

Abstract
Heart failure with preserved ejection fraction (HFpEF) remains a medical anomaly that baffles researchers and physicians alike. The overall phenotypical changes of diastolic function and left ventricular hypertrophy observed in HFpEF are definable; however, the metabolic and molecular alterations that ultimately produce these changes are not well established. Comorbidities such as obesity, hypertension, and diabetes, as well as general aging, play crucial roles in its development and progression. Various animal models have recently been developed to better understand the pathophysiological and metabolic developments in HFpEF and to illuminate novel avenues for pharmacotherapy. These models include multi‐hit rodents and feline aortic constriction animals. Recently, genomic, proteomic, and metabolomic approaches have been used to define altered signaling pathways in the heart associated with HFpEF, including those involved in inflammation, cGMP‐related, Ca2+ handling, mitochondrial respiration, and the unfolded protein response in endoplasmic reticulum stress. This article aims to present an overview of what has been learnt by these studies, focusing mainly on the findings in common while highlighting unresolved issues. The knowledge gained from these research models will not simply be of benefit for treating HFpEF but will undoubtedly provide new insights into the mechanisms by which the heart deals with external stresses and how the processes involved can fail.
Keywords: cardiac remodeling, diastolic dysfunction, endoplasmic reticulum stress, metabolomics, preclinical model, proteomics, transcriptomics
Subject Categories: Heart Failure, Hypertrophy, Hypertension, Genetically Altered and Transgenic Models, Animal Models of Human Disease
Nonstandard Abbreviations and Acronyms
- AIU
aldosterone‐infused uninephrectomy
- Fstl1
follistatin like 1
- SAHA
suberoylanilide hydroxamic acid
- UPR
unfolded protein response
…And you know something's happening but you do not know what it is…Do you, Mr. Jones.
Ballad of a Thin Man, by Bob Dylan.
Cardiovascular disease remains one of the cardinal areas of health care research and plagues humankind worldwide (https://www.who.int/news‐room/fact‐sheets/detail/cardiovascular‐diseases‐(cvds)). Of the various types of cardiovascular disease, heart failure accounts for roughly 26 million cases of disease burden. Heart failure can be broadly divided into 2 categories: heart failure with reduced ejection fraction (HFrEF) that is governed by systolic dysfunction and ventricular dilation with ejection fractions <40%, and heart failure with preserved ejection fraction (HFpEF) that is governed by diastolic dysfunction and ventricular hypertrophy with normal ejection fractions of >55%. 1 , 2 HFrEF is typically the result of direct insult to the myocardial tissue by factors such as myocardial infarction and volume overload. In contrast, HFpEF is initiated by a variety of metabolic altering factors such as hypertension, diabetes, obesity, and aging. With the population on average becoming progressively older, as well as the increased prevalence of common comorbidities, such as obesity and diabetes, it is no surprise that HFpEF is a chief concern in the medical and research communities. 3
Over the course of medical advancement, HFrEF has been successfully managed by numerous neurohormonal interventions, such as β‐adrenergic receptor blockers, angiotensin‐converting enzyme inhibitors, angiotensin II type 1 receptor blockers, and mineralocorticoid receptor agonists. 4 However, there are currently no cause‐based treatments to reduce morbidity and mortality of HFpEF, although a sodium‐glucose cotransporter 2 (SGLT2) inhibitor was recently demonstrated to be effective regardless of the presence or absence of diabetes. 5 In addition, certain patients (females or those in the lower strata for “preserved” ejection fraction, ie, <≈60%) may benefit from treatment with the angiotensin receptor–neprilysin inhibitor sacubitril–valsartan, particularly for heart failure hospitalization. 6
Even though the overall phenotypical changes involving HFpEF are generally well characterized, the underlying genomic, proteomic, and molecular alterations that ultimately lead to HFpEF are rather undetermined. HFpEF has become an object of various preclinical, experimental rodent models in attempts to understand its complex cause. These experimental models use various approaches, including models that are “multi‐hit”, diet‐induced, physiological age‐induced, or involve altered gene expression. Recently, an experimental feline model has been described to understand the molecular pathogenesis of HFpEF better. Besides determining the underlying physiological mechanisms and pathological progression of HFpEF, these models could provide insight into whether HFpEF is one collective pathology or whether there are distinct biological “phenogroups” as recently suggested. 7
Our review aims to analyze the currently accepted HFpEF rodent and feline models and compare their similarities and differences (Figure 1). All 5 of the models discussed are reported to exhibit left ventricular (LV) concentric hypertrophy, diastolic dysfunction with preserved systolic function, pulmonary congestion, and (with the possible exception of the aldosterone‐infused uninephrectomy [AIU] mouse and feline aortic constriction model) exercise intolerance. With the possible exception of the AIU mouse model, diastolic dysfunction has been assessed by both echocardiography and invasive hemodynamic pressure–volume measurements. 8 , 9 , 10 , 11 , 12 Diastolic dysfunction has been attributed to increased fibrosis and/or increased stiffness of the sarcomere structural protein titin, impaired calcium handling, or mitochondrial dysfunction. The molecular basis for these alterations is not firmly established. Still, it is postulated to include reduced NO/cGMP/PKG signaling (which is antifibrotic and antihypertrophic) because of increased oxidative stress (in part from inducible NOS [iNOS], aka NOS2) and/or endoplasmic reticulum (ER) stress. A comprehensive overview of the various models could provide insight into the next era of pharmacological techniques in the battle against HFpEF and determine if understanding this syndrome could provide insight into countering ordinary “wear and tear” on the heart.
Figure 1. Comparisons among heart failure with preserved ejection fraction models for mechanisms linked to diastolic dysfunction.
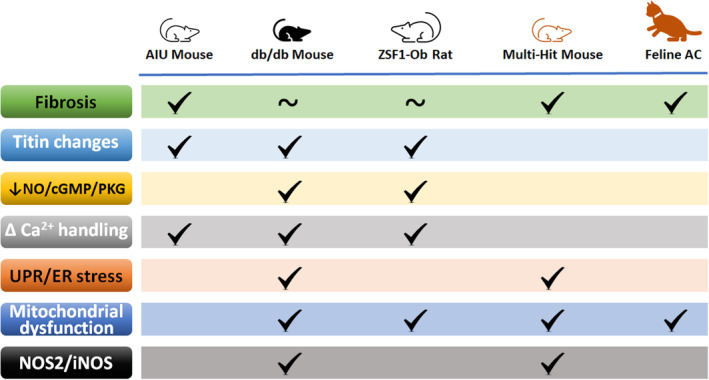
Prominent among these are increased fibrosis and altered titin isoform expression and/or posttranslational modifications. Reduced NO/cGMP/PKG signaling, which has antifibrotic and antihypertrophic actions and also affects titin phosphorylation, may be linked to enhanced NOS2/iNOS expression. The latter may affect Ca2+ handling, which is linked as well to unfolded protein response/endoplasmic reticulum stress and mitochondrial dysfunction. ~ indicates not prominent/involved or conflicting reports. AC indicates aortic constriction; AIU, aldosterone‐infused uninephrectomy; cGMP, cyclic guanosine monophosphate; ER, endoplasmic reticulum; iNOS, inducible nitric oxide synthase; IRE1α, inositol requiring transmembrane kinase endoribonuclease 1α; NO, nitric oxide; NOS2, nitric oxide synthase 2; PKG, protein kinase G; and UPR, unfolded protein response.
CLINICAL ASPECTS
More than half of patients with heart failure have a normal ejection fraction. This heart failure subtype disproportionately affects women and the elderly and commonly is associated with other cardiovascular comorbidities, such as hypertension, obesity, and diabetes. 13 In Europe, almost 5% of those aged ≥60 years, representing several million, were identified to have HFpEF. 14 In the United States, the prevalence of HFpEF is estimated to be 2.4 to 3.4 million. 15 The number of people with HFpEF will increase further as people live longer and obesity and diabetes become more common. HFpEF already accounts for more than half the heart failure hospitalities. 16 Unlike for HFrEF, cardiovascular and noncardiovascular mortality for HFpEF remained unchanged over the decades 2005 to 2014. 15 A recent study indicated that 1‐year survival for HFpEF after hospital reviles that of HFrEF. 17 An older study showed that mortality and morbidity for HFpEF is high with 5‐year‐survival of only 35% to 40% after hospitalization for HF. 18 As of today, HFpEF remains a major unmet clinical need. 16 , 19
Diagnosing HFpEF can be challenging to a physician, particularly at early stages that may be characterized by only dyspnea in patients who are euvolemic. Two multiparametric diagnostic algorithms have been devised to address this issue, as well as to define the stage of HFpEF in terms of severity. The H2FPEF score uses 4 clinical variables (age, body mass index, use of ≥2 antihypertensive medicines, and atrial fibrillation) and 2 echocardiographic variables (ratio of the early diastolic mitral inflow velocity to early diastolic mitral annular tissue velocity [E/e′] and pulmonary artery systolic pressure). 20 The HFA‐PEFF score is more complicated and evaluates natriuretic peptide levels and echocardiographic findings of cardiac function and structure. 16 Both appear to have diagnostic and prognostic value, 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 although H2FPEF may be better at predicting exercise intolerance. 32 Recently, the HFA‐PEFF score was used to identify 4 distinct phenogroups in “early‐HFpEF” patients of increasing severity: (1) no LV diastolic dysfunction, (2) LV diastolic dysfunction with functional LV abnormalities; (3) LV diastolic dysfunction with functional and structural LV abnormalities, and (4) LV diastolic dysfunction with functional and structural LV abnormalities and elevated B‐type natriuretic peptide. 33 The 4 groups were further distinguished by increasing circulating levels with severity of biomarkers mainly involved in inflammation and extracellular matrix remodeling.
