

Abstract
The cell cycle is a progression of 4 distinct phases (G1, S, G2, and M), with various cycle proteins being essential in regulating this process. We aimed to develop a radiolabeled cyclin-dependent kinase 4/6 (CDK4/6) inhibitor for breast cancer imaging. Our transfluorinated analog (18F-CDKi) was evaluated and validated as a novel PET imaging agent to quantify CDK4/6 expression in estrogen receptor (ER)–positive human epidermal growth factor receptor 2 (HER2)–negative breast cancer. Methods: 18F-CDKi was synthesized and assayed against CDK4/6 kinases. 18F-CDKi was prepared with a 2-step automated synthetic strategy that yielded the final product with remarkable purity and molar activity. In vitro and in vivo biologic specificity was assessed in a MCF-7 cell line and in mice bearing MCF-7 breast tumors. Nonradioactive palbociclib was used as a blocking agent to investigate the binding specificity and selectivity of 18F-CDKi. Results: 18F-CDKi was obtained with an overall radiochemical uncorrected yield of 15% and radiochemical purity higher than 98%. The total time from the start of synthesis to the final injectable formulated tracer is 70 min. The retention time reported for 18F-CDKi and 19F-CDKi is 27.4 min as demonstrated by coinjection with 19F-CDKi in a high-pressure liquid chromatograph. In vivo blood half-life (weighted, 7.03 min) and octanol/water phase partition coefficient (1.91 ± 0.24) showed a mainly lipophilic behavior. 18F-CDKi is stable in vitro and in vivo (>98% at 4 h after injection) and maintained its potent targeting affinity to CDK4/6. Cellular uptake experiments performed on the MCF-7 breast cancer cell line (ER-positive and HER2-negative) demonstrated specific uptake with a maximum intracellular concentration of about 65% as early as 10 min after incubation. The tracer uptake was reduced to less than 5% when cells were coincubated with a molar excess of palbociclib. In vivo imaging and ex vivo biodistribution of ER-positive, HER2-negative MCF-7 breast cancer models showed a specific uptake of approximately 4% injected dose/g of tumor (reduced to ∼0.3% with a 50-fold excess of cold palbociclib). A comprehensive biodistribution analysis also revealed a significantly lower activation of CDK4/6 in nontargeting organs. Conclusion: 18F-CDKi represents the first 18F PET CDK4/6 imaging agent and a promising imaging agent for ER-positive, HER2-negative breast cancer.
Keywords: cell cycle, CDK4/6, PET/CT imaging, breast cancer, MCF-7
The cell cycle is a progression of 4 distinct phases (G1, S, G2, and M) that cells use to maintain the integrity of their genomes (1–5). Sophisticated cell cycle pathways and cell cycle arrest proteins are used to remediate DNA damage, efficiently control the cell cycle and DNA replication (5,6), and trigger DNA repair. Alterations in the cell cycle and uncontrolled proliferation are well-known hallmarks of cancer (7,8). Cyclin-dependent kinases 4/6 (CDK4/6) are 2 apical kinases that control the cell cycle by arresting the progression in case of DNA damage (9–12). Subsequent to cell damage, the cyclin D1 (cycD1)–CDK4/6 complex arrests the cell cycle progression in G1 to limit the proliferation of the DNA-damaged cells (13). Dysregulation of the cycD1-CDK4/6 axis appears to be an early step in cancer pathogenesis, and cycD1 overexpression is shown as early as ductal carcinoma in both in situ and metastatic lesions (7,13–17). Notably, amplification of genes encoding D-type cyclins is commonly observed in human cancer and correlates with increased levels of cyclin D protein. A major target of CDK4 and CDK6 during cell cycle progression is the retinoblastoma protein. CDK4/6-cycD1 complexes phosphorylate retinoblastoma. When retinoblastoma is phosphorylated, it dissociates from the E2F family, which enables cell cycle progression to the S phase. Selective inhibitors act on CDK4/6 kinases, dephosphorylate retinoblastoma, and stall the cell cycle progression in G1. This action inhibits the proliferation of cancer cells and triggers the DNA-damage repair. Hence, it is intuitive that efficient inhibition of CDK4/6 can enhance and amplify the chemotherapeutic effects of therapies aimed at targeting the cell cycle proliferation, checkpoints, and arrest.
