

Abstract
During translation of the Bacillus subtilis cdd gene, encoding cytidine deaminase (CDA), a ribosomal −1 frameshift occurs near the stop codon, resulting in a CDA subunit extended by 13 amino acids. The frequency of the frameshift is approximately 16%, and it occurs both when the cdd gene is expressed from a multicopy plasmid in Escherichia coli and when it is expressed from the chromosomal copy in B. subtilis. As a result, heterotetrameric forms of the enzyme are formed in vivo along with the dominant homotetrameric species. The different forms have approximately the same specific activity. The cdd gene was cloned in pUC19 such that the lacZ′ gene of the vector followed the cdd gene in the −1 reading frame immediately after the cdd stop codon. By using site-directed mutagenesis of the cdd-lacZ′ fusion, it was shown that frameshifting occurred at the sequence CGA AAG, 9 bp upstream of the in-frame cdd stop codon, and that it was stimulated by a Shine-Dalgarno-like sequence located 14 bp upstream of the shift site. The possible function of this frameshift in gene expression is discussed.
Some mRNAs contain signals that direct a high proportion of ribosomes to change reading frame at a specific shift site. These “programmed” events can occur at levels that are 1,000- to 10,000-fold above the low background of error frameshifting. Their function can be either as a sensor for regulatory circuits, as in the decoding of the genes for Escherichia coli polypeptide chain release factor 2 (prfB) or mammalian antizyme, or to give a set ratio of two different products that have some identical sequences, as in the decoding of E. coli dnaX or human immunodeficiency virus gag-pol. Quite a number of cases of programmed frameshifting are known in the expression of viruses and in mobile chromosomal elements such as yeast Ty elements and IS elements of the IS3 family (for reviews, see references 5, 11 and 13). However, very few cases are known for nonmobile chromosomal genes. In mammals antizyme is the only known case (26, 29), and in bacteria the list is restricted to prfB (8, 40) and dnaX, which encode two subunits of DNA polymerase III (4, 12, 39).
Early studies showed that disruption of codon-anticodon pairing and re-pairing of the anticodon to an overlapping codon explained many cases of frameshifting in model systems, and the involvement of a single tRNA in such a process is the basis for −1 frameshifting in potato virus M (14). However, studies on retroviruses showed that for −1 frameshifting, tandem shifts of two tRNAs on a sequence of the general form X XXY YYZ was very effective (15). In E. coli, a very slippery form of this heptanucleotide was found to be A AAA AAG, which occurs at the shift sequence for dnaX (4, 12, 39) and IS911 (32). The majority of these tandem shift sites were found to require secondary mRNA structures such as pseudoknots or stem-loop structures downstream of the slippery heptanucleotide to achieve maximal efficiency (5, 22, 38).
Frameshifting studies showed that the anti-Shine-Dalgarno (anti-SD) sequence close to the 3′ end of 16S rRNA within translating ribosomes must be scanning mRNA for potential complementarity. An SD-like sequence 3 bases 5′ of the shift site is important for the obligatory +1 frameshifting in decoding release factor 2 and its spacing has to be precise (9, 40). An SD-like sequence 10 bases 5′ of the shift site is important for the −1 frameshifting in dnaX, with flexibility between 9 and 15 nucleotides for the spacer length (21).
The Bacillus subtilis cdd gene, encoding the pyrimidine salvage enzyme cytidine deaminase (CDA), was cloned and sequenced by Song and Neuhard (37). The deduced amino acid sequence indicated a subunit size of 14.9 kDa, and preliminary studies suggested that the native enzyme was a homotetramer. In the present work, we observed that expression of the gene, both from a plasmid-borne copy in E. coli and from the chromosome in B. subtilis, yielded two types of subunits: the predicted 14.9-kDa subunit and smaller amounts of a 16-kDa subunit. As a result, measurable amounts of heterotetrameric forms of the enzyme were detected. We showed that the heterogeneity in subunit size is due to a −1 ribosomal frameshift occurring during translation of the 3′ end of the coding sequence and identified a new type of shift site with an upstream stimulatory SD-like sequence.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The E. coli strains used were JF611 (cdd pyrE argE his proA thr leu thi), obtained from Jim Friesen, and SØ5299, a cdd::Tn10 derivative of JM83. Both strains are defective in CDA due to cdd mutations. They were grown at 37°C in Luria broth (2) or AB minimal medium (6) supplemented with 0.2% glucose, 0.2% Casamino Acids, and 1 μg of thiamine per ml. When required, ampicillin was present at 100 μg per ml. B. subtilis 168 (trpC) was grown at 37°C in a modified Spizizen salts minimal medium supplemented with 0.4% glucose, 0.2% glutamate, and 1 μg of thiamine per ml (36).
DNA techniques.
DNA manipulations, transformations, and isolation of plasmid DNA were performed by standard procedures as described by Sambrook et al. (34). PCR products were purified with the Qiagen PCR purification kit. Endonuclease digestion and ligation of DNA were done as specified by the suppliers. DNA sequencing was performed by the chain-termination method (35) with double-stranded DNAs as templates.
Plasmids.
