

Abstract
Background
Precision medicine incorporating genetic profiling is becoming a standard of care in medical oncology. However, in the field of radiation oncology there is limited use of genetic profiling and the impact of germline genetic biomarkers on radiosensitivity, radioresistance, or patient outcomes after radiation therapy is poorly understood. In HNSCC, the toxicity associated with treatment can cause delays or early cessation which has been associated with worse outcomes. Identifying potential biomarkers which can help predict toxicity, as well as response to treatment, is of significant interest.
Methods
Patients with HNSCC who received RT and underwent next generation sequencing of somatic tumor samples, transcriptome RNA-seq with matched normal tissue samples were included. Patients were then grouped by propensity towards increased late vs. early toxicity (Group A) and those without (Group B), assessed by CTCAE v5.0. The groups were then analyzed for association of specific germline variants with toxicity and clinical outcomes.
Results
In this study we analyzed 37 patients for correlation between germline variants and toxicity. We observed that TSC2, HLA-A, TET2, GEN1, NCOR2 and other germline variants were significantly associated with long term toxicities. 34 HNSCC patients treated with curative intent were evaluated for clinical outcomes. Group A had significantly improved overall survival as well as improved rates of locoregional recurrence and metastatic disease. Specific variants associated with improved clinical outcomes included TSC2, FANCD2, and PPP1R15A, while the HLA-A and GEN1 variants were not correlated with survival or recurrence. A group of five HLA-DMA/HLA-DMB variants was only found in Group B and was associated with a higher risk of locoregional recurrence.
Conclusions
This study indicates that germline genetic biomarkers may have utility in predicting toxicity and outcomes after radiation therapy and deserve further investigation in precision radiation medicine approaches.
Keywords: Predictive biomarkers of radiation toxicity, Head and neck squamous cell carcinoma, Radiogenomics, Germline variants, TSC2, HLA-A, TET2, GEN1, NCOR2
Background
Radiation therapy (RT) contributes to 40% of all cancer cures world-wide and improves the quality of life for many others [1]. Some patients tolerate treatments extremely well, but others experience severe RT adverse events (rtAEs) that can have lasting and debilitating characteristics and negatively impact patient quality of life. In the clinic, radiation doses for patients are currently prescribed in a “one-size-fits-all” approach and are independent of their genetics profile. Personalizing RT for each patient, based on a radiosensitivity profile determined from their individual genetics and the genetic characteristics of their cancer, could significantly improve cancer patients’ quality of life in the long term.
In this work we chose to study head and neck squamous cell carcinoma (HNSCC) as a model cancer type in which to identify genetic biomarkers that could be used to personalize a course of radiotherapy to improve patient outcomes and quality of life. In HNSCC, frequent rtAEs include dysphagia, xerostomia, and cutaneous fibrosis [2], which are debilitating to the patients who otherwise benefit from curative RT of their tumor. Significant technological advancements including intensity modulated RT (IMRT) have allowed for meaningful reductions in dose to uninvolved organs at risk (OAR) [3–5]. Despite these innovations, HNSCC RT toxicity continues to have a significant impact on patient recovery and quality of life, often resulting in delays or premature termination of treatment, which are both associated with higher rates of local recurrence [6–8]. Specifically, missing two or more treatments has been associated with increased recurrence risk and inferior overall survival (OS), with the decrement to OS estimated at 1% per 1 missed day [9]. Several reports including surveillance, epidemiology, and end results (SEER) analysis of more than 300,000 head and neck cancer (HNC) patients, have shown excessive rates of suicide in survivors of HNSCC, second only to survivors of pancreatic cancer [10, 11].
There is substantial literature implicating patient germline variants as factors in influencing patient-specific radiosensitivity. For example, deoxyribonucleic acid (DNA) repair genes, such as ATM serine/threonine kinase (ATM) germline variants, are known to have significant effects on radiosensitivity [12–14]. Other clinical conditions such as Nijmegen breakage syndrome, Fanconi anemia, retinoblastoma and Riddle syndrome are characterized for genetic variants that contribute to cellular and clinical radiosensitivity [15–21]. Similar associations of specific germline variants and toxicity outcomes have been described in prostate and non-small cell lung cancer (NSCLC) [22]. In a recent study an association with mucositis RT induced toxicity in HNSCC and a specific locus on chromosome 5 was reported [23]. Here, we proceed to analyze the association of germline variants with HNSCC rtAEs.
As a benchmark for assessment, in this exploratory study patients were evaluated based on their overall toxicity profiles as well as by assessing increase in late versus early toxicity symptoms. Here, we report the germline variants associated with RT toxicity in HNSCC.
Methods
Data source and patient inclusion criteria
This retrospective analysis was approved by our institutional review board (UCSD HRPP#200495). Thirty-seven HNSCC patients who underwent Tempus xT somatic tumor testing paired with normal matched specimens and received RT with available dosimetric data were selected for this study. All selected patients in our study cohort (n = 37) were treated at the Moores Cancer Center at the University of California San Diego between 2009 and 2021.
The CAP/CLIA validated Tempus xT test is ordered by a clinician to provide predictive, prognostic, and therapeutic management for patients. Patients were consented for the test in accordance with federal, state, University, and UCSD Human Research Protection Program policies. Normal matched specimen sources accepted for testing include blood or saliva, collected at time of ordering the test.
Patient demographics and treatment variables
We acquired patient characteristics and radiation data for these 37 patients including age at diagnosis, gender, smoking history, and human papillomavirus (HPV) status. Treatment parameters including pre-RT surgical resection, radiation dose, and induction/concurrent systemic therapy were recorded as well. Staging information was collected according to the American Joint Committee on Cancer (AJCC) classification edition in effect at the time of diagnosis, ranging from 6 to 8th edition [24].
Collection of toxicity data
Patient charts were utilized to report early and late rtAE endpoints for mucositis, dysphagia and xerostomia. Toxicities were recorded using Common Terminology Criteria for Adverse Events (CTCAE) v.5.0, which were scored and reported by the treating physician on the day of service during therapy and in follow up. Early toxicity endpoints were recorded as the highest CTCAE grade experienced during therapy or within 6 weeks of completing therapy. Late toxicity endpoints were recorded as the highest CTCAE grade experienced from 6 months post-RT to the time of most recent follow up.
