

Summary
A key challenge in psoriasis therapy is the tendency for lesions to recur in previously affected anatomical locations after treatment discontinuation following lesion resolution. Available evidence supports the concept of a localized immunological ‘memory’ that persists in resolved skin after complete disappearance of visible inflammation, as well as the role of a specific subpopulation of T cells characterized by the dermotropic CCR4+ phenotype and forming a local memory. Increasing knowledge of the interleukin (IL)‐23/T helper 17 (Th17) cell pathway in psoriasis immunopathology is pointing away from the historical classification of psoriasis as primarily a Th1‐type disease. Research undertaken from the 1990s to the mid‐2000s provided evidence for the existence of a large population of CD8+ and CD4+ tissue‐resident memory T cells in resolved skin, which can initiate and perpetuate immune responses of psoriasis in the absence of T‐cell recruitment from the blood. Dendritic cells (DCs) are antigen‐presenting cells that contribute to psoriasis pathology via the secretion of IL‐23, the upstream regulator of Th17 cells, while plasmacytoid DCs are involved via IL‐36 signalling and type I interferon activation. Overall, the evidence discussed in this review indicates that IL‐23‐driven/IL‐17‐producing T cells play a critical role in psoriasis pathology and recurrence, making these cytokines logical therapeutic targets. The review also explains the clinical efficacy of IL‐17 and IL‐23 receptor blockers in the treatment of psoriasis.
![]()
Plaque‐type psoriasis is a chronic inflammatory skin disease characterized by thickened red plaques and silvery lamellar scales predominantly on the scalp, trunk and extensor surfaces. 1 The prevalence is about 2–3% in Western countries. 2 , 3 An increased understanding of the immune mechanisms underlying plaque formation has driven recent advances in psoriasis treatment. 4 , 5 , 6 However, an intriguing characteristic of psoriasis is the tendency of lesions to recur in the same anatomical locations after therapy is discontinued following plaque resolution.
Available evidence supports the concept of a local immunological ‘memory’ in resolved sites after disappearance of visible inflammation. 7 This review provides a historical narrative of the immunopathology of psoriasis lesion recurrence, including insights from emerging data on the molecular and cellular basis of disease memory in resolved psoriatic plaques.
Role of T cells in psoriasis immunopathology
The involvement of T cells in psoriasis was already demonstrated in 1986, when a study published by Fry and colleagues demonstrated clearance of psoriasis following administration of low‐dose ciclosporin. 8 However, psoriasis was previously considered a primarily keratinocyte‐driven disease for decades, until approaches involving anti‐CD3 and anti‐CD4 monoclonal antibodies specifically targeting T cells 9 , 10 produced a paradigm shift in our understanding of its immunopathology. In 1995, a landmark study by Gottlieb et al. 11 demonstrated psoriatic plaque reduction after the targeted depletion of activated interleukin (IL)‐2 receptor (R)‐expressing T cells by treatment with a fusion protein composed of IL‐2 and diphtheria toxin. Although the observed mean reduction of 32% in Psoriasis Area and Severity Index (PASI 32) is low by present‐day therapeutic standards, it was considerable at the time. This research suggested the primary role of T cells in psoriasis pathogenesis, and was further supported by studies identifying large numbers of activated CD4+ and CD8+ T cells in the skin lesions and peripheral blood of patients with psoriasis. 12 , 13 Xenograft studies on an immunosuppressed AGR129 mouse model demonstrated that T‐cell proliferation and migration into the epidermis precedes the onset of psoriasis and is essential for disease development. Therefore, accumulating epidermal T cells are now seen as effectors in psoriasis pathogenesis rather than triggers. 14 Such T cells show oligoclonal expansion in psoriatic skin, 15 , 16 particularly in the epidermis, 17 indicating the potential for a common (epidermal) antigen for autoimmune T cells in psoriasis.
In the 1990s, characterization of the cells and cytokines involved in psoriasis pathogenesis demonstrated overexpression of the T helper (Th)1 cytokines interferon gamma (IFN‐γ), tumour necrosis factor alpha (TNF‐α) and interleukins in psoriatic lesions, 18 , 19 and led to the description of psoriasis as a Th1‐type disease. However, when IL‐17‐producing CD4+ T cells (Th17 cells) were isolated from psoriatic plaques, focus shifted towards a novel T‐cell subset. 20 , 21 , 22 , 23 , 24 , 25 In 2014–2015, specific T‐cell responses against the autoantigen keratin 17, the cathelicidin antimicrobial peptide LL‐37 and the melanocyte antigen ADAMTSL5 were identified in patients with psoriasis, supporting the hypothesis of plaque‐type psoriasis as a T‐cell mediated autoimmune disease. 26 , 27 Moreover, epidermal CD8+ T cells, which express the Th17 cytokines IL‐17 and IL‐22, were shown to play an essential role in the pathogenesis of psoriasis. 28 , 29 In fact, IL‐17 and IL‐22 represent key disease mediators, linking the adaptive immune response and epithelial dysregulation in psoriasis by inducing keratinocyte hyperproliferation via Th17 and LL‐37 upregulation from activated keratinocytes. 30
Whereas Th1 cell development is driven by IL‐12, development and maintenance of Th17 cells is linked to IL‐23. IL‐12 is composed of a p35 and a p40 subunit, while IL‐23 is composed of a p19 and a p40 subunit. Both cytokines therefore share the p40 subunit, and a therapeutic agent targeting the p40 subunit (ustekinumab) has shown clinical efficacy in patients with psoriasis. 31 In 2009, McGeachy et al. demonstrated that IL‐23 was required for in vivo Th17 cell differentiation in autoimmunity, inflammation and infection, thus indicating a more central role for IL‐23 in Th17 function than previously thought. 32 Interference with correct Th17 cell development inhibits psoriasis plaque formation, as shown in a psoriasis xenograft mouse model where antibodies directed against IL‐21, a key cytokine in Th17 early development, had therapeutic effects. 33 In 2010, a study showed that specific IL‐23 p19 inhibition blocked psoriasis development in the immunosuppressed AGR129 mouse model, suggesting that IL‐23 plays a major role in the pathogenesis of psoriasis. 34 The relevance of the IL‐23/Th17 pathway has been further supported by genome‐wide association studies. 35 , 36 IL‐23 appears to be critical in promoting the differentiation of pathogenic tissue‐resident memory (TRM) T cells; this pathogenic subset of TRM cells has an important role in autoimmune inflammation in human skin. 37 This TRM cell differentiation via serum glucocorticoid‐regulated kinase‐1 induction of FOXO1 deactivation then leads to further IL‐23R expression. 38
In summary, the IL‐23/Th17 axis has a central role in the pathogenesis of psoriasis. 39 (Figure 1), as well as in psoriatic arthritis and other spondyloarthritides. 40
Figure 1.
