

Abstract

Copper(I) [Cu2(μ-Br)2(tBuImCH2pyCH2L)]n (L = OMe, NEt2, NHtBu) compounds supported by flexible functionalized NHC-based polydentate ligands have been prepared in a one-pot procedure by reacting the corresponding imidazolium salt with an excess of copper powder and Ag2O. An X-ray diffraction analysis has revealed that [Cu2(μ-Br)2(tBuImCH2pyCH2NEt2)]n is a linear coordination polymer formed by bimetallic [Cu(μ-Br)]2 units linked by the lutidine-based NHC-py-NEt2 ligand, which acts as a heteroditopic ligand with a 1κC-2κ2N,N′ coordination mode. We propose that the polymeric compounds break down in the solution into more compact tetranuclear [Cu2(μ-Br)2(tBuImCH2pyCH2L)]2 compounds with a coordination mode identical to the functionalized NHC ligands. These compounds have been found to exhibit high catalytic activity in the Cu-catalyzed azide–alkyne cycloaddition (CuAAC) reaction. In particular, [Cu2(μ-Br)2(tBuImCH2pyCH2NEt2)]2 efficiently catalyzes the click reaction of a range of azides and alkynes, under an inert atmosphere at room temperature in neat conditions at a very low catalyst loading, to quantitatively afford the corresponding 1,4-disubstituted 1,2,3-triazole derivatives in a few minutes. The cycloaddition reaction of benzyl azide to phenylacetylene can be performed at 25–50 ppm catalyst loading by increasing the reaction time and/or temperature. Reactivity studies have shown that the activation of the polynuclear catalyst precursor involves the alkyne deprotonation by the NHC moiety of the polydentate ligand to afford a copper(I)-alkynyl species bearing a functionalized imidazolium ligand. DFT calculations support the participation of the dinuclear species [(CuBr)2(μ-tBuImCH2pyCH2NEt2)], resulting from the fragmentation of the tetranuclear compound, as the catalytically active species. The proposed reaction pathway proceeds through zwitterionic dinuclear intermediates and entails the active participation of both copper atoms, as well as the NHC moiety as an internal base, which activates the reacting alkyne via deprotonation.
Introduction
The copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction has greatly expanded the toolbox of synthetic organic chemistry.1 This process selectively transforms organic azides and terminal alkynes into the corresponding 1,4-disubstituted 1,2,3-triazoles under mild reaction conditions, thus fulfilling the criteria of ″click chemistry″ defined by Sharpless et al.2 In contrast, the noncatalyzed version, i.e., the Huisgen thermal reaction, proceeds at much higher temperatures and affords mixtures of 1,4- and 1,5-disubstituted triazole regioisomers.3 This synthetic protocol, which provides access to complex organic architectures from readily available building blocks, has found numerous applications in such important disciplines as modern organic chemistry, nanotechnology, chemical biology, medicinal chemistry, drug discovery, and materials science.4
It is well established that the CuAAC reaction proceeds through copper(I) active species that have a limited stability. Thus, the addition of ligands with the ability to stabilize the copper(I) intermediates or, alternatively, the use of well-defined copper(I) catalysts generally results in the improvement of the catalytic efficiency. In fact, a number of N-, P-, and S-based mono- and polydentate ligands with distinct stereoelectronic properties have been thoroughly investigated.5 Research in CuAAC has experienced a spectacular growth in recent years as a consequence of (i) the preparation of well-defined copper(I) catalysts, (ii) the availability and broad applicability of straightforward protocols to access 1,2,3-triazole derivatives, and (iii) experimental and theoretical studies to unravel the reaction mechanism.6
N-Heterocyclic carbenes (NHCs) have become essential ligands for transition-metal catalysis owing to their strong coordination ability as well as electronic and steric modularity.7 In fact, Cu(NHC)-mediated catalysis has experienced an intense growth with an increase in the number of applications.8 In this regard, N-heterocyclic carbene-based copper complexes of type [(NHC)CuX]9 and [(NHC)2Cu]X10 have been proven to be very efficient for the click cycloaddition reaction between azides and alkynes under very mild conditions. Remarkably, NHC ligands significantly increase the stability of copper(I) intermediates, allowing for the reduction of the catalyst loading while maintaining a notable catalytic activity. In addition, copper(I) complexes bearing mesoionic NHC carbenes11 and related dinuclear counterparts12 have also been found to be excellent catalysts for the CuAAC reaction. In sharp contrast, copper catalyst precursors based on functionalized NHC ligands are, to our knowledge, much more scarce and limited to NHC ligands having thioether, sulfoxide, or sulfone wingtips13 or polar groups to impart water solubility.14
The first mechanistic studies of such processes were performed by Sharpless and co-workers in 200215 and were complemented later by several theoretical and experimental studies.16 Based on DFT studies, Fokin et al. proposed that the presence of a second copper atom favors the reaction,17 which was further supported from an experimental point of view.18,19 This information led Fokin to propose the reaction mechanism depicted in Figure 1, which highlights the active participation of both copper atoms. The isolation of several dinuclear σ,π-bis(copper)alkynyl species that had been proposed as reaction intermediates further supported the mechanism.19,20
Figure 1.

Mechanism of the CuAAC reaction proposed by Fokin et al.19
These findings have increased the interest in mechanistic studies of CuAAC processes.21 As a result, several groups have reported very significant experimental22 and theoretical studies,23 which mainly focus on identifying, isolating, and characterizing resting states24 with special interest in the nuclearity and the explicit role of the copper atoms involved in the process.22a,25 This has led to a relatively well-established Fokin-like catalytic cycle, although some authors have proposed some modifications, for example, the coordination of the azide to the Cu atom σ-bound to the alkynyl group instead of to the ligand-bound Cu center (Cu′ in Figure 1).23f
In this context, herein we report the synthesis of copper(I) coordination polymers supported by tridentate NHC-py-L (L = OMe, NEt2, NHtBu) ligands and their application in the base-free CuAAC reaction. In addition, we have performed DFT and reactivity studies to determine the possible reaction mechanism as a further step to the complete understanding of the CuAAC processes.
Results and Discussion
Synthesis of Imidazolium Salt Precursors for Tridentante NHC-py-L (L = OMe, NEt2, NHtBu) Ligands
The functionalized imidazolium salt precursors of flexible lutidine-derived polydentate ligands NHC-py-L (L = OMe, NEt2, NHtBu) have been prepared from the imidazolium salt 1-((6-(bromomethyl)pyridin-2-yl)methyl)-3-(tert-butyl)-1H-imidazol-3-ium bromide.26 Thus, nucleophilic substitution at the bromomethyl group by sodium methoxide, dimethylamine, or tert-butylamine afforded the corresponding methoxy (1), diethylamino (2), and tert-butylamino (3) functionalized imidazolium salts, which were isolated as pale yellow solids in 80–95% yield (Scheme 1). Stoichiometric amounts of sodium methoxide and diethylamine were used in the synthesis of 1 and 2, which were carried out at room temperature in methanol and acetonitrile, respectively. However, an excess of tert-butylamine was required in the synthesis of 3, which was carried out in acetonitrile at 373 K.
Scheme 1. Synthetic Pathway for the Preparation of Copper(I) Complexes Supported by Lutidine-Based NHC/L Functionalized Ligands.

The functionalized imidazolium salts 1–3 have been characterized by elemental analysis, mass spectrometry (HRMS-ESI), and 1H and 13C{1H} NMR spectroscopy. Particularly, the mass spectra show a peak that corresponds to the molecular ion. The 1H NMR spectra of imidazolium salts exhibit a series of characteristic resonances. Namely, a low-field signal at δ ≈ 11.0 ppm corresponding to the acidic proton of the imidazolium moiety, the =CH resonances of the pyridine and imidazole rings in the aromatic region, a signal at δ ≈ 1.8 ppm for the −tBu substituent, and two singlets for the bridging >CH2 protons at δ ≈ 5.8 ppm (NHC-CH2-py) and 4.5–3.5 ppm (py-CH2-L). Furthermore, the spectra show the characteristic signals of the corresponding functional groups OMe, δ = 3.45 ppm (s); NEt2, δ = 2.54 (q), 1.03 ppm (t); and NHtBu, δ = 1.69 (s, NH), 1.38 ppm (s, tBu).
Synthesis of Copper(I) Complexes [Cu2(μ-Br)2(tBuImCH2pyCH2L)]n (L = OMe, NEt2, NHtBu)
The synthesis of copper(I) compounds based on the potentially tridentate ligands tBuImCH2pyCH2L (L = OMe, NEt2, NHtBu) has been approached following the synthetic strategy described by Chen et al.27 This methodology allows one to directly obtain organometallic compounds bearing NHC ligands from the metal powder of interest. This way, the one-pot reaction of the imidazolium salt with an excess of copper powder and Ag2O in acetonitrile for 30 h at 323 K afforded yellow solutions of complexes [Cu2(μ-Br)2(tBuImCH2pyCH2L)]n (L = OMe, 4; NEt2, 5; NHtBu, 6) after removing any amount of unreacted copper powder and the newly formed metallic silver (Scheme 1). The compounds were isolated as pale brown (4) or pale green (5 and 6) air sensitive microcrystalline solids with yields close to 50% with respect to the imidazolium salt precursors. In this regard, it should be noted that compounds 4–6 incorporate two bromido ligands for each lutidine-based NHC/L scaffold, showing a mismatch in the stoichiometry of the reactions. We believe that this is partially responsible for the moderate yield achieved in the synthesis of the complexes. To improve this, the synthesis of 5 was carried out by adding 2.2 equiv of KBr to the reaction mixture, which allowed it to be increased to 68%.
Single-crystal X-ray diffraction analysis has revealed that 5 is a linear coordination polymer of formula [Cu2(μ-Br)2(tBuImCH2pyCH2NMe2)]n (Figures 2 and 3). Bimetallic units [Cu(μ-Br)]2 are linked by the lutidine-based ligand NHC-py-NEt2, which acts as a heteroditopic ligand with a 1κC-2κ2N,N′ coordination. The copper centers exhibit different coordination geometries, one being tricoordinate (Cu1) and the other tetracoordinate (Cu2). The intermetallic distance [Cu1···Cu2 2.8185(6) Å] is larger than twice the van der Waals radius of copper (rCu = 1.40 Å), thus ruling out any bond between both metal centers. The [Cu(μ-Br)]2 core deviates from planarity, featuring a puckering angle of 21.0° between the Cu2-Br1-Br2 and the Cu1-Br1-Br2 planes. On the one hand, the coordination sphere of Cu1 is completed by the C1 carbon atom of the NHC moiety, rendering a distorted trigonal planar geometry [C1-Cu1-Br2 129.27(10)°, C1-Cu1-Br1 123.28(10)°, Br2-Cu1-Br1 107.45(2)°] with the NHC core slightly deviating from the perpendicular arrangement with respect to the coordination plane [N2-C1-Cu1-Br1–73.9(3)°]. On the other hand, the copper center Cu2 is bonded to the pyridine nitrogen atom N12 and the amine nitrogen atom N18, exhibiting a distorted tetrahedral geometry (Figure 3). Finally, it is worth mentioning that the five-member ring Cu2′-N12-C13-C17-N18 adopts an envelope configuration [Cremer–Pople28 parameters: q 0.450(3) Å, Φ 141.3(4)°] and the pyridine fragment deviates from the ideal arrangement with respect to the Cu2′-N12 bond (pitch, θ 2.4°; yaw, ψ 9.5°).29 This is surely a consequence of the small bite angle of the pyridine-amine ligating site [N12″-Cu2-N18″ 82.44(11)°].
Figure 2.