Statistical analysis
The relationship between categorical variables and each outcome was analyzed with a chi-square test where p-values ≤ 0.05 were significant. Differences in patient characteristics were compared using Fisher’s exact test or chi-square as appropriate. The effect size was calculated as the ratio of variant to wild type samples in group A divided by the ratio of variant to wild type samples in group B: (Mut A/WT A)/(Mut B/WT B). Differences in OS, locoregional recurrence (LRR), locoregional failure free survival (LFS), progression free survival (PFS), and metastasis free survival (MFS) were compared between Group A and Group B as well as Group C and group D by Kaplan–Meier (KM) survival analysis with log-rank testing for significance. Analysis was performed using SPSS V22.0 [25].
RNA-seq analysis
Ribonucleic acid (RNA) sequencing (RNA-seq) FastQ files were processed for alignment and quality control. The reads were trimmed, and low-quality reads were removed using Trimgalore v 0.6.3_dev [26] with the “paired” parameter and length of 76 bps. Trimmed FastQ sequences were aligned to the human reference genome (GRCh38) using STAR aligner v2.7.1a. Bam files were sorted by coordinate by using option “–outSAMtype BAM SortedByCoordinate”. Alignment quality control (QC) and read mapping statistics were obtained from Picard v2.20.3 tools using function “CollectMultipleMetrics” [27]. FastQC v0.11.8 was used to perform QC checks on the raw sequencing data.
SNP genotype validation
Variant genotypes per patient were first validated in DNA sequencing samples using IGV [28]. Each patient’s total depth and allele frequency were recorded and an empirical assessment of genotype was made. If 100% of reads supported an alternative variant in germline DNA sequencing then the patient was deemed homozygous for that particular variant. If < 100% and > 0% of germline reads supported the variant, the patient was deemed heterozygous. Otherwise, the patient was deemed homozygous for the reference allele. This process overrode variant caller germline categorizations where disagreement was present.
RNA-Seq expression measurement
Each genotype was annotated with total RNA read depth and RNA variant allelic fraction (VAF). Bam-readcount [29] was used to measure the depth and VAF of alternate alleles at the locus of each respective variant for each patient.
Population minor allele frequencies
GnomAD [30] was queried to determine the population minor allele frequency (MAF) of variants of interest. Population MAF was then broken into major ethnic groups as follows: European non-Finnish, African/African American, Latino/Admixed American, Ashkenazi Jewish, South Asian, and East Asian.
Protein structure modeling
The variant protein sequence was deposited in the Baker lab folding algorithm [31]. The deduced model was presented by ChimeraX [32].
Results
NGS sequencing and patient study cohort characteristics
For optimal assessment of normal tissue function associated with RT toxicity, we build a next generation sequencing (NGS) database that included somatic tumor and paired normal tissue [33, 34]. NGS was performed using the same sequencing platform for all samples to maximize consistent data output for discovery of germline variants implicated in RT toxicity. Our dataset included 37 HNSCC patients (n = 37) with NGS data; 34 of which were HNSCC patients treated with curative intent and analyzed for clinical outcomes. Their characteristics are summarized in Table 1. Excluded patients include one with metastatic disease at diagnosis and two with locoregionally advanced cutaneous SCC. Metastatic disease was excluded due to inability to evaluate time to certain outcomes, such as metastasis free survival. Patients with cutaneous SCC were excluded due to differences in treatment paradigm, disease course and prognosis. While these patients were not evaluated for clinical outcomes they were retained for NGS analyses to assess for potential biomarkers associated with differing radiation AEs.
Table 1.
Patient characteristics
| Characteristics | Group A n = 16 |
Group B n = 18 |
Total n = 34a |
|---|---|---|---|
| n (%) | n (%) | n (%) | |
| Age at diagnosis | |||
| < 65 | 10 (62.5) | 8 (44.4) | 18 (52.9) |
| ≥ 65 | 6 (37.5) | 10 (55.6) | 16 (47.1) |
| Gender | |||
| Male | 15 (93.8) | 16 (88.9) | 31 (91.2) |
| Female | 1 (6.2) | 2 (11.1) | 3 (8.8) |
| Smoking | |||
| ≤ 10 pack years | 11 (68.8) | 14 (77.8) | 25 (73.5) |
| > 10 pack years | 5 (31.2) | 4 (22.2) | 9 (26.5) |
| T stage | |||
| 0 | 1 (6.2) | 0 (0.0) | 1 (2.9) |
| 1 | 3 (18.8) | 3 (16.7) | 6 (17.6) |
| 2 | 5 (31.2) | 5 (27.8) | 10 (29.4) |
| 3 | 4 (25.0) | 3 (16.7) | 7 (20.6) |
| 4 | 3 (18.8) | 7 (38.9) | 10 (29.4) |
| N stage | |||
| 0 | 4 (25.0) | 3 (16.7) | 7 (20.6) |
| 1 | 4 (25.0) | 3 (16.7) | 7 (20.6) |
| 2 | 8 (50.0) | 6 (33.3) | 14 (41.2) |
| 3 | 0 (0.0) | 6 (33.3) | 6 (17.6) |
| Overall stage | |||
| I | 3 (18.8) | 2 (11.1) | 5 (14.7) |
| II | 1 (6.2) | 2 (11.1) | 3 (8.8) |
| III | 2 (12.5) | 2 (11.1) | 4 (11.8) |
| IVa | 8 (50.0) | 6 (33.3) | 14 (41.2) |
| IVb | 2 (12.5) | 6 (33.3) | 8 (23.5) |
| p16 status | |||
| Positive | 5 (31.2) | 8 (44.4) | 13 (38.2) |
| Negative | 7 (43.8) | 9 (50.0) | 16 (47.1) |
| Unknown | 4 (25.0) | 1 (5.6) | 5 (14.7) |
| Primary site | |||
| Oral cavity | 4 (25.0) | 6 (33.3) | 10 (29.4) |
| Oropharynx | 8 (50.0) | 7 (38.9) | 15 (44.1) |
| Larynx | 2 (12.5) | 3 (16.7) | 5 (14.7) |
| Hypopharynx | 0 (0.0) | 1 (5.6) | 1 (2.9) |
| Nasopharynx | 1 (6.2) | 0 (0.0) | 1 (2.9) |
| Nasal cavity | 0 (0.0) | 1 (5.6) | 1 (2.9) |
| Unknown | 1 (6.2) | 0 (0.0) | 1 (2.9) |
| Primary treatment | |||
| Definitive | 12 (75.0) | 8 (44.4) | 20 |
| Post-operative | 4 (25.0) | 10 (55.6) | 14 |
| Concurrent chemotherapy | |||
| Yes | 13 (81.2) | 14 (77.8) | 27 (79.4) |
| No | 3 (18.8) | 4 (22.2) | 7 (20.6) |
a3 patients were excluded from clinical analysis (one was treated palliatively and was metastatic at diagnosis, two had cutaneous SCC
RT patient germline variants associated with increased late toxicity
There are previously reported findings in HNSCC pointing to differential tissue response in acute and late toxicity, where acute toxicity is associated with inflammation while necrotic developments may drive late toxicity outcomes [35]. To isolate contributing genetic factors in early and late responses, the patient cohort was divided into two groups (Group A and Group B) based on their toxicity profile. Specifically, for this grouping, patients in Group A had significant increase in late toxicity while patients in Group B did not experience a late stage change in toxicity. Based on these criteria 18 patients were designated as Group A and 19 patients were designated as Group B.