Amplification of CDK4/6 and cycD1 has been reported in a significant number of cancers, and overexpression of cycD1 was observed in more than 60% of all breast cancers (14,18–20). Furthermore, amplification and overexpression of cycD1 and CKD4/6 has been described in patients with head and neck cancer (21), non–small cell lung cancer (22), melanoma (23–25), and glioblastoma (26). Gene amplification of cycD1 was found to be most frequent in luminal A, luminal B, and human epidermal growth factor receptor 2 (HER2)–enriched breast cancer subtypes (frequencies of alteration: 29%, 58%, and 38%, respectively) (7). Similarly, amplifications of CDK4/6 were more common in the luminal A, luminal B, and HER2-enriched subgroups (14%, 25%, and 24%, respectively) (7). Moreover, in patients with luminal estrogen receptor (ER)–positive breast cancer, which represents approximately 75% of all breast cancers, cycD1-CDK4/6 is expressed at a high level (15). Thus, ER-positive, luminal breast cancer is the typical and the most indicated model for investigating the effectiveness of CDK4/6 inhibitors and served as a platform for our initial biologic evaluation (7).
We developed 18F-CDKi, a PET-radiolabeled version of palbociclib (Ibrance; Pfizer) to noninvasively image kinase expression in breast cancer models. Specifically, we introduced an 18F prosthetic group (18F-fluorobenzoic acid) on the terminal piperazine (27) and synthesized a novel PET-active functional molecule. We aimed to determine whether 18F-CDKi has suitable pharmacokinetic properties for noninvasive PET imaging and whether the tracer is selective for CDK4/6. For both in vitro and in vivo evaluation, a breast cancer ER-positive, HER2-negative cell line (MCF-7) was used.
To our knowledge, this is a first-in-class radiolabeled inhibitor able to target specifically CDK4/6. Because of these promising results in mouse models, we anticipate 18F-CDKi to have a high prognostic value for tumor imaging and treatment response monitoring.
MATERIALS AND METHODS
Materials
No-carrier-added 18F-fluoride was produced by the (p,n) reaction of 18O-H2O in an RDS-112 cyclotron (Siemens). Potassium carbonate, Kryptofix, ethyl 4-nitrobenzoate, N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU), triethylamine, ammonium formate, sodium hydroxide, hydrochloric acid, acetonitrile, and dimethyl sulfoxide were purchased from Millipore-Sigma and used without further purification unless otherwise stated. Palbociclib hydrochloride was purchased from Selleck. Adenosine triphosphate was obtained from PerkinElmer. Ethanol was purchased from Thermo Fisher. Sep-Pak Accell Plus quaternary methyl ammonium and C18 Sep-Pak cartridges were purchased from Waters. Semipreparative high-pressure liquid chromatography (HPLC) was conducted in a TRACERlab FX2N (GE Healthcare) under the following HPLC conditions: Phenomenex Gemini C6 phenyl column (10 × 250 mm, 10 μm); mobile phase, 64% 75 mM ammonium fluoride and 36% MeCN; 5 mL/min (method A). Analytic HPLC was performed using a Shimadzu binary LC-20AR HPLC gradient pump with an inline SPD-20A variable wavelength ultraviolet/visible-light detector and a flow count unit with a PMT detector using a Phenomenex C6 Gemini reverse-phase column (4.6 × 250 mm, 5 μm) with a mobile phase of 64% 75 mM ammonium fluoride and 36% MeCN at a rate of 2 mL/min (method B). Analytic chromatograms were collected by an analog-to-digital converter using Lab Solutions software. Preparative and analytic HPLC analyses of 18F-labeled compounds were calibrated with the corresponding 19F analogs. Radioactivity in blood half-life, cell uptake, and biodistribution studies was quantified with a WIZARD2 automatic γ-counter (PerkinElmer). All in vivo experiments were carried out with Inveon PET/CT (Siemens Medical Solutions) and reconstructed using Inveon Research (Siemens Medical Solutions). All 18F and 19F CDKi final products were formulated in 10% ethanol/90% saline (0.9%).
Cell Culture
Phosphate-buffered saline and Dulbecco modified Eagle medium were purchased from Thermo Fisher. MCF-7, a human ER-positive, HER2-negative breast cancer cell line, was purchased from ATCC. Dulbecco modified Eagle medium contained 10% (v/v) heat-inactivated fetal bovine serum, 100 IU penicillin, and a 100 μg/mL concentration of streptomycin.
Mouse Model
Female athymic nude CrTac:NCr-Foxn1nu mice (n = 35) were purchased from Taconic Laboratories. Twenty mice received subcutaneous injections with 2 × 106 human MCF-7 cancer cells in Matrigel (BD Biosciences) into each right shoulder and were allowed to grow for approximately 2 mo until the tumors reached about 10 mm in diameter. All mice were supplemented with a 0.72-mg slow-release estradiol pellet (left for 60 d) implanted in the left flank (Innovative Research of America, Sarasota, FL). Mice were anesthetized (isoflurane, 1.5%; 2 L/min flow of medical air) during tumor implantation and small-animal PET imaging.