ORTEP views of the polymeric array of 5. Thermal ellipsoids are at 50% probability. Hydrogen atoms are omitted for clarity.
Figure 3.
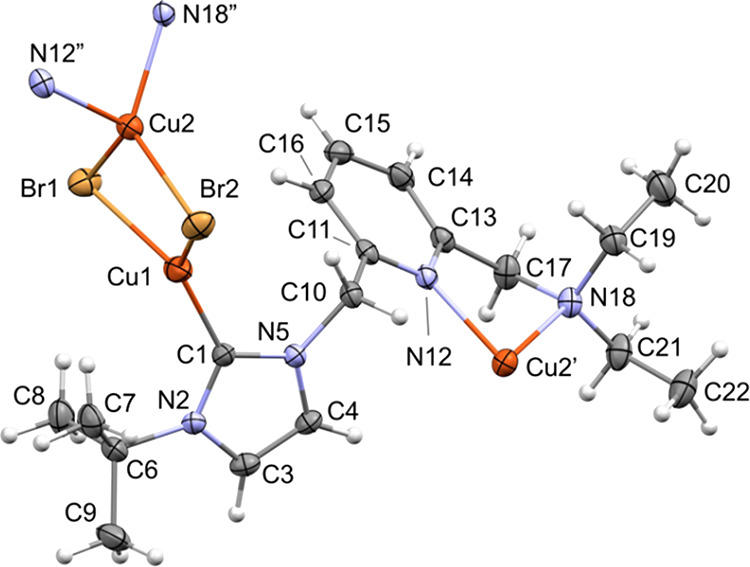
ORTEP view of the repeating unit of 5. Thermal ellipsoids are at 50% probability. Selected bond lengths (Å) and angles (°) are as follows: Cu1-Br2 2.4226(6), Cu1-Br1 2.4603(6), Cu2-N12″ 2.064(3), Cu2-N18″ 2.174(3), Cu2-Br1 2.4133(5), Cu2-Br2 2.4430(6), Cu1···Cu2 2.8185(6), C1-Cu1-Br2 129.27(10), C1-Cu1-Br1 123.28(10), Br2-Cu1-Br1 107.45(2), N12″-Cu2-N18″ 82.44(11), N12″-Cu2-Br1 118.78(8), N18″-Cu2-Br1 115.65(8), N12″-Cu2-Br2 122.22(7), N18″-Cu2-Br2 106.25(8), and Br1-Cu2-Br2 108.31(2). Symmetry transformations used to generate equivalent atoms Cu2′, N12″, and N18″: ′ −x + 1/2, y + 1/2, −z + 1/2; ″ −x + 1/2, y – 1/2, −z + 1/2.
The compounds have been fully characterized by elemental analysis, mass spectrometry, and multinuclear NMR spectroscopy. The most noticeable feature of the 1H NMR spectra is the absence of the characteristic low field resonance of the imidazolium fragment, which confirms the formation of the Cu-NHC bond. Also, the carbenic carbon atom (NCN) is observed at around δ 178 ppm in the 13C{1H} NMR spectra. In view of the similarities of the NMR spectra, the three complexes seem to be isostructural in solution. In this regard, their 1H and 13C NMR spectra show the same signals for the pyridine and imidazol-2-ylidene moieties, the only differences arising from the characteristic resonances of the functional group L (L = OMe, NEt2, or NHtBu), which appear at the expected chemical shifts in each case. Surprisingly, the >CH2 protons joining the pyridine ring with the imidazol-2-ylidene fragment appear as singlets at around δ 5.4–5.7 ppm despite the coordination of both groups. Besides, the methylene protons of py-CH2-L fragment give rise to a singlet at δ 4.51 and 3.75 ppm for 4 and 5, respectively, and a multiplet at δ 4.07 ppm for 6.
The high solubility of the aforementioned coordination polymers in conjunction with the simplicity of the NMR spectra as well as the sharp profile of the signals suggests that polymers are prone to fragment in solutions, yielding smaller subunits that likely recombine in a dynamic equilibrium. Fragmentation of the polymer [Cu2(μ-Br)2(tBuImCH2pyCH2L)]n into its constituents should give rise to dinuclear species of composition [(CuBr)2(μ-tBuImCH2pyCH2L)] having di- and tri-coordinated Cu(I) centers (Scheme 2).30 The high-resolution ESI+ mass spectra of the compounds show both the peak of the metal fragment [L + Cu]+ and that of the imidazolium salt (LH+). In addition, the fragments [L + CuBr+H]+ and [L + Cu2 + Br]+ were observed in the mass spectrum of 6. Interestingly, a peak at m/z 1012.0384 [M-2Br-2H]+ derived from a copper tetranuclear species was also observed in the HRMS of 5.
Scheme 2. Fragmentation of the Coordination Polymers and Reversible Formation of the Tetranuclear Species in the Solution (L = OMe, NEt2, NHtBu).

In this regard, DFT calculations have shown that the related tetranuclear species [Cu2(μ-Br)2(tBuImCH2pyCH2L)]2 are more stable than the dinuclear ones [(CuBr)2(μ-tBuImCH2pyCH2L)]. However, the fragmentation process of the tetranuclear species [Cu2(μ-Br)2(tBuImCH2pyCH2NEt2)]2 to yield two dinuclear species is affordable, the dinuclear fragments being 6.6 kcal·mol–1 higher in energy than the original tetramer. In addition, this process is less unfavorable for 4 (L = OMe) and 6 (L = NHtBu), with ΔG being 0.1 and 0.9 kcal·mol–1, respectively. Although the coordination mode of each functionalized NHC ligand in the proposed tetranuclear species is identical to that shown in the polymer structure, only two Cu2(μ-Br)2 moieties are involved, thereby resulting in a compact tetranuclear structure of C2 symmetry compatible with the NMR spectroscopic data. At this point, it is worth mentioning that Cu(I) complexes having 2-pyridylmethyl-functionalized NHC ligands exhibit a considerable structural diversity ranging from mononuclear31 to dinuclear structures32 or coordination polymers.33 Taking this information into account, we propose that compounds 4–6 are coordination polymers in the solid state that fragment in the solution to afford the corresponding tetranuclear species, which are likely in equilibrium with the dinuclear analogs (Scheme 2).
In terms of structural parameters, the DFT-computed Cu–Cu distance of the Cu2(μ-Br)2 moiety in the tetranuclear species resulting from 5 is shorter than that determined crystallographically in the polymer structure of 5, 2.652 Å vs 2.8185(6) Å (Figure 4a). This is a consequence of the compact structure of the tetranuclear species, which allows for a Cu–Cu bonding interaction. In addition, we found that the dinuclear species also exhibit a bonding interaction with a computed Cu–Cu distance of 2.599 Å (Figure 4b), which may contribute to the stabilization of the structure.
Figure 4.

Optimized structure for (a) the tetranuclear compound [Cu2(μ-Br)2(tBuImCH2pyCH2NEt2)]2 (5) and (b) the dinuclear [(CuBr)2(μ-tBuImCH2pyCH2NEt2)]. Hydrogen atoms have been omitted for clarity.
Azide–Alkyne Cycloaddition Reactions Catalyzed by [Cu2(μ-Br)2(tBuImCH2pyCH2L)]2
[Cu2(μ-Br)2(tBuImCH2pyCH2L)]2 (L = OMe, NEt2, NHtBu) are efficient catalysts for the [3 + 2] azide–alkyne cycloaddition reaction that selectively affords the 1,4-regioisomer even at low catalytic loadings. The catalytic activity of the copper(I) polynuclear complexes was first examined for the cycloaddition of benzyl azide to phenylacetylene as a benchmark click reaction. The catalysis was carried out at room temperature under an argon atmosphere, in the absence of a base and solvent, using a catalyst loading of 0.5 mol % (Table 1). Compounds 5 and 6, featuring an amino functional group, were found to be more active than 4 (L = OMe), which achieves 79% conversion in 5 min (entries 6–8). Quantitative conversion to 1-benzyl-4-phenyl-1H-1,2,3-triazole was attained with catalyst 5 (L = NEt2) in 5 min (entry 7). Under these conditions, the reaction does not proceed without a catalyst, and when 5 is replaced by CuBr or an equimolecular CuBr/NEt3, the reaction efficiency decreases significantly even after prolonged reaction times (entries 1 and 2–5). We also considered acetonitrile as the solvent. However, the 5-catalyzed cycloaddition reaction is slower, with roughly 70% conversion in 5 min, which increases to 98% after 30 min (entries 9–11). This activity decrease was also observed in dichloromethane and becomes more pronounced in polar solvents such as methanol and water (entries 12–14).
Table 1. Catalyst Screening and Solvent Optimization for the Cycloaddition of Benzyl Azide and Phenylacetylenea.

| entry | catalyst | solventb | t (min) | conversion (%)c |
|---|---|---|---|---|
| 1 | none | neat | 5 | 0 |
| 2 | CuBr | neat | 5 | 9 |
| 3 | CuBr | CH3CN | 60 | 27 |
| 4 | CuBr + NEt3 (1:1) | neat | 5 | 21 |
| 5 | CuBr + NEt3 (1:1) | CH3CN | 60 | 41 |
| 6 | 4 | neat | 5 | 79 |
| 7 | 5 | neat | 5 | 100 |
| 8 | 6 | neat | 5 | 94 |
| 9 | 5 | CH3CN | 5 | 69 |
| 10 | 5 | CH3CN | 20 | 92 |
| 11 | 5 | CH3CN | 30 | 98 |
| 12 | 5 | CH2Cl2 | 5 | 68 |
| 13 | 5 | MeOH | 5 | 51 |
| 14 | 5 | H2O | 5 | 36 |
Reaction conditions: benzyl azide (0.5 mmol), phenylacetylene (0.5 mmol), and catalyst CuBr or [Cu2(μ-Br)2(tBuImCH2pyCH2L)]2 (0.0025 mmol, 0.5 mol %) at 298 K.
Solvent (0.5 mL).
Conversions, relative to benzyl azide, determined by GC using mesitylene as internal standard.
As for the scope of the cycloaddition reaction, the performance of 5 was investigated by using benzyl azide and phenyl azide, as representative azides, and a variety of aromatic and aliphatic alkynes under optimized catalytic conditions (Table 2). The reactions were carried out at room temperature under argon in the absence of solvent and a catalyst loading of 0.5 mol %. Ring-substituted phenylacetylene derivatives bearing electron-rich substituents at the para position (−Me, −OMe, −tBu) proceeded efficiently to quantitatively afford the corresponding 1,2,3-triazoles 9b–9d in 5 min. Electron-poor alkynes (−CF3) reacted slightly slower, requiring 10 min to achieve complete benzyl azide conversion. Sterically hindered alkynes, such as o-substituted phenylacetylene (−OMe), reacted at the same rate as the m- and p-substituted derivatives and reached full conversion in 5 min. Besides, the functionalized alkyne 2-ethynylpyridine was efficiently transformed into 9h in 5 min. As expected, aliphatic alkynes are less reactive than aromatic ones. Namely, a 42% conversion was achieved in the cycloaddition reaction of benzyl azide to hex-1-yne in 5 min, although conversion increased to 63% in 30 min, and full conversion to the target 1,2,3-triazole 9i took 3 h. Similarly, 1,7-octadiyne was efficiently transformed into the corresponding bis-triazole product 9j in 3 h. On the other hand, internal alkynes such as but-2-yne or diphenylacetylene exhibited much slower reaction rates, affording around 25% conversion to the desired 1,2,3-triazoles 9k and 9l in 72 h at 343 K.
Table 2. [3 + 2] Cycloaddition of Azides and Alkynes Catalyzed by [Cu2(μ-Br)2(tBuImCH2pyCH2NEt2)]2 (5)i.