HUMAN GENE, PROTEOMIC, AND METABOLOMIC PROFILES
RNA‐seq analysis of human endomyocardial biopsies showed that patients with HFpEF exhibit a gene expression profile distinct from normal controls and those with HFrEF. 34 Pathway analysis highlighted processes that distinguished HFpEF from HFrEF. Notably, these differences did not involve processes typically associated with HFpEF, such as hypertrophy, fibrosis, inflammation, and oxidant stress. Pathways more unique to HFpEF encompass ER stress, protein hemostasis, and angiogenesis. Compared with those with HFrEF, patients with HFpEF had enhanced expression of genes associated with oxidative phosphorylation, but lower expression of autophagy, fibrosis, hypertrophy‐related, ER (processing and stress), angiogenesis, and cGMP‐related genes. The increase in genes associated with oxidative phosphorylation was attributed to a normal aspect of obesity. Further analysis revealed that patients with HFpEF fell into 3 transcriptomic subgroups with distinct pathways and clinical correlates: (1) one of higher mortality closest to HFrEF, (2) a primarily female group with smaller hearts and proinflammatory signaling, and (3) a heterogeneous phenotype with worse heart failure symptoms, but lower NT‐proBNP (N‐terminal proB‐type natriuretic peptide) and smaller hearts also.
Circulating metabolites and lipids may provide etiological and diagnostic utility to HFpEF. Analysis of metabolites from participants of the Framingham Heart Study and Women's Health Initiative revealed an association of HFpEF with sleep apnea because of the proteinogenic amino acid glycine and ribose, which can feed into the pentose phosphate pathway. 35 After multivariable adjustment for age, sex, and body mass index, 11 metabolites were associated with incident HFpEF. The top associated metabolite was ornithine, likely from perturbed arginine and NO metabolism. The competitive inhibitor of NOS and marker of abnormal NO‐mediated vascular tone, NG‐monomethyl‐l‐arginine was also significantly associated with incident HFpEF. The second most associated metabolite with incident HFpEF was cardiac energy substrate glycerol, followed by the cardiovascular disease mortality risk marker, dimethylglycine. Both the proteinogenic asparagine and pro‐hypertrophic metabolite 2‐hydroxyglutarate partly mediated the association of LV wall thickness with HFpEF.
A smaller exploratory study analyzed plasma metabolites of patients with new onset HFpEF to those with new onset HFrEF. 36 Overall, the metabolomic profile of those with HFpEF was consistent with impaired lipid metabolism, enhanced inflammation and oxidative stress, increased collagen synthesis, and downregulated NO signaling. Compared with HFrEF, HFpEF was associated with increased symmetric dimethyl arginine, hydroxyproline, cysteine, alanine, and kynurenine, and decreased cGMP, cAMP, l‐carnitine, lysophosphatidylcholine (18:2), serine, lactate, and arginine. In another study, accumulation of epicardial adipose tissue was associated with worse hemodynamic and metabolic profile, as well as survival, in patients with HFpEF. 37
Proteome analysis of autopsied LV myocardium of patient with HFpEF, at the early stage of LV diastolic dysfunction and without major comorbidities except for hypertension, revealed 57 differentially expressed proteins. 38 Molecular network analysis indicated the importance of ER stress. Notably, the expression of proteins associated with the ER stress response was impaired. Obesity in HFpEF may represent a distinct phenotype. Compared with non‐obese patients with HFpEF or obese patients without HFpEF, obese patients with HFpEF showed higher circulating levels of biomarkers consistent with volume expansion, myocardial fibrosis, and systemic inflammation. 39
While eloquent and insightful, these studies have limitations such as not discerning cause and effect, nor addressing the issues of compensation, role of posttranslational modifications of protein, or functional assessments at the cellular or subcellular level. Although applying newer sequencing technology and updated proteomics might eventually be a robust means of improving care for patients with HFpEF, these limitations support the need for preclinical models of HFpEF.
LARGE ANIMAL MODELS OF HFpEF
Notable differences in energetics, excitation, and contractility exist between hearts of humans and small rodents, although there are remarkable similarities as well. 40 , 41 , 42 , 43 Moreover, small animals in research offer the possibility of ready genetic manipulations, efficacy, convenience, a narrower time frame, and lower costs. Nonetheless, a recent working group of the National Heart, Lung, and Blood Institute on HFpEF identified the need for “improved animal models, including large animal models, which incorporate the effects of aging and associated comorbid conditions.” 44 Several large animal models of HFpEF have been introduced of late. These include the Göttingen minipig administered a Western diet and subjected to deoxycorticosterone acetate‐salt pressure/volume stress, 45 aortic banding, or progressive aortic constriction in dogs and pigs, 46 , 47 and the Yorkshire×landrace swine with induction of diabetes (streptozotocin), hypercholesterolemia (high‐fat and high‐sugar diet with increasing salt), and chronic kidney disease (microembolization of the global right kidney). 48 As discussed here, small animal models are arguably perhaps better suited for an “omics” approach, involving genomics, proteomics, and metabolomics, to profile the changes in the heart occurring with HFpEF in a temporal and comorbidity‐dependent manner.
CONVENTIONAL PRECLINICAL MODELS
Valero‐Munoz and colleagues identified 3 small rodent models that fulfill critical determinants of HFpEF, namely left ventricular concentric hypertrophy, diastolic dysfunction with preserved systolic function, pulmonary congestion, and exercise intolerance. 49 Diastolic dysfunction has been assessed using Doppler echocardiography and tissue Doppler imaging by changes in the ratio between early mitral inflow velocity and mitral annular early diastolic velocity (E/e′), and the ratio between early (E) and late (atrial‐A) ventricular filling velocity (E/A). 50 , 51 , 52 , 53 , 54 The models are the AIU mouse, the db/db mouse, and the ZSF1 rat. Alterations in signaling events, particularly with regard to inflammation, have been reported for the first 2 models.
AIU Mouse
Strong evidence has demonstrated that the combination of aldosterone infusion, 1% NaCl intake, and uninephrectomy in mice/rats for 4 weeks recapitulates many characteristics of HFpEF in humans. These animals develop elevated blood pressure, LV hypertrophy, cardiac fibrosis, echocardiographic evidence of diastolic dysfunction with preserved systolic function, characterized by increased E/A ratio, pulmonary congestion, and enhanced proinflammatory response. 55 , 56 , 57 , 58 , 59 , 60 , 61 There is also evidence for impaired Ca2+ handling in the heart 55 , 59 and increased expression of the stiffer isoform of titin. 61
LV hypertrophy is a common morphological alteration associated with HFpEF. A broad range of molecular pathways is thought to be involved in the progression and development of LV hypertrophy and, consequently HFpEF. For instance, it has been reported that LV tissues from AIU mice exhibit increased atrial natriuretic peptide and titin transcript variant n2ba and n2b mRNA expression levels, both of which are involved in exacerbated LV hypertrophy and aggravated diastolic stiffness. 51 , 61 Similarly, adult rat ventricular cardiomyocytes treated with aldosterone showed a significant increase in atrial natriuretic peptide mRNA expression levels. 62 Additionally, an increase in endothelin‐1 and monocyte enhancer factor 2a, mediators of cardiomyocyte hypertrophy, were observed in an in vivo and in vitro model of AIU‐induced HFpEF. 61 As a compensatory mechanism, Tanaka and coworkers observed increased levels of Fstl1 (follistatin like 1), an extracellular glycoprotein, in plasma of patients with HFpEF, AUI mice, and adult rat ventricular cardiomyocytes stimulated with aldosterone. 62 It has been shown that elevated Fstl1 levels in the setting of cardiac injury is associated with decreased apoptosis and hypertrophy, which could be mediated by the activation of AMP‐activated protein kinase pathways. 63 , 64 Fst1l has also been described as an anti‐inflammatory marker following myocardial and vascular injury 64 , 65 ; however, this role was not addressed in the study by Tanaka and colleagues. 62
Increased cardiomyocyte cross‐sectional area is usually accompanied by an accumulation of fibrosis, resulting in exacerbated LV diastolic stiffness and dysfunction. 66 In that regard, a study performed by LeBrasseur et al, indicated that AIU‐induced HFpEF increased matrix metalloproteinase‐2/tissue inhibitor of metalloproteinases 2 ratio. 58 Elevated matrix metalloproteinase‐2/tissue inhibitor of metalloproteinases 2 ratio is tightly linked to enhanced fibrosis. 58 , 67 , 68 , 69 Additionally, aldosterone administration substantially increased collagen I and III mRNA expression and protein levels, suggesting enhanced cardiac fibrosis, consequently worsening diastolic stiffness. 57 Furthermore, Valero‐Munoz et al demonstrated elevated transforming growth factor‐beta mRNA expression levels, a well‐known mediator of cardiac fibrosis and remodeling, in the left ventricle of AIU mice. 61
Increased oxidative stress and inflammation are major contributors to LV hypertrophy and cardiac fibrosis. 70 , 71 A study by Tanaka et al demonstrated increased myocardial 3‐nitrotyrosine production, a marker of oxidative stress, in the left ventricle of AIU mice. 55 Other investigators showed macrophage infiltration in the LV interstitial area, along with elevated LV tumor necrosis factor‐alpha, interferon‐γ, and IL‐6 (interleukin‐6) mRNA expression levels in AIU‐induced HFpEF. 51 Of note, IL‐6 is not only involved in the enhanced proinflammatory response, but also exacerbated cardiomyocyte hypertrophy. 72 Interestingly, another preclinical study observed that interferon‐γ decreases LV hypertrophy in aldosterone induced‐HFpEF, challenging, therefore, the notion that proinflammatory cytokines are involved only in adverse effects in the setting of HFpEF. 57 In addition, plasma soluble vascular cell adhesion molecule levels have been observed to be significantly increased in AIU mice. 51 It has been reported that elevated soluble vascular cell adhesion molecule level is associated with increased endogenous NO synthase (NOS) inhibitor, exerting consequently deleterious effects on the vasculature. 73 Diastolic intracellular calcium handling plays a crucial role in LV relaxation. 71 , 74 Aldosterone‐induced HFpEF has been associated with decreased PKA (protein kinase A)‐mediated phosphorylation of phospholamban at Ser16, and decreased Ca2+/CaMKII (calmodulin‐dependent protein kinase II)‐dependent phospholamban phosphorylation at Thr17, resulting in depressed sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2a (SERCA2a) activity (protein levels are also decreased), a major regulator of Ca2+ concentration during the cardiac cycle, worsening thereafter diastolic dysfunction. 55
The AIU mouse model is not well characterized as far as sex differences from the perspective of cardiac changes and HFpEF. The model is focused mainly on hypertension and kidney disease. In addition, it involves severe surgical manipulation that is not trivial.
db/db Mouse Model
The db/db mouse model is used to embody the phenotype of obesity, insulin resistance, and type 2 diabetes conferred by mutational inactivation of the leptin receptor. This well‐characterized model exhibits an early systemic inflammatory signature, develops cardiac hypertrophy and diastolic dysfunction with age, and capillary rarefaction in the heart. 75 Both male and female db/db mice develop hypertrophic ventricular remodeling with diastolic dysfunction. 52 Females exhibit a more significant increase in left ventricular mass, likely because of a rise in both systolic and diastolic blood pressure in contrast to males. Hearts exhibit increased interstitial and perivascular fibrosis in both sexes, although there are variable reports on its magnitude. 76 , 77 , 78 Other cardiac features include altered titin isoform expression and/or modifications, 76 , 79 , 80 impaired NO/cGMP/PKG signaling 78 , 79 , 80 and Ca2+ handling, 76 , 78 increased ER stress, 81 NOS2 expression, 82 and mitochondrial dysfunction. 83 , 84 , 85 Transcriptomic, metabolic, epigenetic, and posttranslational modifications in the db/db model are emerging as fundamentals contributing to the development and progression of diabetes.
