

Abstract
Sorafenib tosylate (SFB) is a multikinase inhibitor that inhibits tumour growth and proliferation for the management of breast cancer but is also associated with issues like toxicity and drug resistance. Also, being a biopharmaceutical class II (BCS II) drug, its oral bioavailability is the other challenge. Henceforth, this report intended to encapsulate SFB into a biocompatible carrier with biodegradable components, i.e., phospholipid. The microemulsion of the SFB was prepared and characterized for the surface charge, morphology, micromeritics and drug release studies. The cell viability assay was performed on 4T1 cell lines and inferred that the IC50 value of sorafenib-loaded microemulsion (SFB-loaded ME) was enhanced compared to the naïve SFB at the concentrations of about 0.75 µM. More drug was available for the pharmacological response, as the protein binding was notably decreased, and the drug from the developed carriers was released in a controlled manner. Furthermore, the pharmacokinetic studies established that the developed nanocarrier was suitable for the oral administration of a drug by substantially enhancing the bioavailability of the drug to that of the free SFB. The results bring forth the preliminary evidence for the future scope of SFB as a successful therapeutic entity in its nano-form for effective and safer cancer chemotherapy via the oral route.
Subject terms: Nanoparticles, Breast cancer
Introduction
According to WHO, 9.6 million people died worldwide in the last 10 years because of cancer1. There are various types of cancer, like liver cancer, skin cancer, prostate cancer, colon cancer, leukaemia. The most common cancer is breast cancer, which is a major cause of worldwide deaths due to cancer2. The etiology behind the incidence of breast cancer are hormonal factors, environmental factors and lifestyle changes. Breast cancer is mainly associated with the complex genetic behaviour of the person3.
Protein kinase plays a vital role in regulating cellular functions like metabolism, differentiation, signal transduction, survival, and programmed cell death4. Sorafenib tosylate (SFB), a multikinase inhibitor drug, was developed by the Bayer and Onyx companies as BAY 43-9006 in 2001. Its patent was issued in 2004 from the United States Patent and Trademark Office (USPTO). SFB possesses anti-proliferative and anti-angiogenic effects5. The structure of SFB is shown in Fig. 1. SFB was clinically approved for treating cancer in December 2006 by US-FDA and in August 2007 by the central drug standard control organisation (CDSCO)6. It is used to treat hepatocellular carcinoma, renal carcinoma, breast cancer, thyroid cancer, and prostate cancer. The mode of action of SFB is the serine/threonine kinases c-Raf (Raf-1) inhibition and B-Raf. SFB also inhibits the tyrosine kinase receptor Flt-3 and RET, involved in the pathogenesis of breast cancer7. Various clinical trials to identify its therapeutic efficacy on breast cancer are in different phases8. Despite its high therapeutic effectiveness in cancer management, it also possesses various side effects on oral administration such as diarrhoea (30%), fatigue (18%), hypertension (8–16%), pancreatitis (< 1%). SFB is also linked with dermatological side effects such as seborrheic dermatitis, hand-foot skin reaction, alopecia, rash, stomatitis, and erythema. The dermatological side effects are the most commonly reported side effects of SFB. It also possesses pharmacokinetic related challenges like less oral bioavailability (< 30%), reduced half-life (25–48 h), a plasma level of 7 days and extensive first-pass metabolism. It also exhibits poor water solubility, a BCS class II drug9. All these challenges are associated with the SFB oral administration provide a scope for the researchers' to focus on this aspect. In recent past, attempts have been made to develop various nanoformulations of SFB such as lipid polymer hybrid nanoparticles10, liposomes11, self-emulsifying drug delivery systems12, cyclodextrin-modified silicon nanoparticles13, nanogels14, diatomite nanoparticles15, nano colloidal carrier16, and pullulan nanoparticles17. Most of the developed nanocarriers are non-oral, and very few studies on the oral delivery of SFB employing nanocarriers are reported.
Figure 1.
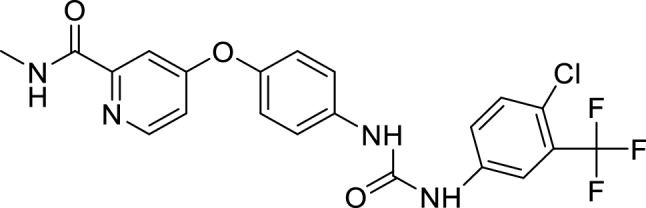
Structure of SFB.
Microemulsions (ME) are biocompatible and thermodynamically stable drug delivery cargoes consisting of aqueous and organic phases stabilized by emulsifiers or surfactants. MEs have proven their efficiency in drug delivery through promising outcomes in drug loading enhancement, controlled drug release, increased scalability, and reduced toxicity18. Microemulsions are not nanoemulsions, though their droplet size is in the range of 10–300 nm, the difference lies in the thermodynamic stability, methodology and composition19–21. Previously, several research groups have explored the efficacy of ME based formulations in the oral delivery of anticancer drugs22–24. Inclusion of phospholipids are known to improve the safety and efficacy of the therapeutic moiety, when incorporated in the nanocarriers by one or other means25.
Considering the challenges in oral delivery of SFB, a ME based formulation was optimized and developed employing phospholipid as one of the major components. Pre-clinical studies including in-vitro and in-vivo experiments were performed to evaluate the therapeutic efficacy of SFB-loaded ME. The developed formulation is novel as the ME of SFB is not reported till date.
Materials and methods
Chemicals and instruments
Sorafenib tosylate was provided as a generous gift from the M/s Cipla Pvt. Ltd., Mumbai, India. Phospholipon 90G was also a gift sample from Lipoid Gembh, Germany via Chemet, New Delhi. Soranib tablets were purchased from local pharmacy store, manufactured by M/s Cipla Ltd., Mumbai, India. Tween 80, Tween 60, Tween 40, Tween 20, methanol, and dialysis membrane was purchased by M/s Fisher Scientific [Pvt] Ltd, Mumbai, India, M/s SDFCL Chem limited, Mumbai, India, and M/s Himedia laboratories [Pvt] Ltd, Mumbai, India, respectively. Isopropyl myristate was purchased from M/s Kempaphore Pvt. Ltd., Mumbai, India. The 4T1 cell lines were provided as a gift sample from Dr. Deepak Chitkara, Assistant Professor, BITS, Pilani, India. Coumarin-6, 4′,6-diamidino-2-phenylindole (DAPI) and Fluorescein IsoThioCyanate (FITC) were purchased from M/s TCI Chemicals Pvt. Ltd., Chennai, M/s Sigma Aldrich, USA and M/s SRL Research Laboratories Pvt. Ltd., Chennai respectively. The Dulbecco’s modified eagle medium and Fetal bovine serum were purchased from M/s Thermo Fisher Scientific, USA. The MTT (3-(4,5-diemthylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (Catalogue no. 298931) was purchased by M/s Sigma Aldrich, USA. The phosphate buffer saline and dimethyl sulfoxide were purchased from M/s Hi-Media Laboratories, India. The solvents acetonitrile, n-octanol, methanol and water used in the study were of HPLC grade and purchased from M/s Spectrochem [Pvt] Ltd, Mumbai, India. All the material reagents and chemicals utilized in this research were pure and of analytical grade and were used as such.