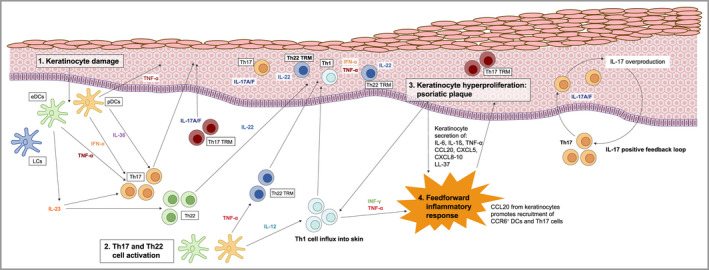
Current model of psoriasis immunopathology 30 , 93 , 94 (1) Activation of plasmacytoid dendritic cells (pDCs) and interleukin (IL)‐23‐producing epidermal DCs due to keratinocyte damage. (2) T helper (Th)17 cell polarization and clonal expansion is triggered. T‐cell activation leads to production of proinflammatory cytokines. IL‐23 promotes Th17 cell clonal expansion and differentiation. (3) IL‐17 +/– tumour necrosis factor alpha (TNF‐α) induces further inflammation with terminal keratinocyte differentiation and proliferation – forming a psoriatic plaque. (4) A feedforward inflammatory response is induced, with IL‐17 inducing psoriasis‐related gene expression in keratinocytes, driving further inflammation. eDCs, epidermal DCs; IFN, interferon; LC, Langerhans cell; LL‐37, cathelicidin antimicrobial peptide; TRM, tissue‐resident memory cell. [Colour figure can be viewed at wileyonlinelibrary.com]
Dendritic cells and mononuclear phagocytes in psoriasis immunopathology
Dendritic cells (DCs) are antigen‐presenting cells that can be recruited from the circulation to modulate local immune responses at sites of skin inflammation or infection; 41 Langerhans cells (LCs) are specialized DCs residing in the epidermal layer of the skin. 42 , 43 Martini et al. 44 demonstrated differences in the phenotypic and functional properties of epidermal DCs (eDCs) and epidermal‐resident LCs in psoriasis where eDCs, which occurred only in psoriasis, were phenotypically different from normal DCs and were completely absent in resolved psoriasis. They also found that eDCs highly expressed genes involved in neutrophil recruitment and keratinocyte and T‐cell activation. Additionally, while epidermal‐resident LCs responded to Toll‐like receptor (TLR) activation with increased IL‐23 production, eDCs produced IL‐23 together with IL‐1β and TNF‐α. This finding is further supported by a recent single‐cell RNA sequencing study of all DC subsets, which found that CD14+ DCs increased in lesional skin and coproduced IL‐1β and IL‐23A. 45
Nakajima et al. 46 used a transgenic mouse model of psoriasis in which Stat3 is constitutively activated in keratinocytes to investigate whether LCs are involved in the pathogenesis of psoriasis lesions. Compared with non‐transgenic mice, the K5.Stat3C mice showed an increased number of LCs in skin‐draining lymph nodes that preceded development of psoriasis‐like lesions. Notably, psoriasis‐like lesions were attenuated when LCs were depleted, suggesting a critical role for LCs in the pathogenesis of psoriasis. Additionally, LCs in the lesions of both K5.Stat3C mice and patients with psoriasis produced IL‐23, which in turn induces IL‐17‐producing Th17 cell differentiation.
Plasmacytoid DCs (pDCs) are a unique, rare subset of DCs that are capable of secreting large amounts of type I IFNs. 47 During infection, pDCs are activated through TLR7 and TLR9 by viruses that enter the cells. Besides their critical role in antiviral immunity, type I IFNs are also implicated in numerous autoimmune diseases, as they link innate and adaptive immunity. Under physiological conditions, skin injury induces transient expression of antimicrobial peptides, which can bind and transport microbial and self nucleic acids into pDCs, enabling their activation. In psoriasis, antimicrobial peptides are overexpressed, thereby leading to prolonged pDC activation and excessive type I IFN production. Type I IFN then triggers chronic activation of dermal DCs, which produce TNF and IL‐23, which in turn drive the pathogenic Th17 response. Thus, the type I IFN‐driven pathway, which is dominant in early acute psoriasis, is relayed by TNF–IL‐23–Th17‐driven inflammation mediated by T cells in chronic plaque psoriasis. Interestingly, IL‐17 and IL‐22 induce the expression of antimicrobial peptides in keratinocytes, leading to a self‐sustained feedback loop and chronification of the disease. Moreover, pDCs have been identified as a mediator of cytokine IL‐36 activity and are implicated in the IL‐36/TLR9 pathway, which upregulates systemic type I IFN activation in psoriasis. 48
Mehta et al. 49 analysed mononuclear phagocytes and T cells of skin from patients before and during treatment with guselkumab or secukinumab using high‐dimensional unsupervised flow cytometry. Among the assessed phagocytes they found that CD64brightCD163–CD14brightCD1c–CD1a– inflammatory monocyte‐like cells were the predominant source of IL‐23 and that these cells, together with CD64–CD163–CD14–IL‐23p19–TNF‐α+ inflammatory dendritic‐like cells, were increased in lesional vs. non‐lesional skin from the same patient. With regard to drug effects, they reported that neither drug modified the frequencies of IL‐17A+IL‐17F+/CD4+ or CD8+ T cells but found that the IL‐23 antagonist guselkumab reduced memory T cells while maintaining regulatory T cells (Tregs), whereas the opposite effect was observed with the IL‐17 antagonist secukinumab.
The Koebner phenomenon refers to the occurrence of psoriatic lesions in a previously unaffected area following another type of skin injury to the area. Epidermal trauma induces expression of antimicrobial peptides in the skin and drives the pathogenic immune response as described above. In addition, induction of scratch injury on keratinocytes showed upregulated mRNA expression and protein secretion of CCL20 and CXCL8, suggesting another potential mechanism for its induction. 50 Another hypothesis is mechanical stretch‐induced adenosine triphosphate release from keratinocytes, which can trigger the Koebner phenomenon in psoriasis. 51 A number of inflammatory mediators, including IL‐6, IL‐8, IL‐17 and IL‐36γ, as well as changes in the ratio of CD4+/CD8+ T cells, have also previously been reported to play a role in the induction of the Koebner phenomenon. 52
Tissue‐resident memory T cells and psoriasis immunopathology
A detailed review on the pathophysiology of TRM cells was recently published; 53 here we focus on specific aspects of TRM cells as they relate to psoriasis. In the 1990s, published evidence supported the concept of a role for skin‐homing T cells in the pathogenesis of inflammatory skin disease, by trafficking from the blood into lesional skin via the vascular addressin E‐selectin. 54 , 55 , 56 However, in 2002 Bhushan et al. 57 reported that an anti‐E‐selectin monoclonal antibody (CDP850) was ineffective in treating chronic plaque psoriasis, and suggested the alternative hypothesis that skin‐resident T cells are critical for psoriasis development. Subsequently, supporting evidence from the immunodeficient AGR129 mouse model showed that skin‐resident T cells in human skin grafts undergo local proliferation and infiltration of the host dermis and epidermis. 58 The T‐cell infiltrate triggered an inflammatory cascade and expression of a psoriatic phenotype in the host. The cytokine TNF‐α was shown to be a key regulator of local T‐cell proliferation and disease development.
