

Abstract
Adoptive T cell therapy (ACT) is a promising strategy for treating cancer, but it often fails due to cell intrinsic regulatory programs that limit the degree or duration of T cell function. In this study, we found that ectopic expression of microRNA-200c (miR-200c) markedly enhanced the anti-tumor activity of CD8+ cytotoxic T lymphocytes (CTLs) during ACT in multiple mouse models. CTLs transduced with miR-200c exhibited reduced apoptosis during engraftment and enhanced in vivo persistence, accompanied by upregulation of the transcriptional regulator T cell factor 1 (TCF1) and the inflammatory cytokine tumor necrosis factor (TNF). miR-200c elicited these changes by suppressing the transcription factor Zeb1 and thereby inducing genes characteristic of epithelial cells. Overexpression of one of these genes, Epcam, was sufficient to augment therapeutic T cell responses against both solid and liquid tumors. These results identify the miR-200c–EpCAM axis as an avenue for improving ACT and demonstrate that select genetic perturbations can produce phenotypically distinct T cells with advantageous therapeutic properties.
Once Sentence Summary:
Upregulation of the miR-200c-EpCAM axis promotes the engraftment, persistence, and anti-tumor activity of transferred T cells in murine models.
INTRODUCTION
Anti-tumor adoptive cell therapy (ACT) involves expanding patient-derived T cells ex vivo and then reinfusing them to treat disease. It is a particularly exciting treatment for cancer because it provides avenues for enhancing the specificity of the therapeutic response (1, 2). T cells that recognize tumor antigens can be expanded selectively or T cells can be transduced with chimeric antigen receptors (CARs) that target tumor antigens. CAR therapy, in particular, has yielded remarkable results against certain B cell leukemias (3). It has been very challenging, however, to apply ACT to other malignancies, particularly solid tumors (4). At least some of this difficulty arises from cell-intrinsic regulatory programs that limit the scope of T cell function. The ability to mount potent cytotoxic and inflammatory responses is largely restricted to short-lived effector T cells, which can neither survive nor self-renew over prolonged periods (5). In addition, T cells become functionally exhausted after sustained antigen exposure, placing strict constraints on their activity (6). Under homeostatic conditions or when the immune system is battling transient infections, this behavior is appropriate. It is counterproductive, however, in the context of cellular immunotherapy, where T cells must mount vigorous responses against established tumors for extended periods of time.
The importance of sustained T cell activity for ACT (7–10) has generated a great deal of interest in methods that increase the effective size of the therapeutic T cell population and prolong its functionality. Patients are typically subjected to conditioning chemotherapy or ablative radiation prior to infusion in order to reduce competition between transferred T cells and endogenous lymphocytes for homeostatic cytokines (11). Although these approaches increase engraftment efficiency, a large fraction of infused T cells still perish within days (12, 13), and strategies for mitigating this death are poorly developed. Maintaining functional persistence after engraftment is also of critical importance. Much recent excitement in this area has focused on the high-mobility group (HMG) family transcription factor T cell factor 1 (TCF1), a critical regulator of T cell survival and pluripotency in vivo. TCF1 and its homolog, Lef1, are both required for the formation of memory T cells capable of long-term self-renewal (14–16), and during chronic infection, TCF1 drives the formation of a progenitor-like population that mediates sustained responses by continuously generating new effector cells (17–19). TCF/Lef family proteins form activating transcriptional complexes with the Wnt signaling adaptor, β-catenin (20), which is constitutively degraded in T cells (21, 22). Accordingly, efforts to enhance persistence via TCF1 have concentrated on stabilizing β-catenin expression by either genetic or pharmacological means (21–23). Although these strategies prolong T cell survival, they also attenuate antigen-induced proliferation and cytokine secretion, further emphasizing the reciprocal relationship between persistence and functional capacity that seems to be hard-wired into T cell physiology.
As an alternative to modulating signaling networks within the established framework of T cell homeostasis, one strategy could be to introduce molecular perturbations that shift T cells into phenotypes where they are less constrained by evolved control mechanisms. Using unbiased screening, we have identified such a strategy, which is based on the ectopic expression of the epithelial cell markers, microRNA-200c (miR-200c) and EpCAM (for epithelial cell adhesion molecule). Both molecules prolonged the engraftment efficiency and persistence of T cells in tumor bearing mice without compromising antigen-driven proliferation and cytokine secretion. Consequently, T cells overexpressing miR-200c or EpCAM displayed strikingly enhanced anti-tumor activity in both T cell receptor (TCR) and CAR driven models of ACT. In addition to providing methods for enhancing ACT, our results highlight the value of looking beyond naturally occurring immune cell lineages for therapeutic applications.
RESULTS
A functional screen for miR modulators of CTL cytotoxicity
To identify strategies for improving anti-tumor ACT, we performed an unbiased screen for genetic modifications that alter CTL functionality. We chose to focus this screen on miRs (table S1), rather than small hairpin RNA (shRNA) or clustered regularly interspaced short palindromic repeat (CRISPR) libraries, because miRs regulate suites of genes (24) and therefore seemed more likely to drive unexpected changes in differentiation state. CTL-mediated killing is often accompanied by the exchange of cell surface material between the CTL and the target cell, a process known as trogocytosis (25). As trogocytosis scales with cytotoxic activity (26, 27), we used it as a proxy for identifying the most lethal CTLs in a population (fig. S1A). Murine CTLs bearing the OT1 T cell receptor (TCR), which is specific for the ovalbumin257–264 (OVA) peptide presented by the major histocompatibility complex (MHC) class I molecule, H-2Kb, were retrovirally transduced with one of five miR pools and then incubated with OVA-loaded EL4 target cells bearing one of three different membrane stains. After two hours, CTLs bearing fluorescent label from all three kinds of targets (trogocytosishi) and CTLs that had not acquired any stain (trogocytosislo) were sorted and subjected to deep sequencing to identify miRs that were enriched in either the trogocytosishi or the trogocytosislo subsets.
In this manner, we were able to identify miRs that reproducibly altered cytotoxic responses in vitro. Two candidates, miR-16 (trogocytosishi) and miR-200c (trogocytosislo), generated the most striking functional phenotypes in validatory cytotoxicity assays and were selected for further analysis (Fig. 1A and B). CTLs transduced with miR-16 (miR-16 CTLs) killed both adherent (B16 melanoma) and non-adherent (EL4 lymphoma) target cells more effectively than control CTLs expressing a scrambled miR (miR-Scr CTLs), whereas CTLs overexpressing miR-200c (miR-200c CTLs) exhibited reduced cytotoxicity in both assays (Fig. 1C and D). We also examined two biological events associated with killing, the formation of CTL-target cell conjugates and the release of cytotoxic proteins, also called degranulation. Both responses were enhanced by miR-16 and suppressed by miR-200c (Fig. 1E and F), consistent with the observed cytotoxicity phenotypes.
Figure 1. miRs that alter in vitro T cell cytotoxicity identified by a trogocytosis screen.
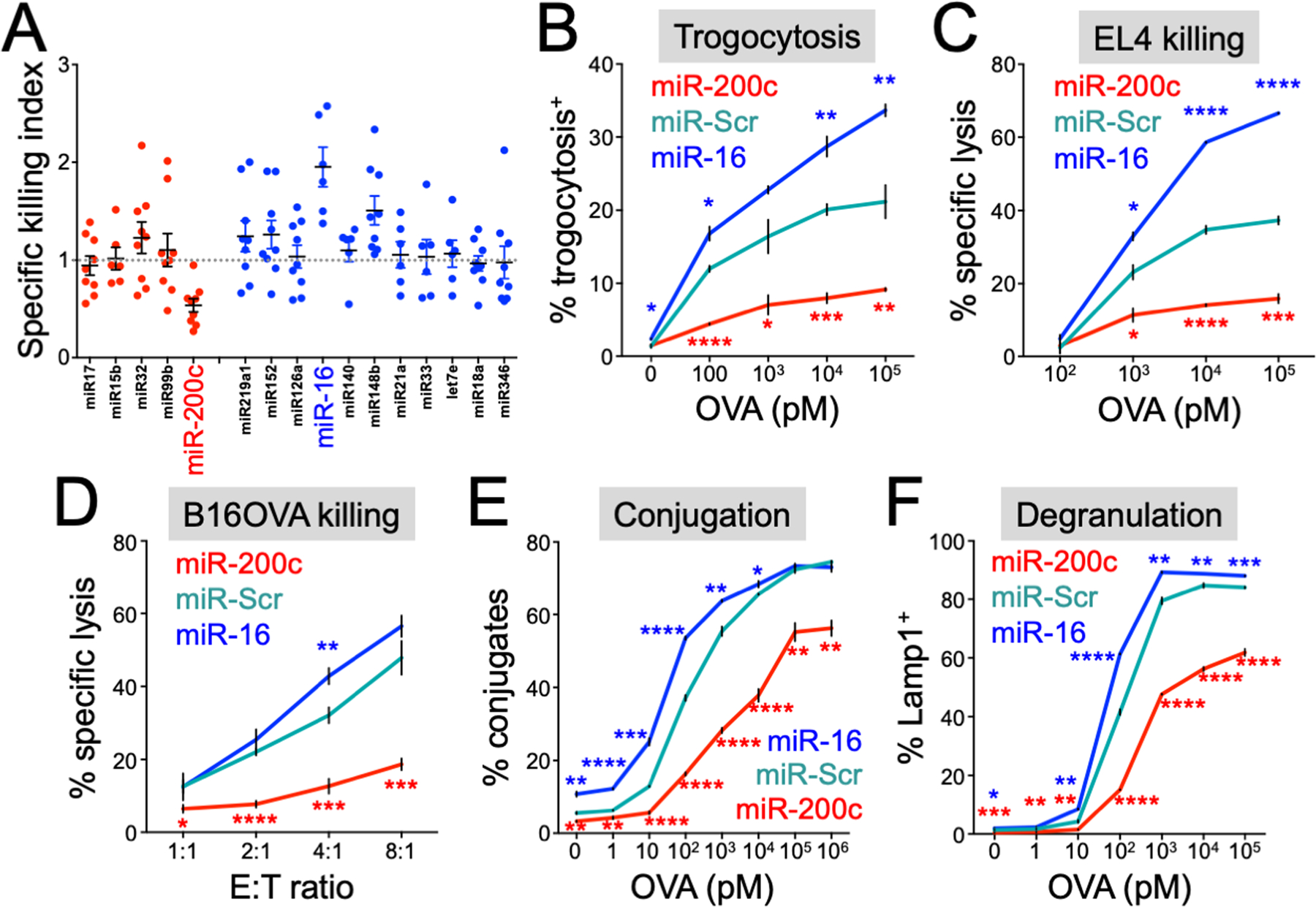
(A) miRs conferring high (blue) or low (red) trogocytosis in the trogocytosis screen were transduced, along with miR-Scr, into OT1 CTLs. The cytotoxicity of the resulting CTLs was then evaluated using OVA-loaded EL4 cells as targets. Data points denote the killing index, defined as the fraction of target cells lysed by CTLs expressing a miR of interest divided by the fraction of target cells lysed by CTLs expressing miR-Scr. miR-200c and miR-16 are highlighted. (B and C) OT1 CTLs expressing the indicated miRs were incubated with EL4 target cells loaded with the indicated concentrations of OVA. (B) Trogocytosis was quantified by flow cytometry after 2 hours. (C) Specific lysis of target cells was assessed after 4 hours. (D) OT1 CTLs expressing the indicated miRs were incubated with B16OVA target cells at various effector:target (E:T) ratios, and target cell lysis assessed after 4 hours. (E and F) OT1 CTLs expressing the indicated miRs were incubated with EL4 target cells loaded with the indicated concentrations of OVA as in (B and C). (E) CTL-target cell conjugate formation was assessed by flow cytometry after 20 minutes. (F) CTL degranulation was assessed by surface exposure of Lamp1 after 2 hours. All error bars denote standard error of the mean (SEM). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001, calculated by Student’s t test. Significance calculations compared miR-200c (red asterisks) or miR-16 (blue asterisks) CTLs with miR-Scr CTLs. All data are representative of at least 2 independent experiments.
