyses. See Fig. 1 legend for details.
Figure S2. PP2 treatments of HepG2 cells reduce cell proliferation without altering OXPHOS expression. HepG2 cells were treated with 0 – 10 μM PP2 for 72 h before collection. Expression of OXPHOS complex subunits, including NDUF8 (complex I), SDHA and SDHB (complex II), UQCRC2 (complex III), COII (complex IV), and ATP5A (complex V) were detected by Western blot analyses. Approximately 30 μg of protein lysates from HepG2 cells treated with PP2 were separated by 12% SDS-PAGE and equal protein loading was assessed by Ponceau S staining and GAPDH antibody probing of the membranes.
Figure S3. Increased tyrosine phosphorylation is correlated with increased c-Src. The overall tyrosine phosphorylation and the expression of citrate synthase (CS) and pyruvate dehydrogenase (PDH) were detected by Western blot analyses in WT, SYF, Src++, and Src cells. See Fig. 4 legend for details.
Figure S4. Inhibition of c-Src with PP2 stimulates OXPHOS expression in Src++ cells. A) Src++ cells were treated with 0 – 5 μM PP2 for 24 h before collection. The expression of OXPHOS complex subunits were detected by Western blot analyses with respect to GAPDH as a loading control. The quantitation of the protein expression of OXPHOS subunits are represented as the mean ± SD of at least three experiments (right panel) and are expressed as a percentage of the control cells (cont = 100%). Significant difference was observed between PP2 treated Src++ cells in mitochondrial-encoded COI (P = 0.0084) and nuclear-encoded NDUFB8 (P = 0.0022) subunits. B) Cell proliferation was measured between the control and PP2 treated Src++ cells and significant difference was observed (P = 0.0003). Results are presented as the mean ± SD of at least three experiments and expressed as a percentage of the control cells (cont = 100%). The statistical significance was analyzed by unpaired student’s t-tests (2-tailed), *P < 0.05. See Fig. 5 legend for details.
Figure S5. PP2 treatments of SYF cells reduce cell proliferation without changing the expression of OXPHOS complexes. SYF cells were treated with 0 – 10 μM PP2 for 48 h before collection. Expression of OXPHOS complex subunits were detected by Western blot analyses with respect to Ponceau S staining of the membrane. See Fig. 5 legend for details.