We tested for germline variant association with RT toxicity outcomes, restricting to germline variants that were present in at least 25% MAF with significant p-value ≤ 0.05 between Groups A and B. Among the top 30 germline variants for Groups A and B, we identified 5 that met our selection criteria: human leukocyte antigen (HLA-A), tuberous sclerosis complex 2 (TSC2), marker of proliferation Ki-67 (MKI67), interferon induced protein with tetratricopeptide repeats 2 (IFIT2), and interleukin 10 receptor subunit alpha (IL10RA).
HLA-A Arg68Lys/Val91Met germline variant
We found 13 patients representing 37.1% of our cohort that carried the HLA-A Arg68Lys/Val91Met variant with 10 patients in Group A and 3 in Group B (p-value = 0.052, Table 2).
Table 2.
Chi-square results: recurrent freebayes mutations expanded beyond tumor suppressors and oncogenes

Patients in Group A experienced a higher CTCAE grade for late dysphagia compared to their early dysphagia. Patients in Group B experienced the same or lower CTCAE grade for early and late dysphagia. The effect value > 1 is the odds ratio for increased proportion of germline variants in Group A
All patients with the HLA-A Val91Met variant (rs79361534) also carried the HLA-A Arg68Lys variant (rs707910). This particular variant is documented in the HLA-A polymorphism database with a low MAF in a population of 1000 [36]. We found new characteristics of this HLA-A variant that have not been reported previously. This particular variant co-caries two isolated single nucleotide changes resulting in two amino acid substitutions at protein location 68 and 91. This has not been reported before since the two changes are listed as occurring separately and resulting in two HLA-A polymorphism genes (rs79361534 and rs707910). To examine the impact of these changes on the structure and function of the encoded protein, we performed a 3D modeling analysis (Fig. 1). We found that the valine substituted by methionine at position 91 is situated in the protein peptide binding groove consisting of two α-helixes and thus it is reasonable to conjecture that this amino acid substitution could be a factor in modified peptide binding affinity resulting in neo-antigen presentation. The Arginine substitution with Lysine at position 68 may introduce a new ubiquitin binding site, in addition, this variant was predicted as deleterious by using the Sorting Intolerant from Tolerant (SIFT) algorithm [37]. This HLA variant was not significantly associated with differences in PFS (p = 0.402), LRR (p = 0.173), MFS (p = 0.769) or OS (p = 0.757, Table 3). The effect size for this variant was high (6.67) signifying higher odds of late RT toxicity and potentially serving as a biomarker for RT stratification.
Fig. 1.
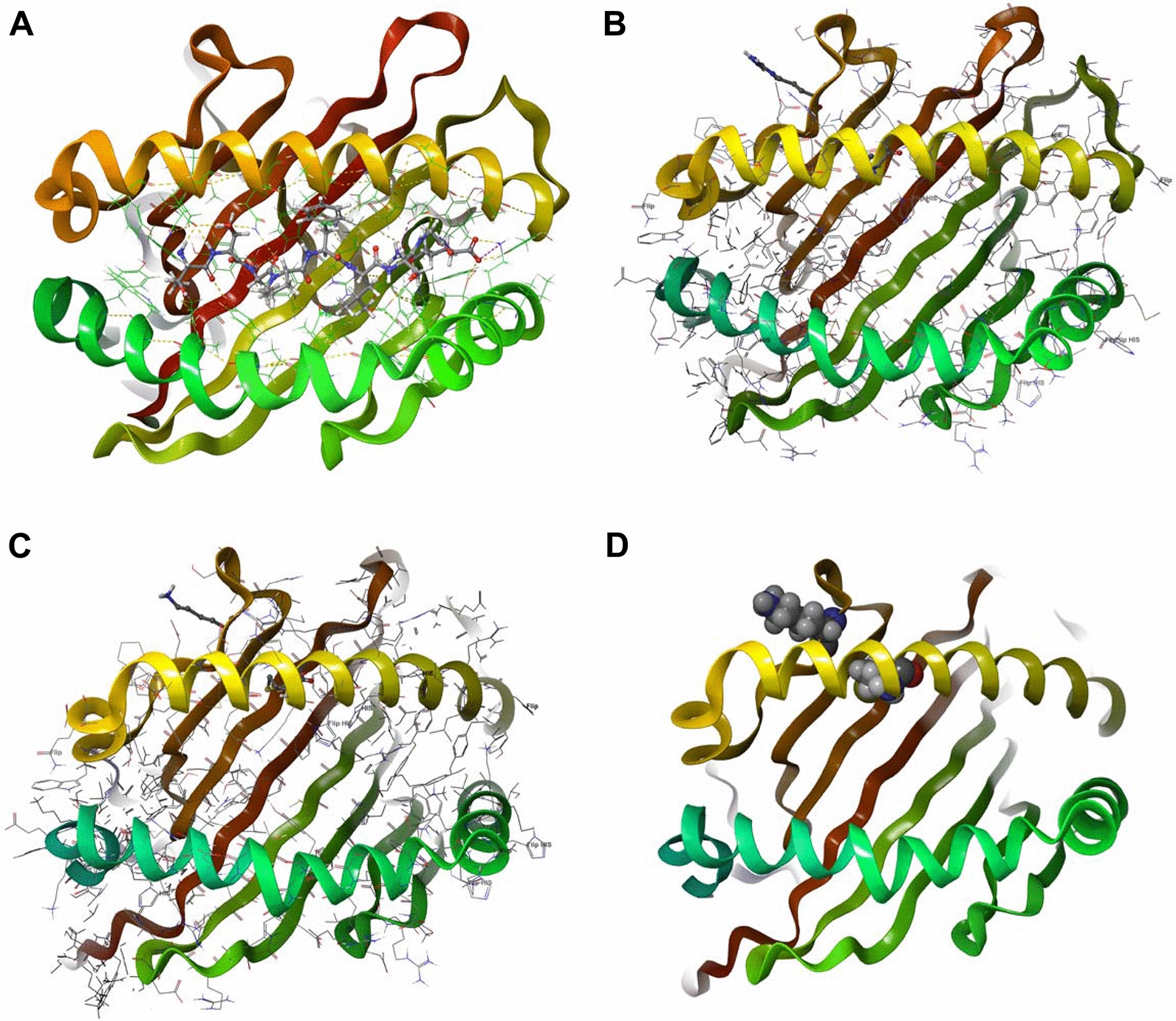
Structural HLA. A 3D structure of human HLA-A (609C) decorated by an epitope. B HLA-A wild type version (arginine to valine substitution). C HLA-A double mutant version (lysine to methionine substitution). D HLA-A double mutant version (lysine methionine) CPK presentation
Table 3.