miR-200c enhances CTL anti-tumor activity in vivo
Having determined that miR-16 and miR-200c altered CTL-mediated killing in vitro, we used an established ACT model to examine their effects on CTL function in vivo. B16 melanoma cells expressing ovalbumin (B16OVA) were implanted subcutaneously in C57BL/6 recipient mice and allowed to form tumors. A week later, the mice were sublethally irradiated to facillitate T cell engraftment. In vitro differentiated OT1 CTLs overexpressing miR-16, miR-200c, or miR-Scr were then adoptively transferred (Fig. 2A). OT1 CTLs typically delay, but do not prevent, tumor outgrowth in this model. Surprisingly, overexpression of miR-16, which induced strong cancer cell killing in vitro, failed to improve anti-tumor responses relative to miR-Scr (Fig. 2A). Conversely, miR-200c profoundly enhanced tumor suppression (Fig. 2A), in stark contrast to its inhibitory effects on cytotoxicity in vitro (Fig. 1C and D). To assess the specificity of this surprising anti-tumor phenotype, we developed a “sponge” RNA construct that abrogated the activity of miR-200c (fig. S1B and C). Coexpression of this sponge reversed the effects of miR-200c on in vitro cytotoxicity and in vivo B16OVA tumor growth (fig. S1D and E), indicating that these phenotypes did indeed result from miR-200c dependent gene regulation.
Figure 2. miR-200c enhances CTL persistence and anti-tumor function in vivo.
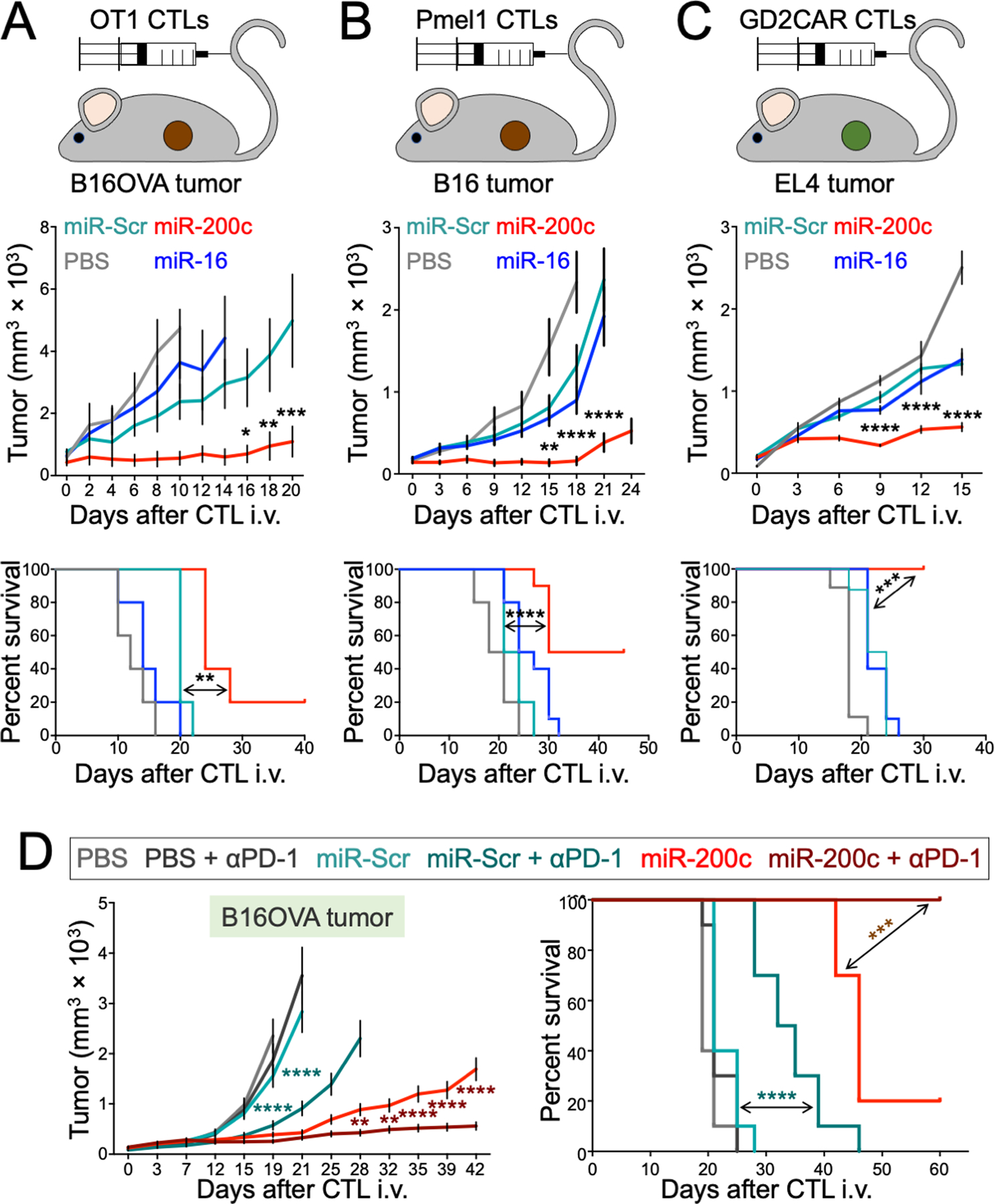
(A to C) Mice bearing subcutaneous (s.c.) B16OVA (A), B16 (B), or EL4 (C) tumors received transfers of OT1 (A) or Pmel1 (B) CTLs expressing the indicated miRs or OT1 CTLs expressing the indicated miRs together with GD2CAR (C). An additional group received vehicle control (PBS). Above, schematic diagrams of each model are shown. Middle, mean tumor volume is plotted against time. Below, Kaplan-Meier plots show overall survival. n = 5 per group for (A), n = 10 per group for (B) and (C). (D) Mice bearing s.c. B16OVA tumors received adoptive transfer of OT1 CTLs expressing the indicated miRs and treated with either anti-PD-1 antibody (αPD-1) or isotype control. Mean tumor volume is plotted on the left, with survival on the right. n = 10 per group. All error bars denote SEM. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001, calculated by two-way ANOVA for tumor growth curves and Log-rank test for survival plots. In (A to C), significance calculations compared the miR-200c and miR-Scr groups, whereas in (D), significance calculation compared the miR-200c + αPD-1 and miR-200c groups (maroon asterisks) or the miR-Scr + αPD-1 and miR-Scr groups (aqua asterisks). All data are representative of at least 2 independent experiments.
The OT1 TCR binds its cognate antigen with unusually high affinity, raising the possibility that the effects of miR-200c on ACT might not apply to T cells with more typical TCRs. To address this issue, we applied ectopic miR-200c and miR-16 expression to a second ACT system in which B16 tumor-bearing mice were treated with CTLs expressing the Pmel1 TCR, which recognizes an endogenous melanoma antigen (Fig. 2B). miR-200c, but not miR-16, improved anti-tumor responses in this model, demonstrating that the capacity of miR-200c to augment ACT was not restricted to the OT1 TCR. We also investigated whether miR-200c could improve CAR-driven ACT, using a model where CTLs expressing a CAR against the glycolipid tumor antigen, GD2 (28), were used to treat mice bearing subcutaneous tumors of GD2+ EL4 cells (Fig. 2C). miR-200c CTLs outperformed both miR-16 and miR-Scr CTLs in this system as well, indicating that the approach is applicable to both CAR- and TCR-driven ACT.
Finally, we examined whether miR-200c overexpression could combine effectively with immune checkpoint blockade (ICB), a class of antibody-based therapies targeting inhibitory lymphocyte immunoreceptors such as programmed cell death protein 1 (PD-1) (29). Mice bearing B16OVA tumors received miR-200c or miR-Scr OT1 T cells and were then treated twice a week with anti-PD-1 or isotype control antibodies. Although PD-1 blockade enhanced the anti-tumor efficacy of both miR-200c and miR-Scr CTLs, the combination of miR-200c and anti-PD-1 was particularly effective, suppressing tumor growth profoundly and extending the survival of treated mice past 60 days (Fig. 2D). Collectively, our results demonstrate that miR-200c boosts therapeutic T cell function in a variety of contexts and combines additively with ICB.
miR-200c augments CTL engraftment and persistence in vivo
The reduced killing potential of miR-200c CTLs implied that their enhanced in vivo functionality resulted from phenotypic changes unrelated to cytotoxicity. Accordingly, we examined the persistence of miR expressing CTLs after transfer into tumor bearing mice. OT1 CTLs expressing either miR-200c or miR-16 were mixed 1:1 with OT1 miR-Scr controls and then transferred into irradiated, congenically-marked recipients bearing B16OVA tumors (Fig. 3A). After one week, OT1 T cells were quantified in the tumor, blood, spleen, lymph nodes, and peripheral organs. miR-200c, but not miR-16, increased CTL persistence in all locations (Fig. 3A and fig. S2A). This phenotype did not depend entirely on antigen, as miR-200c OT1 CTLs also outcompeted controls in mice carrying B16 tumors without OVA, albeit to a lesser extent (fig. S2B). To confirm these observations, we examined B16OVA tumor sections from recipient mice treated with either miR-200c CTLs or miR-Scr CTLs. This histological analysis revealed markedly higher densities of miR-200c CTLs than miR-Scr controls in the tumor microenvironment (Fig. S2C), consistent with our flow cytometry-based results.
Figure 3. miR-200c promotes CTL survival and effector function in vivo.
