

Abstract
Exosomes are nanometer scale, membrane-enclosed vesicles that can potentially be used to detect nephrotoxicity, and reveal the subsequent response of the kidney. Epithelial cells of every nephron segment can contribute to the urinary exosome population, which is rich in potential biomarkers, including membrane proteins such as transporters and receptors, transcription factors, and microRNAs. These exosomal biomarkers may be up- or down-regulated upon nephrotoxicant exposure. Exosome isolation is an area of ongoing research. Although faster and simpler methods have been developed, ultracentrifugation remains a mainstay for purification. A single ultracentrifugation step provides an enriched preparation suitable for biomarker discovery, and a second ultracentrifugation on a sucrose/D2O cushion provides the purest exosome preparation currently available and may be preferred for bioactivity assays. The concentration of exosomes can be determined using Nanosight Nanoparticle Tracking Analysis and their contents studied with a variety of approaches including western blots for proteins and RT-qPCR for microRNAs.
Keywords: exosomes, biomarker, urine, TSG101, ultracentrifugation, Nanosight, nephrotoxicity
1. Introduction
Urinary exosomes are extracellular vesicles that originate from glomerular podocytes, epithelial cells lining the renal tubules, and the rest of the epithelial lining of the genitourinary tract, including the bladder and prostate (1). Exosomes are formed from the endocytic pathway, a process that involves two sequential membrane invaginations. First, plasma membrane-derived endocytosis forms endocytic vesicles with an inside-out orientation that contain extracellular fluid. The fusion of multiple endocytic vesicles results in the formation of an early endosome, which matures into a multivesicular body (MVB). Then, the MVB outer membrane can generate intraluminal vesicles by a subsequent invagination, resulting in a right-side out orientation with cytoplasm trapped inside. The MVB can then follow one of two alternate paths. It either fuses with a lysosome to degrade and recycle its contents, or it is routed to the cell surface. When the MVB fuses with the plasma membrane of a renal/genitourinary tract epithelial cell, the right-side out intraluminal vesicles are released into the urinary lumen as exosomes. Exosomes are distinct from microvesicles/microparticles/ectosomes, which are right-side out extracellular vesicles that are released directly from plasma membrane outpouching, and are typically not found in urine. Proteins that were identified through the proteomic analysis of human urinary exosomes (2) and are known to be involved in the biogenesis of MVB, such as TSG101, can be used to estimate exosomal mass by western blot, which may be useful as a denominator for exosomal biomarker normalization.
Exosome isolation is a straightforward enrichment strategy of potential nephrotoxicant biomarkers by reducing the complexity of urine composition, with minimal loss of yield. By isolating exosomes, high-abundance constituents may be removed in non-exosomal urine fractions, while low-abundance constituents that may have toxicological significance may be simultaneously enriched.
Collection, storage and handling methods can greatly affect the quality and quantity of urinary exosomal protein biomarkers (3). Protease inhibitors are needed to preserve some proteins exposed to exosome exteriors such as NCC or NKCC2. On the other hand, exosomal miRNA or mRNA do not need protection from RNases found in urine. When shipping and/or storage of urine is necessary, it is recommended to freeze fresh urine samples at −80°C instead of −20°C. During the final stages of thawing, samples should be vortexed extensively to maximize the yield of exosomes from frozen urine (3).
We describe an ultracentrifugation protocol for isolation of urinary exosomes, and comment on alternatives. Methods for exosomal RNA isolation, exosome characterization by Nanosight Nanoparticle Tracking Analysis and preparation of exosome samples for western blot are described. Electron microscopy and proteomics were described previously (4) and will not be covered here.
2. Materials
2.1. Purification of urinary exosomes
Urine collection additive cOmplete, Mini Protease Inhibitor Cocktail (Roche): add 1 Roche cOmplete Mini tablet (stored at 4°C) per ≤50 ml of fresh urine. When dissolving the tablet, swirl the sample until no longer visible (see Note 1).
Sterile 1X Phosphate Buffered Saline (PBS).
15 or 50 ml conical centrifuge tubes.
Centrifuge with swinging bucket rotor.
Clean Crystalgen 23-2589 (or equivalent) tube(s).