Reaction conditions: azide (0.5 mmol), alkyne (0.5 mmol), and catalyst 5 (0.0025 mmol, 0.5 mol %) at 298 K.
Conversion relative to azide determined by GC using mesitylene as an internal standard.
Isolated yields in parenthesis.
Azide (1.0 mmol).
Temperature 343 K.
The cycloaddition reaction involving the sterically demanding phenyl azide proceeded more slowly than benzyl azide. Even so, ring-substituted phenylacetylene derivatives were efficiently transformed into the corresponding triazoles 9m–9p in 30 min regardless of the electronic character of the substituent at the para position. Gratifyingly, hex-1-yne produced quantitatively 9q in only 30 min. The increased reaction rate compared to benzyl azide can be tentatively attributed to the electron-delocalization enabled by the azide, which allows overcoming the steric penalty.
The 1,4-disubstituted-1,2,3-triazole derivatives 9 were isolated as solids in excellent yields for any azide–alkyne combination, typically 90–95%, by washing the crude product with pentane.
Azide–Alkyne Cycloaddition Reactions Catalyzed by [Cu2(μ-Br)2(tBuImCH2pyCH2NEt2)]2 (5) at Low Catalyst Loading
The excellent catalytic activity of 5 at a relatively low catalyst loading (0.5 mol %) prompted us to explore the possibility of decreasing it even more. This way, the performance of 5 was investigated using benzyl azide and phenylacetylene as model substrates at lower catalyst loading under neat conditions (Table 3).
Table 3. Cycloaddition of Benzyl Azide and Phenylacetylene Catalyzed by [Cu2(μ-Br)2(tBuImCH2pyCH2NEt2)]2 (5) at Low Catalyst Loadinga.

| entry | T (K) | solvent | 5 (mol %) | t (h) | conversion (%)b | TON | TOF (h–1) |
|---|---|---|---|---|---|---|---|
| 1 | 298 | neat | 0.25 | 0.08 | 56 | 224 | 2800 |
| 2 | 298 | neat | 0.05 | 0.5 | 50 | 1000 | 2000 |
| 3 | 298 | neat | 0.05 | 3 | 88 | 1760 | 600 |
| 4 | 298 | neat | 0.005 | 24 | 96 | 19,200 | 800 |
| 5 | 323 | neat | 0.005 | 24 | 99 | 19,800 | 820 |
| 6 | 298 | CH3CN | 0.005 | 24 | 0 | 0 | 0 |
| 7 | 323 | CH3CN | 0.005 | 48 | 80 | 16,000 | 300 |
| 8 | 298 | neat | 0.0025 | 24 | 66 | 26,400 | 1100 |
| 9 | 298 | neat | 0.0025 | 48 | 82 | 32,800 | 680 |
| 10 | 323 | neat | 0.0025 | 24 | 90 | 36,000 | 1500 |
| 11 | 323 | neat | 0.0025 | 48 | 98 | 39,200 | 820 |
Reaction conditions: benzyl azide (0.5 mmol), phenylacetylene (0.5 mmol), solvent (0.5 mL).
Conversion relative to benzyl azide and selectivities determined by GC using mesitylene as internal standard.
Decreasing the catalyst loading to 0.25 mol % resulted in a conversion of phenyl azide of 56% at 298 K (entry 1). Similar conversion was attained after 0.5 h by decreasing the catalyst loading to 0.05 mol %, which increased to 88% after 3 h (entries 2 and 3). Further decreasing the catalyst loading to 0.005 mol % (50 ppm) gave 96% conversion in 24 h at 298 K. Increasing the temperature to 323 K allowed to quantitatively afford triazole 9a in 24 h (TOF = 820 h–1) (entries 4 and 5). The reaction profile (conversion vs time) for the cycloaddition of benzyl azide to phenylacetylene catalyzed by 5 (0.005 mol %) at 298 and 323 K is shown in Figure 5. The process is faster at 323 K at the early reaction stage, although similar reaction times are required to achieve complete conversion of benzyl azide. Although no reaction was observed in acetonitrile at 298 K at the same catalyst loading, 80% conversion was attained at 323 K after 48 h (entries 6 and 7). Remarkably, it is possible to decrease the catalyst loading up to 0.0025 mol % (25 ppm) working under neat conditions at reasonable reaction times. At 298 K, a benzyl azide conversion of 82% was achieved in 48 h, increasing to 90% (24 h, TOF = 1500 h–1) and 98% (48 h, TOF = 820 h–1) at 323 K (entries 8–11).
Figure 5.

Reaction profile of conversion vs time for the cycloaddition of neat benzyl azide (0.5 mmol) and phenylacetylene (0.5 mmol) catalyzed by 5 (0.005 mol %) at 298 and 323 K.
Catalytic studies performed at low catalyst loading further highlight the efficiency of catalyst 5 (Table 4). CuAAC of benzyl azide with para-substituted phenylacetylene derivatives proceeded efficiently at 0.005 mol % (50 ppm) at room temperature regardless of the electronic character of the substituent (−OMe and −CF3), with conversions to 9c and 9e higher than 90% in 24 h. Similar conversion was attained when the reactions were performed at 0.0025 mol % (25 ppm) at 323 K. Interestingly, the formation of the 1,5-disubstituted-1,2,3-triazole regioisomers was not observed in any case under these conditions. Unfortunately, aliphatic alkynes such as hex-1-yne were not transformed under low catalyst loading conditions even at 323 K.
Table 4. Cycloaddition of Benzyl Azide and Alkynes Catalyzed by [Cu2(μ-Br)2(tBuImCH2pyCH2NEt2)]2 (5) at Low Catalyst Loadinga,b.


Reaction conditions: benzyl azide (0.5 mmol) and alkyne (0.5 mmol).
Conversion relative to benzyl azide determined by GC using mesitylene as internal standard.
Mechanistic Insights on the [3 + 2] Azide–Alkyne Cycloaddition Reaction Catalyzed by [Cu2(μ-Br)2(tBuImCH2pyCH2NEt2)]2 (5)
To ascertain the reaction mechanism of the azide–alkyne cycloaddition catalyzed by 5 and attain an in-depth understanding of the origin of its high activity and selectivity, we performed a series of reactivity experiments and a DFT-based theoretical study.
Reactivity Studies
First, we studied the cycloaddition reaction by NMR using a high catalyst loading. Reaction of benzyl azide and phenylacetylene (20 equiv, 1:1 molar ratio) catalyzed by 5 (2.5 mol %) in CD3CN at 298 K resulted in the formation of the cycloaddition reaction product 1-benzyl-4-phenyl-1H-1,2,3-triazole in 5 min. In addition, the 1H NMR of the reaction mixture showed the disappearance of 5 and the formation of a new species, 10. It shows a new set of resonances for the lutidine-derived polydentate ligand, including a downfield resonance at δ 9.19 ppm suggesting the presence of an imidazolium fragment in the compound. Remarkably, the addition of a new load of benzyl azide and phenylacetylene (20 equiv) to the solution afforded the 1,2,3-triazole reaction product within a few minutes, thus suggesting that 10 is catalytically competent for the cycloaddition reaction.
Compound 10 was formed immediately by reaction of 5 with a moderate excess of phenylacetylene (1.2 equiv relative to the dinuclear derivative of 5) in CD3CN. However, NMR spectra of the reaction mixture at 243 K evidenced the presence of unreacted 5 and phenylacetylene. The amount of 10 in the reaction mixture increased upon increasing the amount of phenylacetylene added (5 equiv), which eventually made it possible to record the 13C{1H}-APT NMR spectrum (see the Supporting Information). Compound 10 has been identified as a zwitterionic copper(I) dinuclear alkynyl species [(CuBr)2(C≡CPh)(tBuHImCH2pyCH2NEt2)] bearing the functionalized imidazolium ligand. The proposed structure for 10 (shown in Scheme 3) is based on DFT calculations (see below) and the experimental evidence. Namely, the existence of an imidazolium group in 10 was confirmed by bidimensional HSQC and HMBC spectra. The broad singlet at δ 9.19 ppm, corresponding to the acidic imidazolium proton NCHN, correlates with the CH signal at δ 139.2 ppm in the 13C{1H}-APT NMR spectra. Moreover, the long-range 1H–13C HMBC spectrum showed cross-peaks between the NCHN proton and the =CH carbons of imidazole at δ 124.1 and 121.0 ppm. In addition, the 13C NMR spectra showed two resonances at δ 126.7 and 98.8 ppm that are assigned to the Cα and Cβ atoms of the alkynyl ligand, respectively.19,20
Scheme 3. Reaction of 5 with Phenylacetylene: Formation of [(CuBr)2(C≡CPh)(tBuHImCH2pyCH2NEt2)] (10) (Structure Based on DFT Calculations).

In short, intermediate 10 is the result of the deprotonation of phenylacetylene mediated by the imidazole-2-ylidene scaffold, which leads to the formation of an imidazolium-alkynyl species. Although it might seem somewhat counterintuitive, this kind of result is not unprecedented, as the formation of alkynyl intermediates by protonation of the NHC groups dissociated from the Cu atom has already been reported in several CuAAC processes.10a,10,25d
To shed more light on the reaction mechanism, we complemented the previous studies with deuterium labeling experiments. In this regard, treatment of benzylazide (0.5 mmol) with phenylacetylene-d1 (0.5 mmol) in the presence of a solution of catalyst 5 (0.025 mol %) in CD3CN (0.5 mL) resulted in the formation of the 1,2,3-triazole after 5 h at room temperature. Importantly, the position 5 of the N-heterocyclic ring was fully deuterated, with no deuteration observed at any other position.
Theoretical Study on the [3 + 2] Azide–Alkyne Cycloaddition Reaction Catalyzed by 5
We have found experimental and theoretical evidence that coordination polymer 5 likely breaks down into tetranuclear [Cu2(μ-Br)2(tBuImCH2pyCH2NEt2)]2 species. In addition, fragmentation of the tetranuclear structure into the dinuclear species is an affordable process, which is favored from an entropic point of view, as once the tetramer breaks apart, the feasibility of a molecular encounter that regenerates the original structure is very low under catalytic conditions. Thus, we hypothesized that the dinuclear derivative of complex 5 (see Scheme 2) is the active species for the catalytic cycle and computed the reaction profile on the basis of this structure. In addition, as previously explained, activation of the catalyst precursor 5 involves the reaction with phenylacetylene, leading to protonation of the NHC moiety. The resulting alkynyl compound 10 corresponds to a zwitterionic copper(I) dinuclear complex with a functionalized imidazolium ligand, as discussed in the previous subsection and shown in Scheme 3 (10 is named as C in the reaction mechanism), which arises in a direct way in the proposed reaction mechanism. Notice that we selected phenylacetylene and benzyl azide as models for the alkyne and the azide, respectively. For the sake of clarity, the reaction intermediates and transition states are referred to by capital letters starting with A (which corresponds to the dinuclear derivative of 5).
As an initial step, we computed the reaction profile for the classical, noncatalyzed, Huisgen 1,3-dipolar cycloaddition reaction, which is known to yield a mixture of the corresponding 1,4- and 1,5-substituted 1,2,3-triazoles.3 This general result is consistent with our calculations, as the Gibbs energy difference between the transition structures that lead to the 1,4- and 1,5-regiosiomers is only 2.0 kcal·mol–1, thus explaining the lack of selectivity of the process (see Figure 6). In addition, the energy barrier would be 28.4 kcal·mol–1 (for the 1,4-disubstituted-1,2,3-triazole), which is too high for the reaction to take place at room temperature.
Figure 6.