Instrumentation
The types of equipment used in this report are ultra-high performance liquid chromatography (UHPLC, Agilent 1290 Infinity LC System, USA) for the drug quantification at various preformulation stages. The particle size, size distribution, polydispersity index (PDI) and zeta potential of the SFB-loaded ME and blank ME were carried out through Malvern Zetasizer (M/s Malvern, Worcestershire, UK). The shape and surface morphology of the ME were observed using transmission electron microscopy (TEM, M/s FEI Tecnai, Europe). The Fourier-transform infrared spectroscopy (FT-IR, M/s Brukers Alpha II, USA) was performed for the drug's structural analysis and physical compatibility of tween 80, IPM and PL 90G over a wavenumber of 4000–400 cm−1. The ELISA plate reader (Biotek Epoch Microplate Reader) was used for the determination of percent cell viability.
Physicochemical characterization of SFB
The SFB was examined for partition coefficient (log P) by shake flask method using pre-saturated n-octanol and water mixture26. Melting point (M.P) was determined using M-560 BUCHI Machine, Switzerland, as per the standard protocol.
Construction of pseudo ternary phase diagram
On the basis of solubility of SFB in surfactant and Isopropyl myristate (IPM), IPM was selected as the oil phase27. Tween 80 (T80), Tween 60 (T60), Tween 40 (T40) and Tween 20 (T20) were employed as the surfactants, whereas phospholipid 90G (PL 90G) and ethanol were utilized as the cosurfactants. The distilled water was used as the aqueous phase to design phase diagrams. The eight different groups for the phase diagram were formed considering oil, surfactants and cosurfactants. As mentioned in Table 1, 2.75: 1 and 1: 1 were selected as the ratio of surfactant and cosurfactant (Smix ratios) for each surfactant. The ratio of cosurfactants i.e., PL 90G and ethanol was maintained at 1: 10 throughout the experiment to construct the pseudo ternary phase diagram. The ratio of Smix and aqueous phase was varied from 1: 9–9: 1, which were titrated with oil until they reached the turbidity. Likewise, the ratio of Smix and oil was varied in between 1:9 to 9:1 followed by titration with an aqueous phase until the turbidity appeared. The pseudo ternary phase diagram was constructed for obtaining the ME physical state by marking one axis as oil phase, the other as aqueous phase and the third one as a fixed Smix ratio. Evaluating the diagram through visual inspection, the area which corresponds to the formation of clear and transparent formulation was identified as the ME region in the pseudo ternary phase diagram19.
Table 1.
The oil, surfactant, and cosurfactant group in various predetermined combinations.
| Group | Oil | Surfactant | The ratio of surfactant: cosurfactant (Smix) |
|---|---|---|---|
| A | IPM | T20 | 1:1 |
| B | IPM | T20 | 2.7:1 |
| C | IPM | T40 | 1:1 |
| D | IPM | T40 | 2.7:1 |
| E | IPM | T60 | 1:1 |
| F | IPM | T60 | 2.7:1 |
| G | IPM | T80 | 1:1 |
| H | IPM | T80 | 2.7:1 |
Selection of formulation based on phase diagram
After constructing the phase diagram, the minimum quantity of surfactant to a higher fixed value was chosen to prepare different formulations. The selected ratio was supposed to represent the whole pseudo ternary phase diagram.
Preparation of microemulsion
The ME was developed based on the selected ME areas in the ternary phase diagram at different ratios of components. All MEs were prepared to employ the emulsification method. Firstly, the PL 90G was mixed with IPM at a temperature of 50–60 °C with continuous stirring (700 rpm for 10 min). Then, the surfactant was poured to the mixture while reducing the system's temperature. After that, SFB was incorporated into the system with gentle stirring, and the required amount of aqueous phase was poured dropwise in the above ME with continuous stirring. A clear and transparent ME, having yellowish colour, was obtained upon the addition of ethanol. A pictorial representation of the preparation method has been described in Fig. 2.
Figure 2.

Schematic representation for the preparation of SFB-loaded microemulsion.
Drug content
To determine the drug content in the prepared SF-loaded ME, SFB was excerpted in methanol and quantified using UHPLC. Blank ME without drug was used as the blank formulation.
Percent drug loading (DL) and drug entrapment (EE) studies
EE and DL, the formulation studies were performed using the dialysis method. The SFB-loaded ME (eq. to 1 mg of SFB) was weighed and packed carefully in dialysis bag. The dialysis bag was then placed on magnetic stirrer for continuous stirring in 100 mL beaker containing 50 mL of methanol for 2 h. After 2 h, the 1mL sample was withdrawn from the beaker, and the sample was quantified for SFB using UHPLC28. Further, the percent drug entrapment and percent drug loading were calculated using the following formula29.
The drug amounts in the dialysis bags were also quantified to confirm the results.
In-vitro drug release and release kinetics
Drug release and drug kinetic studies were performed by placing free SFB (1 mg), SFB tablet (marketed formulation; batch no. GJ10330) (equivalent to 1 mg of free SFB) and SFB-loaded ME (equivalent to 1 mg of free SFB) in dialysis bags. In brief, the SFB and SFB tablet suspension and the ME were packed in separate dialysis bags. Each bag was suspended separately in 50 mL of 0.1 N HCl (pH 1.2) containing 1% Tween 80 for the first two hours with continuous stirring and then dipped individually into the 50 mL of phosphate buffer saline (PBS) 7.4 containing 1% Tween 80 for a total of 24 h30. The 2 mL samples were withdrawn from each beaker at regular time intervals and adequately maintained the sink conditions. Then cumulative drug release was determined using the UHPLC31. The data of percentage drug release were fitted on all the release models such as first-order, zero-order, and Higuchi model with their details enlisted in Table 232.
Table 2.
Details of various drug release models.
| Release model | Parameter on x-axis | Parameter on y-axis | Equation |
|---|---|---|---|
| Zero-order | Time | % Drug release | Ct = C0 + K0t |
| First-order | Time | Log % drug release | Log Ct = log C0 + Ktt/2.303 |
| Higuchi | The square root of time | % Drug release | Ct = A√D (2C0 – Cs) Cst |
where, Ct = amount of drug dissolved in time t, C0 = initial amount of drug in the solution, K0 = zero order release constant, Kt = first order release constant, Cs = drug solubility in the matrix media, D = diffusivity of drug molecules in the matrix, Mt/M∞ = fraction of drug released at time t, Kp = release rate constant, n = release exponent.
Protein binding studies
The PBS of pH 7.4 was prepared and mixed with human serum albumin (HSA), 4% w/v. The pure drug (SFB, 2.5 mg) was dispersed in PBS 7.4 (1 mL) and SFB-loaded ME containing an equal amount of drug were packed in dialysis membrane bags. To the previously prepared HSA dispersion (4% w/v), the dialysis bags containing different samples were immersed for 12 h with constant stirring. The samples were withdrawn after 12 h followed by centrifugation at 14,000 rpm (21.036×g) at 4 °C. After centrifugation, the supernatants were removed, mixed with an equal amount of acetonitrile (ACN) and filtered through a 0.22 µm filter. The samples were then analysed through UHPLC at the respective wavelength for the quantification of free SFB33,34.