Fifteen mice were used for blood half-life measurements and in vivo stability studies. All animal experiments were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee of NYU Langone Health and followed National Institutes of Health guidelines for animal welfare.
Preparation of 19F-CDKi
To a solution of palbociclib in dimethylsulfoxide (20 mg, 0.04 mmol), fluorobenzoic acid (15 mg, 0.1 mmol), HBTU (45 mg, 0.1 mmol), and triethylamine (12 mg, 0.1 mmol) were added and the solution reacted at 37°C for 24 h. The resulting solution was filtered and purified by reverse-phase HPLC to the desired derivative (yield, 70%), dried, and lyophilized to yield the final product. Electrospray ionization mass spectrometry spectra were recorded with a Shimadzu LC-2020 with electrospray ionization single-quad detector.
Radiochemistry
No-carrier-added 18F-fluoride was obtained via the 18O(p,n)18F nuclear reaction of 11-MeV protons in an RDS Eclipse (Siemens Medical Solutions). Synthesis was automated using an FX2N module (GE Healthcare). Briefly, a quaternary methyl ammonium cartridge containing cyclotron-produced 18F-fluoride ion was eluted with a solution containing 9 mg of Kryptofix [2.2.2] (4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane), 0.08 mL of 0.15 M K2CO3, and 1.92 mL of MeCN into a 5-mL reaction vial. Water was removed azeotropically at 120°C. One milligram of ethyl-4-nitrobenzoate was dissolved in 300 μL of dimethylsulfoxide and added to the reaction vial, heated to 150°C for 15 min, and then cooled to room temperature. Afterward, 150 μL of 1 M NaOH were added. The reaction mixture was stirred for 1 min, and 150 μL of 1 M HCl were added to quench. Then, 2 mg of palbociclib dissolved in 200 μL of dimethylsulfoxide were added, followed by 10 mg of HBTU dissolved in 200 μL of dimethylsulfoxide and 30 μL of Et3N. MeCN (400 μL) followed by 700 μL of H2O was then added, and the solution was injected onto a C6-phenyl analytic HPLC column and eluted under isocratic conditions. Then, the collected fraction was passed through a C18 light-SepPak cartridge and 18F-CDKi was eluted using EtOH (400 μL). The solution was then diluted with 0.9% saline to 10% EtOH. Molar activity was determined by dividing the activity present in the final formulated product (GBq) by the material remaining in the formulated product after purification (moles) and was determined using an ultraviolet calibration curve (λ = 254 nm).
Chemical Hydrophobicity Index and Octanol/Water Partition Coefficient
The chemical hydrophobicity indices were measured using a previously developed procedure (28,29). Briefly, reverse-phase HPLC was used to measure the retention times of a set of standards with known chemical hydrophobicity index. A standard curve was then created to calculate the chemical hydrophobicity index of 19F-CDKi based on the HPLC retention time. The lipophilicity of the 18F-CDKi was acquired by adding 0.09 MBq to a mixture of 0.5 mL of 1-octanol and 0.5 mL of 25 mM phosphate-buffered saline (pH 7.4) and mixed for 5 min. Then, the mixture was centrifuged at 15,000 rpm for 5 min. Samples (100 μL) were obtained from organic and aqueous layers, and the radioactivity of the samples was measured in a WIZARD2 automatic γ-counter. The experiment was performed in triplicate, and the resulting octanol–water partition coefficient was calculated as the mean ± SD.
Blood Half-Life
The blood half-life of 18F-CDKi was calculated by measuring the activity of blood samples collected at different time points after injection (5, 15, 30, 45, 60, 90, and 120 min). Female nude mice (n = 3) were injected via the lateral tail vein with 18F-CDKi, and blood samples were obtained by a retroorbital bleed using tared capillary tubes. Samples were weighed, and activity was measured by γ-counter. The blood half-life was calculated by Prism 7 (GraphPad Software) using a 2-phase decay least-squares fitting method and expressed as percentage injected dose (%ID)/g.
In Vivo and Ex Vivo Blood Stability
18F-CDKi (7.4 MBq) was injected in healthy athymic nude mice (n = 15) via tail injection. The mice were killed at different time points (0, 60, 120, 180, and 240 min after injection), and blood was collected. MeCN (750 μL) was added to the collected blood and then centrifuged (5 min at 5,000 rpm) to pellet blood cells and proteins. The supernatant was collected, diluted with 750 μL of H2O, and injected onto an HPLC column. The blood stability was measured by HPLC analysis (method B).