P-values for PFS, LRR, MFS, and OS for patients with these variants vs. without
| Variant | PFS | LRR | MFS | OS |
|---|---|---|---|---|
| TSC2 | 0.216 | 0.018 | 0.702 | 0.685 |
| HLA-A V91Ma | 0.402 | 0.173 | 0.769 | 0.757 |
| HLA-A R68Ka | ||||
| FANCD2 | 0.039 | 0.774 | 0.018 | 0.448 |
| IFIT2 | 0.163 | 0.127 | 0.852 | 0.202 |
| GEN1 | 0.164 | 0.154 | 0.750 | 0.138 |
| NCOR2 | 0.712 | 0.564 | 0.660 | 0.503 |
| TET2 | 0.067* | 0.306 | 0.266 | 0.495 |
| MKI67 | 0.025 | 0.480 | 0.151 | 0.195 |
| FANCI | 0.189 | 0.910 | 0.376 | 0.687 |
| RAD51D | 0.590 | 0.654 | 0.807 | 0.280 |
| IL10RA | 0.340 | 0.084 | 0.600 | 0.972 |
| IDH1 | 0.861 | 0.607 | 0.831 | 0.257 |
| HLA-F N353L | 0.266 | 0.901 | 0.046 | 0.262 |
| FANCA V6Db | 0.157 | 0.270 | 0.722 | 0.839 |
| FANCA 5c | 0.346 | 0.623 | 0.244 | 0.070 |
| HLA-B Y140F | 0.043 | 0.283 | 0.165 | 0.123 |
| HLA-DMA/B 5d | 0.456 | 0.041 | 0.966 | 0.213 |
| DYNC2H1 Q304L | 0.531 | 0.773 | 0.245 | 0.120 |
| DYNC2H1 N1576K | 0.231 | 0.511 | 0.926 | 0.243 |
| PPP1R15A | 0.008 | 0.028 | 0.191 | 0.255 |
| SLX4 | 0.100 | 0.195 | 0.219 | 0.552 |
| HLA-DQA2 | 0.507 | 0.308 | 0.662 | 0.887 |
P-values with bold text are < 0.05 with the variant conferring improved outcomes, those with underline text are associated with worse outcomes, and those marked with an asterisk are approaching significance
aThese two variants appear together in the same group of patients
bOverlap with patients with MC1R R160W
cGroup of patients with the same 5 changes in FANCA
d5 HLA mutations present in the same group (HLA-DMB S71N, D49V, T28A and HLA-DMA R210H, G181A)
TSC2 splice variant
Our data analysis identified an intronic C>T TSC2 variant (rs1800720) flanking a splice donor site and occurring in 10 patients (28.6%) in our n37 cohort, with a trend towards a significant enrichment in Group A. The variant was detected in 8 Group A patients versus 2 in Group B (p = 0.0577, Table 2). This TSC2 variant was particularly enriched in our cohort (MAF = 0.189) relative to the gnomAD data (MAF = 0.0968) [30]. Interestingly, the MAF in our cohort closely aligns with the African/African American gnomAD population (MAF = 0.202).
This specific TSC2 splice region is identified as an associated germline variant in tuberous sclerosis syndrome. TSC2 in complex with TSC1 has tumor suppressor functions via regulation of the mechanistic target of rapamycin kinase (mTOR) signaling pathway [38]. Aberrant activation of the mTOR pathway has been widely implicated in HNSCC [39] and it is possible that this TSC2 variant is associated with HNSCC via effects on the mTOR pathway. Clinical outcome KM analysis for this variant is associated with lower risk of LRR (p = 0.018), however no association was found with PFS (p = 0.216), MFS (p = 0.702) and OS (p = 0.685, Table 3). This is the first time to our knowledge that the finding of this germline variant is reported in HNSCC.
Other, low frequency germline variants in HNSCC
We detected 9 germline variants of significance associated with early versus late toxicity that did not meet our biomarker selection criteria due to less than 25% frequency of occurrence (Table 2). These were Fanconi anemia complementation group D2 (FANCD2) pPro714Leu (rs3864017); dynein cytoplasmic 2 heavy chain 1 (DYNC2H1) pGly304Leu (rs12146610; rs72989734); protein phosphatase 1 regulatory subunit 15A (PPP1R15A) pAla32Thr (rs3786734); SLX4 structure-specific endonuclease subunit (SLX4) pSer71Asn, pAsn457Lys, pArg204Cys (rs77985244, rs74319927, rs97842542); major histocompatibility complex, class II, DQ alpha 2 (HLA-DQA2) splice site (rs4398729); major histocompatibility complex, class II, DM beta (HLA-DMB) pSer71Asn, pAsp49Val, pThr28Ala (rs17617321, rs17617333, rs17583782); major histocompatibility complex, class II, DM alpha (HLA-DMA) pArg210His, pGly181Ala (rs41555121, rs6926628); Fanconi anemia complementation group A (FANCA) pVal6Asp, (rs1800282); and melanocortin 1 receptor (MC1R) pArg160Trp (rs1805008). The known functions of these genes cluster into two main functional families, 3 out of 8 are DNA repair genes associated with Fanconi anemia: FANCD2, SLX4, and FANCA [40–45], while 3 out the 8 are HLA genes comprising the major histocompatibility complex (MHC) involving immunity. The remaining genes are associated with skeletal neogenesis [46–48], protein synthesis homeostasis [49] and skin pigmentation [50]. All these germline variants were previously reported in the general population validating our findings, however their structure function-relationship in their individual encoded proteins awaits characterization. We report here that these gene variants occur in HNSCC patients and could be associated with late toxicity. Based on our study design, we found that FANCD2, DYNC2H1, PPP1R15A and HLA-A variants are associated with a significant increase in late toxicity, while SLX4, HLA-DQA2, HLA-DMB, HLA-DMA, FANCA, and MC1R variants are associated with fewer late RT toxicity events.