Ultracentrifuge.
Clean ultracentrifuge tubes.
30% Sucrose/D2O for Density Cushion. Volume for 50 mL solution: mix 15 g of Ultrapure Sucrose with D2O to 50 mL (see Note 2).
2.2. Preparation of exosomes for western blot
SDS-Laemmli-DTT sample buffer
2X Laemmli Sample Buffer (Bio-Rad 1610737) is supplemented with 200 mM dithiothreitol (30.86 mg/ml) and stored at −20°C.
Heat block.
1.5 ml microfuge tubes
2.3. Nanosight Nanoparticle Tracking Analysis (NTA)
Nanosight LM-10 instrument (Malvern Instruments Ltd.) with NTA version 3.1 software.
Syringe pump.
2.4. RNA extraction materials
TRIzol Reagent (Invitrogen).
Chloroform.
100% ethanol.
Phase Lock Gel Heavy 1.5 mL Tubes (5 Prime).
miRNeasy Mini Kit (Qiagen): contains sufficient quantities of the following reagents for 50 samples: RNeasy Mini Spin Columns, 1.5 mL and 2 mL collection tubes, QIAzol lysis reagent, concentrated stock RWT and RPE buffers, RNase-free water.
Ultrapure water.
3. Methods
Exosomes were originally purified by ultracentrifugation, which enabled their initial characterization, and this technique remains the most common approach for their isolation. The high capital costs and time consuming nature of ultracentrifugation are recognized as impediments to the widespread study of exosomes, beginning with preclinical toxicology studies, translation to clinical trials and perhaps to postapproval monitoring. As such, there is significant interest in alternative isolation methods. Although our previously described nanomembrane concentrator method is a viable option (5), we eventually concluded that ultracentrifugation is preferable when available. We have limited experience with other newer techniques now available, and the increasing availability of commercial kits will allow higher throughput screening of large sample sets; biomarkers identified from this process should be validated as exosomal by using the best available methods for purification and validation. For this chapter we focus on ultracentrifugation, as it enables the greatest breadth of post-purification analyses. A single ultracentrifugation step is usually sufficient for biomarker discovery studies, and greater purity for bioactivity assays can be achieved with an additional ultracentrifugation step using a sucrose/D2O cushion (6,7).
Analysis of purified exosomes falls in four main categories: size, number, purity, and content. For size and purity, transmission electron microscopy remains the standard, but it is a very low throughput method to perform and analyze data (4). More recently the Nanosight system (8) has been used to determine size and number. A promising approach to assessing purity uses Nanosight combined with protein assay to determine a value analogous to specific activity (9), but would require that standards be determined for each biofluid source of exosomes. Content can be surveyed by –omics methods such as proteomics, transcriptomics (miRNA or mRNA), lipidomics, or glycomics (glycoproteins), or analyzed by more focused methods such as western blot, RT-qPCR, or GC-MS.
3.1. Purification of urinary exosomes (Fig. 1)
Figure 1.

Flow chart outlining the ultracentrifugation protocol for urinary exosome isolation. A single ultracentrifugation is typical for biomarker discovery studies (left). If a higher level of purity is required, such as for bioactivity assays, an additional ultracentrifugation step using a Sucrose/D2O cushion may be added (right).
3.1.1. Ultracentrifugation
Add protease inhibitors to a fresh urine sample if detecting proteins.
Process whole urine immediately or store at −80°C for future processing.
If thawing frozen samples, vigorous vortexing should be used prior to complete melting of sample to maximize exosome recovery.
Centrifuge urine sample in a 15 or 50 ml conical centrifuge tube at 1,000 x g for 10 minutes at 4°C (see Note 3).
Transfer the 1,000 x g supernatant to a clean Crystalgen 23-2589 (or equivalent) tube(s).
Centrifuge 1,000 x g supernatant at 17,000 x g for 15 minutes at 4°C (see Note 4).
Transfer the 17,000 x g supernatant into a clean ultracentrifuge tube(s) (see Note 5).
Ultracentrifuge 17,000 x g supernatant at 200,000 x g for 1 hour at 4°C.
Discard the 200,000 x g supernatant. If more 17,000 x g supernatant is available, add additional 17,000 x g supernatant to tubes used for step 8 and then repeat step 8 (see Notes 6, 7).