The cause of obesity and diabetes resides primarily in the metabolic and energetic perturbations that significantly overlap with those observed in HFpEF, involving impaired fuel utilization and induced conditions of substrate toxicity. Unlike HFrEF, the decrease in glucose aerobic oxidation in HFpEF is accompanied by an increase in fatty acid (FA) oxidation. 86 However, cardiac impairment seems to be associated with the dissonance between the supply of FAs and their oxidation, which reflects on the pattern of cardiac transcriptomic profile. Metabolic phenotyping analysis using untargeted ultra‐high‐performance liquid chromatography‐mass spectrometry identified 493 differentially modulated metabolites in the heart of diabetic db/db mice with a prominent contribution for a wide range of lipid species, including upregulation in acyl‐carnitine, ceramides, and sphingolipid, diacylglycerides, fatty acids, and triacylglycerides. 87 The accumulation of lipids can contribute to oxidative stress, mitochondrial damage, and cardiomyocytes death, a phenomenon that is called lipotoxicity. 88 , 89 In humans, multiple studies have demonstrated that patients with HFpEF have higher myocardial lipid content than nonheart failure and patients with HFrEF, which independently is correlated with diastolic dysfunction. 90 , 91 , 92 Of the differentially upregulated genes in high‐fat diet‐fed and db/db mice, pathway enrichment analysis showed an interesting signature for peroxisome proliferator‐activated receptors (PPARs) signaling and downstream FA degradation pathways. 93 PPAR signaling has a major transcriptional control on cardiac energy metabolism. 94 It is likely that in the lipotoxic models, the profound lipid uptake exceeds the increased flux through β‐oxidation. As FAs and their derivatives can serve as ligands for PPARs, this can stimulate PPAR signaling, blunting thereby metabolic flexibility. In support, according to a meta‐analysis of randomized clinical trials, PPAR agonists increased the risk of heart failure in patients with type 2 diabetes despite the hypoglycemic and hypolipidemic effect. 95
Evidence suggests that diabetic mitochondria are less able to generate ATP, are metabolically inflexible, and trigger oxidative stress in cardiac myocytes. At the posttranscriptional level, miRNAs can modulate mitochondrial processes via genomic regulation. In db/db mice, microarray analysis indicated a downregulation in 14 cardiac mitochondrial RNAs. A decrease in the expression of mitochondrial Cytb (cytochrome b) is featured in the cardiac tissue of these mice. 96 The downregulation of mitochondrial miR‐92a‐2‐5p and let‐7b‐5p was shown to inhibit the translation of Cytb and increase mitochondrial reactive oxygen species production. In addition, the expression of the insulin receptor substrate 1 in the cytosol was shown to be controlled by let‐7b‐5p. This finding has further denoted a mechanism that fortifies lipid deposition and cardiac dysfunction in the db/db model. 96 The involvement of epigenetics in diabetic db/db mice has also been underscored by Shepherd et al, who addressed the presence of impaired nuclear‐mitochondrial communication via disruption in mitochondrial protein import. 97 The downregulation in mtHsp70 (mitochondrial heat shock protein 70) was defined as a critical component in the process by affecting mitochondrial proteomic signature and function. Proteomic decrements were involved in electron transport function and FA metabolism. The impaired expression of mtHSP70 was previously demonstrated in both diabetic animals and humans. 98 , 99 Transcriptional repression is thought to be epigenetically caused by increased histone 3 lysine 27 trimethylation at the Hspa9 genetic loci of the promotor. 97
Apart from the transcriptional modifications, mitochondrial function in db/db mice was also shown to deteriorate as a result of posttranslational hyperacetylation. Cardiac mitochondria were shown to be affected by acetylation‐mediated modification of crucial metabolic enzymes for mitochondrial respiration, such as ND1, UQCRQ, and ATPAF. 100 Concomitantly, mitochondrial bioenergetics was compromised, as evidenced by decreased SIRT3 (Sirtuin3) activity, nicotinamide adenine dinucleotide oxidized/reduced (NAD+/NADH) ratio, and ATP production. The NAD+/SIRT3 pathway appeared to modulate these alterations in mitochondrial metabolic capacity and, ultimately, cardiac function. 100 Strong evidence defined the importance of this pathway in heart failure, which has started to emerge as a therapeutic target. 101 , 102 , 103 , 104
At the functional phenotype level, the db/db model is characterized by a profound cardiac dysfunction that is not seen in other animal models of insulin resistance and obesity, such as obese Zucker rats or ob/ob mice. 105 , 106 , 107 This disparity might be attributed to the severe signs of diabetes that are associated with the db/db model. Circulating levels of tumor necrosis factor‐alpha IL‐1 (interleukin‐1) are increased in db/db mice. 108 The low‐grade systemic inflammation can initiate detrimental microvascular effects. Superperoxide production has been observed in the db/db heart vessels, which may cause endothelial dysfunction altering cGMP/PKC signaling. These altered pathways transform a normal heart into a hypertrophied stiff muscle with impaired diastolic function. 108 This paradigm has also been supported in HFpEF which seems more profuse than that in HFrEF. 109 In line with this, systemic proinflammatory cytokines, such as C‐reactive protein, IL‐1, and tumor necrosis factor‐alpha, were more pronounced and predictive in HFpEF than in HFrEF. 110 , 111 , 112
In db/db mice, impaired relaxation is attributed to cardiac fibrosis and contractile stiffness. Increased expression of the stiffer N2B‐isoform of titin and decreased phosphorylation at threonine 17 of phospholamban was consistent with the presence of excessive passive elastance in the db/db mice. Therefore, titin isoform switching and calcium mishandling because of SERCA2a inhibition have been postulated to drive the diastolic dysfunction. 76 Similarly, biopsy samples from patients with HFpEF showed a substantial rise in passive force and high N2B‐isoform expression. In contrast, the passive ventricular force detected in patients with HFrEF was less potentiated and the flexible N2BA isoform was increased. 113 These changes might be additionally affected by several other mechanisms that are alternatively regulated between HFrEF and HFpEF, such as posttranslational modifications of titin and its proteolysis. 114 , 115 , 116 In addition, while ventricular stiffness leading to diastolic dysfunction serves as a hallmark of HFpEF, changes in ventricular stiffness in HFrEF vary depending on the underlying risk factors. 117 , 118
The db/db mouse develops diastolic dysfunction in both sexes. The strong diabetic component may skew the model towards that of diabetic cardiomyopathy, which is defined as “the existence of abnormal myocardial structure and performance in the absence of other cardiac risk factors, such as coronary artery disease, hypertension, and significant valvular disease, in individuals with diabetes mellitus.” 119 As noted elsewhere, there are significant differences between HFpEF and diabetic cardiomyopathy, and between the db/db mouse and the ZSF1 rat, particularly with regard to mitochondrial function and metabolism. 120
ZSF1‐Obese Rat
The obese ZSF1 rat (ZSF1‐Ob) model is a cross between a Zucker diabetic fatty female with a mutation in the leptin receptor and a spontaneously hypertensive heart failure male rat. 121 Male offspring exhibit aspects of metabolic syndrome, such as impaired glucose metabolism, obesity, and hypertension. This unique model offers insight into a combination of comorbidities that have been on the rise in the United States. Hyperglycemia, hyperlipidemia, and hypertension associated with the ZSF1‐Ob model ultimately play a crucial role in contributing to endothelial dysfunction and atherosclerotic disease, which is widely accepted as a significant contributing factor for HFpEF. 122 The ZSF1‐Ob rat model has been shown to have significant increases in left ventricular mass, left ventricular wall thickness, and left ventricular end‐diastolic diameter as early as 20 weeks, along with diastolic dysfunction. 53 Prominent cardiac features include altered titin isoform expression and/or posttranslational modification, 8 , 123 , 124 impaired NO/cGMP/PKG signaling 124 , 125 , 126 and Ca2+ handling, 8 , 127 , 128 and mitochondrial dysfunction. 127 , 128 Cardiac fibrosis would seem to be a less prominent feature of this model. 124 , 129 , 130
Obese female ZSF‐1 exhibit somewhat greater dyslipidemia attributable to estrogen, but do not show hyperglycemia, which is unrelated to the presence of estrogen. 130 Both sexes exhibit comparable diastolic dysfunction and cardiac remodeling, with the exception that obese females exhibit a body size‐related increase in end diastolic volume. Notably, unlike male db/db mice, neither male nor female ZSF1‐Ob rats show decreased intramyocardial capillary density. 49 , 52 , 130
RNA‐seq results of the ZSF1‐Ob rat show changes in the expression LV genes associated with FA and branched chain amino acid metabolism, as well as cardiac hypertrophy cardiomyopathy, and heart failure. 131 Summer et al recently used deep sequencing RNA data together with 11 preselected cardiac‐specific processes and the STRING protein–protein interaction database to perform network analysis comparing ZSF1‐Ob rats to their lean counterparts. 132 Of the 11 processes, 5 core processes that showed strong interaction in shared and differentially expressed genes stood out, specifically endothelial function, inflammation, apoptosis/autophagy, reactive oxygen species, and extracellular matrix. This finding supports the hypothesis that HFpEF is caused by low‐grade systemic inflammation affecting the endothelium, leading to coronary microvascular dysfunction that causes myocardial stiffness in part via the extracellular matrix. 109 Notably, 2 other processes, namely mitochondrial respiration and substrate metabolism, had the highest percentage of differentially expressed genes between ZSF1‐Ob and ZSF1‐lean rats. 132
However, the ZSF1‐Ob rat does not show much cardiac fibrosis, 129 while Ca2+ handling in cardiac myocytes was reported to be impaired and T‐tubule structure normal. 133 Cardiac myocytes of patients with HFpEF with diabetes showed similar features. In contrast, in patients with HFrEF or in HFrEF animal models, T‐tubule remodeling occurs with a reduced density and infiltration of collagen. Compared with lean controls, diastolic Ca2+ homeostasis was found to be impaired in ZSF1‐Ob rats, which was attributed to reduced SERCA2a and NCX (sodium‐calcium exchanger) activities. In addition, sarcoplasmic reticulum Ca2+ content was reduced, while diastolic Ca2+ levels were elevated at high stimulation frequencies. Another study also reported impaired Ca2+ uptake by the sarcoplasmic reticulum in ZSF1‐Ob rats. 128 In this case, the data supported a decrease in SERCA2a/phospholamban ratio and reduced phospholamban phosphorylation. Increased basal Ca2+ levels in mitochondria correlated with cytosolic Ca2+ levels. The increased mitochondrial Ca2+ was postulated to be a compensatory mechanism for deficient complex 1 activity and reduced oxidative phosphorylation, as it enhances the activity of certain dehydrogenases. 134 Moreover, mitochondria of cardiac myocytes from ZSF1‐Ob rats showed more significant swelling at a similar external Ca2+ concentration because of greater Ca2+ uptake, which might increase the risk for mitochondrial permeability transition pore opening with cell damage or death.