Cumulative RT toxicity variants
In addition to early versus late toxicity gene variant effects, we analyzed cumulative toxicity associations, Group C (Grade 2–4) and Group D (Grade 0–1) in the same cohort of patients (n = 37, Table 4).
Table 4.
Chi-square results: recurrent freebayes mutations expanded beyond tumor suppressors and oncogenes

Patients in Group C experienced high toxicity for late dysphagia (CTCAE grade 2 +), while patients in Group D experienced a low toxicity for late dysphagia (CTCAE grade 0–1)
Not surprisingly, the TSC2 splice site variant (rs1800720) detected in our previous study design displayed significant cumulative toxicity association. In addition, we detected a new variant GEN1, a Holliday junction 5′ flap endonuclease (GEN1) pLys839Glufs (rs149936944) that was associated with significant protection from RT therapy induced toxicity. We found that 9 patients carried this variant and that 2 of them were bi-allelic (Fig. 2). We confirmed this finding by analyzing the RNA-seq data. The reference GEN1 protein is 908 amino acids long. The variant we detected has a frame shift mutation introducing a stop codon at position 839 resulting in truncated messenger RNA (mRNA) missing 69 amino acids at the C-terminus of the GEN1 protein with potential for nonsense-mediated RNA decay. The presence of homozygosity (Fig. 2) of this variant implicates GEN1 redundancy, however, it is possible that this change leads to an alternate biological function. Indeed, GEN1 redundancy has been reported by Wang et.al [51]. They found that GEN1 and essential meiotic structure-specific endonuclease 1 (EME1) play redundant roles in meiotic recombination in a mouse model and that deletion of both genes confer synthetic lethality in mice [51]. In addition, a GEN1 knockout mouse is viable [52, 53].
Fig. 2.

Bulk RNA-Seq derived variant allelic fraction (VAF) was generated for each variant and each patient. VAF values (y-axis) were compared against curated patient germline genotype values (x-axis) for each variant, where 0 = homozygous no variant, 1 = heterozygous variant, 2 = homozygous variant. Point color represents radiation toxicity group and point size represents total read depth at the variant locus of interest
We found that the nuclear receptor corepressor 2 (NCOR2) Gln510dup variant (rs35831183), showed a strong association with Group D (p = 0.008151).
Clinical outcomes associated with selected germline variants
We also examined clinical outcomes between the two sets of groups and the implication of certain variants. For this portion of the analysis only those patients with HNSCC treated with definitive or post-op radiation/chemoRT were included. A total of 34 patients were included with radiation treatment dates ranging from January 2009 to January 2021. There were 14 patients (41.2%) that were treated in the post-operative setting. Radiation dose was available for 31 (91.2%) of patients, ranging 60–70 Gy (Gy). 27 patients (79.4%) received concurrent systemic therapy, most frequently cisplatin (70.4%). Median age at treatment was 62.5 years (range 37–85, Table 1), with a median follow up of 42.5 months (range 7–205). Between Groups A vs. B and Groups C vs. D there were no significant differences in age, stage, site of primary, HPV status, use of surgery or concurrent chemotherapy. Patient characteristics can be found in Table 1. 11.7% of patients had Grade 3 early mucositis or dysphagia and 29.4% of patients had Grade 3 dysphagia; no Grade 4 or 5 AEs were recorded.
Patients in Group A had significantly better OS in comparison to Group B (p = 0.017, Fig. 3). There were also significant differences in PFS (p = 0.001), LFS (p = 0.041) and MFS (0.010). As noted previously, alterations in TSC2 were more prevalent in elevated late toxicity Group A, and patients with variants in TSC2 also had significantly improved LFS (p = 0.018, Table 3). FANCD2 was also more prevalent in Group A and associated with improved PFS (p = 0.039) and MFS (p = 0.018), as was PPP1R15A with PFS (p = 0.008) and LFS (0.028). There were five HLA-DMB/HLA-DMA variants only present in patients in Group B which were associated with a higher risk of LRR (p = 0.041). Within Groups C and D the major HLA-F N353L variant was significantly more prevalent in Group D and associated with more frequent distant metastases (p = 0.046).
Fig. 3.

Analysis of outcomes for Groups A and B by A OS, B PFS, C LFS and D MFS. Log-rank p-values are shown
Discussion
We designed this pilot study to assess the viability of our method for detecting genetic variants associated with RT toxicity in HNSCC. In an earlier published study we found that somatic tumor mutations can be associated with RT toxicity [54]. In this study, we analyzed germline variants to identify biomarkers of RT toxicity that could aid patient stratification for personalizing radiation. Germline genetic biomarkers would be preferred for this purpose due to the ease of sample collection (blood/saliva) for targeted NGS testing.
While we did not aim to study the mechanism of action of these genetic variants, our findings pave the way to further the research deciphering the functional implications of these genes in HNSCC and other cancer types. Importantly, the majority of the variants we identified had several fold higher MAF in HNSCC patients than in the general population, which is suggestive of their potential role in HNSCC. Furthermore, the Fanconi anemia DNA repair genes were widely detected in our study. Since they appeared at a low frequency they did not meet our biomarker selection criteria. However, we found that these germline variants had strong, often opposite associations with RT toxicity outcomes. FANCD2 pPro714Leu and FANCA pVal6Asp correlated with increased toxicity while SLX4 variant comprising 3 different missense changes in this gene had protective effects in toxicity. The triple missense single nucleotide alteration resulted in amino acid changes Ser71Asn, Asp49Val, Thr28Ala suggestive of structural and functional selective pressures, and we are the first to report this finding. Another variant of interest is GEN1 and we verified that this variant transcript is expressed (Fig. 2), and thus may play a functional role in DNA repair. Although it is widely reported that DNA repair mechanisms play an important role in radiation sensitivity the protective effects of specific variants, such as the SLX4 triple variant, cannot be overlooked. Taken together, we observed a large number of DNA repair and MHC polymorphisms consistent with widely reported mechanistic studies of drivers of toxicity.
The clinical outcome data for all the germline variants selected for biomarker development was evaluated to isolate the significance of these variants for clinical outcomes in order to ensure that utilizing these biomarkers in adapting the radiation dose would not diminish treatment benefits. Specifically, GEN1, HLA-A and TSC2 germline variants meet this criteria for biomarker development. All carriers of GEN1 frame shift variant pLys839Glu exhibited significantly lower cumulative toxicity (p = 0.027) as determined by CTCAE grading. We did not detect significant correlation with clinical outcomes for this variant (Table 4). The HLA-A variant was associated with late toxicity in 10 of 13 patients (p = 0.052). The presence of this variant did not affect clinical outcomes while it was found in 35% of our cohort. We consider this variant as a good biomarker candidate.