To achieve overproduction of B. subtilis CDA in E. coli, plasmid pSO143 was used (Fig. 1A) (37). It contains the B. subtilis cdd gene without its promoter but with its native ribosomal binding site on a 740-bp KpnI-EcoRI fragment in pUC19. Immediately downstream of cdd, with a 20-nucleotide overlap, the fragment carries the first third of the bex gene (18). Expression of cdd occurs from the lac promoter on the vector. All other plasmids used in the present study were derived from pSO143 and varied only in the region between the cdd stop codon and lacZ′ of the vector. Plasmid pSO1000 was constructed by subcloning the cdd gene from pSO143 on a 470-bp PstI-BsaBI fragment into PstI- and SmaI-digested pUC19 (Fig. 1B). On pSO1000, lacZ′ is in the 0 reading frame compared to cdd. In unrelated structure-function studies of B. subtilis CDA, we used pSO1000 as a template for PCR-mediated site-directed mutagenesis of the coding region of cdd. One of the constructs obtained, pSO1001, was shown by DNA sequencing to have suffered an unintended deletion of 1 bp (A) immediately 3′ of the cdd stop codon, in addition to the mutation introduced by PCR in the coding region (TG → CA, yielding a C53H mutation in CDA). As a result of the deletion, lacZ′ is fused in the −1 reading frame compared to cdd on pSO1001 (Fig. 1B). Plasmid pSO1001 was opened at the unique EcoRI site in front of lacZ′, digested by mung bean nuclease to remove the 5′ overhangs, and ligated to create pSO1002, which carries lacZ′ in the +1 reading frame (Fig. 1B).
FIG. 1.

Structures of the plasmids used in this study. (A) pSO143. Thin lines, pUC19 DNA; solid bar, coding region of the B. subtilis cdd gene; open bar, leader region of the cdd gene; hatched bar, coding region of the 5′ end of the B. subtilis bex gene. Restriction endonuclease sites: E, EcoRI; Bs, BsaBI; K, KpnI; P, PstI; S, SmaI. The nucleotide sequence in the frameshift region near the end of the cdd gene, as well as the deduced amino acids encoded by the three reading frames, are shown below the graph. Capital letters, C-terminal amino acids of the wild-type CDA subunit; bold capital letters, C-terminal amino acids of the extended subunit (Sext); italicized capital letters, N-terminal amino acids of the Bex protein. (B) Nucleotide sequence of the region between the CDA stop codon and lacZ′ in various plasmids. The CDA stop codon is overlined, and the EcoRI site is underlined. pUC19 sequences are in italics, and lacZ′ sequences are in lowercase letters. Numbers in parentheses refer to the reading frame of lacZ′ relative to cdd.
Plasmid pNMJ62 was used for quantitation of −1 frameshifting. It contains the entire cdd coding region inserted in pUC19 in such a way that the −1 reading frame of cdd continues into lacZ′ of the vector. It was constructed by PCR amplification of cdd from pSO143 with, as the 5′ primer, the 24-mer reverse sequencing primer (−48) of M13/pUC and, as the 3′ primer, the wild-type primer shown in Table 1. This latter primer was complementary to the last 4 codons of cdd and had an EcoRI site introduced 1 bp after the stop codon. The resulting fragment was digested with PstI and EcoRI and inserted into the multiple-cloning site of pUC19.
TABLE 1.
3′ primers for PCR-mediated site-directed mutagenesis
| Sequence (5′ → 3′)a | Resulting construct |
|---|---|
| GGAATTCTTTAAAGCTTTCG | Wild type |
| GGAATTCTGTAAAGCTTTCG | Stop UAA → Tyr AUC |
| GGAATTCTTTACAGCTTTCGTTCG | Leu CUU → Leu CUG |
| GGAATTCTTTAAAGCTTTCGCTCGTCATG | Glu GAA → Glu GAG |
| GGAATTCTTTAAAGTTTTCGTTCG | Lys AAG → Lys AAA |
| GGAATTCTTTAAAGCTTGCGTTCG | Arg CGA → Arg CGC |
| GGAATTCTTTAAAGCTTTCTTTCG | Arg CGA → Arg AGA |
| GGAATTCTTTAAAGCTTTCGTTCGTCATGTAAATCTTCGGATG | GGAGG → CGAAG |
| GGAATTCTTTAAAGCTTTTCGTTCG | AAG → AAAG |
Letters in italics indicate the cdd stop codon. The EcoRI site downstream of the stop codon is underlined. Mutations are shown in bold letters.
Site-directed mutagenesis.
Site-directed mutagenesis of the frameshift region upstream of the cdd stop codon was accomplished by PCR amplification of the entire cdd gene on pNMJ62 with, as the 5′ primer, the 24-mer reverse sequencing primer (−48) of M13/pUC in all cases. The 3′ primers were all complementary to the 3′ end of the cdd gene except for the desired mutation(s) and included the EcoRI site 1 bp downstream of the stop codon (Table 1). The amplified fragments were digested with PstI and EcoRI and cloned into pUC19 digested with the same enzymes. Thus, the vector-borne lacZ′ followed the cdd stop codon in the −1 reading frame of cdd, as in pNMJ62. All plasmid constructs were confirmed by DNA sequencing.
Purification of recombinant CDA from E. coli.
Cells from a 1-liter culture of E. coli JF611/pSO143 grown overnight at 37°C in Luria broth supplemented with ampicillin (100 μg/ml) were harvested by centrifugation, washed with 0.9% NaCl, resuspended in 6 to 8 volumes of 50 mM Tris-HCl (pH 7.2) (buffer A), and disrupted by sonic oscillations at 4°C. All subsequent steps were performed at 4°C. Cellular debris was removed by centrifugation, and streptomycin sulfate was added to the supernatant to a final concentration of 1%. Following centrifugation, the supernatant was applied to a DEAE-cellulose (DE-52) column (2.5 by 24 cm) equilibrated with buffer A. The column was washed with 7 volumes of buffer A, and the enzyme was eluted with a linear gradient of NaCl in buffer A. The fractions containing CDA activity were concentrated by pressure filtration to 5 ml and treated at 68°C for 10 min. The supernatant after heat denaturation was subjected to gel filtration on a Sephadex G-100 column (2.5 by 85 cm) equilibrated and eluted with buffer A. The active fractions were pooled and applied to an ion-exchange column (Q5; Bio-Rad) equilibrated with 20 mM Tris-HCl (pH 7.6) and connected to a Pharmacia fast protein liquid chromatography (FPLC) apparatus. The column was washed with 3 volumes of 20 mM Tris-HCl (pH 7.6), and the enzyme was eluted with a gradient of KCl in the same buffer. For the result of a typical purification, see Table 2.