Gibbs energy profile (kcal·mol–1) for the noncatalyzed Huisgen 1,3-dipolar cycloaddition between phenylacetylene and benzyl azide.
The Gibbs energy landscape obtained for the CuAAC reaction catalyzed by A along with selected calculated transition state structures is shown in Figures 7–9. As depicted in Figure 7, the first step involves the π-coordination of the alkyne to the Cu2 center—notice that we have used the same labeling as for the crystal structure—leading to intermediate B, which is 1.0 kcal·mol–1 higher in energy. As expected, this coordination leads to a subtle increase in the Cu–Cu distance from 2.599 Å (in A) to 2.674 Å (in B). The next step consists of the alkyne deprotonation by the NHC moiety via TSB-C, yielding the zwitterionic intermediate C (see Figure 8a,b), which we find to be consistent with the experimentally characterized species 10. Notice that the structure of C corresponds to a σ,π-bis(copper)acetylide complex, which has extensively been reported in CuAAC processes.20 The process has an effective energy barrier of 23.8 kcal·mol–1 (dictated by the energy difference between TSB-C and A), which is affordable under the reaction conditions, especially considering that the catalytic reaction takes place in neat phenylacetylene. It is also remarkable that C is 7.5 kcal·mol–1 more stable than A, which agrees with the experimental observation of the formation of 10 by the reaction of 5 with phenylacetylene.
Figure 7.

DFT calculated Gibbs energy reaction profile (in kcal·mol–1 relative to A and isolated molecules) for the alkyne activation and triazolyl scaffold formation in the CuAAC reaction catalyzed by A. The labeling used for Cu atoms in the main text is shown in structure A.
Figure 9.

DFT calculated Gibbs energy reaction profile (in kcal·mol–1 relative to A and isolated molecules) for the 1,4-disubstituted 1,2,3-triazole product formation in the CuAAC reaction catalyzed by A.
Figure 8.