Add 100 μl sterile 1X PBS to each ultracentrifuge tube and vigorously vortex until pellet has been resuspended.
3.1.2. Ultracentrifugation with Sucrose/D2O Cushion
Process urine as per steps 1 through 10 of the ultracentrifugation protocol.
Add 1 mL of 30% Sucrose/D2O to clean ultracentrifuge tube(s) before transferring the resuspended 200,000 x g pellet.
Slowly pipet the resuspended 200,000 x g pellet onto the sucrose/ D2O cushion in order to not disturb the interface.
Ultracentrifuge at 200,000 x g for 1hr at 4°C.
Remove the supernatant. Retrieve the interface and transfer to a clean ultracentrifuge tube.
Add 6 mL sterile 1X PBS to each ultracentrifuge tube containing a retrieved interface sample.
Ultracentrifuge the diluted interface at 200,000 x g for 1 hour at 4°C.
Discard the supernatant from the 200,000 x g wash step (see Note 7).
Add 100 μl sterile 1X PBS to each ultracentrifuge tube and vigorously vortex until pellet has been resuspended.
3.2. Preparation of exosomes for western blot
Add an equal volume of 2X SDS-Laemmli-DTT sample buffer to the resuspended exosomes, vortex thoroughly, spin briefly in microfuge to remove bubbles.
Heat samples on heat block at 95°C for 5 minutes, spin briefly in microfuge to consolidate condensation on the lid of the microfuge tube.
Store at −80°C.
3.3. Nanosight Nanoparticle Tracking Analysis (NTA) (see Note 8)
Dilute exosome preparation with sterile 1X PBS, so that particle concentration is within the instrument’s linear dynamic range (~107–109 particles/ml). Load a sample into the Nanosight chamber using a syringe, taking care not to introduce bubbles. If possible, use a syringe pump (~0.2 mL/hr) to allow for the acquisition of more accurate data.
Turn on the camera, adjust the x-y position to center the laser beam, set the camera level using the software, focus the instrument if necessary and confirm on the monitor that there are approximately 20-100 particles in view at any given time (Fig. 2). Note the temperature of the chamber.
Under the Standard Operating Procedure (SOP) tab create and run a new script for 5 acquisitions of 60 seconds each.
Withdraw or push through the rest of the sample and thoroughly rinse with ultrapure water to ensure there is no contamination between samples. Contamination can be assessed by looking at the screen when the chamber is filled with water to see if there are particles still present.
Repeat step 1 and load the next sample until finished (see Note 9).
For data analysis the detection threshold should be set such that the number of visible particles with red crosses is maximized while the number of blue crosses on the screen is minimized (less than five). If the detection threshold is too low, non-particle background noise will be tracked. If the threshold is too high, some particles will be excluded (Fig. 3).
Raw data can be exported to .csv files for further analysis (see Fig. 4 for example).
Figure 2.
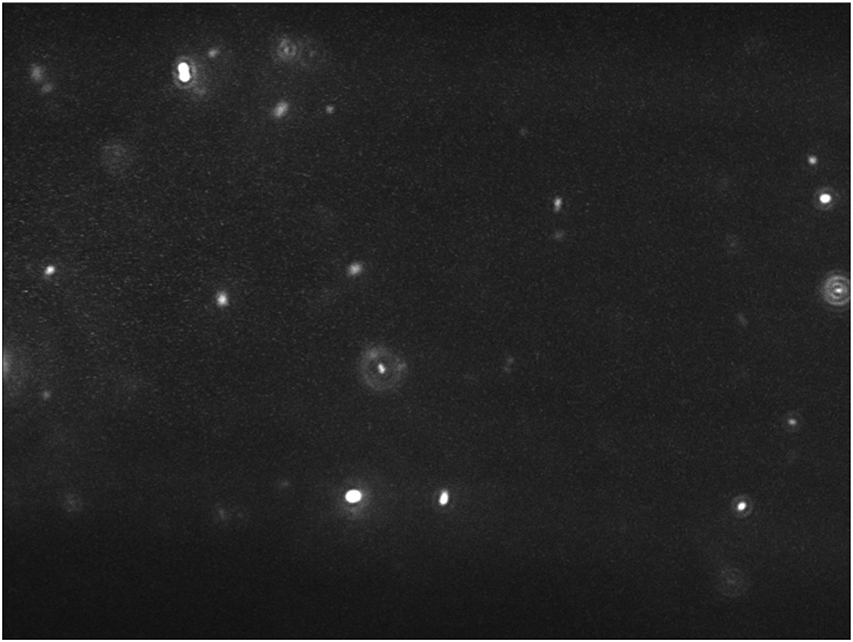
Sample image from NTA 3.1 illustrating a proper concentration of particles present in the field of view (20-100 particles).