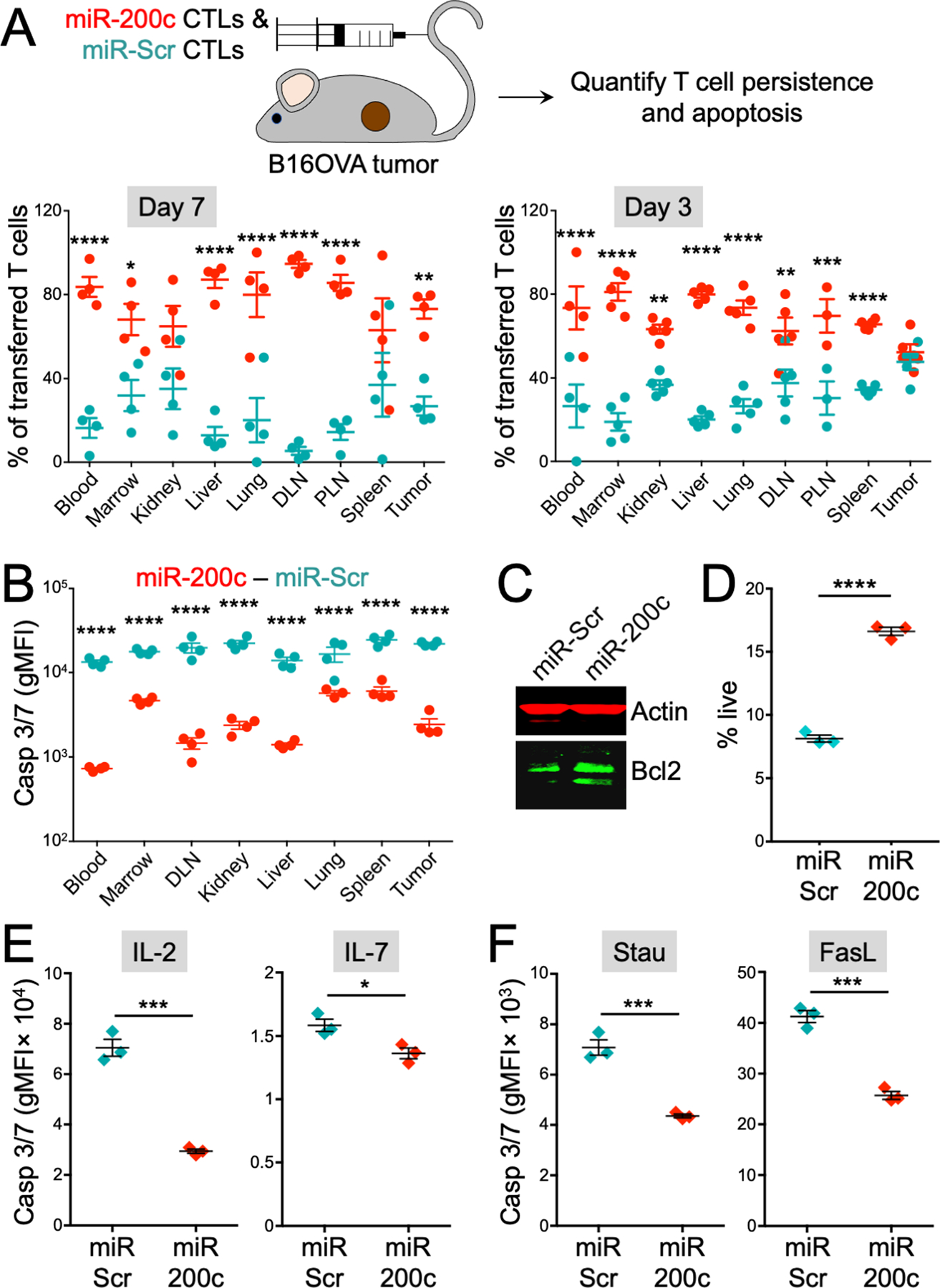
(A) A 1:1 mixture of miR-200c and miR-Scr OT1 CTLs was transferred into B16OVA tumor bearing mice. After 3 and 7 days, CTLs were extracted from various organs and analyzed by flow cytometry. Above, a schematic diagram of the experiment is shown. Below, graphs showing miR-200c and miR-Scr CTL persistence in various organs after 7 (left) and 3 (right) days. n ≥ 3 per group. (B) Mice bearing s.c. B16OVA tumors received either miR-200c or miR-Scr OT1 CTLs. After 3 days, OT1 CTLs were extracted from the indicated organs and assessed for caspase activity using CellEvent Caspase 3/7 staining. gMFI, geometric mean fluorescence intensity. (C) Bcl2 abundance in miR-200c and miR-Scr CTLs was assessed by immunoblot with actin as a loading control. (D) CTLs expressing miR-200c or miR-Scr were transferred into medium lacking IL-2 and survival was assessed 48 hours later by DAPI incorporation. n = 3 for each group. (E) CellEvent Caspase 3/7 staining is shown for miR-200c and miR-Scr CTLs cultured in RPMI-1640 for 48 hours with either 30 international units (IU)/ml IL-2 (left) or 5 ng/ml IL-7 (right). n = 3 for each group. (F) CellEvent Caspase 3/7 staining is shown for miR-200c and miR-Scr CTLs treated with 2 μM staurosporine for 4 hours or 20 ng/ml FasL for 48 hours. n = 3 for each group. All error bars denote SEM. DLN = draining lymph node, PLN = nondraining (peripheral) lymph node. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001, calculated by two-way ANOVA (A and B) and Student’s t test (D to F). All data are representative of at least 2 independent experiments.
Enhanced miR-200c CTL persistence manifested as early as three days post adoptive transfer (Fig. 3A), suggesting that miR-200c might augment survival during the early stages of engraftment. To investigate this hypothesis, we extracted CTLs from tumor bearing mice three days after initial transfer and stained them with CellEvent Caspase 3/7 reagent, a probe for apoptosis. miR-200c CTLs exhibited markedly reduced caspase activation at this early time point (Fig. 3B), suggesting that they can resist apoptosis associated with engraftment more effectively than controls (12). Consistent with this interpretation, miR-200c CTLs expressed the anti-apoptotic protein Bcl2 to a higher degree (Fig. 3C), and they survived better in vitro after transfer into growth medium lacking interleukin (IL)-2 (Fig. 3D). Additional in vitro experiments indicated that miR-200c CTLs were less susceptible to apoptosis at steady state, regardless of whether CTLs were cultured in IL-2 or IL-7 (Fig. 3E). Overexpression of miR-200c also dampened apoptotic responses to the cell death inducers staurosporine and Fas ligand (FasL) (Fig. 3F). Hence, miR-200c transduction promotes the survival of therapeutic T cells.
Next, we investigated whether miR-200c conferred CTLs with enhanced proliferation, as this could also explain the observed persistence phenotype. miR-200c CTLs did divide somewhat more extensively than miR-Scr controls after antigenic stimulation in vitro, and they also expressed slightly more of the proliferation marker Ki67 (fig. S3A and B). We did not observe higher expression of Ki67 in vivo, however, implying little to no proliferative advantage after infusion (fig. S3C). We conclude that the in vivo persistence phenotype of miR-200c CTLs is more likely due to increased survival than enhanced proliferation.
We reasoned that the enhanced persistence miR-200c CTLs in vivo could effectively compensate for their reduced cytotoxic activity. To test this hypothesis, we performed in vivo killing assays in which mice receiving transfer of OVA-loaded splenocytes were treated with miR-200c or miR-Scr OT1 CTLs (fig. S3D and E). Similar degrees of specific killing were observed in both cases (fig. S3F), suggesting that miR-200c expression does not substantially reduced the effective cytotoxicity of the therapeutic T cell pool.
miR-200c promotes TCF1 and TNF expression in tumors
To further explore the functional ramifications of miR-200c in T cells, we activated CTLs in vitro and measured cytokine production. miR-200c CTLs generated much more tumor necrosis factor (TNF) than miR-Scr controls, whereas interferon-γ (IFN-γ) responses were comparable (fig. S4A and B). We observed the same pattern of results after restimulation of CTLs extracted from B16OVA tumors; miR-200c dramatically enhanced TNF production without altering IFN-γ responses (Fig. 4A). Hence, miR-200c augments not only the survival but also the functional capacity of CTLs both in culture and in the tumor microenvironment.
Figure 4. miR-200c promotes CTL survival and effector function in vivo.

A 1:1 mixture of miR-200c and miR-Scr OT1 CTLs was transferred into B16OVA tumor bearing mice. At various time points, CTLs were extracted and analyzed by flow cytometry. (A) Top left, representative flow cytometry plot showing TNF and IFN-γ expression in the indicated tumor-infiltrating CTLs, extracted 1 week after infusion and restimulated with PMA and ionomycin. Right and below, quantification of TNF+ (top right), IFN-γ+ (bottom left), and TNF+IFN-γ+ (bottom right) tumor infiltrating CTLs from unstimulated (un) and PMA and ionomycin stimulated (stim) samples is shown. n = 3 mice per group. (B) Above, representative flow cytometry plot showing TCF1 and T-bet expression in tumor infiltrating CTLs, extracted 1 week after infusion into tumor bearing mice. Below, quantification of TCF1 (left) and T-bet (right) expression in tumor infiltrating CTLs is shown. n = 3 mice per group. (C) Flow cytometric analysis of exhaustion markers in miR-Scr or miR-200c OT1 CTLs, extracted 2 weeks after infusion into B16OVA tumor bearing mice. Quantification of PD-1 (left), LAG3 (middle), and TIM3 (right) expression in CTLs extracted from the tumor and spleen (sp) is shown. n ≥ 6 per group for PD-1 and LAG3, n = 4 per group for TIM3. (D) β-catenin (βcat) abundance in CTLs expressing the indicated miRs was assessed by immunoblot using actin as a loading control. All error bars denote SEM. ns, not significant, *P ≤ 0.05, and **P ≤ 0.01, calculated by Student’s t test. Data in (C) were pooled from two independent experiments. All other data are representative of at least 2 independent experiments.
Next, we examined the expression of critical transcription factors that control T cell differentiation. We were particularly interested in TCF1 because of its links to functional persistence and self-renewal (15–19). Tumor infiltrating miR-200c CTLs displayed markedly higher expression of this protein (Fig. 4B), consistent with their enhanced survival and anti-tumor activity. They also expressed more of the T-box family member T-bet (Fig. 4B), which has been associated with sustained functionality in the context of chronic antigen exposure (30). Conversely, overexpression of miR-200c did not alter the expression of Eomes and Blimp1 (fig. S4C), two transcription factors that characterize differentiated effector subsets. Interestingly, cultured miR-200c and miR-Scr CTLs expressed similar amounts of TCF1 and T-bet in vitro (fig. S4D), implying that the upregulation of these transcription factors by miR-200c also requires some feature of the in vivo environment. Taken together, these results indicate that miR-200c predisposes CTLs to acquire the transcriptional indices of self-renewal and pluripotency in vivo.
Sustained T cell functionality in the presence of antigen is typically limited by the onset of exhaustion (6). To assess whether miR-200c overexpression alters this process, we quantified cell surface abundance of PD-1, lymphocyte-activation Gene 3 (LAG3), and T cell immunoglobulin and mucin domain-containing protein 3 (TIM3), three inhibitory receptors associated with the exhausted state. Compared to miR-Scr controls, tumor-infiltrating miR-200c CTLs expressed equivalent amounts of LAG3 and TIM3 and slightly less PD-1 two weeks after infusion (Fig. 4C). Notably, this modest reduction in PD-1 expression was not apparent at the one-week time point (fig. S4E), when enhanced miR-200c CTL persistence and tumor suppression is already apparent (Fig. 2 and 3). Hence, it seems unlikely that the robust anti-tumor activity of miR-200c CTLs resulted from the suppression of canonical T cell exhaustion.