Half-Maximal Inhibitory Concentration (IC50) Binding Affinity and Competitive Inhibitory Displacement
IC50 values were determined using a competitive adenosine triphosphate quantitative assay as reported elsewhere (30) and 19F-CDKi. MCF-7 cells were cultured at 37°C for 4 h. Then, 19F-CDKi or vehicle was added at different concentrations and incubated at 37°C for 96 h. Cell viability was then assessed using a CellTiter-Glo Luminescent Assay (Promega), according to the manufacturer’s guidelines. Cell viability inhibition (%) was calculated according to the formula [1 − (mean luminosity of treated sample/mean luminosity of vehicle control)] × 100. The IC50 for growth or viability inhibition was calculated using Prism 8. We also used 18F-CDKi and palbociclib for competitive displacement studies. MCF-7 cells were seeded in a 12-well plate (1 × 105 cells 24 h before the experiment). The next day, a fixed concentration of 18F-CDKi (50 nM) was coincubated with linearly doubling concentrations of palbociclib (from 0 to 500 nM) at 37°C for 2 h. Then, the cells were washed twice with phosphate-buffered saline and lysed with 1N NaOH, and the activity (cpm) was measured in a γ-counter for bound 18F radioligand. The percentage of bound radioligand was finally plotted against palbociclib concentration. Competitive displacement curves were fitted using Prism 8.
In Vitro Uptake
MCF-7 cells were seeded in a 6-well plate (5 × 106 cells 24 h before the experiment). The next day, 18F-CDKi was added alone or together with a 50-fold excess of palbociclib and incubated at 37°C for 1 h. After 1 h, cells were first washed 3 times with phosphate-buffered saline and then lysed with 1N NaOH, and finally the medium and cells were counted in a γ-counter. The percentage of bound-to-unbound radioligand at each time point was measured in triplicate and plotted as a function of time.
Small-Animal PET/CT Imaging
Twelve subcutaneous MCF-7 implanted athymic nude mice were divided into 2 groups (blocked and18F-CDKi) and administered 18F-CDKi (∼7.4 MBq) via tail vein injection. Approximately 5 min before PET acquisition, the mice were anesthetized by inhalation of a mixture of isoflurane (2% isoflurane, 2 L/min flow of medical air; Baxter Healthcare) and positioned in the scanner. Anesthesia was maintained using a 1% isoflurane/O2 mixture. PET data for each mouse were recorded and acquired at 30 and 120 min after injection. Blocking studies were performed after preinjection of a 50-fold excess of palbociclib (8.4 nmol, 3.8 μg, 30 min beforehand). The PET/CT sessions were 20 min each.
Biodistribution
The biodistribution of 18F-CDKi was determined in subcutaneous MCF-7–bearing athymic nude mice (n = 8). The mice were divided into blocked and unblocked groups (n = 4/group) (50-fold excess of palbociclib, 2 nmol, 0.95 μg, 30 min beforehand) and administered 18F-CDKi via tail vein injection (1.85 MBq). At 120 min after injection of the radioligand, the mice were killed and organs of interest were collected. Organs were weighed, and activity was measured with a WIZARD2 automatic γ-counter. Radiopharmaceutical uptake was expressed as %ID/g using the following formula: [(activity in the target organ/grams of tissue)/injected dose] × 100%.
Statistical Analysis
All data are expressed as mean ± SD. Differences between mouse cohorts were analyzed with the 2-tailed unpaired Student t test and were considered statistically significant when the P value was less than 0.05.
RESULTS
Chemistry and Radiochemistry
18F-CDKi was synthesized by a 2-step method (Fig. 1A; Supplemental Fig. 1 [supplemental materials are available at http://jnm.snmjournals.org]). The final 18F transfluorinated product (Fig. 1B) was obtained with an overall uncorrected yield of about 15% (n = 4) and a radiochemical purity of more than 98%. Molar activity was 44 GBq/μmol, and the total automated synthesis time was 80–90 min. The retention time reported for 18F-CDKi and 19F-CDKi was about 17 min as demonstrated by coinjection with 19F-CDKi on an HPLC column (Fig. 1C). 19F-CDKi (Fig. 2A; final yield, ∼80%) was obtained with high purity as shown by liquid chromatography–mass spectrometry. The observed mass-to-charge ratio for 19F-CDKi was 566 ([M] + H) and 564 ([M] − H) in positive- and negative-polarity mode (expected mass-to-charge ratio for 19F-CDKi, 565) (Supplemental Fig. 2B). No unreacted palbociclib (molecular weight, 447.5 g/mol) was observed after purification, indicating a high chemical purity of the HPLC-purified 19F-CDKi.