TABLE 2.
Purification of B. subtilis recombinant CDAa
| Purification step | Total amt of protein (mg) | Total activity (U)b | Sp act (U/mg) | Yield (%) |
|---|---|---|---|---|
| 1. Crude extract | 331 | 8,280 | 25 | 100 |
| 2. Streptomycin | 293 | 9,080 | 31 | 110 |
| 3. DEAE | 79 | 5,280 | 67 | 57 |
| 4. Heat treatment | 31 | 4,590 | 149 | 55 |
| 5. G-100 filtration | 25 | 3,690 | 148 | 44 |
| 6. Q5 FPLC | 7 | 1,250 | 176 | 15 |
From 1 liter of bacterial culture.
Units are micromoles of cytidine deaminated per minute at 25°C.
Purification of native CDA from B. subtilis.
Cells from a 5-liter culture of B. subtilis 168 in early stationary phase were harvested by centrifugation, washed with cold 0.9% NaCl, resuspended in 6 to 8 volumes of buffer A, and disrupted by sonication. The enzyme from the sonic extract was partially purified by the same procedure as described for the recombinant enzyme. The final preparation represented a 150-fold purification over crude extract and yielded an enzyme which, according to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), was approximately 30% pure.
Preparation of cell extracts and enzyme assays.
Crude cellular extracts were prepared by sonic disruption of cells suspended in 0.1 M Tris-HCl (pH 7.6), followed by centrifugation to remove cellular debris. β-Galactosidase activity was determined as described by Miller (28), and CDA activity was measured spectrophotometrically (7). Protein concentration was measured by the Lowry method with bovine serum albumin as the standard.
SDS-PAGE.
Protein samples or whole cells were incubated for 2 min at 100°C in 2× SDS loading buffer (100 mM Tris-HCl [pH 6.8], 20% glycerol, 4% SDS, 0.2% bromophenol blue) and run on SDS-polyacrylamide gels (19). Polypeptides were identified on the gels by staining with Coomassie blue G250.
Preparation of antibodies and Western blotting.
The antibodies were prepared by Michael Theisen, State Serum Institute, Copenhagen, Denmark. The antibodies were raised in rabbits with a homogenic preparation of homotetrameric CDA purified from E. coli JF611/pSO143. The antibody preparation (serum) was stored at 4°C. Polypeptides separated by SDS-PAGE were transferred to a nitrocellulose membrane (Schleicher & Schüll, Dassel, Germany) in 48 mM Tris–39 mM glycine–1.3 mM SDS–20% methanol by using a Semi-dry electroblotter (JKA-Biotech) for 1 h at 50 mA. The membrane was incubated with phosphate-buffered saline (PBS) (8 mM Na2HPO4, 20 mM KH2PO4, 130 mM NaCl) plus 0.5% Tween 20 for 10 min and subsequently incubated overnight with PBS containing 0.05% Tween 20, 0.37 M NaCl, and antibodies against CDA diluted 1:50. After incubation with primary antibody, the membrane was washed twice in PBS containing 0.05% Tween 20 and 0.37 M NaCl and incubated with swine anti-rabbit immunoglobulin conjugated with horseradish peroxidase (Dako, Copenhagen, Denmark) for 1 h. The membrane was washed twice for 20 min each in PBS containing 0.05% Tween 20 and 0.37 M NaCl and once for 1 min in citrate-phosphate buffer (pH 5.0) (100 mM Na2HPO4, 50 mM citric acid). Staining was carried out in 10 ml of 0.8% (wt/vol) dioctylsodium phosphosuccinate in ethanol (DONS solution)–0.33 ml of tetramethylbenzidine (7% [wt/vol] in DONS solution)–40 ml of citrate-phosphate buffer (pH 5.0)–20 μl of 30% H2O2. Finally, the membrane was washed in 10 ml of DONS solution–40 ml of H2O.
N-terminal amino acid determination.
Heterotetrameric CDA purified from JF611/pSO143 was subjected to SDS-PAGE (13.5% polyacrylamide), and the bands corresponding to the two subunits were blotted to a polyvinylidene difluoride membrane (Bio-Rad) by using a Semi-dry electroblotter (JKA-Biotech). The N-terminal amino acid sequence of the blotted polypeptides was determined by automated Edman degradation on an Applied Biosystems 477A gas-phase Sequenator by Arne Jensen, Department of Protein Chemistry, University of Copenhagen, Copenhagen, Denmark.
Mass spectrometry.
Liquid chromatography-mass spectrometry analysis, using positive-ion electrospray ionization, of the homotetrameric (S4) and the heterotetrameric (S3Sext) forms of CDA was performed. Aliquots of the homotetramer (1,300 pmol) and the heterotetramer (500 pmol) in 5 mM Tris buffer were loaded onto a C18 reversed-phase high-pressure liquid chromatography column (Brownlee Aquapore RP-300; 7-μm pore size, 100 by 2.1 mm), which was used as a trapping device to desalt the protein products for subsequent electrospray ionization and mass analysis. The proteins were washed (desalted) on the high-pressure liquid chromatography column with 30% acetonitrile–0.2% acetic acid for 3 min and then eluted by increasing the acetonitrile to 70% over a period of 2 min at a flow rate of 200 μl/min. About 33% of the eluent was directed to the electrospray interface of a Quattro II mass spectrometer (Micromass, Inc.). Mass spectra were acquired over the range of 750 to 1,250 Da every 5 s with a spray voltage of 2.7 kV and a sample cone voltage of 33 eV. Protein molecular mass spectra were processed through deconvolution of multiply charged molecular ions by using MaxEnt software (Micromass, Inc.) (see Fig. 6). Molecular mass assignments for the standard translation product (homotetramer, 14,854.6 Da) and the frameshift product (heterotetramer, 16,355.9 Da) are within 0.003% mass error of the predicted masses.