In 2006, a key study showed that there are 2 × 1010 T cells (predominantly Th1 memory effector cells) in healthy human skin (dermis and epidermis), nearly twice as many as those circulating in the blood. 59 These results suggested the existence of a large pool of TRM cells in healthy skin that can initiate and perpetuate immune responses in the absence of T‐cell recruitment from the blood.
TRM cells residing in epithelial barrier tissues in the gastrointestinal, respiratory and reproductive tracts, and skin, provide a rapid adaptive defence against pathogens. 60 The most notable biological characteristics of TRM cells are their longevity and low migration rates away from their resident tissue. 60 TRM cells can be divided into CD8+ and CD4+ subsets. CD8+ TRM cells are localized in the epidermis where they play a critical role in immune responses; CD4+ TRM cells are less well characterized and potentially play a critical role in protective immunity against bacterial and fungal infections in the skin. 60 , 61 The majority of CD8+ TRM cells express the cell marker CD103 (also known as integrin alpha‐E), which is implicated in lodging TRM cells in the epidermis 62 , 63 via binding of the T cells to E‐cadherin, a highly expressed epithelial protein. 64
In 2017, Cheuk et al. 65 differentiated CD49+CD8+ TRM cells from CD49–CD8+ TRM cells. CD49+ TRM cells are characterized by IFN‐γ production and rapidly gain a cytotoxic capacity following IL‐15 stimulation, whereas CD49– TRM cells produce IL‐17. The functional dichotomy between CD49+ and CD49– TRM cells was evident in a comparison of TRM cell characteristics in two distinct immune‐mediated skin diseases. In vitiligo skin, where melanocytes are locally eradicated by cytotoxic activity, skin biopsy specimens showed a predominance of cytotoxic CD49+ TRM cells, compared with skin biopsy specimens from psoriatic lesions, which contained predominantly IL‐17‐producing CD49– TRM cells. In summary, the data from this study showed that CD49 expression delineated CD8+ TRM cell specialization in human skin.
Recently, Vo et al. reported that CD8+ TRM cells were enriched in both psoriatic lesional and non‐lesional skin compared with normal skin. Furthermore, the percentage ratio of IL‐17A‐producing cells to IFN‐γ‐producing cells in the whole CD8 fraction correlated with disease duration. 66
Casciano et al. identified an increased population of circulating CD8+ central memory T (TCM) cells with a CCR4+CXCR3+ phenotype in isolated peripheral blood mononuclear cells from patients with psoriasis compared with healthy controls. This implicates the skin as a key trafficking site for TCM cells, where antigen encounter may occur and, under appropriate conditions, may lead to the generation of non‐circulating TRM cells. The authors suggest that CCR4+CXCR3+ T cells could represent a key population of TCM cells that play a role in disease recurrence or redistribution to distant sites such as joint synovial tissues and entheses. 67
Role of residual disease ‘memory’ in psoriatic lesion recurrence
Effective treatment of psoriasis returns lesional skin to an apparently normal state, with a lack of clinically visible inflammation, normalized epidermal thickness, and reductions in inflammatory cellular infiltrates and disease‐associated gene expression. 22 , 68 However, once therapy is discontinued, psoriatic skin lesions tend to recur at previously affected sites, 7 suggesting that residual disease may persist at the site of apparently healed lesions, predisposing individuals to recurrence in the same locations.
Molecular scarring
The concept of a residual disease gene expression profile in psoriasis was first proposed in 2011 by Suárez‐Fariñas et al. 69 in a landmark study of 20 patients with psoriasis who were treated with the TNF inhibitor etanercept. In post‐treatment biopsy specimens, epidermal thickness improved by 92%, Ki67 staining (a general marker of cellular proliferation) improved by 119%, and all samples were negative for keratin 16 (a marker of keratinocyte hyperproliferation). However, a subset of 248 genes did not return to normal levels, including those coding for proinflammatory molecules IL‐12, IFN‐induced guanosine triphosphate‐binding protein Mx1 (MX1), IL‐22, IL‐17 and IFN‐γ, and genes encoding cutaneous structural or regulatory components such as lymphatic vessel endothelial hyaluronic acid receptor‐1, Wnt‐5a, Ras‐related protein Rab‐31 and aquaporin‐9. Taken together, these findings suggested the concept of a ‘molecular scar’ within the epidermis of healed lesions.
In 2014, Cheuk et al. 70 reported a detailed analysis of gene expression and cytokine production in T cells within the epidermis and dermis of resolved psoriasis lesions after successful treatment with three different therapies: narrowband ultraviolet B, the IL‐12/23 inhibitor ustekinumab and the TNF‐α inhibitor infliximab. A population of epidermal CD8+ T cells expressing CLA, CCR6, CD103 and IL‐23R was highly enriched in resolved psoriasis lesions, where those expressing CD103 responded to ex vivo stimulation with IL‐17 production. The presence of these CD49a‐expressing epidermal CD8+ cells suggested a stable, resting population of TRM cells in resolved psoriasis lesions, where they retain the ability to produce IL‐17. The authors proposed a model in which, following the appropriate trigger in resolved lesions, skin‐residing CD4+ T cells would drive keratinocyte pathology through IL‐22 production, and CD8+ T cells would drive recurrent local inflammation by recruiting circulating leucocytes through IL‐17A production. 70
Matos et al. 71 hypothesized in 2017 that skin‐resident T cells in resolved psoriatic lesions are derived from expanded clonal or oligoclonal populations of the original IL‐17‐producing pathogenic T cells that mediated the disease. Using both high‐throughput screening of T‐cell receptor (TCR) CDR3 regions (antigen recognition domains) and immunostaining to evaluate the clonality and cytokine production of T cells in resolved lesions, clinically resolved psoriatic lesions were found to contain oligoclonal populations of IL‐17‐producing αβ T cells. A total of four TCRα and 15 TCRβ antigen receptor sequences were found to be shared between patients with psoriasis and were not observed in healthy controls or other inflammatory skin conditions.