In-vitro cytotoxicity assay
The cytotoxic effects of the developed nano-formulation (SFB-loaded ME) were evaluated against 4T1 breast cancer cells and were compared to that of the blank formulation and free SFB. The method was performed by the relevant guidelines and regulations. 4T1 cells (mouse mammary carcinoma) were cultured in culture plates of 96 wells using DMEM culture media. Different concentrations of SFB, blank formulation, and SFB-loaded ME (0.25–50 µM) were prepared separately using the same culture media. The prepared samples were transferred into the 96-well culture plates containing 4T1 cells followed by incubation of 24 h at 37 °C. Further, 10 μL of MTT solution was added, and the 4T1 cell culture plates were re-incubated for an additional 4 h. The purple-coloured formazan crystals were dissolved in 200 μL of DMSO. Using an ELISA plate reader, the samples were analysed for optical density at λmax of 570 nm. Percentage cell viability was determined in comparison to untreated cells. The results obtained from optical density values were utilised to calculate the percentage cell viability. The samples were also analysed for microscopic observation using an optical microscope35.
In-vitro cellular uptake studies
The cellular uptake of the developed SFB-loaded ME (ME16) was evaluated using a qualitative and quantitative manner using fluorescence microscopy and flowcytometry, respectively. Briefly, the breast cancer cells (4T1 cells) were seeded in a six-well plate with a density of 1 × 106 cells/well and incubated at 37 °C overnight. The next day, coumarin-6 loaded ME formulation (Coumarin-6-ME16) was added to each well (n = 03), and cells were further incubated for another 6 h at 37 °C. Herein, the free coumarin-6 was added to the control group. After the incubation time, PBS was used to wash the cells at least three times, then fixed the cells with 4% paraformaldehyde solution for 15 min, and lastly, counterstained using DAPI. Further, cells were directly observed under a fluorescence microscope (Vert. A1 ZEISS Axiocam, Germany), and the images were acquired at excitation and emission of 488 nm and 510 nm, respectively. On the other hand, the cells were trypsinised, washed with PBS, and analysed using flowcytometry (Cytoflex, Beckman Coulter, USA) to quantify the cellular uptake of the coumarin-6-ME16. The flowcytometry data were processed and analysed using CytExpert software36,37.
In-vivo pharmacokinetic studies
The pharmacokinetics were implemented on the Unisex Wistar rats (200–300 g; 4–6 weeks old). The Institutional Animal Ethics Committee (Panjab University, Chandigarh, India (PU/IAEC/S/16/18)) approved all the animal protocols. All experiments were performed under the relevant guidelines and regulations. The study in complied with Animal Research: Reporting of In-Vivo Experiments (ARRIVE) guidelines. The rats were separated into two different groups. The group I received free SFB suspended in 0.2% carboxymethyl cellulose, and group II administered the SFB-loaded ME via oral gavage. The human dose of SFB is 200 mg/70 kg which was applied to calculate the dose of SFB for each rat peroral route38. The blood samples (200 µL) were withdrawn at 0.25, 0.5, 1, 2, 4, 8, 12, 24 and 36 h time intervals from the retro-orbital plexus of rats. The plasma proteins were precipitated from the blood samples taken in heparin containing micro-centrifuge using micro-centrifugation at 12,000 rpm (10,866*g) for 10 min. Henceforth, the UHPLC method was incorporated to estimate SFB in the extracted plasma of rats at a wavelength of 265 nm with solvents (ACN and water; 65:35 v/v) and a flow rate of 0.8 mL/min using a C18 column39. Then, all the pharmacokinetic parameters such as Volume of distribution (V/F), maximum plasma concentration (Cmax), absorption rate constant (Ka), time at the maximum concentration (Tmax), the area under the curve (AUC), half-life (t1/2) and elimination rate constant (K) were determined as per 1CBM peroral model using PK solver software40.
Stability studies
The stability studies of the SFB-loaded ME were performed as per the guidelines of the International Council on Harmonization (ICH). To estimate the stability of the formulation, samples were stored under different storage conditions at 4 ± 2 °C, 30 ± 2 °C, 45 ± 2 °C for at least 6 months. The particle size, zeta potential and drug loading were observed at 0, 3rd and 6th months of interval41.
Statistical analysis
Statistical analysis of the data was performed using t-test and analysis of variance (ANOVA) software. All the studies were performed at least three times (n = 3), and the data is shown as the means ± SD.
Results and discussion
Spectroscopic analysis
Fourier transform infrared spectroscopy (FT-IR)
The FT-IR spectra of SFB in Fig. 3A showed amide bond formation indicated peak at 2920.78 cm−1 ascribed presence of amine group in SFB. Various characteristic peaks were observed, in the range 1634.51–1425.93 cm−1. FT-IR spectra of blank ME (Fig. 3B), SFB-loaded ME (Fig. 3C), and excipients like Tween 80 (Fig. 3D) and PL (Fig. 3E), IPM (Fig. 3F) were also observed. The broadband centred at 3320.90 cm−1 was assigned to –OH stretching in PL. The absorption at 2978.18 cm−1 and 2922.60 cm−1 were assigned to –CH stretching of methylene group in blank ME. This absorption rate was decreased from 2922.52 to 2854.67 cm−1, significantly indicating the formation of SFB-loaded ME. There were broad spectra observed at 1106.00 cm−1 of the C–O stretch. Thus, it confirmed the formation of SFB-loaded ME42.
Figure 3.

FTIR spectra of (a) Free SFB (b) Blank ME (c) SFB-loaded ME (d) Tween 80 (e) PL 90G (f) IPM.
Ultra-high performance liquid chromatography (UHPLC)
The chromatogram and calibration curve of SFB using UHPLC has been demonstrated using ACN and water (65:35% v/v) as mobile phase under isocratic conditions with a flow rate of 0.8 mL/min and injection volume of 10 µL39. The Calibration curve and chromatogram are shown in Fig. 4a,b. It was observed that the retention time of SFB was determined to be 2.37 min, and r2 value was found to be 0.999.
Figure 4.

(a) Chromatogram of SFB. (b) Calibration curve of SFB.
Physicochemical characterization of SFB
The melting point of the drug was determined to be 204.5 °C, indicating the stability of SFB at higher temperatures, which was favourable for ME preparation. The practical log P value was estimated at 4.18, suggesting that the drug was highly lipophilic and suitable for oral delivery.
Screening of components of the microemulsion
To improve the aqueous solubility challenge of SFB, the IPM was selected as oil for ME preparation. In terms of toxicity, the nonionic surfactants show less toxicity in comparison to the ionic surfactants and have low critical micellar concentrations. SFB also showed solubility in these non-ionic surfactants. Therefore, four non-ionic surfactants were selected for the optimisation of ME43.