FIG. 6.

Electrospray mass spectra of CDA. Top, homotetramer (S4); bottom, heterotetramer (S3Sext). Measured molecular weights are shown.
RESULTS
CDA is expressed as two different subunits from one DNA sequence.
Recombinant B. subtilis CDA was purified from E. coli JF611 harboring the B. subtilis cdd gene on the multicopy plasmid pSO143 (Table 2). SDS-PAGE of the enzyme preparation following DEAE-cellulose chromatography, heat treatment, and gel filtration (Table 2, step 5) showed two bands corresponding to polypeptides with apparent molecular masses of 14.5 and 16 kDa, respectively (Fig. 2, lane 4). Automated Edman degradation of the two polypeptides showed that both had the N-terminal amino acid sequence MNRQE, which is identical to the sequence of the first five N-terminal amino acids deduced from the nucleotide sequence. The coding region of cdd consists of 408 nucleotides encoding a 136-amino-acid polypeptide with a predicted molecular mass of 14,854 Da. This corresponded to the mass of the major polypeptide of the purified CDA. The 16-kDa polypeptide was present in smaller amounts and presumably represented a CDA subunit with an extended C terminus.
FIG. 2.

SDS-PAGE (16% polyacrylamide) of recombinant B. subtilis CDA. Lanes: 1, size markers; 2, crude cellular extract of JF611; 3, crude cellular extract of JF611/pSO143; 4, purified recombinant CDA from step 5 of the purification in Table 1. The polypeptide bands were visualized by staining with Coomassie brilliant blue G-250.
FPLC of the purified CDA preparation (Table 2, step 5) on a Q5 ion-exchange column resolved the enzyme in three peaks (Fig. 3) of approximately the same specific CDA activity (data not shown). SDS-PAGE of the individual peak fractions (Fig. 3) showed that CDA from peak 1 consisted of the 14.5-kDa subunit (S) only and hence represented the homotetrameric form (S4). In contrast, CDA from peaks 2 and 3 contained both S and the extended subunit (Sext). As judged from the relative intensities of the bands on the gel, peaks 2 and 3 represented heterotetrameric forms of the enzyme with the compositions S3Sext and S2Sext2, respectively.
FIG. 3.


Chromatography of purified recombinant CDA from step 5 of Table 1 on a Q5 FPLC column. (Left) Elution profile. Full line, absorbance at 280 nm (A280) of individual fractions; dashed line, salt gradient percentage of 1 M KCl. (Right) SDS-PAGE (13.5% polyacrylamide) of individual peak fractions from the column. Lanes: 1, peak 1; 2, peak 2; 3, peak 3; 4, size markers.
To assess whether the production of Sext was a result of overexpressing the B. subtilis cdd gene in E. coli, a preparation of CDA, partially purified from B. subtilis 168, was subjected to SDS-PAGE and the electropherogram was immunoblotted with polyclonal antibodies raised against the purified recombinant enzyme. As shown in Fig. 4, the immunoblot revealed two bands with about the same mobilities and relative intensities as observed with the recombinant enzyme produced in E. coli.
FIG. 4.
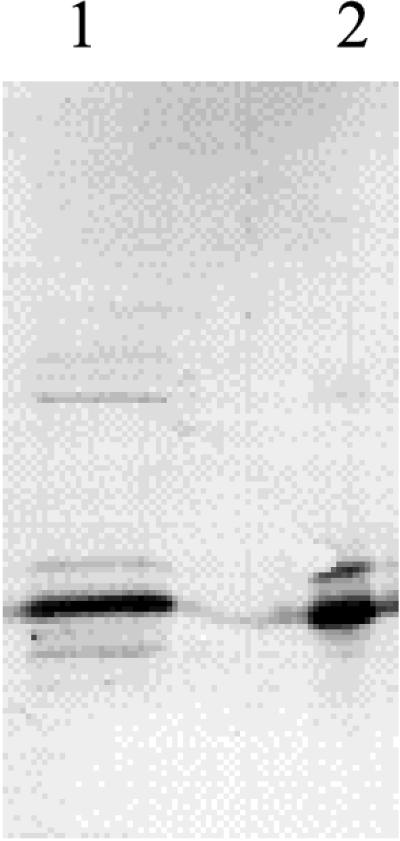
Immunoblot of an SDS-PAGE (13.5% polyacrylamide) electropherogram with antibodies raised against recombinant B. subtilis CDA. Lanes: 1, partially purified CDA from B. subtilis 168; 2, crude extract of JF611/pSO143.
The ribosome slips to the −1 frame in front of the cdd stop codon.
The apparent molecular mass of Sext (16 kDa) in conjunction with analysis of the DNA sequence of pSO143 suggested that a −1 frameshift had occurred late in the cdd gene, resulting in a 13-amino-acid extension of the CDA subunit (Fig. 1A). To examine this further, lacZ′ from pUC19 was fused to cdd in all three reading frames downstream of the stop codon, yielding pSO1001, pSO1000, and pSO1002. A −1 frameshift occurring before the stop codon of cdd carried by these plasmids would result in the synthesis of CDA subunits with 93-, 19-, and 57-amino-acid extensions and molecular masses of 25.6, 17.0, and 20.9 kDa, respectively. E. coli cells harboring pSO1001, pSO1000, and pSO1002 were analyzed by SDS-PAGE and immunoblotting as described above. As shown in Fig. 5, the apparent sizes of the extended subunits were all in accordance with the values predicted as a result of a −1 frameshift occurring in front of the cdd stop codon. Accordingly, the pSO1001 construct, which constituted the in-frame situation for lacZ′ in the −1 reading frame of cdd, mediated β-galactosidase activity when transformed into SØ5299, a lac deletion strain carrying φ80dlacΔ(lacZ)M15.
FIG. 5.