Arginine metabolism and NO turnover in the heart have also been evaluated in the ZSF1‐Ob rat model. 135 NO levels decrease with natural aging and diseases of chronic inflammation, such as is thought to occur in HFpEF. 136 The study provided evidence that an influx of granulocytes and macrophages results in decreased NO production through an increase in arginase 1 and 2 that diminishes the substrate l‐arginine for NOS. In addition, lower l‐arginine uncouples NOS resulting in the production of superoxide and peroxynitrite, further disrupting endothelial function by targeting both NOS signaling and initiating continued inflammatory responses, or by simply quenching bioavailable NO. 137 , 138
Recently, elevating cardiac NAD+ levels by treating ZSF1 rats with its precursor, nicotinamide was reported to improve diastolic function through increased deacetylation of titin and SERCA2a. 8 In both males and females, nicotinamide attenuated cardiac hypertrophy, hypertension, body weight, and lung congestion, and restored energy sources in the heart, which was associated with improved FA oxidation and oxidative phosphorylation. This study documents that restoring metabolic programming improves energy homeostasis in HFpEF.
The Dahl salt‐sensitive (Dahl SS) rat fed a high‐salt diet has provided insights into altered gene expression patterns accompanying the development of diastolic dysfunction. 139 Differently from the ZSF1‐Ob rat, the Dahl SS rat model is mainly driven by high blood pressure, kidney failure, and diastolic dysfunction, thus the metabolic syndrome is almost secondary. However, the Dahl SS on a high‐salt diet may progress towards systolic dysfunction and HFrEF, and over time exhibits clinically non‐relevant increases in blood pressure. 49 Thus, the Dahl SS is perhaps better characterized as a model of early HFpEF progression.
The ZSF1‐Ob rat is a meek animal that is easy to handle, although price and restrictions on maintaining a breeding colony are drawbacks. In addition, hypertension is moderate in this model. Induction of the type 2 diabetic phenotype also requires a special diet.
MULTI‐HIT MOUSE MODELS
As previously stated, HFpEF is often recognized as a multifactorial syndrome of various etiologies, developing and progressing from various factors within the human lifetime. A question then could be asked is what specific comorbidity combination causes the most detrimental progression of HFpEF in terms of time of development and severity. This was the thinking behind the development of multi‐hit mouse models, which are reported to exhibit cardiac fibrosis, 140 ER stress, 10 , 141 , 142 NOS2 expression, 10 , 141 and mitochondrial dysfunction. 11
Schiattarella et al developed a 2‐hit mouse model that combines a high‐fat diet with eNOS inhibition (L‐NG‐nitro arginine methyl ester)‐induced hypertension. 10 Metabolic and hypertensive stress are dominant in this model. These animals exhibit multiple features of HFpEF, including increased LV filling pressure, increased lung weight indicative of pulmonary congestion, cardiomyocyte hypertrophy, cardiac fibrosis, reduced myocardial capillary density, impaired endothelial function in the coronary arteries with reduced coronary flow reserve, and diminished exercise performance. Consistent with the previously mentioned data on humans, evidence was provided that ER stress contributes to the genesis of HFpEF in this model. The impetus for investigating this possibility was a report linking transthyretin amyloidosis to many cases of HFpEF in patients. 143 Specifically, in contrast to a model of HFrEF or to treatments with high‐fat diet or L‐NG‐nitro arginine methyl ester singularly, the authors observed a reduction in multiple markers of the unfolded protein response (UPR) in hearts of their 2‐hit mouse model. UPR acts to dissipate ER stress. Most notable, there was a reduction in the splice form of Xbp1 (Xpb1s), which encodes the active transcription factor and key component of one branch of the ER stress response. This was attributed to impaired inositol‐requiring transmembrane kinase endoribonuclease‐1α splicing activity as evidenced by its diminished phosphorylation. Similar findings were found with the hearts of ZSF1‐Ob rats and endomyocardial biopsies of patients with HFpEF. Impaired IRE1α (inositol requiring transmembrane kinase endoribonuclease 1α) splicing activity was attributed to its S‐nitrosylation because of the upregulation of iNOS. By knocking out the gene for iNOS, the inositol‐requiring transmembrane kinase endoribonuclease‐1α – XBP1 (X‐box binding protein 1) axis was restored in the 2‐hit mouse model, and diastolic dysfunction and exercise performance were improved. Interestingly, acute pharmacological inhibition of iNOS also improved diastolic dysfunction and exercise performance, but without restoring the inositol‐requiring transmembrane kinase endoribonuclease‐1α – XBP1 axis. This observation would suggest additional benefits of iNOS inhibition as well as different targets of iNOS.
The UPR also has a vital role in lipid homeostasis, autophagy, glycosylation, apoptosis, and redox homeostasis. 144 More needs to be known about how these aspects of the UPR are impaired in HFpEF. The UPR helps maintain intracellular redox homeostasis by regulating the expression of amino acid metabolic enzymes involved in glutathione synthesis and antioxidant enzymes. The ER is the most oxidized subcellular compartment and levels in the heart of glutathione peroxidase 4, which protects cells against lipid peroxidation, were recently reported to be reduced in the 2‐hit mouse model. 141
Sex differences need to be better defined in the multi‐hit murine model. There is evidence that female mice are protected somewhat from HFpEF, unlike in women. 145 The role that estrogen might play in that protection is unclear.
RECENT INSIGHTS FROM A FELINE MODEL
A domestic cat model based on slow, progressive banding of the ascending aorta that recapitulates many features of HFpEF in humans was recently described. 12 Two notable cardiac features are fibrosis and mitochondrial dysfunction. 12 , 146 , 147 One advantage of this model (and perhaps limitation) is that it allows for identifying heart‐focused processes in HFpEF development independent of comorbidities, such as age, obesity, and diabetes. 147 Four months post‐banding, the hearts exhibit concentric LV hypertrophy, left atrial enlargement and dysfunction, and LV diastolic dysfunction with a preserved systolic function that progresses to elevated LV end‐diastolic pressures and pulmonary hypertension. LV diastolic dysfunction was associated with increased LV fibrosis, cardiomyocyte hypertrophy, and high plasma levels of NT‐proBNP. In addition, morphological and functional changes are observed in the lungs consistent with impaired respiration. An interventional study starting at 2 months using a pan‐histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), aka vorinostat, was found to rescue the HFpEF phenotype. 146 SAHA improved diastolic function, decreased LV filling pressures, improved myofibril relaxation, and decreased cardiomyocyte size with some improvement in fibrosis. In isolated adult feline cardiomyocytes, SAHA treatment was seen to increase oxygen consumption rate during maximal respiration. At the same time, key mitochondrial enzymes from left ventricles of SAHA‐treated cats exhibited altered acetylation patterns, suggesting that SAHA affected mitochondrial metabolism by posttranslational modifications. Notably, 7 sites in 5 proteins involved in mitochondrial FA oxidation were less acetylated in SAHA‐treated hearts.
Gibb and colleagues used a systems biology approach involving metabolomics and transcriptomics to assess the progression of HFpEF in the feline model from 1 to 4 months of aortic banding. 147 At 1 month, hearts exhibit hypertrophy and fibrosis with no evidence yet of diastolic dysfunction. While a number of genes were altered at 1 month, only a few directly related to metabolism were differentially expressed, including genes associated with the mitochondrial electron transport chain. Notably, cardiac mitochondrial function was impaired, which with the metabolic changes indicated impaired oxidative phosphorylation. Surprisingly, at 4 months, while metabolomic and transcriptomic analysis of banded hearts demonstrated a shift from reliance on oxidative to glycolytic metabolism, mitochondrial function was normalized. Intermediate metabolic pathway regulation was not altered at the transcriptional level; however, factors associated with enzymatic regulation such as protein kinase activity and phosphoprotein interactions were increased. Two conclusions can be drawn from these findings and those involving the histone deacetylase inhibitor. First, unlike with HFrEF, the metabolic reprograming that occurs in HFpEF may reflect more the consequences of posttranslational modifications of proteins. Secondly, the metabolic reprogramming may be susceptible to therapeutic manipulations. Moreover, the study by Gibb et al provided evidence that cardiac‐derived signaling in HFpEF contributes to peripheral tissue (skeletal muscle) maladaptation. 147
The feline model has certain shortcomings. As far as we know, only findings using male cats have been reported. 146 In addition, this model explores the contribution of a single comorbidity, hypertension (increased afterload). Moreover, it is unclear whether or not the model does eventually exhibit a reduction in systolic dysfunction. Finally, this model does not lend itself readily to genetic manipulations.