Additional evidence for the role of IL‐17 expression in lesional memory came from a 2018 study by Gallais Sérézal et al. 72 using transcriptomic analyses to explore whether T cells had different gene expression patterns in psoriatic skin compared with healthy skin. Skin explants treated with the pan‐T‐cell activating antibody OKT‐3 responded with IFN‐γ‐induced pathways regardless of the inflammatory status of the skin, whereas IL‐17‐induced pathways were preferentially activated in psoriatic skin and persisted in the epidermis of resolved lesions. As with healed skin, never‐lesional skin explants from patients with psoriasis contained resident CD8+ T cells with the potential to produce IL‐22, IL‐17 and IFN‐γ. 72
The gene expression studies discussed above are subject to a number of important limitations. For example, the use of specific cut‐offs to define what constitutes increased gene expression or clinical response is arbitrary, and results may vary substantially if different cut‐offs are employed. Moreover, the results reflect gene expression only at the time of biopsy, meaning the timing of the biopsy in relation to the start of treatment may also substantially influence results. Nonetheless, with these limitations in mind, the evidence from these studies overall does suggest that pathogenic TRM cells can create a proinflammatory environment in the skin of patients with psoriasis, and could be responsible for local disease relapse by priming tissue response, driven by IL‐22 and IL‐17A production by skin‐resident CD4+ and CD8+ T cells, respectively. 70 , 72 As previously mentioned, CD103+ CD8 TRM cells are enriched in both lesional and non‐lesional disease‐naïve skin, and increase IL‐17A‐generating potential with disease duration. 66 A current model for disease recurrence in psoriasis is shown in Figure 2.
Figure 2.
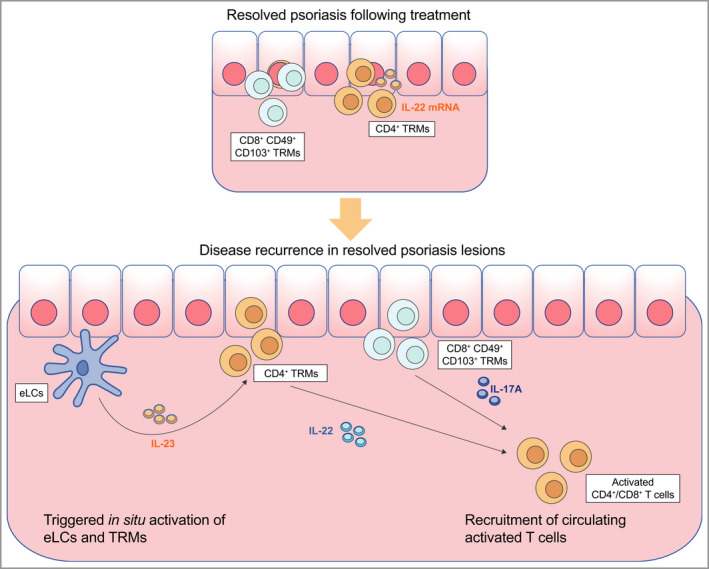
Current model of disease recurrence in resolved psoriatic lesions 70 , 95 In resolved lesions, CD4+ tissue‐resident memory cells (TRMs) remain in the dermis, and CD8+ TRMs and epidermal Langerhans cells (eLCs) remain in the epidermis. Upon the disease trigger, eLCs and TRMs actively produce proinflammatory cytokines [interleukin (IL)‐23, IL‐17A and IL‐22] that induce keratinocyte hyperproliferation and recurrent disease. [Colour figure can be viewed at wileyonlinelibrary.com]
More recently, a number of RNA transcriptomics analyses have provided further insights into the molecular basis of psoriasis pathophysiology. A recent paper reported the re‐emergence of developmental gene programmes by vascular endothelial cells and macrophages in psoriasis pathophysiology, similar to what has been observed in carcinogenesis and metastasis of tumours. 73 Other studies have reported alterations in T‐cell expression of inflammatory markers such as ratios of IL‐17A vs. IL‐17F expression, IFN‐γ vs. IL‐10 expression and disease severity‐dependent expression of the chemokine CXCL13. 74 , 75
Impact of interleukin‐17 and interleukin‐23 antagonists on tissue‐resident memory T cells
A number of IL‐17 and IL‐23 inhibitors have demonstrated efficacy, and are currently approved, in the treatment of psoriasis, 76 , 77 , 78 underscoring the critical role of IL‐17/IL‐23 in psoriasis pathogenesis. 30 Moreover, several clinical studies have shown that clinical response has been maintained with IL‐23 antagonists following treatment discontinuation. In the VOYAGE 2 Phase III trial, the median time to loss of PASI 90 response after withdrawal from the IL‐23 inhibitor guselkumab was 23 weeks. 79 In a pooled analysis of data from the UNCOVER‐1 and UNCOVER‐2 Phase III trials, the median time to relapse after withdrawal from IL‐17A inhibitor ixekizumab was approximately 20 weeks. 80 The maintenance of response with these agents was greater than might be anticipated based on their elimination half‐lives of approximately 18 days 81 and 13 days 82 for guselkumab and ixekizumab, respectively. Similar results have been reported in randomized withdrawal studies of risankizumab and other IL‐17/IL‐23 antagonists. For the IL‐23A inhibitor risankizumab, a median time of 30 weeks from withdrawal of treatment until loss of PASI 90 response was reported. 83 For the IL‐23 inhibitor tildrakizumab, the median time until loss of PASI 90 response was 16 and 20 weeks for patients withdrawing from a 100‐mg and 200‐mg dose, respectively. 84 For the IL‐17A/F inhibitor bimekizumab, the median time to loss of PASI 90 response was approximately 20 weeks after the final dose. 85 A somewhat shorter time to loss of response was reported for the anti‐IL‐17A receptor antibody brodalumab, with loss of response for static Physician’s Global Assessment (score of ≥3) occurring after a median of 11 weeks. 86
A subanalysis of lesional biopsy specimens taken from patients treated with guselkumab and secukinumab in the ECLIPSE study 87 described the frequencies of immune cell populations over 24 weeks of the respective treatments. 49 The frequencies of both CD4+ and CD8+ TRM cells decreased in psoriatic lesions of both treatment groups from baseline up to week 24, while the frequency of CD8+ TRM cells decreased significantly with guselkumab, but not with secukinumab. Conversely, secukinumab treatment decreased the frequency of Tregs, whereas the frequency of Tregs was maintained in biopsies of patients receiving guselkumab. 49 The authors hypothesize that the increased Treg/CD8+ TRM ratio may be related to the superior long‐term control of skin inflammation with guselkumab observed in the ECLIPSE study. However, further studies with larger numbers of samples are needed to confirm these preliminary findings.