FIGURE 1.

(A) 18F-CDKi radiochemistry and production method. (B) Chemical structure of 18F-CDKi. (C) Chromatograms representing coinjection of 18F-CDKi with 19F-CDKi and showing successful synthesis and purity of PET tracer. PDA = photometric diode array.
FIGURE 2.

(A) 18F-CDKi/19F-CDKi shows nanomolar binding affinity to CDK4/6. (B–E) 18F-CDKi displays 7-min blood half-life (B), high in vitro and in vivo stability (C and D), and lipophilic behavior (E). LogCHI = chromatography hydrophobicity index; logDo/w = octanol/water phase partition coefficient.
Cellular Binding Specificity and Cell Internalization
18F-CDKi maintained a potent targeting affinity to CDK4/6 (∼13 nM) (Fig. 2A). The competitive displacement of palbociclib in MCF-7 cells was an indication that 18F-CDKi had an inhibition effect similar to that of 19F-CDKi (∼18 nM) in MCF-7 cells (Supplemental Fig. 3). Cell-associated uptake of 18F-CDKi is shown in Supplemental Figure 4. Cell uptake could be blocked nearly completely (>98%) by the addition of an excess of cold, unlabeled palbociclib (P < 0.0001), suggesting the high cellular specificity of 18F-CDKi. Cellular uptake experiments performed on the MCF-7 breast cancer cell line (ER-positive and HER2-negative) demonstrated a maximum intracellular concentration of about 65% already at 10 min after incubation, when the internalization rate plateaued.
Stability and Pharmacokinetics
The in vivo blood half-life (weighted) was 7.03 min (Fig. 2B). 18F-CDKi was stable both in vitro and in vivo, with more than 98% of the parent compound intact after 4 h of incubation (>98% at 4 h after injection). The octanol-to-water phase partition coefficient was 1.91 ± 0.24, and the chemical hydrophobicity index was 61.23 (Figs. 2C–2E).
In Vivo Imaging and Ex Vivo Biodistribution
PET imaging showed a significant specific uptake in the tumor lesion (Fig. 3). MCF-7 xenografts showed approximately 4 %ID/g when mice were injected with 18F-CDKi. Tracer uptake in the tumor decreased to about 0.3 %ID/g when mice were blocked with a 50-fold excess of palbociclib. PET imaging and biodistribution data showed that 18F-CDKi was cleared via the hepatobiliary route (Figs. 3 and 4A). A high concentration of 18F-CDKi was found in the kidneys (7.99 ± 0.7 or 9.6 ± 0.98 %ID/g), liver (6 ± 0.5 or 6.77 ± 1.15 %ID/g), and small intestines (9.6 ± 2.7 or 10 ± 1.19 %ID/g), with relatively low distribution in other tissues (Fig. 4A) at 2 h after injection. A statistically significant difference in uptake was obtained in the tumor (Fig. 4B). A comprehensive biodistribution analysis also revealed remarkable tumor-to-blood (>5), tumor-to-muscle (>15), and tumor-to-bone (>10) ratios (Fig. 4C). A complete dataset showing a favorable in vivo pharmacokinetic profile for 18F-CDKi is available in Supplemental Figure 5.
FIGURE 3.
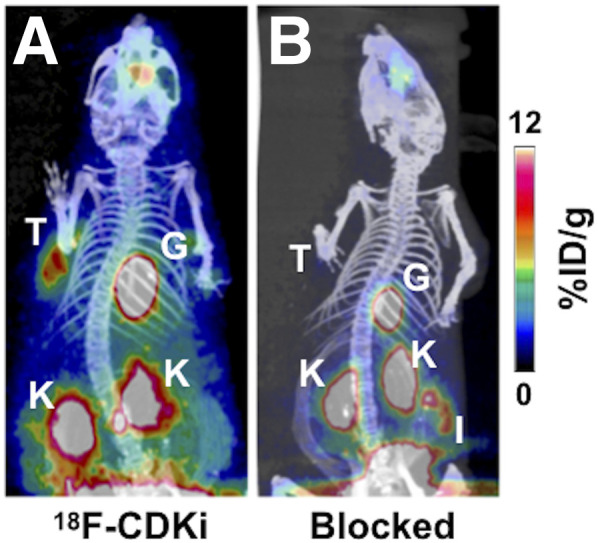
In vivo small-animal PET/CT images of MCF-7–bearing mouse models. (A) Single-bolus injection of 18F-CDKi (7.4 MBq acquired at 2 h after injection). (B) Control MCF-7 mouse (preinjected with excess of palbociclib 30 min before 18F-CDKi, 7.4 MBq, with image acquired 2 h after injection). Active areas in scans are nasal epithelium and Harderian glands, tumor (T), gallbladder (G), kidneys (K), and intestines (I).