Immunoblot of an SDS-PAGE (15% polyacrylamide) electropherogram with antibodies raised against recombinant B. subtilis CDA. Crude cellular extracts of the following strains were used: lane 1, JF611/pSO143; lane 2, SØ5299/pSO1001; lane 3, SØ5299/pSO1000; lane 4, SØ5299/pSO1002.
Identification of the slip site.
Mass spectrometric analysis of purified homotetrameric (Fig. 3, peak 1) and heterotetrameric (peak 2) CDA established that the exact masses of the two subunits, S and Sext, were 14,854.5 and 16,355.9 Da, respectively (Fig. 6). The 14,854.5-Da value agreed exactly with a mass of 14,854 Da for S calculated from the deduced amino acid sequence. Inspection of the nucleotide sequence revealed that a −1 frameshift at the penultimate cdd codon, i.e., AAG to AAA (Fig. 1), would give rise to a subunit with a deduced molecular mass of 16,356 Da, in complete accordance with the determined size of 16,355.9 Da. No other −1 frameshift in the distal end of cdd would give rise to a subunit of that size.
Analysis of the slip site by site-directed mutagenesis.
To examine the effect of the nucleotide sequence context on the −1 frameshift at the putative shift site, A-AAG, mutations were introduced at each of the five last codons of the gene, including the termination codon. The mutations were made in pNMJ62, which contained lacZ′ fused in the −1 frame of cdd immediately following the termination codon. With the exception of one insertion mutation (AAG → AAAG) and the change of the stop codon to a sense codon (TAA → TAC), the mutations did not change the amino acid sequence in the 0 reading frame. The resulting constructs were transformed into SØ5299, and the level of β-galactosidase in the transformants was used as a measure of frameshifting. Table 3 summarizes the results obtained with the different mutants. As an indicator of the intracellular level of functional CDA transcript, the specific CDA activity was also determined in each strain (data not shown). Two of the mutants, i.e., Stop UAA → Tyr UAC and AAG → AAAG, produced no detectable CDA activity. Translation of these constructs in the 0 frame produced CDA subunits with 51- and 86-amino-acid extensions, respectively, which presumably are incapable of forming active enzymes. Correction of the β-galactosidase values for CDA activity did not result in major changes in the measured frameshift levels, as shown by the numbers in parentheses in Table 3.
TABLE 3.
Mutational analysis of the shift region in the 3′ terminus of cdda
| Mutation | DNA sequence and deduced amino acidsb | β-Galactosidase activityc
|
|
|---|---|---|---|
| In crude extract | % (corrected)d | ||
| K A L LacZ′ | |||
| S E D L H D E R K L * | |||
| Wild type | TCG GAG GAT TTA CAT GAC GAA CGA AAG CTT TAA | 7,000 ± 250 | 100 (100) |
| Stop UAA → Tyr UAC | ........................................TAC. | 10,800 ± 450 | 155 (—)e |
| Leu CUU → Leu CUG | ....................................CTG..... | 6,000 ± 600 | 85 (74) |
| Glu GAA → Glu GAG | ........................GAG................. | 6,400 ± 200 | 91 (104) |
| Lys AAG → Lys AAA | ................................AAA......... | 410 ± 40 | 6 (3) |
| Arg CGA → Arg CGC | ............................CGC............. | 81 ± 14 | 1 (2) |
| Arg CGA → Arg AGA | ............................AGA............. | 380 ± 40 | 5 (1) |
| GGAGG → CGAAG | ..C GAA G................................... | 640 ± 40 | 9 (7) |
| AAG → AAAG | ...............................AAAG......... | 44,200 ± 1,600 | 630 (—) |
All mutations were introduced into pNMJ62, and the resulting plasmids were transformed into SØ5299.
The deduced amino acid sequence is shown above the DNA sequence in the 0 (bottom) and −1 (top) reading frames with respect to CDA. The CDA stop codon is in italics, the shift site is underlined, and the SD-like sequence is overlined. Mutations are indicated in bold letters, and the insertion is in bold italics.
Enzyme activities were measured in sonic extracts of cells from exponentially growing cultures. β-Galactosidase activities are expressed in nanomoles per minute per milligram of protein and are the means of nine measurements.
Percentage of the activity measured in the wild type. The numbers in parentheses are corrected for the level of CDA activity in the extracts (β-galactosidasemutant × CDAwild type × 100/CDAmutant × β-galactosidasewild type).
—, no detectable CDA activity in extracts.
Changing the UAA stop codon to the tyrosine codon UAC, the leucine codon CUU to CUG, or the 5′ glutamic acid codon GAA to GAG had only a minor effect on the β-galactosidase level, resulting in a frameshift level of 155, 85, or 91%, respectively, compared to that of the wild type. In contrast, mutations in the lysine and arginine codons that changed the sequence A-AAG had dramatic effects. Thus, the frameshift level dropped to 6% when the lysine codon AAG was changed to AAA, and modifications of the rare CGA codon for arginine to the common CGC codon resulted in barely detectable frameshift levels. Mutating the CGA codon to arginine AGA also resulted in very low levels of frameshifting, indicating that the minimal sequence requirement for efficient frameshifting was CGA AAG.
Stimulatory mRNA elements.
An SD-like sequence, capable of forming five Watson-Crick base pairs with the 3′ end of 16S rRNA, is located 14 nucleotides 5′ of the slip region CGA AAG. Since an SD sequence has been shown to stimulate −1 frameshifting in the E. coli dnaX gene (21), this region was mutated such that base pairing with the 16S rRNA was weakened without changing the corresponding protein product. Modifying the sequence G GAG GA to C GAA GA reduced the frameshift level to 9% (Table 3), indicating that this region on the mRNA is a 5′ stimulatory element for the frameshift.