Mashiko et al. 88 investigated potential mechanisms involved in psoriasis plaque persistence (existence of residual plaques despite ongoing treatment) by comparing patients with psoriasis treated with adalimumab, ustekinumab and secukinumab with a group of untreated patients. A cytokine signature analysis showed that TNF, IL‐23, IL‐17 and IFN‐1 were elevated in lesional vs. non‐lesional skin in all patient groups. T‐cell subsets and their cytokine production were also investigated. Cells were stained with CD103 (a TRM cell marker) and CD161 (an IL‐17‐producing T‐cell marker). Percentages of CD103+ and CD103– cells were similar across groups, as were those of IL‐17‐producing cells. Epidermal CD103+ T cells showed no changes in cytokine staining, while dermal CD161–CD103– T cells and CD161+CD103– Th17 cells showed decreased IL‐17A production in treated plaques, although not significantly in all groups. Overall, their results suggest that aside from subtle differences in drug‐treated plaques, underlying biological mechanisms are similar to those present in untreated lesions. 88
Based on preliminary observations and experiences from other immune‐mediated inflammatory diseases such as rheumatoid arthritis, Eyerich et al. 89 hypothesized that patients with a short disease duration (≤2 years) may show a more rapid and pronounced response to guselkumab treatment. These patients may also be able to maintain longer drug‐free control of disease after guselkumab withdrawal. This hypothesis is currently a subject of research in the phase III GUIDE study, which is also investigating whether patients who demonstrate a rapid and strong initial response to guselkumab treatment can maintain disease control with less frequent dosing (every 16 weeks instead of every 8 weeks). 89
Conclusions and unresolved questions
The current model of psoriasis immunopathogenesis is presented in Figure 1. Following keratinocyte damage, IL‐23 plays a major role in the activation of Th17; clonal expansion and differentiation of these cells stimulates the production of proinflammatory cytokines, leading to keratinocyte hyperproliferation and a feedforward inflammatory response. The model incorporates an IL‐17 positive feedback loop, in which Th17‐derived IL‐17 induces overproduction of IL‐17 from psoriatic keratinocytes, which in turn induces Th17 cells to secrete more IL‐17. 90 , 91
IL‐17‐producing T cells, epidermal‐resident LCs and TRM cells all play an important role in the pathogenesis of psoriasis and the recurrence of disease in resolved psoriatic lesions. A ‘residual disease genomic profile’ exists in resolved lesions, characterized by the elevated expression of proinflammatory genes, including those encoding IL‐12, MX1, IL‐22, IL‐17 and IFN‐γ. IL‐17‐producing CD8+ TRM cells have been found in clinically resolved lesions, suggesting that TRM cells represent a nidus of latent inflammation that can predispose to reactivation of overt inflammation and lesion recurrence. Therefore, novel treatments for psoriasis that aim at sustained disease remission 92 should target the depletion of pathogenic CD8+ TRM cells.
The clinical efficacy of IL‐23 and IL‐17 antagonists in the treatment of psoriasis underscores the critical role of these cytokines and their cellular targets in the pathogenesis of this disease. TRM cells initiate and perpetuate immune responses, play an important role in lesion recurrence and may be differentially regulated by IL‐23 and IL‐17 antagonism. Further studies investigating the effect of approved IL‐23 and IL‐17 antagonists on lesion recurrence and long‐term disease control are warranted.
Acknowledgements
Medical writing support was provided by Cello Health MedErgy, under the direction of the authors.
Conflicts of interest: L.P. has received honoraria for consultancy from, and/or served as an investigator in clinical trials for AbbVie, Almirall, Amgen, Baxalta, Biogen, Boehringer Ingelheim, Celgene, Eli Lilly, Gebro Pharma, Janssen, BIOCAD, LEO Pharma, Merck Serono, MSD, Mylan, Novartis, Pfizer, Regeneron, Roche, Samsung‐Bioepis, Sandoz, Sanofi and UCB. A.C. has received honoraria for consultancy from AbbVie, Almirall, Amgen, Celgene, Eli Lilly, Galderma, LEO Pharma, Novartis and UCB. E.J.M.‐E. was a full‐time employee of Janssen at the time of manuscript conception and development. M.J. is a full‐time employee of Janssen. S.W. is a full‐time employee of Janssen, and a shareholder of Johnson & Johnson. C.F.P. received research grants from AbbVie, Boehringer Ingelheim, Eli Lilly, Janssen, Pfizer, Pierre Fabre, Sanofi and UCB, and received honoraria for consultancy from AbbVie, Almirall, Amgen, Boehringer Ingelheim, Celgene, Eli Lilly, Janssen, LEO Pharma, Merck, Pfizer, Pierre Fabre, Regeneron, Sanofi and UCB. C.C. served as a scientific adviser and/or clinical study investigator for AbbVie, Actelion, Amgen, Boehringer Ingelheim, Bristol‐Myers Squibb, Celgene, Eli Lilly, Galderma, Incyte, Janssen, LEO Pharma, MSD, Novartis, Pfizer, Samsung and UCB. All authors received non‐financial support from Janssen during the drafting of the manuscript.
E.J.M.‐E. was a full‐time employee at the time of manuscript conception and development.
Funding sources This article was sponsored by Janssen. Medical writing and editorial support were funded by Janssen.
Conflicts of interest Conflicts of interest statements can be found in the Appendix.
Data availability statement Data sharing is not applicable as no datasets were generated and/or analysed for this study.