FIGURE 4.

(A) Ex vivo biodistribution (2 h after injection) with single 18F-CDKi injection or with preinjection of palbociclib (blocked). (B) Significance of tumor %ID/g between 18F-CDKi and blocked control group. (C) Tumor-to-nontarget (NT) ratio for blood, muscle, and bone (n = 4). Int = intestine.
DISCUSSION
In this article, we present the first report on an in vivo CDK4/6 imaging agent with strong similarities to palbociclib, the first clinically Food and Drug Administration–approved CDK4/6 inhibitor (31).
CDK4/6 inhibitors are being intensively studied, and several clinical trials were established to study the effects of various compounds in synthetic lethal combination settings (14,32,33). A common characteristic to those treatments is the development of resistance (32,34,35). Poor oral availability, poor clearance and metabolism, biological feedback from other cyclins, and lack of target enzyme are all hypotheses for innate and acquired resistance to CDK4/6 inhibitors (34). However, none of the potential resistance mechanisms demonstrated in preclinical settings could be further confirmed in clinical studies (32–34). For this reason, we synthesized a radioligand that can reveal whether the drug accumulation in the tumor is limiting the therapeutic efficiency. 18F-CDKi might help to select patient responders or nonresponders to CDK4/6 inhibitors. 18F-CDKi might assess whether CDK or palbociclib treatment combinations are well tolerated, even without an increase of febrile neutropenia, which is significantly more common in the palbociclib-containing combinations than in hormonal therapy alone. Moreover, 18F-CDKi can indicate whether treatments necessitate dose interruptions and reductions. Finally, 18F-CDKi may allow the study of the pharmacodynamic effects of lower doses of palbociclib and thus help to tailor a specific line of treatment in ER-positive, HER2-negative breast cancer patients.
Structurally, the introduction of an 18F prosthetic synthon to the piperazine group appeared to be a viable approach for generating a labeled CDK4/6 inhibitor. First, we synthesized and profiled 19F-CDKi, the cold fluorinated analog of the radiolabeled counterpart. 19F-CDKi was key to assessing whether the fluorobenzoic tag would affect the compound’s biologic activity. 18F-CDKi was readily synthesized using a modified published synthetic approach (36,37). Minor modifications for automation purposes were included as well. Synthesis started from conversion of 4-ethyl-nitrobenzoate to 4-18F-fluorobenzoic acid. Subsequent reaction with palbociclib was rapidly accomplished in basic conditions to yield the final 18F-CDKi imaging agent. After scale-up synthesis and characterization of the radiotracer, we next tested the radiolabeled tracer 18F-CDKi both in vitro and in vivo. In light of the relevance of palbociclib in treating ER-positive, HER2-negative breast cancer, we chose MCF-7 as a mouse model for our in vitro and in vivo studies.
Initially, we determined the in vivo blood half-life and plasma stability of 18F-CDKi. The tracer displayed a specific intracellular uptake in MCF-7 cells and an IC50 in the nanomolar range, similar to the parent compound, palbociclib (27). The insertion of an 18F prosthetic group did not modify the binding properties of CDKi. Furthermore, like other CDK4/6 inhibitors, our molecule is also an adenosine triphosphate–competitive ligand of CDK4 and CDK6. Herein, as previously reported elsewhere, the IC50 is reported with single-digit nanomolar potency against both CDK4 and CDK6 kinases (38). The blood half-life, after a single-bolus intravenous injection, showed a biphasic pharmacokinetic profile with rapid elimination of 18F-CDKi during the first 10 min, similar to other small-molecule inhibitors (37,39). 18F-CDKi shows a marked lipophilic behavior, and more than 98% of the tracer was stable in vivo at 4 h after injection. In subcutaneous MCF-7 xenografts, 18F-CDKi was rapidly washed out from nontarget organs, resulting in a remarkable ratio of tumor to nontarget tissue at 2 h after injection. Consistent with CDKi activation and cycD1 overexpression in breast tumors, 18F-CDKi was observed to specifically accumulate in the tumor as proven by the blocking experiment. MCF-7–bearing mice that received an injection of palbociclib before the radiotracer showed negligible 18F-CDKi tumor uptake. There is significant promise that more positive clinical outcomes will emerge through a detailed understanding of the biology of CDK4/6 inhibitors (14,15,33).