Prolonged in vivo persistence and high expression of TCF1 and Bcl2 are hallmarks of stem cell memory T cells (Tscm), which are known to mediate enhanced anti-tumor responses in ACT models (22, 23). The phenotypic and functional similarities between miR-200c CTLs and Tscm cells raised the possibility that they might be the same cell type. Tscm cells can be generated in vitro by pharmacological stabilization of β-catenin, which drives elevated Wnt signaling. Like Tscm cells, miR-200c CTLs expressed abundant β-catenin protein (Fig. 4D). They did not, however, exhibit reduced proliferation and cytokine secretion (Fig. 4 and fig. S3 and S4), which are both Tscm hallmarks (22). In addition, whereas Tscm cells express high CD62L and low CD44, indicative of attenuated effector differentiation, miR-200c transduction failed to alter the expression of either marker (fig. S4F). We conclude that, despite some similarities with Tscm cells and other memory subsets, miR-200c CTLs exhibit a distinct phenotype.
miR-200c drives epithelial genes via suppression of Zeb1
T cells typically contain modest amounts of miR-200c. Retroviral transduction with miR-200c achieved far higher expression (fig. S5A), raising the possibility of supraphysiological, gain-of-function effects. To explore this hypothesis, we performed whole transcriptome RNA sequencing (RNA-seq) comparing miR-200c and miR-Scr CTLs (fig. S5B). Gene set enrichment analysis (GSEA) of the resulting data did not reveal obvious links between miR-200c CTLs and established T cell differentiation states. Genes associated with T cell memory and exhaustion, for instance, were not markedly enriched relative to those of naïve and effector subsets (fig. S5C). By contrast, genes characteristic of differentiated epithelial cells were strongly induced by miR-200c (fig. S5B). Two of these genes, Epcam and Cdh1, encode cell surface proteins (EpCAM and E-cadherin, respectively) that are not typically found on lymphocytes. miR-200c CTLs exhibited higher expression of both molecules, with EpCAM being particularly abundant (Fig. 5A and fig. S4G). Ectopic miR-200c also induced EpCAM expression in CD4+ T cells (fig. S4H). miR-200c and its homologs (miR200a, miR200b, miR141, miR429) have been shown to restrain malignant transformation in epithelial tissues (31, 32). Consistent with this prior work, we documented strong correlations between miR-200c induced transcription and gene sets previously implicated in the inhibition of epithelial-to-mesenchymal transition (EMT) and metastasis (Fig. 5B and fig. S5D). A link with EMT inhibition was not apparent in all gene sets, however; indeed, miR-200c induced gene expression changes were positively correlated with the Hallmark EMT gene set (fig. S5D). Hence, miR-200c drives a gene expression program in CTLs that includes a subset of characteristic epithelial markers.
Figure 5. miR-200c promotes epithelial gene expression and anti-tumor function by suppressing Zeb1.
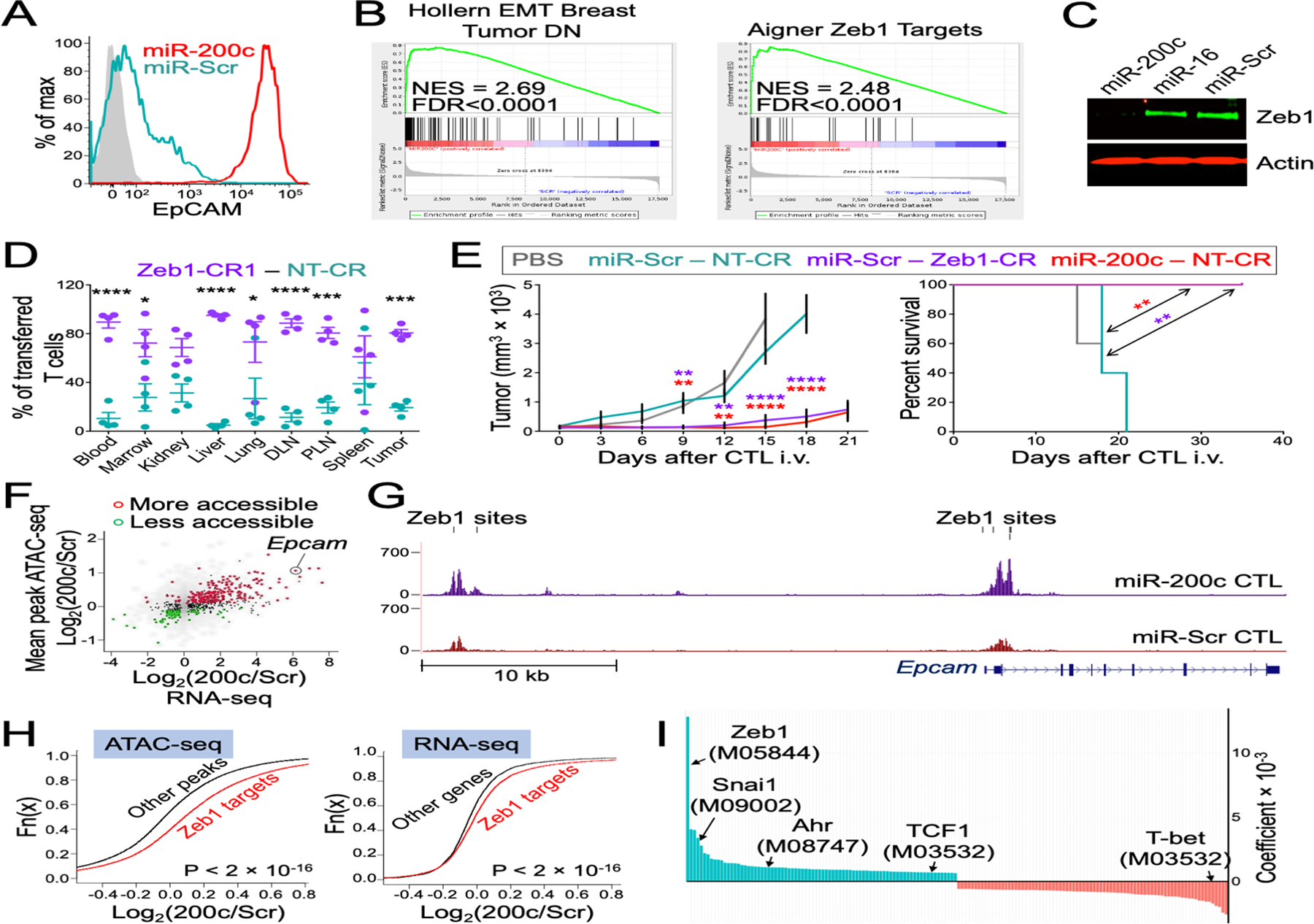
(A) A representative histogram shows upregulation of EpCAM in miR-200c CTLs. Isotype control staining is shown in gray. (B) GSEA of RNA-seq data shows correspondence between miR-200c-induced genes and genes downregulated during EMT (54), and genes repressed by Zeb1 (55). NES = Normalized Enrichment Score. (C) The immunoblot shows downregulation of Zeb1 in miR-200c CTLs. Actin served as a loading control. (D) A 1:1 mixture of OT1 Cas9 CTLs expressing Zeb1 specific gRNA (Zeb1-CR1) and controls expressing nontargeting gRNA (NT-CR) CTLs was transferred into B16OVA tumor bearing mice. After 7 days, CTLs were extracted from indicated organs and quantified by flow cytometry. n = 4 for each group. (E) Mice bearing s.c. B16OVA tumors were treated with OT1 Cas9 CTLs expressing the indicated miRs and gRNAs. Left, tumor size was graphed against time. Right, Kaplan-Meier plot show overall survival. PBS denotes vehicle control. n = 5 for each group. All error bars in (D to E) denote SEM. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001, calculated by two-way ANOVA (D and the left graph in E), and Log-rank test for the right graph in (E). In (E), significance calculations compared miR-200c-NT-CR with miR-Scr-NT-CR (red asterisks) and miR-Scr-Zeb1-CR1 with miR-Scr-NT-CR (purple asterisks). (F) Scatter plot of differential expression (x-axis, determined by RNA-seq) and differential accessibility (y-axis, determined by ATAC-seq) between miR-200c CTLs and miR-Scr CTLs. Significantly differentially accessible genes highlighted with red or green color. (G) ATAC-seq signal profiles are shown for the Epcam locus, taken from miR-200c and miR-Scr CTLs. Vertical black lines indicate Zeb1 binding sites within putative regulatory regions. (H) Cumulative distribution functions highlighting the increased accessibility of peaks (left) and expression of genes (right) containing Zeb1 binding motifs in miR-200c CTLs. P-value were calculated by Kolmogorov-Smirnov test. (I) Enrichment of transcription factor binding motifs in ATAC-seq peaks more accessible in miR-200c CTLs (cyan) or miR-Scr CTLs (red).
miR-200 family members inhibit EMT and metastasis in epithelial tissues by suppressing the transcriptional repressors Zeb1 and Zeb2 (33, 34). In our hands, miR-200c downregulated Zeb1 protein and induced a gene expression signature in CTLs that strongly overlapped with a curated set of Zeb1 targets (Fig. 5B and C). To determine the importance of Zeb1 for the miR-200c CTL phenotype, we transduced OT1 T cells expressing the Cas9 nuclease with guide RNAs (gRNAs) that either completely (CR1 and CR4) or partially (CR6) eliminated Zeb1 protein (fig. S6A). Zeb1 depletion mimicked the effects of miR-200c on EpCAM and E-cadherin expression (fig. S6B and C), in vitro survival and proliferation (fig. S6D and E), and in vivo persistence (Fig. 5D). In many cases, the severity of the phenotype scaled with the degree of Zeb1 suppression, further supporting a causal link between the latter and the former. Interestingly, Zeb1 depletion did not affect cancer cell killing in vitro (fig. S6F), implying that miR-200c inhibits this response through a distinct mechanism. To assess the extent to which Zeb1 inhibition accounts for in vivo anti-tumor responses, OT1 Cas9 CTLs were retrovirally transduced with a bicistronic vector expressing a miR (Scr or 200c) and a gRNA (Zeb1-CR1 or nontargeting (NT) control). This design enabled us to compare the activity of miR-200c- and Zeb1-deficient CTLs against the same shared control. Both groups of cells enhanced anti-tumor function to a similar extent (Fig. 5E), strongly suggesting that Zeb1 inhibition accounts for most, if not all, of the therapeutic benefit conferred by miR-200c.
miR-200c CTLs also exhibited marked upregulation of the aryl hydrocarbon receptor (AhR) (fig. S7A), a transcription factor that has been implicated in the differentiation of both T cells and B cells (35). This phenotype was Zeb1-independent, as it was not recapitulated in Zeb1 deficient CTLs (fig. S7A). Interestingly, ectopic expression of AhR inhibited CTL-mediated killing in vitro (fig. S7A and B), suggesting that the AhR branch of the miR-200c network, rather than the Zeb1 branch, regulates cytotoxicity. AhR overexpression had no effect on in vivo anti-tumor responses (fig. S7C), however, indicating it is dispensable for miR-200c-induced potentiation of ACT. Consistent with this interpretation, homozygous deletion of the Ahr gene did not alter the capacity of miR-200c to enhance CTL anti-tumor function (fig. S7A and S7D). Hence, unlike Zeb1, AhR does not appear to play a therapeutically relevant role in CTLs downstream of miR-200c.