PL 90G and ethanol was selected as a cosurfactant for the ME in which PL 90G exhibited high biocompatibility and safety profile along with high solubility of SFB in ethanol. The selected ratio of PL 90G and ethanol was 1:10 to construct the phase diagram due to its maximum solubility with the drug and cost-effective nature. Solubility studies were performed in predetermined Smix ratios i.e., 1:1, 2.75:1, 5:1, 10:1 and 20:1. Based on the outcomes of solubility studies, the Smix ratio was chosen for the effective formulation development. The maximum solubility was manifested in the ratio 2.7:1 followed by 1:1 compared to other Smix ratios.
Construction of pseudo-ternary phase diagram
The phase behaviour of a mixture and its components relation can be easily identified by forming a phase diagram. The pseudo ternary phase diagrams of o/w ME of all the eight groups comprising IPM, ethanol, PL 90G, Tween 20/40/60/80 and distilled water are shown in Figs. 5 and 6. The ratio of surfactant and cosurfactant along with the oil played a significant role in enhancing the phase properties of the ME region. It can be observed in Fig. 5A,B that on increasing the concentration of surfactant (2.7:1), the ME area increased. A similar observation was also seen in other ternary phase diagrams. Also, the ME area was considerably smaller when T20, T40, and T60 were used as surfactants instead of T80, which offered a better emulsification region (32.45%). Hence, there were no miscibility issues of drug with the surfactants. The maximum emulsification property of T80 imposed its selection as the surfactant. Additionally, the loading of SFB was also assumed to be enhanced by the use of T80 as emulsifier. The stability of MEs was also enhanced by the presence of co-surfactant, which reduced the interfacial tension. The phospholipid 90G along with ethanol showed biocompatibility and reduced the phospholipid films rigidity needed for the ME globule formation44.
Figure 5.

Pseudo-ternary phase diagrams; IPM as oil and T20 as a surfactant (A and B), T40 as a surfactant (C and D) at two Smix ratios (1:1 and 2.7:1).
Figure 6.

Pseudo-ternary phase diagrams; IPM as oil and T60 as a surfactant (A and B), T80 as a surfactant (C and D) at two Smix ratios (1:1 and 2.7:1).
Selection of formulation based on phase diagram
Several MEs can be prepared from the ME region of a single-phase diagram. Due to the different surfactants used for the phase diagram construction, the number of formulations was also varied. From all eight pseudo-ternary phase diagrams, formulations were selected from each phase diagram. The composition of all of the selected sixteen ME formulations is given in Table 3.
Table 3.
Composition of selected microemulsions.
| Formulation code | Oil phase (%) | Smix ratio | Smix (%) |
|---|---|---|---|
| ME1 | IPM (6%) | 1:1 | T20: (PL90G: ethanol) (88%) |
| ME2 | IPM (10%) | 1:1 | T20: (PL90G: ethanol) (70%) |
| ME3 | IPM (7%) | 2.7:1 | T20: (PL90G: ethanol) (70%) |
| ME4 | IPM (2%) | 2.7:1 | T20: (PL90G: ethanol) (50%) |
| ME5 | IPM (6%) | 1:1 | T40: (PL90G: ethanol) (40%) |
| ME6 | IPM (3%) | 1:1 | T40: (PL90G: ethanol) (48%) |
| ME7 | IPM (4%) | 2.7:1 | T40: (PL90G: ethanol) (46%) |
| ME8 | IPM (10%) | 2.7:1 | T40: (PL90G: ethanol) (60%) |
| ME9 | IPM (4%) | 1:1 | T60: (PL90G: ethanol) (56%) |
| ME10 | IPM (8%) | 1:1 | T60: (PL90G: ethanol) (47%) |
| ME11 | IPM (18%) | 2.7:1 | T60: (PL90G: ethanol) (66%) |
| ME12 | IPM (4%) | 2.7:1 | T60: (PL90G: ethanol) (50%) |
| ME13 | IPM (3%) | 1:1 | T80: (PL90G: ethanol) (74%) |
| ME14 | IPM (2%) | 1:1 | T80: (PL90G: ethanol) (83%) |
| ME15 | IPM (9%) | 2.7:1 | T80: (PL90G: ethanol) (69%) |
| ME16 | IPM (9%) | 2.7:1 | T80: (PL90G: ethanol) (45%) |
Drug content
All prepared formulations had a drug content of 99.42% to 99.83%, with an average value of 99.62% (Table 4). As the drug content was observed to be so high, it thereby confirmed minimal drug loss during the preparation of MEs and assured the authenticity of the preparation method.
Table 4.
Characterization studies of microemulsion formulations.
| Formulation code | Globule size (nm) | PDI | Zeta potential (mV) | Drug content (%) |
|---|---|---|---|---|
| ME1 | 1081.3 ± 0.4 | 0.69 ± 0.5 | − 3.15 ± 0.6 | 99.49 |
| ME2 | 862.1 ± 4.2 | 0.20 ± 0.03 | − 0.22 ± 0.05 | 99.62 |
| ME3 | 137.0 ± 3.8 | 0.32 ± 0.14 | − 0.42 ± 0.21 | 99.68 |
| ME4 | 513.5 ± 7.3 | 0.92 ± 0.11 | 0.31 ± 0.43 | 99.77 |
| ME5 | 90.0 ± 6.6 | 0.23 ± 0.02 | 0.021 ± 0.15 | 99.53 |
| ME6 | 122.6 ± 2.7 | 0.21 ± 0.03 | − 0.36 ± 0.10 | 99.59 |
| ME7 | 81.30 ± 0.49 | 0.21 ± 0.32 | 0.15 ± 0.07 | 99.67 |
| ME8 | 79.09 ± 0.03 | 0.20 ± 0.05 | 0.16 ± 0.05 | 99.81 |
| ME9 | 648.7 ± 1.02 | 0.57 ± 0.02 | − 4.03 ± 0.02 | 99.46 |
| ME10 | 512.8 ± 7.3 | 0.53 ± 0.51 | − 1.67 ± 0.03 | 99.75 |
| ME11 | 244.3 ± 0.5 | 0.65 ± 0.61 | − 3.78 ± 0.12 | 99.62 |
| ME12 | 83.2 ± 1.4 | 0.26 ± 0.14 | − 2.11 ± 0.23 | 99.51 |
| ME13 | 266.0 ± 2.5 | 0.65 ± 0.06 | − 1.82 ± 0.04 | 99.42 |
| ME14 | 319.3 ± 4.7 | 0.53 ± 0.05 | − 2.57 ± 0.41 | 99.56 |
| ME15 | 75.6 ± 0.2 | 0.20 ± 0.03 | 0.13 ± 0.11 | 99.71 |
| ME16 | 58.8 ± 0.02 | 0.19 ± 0.14 | 0.05 ± 0.03 | 99.83 |
Micromeritics and zeta potential
The zeta potential of several formulations was near to zero (Table 4), which attributed to the non-ionic nature of the surfactants used and imparted better stability to the MEs against any ionic reactions during the long-term storage. Out of all the formulations, the smallest globule size of the ME was of formulation ME16, i.e., 58.8 ± 0.02 nm (Fig. 7) with a zeta potential of 0.05 ± 0.03 (Table 4). The PDI of the ME16 formulation (0.19 ± 0.14) also showed the homogeneity of the ME system. Hence, these results ensured that ME16 was the optimized formulation and selected for further evaluation studies.