Figure 3.
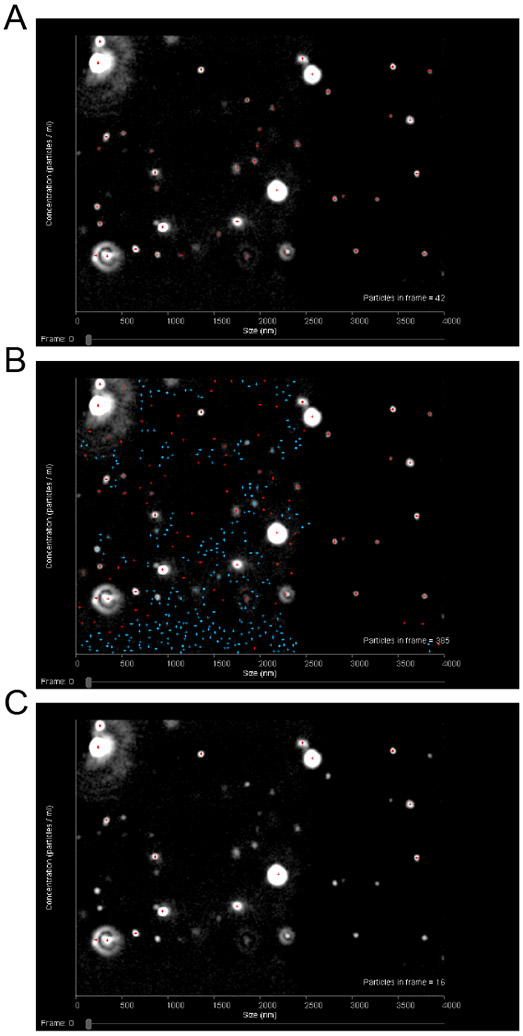
Sample images from NTA 3.1 illustrating how to choose a proper detection threshold setting for analysis. When the detection threshold is set properly (A), there should be red crosses on each particle in the field of view and no crosses on other parts of the field. The number of blue crosses present should also be minimized (less than five). When the detection threshold is set too low (B), there will be crosses on non-particle background as well as an abundance of blue crosses visible. When the detection threshold is set too high (C), there will be particles visible but without red crosses on them.
Figure 4.
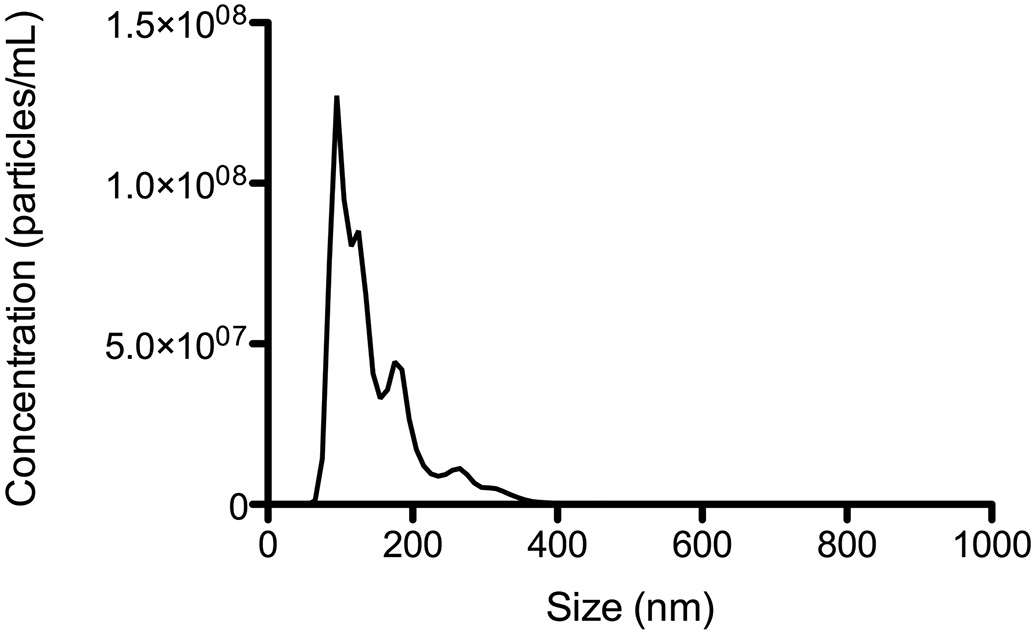
Sample size-concentration distribution of urine exosomes isolated from a healthy patient obtained using Nanosight NTA. Exosomes were resuspended in 1X PBS. The sample was diluted in order for the measured concentration to fall within the dynamic range of the instrument.