This novel tracer might further be used as a tool to improve patients outcome or design more personalized combination strategies (32,34).
CONCLUSION
18F-CDKi combines high stability, fast penetration, high contrast, and rapid washout properties with a remarkable in vitro and in vivo binding to CDK4/6. These results suggest that this probe could offer an early prognostic assessment of treatment response or acquired resistance to CDK4/6 inhibitors. Moreover, with a hybrid imaging technique, such as PET 18F-CDKi in combination with MRI, clinicians might have an alternative approach to tailor precise medical treatments and therapies on the basis of patient CDK4/6 biomarker expression.
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Can 18F-CDKi provide an exact and accurate assessment of functional CDK4/6 expression in breast cancer development and progression and be used to monitor CDK4/6-based treatment?
PERTINENT FINDINGS: The first in vitro experiment aiming to analyze pharmacokinetics and in vitro activity revealed that 18F-CDKi (based on palbociclib, a Food and Drug Administration–approved drug) can be a successful PET agent with nearly ideal imaging characteristics. We demonstrated that 18F-CDKi is stable in vitro and in vivo and maintained a potent targeting affinity to CDK4/6. Cellular uptake experiments performed on a MCF-7 breast cancer cell line (ER-positive, HER2-negative) demonstrated specific uptake. Similar significant uptake values were also observed in MCF-7–bearing mouse models. The strong activation of CDK4/6 in cancer cells in concert with its low activation in untransformed healthy cells (as resulted from our tumor–to–nontarget-tissue ratio calculations) makes 18F-CDKi an ideal imaging agent, first in its class, for CDK4/6 assessment.
IMPLICATIONS FOR PATIENT CARE: 18F-CDKi can improve screening for breast cancer and selection of responders to CDK4/6 therapy. 18F-CDKi allows quantification of CDK4/6 activation (at a cellular level) and assessment of CDK4/6 protein status in current translational cancer research.
Acknowledgments
We thank the Education and Research Foundation for Nuclear Medicine and Molecular Imaging (2017 ERF-SNMMI for Giuseppe Carlucci) for its generous support. This work was supported by National Institutes of Health grants P41 EB017183 (Fernando Boada and Giuseppe Carlucci) and R01 CA204441 and P30 CA008748 (Thomas Reiner). We thank the Tow Foundation and MSK’s Center for Molecular Imaging & Nanotechnology and Imaging and Radiation Sciences Program.
REFERENCES
- 1.Jackson SP, Helleday T. Drugging DNA repair. Science. 2016;352:1178–1179. [DOI] [PubMed] [Google Scholar]
- 2.Da-Rè C, Halazonetis TD. DNA replication stress as an Achilles’ heel of cancer. Oncotarget. 2015;6:1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartek J, Mistrik M, Bartkova J. Thresholds of replication stress signaling in cancer development and treatment. Nat Struct Mol Biol. 2012;19:5–7. [DOI] [PubMed] [Google Scholar]
- 4.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. [DOI] [PubMed] [Google Scholar]
- 6.Benada J, Macurek L. Targeting the checkpoint to kill cancer cells. Biomolecules. 2015;5:1912–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Finn RS, Aleshin A, Slamon DJ. Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res. 2016;18:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. [DOI] [PubMed] [Google Scholar]
- 9.Manic G, Obrist F, Sistigu A, Vitale I. Trial watch: targeting ATM–CHK2 and ATR–CHK1 pathways for anticancer therapy. Mol Cell Oncol. 2015;2:e1012976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weber AM, Ryan AJ. ATM and ATR as therapeutic targets in cancer. Pharmacol Ther. 2015;149:124–138. [DOI] [PubMed] [Google Scholar]
- 11.Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. [DOI] [PubMed] [Google Scholar]
- 12.Pleasance ED, Cheetham RK, Stephens PJ, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11:558–572. [DOI] [PubMed] [Google Scholar]
- 14.Hamilton E, Infante JR. Targeting CDK4/6 in patients with cancer. Cancer Treat Rev. 2016;45:129–138. [DOI] [PubMed] [Google Scholar]
- 15.VanArsdale T, Boshoff C, Arndt KT, Abraham RT. Molecular pathways: targeting the cyclin D-CDK4/6 axis for cancer treatment. Clin Cancer Res. 2015;21:2905–2910. [DOI] [PubMed] [Google Scholar]