Other gene variants we identified present a rationale for new therapeutic targets in HNSCC. In particular, the TSC2 splice site variant found in 27% of patients was significantly associated with cumulative as well as late stage toxicity. This particular variant (rs1800720) is implicated in Tuberous Sclerosis syndrome. TSC2 in complex with TSC1 has a regulatory role in the mTOR signaling pathway in HNSCC [38]. We detected the variant transcript expression as heterozygous in 8 patients and homozygous in 2 patients (Fig. 2). Characterizing this variant in experimental models may further aid in decoding the TSC2/mTOR function in HNSCC. Other variants we identified, while occurring at a lower frequency, can further aid in investigating their functional implications, thus shedding light on their “normal” function in HNSCC. An additional notable result of our study which deserves further investigation is the finding that patients exhibiting higher RT toxicities show significant beneficial clinical outcomes. A similar pattern has been observed in patients with ATM mutations, where it has been suggested as a potential biomarker for rtAEs with several studies also showing it conferred improved treatment response to radiation [55–57].
There are limitations to our study including the limited sample size and retrospective nature. Based on this pilot data we are designing a larger study with prospective validation cohorts to further investigate these findings.
Conclusions
In summary we identified unique germline variants which were associated with various patient outcomes including long term toxicity, LRR, and OS after radiation therapy in HNSCC. These findings provide rationale for larger studies incorporating genetic biomarkers to better predict responses to radiation therapy and advance personalized radiation medicine.
Acknowledgements
The authors wish to thank Napoleone Ferrara, M.D., J. Silvio Gutkind, Ph.D., and Milan Mikale, Ph.D., for reviewing the manuscript and providing constructive feedback.
Abbreviations
- AJCC
American Joint Committee on Cancer
- ATM
ATM serine/threonine kinase
- CTCAE
Common Terminology Criteria for Adverse Events
- DNA
Deoxyribonucleic acid
- DYNC2H1
Dynein cytoplasmic 2 heavy chain 1
- EME1
Essential meiotic structure-specific endonuclease 1
- FANCA
Fanconi anemia complementation group A
- FANCD2
Fanconi anemia complementation group D2
- GEN1
GEN1 Holliday junction 5′ flap endonuclease
- Gy
Gray
- HLA-A
Major histocompatibility complex, class I, A
- HLA-DMA
Major histocompatibility complex, class II, DM alpha
- HLA-DMB
Major histocompatibility complex, class II, DM beta
- HLA-DQA2
Major histocompatibility complex, class II, DQ alpha 2
- HNC
Head and neck cancer
- HNSCC
Head and neck squamous cell carcinoma
- HPV
Human papillomavirus
- IFIT2
Interferon induced protein with tetratricopeptide repeats 2
- IL10RA
Interleukin 10 receptor subunit alpha
- IMRT
Intensity modulated RT
- KM
Kaplan–Meier
- LFS
Locoregional failure free survival
- LRR
Locoregional recurrence
- MAF
Minor allele frequency
- MC1R
Melanocortin 1 receptor
- MFS
Metastasis free survival
- MHC
Major histocompatibility complex
- MKI67
Marker of proliferation Ki-67
- mRNA
Messenger RNA
- mTOR
Mechanistic target of rapamycin kinase
- NCOR2
Nuclear receptor corepressor 2
- NGS
Next generation sequencing
- NSCLC
Non-small cell lung cancer
- OAR
Organs at risk
- OS
Overall survival
- PFS
Progression free survival
- PPP1R15A
Protein phosphatase 1 regulatory subunit 15A
- QC
Quality control
- RNA
Ribonucleic acid
- RNA-seq
RNA-sequencing
- RT
Radiation therapy
- rtAE
RT adverse event
- SEER
Surveillance, epidemiology, and end results
- SIFT
Sorting Intolerant from Tolerant
- SLX4
SLX4 structure-specific endonuclease subunit
- TET2
Tet methylcytosine dioxygenase 2
- TSC2
TSC complex subunit 2
- VAF
Variant allelic fraction
Author contributions
ID prepared and wrote the manuscript, designed the study, collected and analyzed data, and contributed to manuscript review and preparation. AH collected and analyzed clinical and dose data, and contributed to manuscript writing and review. ID and AH contributed equally to this work. LK analyzed genomics data, and contributed to manuscript writing. TJS analyzed genomics data, and contributed to manuscript writing. LS collected clinical and genomics data, build the database, and contributed to manuscript review and preparation. XR contributed to manuscript writing and review. AS aided in clinical aspects of the study and contributed to manuscript writing and review. PS aided in clinical aspects of the study and reviewed the manuscript. HC analyzed genomics data, aided in study design, and contributed to manuscript writing and review. VM contributed to study design, collected radiation therapy data, and contributed to manuscript writing and review. All authors read and approved the final manuscript.
Funding
This work has been generously supported by the UC San Diego Department of Radiation Medicine and Applied Sciences as well as NIH Grant R01CA269919 to HC.
Availability of data and materials
The datasets used, generated, and analyzed during the current study are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
This study was performed in accordance with federal, state and University policies, and approved by UCSD Human Research Protection Program IRB#200495. This protocol was reviewed and approved by the UCSD Human Research Protection Program in accordance with the requirements of the Code of Federal Regulations on the Protection of Human Subjects (45 CFR 46), including its relevant Subparts, for federally funded/support research studies. It was determined that a waiver of informed consent was granted for historic subjects (both clinical and research) as it meets the requirements outlined in 45 CFR 46.116(f).
Consent for publication
Not applicable.