DFT-optimized structures and selected distances of (a) TSB-C, (b) C, (c) TSD-E, and (d) TSD-E′. Nonrelevant hydrogen atoms have been omitted for clarity.
Then, the azide coordinates to the Cu atom bonded to the alkynyl moiety,23f yielding intermediate D, with a relative Gibbs energy of −3.2 kcal·mol–1. As a result, the alkynyl ligand changes its coordination mode to μ2-η1 and the Cu–Cu distance decreases from 3.016 Å (in C) to 2.508 Å (in D). The next step involves the cycloaddition between the azide and the alkyne, which is mediated by the Cu active center, forming the six-membered metallacycle E. This step proceeds via TSD-E and determines the regioselectivity of the process, as the relative orientation of the azide and the alkyne within the transition structure determines whether the 1,4- or 1,5-disubstituted 1,2,3-triazole product will form. In this regard, the transition structure leading to the 1,4-substituted triazole, TSD-E, is 8.9 kcal·mol–1 lower in energy than that leading to the 1,5-product, TSD-E′, and this result explains the complete process regioselectivity (see Figure 8c,d). Thereby, the first C–N bond is formed, overcoming an effective energy span of 15.1 kcal·mol–1 (with respect to C), thus being affordable at the reaction conditions. As expected, both Cu atoms actively participate in this step, as highlighted by Cu–C distances of 1.921 and 1.951 Å, as well as a Cu–Cu distance of 2.492 Å (Figure 8c), which are similar to those reported by other authors for related CuAAC processes.23b As previously introduced, TSD-E leads to the formation of Cu-based metallacycle E, with a relative energy of −1.0 kcal·mol–1 and in which both Cu atoms are bonded to the C atom at position 5 (as shown by Cu–C distances of 1.868 and 1.925 Å).
The following reaction step involves the contraction of the six-membered metallacycle E through the formation of the second C–N bond to yield the Cu-triazolyl species F. This process takes place via TSE-F, bearing an effective energy barrier of 10.9 kcal·mol–1 (with respect to C), and is highly exergonic, as F is 41.8 kcal·mol–1 more stable than E, having a relative Gibbs energy of −42.8 kcal·mol–1. Also note that the Cu–Cu distance in F is 2.470 Å, indicative of a strong cuprophilic interaction between both metal centers. In addition, the triazolyl moiety is coordinated in a bridging fashion to both Cu atoms, with Cu–C distances of 1.959 and 2.115 Å, in line with DFT calculations reported by other authors.25c
As a final step, the 1,4-disubstituted-1,2,3-triazole product is released, which might proceed by two different, although related, reaction pathways (see Figure 9). The first possibility consists in the proton transfer from the protonated NHC scaffold to the triazolyl moiety in F, via TSF-A, which leads to the final product and recovers the initial dinuclear structure (A). This pathway requires to overcome an energy barrier of 22.7 kcal·mol–1, determined by the Gibbs energy difference between TSF-A and intermediate F. Alternatively, the triazolyl fragment could be protonated by an additional phenylacetylene molecule, also releasing the final product and recovering intermediate C (which has been detected experimentally). This second route bears an energy barrier of 22.9 kcal·mol–1 (Gibbs energy difference between TSF-C and C), which is only 0.2 kcal·mol–1 higher than that involving TSF-A. Hence, both possibilities are expected to be operative under the reaction conditions.
Overall, the calculated effective energy span (ΔG⧧) for the catalytic cycle in terms of the framework proposed by Kozuch and Shaik is 23.8 kcal·mol–1,34 the rate-determining step (RDS) being the alkyne deprotonation of the initial complex (A) to afford Cu(I) acetylide species C via TSB-C. At this point, it should be noted that if the final reaction step proceeds through TSF-C instead of TSF-A (whose relative energy difference is only 0.2 kcal·mol–1), the subsequent reaction cycle would start directly in C, avoiding the need for overcoming TSB-C again. In this situation, the RDS would be the final protonation of the triazolyl moiety (which also implies the alkyne activation). This corresponds to a very similar process, and the effective energy span would be 22.9 kcal·mol–1 (determined by TSF-C). It is remarkable that some authors have proposed the C–N formation as the RDS,10b,16c,23b,24b,35 while others propose that the RDS corresponds to alkyne deprotonation processes.20,36,37 In this regard, our results are in line with the latter observations, although it should be considered that the medium acidity will play a crucial role in this difference.16d In this regard, it is worth mentioning that some reports indicate that substrate selection may switch the RDS step,35,36a,38 which provides additional evidence of the possibility of switching between both alternatives. At this point, it is also remarkable to note that we found the azide to coordinate to the σ-alkynyl bound Cu atom (Cu2 in Figure 7) instead of to the typically proposed ligand-bound Cu center (Cu1 in Figure 7), in agreement with reports from other authors.23f,25d
According to the reaction mechanism proposed on the basis of DFT calculations, the deprotonation of the alkyne in the RDS of both reaction pathways should lead to the observation of a primary deuterium kinetic isotope effect. However, the KIE (kH/kD) of 0.97 ± 0.01 determined in acetonitrile (see the Supporting Information) suggests that the solvent might be involved in the mechanism. In this regard, we note that the cycloaddition reaction is much slower in acetonitrile than under neat conditions (see Table 1), and a significant dependency of the reaction kinetics due to solvent effects for these kinds of processes has already been reported.36a
This way, we expect that solvent effects might be responsible for the lack of agreement between DFT and experimental results, without excluding other potential processes that may take place in the reaction medium and are likely to affect the reaction mechanism.
Conclusions
Copper(I) [Cu2(μ-Br)2(tBuImCH2pyCH2L)]n (L = OMe, NEt2, NHtBu) compounds have been prepared from the corresponding functionalized imidazolium salt by reacting the in situ generated Ag-NHC complexes with copper powder. The compounds are coordination polymers in the solid state that very likely break down in solutions into tetranuclear [Cu2(μ-Br)2(tBuImCH2pyCH2L)]2 species in equilibrium with dinuclear ones. DFT calculations have shown that the fragmentation of the tetranuclear compounds into dinuclear [(CuBr)2(μ-tBuImCH2pyCH2L)] species is an affordable process. These compounds have been proven to be efficient catalysts in the base-free [3 + 2] cycloaddition reactions of azides and alkynes at room temperature under an inert atmosphere without solvent and a catalyst loading of 0.5 mol %, with compound [Cu2(μ-Br)2(tBuImCH2pyCH2NEt2)]2 being the most active. This system efficiently catalyzes the cycloaddition reaction of benzyl azide and phenyl azide for a wide range of terminal alkynes with different properties to quantitatively afford the corresponding 1,4-disubstituted 1,2,3 triazole derivatives in a few minutes. The cycloaddition reaction of benzyl azide to phenylacetylene can be performed with a catalyst loading of 25–50 ppm by increasing the reaction time and/or temperature.
Reactivity studies have shown that the activation of the catalyst precursor [Cu2(μ-Br)2(tBuImCH2pyCH2NEt2)]2 involves the alkyne deprotonation by the NHC moiety of the polydentate ligand to afford a copper(I)-alkynyl species bearing a functionalized imidazolium ligand. We have explored a possible reaction mechanism involving the dinuclear compound [Cu2(μ-Br)2(tBuImCH2pyCH2NEt2)] as the catalytic active species or the active catalyst. DFT calculations have shown that the reaction proceeds through zwitterionic dinuclear intermediates with participation of both copper atoms following, in general terms, a Fokin mechanism. Nonetheless, there is a notable difference with respect to the original proposal, as the azide coordinates to the copper atom bonded to the alkynyl moiety rather than to the ligand-coordinated Cu center. The DFT results point to the RDS being the alkyne activation, via deprotonation by the NHC moiety, which is a consequence of the lability of the Cu-NHC bond and the basicity of the carbene. In addition, two possibilities that are very close in energy were identified for the last reaction step (triazolyl moiety protonation). Namely, it can proceed by proton transfer from the imidazolium moiety or from a phenylacetylene molecule, both being operative in the reaction conditions.
Experimental Section
General Considerations
All the experimental procedures were performed under an argon atmosphere using Schlenk or glovebox techniques. Solvents were distilled immediately prior to use from the appropriate drying agents or obtained from a Solvent Purification System (Innovative Technologies). Oxygen-free solvents were employed throughout. CDCl3 and CD3CN were dried using activated molecular sieves and degassed by three freeze–pump–thaw cycles. The functionalized lutidine-derived imidazolium salts [tBuHImCH2pyCH2Br]Br and [tBuHImCH2pyCH2OMe]Br (1) were prepared following the procedure recently reported by us.26 Phenyl azide39 and benzyl azide40 were prepared according to methods described in the literature. The organic substrates were obtained from common commercial sources and used as received or distilled prior to use depending on their purity.
Scientific Equipment
C, H, and N analyses were carried out in a PerkinElmer 2400 Series II CHNS/O analyzer. 1H and 13C{1H} NMR spectra were recorded on a Bruker Avance 300 (300.1276 and 75.4792 MHz). NMR chemical shifts are reported in ppm relative to tetramethylsilane and are referenced to partially deuterated solvent resonances. Coupling constants (J) are given in hertz. Spectral assignments were achieved by combination of 1H–1H COSY, 13C APT, 1H–13C HSQC, and 1H–13C HMBC experiments. High-resolution electrospray ionization mass spectra (HRMS-ESI) were recorded on a Bruker MicroToF-Q equipped with an API-ESI source and a Q-ToF mass analyzer, which leads to a maximum error in the measurement of 5 ppm, using sodium formate as reference. The catalytic reactions were analyzed on an Agilent 4890D system equipped with an HP-INNOWax capillary column (0.4 μm film thickness, 25 m × 0.2 mm i.d.) using mesitylene as internal standard. Organic compounds were identified by gas chromatography–mass spectrometry (GC/MS) using an Agilent 6890 GC system with an Agilent 5973 MS detector equipped with an HP-5MS polar capillary column (0.25 μm film thickness, 30 m × 0.25 mm i.d.).
Synthesis of Functionalized Imidazolium Salts and Copper(I) Complexes: Synthesis of [tBuHImCH2pyCH2NEt2]Br (2)