Figure 7.

(a) Particle size of ME16 and (b) Zeta potential of ME16.
Percent drug loading (DL) and entrapment efficacy (EE) of ME
The percentage DL and EE of the selected SFB-loaded ME (ME16) was observed to be 21.07 ± 2.16% and 72.64 ± 0.84%, respectively. The value of EE depicted the higher amount of entrapment of SFB in ME with the help of an emulsifying agent (Tween 80). The drug-carrying capacity of the nano-system was quite favourable and assured high loading of SFB for better delivery to the target site.
Surface morphology
The photomicrographs (Fig. 8) of TEM exhibited a spherical shape of the ME16 globules with homogeneity in size. The TEM images showed the smooth surface of the developed ME deprived of any agglomeration45.
Figure 8.
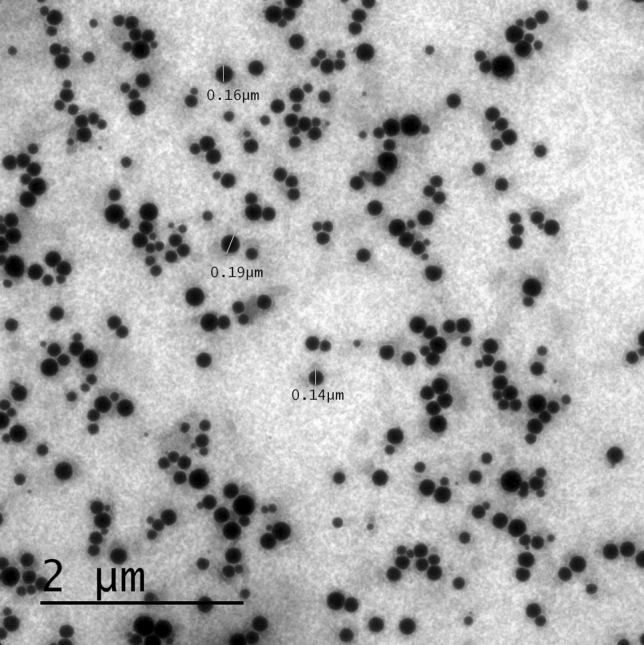
TEM image of SFB-loaded ME (ME16).
In-vitro drug release studies and release kinetics
The drug release studies of free SFB, ME16 and SFB-marketed formulation (Soranib tablet, Batch no. GJ10330) was carried out in 0.1 N HCl for 2 h, followed by pH 6.8 for the further time period46. The graph (Fig. 9) depicted that the drug release was pH-dependent, and ME16 exhibited a sustained release pattern. Free SFB showed more than 90% of drug release within 2 h in simulated acidic medium (0.1 N HCl). The release of SFB from the marketed product across the dialysis membrane was quite less as shown in the Fig. 9. On the other hand, the drug dissolution profile of the marketed product (Soranib tablet, Batch no. GJ10330) using the USP II apparatus following a USFDA approved protocol47 was well within the acceptable range (Supplementary data, Fig. S1). The reason for slow release by the marketed product might be the presence of dialysis membrane. The developed microemulsion system was able to sustain the drug release over 24 h, however, the drug dissolution of marketed product released the drug in 1 h. The results unequivocally vouch the sustained and controlled drug release behavior of the developed system. On fitting the values of % drug release versus time into the various release kinetic models, it was inferred that ME16 followed the Higuchi kinetic model. The r2 values of multiple models are depicted in Table 548.
Figure 9.

Graph of drug release study of free SFB, SFB-loaded ME (ME16) and SFB-marketed Formulation.
Table 5.
Parameters of various release kinetic models.
| Release kinetic models | Formulations | Parameters | ||
|---|---|---|---|---|
| Slope | Intercept | r2 | ||
| Zero order | Pure drug | 0.0296 | 18.769 | 0.4996 |
| SFB-loaded ME | 0.055 | 20.834 | 0.662 | |
| SFB-marketed formulation | 0.0124 | 4.9346 | 0.4992 | |
| First order | Pure drug | 0.007 | 1.067 | 0.256 |
| SFB-loaded ME | 0.0009 | 1.07 | 0.334 | |
| SFB-marketed formulation | 0.0007 | 0.5248 | 0.4308 | |
| Higuchi | Pure drug | 0.565 | 3.004 | 0.754 |
| SFB-loaded ME | 0.379 | 1.361 | 0.889 | |
| SFB-marketed formulation | 1.3215 | 0.8616 | 0.7364 | |
Protein binding studies
Protein binding studies in the equivalent doses of 10 mg/kg for the free SFB at a specific 1 mg/mL concentration is > 90%49. The study revealed that after loading onto the ME, the protein binding was substantially decreased (p < 0.05), as shown in Fig. 10. The protein binding of free SFB was more than the ME16. It stated that the developed formulation could provide better affinity of SFB to the target site as compared to the free SFB.
Figure 10.
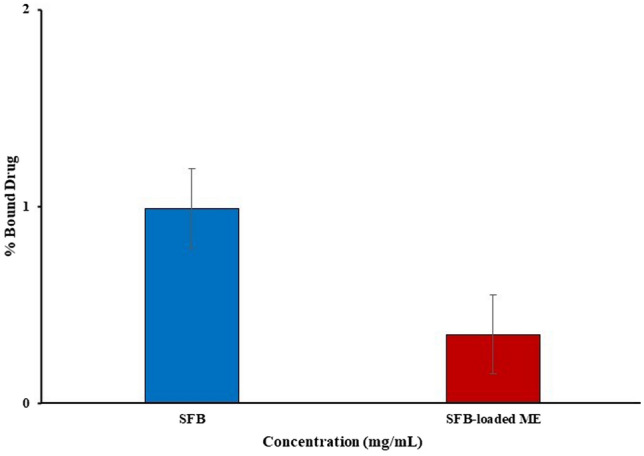
Graph of protein binding of free SFB and SFB-loaded ME (ME16).