References
- 1. Boehncke W‐H, Schön MP. Psoriasis. Lancet 2015; 386:983–94. [DOI] [PubMed] [Google Scholar]
- 2. Gelfand JM, Weinstein R, Porter SB et al. Prevalence and treatment of psoriasis in the United Kingdom: a population‐based study. Arch Dermatol 2005; 141:1537–41. [DOI] [PubMed] [Google Scholar]
- 3. Rachakonda TD, Schupp CW, Armstrong AW. Psoriasis prevalence among adults in the United States. J Am Acad Dermatol 2014; 70:512–16. [DOI] [PubMed] [Google Scholar]
- 4. Lowes MA, Suárez‐Fariñas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol 2014; 32:227–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chiricozzi A, Romanelli P, Volpe E et al. Scanning the immunopathogenesis of psoriasis. Int J Mol Sci 2018; 19:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ogawa E, Sato Y, Minagawa A, Okuyama R. Pathogenesis of psoriasis and development of treatment. J Dermatol 2018; 45:264–72. [DOI] [PubMed] [Google Scholar]
- 7. Clark RA. Gone but not forgotten: lesional memory in psoriatic skin. J Invest Dermatol 2011; 131:283–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Griffiths CE, Powles AV, Leonard JN et al. Clearance of psoriasis with low dose cyclosporin. Br Med J (Clin Res Ed) 1986; 293:731–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weinshenker BG, Bass BH, Ebers GC, Rice GP. Remission of psoriatic lesions with muromonab‐CD3 (orthoclone OKT3) treatment. J Am Acad Dermatol 1989; 20:1132–3. [DOI] [PubMed] [Google Scholar]
- 10. Bachelez H, Flageul B, Dubertret L et al. Treatment of recalcitrant plaque psoriasis with a humanized non‐depleting antibody to CD4. J Autoimmun 1998; 11:53–62. [DOI] [PubMed] [Google Scholar]
- 11. Gottlieb SL, Gilleaudeau P, Johnson R et al. Response of psoriasis to a lymphocyte‐selective toxin (DAB389IL‐2) suggests a primary immune, but not keratinocyte, pathogenic basis. Nat Med 1995; 1:442–7. [DOI] [PubMed] [Google Scholar]
- 12. Bos JD, Hagenaars C, Das PK et al. Predominance of “memory” T cells (CD4+, CDw29+) over “naïve” T cells (CD4+, CD45R+) in both normal and diseased human skin. Arch Dermatol Res 1989; 281:24–30. [DOI] [PubMed] [Google Scholar]
- 13. Ferenczi K, Burack L, Pope M et al. CD69, HLA‐DR and the IL‐2R identify persistently activated T cells in psoriasis vulgaris lesional skin: blood and skin comparisons by flow cytometry. J Autoimmun 2000; 14:63–78. [DOI] [PubMed] [Google Scholar]
- 14. Conrad C, Boyman O, Tonel G et al. α1β1 integrin is crucial for accumulation of epidermal T cells and the development of psoriasis. Nat Med 2007; 13:836–842. [DOI] [PubMed] [Google Scholar]
- 15. Prinz JC, Vollmer S, Boehncke WH et al. Selection of conserved TCR VDJ rearrangements in chronic psoriatic plaques indicates a common antigen in psoriasis vulgaris. Eur J Immunol 1999; 29:3360–8. [DOI] [PubMed] [Google Scholar]
- 16. Vollmer S, Menssen A, Prinz JC. Dominant lesional T cell receptor rearrangements persist in relapsing psoriasis but are absent from nonlesional skin: evidence for a stable antigen‐specific pathogenic T cell response in psoriasis vulgaris. J Invest Dermatol 2001; 117:1296–301. [DOI] [PubMed] [Google Scholar]
- 17. Chang JC, Smith LR, Froning KJ et al. CD8+ T cells in psoriatic lesions preferentially use T‐cell receptor V beta 3 and/or V beta 13.1 genes. Proc Natl Acad Sci U S A 1994; 91:9282–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schlaak JF, Buslau M, Jochum W et al. T cells involved in psoriasis vulgaris belong to the Th1 subset. J Invest Dermatol 1994; 102:145–9. [DOI] [PubMed] [Google Scholar]
- 19. Austin LM, Ozawa M, Kikuchi T et al. The majority of epidermal T cells in psoriasis vulgaris lesions can produce type 1 cytokines, interferon‐γ, interleukin‐2, and tumor necrosis factor‐α, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: a type 1 differentiation bias. J Invest Dermatol 1999; 113:752–9. [DOI] [PubMed] [Google Scholar]
- 20. Wilson NJ, Boniface K, Chan JR et al. Development, cytokine profile and function of human interleukin 17‐producing helper T cells. Nat Immunol 2007; 8:950–7. [DOI] [PubMed] [Google Scholar]
- 21. Lowes MA, Kikuchi T, Fuentes‐Duculan J et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol 2008; 128:1207–11. [DOI] [PubMed] [Google Scholar]
- 22. Zaba LC, Cardinale I, Gilleaudeau P et al. Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J Exp Med 2007; 204:3183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kryczek I, Bruce AT, Gudjonsson JE et al. Induction of IL‐17+ T cell trafficking and development by IFN‐γ: mechanism and pathological relevance in psoriasis. J Immunol 2008; 181:4733–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kagami S, Rizzo HL, Lee JJ et al. Circulating Th17, Th22, and Th1 cells are increased in psoriasis. J Invest Dermatol 2010; 130:1373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rizzo HL, Kagami S, Phillips KG et al. IL‐23‐mediated psoriasis‐like epidermal hyperplasia is dependent on IL‐17A. J Immunol 2011; 186:1495–502. [DOI] [PubMed] [Google Scholar]
- 26. Lande R, Botti E, Jandus C et al. The antimicrobial peptide LL37 is a T‐cell autoantigen in psoriasis. Nat Commun 2014; 5:5621. [DOI] [PubMed] [Google Scholar]
- 27. Arakawa A, Siewert K, Stöhr J et al. Melanocyte antigen triggers autoimmunity in human psoriasis. J Exp Med 2015; 212:2203–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Di Meglio P, Villanova F, Navarini AA et al. Targeting CD8+ T cells prevents psoriasis development. J Allergy Clin Immunol 2016; 138:274–6. [DOI] [PubMed] [Google Scholar]
- 29. Res PCM, Piskin G, de Boer OJ et al. Overrepresentation of IL‐17A and IL‐22 producing CD8 T cells in lesional skin suggests their involvement in the pathogenesis of psoriasis. PLOS ONE 2010; 5:e14108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hawkes JE, Yan BY, Chan TC, Krueger JG. Discovery of the IL‐23/IL‐17 signaling pathway and the treatment of psoriasis. J Immunol 2018; 201:1605–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leonardi CL, Kimball AB, Papp KA et al. Efficacy and safety of ustekinumab, a human interleukin‐12/23 monoclonal antibody, in patients with psoriasis: 76‐week results from a randomised, double‐blind, placebo‐controlled trial (PHOENIX 1). Lancet 2008; 371:1665–74. [DOI] [PubMed] [Google Scholar]
- 32. McGeachy MJ, Chen Y, Tato CM et al. Interleukin 23 receptor is essential for terminal differentiation of effector T helper type 17 cells in vivo . Nature 2009; 10:314–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Caruso R, Botti E, Sarra M et al. Involvement of interleukin‐21 in the epidermal hyperplasia of psoriasis. Nat Med 2009; 15:1013–15. [DOI] [PubMed] [Google Scholar]