HNEt2 (468 μL, ρ = 0.707 g mL–1, 4.523 mmol) and K2CO3 (2.814 g, 20.360 mmol) were added to a solution of [tBuHImCH2pyCH2Br]Br (1.600 g, 4.112 mmol) in acetonitrile (10 mL), and the mixture was stirred for 60 h at room temperature. The resulting suspension was brought to dryness under a vacuum, and the residue was extracted with dichloromethane (10 mL) to give a suspension that was filtered and washed with dichloromethane (2 × 5 mL). The solution was brought to dryness under a vacuum to afford a pale-yellow oil that was disaggregated by stirring with cold diethyl ether. The obtained yellow solid was washed with diethyl ether (2 × 5 mL) and dried under a vacuum. Yield: 1.241 g, 79%. Anal. calcd for C18H29BrN4: C, 56.69; H, 7.66; N, 14.69. Found: C, 56.08; H, 7.85; N, 13.74. HRMS (ESI+, MeOH, m/z): calcd for C18H29N4 [M]+: 301.2392, found: 301.2399. 1H NMR (298 K, 300 MHz, CDCl3): δ 11.03 (s, 1H, NCHN), 7.78 (d, JH-H = 7.5 Hz, 1H, Hm py), 7.72–7.65 (m, 2H, Hp py, =CH Im), 7.45 (d, JH-H = 7.7 Hz, 1H, Hm py), 7.23 (s, 1H, =CH Im), 5.84 (s, 2H, CH2Im), 3.67 (s, 2H, CH2NEt2), 2.54 (q, JH-H = 7.1 Hz, 4H, CH2 Et), 1.72 (s, 9H, tBu), 1.03 (t, JH-H = 7.1 Hz, 6H, CH3 Et). 13C{1H} NMR (298 K, 75 MHz, CDCl3): δ 161.1, 151.8 (Cq py), 137.9 (Cp py), 136.0 (NCHN), 123.3 (Cm py), 122.8 (=CH Im), 122.4 (Cm py), 118.9 (=CH Im), 60.5 (C tBu), 59.2 (CH2NEt2), 53.9 (CH2Im), 47.4 (CH2 Et), 30.2 (CH3tBu), 12.0 (CH3 Et).
Synthesis of [tBuHImCH2pyCH2NHtBu]Br (3)
A thick glass reaction tube fitted with a greaseless high-vacuum stopcock was charged with [tBuHImCH2pyCH2Br]Br (1.000 g, 2.570 mmol), tBuNH2 (6 mL), and acetonitrile (3 mL). The reaction mixture was stirred for 15 h at 373 K to give a light brown solution. The solution was transferred to a Schlenk tube and brought to dryness under a vacuum to give a solid residue that was dried under a vacuum at 373 K for 4 h. The pale-brown solid was washed with diethyl ether (3 × 10 mL) and dried under a vacuum. Yield: 931 mg, 95%. Anal. calcd for C18H29BrN4: C, 56.69; H, 7.66; N, 14.69. Found: C, 56.61; H, 7.43; N, 14.44. HRMS (ESI+, MeOH, m/z): calcd for C18H29N4 [M]+: 301.2392, found: 301.2380. 1H NMR (298 K, 300 MHz, CDCl3): δ 10.50 (s, 1H, NCHN), 7.78 (t, JH-H = 1.7 Hz, 1H, =CH Im), 7.72–7.60 (m, 2H, Hp py, Hm py), 7.35–7.27 (m, 2H, Hp py, =CH Im), 5.72 (s, 2H, CH2Im), 4.03 (s, 2H, CH2NHtBu), 1.71 (s, 9H, tBuIm), 1.69 (s, 1H, NH), 1.38 (s, 9H, tBuNH). 13C{1H} NMR (298 K, 75 MHz, CDCl3): δ 156.4, 151.9 (Cq py), 138.3 (Cp py), 137.9 (NCHN), 123.3 (=CH Im), 122.8, 122.0 (Cm py), 118.8 (=CH Im), 60.5 (C tBuIm), 54.4 (C tBuNH), 53.7 (CH2Im), 46.7 (CH2NHtBu), 30.3 (CH3tBuIm), 27.8 (CH3tBuNH).
Synthesis of [Cu2(μ-Br)2(tBuImCH2pyCH2L)]n (L = OMe, NEt2, NHtBu)
General Method
Ag2O and copper powder were added to a solution of [tBuHImCH2pyCH2L]Br (L = OMe, NEt2, NHtBu) in acetonitrile (5 mL). The suspension was stirred for 30 h at 323 K and then filtered through Celite. The resulting solution was brought to dryness under a vacuum to give an oily residue that was disaggregated by stirring with cold diethyl ether. The solid was filtered, washed with diethyl ether (2 × 5 mL), and dried under a vacuum.
[Cu2(μ-Br)2(tBuImCH2pyCH2OMe)]n (4)
[tBuHImCH2pyCH2OMe]Br (1) (200 mg, 0.588 mmol), Ag2O (286 mg, 1.235 mmol), and Cu powder (224 mg, 3.528 mmol). Yield: 170 mg, 53% (light brown solid). Anal. calcd for C15H21Br2N3OCu2: 32.98 C, 3.87 H, 7.69 N. Found: 32.90 C, 3.66 H, 7.79 N. HRMS (ESI+, CH3CN, m/z): calcd for C15H21N3OCu [M-Cu-2Br]+: 322.0981, found: 322.0946; calcd for C15H22N3O [M-2Cu-2Br + H]+: 260.1763, found: 260.1704. 1H NMR (298 K, 300 MHz, CD3CN): δ 7.75 (t, JH-H = 7.7 Hz, 1H, Hp py), 7.36 (d, JH-H = 7.8 Hz, 1H, Hm py), 7.28 (d, JH-H = 1.9 Hz, 1H, =CH Im), 7.18 (d, JH-H = 1.9 Hz, 1H, =CH Im), 7.14 (d, JH-H = 7.8 Hz, 1H, Hm py), 5.40 (s, 2H, CH2Im), 4.51 (s, 2H, CH2OMe), 3.39 (s, 3H, OCH3), 1.70 (s, 9H, tBu). 13C{1H} NMR (298 K, 75 MHz, CD3CN): δ 177.1 (CNCN), 159.8, 156.4 (Cq py), 138.9 (Cp py), 121.8, 121.7 6 (Cm py), 121.4, 119.8 (=CH, Im), 75.8 (CH2OMe), 58.9 (OCH3), 58.6 (C tBu), 57.8 (CH2Im), 32.0 (CH3tBu).
[Cu2(μ-Br)2(tBuImCH2pyCH2NEt2)]n (5)
[tBuHImCH2pyCH2NEt2]Br (2) (200 mg, 0.524 mmol), Ag2O (255 mg, 1.100 mmol), and Cu powder (200 mg, 3.144 mmol). Yield: 166 mg, 54% (pale green solid). Anal. calcd for C18H28Br2N4Cu2: 36.81 C, 4.81 H, 9.54 N. Found: 36.42 C, 5.15 H, 9.84 N. HRMS (ESI+, CH3CN, m/z): calcd for C36H58Br2Cu4N8 [2M + 2H-2Br]+: 1012.0335, found: 1012.0384; calcd for C18H31BrCuN4 [M + 2H-Br-Cu]+: 445.1028, found: 445.0860; calcd for C18H28CuN4 [M-2Br-Cu]+: 363.1610, found: 363.1619; calcd for C18H28N4 [M-2Br-2Cu + H]+: 301.2392, found: 301.2392. 1H NMR (298 K, 300 MHz, CD3CN): δ 7.75 (t, JH-H = 7.7 Hz, 1H, Hp py), 7.40 (d, JH-H = 7.7 Hz, 1H, Hm py), 7.26 (d, JH-H = 1.8 Hz, 1H, =CH Im), 7.22–7.16 (m, 2H, Hm py, =CH Im), 5.42 (s, 2H, CH2Im), 3.75 (s, 2H, CH2NEt2), 2.59 (q, JH-H = 7.1 Hz, 4H, CH2 Et), 1.70 (s, 9H, tBu), 1.01 (t, JH-H = 7.1 Hz, 6H, CH3 Et). 13C{1H} NMR (298 K, 75 MHz, CD3CN): δ 178.1 (CNCN), 160.5, 156.3 (Cq py), 139.3 (Cp py), 123.4 (Cm py), 122.3 (=CH Im), 121.5 (Cm py), 119.6 (=CH Im), 60.2 (CH2NEt2), 58.5 (C tBu), 57.7 (CH2Im), 48.3 (CH2 Et), 31.8 (CH3tBu), 11.7 (CH3 Et).
[Cu2(μ-Br)2(tBuImCH2pyCH2NHtBu)]n (6)
[tBuHImCH2pyCH2NHtBu]Br (3) (200 mg, 0.524 mmol), Ag2O (255 mg, 1.100 mmol), and Cu powder (200 mg, 3.144 mmol). Yield: 160 mg, 52% (pale green solid). Anal. calcd for C18H28Br2N4Cu2: 36.81 C, 4.81 H, 9.54 N. Found: 36.56 C, 4.85 H, 9.48 N. HRMS (ESI+, CH3CN, m/z): calcd for C18H29BrCu2N4 [M-Br + H]+: 506.0168, found: 506.5417; calcd for C18H28N4 [M-2Br-2Cu + H]+: 301.2392, found: 301.2691. 1H NMR (298 K, 300 MHz, CD3CN): δ 7.80 (t, JH-H = 7.8 Hz, 1H, Hp py), 7.38–7.28 (m, 3H, Hm py, 2 =CH Im), 7.17 (d, JH-H = 7.7 Hz, 1H, Hm py), 5.76 (s, 2H, CH2Im), 4.07 (m, 2H, CH2NHtBu), 3.35 (t, JH-H = 7.2 Hz, 1H, NH), 1.69 (s, 9H, tBuIm), 1.27 (s, 9H, tBuNH). 13C{1H} NMR (298 K, 75 MHz, CD3CN): δ 177.9 (CNCN), 160.7, 156.4 (Cq py), 140.1 (Cp py), 123.3 (Cm py), 121.7, 119.6 (=CH Im), 58.7 (C tBuIm), 57.7 (CH2Im), 54.3 (C tBuNH), 48.7 (CH2NHtBu), 31.9 (CH3tBuIm), 29.1 (CH3tBuNH).
[(CuBr)2(C≡CPh)(tBuHImCH2pyCH2NEt2)] (10)
A moderate excess of phenylacetylene (0.125 mmol) was added to a solution of 5 (15 mg, 0.025 mmol) in CD3CN (0.5 mL) at 243 K to give immediately a yellow-brown solution. The acetylide compound formed was characterized spectroscopically in the solution. 1H NMR (298 K, 300 MHz, CD3CN): δ 9.19 (s, 1H, NCHN), 8.18 and 7.26 (d, 1H, JH-H = 7.7, Hm py), 7.84 (t, JH-H = 7.7, 1H, Hp py), 7.54 and 7.47 (br, 1H, =CH Im), 7.5–7.0 (m, 5H, Ph), 5.53 (s, 2H, CH2Im), 3.69 (s, 2H, CH2NEt2), 2.55 (q, JH-H = 7.0, 4H, CH2 Et), 1.68 (s, 9H, tBu), 1.02 (t, JH-H = 7.0, 6H, CH3 Et). 13C{1H}-APT NMR (243 K, 100.0 MHz, CD3CN): δ 161.70 and 153.4 (Cq py), 139.2 (NCHN), 132.5 (Cp py), 131.6, 130.4 and 129.1 (CH Ph), 129.6 and 124.3 (both Cm py), 128.1 (Cq Ph), 126.7 (Cu–C≡C), 124.1 and 121.0 (=CH Im), 98.8 (Cu–C≡C), 61.2 (C tBu), 59.9 (CH2NEt2), 54.4 (s, CH2Im), 47.7 (CH2 Et), 29.9 (CH3tBu). 12.0 (CH3 Et).
General Procedure for the Copper-Catalyzed Azide–Alkyne Cycloaddition Reactions
The catalytic reactions were carried out under an argon atmosphere under solvent-free conditions. First, the catalyst (0.005 mmol) was weighted in a Schlenk tube in a glovebox. Alternatively, in the experiments with a low catalyst load, a stock 8.5 × 10–4 M solution of the catalyst in acetonitrile was prepared in the glovebox from which the required volume was taken using a precision microsyringe, transferred to a Schlenk tube, and then brought to dryness under a vacuum. At this point, azide (0.5 mmol), alkyne (0.5 mmol), and mesitylene (0.25 mmol) as internal standard were sequentially added, and the Schlenk tube was introduced in a thermostatic bath at the desired temperature. Caution: CuAAC is a highly exothermic reaction, and scale-up of the reaction without a solvent can have a significant effect on the likelihood of runaway.41
Conversions and selectivities were determined by gas chromatography analysis of aliquots of the reaction mixture or by dissolving the solid formed in dichloromethane under the following conditions: column temperature of 80 °C (4 min) to 250 °C at a heating rate of 20 °C min–1 by using ultrapure He as carrier gas, and temperatures of 250 °C for the injector and the FID detector.
The 1,4-substituted 1,2,3-triazol reaction products were isolated in yields close to or greater than 90%. Typically, the reaction mixture was brought to dryness under a vacuum to give a white residue that was washed with pentane (3 × 10 mL) and dried under a vacuum.10b
Crystal Structure Determination
Single crystals of 5 suitable for the X-ray diffraction studies were grown by slow diffusion of n-hexane into a dichloromethane solution of the compound. X-ray diffraction data were collected at 100(2) K on a Bruker APEX SMART CCD diffractometer with graphite-monochromated Mo–Kα radiation (λ = 0.71073 Å) using <1° ω rotations. Intensities were integrated and corrected for absorption effects with SAINT-PLUS42 and SADABS43 programs, both included in the APEX2 package. The structures were solved by the Patterson method with SHELXS-9744 and refined by full matrix least squares on F2 with SHELXL-201445 under WinGX.46
Crystal Data and Structure Refinement for 5
C18H28Br2Cu2N4, 587.34 g·mol–1, monoclinic, C2/c, a = 28.8644(18) Å, b = 10.0279(6) Å, c = 15.8988(10) Å, β = 108.8500(10)°, V = 4355.1(5) Å3, Z = 8, Dcalc = 1.792 g·cm3, μ = 5.633 mm–1, F(000) = 2336, 0.280 × 0.100 × 0.060 mm3, θmin/θmax 2.163/26.372°, −36 ≤ h ≤ 36, −12 ≤ k ≤ 12, −19 ≤ l ≤ 19, reflections collected/independent 33,953/4445 [R(int) = 0.0349], Tmax/Tmin 0.4920/0.3044, data/restraints/parameters 4445/0/240, GooF(F2) = 1.032, R1 = 0.0321 [I > 2·σ(I)], wR2 = 0.0892 (all data), largest diff. peak/hole 1.350/–1.129 e·Å–3, CCDC deposition number 2091444.
Computational Details
DFT calculations were performed by means of the Gaussian 09 software package, revision D01.47 We selected the B3LYP exchange-correlation functional48 in conjunction with the D3BJ empirical dispersion correction scheme,49 which has been recently applied to similar processes.23e For geometry optimizations and transition state search, we applied the Ahlrichs def2-SVP basis set, while energy results were further refined via single point calculations with the triple-zeta def2-TZVP basis set.50 Solvent effects were modeled through the polarizable continuum model (PCM) approach, as implemented in the Gaussian 09 suite, and were considered in both gradients and energy calculations.51 We selected acetonitrile as the solvent as many experiments were performed in such solvent and it is much easier to model than neat conditions, which would involve a mixture of the azide and the alkyne. Notice that we applied an ″ultrafine″ grid in all the calculations. The nature of the stationary points has been confirmed by analytical frequency analysis, which was also applied for the calculation of Gibbs energy corrections (at 298.15 K and considering a reference concentration of 1 M). The CylView software was used for structure graphical representations.52
Acknowledgments