Competing interests
A.S. reports being a consultant/advisory board member for AstraZeneca, Varian Medical Systems, Primmune, and Jounce Therapeutics; reports receiving commercial research grants from Varian Medical Systems and Pfizer; holds ownership interest in Toragen Inc. outside of submitted work.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Ida Deichaite and Austin Hopper contributed equally to this work
Hannah Carter and Vitali Moiseenko contributed equally to this work
References
- 1.Cancer ROT. Benefits and effectiveness. 2022. Updated October 10, 2020. https://www.targetingcancer.com.au/about-radiation-oncology/benefits-and-effectiveness/. Accessed May 24
- 2.Siddiqui F, Movsas B. Management of radiation toxicity in head and neck cancers. Semin Radiat Oncol. 2017;27(4):340–349. doi: 10.1016/j.semradonc.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 3.Baumann M, Krause M, Overgaard J, et al. Radiation oncology in the era of precision medicine. Nat Rev Cancer. 2016;16(4):234–249. doi: 10.1038/nrc.2016.18. [DOI] [PubMed] [Google Scholar]
- 4.Lee J-H, Lee JCW, Leung W, et al. Polarization engineering of thermal radiation using metallic photonic crystals. Adv Mater. 2008;20(17):3244–3247. doi: 10.1002/adma.200703160. [DOI] [Google Scholar]
- 5.Nutting CM, Morden JP, Harrington KJ, et al. Parotid-sparing intensity modulated versus conventional radiotherapy in head and neck cancer (PARSPORT): a phase 3 multicentre randomised controlled trial. Lancet Oncol. 2011;12(2):127–136. doi: 10.1016/s1470-2045(10)70290-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghanem AI, Schymick M, Bachiri S, et al. The effect of treatment package time in head and neck cancer patients treated with adjuvant radiotherapy and concurrent systemic therapy. World J Otorhinolaryngol Head Neck Surg. 2019;5(3):160–167. doi: 10.1016/j.wjorl.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suwinski R, Sowa A, Rutkowski T, Wydmanski J, Tarnawski R, Maciejewski B. Time factor in postoperative radiotherapy: a multivariate locoregional control analysis in 868 patients. Int J Radiat Oncol Biol Phys. 2003;56(2):399–412. doi: 10.1016/s0360-3016(02)04469-3. [DOI] [PubMed] [Google Scholar]
- 8.Langendijk JA, de Jong MA, Leemans CR, et al. Postoperative radiotherapy in squamous cell carcinoma of the oral cavity: the importance of the overall treatment time. Int J Radiat Oncol Biol Phys. 2003;57(3):693–700. doi: 10.1016/s0360-3016(03)00624-2. [DOI] [PubMed] [Google Scholar]
- 9.Ohri N, Rapkin BD, Guha C, Kalnicki S, Garg M. Radiation therapy noncompliance and clinical outcomes in an urban academic cancer center. Int J Radiat Oncol Biol Phys. 2016;95(2):563–570. doi: 10.1016/j.ijrobp.2016.01.043. [DOI] [PubMed] [Google Scholar]
- 10.Osazuwa-Peters N, Simpson MC, Zhao L, et al. Suicide risk among cancer survivors: head and neck versus other cancers. Cancer. 2018;124(20):4072–4079. doi: 10.1002/cncr.31675. [DOI] [PubMed] [Google Scholar]
- 11.Zeller JL. High suicide risk found for patients with head and neck cancer. JAMA. 2006;296(14):1716–1717. doi: 10.1001/jama.296.14.1716. [DOI] [PubMed] [Google Scholar]
- 12.Dong L, Cui J, Tang F, Cong X, Han F. Ataxia telangiectasia-mutated gene polymorphisms and acute normal tissue injuries in cancer patients after radiation therapy: a systematic review and meta-analysis. Int J Radiat Oncol Biol Phys. 2015;91(5):1090–1098. doi: 10.1016/j.ijrobp.2014.12.041. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Liu Z, Wang M, et al. Single nucleotide polymorphism rs1801516 in ataxia telangiectasia-mutated gene predicts late fibrosis in cancer patients after radiotherapy: a PRISMA-compliant systematic review and meta-analysis. Medicine (Baltimore) 2016;95(14):e3267. doi: 10.1097/MD.0000000000003267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andreassen CN, Rosenstein BS, Kerns SL, et al. Individual patient data meta-analysis shows a significant association between the ATM rs1801516 SNP and toxicity after radiotherapy in 5456 breast and prostate cancer patients. Radiother Oncol. 2016;121(3):431–439. doi: 10.1016/j.radonc.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stewart GS, Stankovic T, Byrd PJ, et al. RIDDLE immunodeficiency syndrome is linked to defects in 53BP1-mediated DNA damage signaling. Proc Natl Acad Sci USA. 2007;104(43):16910–16915. doi: 10.1073/pnas.0708408104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pollard JM, Gatti RA. Clinical radiation sensitivity with DNA repair disorders: an overview. Int J Radiat Oncol Biol Phys. 2009;74(5):1323–1331. doi: 10.1016/j.ijrobp.2009.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waltes R, Kalb R, Gatei M, et al. Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder. Am J Hum Genet. 2009;84(5):605–616. doi: 10.1016/j.ajhg.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marcou Y, D'Andrea A, Jeggo PA, Plowman PN. Normal cellular radiosensitivity in an adult Fanconi anaemia patient with marked clinical radiosensitivity. Radiother Oncol. 2001;60(1):75–79. doi: 10.1016/s0167-8140(01)00370-x. [DOI] [PubMed] [Google Scholar]
- 19.Kleinerman RA. Radiation-sensitive genetically susceptible pediatric sub-populations. Pediatr Radiol. 2009;39(Suppl 1):S27–S31. doi: 10.1007/s00247-008-1015-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson PF, Nagasawa H, Fitzek MM, Little JB, Bedford JS. G2-phase chromosomal radiosensitivity of primary fibroblasts from hereditary retinoblastoma family members and some apparently normal controls. Radiat Res. 2010;173(1):62–70. doi: 10.1667/RR1943.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergom C, West CM, Higginson DS, et al. The implications of genetic testing on radiation therapy decisions: a guide for radiation oncologists. Int J Radiat Oncol Biol Phys. 2019;105(4):698–712. doi: 10.1016/j.ijrobp.2019.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenstein BS. Radiogenomics: identification of genomic predictors for radiation toxicity. Semin Radiat Oncol. 2017;27(4):300–309. doi: 10.1016/j.semradonc.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schack LMH, Naderi E, Fachal L, et al. A genome-wide association study of radiotherapy induced toxicity in head and neck cancer patients identifies a susceptibility locus associated with mucositis. Br J Cancer. 2022;126(7):1082–1090. doi: 10.1038/s41416-021-01670-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amin MB, Greene FL, Edge SB, et al. The Eighth edition AJCC cancer staging manual: continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J Clin. 2017;67(2):93–99. doi: 10.3322/caac.21388. [DOI] [PubMed] [Google Scholar]
- 25.SPSS. Version 22.0. IBM Corporation. https://www.ibm.com/support/pages/spss-statistics-220-available-download.
- 26.Trim Galore. Version 0.6.3_dev. Babraham Institute. https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/.