A number of frameshift events were found to be stimulated by secondary mRNA structures such as pseudoknots or stem-loop structures located 3′ of the shift site. A computer search for stable downstream secondary mRNA structures did not identify any putative stimulatory element. Since dramatic changes in the sequence of the downstream mRNA, such as those present in pSO1000, pSO1001, and pSO1002, did not affect the level of frameshifting significantly (Fig. 5), it was inferred that no shift-stimulating structure was located downstream of the shift site.
Frameshift level.
As judged by SDS-PAGE, the number of Sext subunits was roughly 10 to 20% of that of the number of S subunits (Fig. 2, 4, and 5). To make a more accurate measurement of the wild-type frameshift level, we inserted an additional A at the shift site in pNMJ62, i.e., AAG → AAAG. In this mutant, lacZ′ was in frame with the 0 reading frame of cdd. As shown in Table 3, this mutant mediated a β-galactosidase level 6.3 times higher than that of the wild-type construct, indicating that ribosome frameshift was about 16%. In the absence of the upstream SD-like sequence, frameshifting was reduced to about 1.5%.
DISCUSSION
Studies of B. subtilis CDA expression in E. coli have revealed a new frameshift site, CGA AAG, and probably a new type of disposition of the tRNAs involved. The intrinsic shiftiness of this shift site on its own is 1.5% (Table 3), compared to 2% seen with A AAA AAG (21), which rates it as a good shift site. Simultaneous slippage of tandem tRNAs, as has been observed on sequences of the general form X XXY YYZ, is not involved, as evidenced by the lack of re-pairing possibilities for the cognate CGA-decoding arginine tRNA, whose anticodon is 3′GCI5′, where I is inosine. A and I are the only two purines that face each other in decoding. If they were to pair, it would require nonstandard geometry that may destabilize the codon-anticodon complex (10, 23). It seems probable that the I frequently occludes the A and triplet coding ensues. However, occasionally the A may be available for pairing as the first base of the next codon, resulting in doublet translocation and −1 frameshifting (Fig. 7). A somewhat similar scenario was previously observed for decoding of the glycine GGA codon. It was shown that normal levels of tRNA-Gly2 with a single substitution that gave a CCC anticodon mediated both doublet and triplet decoding of GGA. Due to several differences elsewhere in the tRNA, it requires overexpression of E. coli tRNA-Gly1, which has the same anticodon, to mediate detectable doublet decoding (30). Previously, Lagerkvist and colleagues showed that C at position 32 of Mycoplasma mycoides tRNA-Gly was substantially responsible for its decoding all four glycine GGN codons (20, 24). Replacement of the base at position 32 of E. coli tRNA-Gly1 with C led to enhanced efficiency of doublet decoding.
FIG. 7.

Model of −1 frameshifting in CDA. U*, 5-methylaminomethyl-2-thiouridine.
Pairing of the third base of the second codon, AAG, within the shift site to its cognate lysine-tRNA is also compromised. In E. coli, the lysine codons AAG and AAA are both decoded by a single tRNA with the anticodon 5′-mnm5s2UUU-3′ containing 5-methylaminomethyl-2-thiouridine at the wobble position. The modification causes more efficient binding of the tRNA to AAA than to AAG in vitro (25, 42). In the present case, frameshifting involves dissociation from the AAG codon and re-pairing to the overlapping AAA codon, which includes the A which previously flirted with inosine (Fig. 7). The observation that replacing AAG by AAA caused a strong reduction in frameshifting indicates that binding of tRNA-Lys to AAA is favored over binding to AAG in vivo. Interestingly, replacing the rare CGA codon with another rare arginine codon, AGA, resulted in a very low level of frameshifting, despite conservation of the shifty A AAG sequence. Most probably this is because the AGA codon is decoded by tRNA-Arg with the anticodon UCU, which forms three Watson-Crick base pairs in the codon-anticodon complex, thereby making A at the third position unavailable for re-pairing with the tRNA-Lys in the −1 frame.
The presence of an SD-like sequence 14 nucleotides upstream of the slip site resulted in an 11-fold stimulation of −1 frameshifting (Table 3). Previously, SD interactions by translating ribosomes were found to stimulate −1 frameshifting in decoding of the dnaX gene of E. coli (41), and it was shown by Larsen et al. (21) that the optimal spacing between the SD sequence and the slip site was 10 to 13 nucleotides, i.e., slightly shorter than in the present situation. Mutations in regions of the 16S rRNA near the 3′ end have been shown to promote stop codon readthrough and frameshifting during elongation, indicating that these regions are in proximity to the site of codon-anticodon interaction in the ribosome (31). It seems likely that when the spacing between the SD sequence and the site of action is significantly larger than the optimal spacing for translation initiation, the ribosome will be strained and will tend to pull the peptidyl-tRNA toward the −1 frame.
The frameshift in the 3′ end of the B. subtilis cdd gene occurs not only when the gene is expressed from a multicopy plasmid in E. coli but also in the native condition when the gene is expressed from the chromosome in B. subtilis. Thus, slippage is not due to overexpression of an alien sequence with exotic codon usage. The tRNA anticodons of E. coli (17) and B. subtilis (18) are very similar, and no tRNAs with perfectly matching anticodons exist for the arginine CGA and the lysine AAG codons in either organism. The 3′ sequences of the 16S rRNA from the two bacteria are similar and will presumably base pair equally well with the SD sequence upstream of the cdd stop codon (27). Therefore, it seems likely that the same mechanism is responsible for the frameshifting in both organisms.