CONCLUSIONS AND FUTURE DIRECTIONS
Timing is vital for determining the basis for HFpEF and therapeutic strategy, as compensation likely has an impact. A complication of the findings of the various models discussed in this review is the point at which the analyses were performed, as early events will likely be different from later ones. In addition, while an “omic” approach that incorporates genomics, proteomic, and metabolomics is valuable, combining that with functional analyses is important. A developing theme among the HFpEF model is ER stress related to an impaired UPR (Figure 2). Alterations in mitochondrial function and metabolism have been highlighted as well. Underscoring these developments is coronary microvascular endothelial inflammation and dysfunction. Understanding the processes driving these changes remains challenging at both the cell–cell and subcellular levels.
Figure 2. Small animal models are beginning to identify key cellular events in the progression of adverse cardiac remodeling linked to diastolic dysfunction in heart failure with preserved ejection fraction.
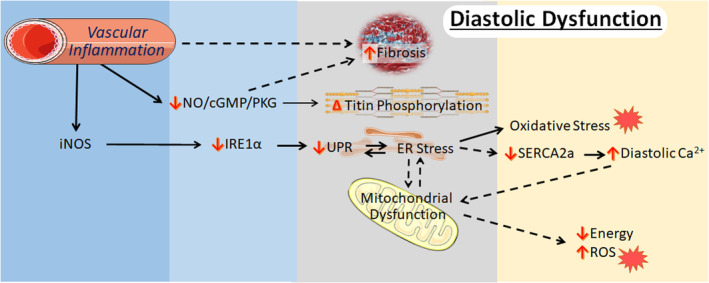
A key initiating factor is thought to be microvascular inflammation, an event linked to diastolic dysfunction or impaired contractile relaxation by 2 means: via increased fibrosis and by impairing normal NO/cGMP/PKG signaling and altering titin phosphorylation. In addition, the upregulation of iNOS leads to an impaired unfolded protein response, resulting from impaired IRE1α (inositol requiring transmembrane kinase endoribonuclease 1α) splicing activity and diminished Xpb1s levels, in cardiomyocytes that are subjected to endoplasmic reticulum stress attributable to hypertrophic pressures. Exacerbation of endoplasmic reticulum stress leads to oxidative stress, mitochondrial dysfunction, and impaired Ca2+ handling that can feedback to worsen diastolic function. Some content is adapted from Servier Medical Art (https://smart.servier.com/) under the terms of the Creative Commons Attributions 3.0 Unported License. AIU, aldosterone‐infused uninephrectomy; cGMP, cyclic guanosine monophosphate; ER, endoplasmic reticulum; iNOS, inducible nitric oxide synthase; NO, nitric oxide; NOS2, nitric oxide synthase 2; PKG, protein kinase G; and UPR, unfolded protein response.
Of necessity, animal models of HFpEF have taken an outward‐to‐inward approach in defining the basis for its cause. In other words, these studies have adopted the strategy of mimicking several comorbidities to assess the consequences on gene, protein, and metabolites. As long as the comorbidities replicate the cardinal features of HFpEF, including diastolic dysfunction, this approach is the most logical and expeditious. Posing a more difficult task are the attempts to understand how events at the cellular and subcellular level predispose an individual to the HFpEF phenotype, including cell–cell (mis‐)communication, cellular adaptations, posttranslational modifications, inflammation, non‐coding RNAs, and mitochondrial dynamics. Of note, a recent study suggested that derangements in ventricular remodeling and fibrosis play a more critical role as a driver of microvascular dysfunction in women (although sex differences are still inadequately addressed in preclinical studies). 148 In this regard, the small animal models are critical for gaining such insights. For instance, in the 2‐hit mouse model, early intervention with a novel drug (imeglimin) that (purportedly) targets complex I and improves mitochondrial function prevented increased iNOS expression, impaired UPR, and diastolic dysfunction. This finding would suggest that mitochondria are early sentinels in the heart that help determine its response to stress. 141 The feline model also supports an early role for mitochondria in HFpEF development with adaptation occuring. 147 Unclear, is what determines a normal versus abnormal response of the mitochondria and ER. A relevant question is whether it is simply a matter of magnitude regarding the imposing stimuli. In that context, a better understanding of how mitochondria and ER communicate with one another, and their environment is needed.
In summary, several small animal models and a feline model that more closely recapitulate the HFpEF phenotype have come to the forefront recently. Time will tell if these models prove their value in revealing the patho‐cause of HFpEF in humans and defining new therapeutic approaches. Regardless, these models will undoubtedly provide new insights into the mechanisms by which the heart deals with comorbidities and how the processes involved can go awry in time or with aging.
Sources of Funding
This work was supported by grants to F.A.Z. from the American University of Beirut Faculty of Medicine (grant number MPP – 320145/320095; URB – 103949) and by Centre National de la Recherche Scientifique (grant number 103507/103487/103941/103944); and Collaborative Research Stimulus (grant number 103556). Booz gratefully acknowledges the support of the Pharmacology Clinical Research Core and the Department of Pharmacology and Toxicology at University of Mississippi Medical Center.
Disclosures
None.
Acknowledgments
The authors thank Dr Nazareno Paolocci for his insightful reading of the manuscript.
For Sources of Funding and Disclosures, see page 10.
Contributor Information
Raffaele Altara, Email: raffaele.altara@gmail.com.
Fouad A. Zouein, Email: fz15@aub.edu.lb.
REFERENCES
- 1. Upadhya B, Kitzman DW. Heart failure with preserved ejection fraction: new approaches to diagnosis and management. Clin Cardiol. 2020;43:145–155. doi: 10.1002/clc.23321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Murphy SP, Ibrahim NE, Januzzi JL Jr. Heart failure with reduced ejection fraction: a review. JAMA. 2020;324:488–504. doi: 10.1001/jama.2020.10262 [DOI] [PubMed] [Google Scholar]
- 3. Dunlay SM, Roger VL, Redfield MM. Epidemiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2017;14:591–602. doi: 10.1038/nrcardio.2017.65 [DOI] [PubMed] [Google Scholar]
- 4. Reed BN, Sueta CA. A practical guide for the treatment of symptomatic heart failure with reduced ejection fraction (HFrEF). Curr Cardiol Rev. 2015;11:23–32. doi: 10.2174/1574884708666131117125508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Anker SD, Butler J, Filippatos G, Ferreira JP, Bocchi E, Bohm M, Brunner‐La Rocca HP, Choi DJ, Chopra V, Chuquiure‐Valenzuela E, et al. Empagliflozin in heart failure with a preserved ejection fraction. N Engl J Med. 2021;385:1451–1461. doi: 10.1056/NEJMoa2107038 [DOI] [PubMed] [Google Scholar]
- 6. Solomon SD, Vaduganathan M, L Claggett B, Packer M, Zile M, Swedberg K, Rouleau J, A Pfeffer M, Desai A, Lund LH, et al. Sacubitril/valsartan across the spectrum of ejection fraction in heart failure. Circulation. 2020;141:352–361. doi: 10.1161/CIRCULATIONAHA.119.044586 [DOI] [PubMed] [Google Scholar]
- 7. Nair N. Epidemiology and pathogenesis of heart failure with preserved ejection fraction. Rev Cardiovasc Med. 2020;21:531–540. doi: 10.31083/j.rcm.2020.04.154 [DOI] [PubMed] [Google Scholar]
- 8. Abdellatif M, Trummer‐Herbst V, Koser F, Durand S, Adao R, Vasques‐Novoa F, Freundt JK, Voglhuber J, Pricolo MR, Kasa M, et al. Nicotinamide for the treatment of heart failure with preserved ejection fraction. Sci Transl Med. 2021;13:eabd7064. doi: 10.1126/scitranslmed.abd7064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mori J, Patel VB, Abo Alrob O, Basu R, Altamimi T, Desaulniers J, Wagg CS, Kassiri Z, Lopaschuk GD, Oudit GY. Angiotensin 1‐7 ameliorates diabetic cardiomyopathy and diastolic dysfunction in db/db mice by reducing lipotoxicity and inflammation. Circ Heart Fail. 2014;7:327–339. doi: 10.1161/CIRCHEARTFAILURE.113.000672 [DOI] [PubMed] [Google Scholar]
- 10. Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, Luo X, Jiang N, May HI, Wang ZV, et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. 2019;568:351–356. doi: 10.1038/s41586-019-1100-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu X, Zhang Y, Deng Y, Yang L, Ou W, Xie M, Ding L, Jiang C, Yu H, Li Q, et al. Mitochondrial protein hyperacetylation underpins heart failure with preserved ejection fraction in mice. J Mol Cell Cardiol. 2022;165:76–85. doi: 10.1016/j.yjmcc.2021.12.015 [DOI] [PubMed] [Google Scholar]
- 12. Wallner M, Eaton DM, Berretta RM, Borghetti G, Wu J, Baker ST, Feldsott EA, Sharp TE III, Mohsin S, Oyama MA, et al. A feline HFpEF model with pulmonary hypertension and compromised pulmonary function. Sci Rep. 2017;7:16587. doi: 10.1038/s41598-017-15851-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zouein FA, de Castro Bras LE, da Costa DV, Lindsey ML, Kurdi M, Booz GW. Heart failure with preserved ejection fraction: emerging drug strategies. J Cardiovasc Pharmacol. 2013;62:13–21. doi: 10.1097/FJC.0b013e31829a4e61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van Riet EE, Hoes AW, Wagenaar KP, Limburg A, Landman MA, Rutten FH. Epidemiology of heart failure: the prevalence of heart failure and ventricular dysfunction in older adults over time. A systematic review. Eur J Heart Fail. 2016;18:242–252. doi: 10.1002/ejhf.483 [DOI] [PubMed] [Google Scholar]