The authors express their appreciation for the financial support from the Spanish Ministerio de Ciencia e Innovación (MICINN/FEDER) under Projects PID2019-103965GB-I00/AEI/10.13039/501100011033 and PGC2018-095953-B-I00 (MCIU/AEI/FEDER, UE), as well as the ″Departamento de Ciencia, Universidad y Sociedad del Conocimiento del Gobierno de Aragón″ (group E42_20R) and ″Fundación para el Fomento en Asturias de la Investigación Científica Aplicada y Tecnológica″ (grant IDI-2021-000054). M.G.-L. thanks the Spanish Ministerio de Economía y Competitividad (MINECO) for a predoctoral fellowship (BES-2014-069624). M.G. thankfully acknowledges the Spanish Ministerio de Ciencia, Innovación y Universidades (MICIU) for a predoctoral FPU fellowship (FPU19/02903).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.organomet.2c00246.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- a Meldal M.; Tornøe C. W. Cu-Catalyzed Azide–Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]; b Hein J. E.; Fokin V. V. Copper-catalyzed azide–alkyne cycloaddition (CuAAC) and beyond: new reactivity of copper(I) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. 10.1039/b904091a. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Liang L.; Astruc D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. 10.1016/j.ccr.2011.06.028. [DOI] [Google Scholar]; d Haldón E.; Nicasio M. C.; Pérez P. J. Copper-catalysed azide–alkyne cycloadditions (CuAAC): an update. Org. Biomol. Chem. 2015, 13, 9528–9550. 10.1039/C5OB01457C. [DOI] [PubMed] [Google Scholar]; e Nebra N.; García-Álvarez J. Recent Progress of Cu-Catalyzed Azide-Alkyne Cycloaddition Reactions (CuAAC) in Sustainable Solvents: Glycerol, Deep Eutectic Solvents and Aqueous Media. Molecules 2020, 25, 2015. 10.3390/molecules25092015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. . [DOI] [PubMed] [Google Scholar]
- a Huisgen R. Kinetics and reaction mechanisms: selected examples from the experience of forty years. Pure Appl. Chem. 1989, 61, 613–628. 10.1351/pac198961040613. [DOI] [Google Scholar]; b Breugst M.; Reissig H.-U. The Huisgen Reaction: Milestones of the 1,3-Dipolar Cycloaddition. Angew. Chem., Int. Ed. 2020, 59, 12293–12307. 10.1002/anie.202003115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Click Reactions in Organic Synthesis; Chandrasekaran S. Ed. 2016, Wiley-VCH: Weinheim, Germany. [Google Scholar]; b Neumann S.; Biewend M.; Rana S.; Binder W. H. The CuAAC: Principles, Homogeneous and Heterogeneous Catalysts, and Novel Developments and Applications. Macromol. Rapid Commun. 2020, 41, 1900359. 10.1002/marc.201900359. [DOI] [PubMed] [Google Scholar]; c Meldal M.; Diness F. Recent Fascinating Aspects of the CuAAC Click Reaction. Trends Chem. 2020, 2, 569–584. 10.1016/j.trechm.2020.03.007. [DOI] [Google Scholar]
- a Diez-Gonzalez S. The Use of Ligands in Copper-Catalyzed [3+2] Azide-Alkyne Cycloaddition: Clicker than Click Chemistry?. Curr. Org. Chem. 2011, 15, 2830–2845. 10.2174/138527211796378488. [DOI] [Google Scholar]; b Díez-González S. Well-defined copper(I) complexes for Click azide–alkyne cycloaddition reactions: one Click beyond. Catal. Sci. Technol. 2011, 1, 166–178. 10.1039/c0cy00064g. [DOI] [Google Scholar]
- a Berg R.; Straub B. F. Advancements in the mechanistic understanding of the copper-catalyzed azide-alkyne cycloaddition. Beilstein J. Org. Chem. 2013, 9, 2715–2750. 10.3762/bjoc.9.308. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang C.; Ikhlef D.; Kahlal S.; Saillard J.-Y.; Astruc D. Metal-catalyzed azide-alkyne ″click″ reactions: Mechanistic overview and recent trends. Coord. Chem. Rev. 2016, 316, 1–20. 10.1016/j.ccr.2016.02.010. [DOI] [Google Scholar]; c Neto J. S. S.; Zeni G. A decade of advances in the reaction of nitrogen sources and alkynes for the synthesis of triazoles. Coord. Chem. Rev. 2020, 409, 213217 10.1016/j.ccr.2020.213217. [DOI] [Google Scholar]
- a Herrmann W. N-Heterocyclic Carbenes: A New Concept in Organometallic Catalysis. Angew. Chem., Int. Ed. 2002, 41, 1290–1309. . [DOI] [PubMed] [Google Scholar]; b Díez-González S.; Marion N.; Nolan S. P. N-Heterocyclic Carbenes in Late Transition Metal Catalysis. Chem. Rev. 2009, 109, 3612–3676. 10.1021/cr900074m. [DOI] [PubMed] [Google Scholar]; c Peris E. Smart N-Heterocyclic Carbene Ligands in Catalysis. Chem. Rev. 2018, 118, 9988–10031. 10.1021/acs.chemrev.6b00695. [DOI] [PubMed] [Google Scholar]
- a Egbert J. D.; Cazin C. S. J.; Nolan S. P. Copper N-heterocyclic carbene complexes in catalysis. Catal. Sci. Technol. 2013, 3, 912–926. 10.1039/c2cy20816d. [DOI] [Google Scholar]; b Lazreg F.; Nahra F.; Cazin C. S. J. Copper–NHC complexes in catalysis. Coord. Chem. Rev. 2015, 293–294, 48–79. 10.1016/j.ccr.2014.12.019. [DOI] [Google Scholar]
- a Díez-González S.; Correa A.; Cavallo L.; Nolan S. P. (NHC)Copper(I)-Catalyzed [3+2] Cycloaddition of Azides and Mono- or Disubstituted Alkynes. Chem. – Eur. J. 2006, 12, 7558–7564. [DOI] [PubMed] [Google Scholar]; b Díez-González S.; Stevens E. D.; Nolan S. P. A [(NHC)CuCl] complex as a latent Click catalyst. Chem. Commun. 2008, 4747–4749. 10.1039/b806806b. [DOI] [PubMed] [Google Scholar]; c Teyssot M. L.; Chevry A.; Traikia M.; El-Ghozzi M.; Avignaut D.; Gautier A. Improved Copper(I)–NHC Catalytic Efficiency on Huisgen Reaction by Addition of Aromatic Nitrogen Donors. Chem. – Eur. J. 2009, 15, 6322–6326. 10.1002/chem.200900727. [DOI] [PubMed] [Google Scholar]; d Wang W.; Wu J.; Xia C.; Li F. Reusable ammonium salt-tagged NHC–Cu(I) complexes: preparation and catalytic application in the three component click reaction. Green Chem. 2011, 13, 3440–3445. 10.1039/c1gc15871f. [DOI] [Google Scholar]
- a Díez-González S.; Nolan S. P. [(NHC)2Cu]X Complexes as Efficient Catalysts for Azide–Alkyne Click Chemistry at Low Catalyst Loadings. Angew. Chem., Int. Ed. 2008, 47, 8881–8884. 10.1002/anie.200803289. [DOI] [PubMed] [Google Scholar]; b Lazreg F.; Slawin A. M. Z.; Cazin C. S. J. Heteroleptic Bis(N-heterocyclic carbene)Copper(I) Complexes: Highly Efficient Systems for the [3+2] Cycloaddition of Azides and Alkynes. Organometallics 2012, 31, 7969–7975. 10.1021/om3006195. [DOI] [Google Scholar]; c Guo S.; Lim M. H.; Huynh H. V. Copper(I) Heteroleptic Bis(NHC) and Mixed NHC/Phosphine Complexes: Syntheses and Catalytic Activities in the One-Pot Sequential CuAAC Reaction of Aromatic Amines. Organometallics 2013, 32, 7225–7233. 10.1021/om400911u. [DOI] [Google Scholar]
- a Nakamura T.; Terashima T.; Ogata K.; Fukuzawa S.-I. Copper(I) 1,2,3-Triazol-5-ylidene Complexes as Efficient Catalysts for Click Reactions of Azides with Alkynes. Org. Lett. 2011, 13, 620–623. 10.1021/ol102858u. [DOI] [PubMed] [Google Scholar]; b Hohloch S.; Scheiffele D.; Sarkar B. Activating azides and alkynes for the Click reaction with [Cu(aNHC)2I] or [Cu(aNHC)2]+ (aNHC = Triazole Derived Abnormal Carbenes): Structural Characterization and Catalytic Properties. Eur. J. Inorg. Chem. 2013, 3956–3965. 10.1002/ejic.201300150. [DOI] [Google Scholar]; c Sau S. C.; Roy S. R.; Sen T. K.; Mullangi D.; Mandala S. K. An Abnormal N-Heterocyclic Carbene–Copper(I) Complex in Click Chemistry. Adv. Synth. Catal. 2013, 355, 2982–2991. 10.1002/adsc.201300343. [DOI] [Google Scholar]; d Bidal Y. D.; Lesieur M.; Melaimi M.; Nahra F.; Cordes D. B.; Athukorala Arachchige K. S.; Slawin A. M.; Bertrand G.; Cazin C. S. Copper(I) Complexes Bearing Carbenes Beyond Classical N-Heterocyclic Carbenes: Synthesis and Catalytic Activity in “Click Chemistry”. Adv. Synth. Catal. 2015, 357, 3155–3161. 10.1002/adsc.201500453. [DOI] [Google Scholar]
- a Hohloch S.; Suntrup L.; Sarkar B. Exploring Potential Cooperative Effects in Dicopper(I)-Di-Mesoionic Carbene Complexes: Applications in Click Catalysis. Inorg. Chem. Front. 2016, 3, 67–77. 10.1039/C5QI00163C. [DOI] [Google Scholar]; b Beerhues J.; Fauché K.; Cisnetti F.; Sarkar B.; Gautier A. A dicopper(I)-dimesoionic carbene complex as a click catalyst: mechanistic implications. Dalton Trans. 2019, 48, 8931–8936. 10.1039/C9DT01882D. [DOI] [PubMed] [Google Scholar]
- a Szadkowska A.; Zaorska E.; Staszko S.; Pawłowski R.; Trzybiński D.; Woźniak K. Synthesis, Structural Characterization and Catalytic Activities of Sulfur-Functionalized NHC–Copper(I) Complexes. Eur. J. Org. Chem. 2017, 4074–4084. 10.1002/ejoc.201700523. [DOI] [Google Scholar]; b Szadkowska A.; Pawłowski R.; Zaorska E.; Staszko S.; Trzybiński D.; Woźniak K. NHC copper complexes functionalized with sulfoxide and sulfone moieties. Appl. Organomet. Chem. 2019, 33, e4983 10.1002/aoc.4983. [DOI] [Google Scholar]
- a Gaulier C.; Hospital A.; Legeret B.; Delmas A. F.; Aucagne V.; Cisnetti F.; Gautier A. A water soluble CuI–NHC for CuAAC ligation of unprotected peptides under open air conditions. Chem. Commun. 2012, 48, 4005–4007. 10.1039/c2cc30515a. [DOI] [PubMed] [Google Scholar]; b Díaz Velázquez H.; Ruiz García Y.; Vandichel M.; Madder A.; Verpoort F. Water-soluble NHC-Cu catalysts: applications in click chemistry, bioconjugation and mechanistic análisis. Org. Biomol. Chem. 2014, 12, 9350–9356. 10.1039/C4OB01350F. [DOI] [PubMed] [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. . [DOI] [PubMed] [Google Scholar]
- a Himo F.; Lovell T.; Hilgraf R.; Rostovtsev V. V.; Noodleman L.; Sharpless K. B.; Fokin V. V. Copper(I)-Catalyzed Synthesis of Azoles. DFT Study Predicts Unprecedented Reactivity and Intermediates. J. Am. Chem. Soc. 2005, 127, 210–216. 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]; b Rodionov V. O.; Fokin V. V.; Finn M. G. Mechanism of the ligand-free CuI-catalyzed azide-alkyne cycloaddition reaction. Angew. Chem., Int. Ed. 2005, 44, 2210–2215. 10.1002/anie.200461496. [DOI] [PubMed] [Google Scholar]; c Straub B. F. μ-Acetylide and μ-alkenylidene ligands in “click” triazole syntheses. Chem. Commun. 2007, 3868–3870. 10.1039/b706926j. [DOI] [PubMed] [Google Scholar]; d Nolte C.; Mayer P.; Straub B. F. Isolation of a copper(I) triazolide: a ″click″ intermediate. Angew. Chem., Int. Ed. 2007, 46, 2101–2103. 10.1002/anie.200604444. [DOI] [PubMed] [Google Scholar]
- Ahlquist M.; Fokin V. V. Enhanced Reactivity of Dinuclear Copper(I) Acetylides in Dipolar Cycloadditions. Organometallics 2007, 26, 4389–4391. 10.1021/om700669v. [DOI] [Google Scholar]
- Buckley B. R.; Dann S. E.; Heaney H. Experimental Evidence for the Involvement of Dinuclear Alkynylcopper(I) Complexes in Alkyne–Azide Chemistry. Chem. – Eur. J. 2010, 16, 6278–6284. 10.1002/chem.201000447. [DOI] [PubMed] [Google Scholar]
- Worrell B. T.; Malik J. A.; Fokin V. V. Direct Evidence of a Dinuclear Copper Intermediate in Cu(I)-Catalyzed Azide-Alkyne Cycloadditions. Science 2013, 340, 457–460. 10.1126/science.1229506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L.; Tolentino D. R.; Melaimi M.; Bertrand G. Isolation of bis(copper) key intermediates in Cu-catalyzed azide-alkyne “click reaction.”. Sci. Adv. 2015, 1, e1500304 10.1126/sciadv.1500304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chung R.; Vo A.; Fokin V. V.; Hein J. E. Catalyst Activation, Chemoselectivity, and Reaction Rate Controlled by the Counterion in the Cu(I)-Catalyzed Cycloaddition between Azide and Terminal or 1-Iodoalkynes. ACS Catal. 2018, 8, 7889–7897. 10.1021/acscatal.8b01342. [DOI] [Google Scholar]; b Haugland M. M.; Borsley S.; Cairns-Gibson D. F.; Elmi A.; Cockroft S. L. Synthetically Diversified Protein Nanopores: Resolving Click Reaction Mechanisms. ACS Nano 2019, 13, 4101–4110. 10.1021/acsnano.8b08691. [DOI] [PubMed] [Google Scholar]