The frameshift appears to be irrelevant to CDA expression since no significant difference in CDA activity of the homotetrameric and the heterotetrameric forms of the enzyme was found. However, the 5′ end of the bex gene overlaps the 3′ end of cdd by 20 nucleotides and the SD-like sequence discussed above is part of the ribosome-binding site for initiation of bex translation (Fig. 1A). The bex gene encodes, in the +1 reading frame relative to cdd, a polypeptide of 301 amino acids. The deduced amino acid sequence of Bex has 39% identity to the gene product of the E. coli era gene, which encodes a membrane-bound G-protein essential for cell growth (1). The function of bex is unknown, but it has been shown to complement an era mutation in E. coli (33). The frameshift event may be expected to cause a translating ribosome to pause in the SD region, thereby preventing initiation of bex translation. Accordingly, any physiological condition that affects frameshifting at this site would be expected to influence bex expression. In that context, it should be recalled that B. subtilis is a differentiating eubacterium and that developmental changes as well as changes in the growth conditions strongly influence the modification of tRNA in this organism (3). A search of the current databases revealed that of the nine putative prokaryotic CDA genes recovered, only the B. subtilis cdd gene displayed the shifty CGA AAG motif at the 3′ end of the gene and only B. subtilis showed the overlapping organization of cdd and bex (era). Furthermore, the search revealed in Archaeoglobus fulgidus, where the gene for 16S rRNA has the sequence CCTCCT at its 3′ end, a GGAGG at nucleotide 7169 (16) followed 11 nucleotides 3′ by CGA AAG. There is a potential initiation codon in the preshift open reading frame followed by 61 codons before the CGA AAG and then 101 codons in the −1 frame before a stop codon. While in this case there is no indication that this is even a real gene, an extensive search for the utilization of CGA AAG-programmed frameshifting in gene expression seems merited.
ACKNOWLEDGMENTS
We thank Frode Engbæk for facilitating the collaboration and Chad Nelson for mass spectrometry work. The help of Barry Moore and Lisbeth Stauning is gratefully acknowledged.
This work was funded by the Danish National Research Foundation to J.N. J.F.A. is funded by NIH grant GM48152 and was supported by the Howard Hughes Medical Institute for part of this work.
REFERENCES
- 1.Ahnn J, March P E, Takiff H E, Inouye M. A GTP-binding protein of Escherichia coli has homology to yeast RAS proteins. Proc Natl Acad Sci USA. 1986;83:8849–8853. doi: 10.1073/pnas.83.23.8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertani G. Studies of lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol. 1951;62:293–300. doi: 10.1128/jb.62.3.293-300.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Björk G R, Ericson J U, Gustafsson C E, Hagervall T G, Jönsson Y H, Wikström P M. Transfer RNA modification. Annu Rev Biochem. 1987;56:263–287. doi: 10.1146/annurev.bi.56.070187.001403. [DOI] [PubMed] [Google Scholar]
- 4.Blinkova A L, Walker J R. Programmed ribosomal frameshifting generates the Escherichia coli DNA polymerase III γ subunit from within the τ subunit reading frame. Nucleic Acids Res. 1990;18:1725–1729. doi: 10.1093/nar/18.7.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chandler M, Fayet O. Translational frameshifting in the control of transposition in bacteria. Mol Microbiol. 1993;7:497–503. doi: 10.1111/j.1365-2958.1993.tb01140.x. [DOI] [PubMed] [Google Scholar]
- 6.Clark D J, Maaløe O. DNA replication and the cell division cycle in Escherichia coli. J Mol Biol. 1967;2:99–112. [Google Scholar]
- 7.Cohen R M, Wolfenden R. Cytidine deaminase from Escherichia coli. Purification, properties, and inhibition by the potential transition state analog 3,4,5,6-tetrahydrouridine. J Biol Chem. 1971;246:7561–7565. [PubMed] [Google Scholar]
- 8.Craigen W J, Caskey C T. Expression of peptide chain release factor 2 requires high-efficiency frameshift. Nature. 1986;322:273–275. doi: 10.1038/322273a0. [DOI] [PubMed] [Google Scholar]
- 9.Curran J, Yarus M. Use of the tRNA suppressors to probe regulation of Escherichia coli release factor 2. J Mol Biol. 1988;203:75–83. doi: 10.1016/0022-2836(88)90092-7. [DOI] [PubMed] [Google Scholar]
- 10.Curran J F. Decoding with the A:I wobble pair is inefficient. Nucleic Acids Res. 1995;23:683–688. doi: 10.1093/nar/23.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farabaugh P J. Programmed translational frameshifting. Microbiol Rev. 1996;60:103–134. doi: 10.1128/mr.60.1.103-134.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flower A M, McHenry C S. The gamma subunit of DNA polymerase III holoenzyme of Escherichia coli is produced by ribosomal frameshifting. Proc Natl Acad Sci USA. 1990;87:3713–3717. doi: 10.1073/pnas.87.10.3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gesteland R F, Atkins J F. Recoding: dynamic reprogramming of translation. Annu Rev Biochem. 1996;65:741–768. doi: 10.1146/annurev.bi.65.070196.003521. [DOI] [PubMed] [Google Scholar]
- 14.Gramstat A, Prüfer D, Rohde W. The nucleic acid-binding zinc finger protein of potato virus M is translated by internal initiation as well as by ribosomal frameshifting involving a shifty stop codon and a novel mechanism of P-site slippage. Nucleic Acids Res. 1994;22:3911–3917. doi: 10.1093/nar/22.19.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacks T, Madhani H D, Masiarz F R, Varmus H E. Signals for ribosomal frameshifting in the Rous sarcoma virus gag-pol region. Cell. 1988;55:447–458. doi: 10.1016/0092-8674(88)90031-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klenk H-P, Clayton R A, Tomb J-F, White O, Nelson K E, et al. The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature. 1997;390:364–370. doi: 10.1038/37052. [DOI] [PubMed] [Google Scholar]