- 34. Tonel G, Conrad C, Laggner U et al. Cutting edge: a critical functional role for IL‐23 in psoriasis. J Immunol 2010; 185:5688–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tsoi LC, Spain SL, Knight J et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet 2012; 44:1341–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tsoi LC, Spain SL, Ellinghaus E et al. Enhanced meta‐analysis and replication studies identify five new psoriasis susceptibility loci. Nat Commun 2015; 6:7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hirai T, Whitley SK, Kaplan DH. Migration and function of memory CD8+ T cells in skin. J Invest Dermatol 2020; 140:748–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu C, Yosef N, Thalhamer T et al. Induction of pathogenic TH17 cells by inducible salt‐sensing kinase SGK1. Nature 2013; 496:513–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Girolomoni G, Strohal R, Puig L et al. The role of IL‐23 and the IL‐23/TH17 immune axis in the pathogenesis and treatment of psoriasis. J Eur Acad Dermatology Venereol 2017; 31:1616–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Steel KJA, Srenathan U, Ridley M et al. Polyfunctional, proinflammatory, tissue‐resident memory phenotype and function of synovial interleukin‐17A+CD8+ T cells in psoriatic arthritis. Arthritis Rheumatol 2020; 72:435–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Johnson‐Huang LM, McNutt NS, Krueger JG, Lowes MA. Cytokine‐producing dendritic cells in the pathogenesis of inflammatory skin diseases. J Clin Immunol 2009; 29:247–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ginhoux F, Merad M. Ontogeny and homeostasis of Langerhans cells. Immunol Cell Biol 2010; 88:387–92. [DOI] [PubMed] [Google Scholar]
- 43. Merad M, Sathe P, Helft J et al. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol 2013; 31:563–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Martini E, Wikén M, Cheuk S et al. Dynamic changes in resident and infiltrating epidermal dendritic cells in active and resolved psoriasis. J Invest Dermatol 2017; 137:865–73. [DOI] [PubMed] [Google Scholar]
- 45. Nakamizo S, Dutertre C‐A, Khalilnezhad A et al. Single‐cell analysis of human skin identifies CD14+ type 3 dendritic cells co‐producing IL1B and IL23A in psoriasis. J Exp Med 2021; 218:e.20202345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nakajima K, Kataoka S, Sato K et al. Stat3 activation in epidermal keratinocytes induces Langerhans cell activation to form an essential circuit for psoriasis via IL‐23 production. J Dermatol Sci 2019; 93:82–91. [DOI] [PubMed] [Google Scholar]
- 47. Conrad C, Meller S, Gilliet M. Plasmacytoid dendritic cells in the skin: to sense or not to sense nucleic acids. Semin Immunol 2009; 21:101–9. [DOI] [PubMed] [Google Scholar]
- 48. Catapano M, Vergnano M, Romano M et al. IL‐36 promotes systemic IFN‐I responses in severe forms of psoriasis. J Invest Dermatol 2020; 140:816–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mehta H, Mashiko S, Angsana J et al. Differential changes in inflammatory mononuclear phagocyte and T‐cell profiles within psoriatic skin during treatment with guselkumab versus secukinumab. J Invest Dermatol 2021; 141:1707–18. [DOI] [PubMed] [Google Scholar]
- 50. Furue K, Ito T, Tanaka Y et al. Cyto/chemokine profile of in vitro scratched keratinocyte model: implications of significant upregulation of CCL20, CXCL8 and IL36G in Koebner phenomenon. J Dermatol Sci 2019; 94:244–51. [DOI] [PubMed] [Google Scholar]
- 51. Okamoto T, Ogawa Y, Kinoshita M et al. Mechanical stretch‐induced ATP release from keratinocytes triggers Koebner phenomenon in psoriasis. J Dermatol Sci 2021; 103:60–2. [DOI] [PubMed] [Google Scholar]
- 52. Ji Y‐Z, Liu S‐R. Koebner phenomenon leading to the formation of new psoriatic lesions: evidences and mechanisms. Biosci Rep 2019; 39:BSR20193266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tokura Y, Phadungsaksawasdi P, Kurihara K et al. Pathophysiology of skin resident memory T cells. Front Immunol 2021; 11:618897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Picker LJ, Kishimoto TK, Smith CW et al. ELAM‐1 is an adhesion molecule for skin‐homing T cells. Nature 1991; 349:796–9. [DOI] [PubMed] [Google Scholar]
- 55. Berg EL, Yoshino T, Rott LS et al. The cutaneous lymphocyte antigen is a skin lymphocyte homing receptor for the vascular lectin endothelial cell‐leukocyte adhesion molecule 1. J Exp Med 1991; 174:1461–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Austrup F, Vestweber D, Borges E et al. P‐ and E‐selectin mediate recruitment of T‐helper‐1 but not T‐helper‐2 cells into inflamed tissues. Nature 1997; 385:81–3. [DOI] [PubMed] [Google Scholar]
- 57. Bhushan M, Bleiker TO, Ballsdon AE et al. Anti‐E‐selectin is ineffective in the treatment of psoriasis: a randomized trial. Br J Dermatol 2002; 146:824–31. [DOI] [PubMed] [Google Scholar]
- 58. Boyman O, Hefti HP, Conrad C et al. Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor‐α. J Exp Med 2004; 199:731–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Clark RA, Chong B, Mirchandani N et al. The vast majority of CLA+ T cells are resident in normal skin. J Immunol 2006; 176:4431–9. [DOI] [PubMed] [Google Scholar]
- 60. Chen L, Shen Z. Tissue‐resident memory T cells and their biological characteristics in the recurrence of inflammatory skin disorders. Cell Mol Immunol 2020; 17:64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ho AW, Kupper TS. T cells and the skin: from protective immunity to inflammatory skin disorders. Nat Rev Immunol 2019; 19:490–502. [DOI] [PubMed] [Google Scholar]
- 62. Watanabe R, Gehad A, Yang C et al. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci Transl Med 2015; 7:279ra39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mackay LK, Rahimpour A, Ma JZ et al. The developmental pathway for CD103+CD8+ tissue‐resident memory T cells of skin. Nat Immunol 2013; 14:1294–1301. [DOI] [PubMed] [Google Scholar]
- 64. Cepek KL, Shaw SK, Parker CM et al. Adhesion between epithelial cells and T lymphocytes mediated by E‐cadherin and the αEβ7 integrin. Nature 1994; 372:190–3. [DOI] [PubMed] [Google Scholar]
- 65. Cheuk S, Schlums H, Gallais Sérézal I et al. CD49a expression defines tissue‐resident CD8+ T cells poised for cytotoxic function in human skin. Immunity 2017; 46:287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vo S, Watanabe R, Koguchi‐Yoshioka H et al. CD8 resident memory T cells with interleukin 17A‐producing potential are accumulated in disease‐naïve nonlesional sites of psoriasis possibly in correlation with disease duration. Br J Dermatol 2019; 181:410–12. [DOI] [PubMed] [Google Scholar]