To further explore the transcriptional consequences of ectopic miR-200c, we performed comparative assay for transposase-accessible chromatin using sequencing (ATAC-seq) analysis of miR-200c CTLs and miR-Scr controls. Data were analyzed by building upon a previously constructed atlas of chromatin accessibility sites in CD8+ T cells (36), which facilitated the identification of putative promoter and enhancer elements. In agreement with prior research (37, 38), we found that increased chromatin accessibility was strongly correlated with increased transcription (Fig. 5F). Of the 406 genes whose expression was enhanced by ectopic miR-200c, 189 were more accessible in miR-200c CTLs, whereas only 18 were more accessible in miR-Scr CTLs. Conversely, of the 158 genes whose expression was suppressed by miR200c, 60 exhibited higher accessibility in miR-Scr CTLs and only 9 were more accessible in miR-200c CTLs. Some of the most obvious miR-200c induced accessibility changes mapped to genes defining the miR-200c “epithelialization” phenotype, including Epcam and Cdh1 (Fig. 5F and G and fig. S8. Both of these genes contain high-affinity Zeb1 binding motifs (also called E-boxes) in their putative promoter and enhancer regions, implying that direct recognition of these elements by Zeb1 leads to chromatin compaction. Indeed, genome wide analysis revealed that regulatory elements containing Zeb1 sites were more accessible in miR-200c CTLs and also that the genes linked to these domains were more highly expressed (Fig. 5H and I). Binding sites for Snai1, another transcriptional regulator of EMT (39), were also associated with increased accessibility (Fig. 5I and fig. S9), further corroborating the link between miR-200c and epithelial gene expression. By contrast, consensus motifs for Ahr, TCF1, and T-bet exhibited little to no correlation with differential ATAC-seq signals. These results further implicate Zeb1 as the critical target of miR-200c in T cells and suggest that relieving Zeb1-dependent chromatin remodeling can promote in vivo persistence and anti-tumor function in T cells.
EpCAM overexpression recapitulates the effects of miR-200c
The capacity of Zeb1 deletion to phenocopy the effects of miR-200c in CTLs motivated us to explore potential roles for Zeb1 target genes. We were particularly interested in EpCAM because of its dramatic upregulation in miR-200c CTLs and also because EpCAM expression promotes proliferation and survival in cancer cell lines (40, 41). Using CRISPR/Cas9 targeting, we were able to partially reverse the upregulation of EpCAM induced by miR-200c overexpression (fig. S10A). Remarkably, miR-200c OT1 CTLs lacking EpCAM were considerably less effective than their EpCAM-sufficient counterparts at suppressing B16OVA tumors (fig. S10B). EpCAM was also required for the potentiation of in vivo persistence by miR-200c (fig. S10C). To determine whether EpCAM alone was sufficient to enhance therapeutic T cell function, we retrovirally transduced CTLs with full-length EpCAM fused to green fluorescent protein (GFP) (Fig. 6A). Ectopic EpCAM expression increased the persistence of transferred CTLs in vivo (Fig. 6B), similar to the effects of miR-200c overexpression and Zeb1 deletion. CTLs transduced with EpCAM (EpCAM CTLs) also exhibited higher Bcl2 expression, along with enhanced survival and reduced apoptosis (fig. S11A to D). As with Zeb1 deletion, EpCAM transduction did not alter cytotoxicity (fig. S11E). Furthermore, EpCAM CTLs were CD44hi and CD62Llo, similar to miR-200c CTLs and unlike Tscm cells (fig. S11F). EpCAM CTLs also contained normal amounts of both Zeb1 and miR-200c (Fig. S5A and fig. S11G), consistent with the idea that EpCAM operates downstream of both molecules. EpCAM transduction did not alter E-cadherin expression (fig. S11H), suggesting that EpCAM is dispensable for the induction of E-cadherin by miR-200c. Conversely, ectopic expression of E-cadherin did lead to a modest increase in EpCAM expression (fig. S11I), although this phenotype was not associated with enhanced survival (fig. S11J). Hence, overexpression of EpCAM, but not E-cadherin, recapitulated the phenotypic properties of miR-200c and Zeb1-CR CTLs.
Figure 6. EpCAM promotes β-catenin expression and CTL persistence.

(A) EpCAM expression was measured in OT1 CTLs transduced with EpCAM or control (Ctrl) retrovirus. (B) A 1:1 mixture of EpCAM and Ctrl CTLs was transferred into B16OVA tumor bearing mice. After 1 week, CTLs were extracted from various organs and quantified by flow cytometry. n ≥ 3 for each group ***P ≤ 0.001 and ****P ≤ 0.0001, calculated by two-way ANOVA. (C) β-catenin (βcat) in CTLs expressing EpCAM or empty vector (Ctrl), assessed by immunoblot using actin as a loading control. (D and E) OT1 CTLs expressing the indicated miRs were fixed and stained for β-catenin together with DAPI to visualize the nucleus. (D) representative z-projection images (scale bars = 5 μm). (E) quantification of mean nuclear β-catenin intensity. n ≥ 13 cells for each group. (F and G) Intracellular localization of GFP (F) and EpCAM-GFP (G) in OT1 CTLs is shown. Representative images of GFP fluorescence and nuclear DAPI staining are shown to the left (scale bars = 7 μm). Normalized intensity linescans, derived from the yellow lines in the images, are shown to the right. (H and I) CTLs expressing EpCAM-GFP or GFP alone were subjected to PLA using antibodies against GFP and β-catenin. (H) Representative images of PLA signal together with nuclear DAPI staining (scale bars = 7 μm). To the left of each image, schematic diagrams illustrate predicted PLA results assuming a specific interaction between EpCAM and β-catenin. (I) Nuclear PLA puncta in EpCAM-GFP (EpC) and GFP transduced CTLs were quantified. n ≥ 26 cells for each group. All error bars indicate SEM. P-values in (E) and (I) were calculated by Student’s t test. All data are representative of at least 2 independent experiments.
EpCAM is known to undergo regulated proteolysis that releases its C-terminal tail into the cytoplasm (41). In tumor cells, this intracellular fragment associates with β-catenin and other Wnt signaling components, forming a transcriptionally active complex that promotes proliferation. Given that both EpCAM and miR-200c CTLs expressed abundant β-catenin (Fig. 4D and 6C), we investigated whether an analogous mechanism might be operating in T cells. Immunocytochemical analysis of miR-200c CTLs revealed that a substantial portion of β-catenin protein localized to the nucleus, consistent with a role in gene regulation (Fig. 6D and E). By contrast, ectopic EpCAM (labeled C-terminally with GFP) accumulated primarily on the plasma membrane and in vesicular compartments (Fig. 6F and G), as one might expect for a transmembrane protein. A small but detectable pool of EpCAM protein, however, was also observed in the nucleus. To assess whether this nuclear EpCAM was bound to β-catenin, we performed a proximity ligation assay (PLA) using antibodies against GFP (to detect EpCAM-GFP) and β-catenin. We observed strong nuclear PLA signals in EpCAM-GFP expressing CTLs, but not in GFP-expressing controls, indicative of complex formation between EpCAM and β-catenin (Fig. 6H and I). These results suggest that EpCAM modulates CTL function through formation of a nuclear Wnt signaling complex.
Finally, we evaluated the therapeutic function of EpCAM CTLs using both TCR-driven (OT1-B16OVA and Pmel1-B16) and CAR-driven (GD2CAR-EL4) solid tumor models of ACT (Fig. 7A to C). EpCAM boosted anti-tumor activity in all three contexts, and this augmented in vivo functionality was associated with increased TCF1, T-bet, and TNF expression in tumor infiltrating CTLs (Fig. 7D to F). By contrast, EpCAM overexpression had no effect on cell surface abundance of PD-1, LAG3, and TIM3 (fig. S12A). To assess the capacity of EpCAM to potentiate anti-tumor responses in human T cells, we used an established system in which human T cells expressing a CAR against the B cell antigen CD19 are used to treat NOD-scid il2rg−/− (NSG) mice bearing NALM6 B cell leukemias. EpCAM overexpression enhanced tumor suppression in this model (Fig. 8A and B), leading to increased survival (Fig. 8C). Comparative analysis of EpCAM and control CAR T cells extracted from the spleen and bone marrow 17 days after infusion revealed no differences in the expression of PD-1, LAG3, and TIM3 (Fig. 8D and fig. S12B and C), implying that the enhanced activity of EpCAM T cells in this model did not result from suppressed exhaustion. We also examined the effects of EpCAM on CAR T cell persistence by adoptively transferring a 1:1 mixture of EpCAM and control CAR T cells to NALM6-bearing NSG mice. After seven days, EpCAM T cells were over-represented in the blood, bone marrow, and liver (Fig. 8E), suggesting that EpCAM transduction boosts the engraftment and persistence of human T cells comparably to murine T cells. We conclude that EpCAM recapitulates critical features of the miR-200c CTL phenotype and that it improves ACT against both solid and liquid malignancies.
Figure 7. EpCAM expression enhances anti-tumor ACT in multiple tumor models.

(A to C) Mice bearing s.c. B16OVA (A), B16 (B), or EL4 (C) tumors received adoptive transfers of OT1 (A) or Pmel1 (B) CTLs expressing EpCAM or empty vector (Ctrl) or OT1 CTLs expressing EpCAM or empty vector (Ctrl) together with GD2CAR (C). An additional group received vehicle control (PBS). Above, mean tumor volume is plotted against time. Below, Kaplan-Meier plots show overall survival. n = 10 for A, n = 10 for B, n ≥ 8 for C. (D to F) A 1:1 mixture of EpCAM and Ctrl CTLs was transferred into B16OVA tumor bearing mice. After 1 week, tumor infiltrating CTLs were stained for TCF1 (D) and T-bet (E), or restimulated and stained for TNF (F). n = 3 mice per group. All error bars indicate SEM. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001, calculated by Student’s t test (D to F), two-way ANOVA (top graphs in A to C), or Log-rank test (bottom graphs in A to C). In (A to C), significance calculations compared the EpCAM and Ctrl groups. Data are representative of at least 2 independent experiments.
Figure 8. EpCAM promotes CD19CAR ACT of B cell leukemia.