In-vitro cytotoxicity assay
The cytotoxicity of SFB, blank ME, and SFB-loaded ME against 4T1 breast cancer cells were evaluated through microscopic observations and IC50 value determination. The microscopic images of the treated 4T1 cells were captured at 1 μM concentration, considering DMSO as control (Fig. 11a)50. Through these microscopic observations, blank ME exhibited no cytotoxic effect on 4T1 breast cancer cells. A significant decrease in cell viability was observed in the case of SFB-loaded ME than free SFB at 1 µM concentration. The cytotoxic effects of different samples are also represented as % cell viability versus concentration plot in the Fig. 11b). Though at lower concentration, the efficacy of both free SFB and SFB-loaded ME are comparable, but at the concentration above 1 µM, the difference is conspicuous (p < 0.05). The IC50 values of free SFB and SFB-loaded ME were calculated to be 3.47 ± 0.21 and 2.15 ± 0.11 µM, respectively. These outcomes confirmed the therapeutic efficacy of the drug loaded ME. Considering the IC50 values, the lethal dose 50 (LD50) values of free SFB and SFB-loaded ME were calculated to be 8.99 and 7.52 µM, respectively. The large therapeutic window provided by SFB-loaded ME supported the safety profile of the formulation. The findings are of greater significance as the dose of the drug for human is 200 mg and the plasma drug concentration for 100% bioavailability (theoretically) comes out to be 350 ng/mL. It implies that 0.136% of the centrally available drug concentration has the potential to kill the cancer cells, when loaded in the developed ME.
Figure 11.

Cytotoxicity evaluation of SFB-loaded ME concerning free SFB determined via (a) microscopic evaluation and (b) MTT based cell viability assay.
In-vitro cellular uptake study
The cellular uptake study was performed and analysed qualitatively and quantitatively on 4T1 breast cancer cells. For the qualitative determination of cellular uptake, coumarin-6-ME16 and free coumarin was observed using a fluorescence microscope (Zeiss, Germany). As shown in Fig. 12, the results revealed higher cellular internalization in case of coumarin-6-ME16 in overlay images. On the other hand, free coumarin did not exhibit significant cellular uptake through microscopic observation. To further confirm the qualitative results, the uptake was quantified using flowcytometry (Cytoflex, Beckman Coulter, USA). The amount of coumarin-6 internalised by the 4T1 cells was assumed to impart the fluorescence intensity. The findings obtained from CytExpert software showed that 9.79% of free coumarin was internalized by the cells, whereas the uptake was 88.96% in the case of coumarin-6-ME16. Qualitative and quantitative analysis confirmed higher 4T1 cell uptake in the case of coumarin-6-ME16, which also supported the cytotoxicity results.
Figure 12.

Cellular uptake study of free coumarin and coumarin-6-ME16.
In-vivo pharmacokinetic studies
The free SFB and SFB-loaded ME was evaluated for further in-vivo pharmacokinetic studies. The study’s graph was plotted between the plasma drug concentration versus time, as shown in Fig. 13. The graph estimated that the rats of group II (ME16) exhibited a higher concentration of plasma at each time point than of the group I receiving free SFB (p < 0.05). The pharmacokinetic parameters were determined by fitting the plasma concentration–time profile in PK solver software. A comparison of various pharmacokinetic parameters has been described in Table 6. The mean residence time (MRT) value of ME16 (21.82 h) was found to be significantly higher than the free SFB (16.49 h; p < 0.05). This result revealed a reduction in the clearance rate of the drug. The clearance rate (CL) of the free drug was found to be 0.09 μg/ml compared to the SFB-loaded ME (0.05 μg/ml). The high entrapment of SFB into the ME could be justified because of the predicted low level of volume of distribution of ME16 (1.08 μg/ml). It is evident from the value of pharmacokinetic parameters that fabrication of SFB in the form of ME resulted in the formulation with a sustained effect. The Tmax was almost safe. However, the bioavailability of SFB was 1.5 times increased by the ME system. The findings provide hope for a formulation with the potential to enhance the biological stay period of the drug and increase the bioavailability fraction. It has immense promises in better efficacy and dose reduction.
Figure 13.
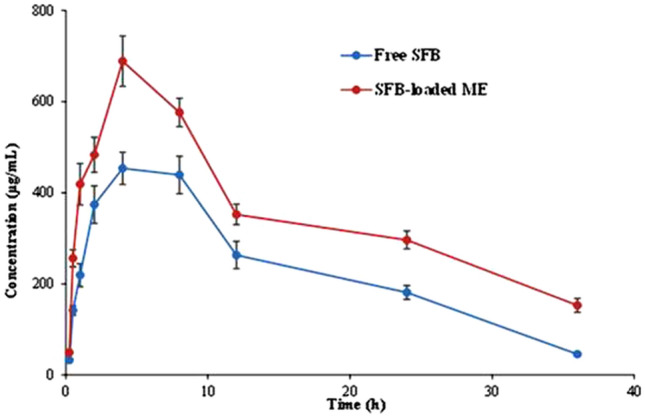
Plasma-concentration time graph of Free SFB and SFB-loaded ME (ME16).
Table 6.
Various pharmacokinetic parameters of ME16 and free SFB.
| Parameters | Unit | SFB-loaded ME | Free SFB |
|---|---|---|---|
| V/F | (mg)/(μg/ml) | 1.08 | 1.30 |
| CL/F | (mg)/(μg/ml)/h | 0.05 | 0.09 |
| Tmax | h | 4.02 | 4.70 |
| Cmax | μg/ml | 625.31 | 456.47 |
| AUC 0-t | μg/ml h | 12,686.98 | 8238.03 |
| AUC 0-inf | μg/ml h | 15,550.59 | 9113.58 |
| MRT | h | 21.82 | 16.49 |
Stability studies
The stability studies of the developed ME16 were carried out for 6 months51; It was observed that 30 ± 2 °C was the most favourable storage conditions for the SFB-loaded ME as no significant changes in particle size, zeta potential and drug loading had been found in this temperature (Table 7).
Table 7.
Stability studies of SFB-loaded ME (ME16) at different temperature conditions.
| Parameters | Particle size (nm) | Zeta potential (mV) | Drug loading (%) |
|---|---|---|---|
| Months | 4 ± 2 °C | ||
| 0 | 55.2 ± 0.13 | − 0.03 ± 0.09 | 22.07 ± 1.18 |
| 3rd | 65.8 ± 0.04 | − 0.12 ± 0.31 | 21.41 ± 0.32 |
| 6th | 56.2 ± 0.02 | − 0.21 ± 0.37 | 23.07 ± 1.8 |
| 30 ± 2 °C | |||
|---|---|---|---|
| 0 | 54.2 ± 0.04 | 0.05 ± 0.08 | 21.07 ± 2.16 |
| 3rd | 56.3 ± 0.06 | 0.06 ± 0.02 | 21.04 ± 1.86 |
| 6th | 56.8 ± 0.02 | 0.05 ± 0.05 | 21.14 + 0.96 |
| 45 ± 2 °C | |||
|---|---|---|---|
| 0 | 55.8 ± 1.02 | 0.06 ± 0.04 | 21.12 ± 2.01 |
| 3rd | 57.1 ± 1.89 | 0.08 ± 1.3 | 19.02 ± 0.04 |
| 6th | 59.07 ± 0.34 | 0.10 ± 0.35 | 19.32 ± 1.87 |
Conclusion
The challenges of SFB related to oral delivery are limiting its efficacy towards breast cancer. The present reports, i.e., in-vitro and in-vivo studies, manifested a new insight towards the SFB oral administration by developing SFB-loaded ME. The developed formulation enhanced the oral bioavailability of the SFB and increased its t1/2 which could result in a once-in-a-day product. The overall performance and cytotoxicity of the SFB to the breast cancer cells were improved after encapsulated into the ME as depicted in cell viability studies. The same can be further investigated in the preclinical studies for confirmation and further exploration.