3.4. RNA extraction using miRNeasy kit (see Notes 10, 11)
Add a 10- (preferable) to 3- (minimum) fold excess (by volume) of TRIzol to the exosome preparation (~50 μl). Vortex and incubate at room temperature (RT) for 5 minutes.
Add one-fifth volume of chloroform, shake tube vigorously and incubate at RT for 2 minutes.
Centrifuge sample at 12,000 x g for 15 minutes at 4 °C.
Transfer aqueous phase to a phase lock tube and add 700 μl QIAzol. Vortex and incubate at RT for 5 minutes.
Add 140 μl chloroform, shake tube vigorously and incubate at RT for 2 minutes.
Centrifuge sample at 12,000 x g for 15 minutes at 4 °C.
Transfer aqueous phase to a 2ml tube and add 1.5 volumes 100% ethanol.
Transfer 700 μl sample, including any precipitate, into an RNeasy mini spin column in a 2 ml collection tube. Centrifuge at > 8,000 x g for 15 seconds at RT and discard flow-through.
Repeat step 8 until the entire sample has been loaded on the column.
Add 700 μl RWT buffer and centrifuge at > 8,000 x g for 15 seconds at RT. Discard flow-through.
Add 500 μl RPE buffer and centrifuge at > 8,000 x g for 15 seconds at RT. Discard flow-through.
Repeat step 11 but centrifuge for 2 minutes.
Transfer mini column to a fresh 2 ml tube and centrifuge for 1 minute at > 8,000 x g.
Transfer mini column to a 1.5 ml tube. Add 30 μl ultrapure water and centrifuge for 1 minute at > 8,000 x g to elute RNA.
Transfer flow-through back to mini column and centrifuge for 1 minute at > 8,000 x g.
Store flow-through at −80 °C.
4. Notes
Alternatively, home-made protease inhibitors can be used. For a volume per ≤50 ml of urine: mix 1.67 ml 100 mM NaN3, 2.5 ml PMSF (2.5 mg/ml in ethanol, store at −20°C) or 5 ml AEBSF (2.75 mg/ml in dd-H2O, store at −20°C) (as an alternative to PMSF, AEBSF offers lower toxicity and improved stability in aqueous solutions), 50 μl Leupeptin (1 mg/ml in dd-H2O, stable at 4°C for 1 week).
The Sucrose/D2O solution should be filtered twice before use in ultracentrifugation. First, filter the solution through a 0.22 μm filter followed by a 0.1 μm filter. This should remove almost all nanoparticle impurities originating from raw materials from the solution.
This step removes whole cells, large aggregates, and other debris. We use the Sorvall RT7Plus centrifuge with a RTH750 swinging bucket rotor.
This step removes organelles, cell fragments, and some Tamm Horsfall protein fibrils. We use the Sorvall RC-6+ refrigerated centrifuge and a SA-600 rotor and Crystalgen 23-2589 tubes.
We use the Beckman Coulter Optima MAX ultracentrifuge with the MLA-55 rotor. The high speed tube we use is the thick-walled, polycarbonate tube #355630 from Beckman, with a practical capacity of ~7.5 ml.