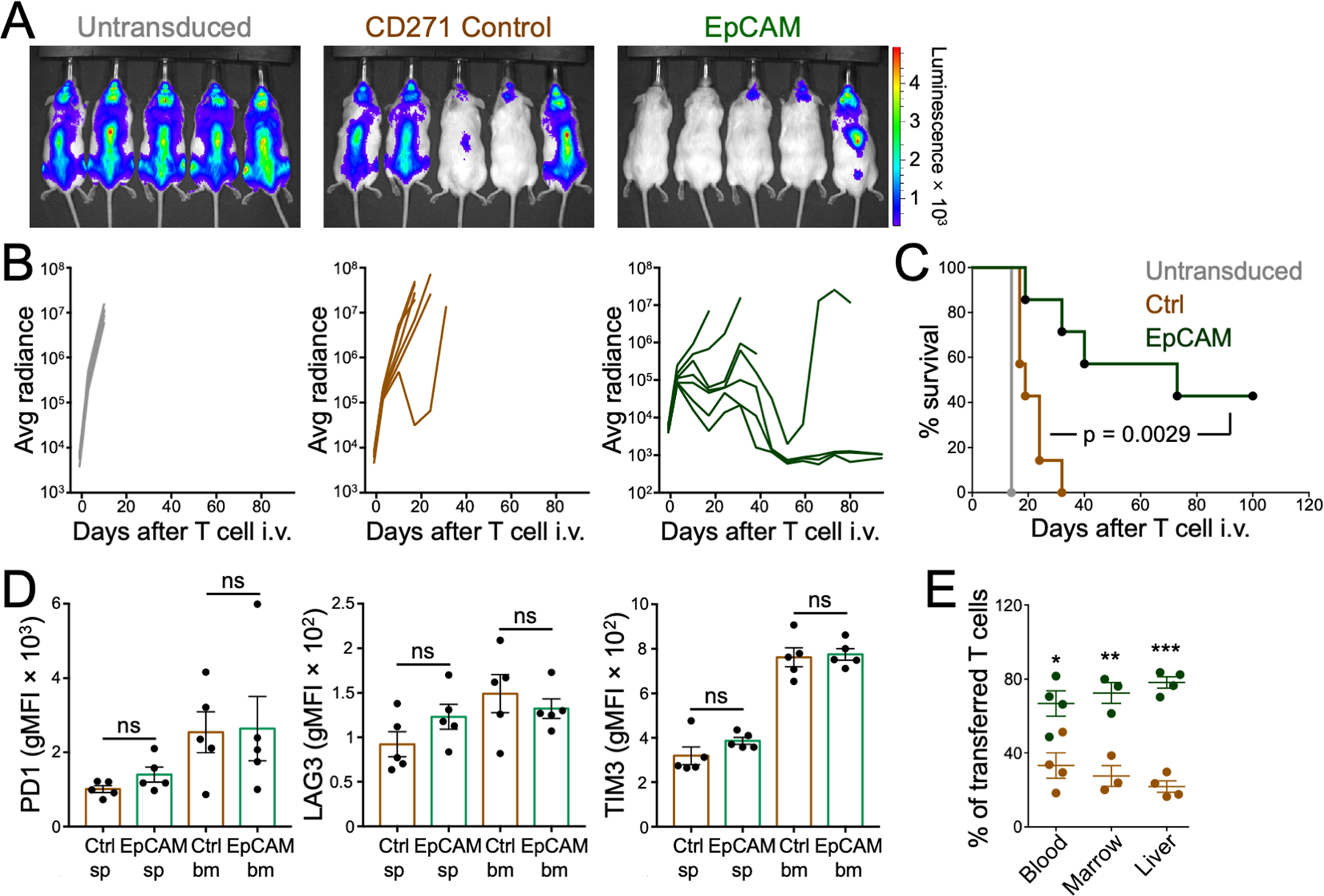
NSG mice bearing NALM6 tumors received human CD19CAR T cells expressing EpCAM or CD271 (Ctrl). (A) Representative BLI images of mice are shown at 2 weeks after the indicated treatments. (B) Tumor growth is shown for mice receiving untransduced T cells (left) and in mice receiving Ctrl (center) or EpCAM (right) CAR T cells. n = 7 mice per group. (C) Survival curves from the same experiment are shown. The p-value was calculated by Log-rank test. (D) Quantification of PD-1 (left), LAG3 (middle), and TIM3 (right) expression is shown for T cells extracted from the bone marrow (bm) and spleen (sp) 17 days after infusion into NALM6 tumor-bearing mice. n = 5 per group. ns denotes not significant, calculated by Student’s t test. (E) NSG mice bearing NALM6 tumors received adoptive transfers of a 1:1 mixture of CD19 CAR T cells transduced with EpCAM or CD271 (Ctrl). After 1 week, CTLs were extracted from indicated organs and quantified by flow cytometry. n ≥ 3 for each group. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001, calculated by two-way ANOVA. All data are representative of at least 2 independent experiments.
DISCUSSION
In this study, we identified the miR-200c–EpCAM axis as a promising avenue for potentiating ACT responses against both solid and liquid tumors. CD8+ CTLs lacking Zeb1 or overexpressing either miR-200c or EpCAM exhibited multiple properties associated with therapeutic efficacy, including enhanced survival, inflammatory cytokine secretion, and expression of TCF1. Although we have not directly examined whether miR-200c-EpCAM signaling augments CD4+ T cell functionality in a similar manner, our results with the CD19CAR model, which employs a mixture of CD4+ and CD8+ T cells derived from human blood, suggest that this may indeed be the case.
Our initial screening approach presupposed that increased in vitro cytotoxicity would translate into improved anti-tumor activity. We found instead that highly cytotoxic miR-16 CTLs were outperformed by weakly cytotoxic miR-200c CTLs in vivo. These results indicate that robust killing capacity alone is insufficient to potentiate anti-cancer ACT, a conclusion that is in line with studies demonstrating that enhanced T cell effector function correlates with low therapeutic efficacy, particularly when coupled with reduced persistence (8, 22, 42). That being said, it is conceivable that in different tumor models, the cytotoxicity defect induced by miR-200c might compromise therapeutic function. Our results indicate that this killing phenotype depends on the upregulation of AhR, rather than the suppression of Zeb1. Accordingly, efforts to further develop this approach therapeutically would be well advised to focus on EpCAM overexpression and possibly Zeb1 depletion, both of which circumvent AhR and therefore recapitulate the miR-200c-induced persistence and anti-tumor phenotypes without reducing cytotoxicity.
The increased therapeutic efficacy of miR-200c and EpCAM CTLs manifested well before observable differences in PD-1, LAG3, and TIM3 expression, implying that the improved performance of these cells did not arise from inhibition of canonical exhaustion. Our results suggest two alternative explanations. First, miR-200c and EpCAM CTLs exhibited enhanced per cell effector function, as evinced by increased antigen-induced TNF expression both in vitro and in vivo. Second, the miR-200c–EpCAM axis improved engraftment efficiency. Indeed, miR-200c overexpression reduced apoptosis and improved persistence within 72 hours of CTL infusion. At least 90% of transferred T cells are thought perish in the first 48 hours (12), likely limiting the efficacy of ACT. The strong association between engraftment efficiency and anti-tumor activity that we observed in our experiments suggests that potentiating the former is an achievable and worthwhile strategy for improving the latter.
Recent studies indicate that systemic or local application of blocking antibodies against PD-1 or its ligand, programmed death ligand 1 (PD-L1), can prolong CAR T cell functionality and attenuate exhaustion, implying that CAR ACT can be effectively combined with ICB (43). Our results indicate that modulation of the miR-200c–EpCAM axis can further boost the efficacy of this approach. ICB enhances anti-tumor immunity by reversing or inhibiting T cell exhaustion, but it is not thought to alter engraftment efficiency. By contrast, both miR-200c and EpCAM promote engraftment and persistence with seemingly minimal effects on exhaustion. They also confer higher expression of TCF1 in vivo, which has been linked to the capacity of T cells to be “reinvigorated” by checkpoint antibodies (44, 45). Hence, there is ample reason to expect that miR-200c–EpCAM signaling would complement ICB-induced tumor suppression, and this is exactly what we observed in the B16OVA system. It will be interesting to explore this strategy in other therapeutic contexts.
The physical interaction we documented between nuclear EpCAM and β-catenin, taken together with the increased β-catenin and TCF1 expression exhibited by miR-200c and EpCAM CTLs, strongly suggest a central role for Wnt signaling in the elaboration of the miR-200c–EpCAM T cell phenotype. In that regard, it is notable that miR-200c and EpCAM CTLs failed to phenocopy Tscm cells, which are also characterized by augmented Wnt signaling (22). Apparently, different strategies for activating the Wnt pathway in T cells can produce qualitatively distinct functional outcomes. In cancer cells, EpCAM drives proliferation by forming a nuclear Wnt signaling complex that contains not only β-catenin but also two additional components, the adaptor molecule four and a half LIM domains 2 (FHL2) and the TCF1-related transcription factor Lef1 (41). Both FHL2 and Lef1 are expressed in T cells, and it will be interesting to investigate their roles in miR-200c and EpCAM induced T cell reprogramming.
Previous chromatin immunoprecipitation (CHIP)-seq and ATAC-seq studies have identified two kinds of Zeb1 regulated genes, a group that contains high-affinity E-boxes and a group that does not (46, 47). Zeb1 is thought to repress the first class via E-box binding and the recruitment of transcriptional corepressors, like the CtBP complex (48), that drive repressive chromatin remodeling. By contrast, Zeb1 tends to activate genes in the second class by associating with other transcription factors that are bound directly to enhancer or promoter sequences (46, 47). Our own data, which demonstrate that genes containing regulatory E-boxes are both more accessible and more highly expressed in miR-200c CTLs, strongly suggest that miR-200c promotes T cell “epithelialization” by relieving Zeb1 dependent chromatin compaction around E-box elements. Whether miR-200c also reverses the activating effects of Zeb1 in this system will require additional experiments that directly profile genome wide Zeb1 binding (such as CHIP-seq). It is interesting to note, however, that Zeb1 dependent gene induction in breast cancer cells involves two transcription factors, Lef1 and activator protein 1 (AP-1) (46, 47), with established roles in T cell activation and differentiation. Accordingly, it is tempting to speculate that Zeb1 controls T cell fate at least in part by modulating AP1 and Lef1 induced transcription.
Our conclusion that miR-200c boosts T cell persistence by suppressing Zeb1 contrasts somewhat with recent work indicating that endogenous expression of miR-200 family members promotes memory T cell differentiation by selectively inhibiting Zeb2, but not Zeb1 (49). Indeed, Zeb1 was actually shown in this prior study to be required for proper memory formation after viral infection. That we were able to inhibit Zeb1 robustly in CTLs using miR-200c likely reflects the supraphysiological expression we achieved by retroviral transduction, which enabled us to access miR-200c dependent regulatory effects more similar to those seen in epithelial tissues rather than what is typically observed in T cells. Indeed, given the degree overexpression we achieved, it is difficult to draw firm conclusions about the role of miR-200c–EpCAM signaling in normal T cell physiology. That being said, our results do demonstrate that prolonged in vivo persistence and high TCF1 expression, two established features of memory T cells, can actually manifest in the absence of Zeb1. Hence, to the extent that Zeb1 is required for memory formation, it likely contributes to other aspects of the phenotype. Finally, it is notable that miR-200c and EpCAM CTLs attain high expression of TCF1 and T-bet only in vivo, implying that the miR-200c–EpCAM axis does not promote T cell memory per se, but rather a state that is poised to acquire memory-like features in the appropriate context.