- a Makarem A.; Berg R.; Rominger F.; Straub B. F. A Fluxional Copper Acetylide Cluster in CuAAC Catalysis. Angew. Chem., Int. Ed. 2015, 54, 7431–7435. 10.1002/anie.201502368. [DOI] [PubMed] [Google Scholar]; b Venderbosch B.; Oudsen J. P. H.; van der Vlugt J. I.; Korstanje T. J.; Tromp M. Cationic Copper Iminophosphorane Complexes as CuAAC Catalysts: A Mechanistic Study. Organometallics 2020, 39, 3480–3489. 10.1021/acs.organomet.0c00348. [DOI] [Google Scholar]
- a Lal S.; Rzepa H. S.; Díez-González S. Catalytic and Computational Studies of N-Heterocyclic Carbene or Phosphine-Containing Copper(I) Complexes for the Synthesis of 5-Iodo-1,2,3-Triazoles. ACS Catal. 2014, 4, 2274–2287. 10.1021/cs500326e. [DOI] [Google Scholar]; b Özkliç Y.; Tüzün N. Ş. A DFT Study on the Binuclear CuAAC Reaction: Mechanism in Light of New Experiments. Organometallics 2016, 35, 2589–2599. 10.1021/acs.organomet.6b00279. [DOI] [Google Scholar]; c Krupicka M.; Dopieralski P.; Marx D. Unclicking the Click: Metal-Assisted Mechanochemical Cycloreversion of Triazoles Is Possible. Angew. Chem., Int. Ed. 2017, 56, 7745–7749. 10.1002/anie.201612507. [DOI] [PubMed] [Google Scholar]; d Silva P. J.; Bernardo C. E. P. Influence of Alkyne and Azide Substituents on the Choice of the Reaction Mechanism of the Cu+-Catalyzed Addition of Azides to Iodoalkynes. J. Phys. Chem. A 2018, 122, 7497–7507. 10.1021/acs.jpca.8b06894. [DOI] [PubMed] [Google Scholar]; e Gan H.; Peng L.; Gu F. L. Mechanistic understanding of the Cu(I)-catalyzed domino reaction constructing 1-aryl-1,2,3-triazole from electron-rich aryl bromide, alkyne, and sodium azide: a DFT study. Catal. Sci. Technol. 2021, 11, 3208–3216. 10.1039/D1CY00123J. [DOI] [Google Scholar]; f Chakraborti G.; Jana R.; Mandal T.; Datta A.; Dash J. Prolinamide plays a key role in promoting copper catalyzed cycloaddition of azides and alkynes in aqueous media via unprecedented metallacycle intermediates. Org. Chem. Front. 2021, 8, 2434–2441. 10.1039/D0QO01150A. [DOI] [Google Scholar]
- a Iacobucci C.; Reale S.; Gal J.-F.; De Angelis F. Dinuclear Copper Intermediates in Copper(I)-Catalyzed Azide–Alkyne Cycloaddition Directly Observed by Electrospray Ionization Mass Spectrometry. Angew. Chem., Int. Ed. 2015, 54, 3065–3068. 10.1002/anie.201410301. [DOI] [PubMed] [Google Scholar]; b Kalvet I.; Tammiku-Taul J.; Mäeorg U.; Tämm K.; Burk P.; Sikk L. NMR and DFT Study of the Copper(I)-Catalyzed Cycloaddition Reaction: H/D Scrambling of Alkynes and Variable Reaction Order of the Catalyst. ChemCatChem 2016, 8, 1804–1808. 10.1002/cctc.201600176. [DOI] [Google Scholar]; c Chen H.; Cai C.; Li S.; Ma Y.; Luozhing S.; Zhu Z. Intermediates Stabilized by Tris(triazolylmethyl)amines in the CuAAC Reaction. Chem. – Eur. J. 2017, 23, 4730–4735. 10.1002/chem.201700555. [DOI] [PubMed] [Google Scholar]
- a Saha S.; Kaur M.; Bera J. K. Fluorinated Anions Promoted “on Water” Activity of Di- and Tetranuclear Copper(I) Catalysts for Functional Triazole Synthesis. Organometallics 2015, 34, 3047–3054. 10.1021/acs.organomet.5b00348. [DOI] [Google Scholar]; b Ziegler M. S.; Lakshmi K. V.; Tilley T. D. Dicopper Cu(I)Cu(I) and Cu(I)Cu(II) Complexes in Copper-Catalyzed Azide–Alkyne Cycloaddition. J. Am. Chem. Soc. 2017, 139, 5378–5386. 10.1021/jacs.6b13261. [DOI] [PubMed] [Google Scholar]; c Sareen N.; Singh A. S.; Tiwarim V. K.; Kant R.; Bhattacharya S. A dinuclear copper(I) thiodiacetate complex as an efficient and reusable ‘click’ catalyst for the synthesis of glycoconjugates. Dalton Trans. 2017, 46, 12705–12710. 10.1039/C7DT02346D. [DOI] [PubMed] [Google Scholar]; d Lin Y.-C.; Chen Y.-J.; Shih T.-Y.; Chen Y.-H.; Lai Y.-C.; Chiang M. Y.; Senadi G. C.; Chen H.-Y.; Chen H. Y. Mechanistic Study in Click Reactions by Using (N-Heterocyclic carbene)Copper(I) Complexes: Anionic Effects. Organometallics 2019, 38, 223–230. 10.1021/acs.organomet.8b00565. [DOI] [Google Scholar]
- González-Lainez M.; Jiménez M. V.; Passarelli V.; Pérez-Torrente J. J. Effective N-methylation of nitroarenes with methanol catalyzed by a functionalized NHC based iridium catalyst: a green approach to N-methyl amines. Catal. Sci. Technol. 2020, 10, 3458–3467. 10.1039/D0CY00707B. [DOI] [Google Scholar]
- Liu B.; Xia Q.; Chen W. Direct synthesis of iron, cobalt, nickel, and copper complexes of N-heterocyclic carbenes by using commercially available metal powders. Angew. Chem., Int. Ed. 2009, 48, 5513–5516. 10.1002/anie.200901850. [DOI] [PubMed] [Google Scholar]
- Cremer D.; Pople J. A. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. 10.1021/ja00839a011. [DOI] [Google Scholar]
- Azpíroz R.; Rubio-Pérez L.; Di Giuseppe A.; Passarelli V.; Lahoz F. J.; Castarlenas R.; Pérez-Torrente J. J.; Oro L. A. Rhodium(I)-N-Heterocyclic Carbene Catalyst for Selective Coupling of N-Vinylpyrazoles with Alkynes via C–H Activation. ACS Catal. 2014, 4, 4244–4253. 10.1021/cs501366q. [DOI] [Google Scholar]
- a Shishkov I. V.; Rominger F.; Hofmann P. Reversible substrate binding at copper centers in neutral copper(I) carbene complexes derived from bis(3-tert-butylimidazole-2-ylidene)methane. Dalton Trans. 2009, 1428–1435. 10.1039/b813790k. [DOI] [PubMed] [Google Scholar]; b Schneider N.; César V.; Bellemin-Laponnaz S.; Gade L. H. Synthesis and structural chemistry of oxazolinyl-carbene copper(I) complexes. J. Organomet. Chem. 2005, 690, 5556–5561. [Google Scholar]; c Casado M. A.; Pérez-Torrente J. J.; Ciriano M. A.; Lahoz F. J.; Oro L. A. Tetranuclear [Rh4(μ-PyS2)2(diolefin)4] complexes as building blocks for new inorganic architectures: Synthesis of coordination polymers and heteropolynuclear complexes with electrophilic d8 and d10 metal fragments. Inorg. Chem. 2004, 43, 1558–1567. 10.1021/ic034893i. [DOI] [PubMed] [Google Scholar]; d Casado M. A.; Pérez-Torrente J. J.; Edwards A. J.; Oro L. A.; Ciriano M. A.; Lahoz F. J. Rhodium tetranuclear complexes as building blocks for the construction of coordination polymers: chiroselectivity in the formation of [ClCuRh4(μ-PyS2)2(cod)4]n (H2PyS2 = 2,6-dimercaptopyridine). CrystEngComm 2000, 2, 125–127. 10.1039/B004750N. [DOI] [Google Scholar]
- a Lake B. R. M.; Willans C. E. Structural Diversity of Copper(I)–N-Heterocyclic Carbene Complexes; Ligand Tuning Facilitates Isolation of the First Structurally Characterised Copper(I)–NHC Containing a Copper(I)–Alkene Interaction. Chem. – Eur. J. 2013, 19, 16780–16790. 10.1002/chem.201301896. [DOI] [PubMed] [Google Scholar]; b Lake B. R. M.; Willans C. E. Remarkable Stability of Copper(II)–N-Heterocyclic Carbene Complexes Void of an Anionic Tether. Organometallics 2014, 33, 2027–2038. 10.1021/om500178e. [DOI] [Google Scholar]
- a Cisnetti F.; Lemoine P.; El-Ghozzi M.; Avignant D.; Gautier A. Copper(I) thiophenolate in copper N-heterocyclic carbene preparation. Tetrahedron Lett. 2010, 51, 5226–5229. 10.1016/j.tetlet.2010.07.124. [DOI] [Google Scholar]; b Riener K.; Pöthig A.; Cokoja M.; Herrmann W. A.; Kühn F. E. Structure and spectroscopic properties of the dimeric copper(I) N-heterocyclic carbene complex [Cu2(CNCt-Bu)2](PF6)2. Acta. Cryst. 2015, C71, 643–646. 10.1107/S2053229615012140. [DOI] [PubMed] [Google Scholar]; c Nitsch J.; Lacemon F.; Lorbach A.; Eichhorn A.; Cisnetti F.; Steffen A. Cuprophilic interactions in highly luminescent dicopper(I)–NHC–picolyl complexes – fast phosphorescence or TADF?. Chem. Commun. 2016, 52, 2932–2935. 10.1039/C5CC09659F. [DOI] [PubMed] [Google Scholar]
- Tulloch A. A.; Danopoulos A. A.; Kleinhenz S.; Light M. E.; Hurtshouse M. B.; Eastman G. Structural diversity in pyridine-N-functionalized carbene copper(I) complexes. Organometallics 2001, 20, 2027–2031. 10.1021/om010014t. [DOI] [Google Scholar]
- Kozuch S.; Shaik S. How to Conceptualize Catalytic Cycles? The Energetic Span Model. Acc. Chem. Res. 2011, 44, 101–110. 10.1021/ar1000956. [DOI] [PubMed] [Google Scholar]
- Larionov V. A.; Stashneva A. R.; Titov A. A.; Lisov A. A.; Medvedev M. G.; Smol’yakov A. F.; Tsedilin A. M.; Shubina E. S.; Maleev V. I. Mechanistic study in azide-alkyne cycloaddition (CuAAC) catalyzed by bifunctional trinuclear copper(I) pyrazolate complex: Shift in rate-determining step. J. Catal. 2020, 390, 37–45. 10.1016/j.jcat.2020.07.010. [DOI] [Google Scholar]
- a Kuang G.-C.; Guha P. M.; Brotherton W. S.; Simmons J. T.; Stankee L. A.; Nguyen B. T.; Clark R. J.; Zhu L. Experimental Investigation on the Mechanism of Chelation-Assisted, Copper(II) Acetate-Accelerated Azide–Alkyne Cycloaddition. J. Am. Chem. Soc. 2011, 133, 13984–14001. 10.1021/ja203733q. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang X.; Liu P.; Zhu L. Structural Determinants of Alkyne Reactivity in Copper-Catalyzed Azide-Alkyne Cycloadditions. Molecules 2016, 21, 1697. 10.3390/molecules21121697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L.; Brassard C. J.; Zhang X.; Guha P. M.; Clark R. J. On the Mechanism of Copper(I)-Catalyzed Azide–Alkyne Cycloaddition. Chem. Rec. 2016, 16, 1501–1517. 10.1002/tcr.201600002. [DOI] [PubMed] [Google Scholar]
- Seath C. P.; Burley G. A.; Watson A. J. B. Determining the Origin of Rate-Independent Chemoselectivity in CuAAC Reactions: An Alkyne-Specific Shift in Rate-Determining Step. Angew. Chem., Int. Ed. 2017, 56, 3314–3318. 10.1002/anie.201612288. [DOI] [PubMed] [Google Scholar]
- Mangione M. I.; Spanevello R. A.; Anzardi M. B. Efficient and straightforward click synthesis of structurally related dendritic triazoles. RSC Adv. 2017, 7, 47681–47688. 10.1039/C7RA09558A. [DOI] [Google Scholar]
- Alvarez S. G.; Alvarez M. T. A Practical Procedure for the Synthesis of Alkyl Azides at Ambient Temperature in Dimethyl Sulfoxide in High Purity and Yield. Synthesis 1997, 413–414. 10.1055/s-1997-1206. [DOI] [Google Scholar]
- Villemur C.; Petit L.; Bianchini N.; Rotureau P. Runaway reaction hazard assessment for chemical processes safety. Chem. Eng. Trans. 2019, 77, 451–456. [Google Scholar]
- SAINT+: Area-Detector Integration Software ; version 6.01; Bruker AXS: Madison, WI. 2001. [Google Scholar]
- Sheldrick G. M.SADABS program; University of Göttingen: Göttingen, Germany, 1999. [Google Scholar]
- Sheldrick G. M.SHELXS 97, Program for the Solution of Crystal Structure; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick G. M. Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrugia L. J. WinGX and ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. 10.1107/S0021889812029111. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V; Cioslowski J.; Fox D. J.. Gaussian 09, revision D.01; Gaussian, Inc.: Wallingford CT, 2009. [Google Scholar]
- Becke A. D. A new mixing of Hartree-Fock and local density functional theories. J. Chem. Phys. 1993, 98, 1372–1377. 10.1063/1.464304. [DOI] [Google Scholar]
- a Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]; b Johnson E. R.; Becke A. D. A. A post-Hartree-Fock model of intermolecular interactions. J. Chem. Phys. 2005, 123, 024101 10.1063/1.1949201. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Scalmani G.; Frisch M. J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110. 10.1063/1.3359469. [DOI] [PubMed] [Google Scholar]
- CYLview, 1.0b. Legault C. Y., Ed.; Université de Sherbrooke, 2009. http://www.cylview.org/ (accessed 2022-06-18).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.