- 67. Casciano F, Diani M, Altomare A et al. CCR4+ skin‐tropic phenotype as a feature of central memory CD8+ T cells in healthy subjects and psoriasis patients. Front Immunol 2020; 11:529. doi: 10.3389/fimmu.2020.00529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chamian F, Lowes MA, Lin SL et al. Alefacept reduces infiltrating T cells, activated dendritic cells, and inflammatory genes in psoriasis vulgaris. Proc Natl Acad Sci U S A 2005; 102:2075–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Suárez‐Fariñas M, Fuentes‐Duculan J, Lowes MA, Krueger JG. Resolved psoriasis lesions retain expression of a subset of disease‐related genes. J Invest Dermatol 2011; 131:391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cheuk S, Wikén M, Blomqvist L et al. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol 2014; 192:3111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Matos TR, O’Malley JT, Lowry EL et al. Clinically resolved psoriatic lesions contain psoriasis‐specific IL‐17‐producing αβ T cell clones. J Clin Invest 2017; 127:4031–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gallais Sérézal I, Classon C, Cheuk S et al. Resident T cells in resolved psoriasis steer tissue responses that stratify clinical outcome. J Invest Dermatol 2018; 138:1754–63. [DOI] [PubMed] [Google Scholar]
- 73. Reynolds G, Vegh P, Fletcher J et al. Developmental cell programs are co‐opted in inflammatory skin disease. Science 2021; 371:eaba6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kim J, Lee J, Kim HJ et al. Single‐cell transcriptomics applied to emigrating cells from psoriasis elucidate pathogenic versus regulatory immune cell subsets. J Allergy Clin Immunol 2021; 148:1281–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Liu J, Chang H‐W, Huang Z‐M et al. Single‐cell RNA sequencing of psoriatic skin identifies pathogenic Tc17 cell subsets and reveals distinctions between CD8+ T cells in autoimmunity and cancer. J Allergy Clin Immunol 2021; 147:2370–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Puig L. The role of IL 23 in the treatment of psoriasis. Expert Rev Clin Immunol 2017; 13:525–34. [DOI] [PubMed] [Google Scholar]
- 77. Ly K, Smith MP, Thibodeaux Q et al. Anti IL‐17 in psoriasis. Expert Rev Clin Immunol 2019; 15:1185–94. [DOI] [PubMed] [Google Scholar]
- 78. Erichsen CY, Jensen P, Kofoed K. Biologic therapies targeting the interleukin (IL)‐23/IL‐17 immune axis for the treatment of moderate‐to‐severe plaque psoriasis: a systematic review and meta‐analysis. J Eur Acad Dermatology Venereol 2020; 34:30–8. [DOI] [PubMed] [Google Scholar]
- 79. Reich K, Armstrong AW, Foley P et al. Efficacy and safety of guselkumab, an anti‐interleukin‐23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double‐blind, placebo‐ and active comparator‐controlled VOYAGE 2 trial. J Am Acad Dermatol 2017; 76:418–31. [DOI] [PubMed] [Google Scholar]
- 80. Blauvelt A, Papp KA, Sofen H et al. Continuous dosing versus interrupted therapy with ixekizumab: an integrated analysis of two phase 3 trials in psoriasis. J Eur Acad Dermatol Venereol 2017; 31:1004–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yao Z, Hu C, Zhu Y et al. Population pharmacokinetic modeling of guselkumab, a human IgG1λ monoclonal antibody targeting IL‐23, in patients with moderate to severe plaque psoriasis. J Clin Pharmacol 2018; 58:613–27. [DOI] [PubMed] [Google Scholar]
- 82. European Medicines Agency . Product information. Taltz : EPAR – Product Information. Last updated 2021. Annex I. Summary of product characteristics. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/taltz#product‐information‐section (last accessed 20 January 2022).
- 83. Blauvelt A, Leonardi CL, Gooderham M et al. Efficacy and safety of continuous risankizumab therapy vs treatment withdrawal in patients with moderate to severe plaque psoriasis. JAMA Dermatol 2020; 156:649–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Warren RB, Carrascosa JM, Fumero E et al. Time to relapse after tildrakizumab withdrawal in patients with moderate‐to‐severe psoriasis who were responders at week 28: post hoc analysis through 64 weeks from reSURFACE 1 trial. J Eur Acad Dermatol Venereol 2021; 35:919–27. [DOI] [PubMed] [Google Scholar]
- 85. Gordon KB, Foley P, Krueger JG et al. Bimekizumab efficacy and safety in moderate to severe plaque psoriasis (BE READY): a multicentre, double‐blind, placebo‐controlled, randomised withdrawal phase 3 trial. Lancet 2021; 397:475–86. [DOI] [PubMed] [Google Scholar]
- 86. Papp K, Menter A, Leonardi C et al. Long‐term efficacy and safety of brodalumab in psoriasis through 120 weeks and after withdrawal and retreatment: subgroup analysis of a randomized phase III trial (AMAGINE‐1). Br J Dermatol 2020; 183:1037–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Reich K, Armstrong AW, Langley RG et al. Guselkumab versus secukinumab for the treatment of moderate‐to‐severe psoriasis (ECLIPSE): results from a Phase 3, randomised controlled trial. Lancet 2019; 394:831–9. [DOI] [PubMed] [Google Scholar]
- 88. Mashiko S, Edelmayer RM, Bi Y et al. Persistence of inflammatory phenotype in residual psoriatic plaques in patients on effective biologic therapy. J Invest Dermatol 2020; 140:1015–25.e4. [DOI] [PubMed] [Google Scholar]
- 89. Eyerich K, Weisenseel P, Pinter A et al. IL‐23 blockade with guselkumab potentially modifies psoriasis pathogenesis: rationale and study protocol of a phase 3b, randomised, double‐blind, multicentre study in participants with moderate‐to‐severe plaque‐type psoriasis (GUIDE). BMJ Open 2021; 11:e049822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chang SH, Reynolds JM, Pappu BP et al. Interleukin‐17C promotes Th17 cell responses and autoimmune disease via interleukin‐17 receptor E. Immunity 2011; 35:611–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Amatya N, Childs EE, Cruz JA et al. IL‐17 integrates multiple self‐reinforcing, feed‐forward mechanisms through the RNA binding protein Arid5a. Sci Signal 2018; 11:eaat4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Armstrong AW, Blauvelt A, Crowley JJ et al. Defining drug‐free remission of skin disease in patients with plaque psoriasis. Br J Dermatol 2020; 182:1484–7. [DOI] [PubMed] [Google Scholar]
- 93. Grän F, Kerstan A, Serfling E et al. Current developments in the immunology of psoriasis. Yale J Biol Med 2020; 93:97–110. [PMC free article] [PubMed] [Google Scholar]
- 94. Eyerich S, Eyerich K, Pennino D et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest 2009; 119:3573–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Benezeder T, Wolf P. Resolution of plaque‐type psoriasis: what is left behind (and reinitiates the disease). Semin Immunopathol 2019; 41:633–44. [DOI] [PMC free article] [PubMed] [Google Scholar]