Although our results demonstrate the therapeutic potential of miR-200c/EpCAM-based perturbations, how this group of strategies performs relative to other methods for enhancing therapeutic T cell function awaits dedicated comparative analyses. In addition, the persistence of miR-200c, Zeb1CR, and EpCAM CTLs in peripheral organs raises the possibility of off-target side effects. While we did not observe obvious morbidity in mice receiving these cells, comprehensive toxicology studies will be required to address this issue conclusively. It is also important to note that the beneficial phenotypes conferred by miR-200c (improved engraftment and functional persistence after infusion) were not the focus of our initial miR screen. That we were nonetheless able to identify miR-200c, Zeb1, and EpCAM as therapeutic agents or targets reflects the occasional serendipity of the scientific process. It seems likely that a reconceived screen built around survival and function in vivo would yield additional and potentially more effective candidate perturbations.
Efforts to generate therapeutic immune cells with improved functions (50) have largely been driven by the rational design of synthetic receptors and signaling pathways. This deterministic approach is well suited for developing new receptor specificities and response circuits but is less effective at controlling supportive cellular processes that, although required for response efficacy, are often less well understood. By screening ectopic genetic perturbations, we were able to generate T cells that did not fall into documented lineages or respond normally to homeostatic restraints. We conclude that the unbiased application of molecular genetic or pharmacologic strategies that expand T cell phenotype space can serve as a useful complementary approach for developing “designer” cellular therapies.
MATERIALS AND METHODS
Study design
The goal of this study was to identify and characterize genetic perturbations that could improve the anti-tumor activity of adoptively transferred therapeutic T cells. To this end, we employed transplantable models of both solid and liquid tumors in vivo, and we also performed phenotypic and functional characterization of modified T cells in vitro. Group sizes were determined based on previously observed assay variation. Experiments were not randomized, and investigators were not blinded during acquisition and data analysis. In general, experiments were performed at least twice (two biological replicates).
Mice
The animal protocols used for this study were approved by the Institutional Animal Care and Use Committee of Memorial Sloan Kettering Cancer Center (MSKCC). C57BL/6J mice, CD45.1+ congenic mice (B6.SJL-PtprcaPepcb/Boy), OT1 TCR transgenic mice, Rosa26-Cas9 knockin mice, and NSG mice (NOD.Cg-PrkdcscidIl2rgtmWjl/SzJ) were obtained from Jackson Laboratory. Pmel1 TCR transgenic mice and Ahr−/− mice were obtained from Dr. Jedd Wolchok (MSKCC) and Dr. Jayanta Chaudhuri (MSKCC), respectively. Ahr−/− mice were crossed with OT1 mice to generate Ahr−/− OT1 and Ahr+/− OT1 mice. OT1 mice and Rosa26-Cas9 knockin mice were crossed to generate OT1 Cas9 mice for in vitro CRISPR/Cas9 knockout.
Tumor models
B16 and B16OVA cell lines were provided by Jedd Wolchok (MSKCC). EL4 cells were provided by Gregoire Altan-Bonnet (NIH-NCI). FFLuc-GFP-NALM6 cells were generated in the Sadelain lab. For the OT1-B16OVA model, 2 × 105 B16OVA cells were injected into C57BL/6J mice and allowed to grow for 6 days. Then, tumor-bearing mice were subjected to sublethal irradiation (600 cGy) using an animal Cesium irradiator (Gammacell) to facilitate T cell engraftment. The following day, 105 OT1 CTLs were injected intravenously (i.v., tail vein for all models), after which tumor size was measured by metric caliper every 2–3 days for up to 4 weeks. CD45.1+ congenic mice were used as recipients in some experiments to facilitate identification of OT1 CTLs in tumors and other organs. In certain experiments, 200 μg/ml anti-PD-1 antibody (clone RMP1–14, BioXCell) or isotope control antibody was injected intraperitoneally into recipient mice on the day of CTL injection and twice a week thereafter for the duration of the experiment.
For the Pmel1-B16 model, 5 × 105 B16 cells were injected into C57BL/6J mice and allowed to grow for 6 days. Then, tumor-bearing mice were subjected to sublethal irradiation (600 cGy) using an animal Cesium irradiator (Gammacell) to facilitate T cell engraftment. The following day, 5 × 105 Pmel1 CTLs were injected i.v., after which tumor size was measured by metric caliper every 2 to 3 days for up to 4 weeks.
For the GD2CAR-EL4 model, C57BL/6J recipient mice were sublethally irradiated (600cGy) one day before subcutaneous injection of 5 × 105 EL4 cells. This is because EL4 cells are particularly radiation sensitive. Tumors were allowed to grow for 1 week, and then 5 × 105 GD2CAR CTLs were injected i.v. Tumor size was measured by metric caliper every 3 days for up to 4 weeks.
For the CD19CAR-NALM6 model, 7 to 8 week old NSG mice were injected i.v. with 5 × 105 FFLuc-GFP-NALM6 cells, followed by i.v. injection of 1 × 105 CAR T cells four days later. Animal bioluminescence imaging was performed weekly in the MSKCC Animal Imaging Core Facility. Data were processed and analyzed using the IVIS Imaging System with Living Image software (Xenogen). Tumor burden was assessed as previously described (51).
Cell extraction from mouse tissue
Prior to organ collection, mice were perfused with 10 ml phosphate-buffered saline (PBS) with 2 mM EDTA. Spleens were crushed over 70 μm strainers, and isolated cell suspensions were cleared of red blood cells using Ammonium–chloride–potassium (ACK) lysing buffer. B16 tumor tissues were mechanically dissociated using metal mesh and filtered through a 70 μm strainer (Fisher). The resulting cells were resuspended in 15 ml Hanks buffered saline solution (HBSS, without Mg2+/Ca2+) with 3% fetal calf serum (FCS). After mixing with 20 U/ml heparin and 8 ml 100% Percoll (Sigma-Aldrich), samples were centrifuged at 600 × g for 10 minutes at 4 °C. The pellet was treated with ACK buffer and washed into HBSS. Liver, kidney, and lungs were dissociated with scissors and digested with Collagenase D (1 mg/ml; MilliporeSigma) in HBSS (with Mg2+/Ca2+) for 20 minutes at 37 °C. Samples were then mechanically disrupted, filtered through a 70 μm strainer, and resuspended in 15 ml cold HBSS (without Mg2+/Ca2+) with 3% FCS. After mixing with 20 U/ml heparin and 8 ml 100% Percoll, samples were centrifuged at 600 × g for 10 minutes at 4 °C. Cell pellets were resuspended in 10 ml HBSS (without Mg2+/Ca2+) with 3% FCS and placed over 60% Percoll. After additional centrifugation at 600 × g for 10 minutes at 4 °C, the middle band of cells was collected, washed in HBSS, and maintained in HBSS (without Mg2+/Ca2+) with 3% FCS prior to use. For bone marrow cell isolation, leg bones were ground with a mortar and pestle, and the resulting sample filtered through a 70 μm strainer. Red blood cells in pellets were cleared using ACK buffer, and the remaining cells were washed into HBSS (without Mg2+/Ca2+) with 3% FCS.
Flow cytometry analysis of transcription factors and cytokines
Cell suspensions were first stained for surface markers (such as CD45.2 and CD8) and then fixed and stained for TCF1 (clone C63C9), T-bet (clone 4B10), Eomes (clone Dan11mag), Blimp1 (clone 5E7), or Ki67 (clone B56) using the Foxp3/Transcription Factor Staining Buffer Kit (Tonbo Biosciences) according to manufacturer instructions. To assess IFN-γ and TNF production, live cell suspensions were stimulated by plate bound anti-CD3ε (1 μg/ml coating concentration; 145-2C11; BioXCell) and anti-CD28 (1 μg/ml coating concentration; 37.51; BioXCell) or PMA and Ionomycin (20 ng/ml and 1 μg/ml, respectively, both from Sigma-Aldrich) for 2 hours at 37 °C, after which GolgiPlug was added, and the cells incubated for an additional 4 hours in 37 °C. After staining for surface markers, samples were fixed with IC Fixation Buffer (eBioscience), treated with permeabilization buffer (eBioscience), and stained with fluorescently labeled anti-IFN-γ (clone XMG1.2) and anti-TNF (clone MP6-XT22) antibodies, followed by flow cytometry analysis (BD Biosciences LSRII and Beckman Coulter Cytoflex LX). Details pertaining to antibody staining, including clone, fluorophore, and dilution, are provided in table S2.
Trogocytosis assay and miR library screen
To measure trogocytosis, EL4 target cells were stained with CellVue Maroon (eBioscience) per manufacturer’s protocol, loaded with various doses of OVA for 45 minutes at 37 °C, and then mixed 1:1 with miR-transduced OT1 CTLs (GFP+). After 1 to 2 hours at 37 °C, CellVue Maroon transfer to CTLs was quantified by flow cytometry on an LSRII (BD Biosciences). For miR library screening, EL4 cells were stained separately with CellVue Maroon, PKH26 (Sigma-Aldrich), or EZ-Link Sulfo-NHS-LC-Biotin (Thermo Fisher Scientific), and then loaded with 100 nM OVA (no OVA as negative control to set gate). An equal mixture of CellVue Maroon+, PKH26+, and biotinylated EL4 cells were then mixed 1:1 with OT1 CTLs (GFP+) expressing 1 of 5 miR pools (Table S1) with each miR cloned into an MSCV-based retroviral plasmid. After a 1.5 to 2 hour incubation at 37 °C, samples were stained with streptavidin-Pacific Blue for 10 minutes at room temperature (to label biotinylated material) and FACS sorted. Total GFP+ cells, GFP+ CellVue Maroon+ PKH26+ Pacific Blue+ (trogocytosis+) cells, and GFP+ CellVue Maroon− PKH26− Pacific Blue− (trogocytosis−) cells were isolated and subjected to DNA extraction using the Genomic DNA Purification Kit (Promega). The miR expression cassettes (200–300 base pairs each) were then PCR-amplified using these oligos: Forward: ACCGGTAGGCCTCGTACGCTTA and Reverse: TCCACAGGGTCGACCACTG, followed by Illumina Next Generation Sequencing analysis (MiSeq PE300) at the Integrated Genomics Operation (IGO) at MSKCC. miRNA reads were identified by adapter sequences using a fuzzy match that allowed up to 2 mismatches. In a typical deep sequencing run, 26 to 28% of the read counts corresponded to miRs. Only miRs with read counts ≥ 50 were considered as potential candidates. miRs associated with trogocytosishi and trogocytosislo populations were identified using the enrichment ratio (counts in trogocytosishi)/(counts in trogocytosislo). 11 trogocytosishi and 5 trogocytosislo candidates were analyzed in a secondary cytotoxicity screen using antigen-loaded EL4 cells as targets. Subsequently, miR-200c and miR-16 were subjected to more in-depth analysis.