Minimizing the number of tubes used for ultracentrifugation increases the yield of exosomes because resuspension and pooling is more labor intensive and less reliable when more tubes are used. However, using fewer tubes results in more spins, and this trade-off needs to be considered. For example, a single spin with 5 tubes would be faster, but would result in lower yield. On the other end of the spectrum, 5 consecutive spins with a single tube would maximize recovery, but would be much more time-consuming. Three spins in two tubes or two spins in three tubes would be rational approaches.
To remove the last traces of 200,000 x g supernatant, the nearly empty tube can be recentrifuged for 10 min using the same orientation within the rotor, then using a pipette to remove the remaining supernatant, or ultracentrifuge tubes can be tipped upside-down for ~15-30 sec (to avoid a dry pellet) and aspirated inside the tube opening.
This procedure may vary slightly if other instruments than the Nanosight LM-10 and if other versions of the NTA software than the 3.1 version are used.
Lower temperature and increased viscosity will artificially increase the size of particles during Nanosight Tracking Analysis. Monitoring temperature during data acquisition and ensuring that all sucrose is removed during preparation are essential for accurate sizing data.
This protocol follows the manufacturer’s recommendations with the addition of a second phase extraction step and the use of phase lock tubes to minimize contamination.
To prevent degradation of the sample the extraction should proceed as quickly as possible. Extraction is performed on batches of up to 12 samples. This number can be processed simultaneously using one standard centrifuge.
5. Acknowledgements
This study was supported by the Intramural Research Program of the NIH, The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK).
6. References
- 1.Pisitkun T, Johnstone R, Knepper MA (2006) Discovery of urinary biomarkers. Molecular & cellular proteomics 5 (10):1760–1771. doi: 10.1074/mcp.R600004-MCP200 [DOI] [PubMed] [Google Scholar]
- 2.Pisitkun T, Shen RF, Knepper MA (2004) Identification and proteomic profiling of exosomes in human urine. Proceedings of the National Academy of Sciences of the United States of America 101 (36):13368–13373. doi: 10.1073/pnas.0403453101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou H, Yuen PS, Pisitkun T, Gonzales PA, Yasuda H, Dear JW, Gross P, Knepper MA, Star RA (2006) Collection, storage, preservation, and normalization of human urinary exosomes for biomarker discovery. Kidney international 69 (8):1471–1476. doi: 10.1038/sj.ki.5000273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gonzales PA, Zhou H, Pisitkun T, Wang NS, Star RA, Knepper MA, Yuen PS (2010) Isolation and purification of exosomes in urine. Methods Mol Biol 641:89–99. doi: 10.1007/978-1-60761-711-2_6 [DOI] [PubMed] [Google Scholar]
- 5.Cheruvanky A, Zhou H, Pisitkun T, Kopp JB, Knepper MA, Yuen PS, Star RA (2007) Rapid isolation of urinary exosomal biomarkers using a nanomembrane ultrafiltration concentrator. American journal of physiology Renal physiology 292 (5):F1657–1661. doi: 10.1152/ajprenal.00434.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raj DA, Fiume I, Capasso G, Pocsfalvi G (2012) A multiplex quantitative proteomics strategy for protein biomarker studies in urinary exosomes. Kidney international 81 (12):1263–1272. doi: 10.1038/ki.2012.25 [DOI] [PubMed] [Google Scholar]
- 7.Thery C, Amigorena S, Raposo G, Clayton A (2006) Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Current protocols in cell biology / Chapter 3:Unit 3.22. doi: 10.1002/0471143030.cb0322s30 [DOI] [PubMed] [Google Scholar]
- 8.Gardiner C, Ferreira YJ, Dragovic RA, Redman CW, Sargent IL (2013) Extracellular vesicle sizing and enumeration by nanoparticle tracking analysis. Journal of extracellular vesicles 2. doi: 10.3402/jev.v2i0.19671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Webber J, Clayton A (2013) How pure are your vesicles? Journal of extracellular vesicles 2. doi: 10.3402/jev.v2i0.19861 [DOI] [PMC free article] [PubMed] [Google Scholar]