- 16.Yu Q, Sicinska E, Geng Y, et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell. 2006;9:23–32. [DOI] [PubMed] [Google Scholar]
- 17.Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell. 2006;9:13–22. [DOI] [PubMed] [Google Scholar]
- 18.Bartkova J, Lukas J, Muller H, Lutzhoft D, Strauss M, Bartek J. Cyclin D1 protein expression and function in human breast cancer. Int J Cancer. 1994;57:353–361. [DOI] [PubMed] [Google Scholar]
- 19.Dickson C, Fantl V, Gillett C, et al. Amplification of chromosome band 11q13 and a role for cyclin D1 in human breast cancer. Cancer Lett. 1995;90:43–50. [DOI] [PubMed] [Google Scholar]
- 20.Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature. 2001;411:1017–1021. [DOI] [PubMed] [Google Scholar]
- 21.Akervall JA, Michalides RJ, Mineta H, et al. Amplification of cyclin D1 in squamous cell carcinoma of the head and neck and the prognostic value of chromosomal abnormalities and cyclin D1 overexpression. Cancer. 1997;79:380–389. [PubMed] [Google Scholar]
- 22.Betticher DC, Heighway J, Hasleton PS, et al. Prognostic significance of CCND1 (cyclin D1) overexpression in primary resected non-small-cell lung cancer. Br J Cancer. 1996;73:294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bleeker FE, Lamba S, Rodolfo M, et al. Mutational profiling of cancer candidate genes in glioblastoma, melanoma and pancreatic carcinoma reveals a snapshot of their genomic landscapes. Hum Mutat. 2009;30:E451–E459. [DOI] [PubMed] [Google Scholar]
- 24.Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. [DOI] [PubMed] [Google Scholar]
- 25.Freedman JA, Tyler DS, Nevins JR, Augustine CK. Use of gene expression and pathway signatures to characterize the complexity of human melanoma. Am J Pathol. 2011;178:2513–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toogood PL, Harvey PJ, Repine JT, et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem. 2005;48:2388–2406. [DOI] [PubMed] [Google Scholar]
- 28.Valkó K. Application of high-performance liquid chromatography based measurements of lipophilicity to model biological distribution. J Chromatogr A. 2004;1037:299–310. [DOI] [PubMed] [Google Scholar]
- 29.Valkó K, Bevan C, Reynolds D. Chromatographic hydrophobicity index by fast-gradient RP-HPLC: a high-throughput alternative to log P/log D. Anal Chem. 1997;69:2022–2029. [DOI] [PubMed] [Google Scholar]
- 30.Schaer DA, Beckmann RP, Dempsey JA, et al. The CDK4/6 inhibitor abemaciclib induces a T cell inflamed tumor microenvironment and enhances the efficacy of PD-L1 checkpoint blockade. Cell Reports. 2018;22:2978–2994. [DOI] [PubMed] [Google Scholar]
- 31.McCain J. First-in-class CDK4/6 inhibitor palbociclib could usher in a new wave of combination therapies for HR+, HER2− breast cancer. P&T. 2015;40:511–520. [PMC free article] [PubMed] [Google Scholar]
- 32.Knudsen ES, Witkiewicz AK. The strange case of CDK4/6 inhibitors: mechanisms, resistance, and combination strategies. Trends Cancer. 2017;3:39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13:417–430. [DOI] [PubMed] [Google Scholar]
- 34.Pandey K, An HJ, Kim SK, et al. Molecular mechanisms of resistance to CDK4/6 inhibitors in breast cancer: a review. Int J Cancer. 2019;145:1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herrera-Abreu MT, Palafox M, Asghar U, et al. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 2016;76:2301–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valeur E, Bradley M. Amide bond formation: beyond the myth of coupling reagents. Chem Soc Rev. 2009;38:606–631. [DOI] [PubMed] [Google Scholar]
- 37.Carney B, Carlucci G, Salinas B, et al. Non-invasive PET imaging of PARP1 expression in glioblastoma models. Mol Imaging Biol. 2016;18:386–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Brien N, Conklin D, Beckmann R, et al. Preclinical activity of abemaciclib alone or in combination with antimitotic and targeted therapies in breast cancer. Mol Cancer Ther. 2018;17:897–907. [DOI] [PubMed] [Google Scholar]
- 39.Hendricks JA, Keliher EJ, Marinelli B, Reiner T, Weissleder R, Mazitschek R. In vivo PET imaging of histone deacetylases by 18F-suberoylanilide hydroxamic acid (18F-SAHA). J Med Chem. 2011;54:5576–5582. [DOI] [PMC free article] [PubMed] [Google Scholar]