Duolink Proximity Ligation Assay (PLA)
PLA experiments were carried out using the Duolink proximity ligation assay in situ red kit (Sigma-Aldrich) per manufacturer’s instructions. T cells were plated on Poly-L-lysine coated glass, fixed, permeabilized, and stained with anti-GFP (polyclonal rabbit IgG) and anti-β-catenin (clone 14/Beta-Catenin, mouse IgG). After washing, slides were incubated with anti-mouse and anti-rabbit PLUS and MINUS PLA Probes, followed by DNA ligation and amplification using the manufacturer recommended protocol. After fluorescent labelling, slides were imaged on an Sp5 confocal microscope (Leica) using the Texas Red and 4’,6-diamidino-2-phenylindole (DAPI) channels. PLA was quantified manually by counting nuclear puncta after background correction.
Statistical analysis
Figures show representative experiments. Statistical analyses were carried out using either representative experiments or pooled data as indicated. Statistical tests for differential gene expression and GSEA were performed using Limma (52) and GSEA (53) software, respectively. Statistical analysis of ATAC-seq data was performed using Rsubread, DESeq2, motifmatchr, and custom R codes (see Supplementary materials and methods). All other statistical analyses were carried out using GraphPad Prism. Details for each type of statistical test (Student’s t test, ANOVA, Log-rank) may be found in the figure legends or Supplementary Materials and Methods. Unless otherwise stated, error bars denote SEM.
Supplementary Material
Acknowledgments
We thank A. Kepecs, C. Firl, C. Jeronimo, and G. Gunset for technical assistance; the MSKCC Molecular Cytology Core Facility for assistance with imaging; the MSKCC Integrated Genomics Operation and Bioinformatics Core for sequencing and analysis; the MSKCC Animal Imaging Core for bioluminescence imaging; C. Leslie for guidance with bioinformatics; and members of the M. H., M. O. Li, and A. S. labs for advice.
Funding:
This work was supported in part by the Leukemia and Lymphoma Society (SCOR award 7014 to M. H., M. S., and H. G. W.), the National Institutes of Health (P30-CA008748 to MSKCC), the Steven A. Greenberg Trust (to M. H.), and the Cancer Research Institute (CLIP Award to M. H.).
Footnotes
Competing interests: M. Z. and M. H. were named as inventors on U.S. patents filed by MSKCC (MIR200C-EPCAM axis reprogramed immune cells for enhanced antitumor function, US 63/151206). The other authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are in the paper or supplementary materials. RNA-seq and ATAC-seq data have been deposited with the Gene Expression Omnibus under the accession numbers PRJNA704756, PRJNA704772, PRJNA704775, and PRJNA704954. All reagents used in the study are available upon request to M. H. (husem@mskcc.org) and completion of a materials transfer agreement.
References and Notes
- 1.Grupp SA, June CH, Adoptive cellular therapy. Curr Top Microbiol Immunol 344, 149–172 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Restifo NP, Dudley ME, Rosenberg SA, Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 12, 269–281 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sadelain M, CAR therapy: the CD19 paradigm. J Clin Invest 125, 3392–3400 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shah NN, Fry TJ, Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol 16, 372–385 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang N, Bevan MJ, CD8(+) T cells: foot soldiers of the immune system. Immunity 35, 161–168 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McLane LM, Abdel-Hakeem MS, Wherry EJ, CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol, (2019). [DOI] [PubMed] [Google Scholar]
- 7.Rosenberg SA, Restifo NP, Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, Finkelstein SE, Theoret MR, Rosenberg SA, Restifo NP, Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest 115, 1616–1626 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hinrichs CS, Gattinoni L, Restifo NP, Programming CD8+ T cells for effective immunotherapy. Curr Opin Immunol 18, 363–370 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fridman WH, Pages F, Sautes-Fridman C, Galon J, The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 12, 298–306 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME, Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer 8, 299–308 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blattman JN, Antia R, Sourdive DJ, Wang X, Kaech SM, Murali-Krishna K, Altman JD, Ahmed R, Estimating the precursor frequency of naive antigen-specific CD8 T cells. J Exp Med 195, 657–664 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR, Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest 118, 294–305 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou X, Xue HH, Cutting edge: generation of memory precursors and functional memory CD8+ T cells depends on T cell factor-1 and lymphoid enhancer-binding factor-1. J Immunol 189, 2722–2726 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, Xue HH, Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 33, 229–240 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeannet G, Boudousquie C, Gardiol N, Kang J, Huelsken J, Held W, Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc Natl Acad Sci U S A 107, 9777–9782 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, Anderson SM, Handon R, Kenyon E, Elkahloun A, Wu W, Lang PA, Gattinoni L, McGavern DB, Schwartzberg PL, The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin WW, Nish SA, Yen B, Chen YH, Adams WC, Kratchmarov R, Rothman NJ, Bhandoola A, Xue HH, Reiner SL, CD8(+) T Lymphocyte Self-Renewal during Effector Cell Determination. Cell Rep 17, 1773–1782 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, Pradervand S, Thimme R, Zehn D, Held W, T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 45, 415–427 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Cadigan KM, Waterman ML, TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb Perspect Biol 4, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Driessens G, Zheng Y, Locke F, Cannon JL, Gounari F, Gajewski TF, Beta-catenin inhibits T cell activation by selective interference with linker for activation of T cells-phospholipase C-gamma1 phosphorylation. J Immunol 186, 784–790 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, Wrzesinski C, Boni A, Cassard L, Garvin LM, Paulos CM, Muranski P, Restifo NP, Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med 15, 808–813 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, Wang E, Douek DC, Price DA, June CH, Marincola FM, Roederer M, Restifo NP, A human memory T cell subset with stem cell-like properties. Nat Med 17, 1290–1297 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bartel DP, MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joly E, Hudrisier D, What is trogocytosis and what is its purpose? Nat Immunol 4, 815 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Machlenkin A, Uzana R, Frankenburg S, Eisenberg G, Eisenbach L, Pitcovski J, Gorodetsky R, Nissan A, Peretz T, Lotem M, Capture of tumor cell membranes by trogocytosis facilitates detection and isolation of tumor-specific functional CTLs. Cancer Res 68, 2006–2013 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Uzana R, Eisenberg G, Sagi Y, Frankenburg S, Merims S, Amariglio N, Yefenof E, Peretz T, Machlenkin A, Lotem M, Trogocytosis is a gateway to characterize functional diversity in melanoma-specific CD8+ T cell clones. J Immunol 188, 632–640 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Hoseini SS, Dobrenkov K, Pankov D, Xu XL, Cheung NK, Bispecific antibody does not induce T-cell death mediated by chimeric antigen receptor against disialoganglioside GD2. Oncoimmunology 6, e1320625 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei SC, Duffy CR, Allison JP, Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov 8, 1069–1086 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, Bikoff EK, Robertson EJ, Lauer GM, Reiner SL, Wherry EJ, Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 338, 1220–1225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brabletz S, Brabletz T, The ZEB/miR-200 feedback loop--a motor of cellular plasticity in development and cancer? EMBO Rep 11, 670–677 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Senfter D, Madlener S, Krupitza G, Mader RM, The microRNA-200 family: still much to discover. Biomol Concepts 7, 311–319 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ, The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10, 593–601 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Park SM, Gaur AB, Lengyel E, Peter ME, The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 22, 894–907 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rothhammer V, Quintana FJ, The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat Rev Immunol 19, 184–197 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Pritykin Y, van der Veeken J, Pine AR, Zhong Y, Sahin M, Mazutis L, Pe’er D, Rudensky AY, Leslie CS, A unified atlas of CD8 T cell dysfunctional states in cancer and infection. Mol Cell, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shashikant T, Ettensohn CA, Genome-wide analysis of chromatin accessibility using ATAC-seq. Methods Cell Biol 151, 219–235 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, Hellmann MD, Wolchok JD, Leslie CS, Schietinger A, Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stemmler MP, Eccles RL, Brabletz S, Brabletz T, Non-redundant functions of EMT transcription factors. Nat Cell Biol 21, 102–112 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Schnell U, Cirulli V, Giepmans BN, EpCAM: structure and function in health and disease. Biochim Biophys Acta 1828, 1989–2001 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Maetzel D, Denzel S, Mack B, Canis M, Went P, Benk M, Kieu C, Papior P, Baeuerle PA, Munz M, Gires O, Nuclear signalling by tumour-associated antigen EpCAM. Nat Cell Biol 11, 162–171 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA, Waldmann TA, Restifo NP, Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A 102, 9571–9576 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grosser R, Cherkassky L, Chintala N, Adusumilli PS, Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 36, 471–482 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, Sharpe AH, Freeman GJ, Germain RN, Nakaya HI, Xue HH, Ahmed R, Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, Luther SA, Speiser DE, Held W, Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211 e110 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Feldker N, Ferrazzi F, Schuhwerk H, Widholz SA, Guenther K, Frisch I, Jakob K, Kleemann J, Riegel D, Bonisch U, Lukassen S, Eccles RL, Schmidl C, Stemmler MP, Brabletz T, Brabletz S, Genome-wide cooperation of EMT transcription factor ZEB1 with YAP and AP-1 in breast cancer. EMBO J 39, e103209 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosmaninho P, Mukusch S, Piscopo V, Teixeira V, Raposo AA, Warta R, Bennewitz R, Tang Y, Herold-Mende C, Stifani S, Momma S, Castro DS, Zeb1 potentiates genome-wide gene transcription with Lef1 to promote glioblastoma cell invasion. EMBO J 37, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi Y, Sawada J, Sui G, Affar el B, Whetstine JR, Lan F, Ogawa H, Luke MP, Nakatani Y, Shi Y, Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 422, 735–738 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Guan T, Dominguez CX, Amezquita RA, Laidlaw BJ, Cheng J, Henao-Mejia J, Williams A, Flavell RA, Lu J, Kaech SM, ZEB1, ZEB2, and the miR-200 family form a counterregulatory network to regulate CD8(+) T cell fates. J Exp Med 215, 1153–1168 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roybal KT, Lim WA, Synthetic Immunology: Hacking Immune Cells to Expand Their Therapeutic Capabilities. Annu Rev Immunol 35, 229–253 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, Plotkin J, Sadelain M, Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 28, 415–428 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hollern DP, Swiatnicki MR, Andrechek ER, Histological subtypes of mouse mammary tumors reveal conserved relationships to human cancers. PLoS Genet 14, e1007135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aigner K, Dampier B, Descovich L, Mikula M, Sultan A, Schreiber M, Mikulits W, Brabletz T, Strand D, Obrist P, Sommergruber W, Schweifer N, Wernitznig A, Beug H, Foisner R, Eger A, The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 26, 6979–6988 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are in the paper or supplementary materials. RNA-seq and ATAC-seq data have been deposited with the Gene Expression Omnibus under the accession numbers PRJNA704756, PRJNA704772, PRJNA704775, and PRJNA704954. All reagents used in the study are available upon request to M. H. (husem@mskcc.org) and completion of a materials transfer agreement.