- 17.Komine Y, Adachi T, Inokuchi H, Ozeki H. Genomic organization and physical mapping of the transfer RNA genes in Escherichia coli K12. J Mol Biol. 1990;212:579–598. doi: 10.1016/0022-2836(90)90224-A. [DOI] [PubMed] [Google Scholar]
- 18.Kunst F, Ogasawara N, Moszer I, Albertini A M, Alloni G, Azevedo V, et al. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature. 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- 19.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;277:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 20.Lagerkvist U. “Two out of three”: an alternative method for codon reading. Proc Natl Acad Sci USA. 1978;75:1759–1762. doi: 10.1073/pnas.75.4.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Larsen B, Wills N M, Gesteland R F, Atkins J F. rRNA-mRNA base pairing stimulates a programmed −1 ribosomal frameshift. J Bacteriol. 1994;176:6842–6851. doi: 10.1128/jb.176.22.6842-6851.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larsen B, Gesteland R F, Atkins J F. Structural probing and mutagenic analysis of the stem-loop required for Escherichia coli dnaX ribosomal frameshifting: programmed efficiency of 50% J Mol Biol. 1997;271:47–60. doi: 10.1006/jmbi.1997.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim V, Venclovas C. Codon-anticodon pairing. A model for interacting codon-anticodon duplexes located at the ribosomal A- and P-sites. FEBS Lett. 1992;313:133–137. doi: 10.1016/0014-5793(92)81429-p. [DOI] [PubMed] [Google Scholar]
- 24.Lustig F, Borén T, Claesson C, Simonsen C, Barciszewska M, Lagerkvist U. The nucleotide in position 32 of the tRNA anticodon loop determines ability of anticodon UCC to discriminate among glycine codons. Proc Natl Acad Sci USA. 1993;90:3343–3347. doi: 10.1073/pnas.90.8.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lustig F, Elias P, Axberg T, Samuelsson T, Tittawella I, Lagerkvist U. Codon reading and translational error. Reading of the glutamine and lysine codons during protein synthesis in vitro. J Biol Chem. 1981;256:2635–2643. [PubMed] [Google Scholar]
- 26.Matsufuji S, Matsufuji T, Miyazaki Y, Murakami Y, Atkins J F, Gesteland R F, Hayashi S. Autoregulatory frameshifting in decoding mammalian ornithine decarboxylase antizyme. Cell. 1995;80:51–60. doi: 10.1016/0092-8674(95)90450-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McLaughlin J R, Murray C L, Rabinowitz J C. Unique features in the ribosome binding site sequence of the gram-positive Staphylococcus aureus β-lactamase gene. J Biol Chem. 1981;256:11283–11291. [PubMed] [Google Scholar]
- 28.Miller J. Experiments in molecular genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- 29.Nilsson J, Koskiniemi S, Persson K, Grahn B, Holm I. Polyamines regulate both transcription and translation of the gene encoding ornithine decarboxylase antizyme in mouse. Eur J Biochem. 1997;250:223–231. doi: 10.1111/j.1432-1033.1997.0223a.x. [DOI] [PubMed] [Google Scholar]
- 30.O’Connor M. tRNA imbalance promotes −1 frameshifting via near-cognate decoding. J Mol Biol. 1998;279:727–736. doi: 10.1006/jmbi.1998.1832. [DOI] [PubMed] [Google Scholar]
- 31.O’Connor M, Thomas C L, Zimmermann R A, Dahlberg A E. Decoding fidelity at the ribosomal A and P sites: influence of mutations in three different regions of the decoding domain in 16S rRNA. Nucleic Acids Res. 1997;25:1185–1193. doi: 10.1093/nar/25.6.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Polard P, Prère M F, Chandler M, Fayet O. Programmed translational frameshifting and initiation at an AUU codon in gene expression of bacterial insertion sequence IS911. J Mol Biol. 1991;222:465–477. doi: 10.1016/0022-2836(91)90490-w. [DOI] [PubMed] [Google Scholar]
- 33.Powell, B. December 1994. Accession no. U18532. [Online.] http://www.embl-heidelberg.de/srs5/. [6 April 1999, last date accessed.]
- 34.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 35.Sanger J, Nicklen S, Coulson A R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saxild H H, Nygaard P. Genetic and physiological characterization of Bacillus subtilis mutants resistant to purine analogs. J Bacteriol. 1987;169:2977–2983. doi: 10.1128/jb.169.7.2977-2983.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song B H, Neuhard J. Chromosomal location, cloning and nucleotide sequence of the Bacillus subtilis cdd gene encoding cytidine/deoxycytidine deaminase. Mol Gen Genet. 1989;216:462–468. doi: 10.1007/BF00334391. [DOI] [PubMed] [Google Scholar]
- 38.Tsuchihashi Z. Translational frameshifting in the Escherichia coli dnaX gene in vitro. Nucleic Acids Res. 1991;19:2457–2462. doi: 10.1093/nar/19.9.2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsuchihashi Z, Kornberg A. Translational frameshifting generates the gamma subunit of DNA polymerase III holoenzyme. Proc Natl Acad Sci USA. 1990;87:2516–2520. doi: 10.1073/pnas.87.7.2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weiss R B, Dunn D M, Atkins J F, Gesteland R F. Slippery runs, shifty stops, backward steps, and forward hops: −2, −1, +1, +2, +5, and +6 ribosomal frameshifting. Cold Spring Harbor Symp Quant Biol. 1987;52:687–693. doi: 10.1101/sqb.1987.052.01.078. [DOI] [PubMed] [Google Scholar]
- 41.Weiss R B, Dunn D M, Dahlberg A E, Atkins J F, Gesteland R F. Reading frame switch caused by base-pair formation between the 3′ end of 16S rRNA and the mRNA during elongation of protein synthesis in Escherichia coli. EMBO J. 1988;7:1503–1507. doi: 10.1002/j.1460-2075.1988.tb02969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yokoyama S, Watanabe T, Murao K, Ishikura H, Yamaizumi Z, Nishimura S, Miyazawa T. Molecular mechanism of codon recognition by tRNA species with modified uridine in the first position of the anticodon. Proc Natl Acad Sci USA. 1985;82:4905–4909. doi: 10.1073/pnas.82.15.4905. [DOI] [PMC free article] [PubMed] [Google Scholar]