- 15. Vasan RS, Xanthakis V, Lyass A, Andersson C, Tsao C, Cheng S, Aragam J, Benjamin EJ, Larson MG. Epidemiology of left ventricular systolic dysfunction and heart failure in the Framingham Study: an echocardiographic study over 3 decades. JACC Cardiovasc Imaging. 2018;11:1–11. doi: 10.1016/j.jcmg.2017.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pieske B, Tschope C, de Boer RA, Fraser AG, Anker SD, Donal E, Edelmann F, Fu M, Guazzi M, Lam CSP, et al. How to diagnose heart failure with preserved ejection fraction: the HFA‐PEFF diagnostic algorithm: a consensus recommendation from the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur J Heart Fail. 2020;22:391–412. doi: 10.1002/ejhf.1741 [DOI] [PubMed] [Google Scholar]
- 17. Jentzer JC, Reddy YN, Rosenbaum AN, Dunlay SM, Borlaug BA, Hollenberg SM. Outcomes and predictors of mortality among cardiac intensive care unit patients with heart failure. J Card Fail. 2022;S1071‐9164(22)00448‐1. doi: 10.1016/j.cardfail.2022.02.015 [DOI] [PubMed] [Google Scholar]
- 18. Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355:251–259. doi: 10.1056/NEJMoa052256 [DOI] [PubMed] [Google Scholar]
- 19. Lloji A, Sreenivasan J, Novograd J, Aronow WS, Pan S, Lanier GM. Heart failure with preserved ejection fraction: key stumbling blocks for experimental drugs in clinical trials. Expert Opin Investig Drugs. 2022;31:463–474. doi: 10.1080/13543784.2022.2069009 [DOI] [PubMed] [Google Scholar]
- 20. Reddy YNV, Carter RE, Obokata M, Redfield MM, Borlaug BA. A simple, evidence‐based approach to help guide diagnosis of heart failure with preserved ejection fraction. Circulation. 2018;138:861–870. doi: 10.1161/CIRCULATIONAHA.118.034646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Del Rio‐Pertuz G, Benjanuwattra J, Juarez M, Triana AJ, Argueta‐Sosa E. Meta‐analysis evaluating H2FPEF score as a prognostic tool to predict mortality and heart failure‐related hospitalization in adults with normal left ventricular ejection fraction and dyspnea. Am J Cardiol. 2022;173:154–157. doi: 10.1016/j.amjcard.2022.03.046 [DOI] [PubMed] [Google Scholar]
- 22. Sun Y, Wang N, Li X, Zhang Y, Yang J, Tse G, Liu Y. Predictive value of H2 FPEF score in patients with heart failure with preserved ejection fraction. ESC Heart Fail. 2021;8:1244–1252. doi: 10.1002/ehf2.13187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tada A, Nagai T, Omote K, Iwano H, Tsujinaga S, Kamiya K, Konishi T, Sato T, Komoriyama H, Kobayashi Y, et al. Performance of the H2FPEF and the HFA‐PEFF scores for the diagnosis of heart failure with preserved ejection fraction in Japanese patients: a report from the Japanese multicenter registry. Int J Cardiol. 2021;342:43–48. doi: 10.1016/j.ijcard.2021.08.001 [DOI] [PubMed] [Google Scholar]
- 24. Hwang IC, Cho GY, Choi HM, Yoon YE, Park JJ, Park JB, Park JH, Lee SP, Kim HK, Kim YJ. H2FPEF score reflects the left atrial strain and predicts prognosis in patients with heart failure with preserved ejection fraction. J Card Fail. 2021;27:198–207. doi: 10.1016/j.cardfail.2020.09.474 [DOI] [PubMed] [Google Scholar]
- 25. Suzuki S, Kaikita K, Yamamoto E, Sueta D, Yamamoto M, Ishii M, Ito M, Fujisue K, Kanazawa H, Araki S, et al. H2 FPEF score for predicting future heart failure in stable outpatients with cardiovascular risk factors. ESC Heart Fail. 2020;7:65–74. doi: 10.1002/ehf2.12570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sueta D, Yamamoto E, Nishihara T, Tokitsu T, Fujisue K, Oike F, Takae M, Usuku H, Takashio S, Arima Y, et al. H2FPEF score as a prognostic value in HFpEF patients. Am J Hypertens. 2019;32:1082–1090. doi: 10.1093/ajh/hpz108 [DOI] [PubMed] [Google Scholar]
- 27. Egashira K, Sueta D, Komorita T, Yamamoto E, Usuku H, Tokitsu T, Fujisue K, Nishihara T, Oike F, Takae M, et al. HFA‐PEFF scores: prognostic value in heart failure with preserved left ventricular ejection fraction. Korean J Intern Med. 2022;37:96–108. doi: 10.3904/kjim.2021.272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sun Y, Si J, Li J, Dai M, King E, Zhang X, Zhang Y, Xia Y, Tse G, Liu Y. Predictive value of HFA‐PEFF score in patients with heart failure with preserved ejection fraction. Front Cardiovasc Med. 2021;8:656536. doi: 10.3389/fcvm.2021.656536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sotomi Y, Iwakura K, Hikoso S, Inoue K, Onishi T, Okada M, Fujii K, Okamura A, Tamaki S, Yano M, et al. Prognostic significance of the HFA‐PEFF score in patients with heart failure with preserved ejection fraction. ESC Heart Fail. 2021;8:2154–2164. doi: 10.1002/ehf2.13302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Verbrugge FH, Reddy YNV, Sorimachi H, Omote K, Carter RE, Borlaug BA. Diagnostic scores predict morbidity and mortality in patients hospitalized for heart failure with preserved ejection fraction. Eur J Heart Fail. 2021;23:954–963. doi: 10.1002/ejhf.2142 [DOI] [PubMed] [Google Scholar]
- 31. Barandiaran Aizpurua A, Sanders‐van Wijk S, Brunner‐La Rocca HP, Henkens M, Heymans S, Beussink‐Nelson L, Shah SJ, van Empel VPM. Validation of the HFA‐PEFF score for the diagnosis of heart failure with preserved ejection fraction. Eur J Heart Fail. 2020;22:413–421. doi: 10.1002/ejhf.1614 [DOI] [PubMed] [Google Scholar]
- 32. Amanai S, Harada T, Kagami K, Yoshida K, Kato T, Wada N, Obokata M. The H2FPEF and HFA‐PEFF algorithms for predicting exercise intolerance and abnormal hemodynamics in heart failure with preserved ejection fraction. Sci Rep. 2022;12:13. doi: 10.1038/s41598-021-03974-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Henkens M, van Ommen AM, Remmelzwaal S, Valstar GB, Wang P, Verdonschot JAJ, Hazebroek MR, Hofstra L, van Empel VPM, Beulens JWJ, et al. The HFA‐PEFF score identifies ‘early‐HFpEF’ phenogroups associated with distinct biomarker profiles. ESC Heart Fail. 2022;9:2032–2036. doi: 10.1002/ehf2.13861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hahn VS, Knutsdottir H, Luo X, Bedi K, Margulies KB, Haldar SM, Stolina M, Yin J, Khakoo AY, Vaishnav J, et al. Myocardial gene expression signatures in human heart failure with preserved ejection fraction. Circulation. 2021;143:120–134. doi: 10.1161/CIRCULATIONAHA.120.050498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dutta S, Li D, Wang A, Ishak M, Cook K, Farnham M, Dissanayake H, Cistulli P, Hunyor I, Liu R, et al. Metabolite signatures of heart failure, sleep apnoea, their interaction, and outcomes in the community. ESC Heart Fail. 2021;8:5392–5402. doi: 10.1002/ehf2.13631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hage C, Lofgren L, Michopoulos F, Nilsson R, Davidsson P, Kumar C, Ekstrom M, Eriksson MJ, Lynga P, Persson B, et al. Metabolomic profile in HFpEF vs HFrEF patients. J Card Fail. 2020;26:1050–1059. doi: 10.1016/j.cardfail.2020.07.010 [DOI] [PubMed] [Google Scholar]
- 37. Pugliese NR, Paneni F, Mazzola M, De Biase N, Del Punta L, Gargani L, Mengozzi A, Virdis A, Nesti L, Taddei S, et al. Impact of epicardial adipose tissue on cardiovascular haemodynamics, metabolic profile, and prognosis in heart failure. Eur J Heart Fail. 2021;23:1858–1871. doi: 10.1002/ejhf.2337 [DOI] [PubMed] [Google Scholar]
- 38. Sato M, Tsumoto H, Toba A, Soejima Y, Arai T, Harada K, Miura Y, Sawabe M. Proteome analysis demonstrates involvement of endoplasmic reticulum stress response in human myocardium with subclinical left ventricular diastolic dysfunction. Geriatr Gerontol Int. 2021;21:577–583. doi: 10.1111/ggi.14197 [DOI] [PubMed] [Google Scholar]
- 39. Kresoja KP, Rommel KP, Wachter R, Henger S, Besler C, Kloting N, Schnelle M, Hoffmann A, Buttner P, Ceglarek U, et al. Proteomics to improve phenotyping in obese patients with heart failure with preserved ejection fraction. Eur J Heart Fail. 2021;23:1633–1644. doi: 10.1002/ejhf.2291 [DOI] [PubMed] [Google Scholar]
- 40. Milani‐Nejad N, Janssen PM. Small and large animal models in cardiac contraction research: advantages and disadvantages. Pharmacol Ther. 2014;141:235–249. doi: 10.1016/j.pharmthera.2013.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hamlin RL, Altschuld RA. Extrapolation from mouse to man. Circ Cardiovasc Imaging. 2011;4:2–4. doi: 10.1161/CIRCIMAGING.110.961979 [DOI] [PubMed] [Google Scholar]