Supplementary Information
Acknowledgements
This support from the facilities created under DST-FIST grant (SR/FST/LSI-659/2016) to the Department of Pharmacy, Central University of Rajasthan, Ajmer, India.
Author contributions
N.C., C.M., T.S.: experimental work; R.J. and D.C.: cell line studies; Md.A.B., H.A.B.: organization and review; B.S. and K.R.: conceptualization, supervision, organization, analysis, drafting and proof reading.
Data availability
The data used to contribute the findings of this research are included within the article and supplementary file.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-022-17333-6.
References
- 1.Sung H, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Fahad Ullah M. Breast cancer: Current perspectives on the disease status. Adv. Exp. Med. Biol. 2019;1152:51–64. doi: 10.1007/978-3-030-20301-6_4. [DOI] [PubMed] [Google Scholar]
- 3.Salzman B, Collins E, Hersh L. Common breast problems. Am. Fam. Physician. 2019;99:505–514A. [PubMed] [Google Scholar]
- 4.O’Brien Z, Moghaddam MF. A systematic analysis of physicochemical and ADME properties of all small molecule kinase inhibitors approved by US FDA from January 2001 to October 2015. Curr. Med. Chem. 2017;24:3159–3184. doi: 10.2174/0929867324666170523124441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abdelgalil AA, Alkahtani HM, Al-Jenoobi FI. Sorafenib. Profiles Drug Subst. Excipients Relat. Methodol. 2019;44:239–266. doi: 10.1016/bs.podrm.2018.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Lang L. FDA approves sorafenib for patients with inoperable liver cancer. Gastroenterology. 2008;134:379. doi: 10.1053/j.gastro.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 7.Cheng AL, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- 8.Zafrakas M, Papasozomenou P, Emmanouilides C. Sorafenib in breast cancer treatment: A systematic review and overview of clinical trials. World J. Clin. Oncol. 2016;7:331–336. doi: 10.5306/wjco.v7.i4.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Zhang X, Liu F, Wang M, Qin S. The effects of triptolide on the pharmacokinetics of sorafenib in rats and its potential mechanism. Pharm. Biol. 2017;55:1863–1867. doi: 10.1080/13880209.2017.1340963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tahir N, et al. Microfluidic fabrication and characterization of Sorafenib-loaded lipid-polymer hybrid nanoparticles for controlled drug delivery. Int. J. Pharm. 2020;581:119275. doi: 10.1016/j.ijpharm.2020.119275. [DOI] [PubMed] [Google Scholar]
- 11.Torrieri G, et al. LinTT1 peptide-functionalized liposomes for targeted breast cancer therapy. Int. J. Pharm. 2021;597:120346. doi: 10.1016/j.ijpharm.2021.120346. [DOI] [PubMed] [Google Scholar]
- 12.Sharma T, et al. Supersaturated LFCS type III self-emulsifying delivery systems of sorafenib tosylate with improved biopharmaceutical performance: QbD-enabled development and evaluation. Drug Deliv. Transl. Res. 2020;10:839–861. doi: 10.1007/s13346-020-00772-x. [DOI] [PubMed] [Google Scholar]
- 13.Correia A, et al. Cyclodextrin-modified porous silicon nanoparticles for efficient sustained drug delivery and proliferation inhibition of breast cancer cells. ACS Appl. Mater. Interfaces. 2015 doi: 10.1021/acsami.5b07033. [DOI] [PubMed] [Google Scholar]
- 14.Murphy EA, et al. Targeted nanogels: A versatile platform for drug delivery to tumors. Mol. Cancer Ther. 2011;10:972–982. doi: 10.1158/1535-7163.MCT-10-0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manuscript A. Surface bioengineering of diatomite based nanovectors for efficient intracellular uptake and drug delivery. Nanoscale. 2015 doi: 10.1039/C5NR05173H. [DOI] [PubMed] [Google Scholar]
- 16.Izadiyan Z, et al. Improvement of physicochemical properties of nanocolloidal carrier loaded with low water solubility drug for parenteral cancer treatment by Response Surface Methodology. Mater. Sci. Eng. C. 2018 doi: 10.1016/j.msec.2018.10.015. [DOI] [PubMed] [Google Scholar]
- 17.Manuscript A. Synergistic chemotherapeutic effect of sorafenib-loaded pullulan-Dox conjugate nanoparticles against murine breast carcinoma. Nanoscale. 2017 doi: 10.1039/C6NR09639E. [DOI] [PubMed] [Google Scholar]
- 18.Ramalho ÍMDM, et al. Chrysin-loaded microemulsion: Formulation design, evaluation and antihyperalgesic activity in mice. Appl. Sci. 2022;12:477. doi: 10.3390/app12010477. [DOI] [Google Scholar]
- 19.Raza K, et al. Novel dithranol phospholipid microemulsion for topical application: Development, characterization and percutaneous absorption studies. J. Microencapsul. 2011;28:190–199. doi: 10.3109/02652048.2010.546435. [DOI] [PubMed] [Google Scholar]
- 20.McClements DJ. Nanoemulsions versus microemulsions: Terminology, differences, and similarities. Soft Matter. 2012;8:1719–1729. doi: 10.1039/C2SM06903B. [DOI] [Google Scholar]
- 21.Tiwari P, Ranjan Sinha V, Kaur R. Clinical considerations on micro- and nanodrug delivery systems. Drug Deliv. Trends. 2020 doi: 10.1016/b978-0-12-817870-6.00004-3. [DOI] [Google Scholar]
- 22.Chen Y, et al. A Tf-modified tripterine-loaded coix seed oil microemulsion enhances anti-cervical cancer treatment. Int. J. Nanomedicine. 2018;13:7275–7287. doi: 10.2147/IJN.S182475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qu D, et al. Non-triggered sequential-release liposomes enhance anti-breast cancer efficacy of STS and celastrol-based microemulsion. Biomater. Sci. 2018;6:3284–3299. doi: 10.1039/C8BM00796A. [DOI] [PubMed] [Google Scholar]
- 24.Salata GC, et al. Microemulsion for prolonged release of fenretinide in the mammary tissue and prevention of breast cancer development. Mol. Pharm. 2021;18:3401–3417. doi: 10.1021/acs.molpharmaceut.1c00319. [DOI] [PubMed] [Google Scholar]
- 25.Tartaro G, Mateos H, Schirone D, Angelico R, Palazzo G. Microemulsion microstructure(s): A tutorial review. Nanomaterials. 2020;10:1–40. doi: 10.3390/nano10091657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taka K, Avdeel A. Interlaboratory study of log P determination by shake-flask and potentiometric methods. J. Pharm. Biomed. Anal. 1996;14:1405–1413. doi: 10.1016/0731-7085(96)01773-6. [DOI] [PubMed] [Google Scholar]