- 27.Picard. Version 2.20.3. Broad Institute. http://broadinstitute.github.io/picard/.
- 28.Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khanna A, Larson DE, Srivatsan SN, et al. Bam-readcount—rapid generation of basepair-resolution sequence metrics. ArXiv. arXiv:2107.12817v1. 2021.
- 30.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosetta@home. http://boinc.bakerlab.org/rosetta/.
- 32.Pettersen EF, Goddard TD, Huang CC, et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 2021;30(1):70–82. doi: 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mandelker D, Zhang L, Kemel Y, et al. Mutation detection in patients with advanced cancer by universal sequencing of cancer-related genes in tumor and normal DNA vs guideline-based germline testing. JAMA. 2017;318(9):825–835. doi: 10.1001/jama.2017.11137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang KL, Mashl RJ, Wu Y, et al. Pathogenic germline variants in 10,389 adult cancers. Cell. 2018;173(2):355–370.e14. doi: 10.1016/j.cell.2018.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deneuve S, Bastogne T, Duclos M, et al. Predicting acute severe toxicity for head and neck squamous cell carcinomas by combining dosimetry with a radiosensitivity biomarker: a pilot study. Tumori. 2022 doi: 10.1177/03008916221078061. [DOI] [PubMed] [Google Scholar]
- 36.Krassowski M, Pellegrina D, Mee MW, Fradet-Turcotte A, Bhat M, Reimand J. ActiveDriverDB: interpreting genetic variation in human and cancer genomes using post-translational modification sites and signaling networks (2021 update) Front Cell Dev Biol. 2021;9:626821. doi: 10.3389/fcell.2021.626821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Gorlova OY, Gorlov IP, et al. Association analysis of driver gene-related genetic variants identified novel lung cancer susceptibility loci with 20,871 lung cancer cases and 15,971 controls. Cancer Epidemiol Biomark Prev. 2020;29(7):1423–1429. doi: 10.1158/1055-9965.EPI-19-1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Y, Corradetti MN, Inoki K, Guan KL. TSC2: filling the GAP in the mTOR signaling pathway. Trends Biochem Sci. 2004;29(1):32–38. doi: 10.1016/j.tibs.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 39.Iglesias-Bartolome R, Martin D, Gutkind JS. Exploiting the head and neck cancer oncogenome: widespread PI3K-mTOR pathway alterations and novel molecular targets. Cancer Discov. 2013;3(7):722–725. doi: 10.1158/2159-8290.CD-13-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sasaki MS, Tonomura A. A high susceptibility of Fanconi’s anemia to chromosome breakage by DNA cross-linking agents. Cancer Res. 1973;33(8):1829–1836. [PubMed] [Google Scholar]
- 41.Hussain S, Wilson JB, Medhurst AL, et al. Direct interaction of FANCD2 with BRCA2 in DNA damage response pathways. Hum Mol Genet. 2004;13(12):1241–1248. doi: 10.1093/hmg/ddh135. [DOI] [PubMed] [Google Scholar]
- 42.Fekairi S, Scaglione S, Chahwan C, et al. Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell. 2009;138(1):78–89. doi: 10.1016/j.cell.2009.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muñoz IM, Hain K, Déclais AC, et al. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol Cell. 2009;35(1):116–127. doi: 10.1016/j.molcel.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 44.Svendsen JM, Smogorzewska A, Sowa ME, et al. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell. 2009;138(1):63–77. doi: 10.1016/j.cell.2009.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dokal I. Fanconi’s anaemia and related bone marrow failure syndromes. Br Med Bull. 2006;77–78:37–53. doi: 10.1093/bmb/ldl007. [DOI] [PubMed] [Google Scholar]
- 46.Schmidts M, Arts HH, Bongers EMHF, et al. Exome sequencing identifies DYNC2H1 mutations as a common cause of asphyxiating thoracic dystrophy (Jeune syndrome) without major polydactyly, renal or retinal involvement. J Med Genet. 2013;50(5):309–323. doi: 10.1136/jmedgenet-2012-101284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scholey JM. Intraflagellar transport motors in cilia: moving along the cell’s antenna. J Cell Biol. 2008;180(1):23–29. doi: 10.1083/jcb.200709133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosenbaum J. Intraflagellar transport. Curr Biol. 2002;12(4):R125. doi: 10.1016/s0960-9822(02)00703-0. [DOI] [PubMed] [Google Scholar]
- 49.Hinnebusch AG. Mechanism and regulation of initiator methionyl-tRNA binding to ribosomes. Cold Spring Harb Monogr Ser. 2000;39:185–244. [Google Scholar]
- 50.Wolf Horrell EM, Boulanger MC, D'Orazio JA. Melanocortin 1 receptor: structure, function, and regulation. Front Genet. 2016;7:95. doi: 10.3389/fgene.2016.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang X, Wang H, Guo B, et al. Gen1 and Eme1 play redundant roles in DNA repair and meiotic recombination in mice. DNA Cell Biol. 2016;35(10):585–590. doi: 10.1089/dna.2015.3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Okazaki Y, Furuno M, Kasukawa T, et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002;420(6915):563–573. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- 53.Carninci P, Kasukawa T, Katayama S, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309(5740):1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 54.Sumner W, Ray X, Sutton L, et al. Gene alterations as predictors of radiation-induced toxicity in head and neck squamous cell carcinoma. J Transl Med. 2021;19(1):212. doi: 10.1186/s12967-021-02876-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang X, Li Y, Pan X, et al. Intensity-modulated proton therapy reduces the dose to normal tissue compared with intensity-modulated radiation therapy or passive scattering proton therapy and enables individualized radical radiotherapy for extensive stage IIIB non-small-cell lung cancer: a virtual clinical study. Int J Radiat Oncol Biol Phys. 2010;77(2):357–366. doi: 10.1016/j.ijrobp.2009.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiong H, Liao Z, Liu Z, et al. ATM polymorphisms predict severe radiation pneumonitis in patients with non-small cell lung cancer treated with definitive radiation therapy. Int J Radiat Oncol Biol Phys. 2013;85(4):1066–1073. doi: 10.1016/j.ijrobp.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee JJ, Yang AJ, Chang JS, Kim HS, Yoon HI, Shin SJ, Kim YB, Koom WS, Ahn JB. Genomic analysis reveals somatic mutations of ATM gene in DNA repair confer exceptional target lesion response to radiation therapy. J Glob Oncol. 2019;5(suppl):130–130. doi: 10.1200/JGO.2019.5.suppl.130. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used, generated, and analyzed during the current study are available from the corresponding author on reasonable request.