- 42. Janssen PM, Biesiadecki BJ, Ziolo MT, Davis JP. The need for speed: mice, men, and myocardial kinetic reserve. Circ Res. 2016;119:418–421. doi: 10.1161/CIRCRESAHA.116.309126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kusunose K, Penn MS, Zhang Y, Cheng Y, Thomas JD, Marwick TH, Popovic ZB. How similar are the mice to men? Between‐species comparison of left ventricular mechanics using strain imaging. PLoS One. 2012;7:e40061. doi: 10.1371/journal.pone.0040061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shah SJ, Borlaug BA, Kitzman DW, McCulloch AD, Blaxall BC, Agarwal R, Chirinos JA, Collins S, Deo RC, Gladwin MT, et al. Research priorities for heart failure with preserved ejection fraction: National Heart, Lung, and Blood Institute Working Group Summary. Circulation. 2020;141:1001–1026. doi: 10.1161/CIRCULATIONAHA.119.041886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sharp TE III, Scarborough AL, Li Z, Polhemus DJ, Hidalgo HA, Schumacher JD, Matsuura TR, Jenkins JS, Kelly DP, Goodchild TT, et al. Novel Gottingen Miniswine model of heart failure with preserved ejection fraction integrating multiple comorbidities. JACC Basic Transl Sci. 2021;6:154–170. doi: 10.1016/j.jacbts.2020.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Miyagi C, Miyamoto T, Kuroda T, Karimov JH, Starling RC, Fukamachi K. Large animal models of heart failure with preserved ejection fraction. Heart Fail Rev. 2021;27:595–608. doi: 10.1007/s10741-021-10184-9 [DOI] [PubMed] [Google Scholar]
- 47. Charles CJ, Rademaker MT, Scott NJA, Richards AM. Large animal models of heart failure: reduced vs. preserved ejection fraction. Animals (Basel). 2020;10:1906. doi: 10.3390/ani10101906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van de Wouw J, Steenhorst JJ, Sorop O, van Drie RWA, Wielopolski PA, Kleinjan A, Hirsch A, Duncker DJ, Merkus D. Impaired pulmonary vasomotor control in exercising swine with multiple comorbidities. Basic Res Cardiol. 2021;116:51. doi: 10.1007/s00395-021-00891-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Valero‐Munoz M, Backman W, Sam F. Murine models of heart failure with preserved ejection fraction: a "fishing expedition". JACC Basic Transl Sci. 2017;2:770–789. doi: 10.1016/j.jacbts.2017.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sam F, Duhaney TA, Sato K, Wilson RM, Ohashi K, Sono‐Romanelli S, Higuchi A, De Silva DS, Qin F, Walsh K, et al. Adiponectin deficiency, diastolic dysfunction, and diastolic heart failure. Endocrinology. 2010;151:322–331. doi: 10.1210/en.2009-0806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wilson RM, De Silva DS, Sato K, Izumiya Y, Sam F. Effects of fixed‐dose isosorbide dinitrate/hydralazine on diastolic function and exercise capacity in hypertension‐induced diastolic heart failure. Hypertension. 2009;54:583–590. doi: 10.1161/HYPERTENSIONAHA.109.134932 [DOI] [PubMed] [Google Scholar]
- 52. Alex L, Russo I, Holoborodko V, Frangogiannis NG. Characterization of a mouse model of obesity‐related fibrotic cardiomyopathy that recapitulates features of human heart failure with preserved ejection fraction. Am J Physiol Heart Circ Physiol. 2018;315:H934–H949. doi: 10.1152/ajpheart.00238.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schauer A, Draskowski R, Jannasch A, Kirchhoff V, Goto K, Mannel A, Barthel P, Augstein A, Winzer E, Tugtekin M, et al. ZSF1 rat as animal model for HFpEF: development of reduced diastolic function and skeletal muscle dysfunction. ESC Heart Fail. 2020;7:2123–2134. doi: 10.1002/ehf2.12915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cuijpers I, Carai P, Mendes‐Ferreira P, Simmonds SJ, Mulder P, Miranda‐Silva D, De Giorgio D, Pokreisz P, Heymans S, Jones EAV. The effect of different anaesthetics on echocardiographic evaluation of diastolic dysfunction in a heart failure with preserved ejection fraction model. Sci Rep. 2020;10:15701. doi: 10.1038/s41598-020-72924-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tanaka K, Wilson RM, Essick EE, Duffen JL, Scherer PE, Ouchi N, Sam F. Effects of adiponectin on calcium‐handling proteins in heart failure with preserved ejection fraction. Circ Heart Fail. 2014;7:976–985. doi: 10.1161/CIRCHEARTFAILURE.114.001279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Valero‐Muñoz M, Li S, Wilson RM, Hulsmans M, Aprahamian T, Fuster JJ, Nahrendorf M, Scherer PE, Sam F. Heart failure with preserved ejection fraction induces beiging in adipose tissue. Circ Heart Fail. 2016;9:e002724. doi: 10.1161/CIRCHEARTFAILURE.115.002724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Garcia AG, Wilson RM, Heo J, Murthy NR, Baid S, Ouchi N, Sam F. Interferon‐γ ablation exacerbates myocardial hypertrophy in diastolic heart failure. Am J Physiol Heart Circ Physiol. 2012;303:H587–H596. doi: 10.1152/ajpheart.00298.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. LeBrasseur NK, Duhaney T‐AS, De Silva DS, Cui L, Ip PC, Joseph L, Sam F. Effects of fenofibrate on cardiac remodeling in aldosterone‐induced hypertension. Hypertension. 2007;50:489–496. doi: 10.1161/HYPERTENSIONAHA.107.092403 [DOI] [PubMed] [Google Scholar]
- 59. Shuai W, Kong B, Yang H, Fu H, Huang H. Loss of myeloid differentiation protein 1 promotes atrial fibrillation in heart failure with preserved ejection fraction. ESC Heart Fail. 2020;7:626–638. doi: 10.1002/ehf2.12620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Killingsworth DW, Stern GA. Pseudomonas keratitis associated with the use of disposable soft contact lenses. Case report. Arch Ophthalmol. 1989;107:795–796. doi: 10.1001/archopht.1989.01070010817012 [DOI] [PubMed] [Google Scholar]
- 61. Valero‐Munoz M, Li S, Wilson RM, Boldbaatar B, Iglarz M, Sam F. Dual endothelin‐A/endothelin‐B receptor blockade and cardiac remodeling in heart failure with preserved ejection fraction. Circ Heart Fail. 2016;9:e003381. doi: 10.1161/CIRCHEARTFAILURE.116.003381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tanaka K, Valero‐Muñoz M, Wilson RM, Essick EE, Fowler CT, Nakamura K, van den Hoff M, Ouchi N, Sam F. Follistatin‐like 1 regulates hypertrophy in heart failure with preserved ejection fraction. JACC Basic Transl Sci. 2016;1:207–221. doi: 10.1016/j.jacbts.2016.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shimano M, Ouchi N, Nakamura K, van Wijk B, Ohashi K, Asaumi Y, Higuchi A, Pimentel DR, Sam F, Murohara T. Cardiac myocyte follistatin‐like 1 functions to attenuate hypertrophy following pressure overload. Proc Natl Acad Sci USA. 2011;108:E899–E906. doi: 10.1073/pnas.1108559108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ogura Y, Ouchi N, Ohashi K, Shibata R, Kataoka Y, Kambara T, Kito T, Maruyama S, Yuasa D, Matsuo K. Therapeutic impact of follistatin‐like 1 on myocardial ischemic injury in preclinical models. Circulation. 2012;126:1728–1738. doi: 10.1161/CIRCULATIONAHA.112.115089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Miyabe M, Ohashi K, Shibata R, Uemura Y, Ogura Y, Yuasa D, Kambara T, Kataoka Y, Yamamoto T, Matsuo K. Muscle‐derived follistatin‐like 1 functions to reduce neointimal formation after vascular injury. Cardiovasc Res. 2014;103:111–120. doi: 10.1093/cvr/cvu105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Weber KT. Are myocardial fibrosis and diastolic dysfunction reversible in hypertensive heart disease? Congest Heart Fail. 2005;11:322–326. doi: 10.1111/j.1527-5299.2005.04479.x [DOI] [PubMed] [Google Scholar]
- 67. Sam F, Xie Z, Ooi H, Kerstetter DL, Colucci WS, Singh M, Singh K. Mice lacking osteopontin exhibit increased left ventricular dilation and reduced fibrosis after aldosterone infusion. Am J Hypertens. 2004;17:188–193. doi: 10.1016/j.amjhyper.2003.10.007 [DOI] [PubMed] [Google Scholar]
- 68. Sun Y, Zhang J, Lu L, Chen SS, Quinn MT, Weber KT. Aldosterone‐induced inflammation in the rat heart: role of oxidative stress. Am J Pathol. 2002;161:1773–1781. doi: 10.1016/S0002-9440(10)64454-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rocha R, Rudolph AE, Frierdich GE, Nachowiak DA, Kekec BK, Blomme EA, McMahon EG, Delyani JA. Aldosterone induces a vascular inflammatory phenotype in the rat heart. Am J Physiol Heart Circ Physiol. 2002;283:H1802–H1810. doi: 10.1152/ajpheart.01096.2001 [DOI] [PubMed] [Google Scholar]
- 70. Zile MR, Baicu CF, Gaasch WH. Diastolic heart failure—abnormalities in active relaxation and passive stiffness of the left ventricle. N Engl J Med. 2004;350:1953–1959. doi: 10.1056/NEJMoa032566 [DOI] [PubMed] [Google Scholar]
- 71. Kass DA, Bronzwaer JG, Paulus WJ. What mechanisms underlie diastolic dysfunction in heart failure? Circ Res. 2004;94:1533–1542. doi: 10.1161/01.RES.0000129254.25507.d6 [DOI] [PubMed] [Google Scholar]
- 72. Kuwahara K, Saito Y, Harada M, Ishikawa M, Ogawa E, Miyamoto Y, Hamanaka I, Kamitani S, Kajiyama N, Takahashi N. Involvement of cardiotrophin‐1 in cardiac myocyte‐nonmyocyte interactions during hypertrophy of rat cardiac myocytes in vitro. Circulation. 1999;100:1116–1124. doi: 10.1161/01.cir.100.10.1116 [DOI] [PubMed] [Google Scholar]