- 27.Izadiyan Z, et al. Modeling and optimization of nanoemulsion containing Sorafenib for cancer treatment by response surface methodology. Chem. Cent. J. 2017;11:1–9. doi: 10.1186/s13065-017-0248-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thakur CK, et al. Chitosan-modified PLGA polymeric nanocarriers with better delivery potential for tamoxifen. Int. J. Biol. Macromol. 2016;93:381–389. doi: 10.1016/j.ijbiomac.2016.08.080. [DOI] [PubMed] [Google Scholar]
- 29.Raza K, et al. Nano-lipoidal carriers of tretinoin with enhanced percutaneous absorption, photostability, biocompatibility and anti-psoriatic activity. Int. J. Pharm. 2013;456:65–72. doi: 10.1016/j.ijpharm.2013.08.019. [DOI] [PubMed] [Google Scholar]
- 30.Tunki L, et al. Modulating the site-specific oral delivery of sorafenib using sugar-grafted nanoparticles for hepatocellular carcinoma treatment. Eur. J. Pharm. Sci. 2019;137:104978. doi: 10.1016/j.ejps.2019.104978. [DOI] [PubMed] [Google Scholar]
- 31.Shah N, et al. Oral bioavailability enhancement of raloxifene by developing microemulsion using D-optimal mixture design: Optimization and in-vivo pharmacokinetic study. Drug Dev. Ind. Pharm. 2018;44:687–696. doi: 10.1080/03639045.2017.1408643. [DOI] [PubMed] [Google Scholar]
- 32.Dash S, Murthy PN, Nath L, Chowdhury P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. Drug Res. 2010;67:217–223. [PubMed] [Google Scholar]
- 33.Kroll M. Tietz textbook of clinical chemistry, Third Edition. Carl A. Burtis and Edward R. Ashwood, eds. Philadelphia, PA: WB Saunders, 1998, 1917 pp., $195.00. ISBN 0-7216-5610-2. Clin. Chem. 1999;45:913–914. doi: 10.1093/clinchem/45.6.913. [DOI] [Google Scholar]
- 34.Henry’s Clinical Diagnosis and Management by Laboratory Methods—23rd Edition. https://www.elsevier.com/books/henrys-clinical-diagnosis-and-management-by-laboratory-methods/mcpherson/978-0-323-41315-2.
- 35.Agarwal DS, Mazumdar S, Italiya KS, Chitkara D, Sakhuja R. Bile-acid-appended triazolyl aryl ketones: Design, synthesis, in vitro anticancer activity and pharmacokinetics in rats. Molecules. 2021;26:5741. doi: 10.3390/molecules26195741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mazumdar S, Italiya KS, Sharma S, Chitkara D, Mittal A. Effective cellular internalization, cell cycle arrest and improved pharmacokinetics of Tamoxifen by cholesterol based lipopolymeric nanoparticles. Int. J. Pharm. 2018;543:96–106. doi: 10.1016/j.ijpharm.2018.03.022. [DOI] [PubMed] [Google Scholar]
- 37.Şengel-Türk CT, et al. Preparation and in vitro evaluation of meloxicam-loaded PLGA nanoparticles on HT-29 human colon adenocarcinoma cells. Drug Dev. Ind. Pharm. 2012;38:1107–1116. doi: 10.3109/03639045.2011.641562. [DOI] [PubMed] [Google Scholar]
- 38.Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J. basic Clin. Pharm. 2016;7:27. doi: 10.4103/0976-0105.177703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharma T, Khurana RK, Jain A, Katare OP, Singh B. Development of a validated liquid chromatographic method for quantification of sorafenib tosylate in the presence of stress-induced degradation products and in biological matrix employing analytical quality by design approach. Biomed. Chromatogr. 2018;32:e4169. doi: 10.1002/bmc.4169. [DOI] [PubMed] [Google Scholar]
- 40.Jain A, et al. Novel cationic supersaturable nanomicellar systems of raloxifene hydrochloride with enhanced biopharmaceutical attributes. Drug Deliv. Transl. Res. 2018;8:670–692. doi: 10.1007/s13346-018-0514-8. [DOI] [PubMed] [Google Scholar]
- 41.Raza K, Katare OP, Setia A, Bhatia A, Singh B. Improved therapeutic performance of dithranol against psoriasis employing systematically optimized nanoemulsomes. J. Microencapsul. 2013;30:225–236. doi: 10.3109/02652048.2012.717115. [DOI] [PubMed] [Google Scholar]
- 42.Tiernan H, Byrne B, Kazarian SG. ATR-FTIR spectroscopy and spectroscopic imaging for the analysis of biopharmaceuticals. Spectrochim. Acta. A. Mol. Biomol. Spectrosc. 2020;241:118636. doi: 10.1016/j.saa.2020.118636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawakami K, et al. Microemulsion formulation for enhanced absorption of poorly soluble drugs. I. Prescription design. J. Control. Release. 2002;81:65–74. doi: 10.1016/S0168-3659(02)00049-4. [DOI] [PubMed] [Google Scholar]
- 44.Xi J, et al. Formulation development and bioavailability evaluation of a self-nanoemulsified drug delivery system of oleanolic acid. AAPS PharmSciTech. 2009;10:172. doi: 10.1208/s12249-009-9190-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Negi P, et al. Phospholipid microemulsion-based hydrogel for enhanced topical delivery of lidocaine and prilocaine: QbD-based development and evaluation. Drug Deliv. 2016;23:951–967. doi: 10.3109/10717544.2014.923067. [DOI] [PubMed] [Google Scholar]
- 46.Subongkot T. Development and mechanistic study of a microemulsion containing vitamin E TPGS for the enhancement of oral absorption of celecoxib. Int. J. Nanomedicine. 2019;14:3087–3102. doi: 10.2147/IJN.S201449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dissolution Methods. https://www.accessdata.fda.gov/scripts/cder/dissolution/dsp_SearchResults.cfm.
- 48.Kumar P, et al. Stearic acid based, systematically designed oral lipid nanoparticles for enhanced brain delivery of dimethyl fumarate. Nanomedicine. 2017;12:2607–2621. doi: 10.2217/nnm-2017-0082. [DOI] [PubMed] [Google Scholar]
- 49.Shi JH, Chen J, Wang J, Zhu YY, Wang Q. Binding interaction of sorafenib with bovine serum albumin: Spectroscopic methodologies and molecular docking. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015;149:630–637. doi: 10.1016/j.saa.2015.04.034. [DOI] [PubMed] [Google Scholar]
- 50.Sharma S, et al. Cholesterol and morpholine grafted cationic amphiphilic copolymers for miRNA-34a delivery. Mol. Pharm. 2018;15:2391–2402. doi: 10.1021/acs.molpharmaceut.8b00228. [DOI] [PubMed] [Google Scholar]
- 51.Das S, et al. Development of microemulsion based topical ivermectin formulations: Pre-formulation and formulation studies. Colloids Surf. B. Biointerfaces. 2020;189:110823. doi: 10.1016/j.colsurfb.2020.110823. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used to contribute the findings of this research are included within the article and supplementary file.