- 73. Cooke JP. Does ADMA cause endothelial dysfunction? Arterioscler Thromb Vasc Biol. 2000;20:2032–2037. doi: 10.1161/01.atv.20.9.2032 [DOI] [PubMed] [Google Scholar]
- 74. Zile MR, Gaasch WH. Abnormal calcium homeostasis: one mechanism in diastolic heart failure. J Am Coll Cardiol. 2011;58:155–157. doi: 10.1016/j.jacc.2010.10.068 [DOI] [PubMed] [Google Scholar]
- 75. Altara R, Giordano M, Norden ES, Cataliotti A, Kurdi M, Bajestani SN, Booz GW. Targeting obesity and diabetes to treat heart failure with preserved ejection fraction. Front Endocrinol (Lausanne). 2017;8:160. doi: 10.3389/fendo.2017.00160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Reil JC, Hohl M, Reil GH, Granzier HL, Kratz MT, Kazakov A, Fries P, Muller A, Lenski M, Custodis F, et al. Heart rate reduction by If‐inhibition improves vascular stiffness and left ventricular systolic and diastolic function in a mouse model of heart failure with preserved ejection fraction. Eur Heart J. 2013;34:2839–2849. doi: 10.1093/eurheartj/ehs218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lu J, Liu J, Zhang L, Wang X, Zhang Y, Tang Q. Morphological and functional characterization of diabetic cardiomyopathy in db/db mice following exercise, metformin alone, or combination treatments. Biochem Biophys Res Commun. 2021;584:80–86. doi: 10.1016/j.bbrc.2021.11.018 [DOI] [PubMed] [Google Scholar]
- 78. Monma Y, Shindo T, Eguchi K, Kurosawa R, Kagaya Y, Ikumi Y, Ichijo S, Nakata T, Miyata S, Matsumoto A, et al. Low‐intensity pulsed ultrasound ameliorates cardiac diastolic dysfunction in mice: a possible novel therapy for heart failure with preserved left ventricular ejection fraction. Cardiovasc Res. 2021;117:1325–1338. doi: 10.1093/cvr/cvaa221 [DOI] [PubMed] [Google Scholar]
- 79. Pappritz K, Klein O, Dong F, Hamdani N, Kovacs A, O'Flynn L, Elliman S, O'Brien T, Tschope C, Van Linthout S. MALDI‐IMS as a tool to determine the myocardial response to Syndecan‐2‐selected mesenchymal stromal cell application in an experimental model of diabetic cardiomyopathy. Proteomics Clin Appl. 2021;15:e2000050. doi: 10.1002/prca.202000050 [DOI] [PubMed] [Google Scholar]
- 80. Hamdani N, Hervent AS, Vandekerckhove L, Matheeussen V, Demolder M, Baerts L, De Meester I, Linke WA, Paulus WJ, De Keulenaer GW. Left ventricular diastolic dysfunction and myocardial stiffness in diabetic mice is attenuated by inhibition of dipeptidyl peptidase 4. Cardiovasc Res. 2014;104:423–431. doi: 10.1093/cvr/cvu223 [DOI] [PubMed] [Google Scholar]
- 81. Zhao R, Xie X, Le K, Li W, Moghadasian MH, Beta T, Shen GX. Endoplasmic reticulum stress in diabetic mouse or glycated LDL‐treated endothelial cells: protective effect of Saskatoon berry powder and cyanidin glycans. J Nutr Biochem. 2015;26:1248–1253. doi: 10.1016/j.jnutbio.2015.05.015 [DOI] [PubMed] [Google Scholar]
- 82. Juguilon C, Wang Z, Wang Y, Enrick M, Jamaiyar A, Xu Y, Gadd J, Chen CW, Pu A, Kolz C, et al. Mechanism of the switch from NO to H2O2 in endothelium‐dependent vasodilation in diabetes. Basic Res Cardiol. 2022;117:2. doi: 10.1007/s00395-022-00910-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ni T, Lin N, Huang X, Lu W, Sun Z, Zhang J, Lin H, Chi J, Guo H. Icariin ameliorates diabetic cardiomyopathy through Apelin/Sirt3 signalling to improve mitochondrial dysfunction. Front Pharmacol. 2020;11:256. doi: 10.3389/fphar.2020.00256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Guo Y, Zhang C, Shang FF, Luo M, You Y, Zhai Q, Xia Y, Suxin L. Ketogenic diet ameliorates cardiac dysfunction via balancing mitochondrial dynamics and inhibiting apoptosis in type 2 diabetic mice. Aging Dis. 2020;11:229–240. doi: 10.14336/AD.2019.0510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Baekkerud FH, Salerno S, Ceriotti P, Morland C, Storm‐Mathisen J, Bergersen LH, Hoydal MA, Catalucci D, Stolen TO. High intensity interval training ameliorates mitochondrial dysfunction in the left ventricle of mice with type 2 diabetes. Cardiovasc Toxicol. 2019;19:422–431. doi: 10.1007/s12012-019-09514-z [DOI] [PubMed] [Google Scholar]
- 86. De Jong KA, Lopaschuk GD. Complex energy metabolic changes in heart failure with preserved ejection fraction and heart failure with reduced ejection fraction. Can J Cardiol. 2017;33:860–871. doi: 10.1016/j.cjca.2017.03.009 [DOI] [PubMed] [Google Scholar]
- 87. Faulkner A, Dang Z, Avolio E, Thomas AC, Batstone T, Lloyd GR, Weber RJ, Najdekr L, Jankevics A, Dunn WB, et al. Multi‐omics analysis of diabetic heart disease in the db/db model reveals potential targets for treatment by a longevity‐associated gene. Cell. 2020;9:1283. doi: 10.3390/cells9051283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. van de Weijer T, Schrauwen‐Hinderling VB, Schrauwen P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc Res. 2011;92:10–18. doi: 10.1093/cvr/cvr212 [DOI] [PubMed] [Google Scholar]
- 89. Law BA, Liao X, Moore KS, Southard A, Roddy P, Ji R, Szulc Z, Bielawska A, Schulze PC, Cowart LA. Lipotoxic very‐long‐chain ceramides cause mitochondrial dysfunction, oxidative stress, and cell death in cardiomyocytes. FASEB J. 2018;32:1403–1416. doi: 10.1096/fj.201700300R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wu CK, Lee JK, Hsu JC, Su MM, Wu YF, Lin TT, Lan CW, Hwang JJ, Lin LY. Myocardial adipose deposition and the development of heart failure with preserved ejection fraction. Eur J Heart Fail. 2020;22:445–454. doi: 10.1002/ejhf.1617 [DOI] [PubMed] [Google Scholar]
- 91. Mahmod M, Pal N, Rayner J, Holloway C, Raman B, Dass S, Levelt E, Ariga R, Ferreira V, Banerjee R, et al. The interplay between metabolic alterations, diastolic strain rate and exercise capacity in mild heart failure with preserved ejection fraction: a cardiovascular magnetic resonance study. J Cardiovasc Magn Reson. 2018;20:88. doi: 10.1186/s12968-018-0511-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wei J, Nelson MD, Szczepaniak EW, Smith L, Mehta PK, Thomson LE, Berman DS, Li D, Bairey Merz CN, Szczepaniak LS. Myocardial steatosis as a possible mechanistic link between diastolic dysfunction and coronary microvascular dysfunction in women. Am J Physiol Heart Circ Physiol. 2016;310:H14–H19. doi: 10.1152/ajpheart.00612.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Liu Z, Ding J, McMillen TS, Villet O, Tian R, Shao D. Enhancing fatty acid oxidation negatively regulates PPARs signaling in the heart. J Mol Cell Cardiol. 2020;146:1–11. doi: 10.1016/j.yjmcc.2020.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lee TW, Bai KJ, Lee TI, Chao TF, Kao YH, Chen YJ. PPARs modulate cardiac metabolism and mitochondrial function in diabetes. J Biomed Sci. 2017;24:5. doi: 10.1186/s12929-016-0309-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Han CL, Qu CZ. Cardiovascular risk and safety evaluation of a dual peroxisome proliferator‐activated receptor‐alpha/gamma agonist, aleglitazar, in patients with type 2 diabetes: a meta‐analysis. J Cardiovasc Pharmacol. 2020;75:351–357. doi: 10.1097/FJC.0000000000000796 [DOI] [PubMed] [Google Scholar]
- 96. Li H, Dai B, Fan J, Chen C, Nie X, Yin Z, Zhao Y, Zhang X, Wang DW. The different roles of miRNA‐92a‐2‐5p and let‐7b‐5p in mitochondrial translation in db/db mice. Mol Ther Nucleic Acids. 2019;17:424–435. doi: 10.1016/j.omtn.2019.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Shepherd DL, Hathaway QA, Nichols CE, Durr AJ, Pinti MV, Hughes KM, Kunovac A, Stine SM, Hollander JM. Mitochondrial proteome disruption in the diabetic heart through targeted epigenetic regulation at the mitochondrial heat shock protein 70 (mtHsp70) nuclear locus. J Mol Cell Cardiol. 2018;119:104–115. doi: 10.1016/j.yjmcc.2018.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Croston TL, Thapa D, Holden AA, Tveter KJ, Lewis SE, Shepherd DL, Nichols CE, Long DM, Olfert IM, Jagannathan R, et al. Functional deficiencies of subsarcolemmal mitochondria in the type 2 diabetic human heart. Am J Physiol Heart Circ Physiol. 2014;307:H54–H65. doi: 10.1152/ajpheart.00845.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dabkowski ER, Baseler WA, Williamson CL, Powell M, Razunguzwa TT, Frisbee JC, Hollander JM. Mitochondrial dysfunction in the type 2 diabetic heart is associated with alterations in spatially distinct mitochondrial proteomes. Am J Physiol Heart Circ Physiol. 2010;299:H529–H540. doi: 10.1152/ajpheart.00267.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sun Y, Teng Z, Sun X, Zhang L, Chen J, Wang B, Lu F, Liu N, Yu M, Peng S, et al. Exogenous H2S reduces the acetylation levels of mitochondrial respiratory enzymes via regulating the NAD(+)‐SIRT3 pathway in cardiac tissues of db/db mice. Am J Physiol Endocrinol Metab. 2019;317:E284–E297. doi: 10.1152/ajpendo.00326.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Horton JL, Martin OJ, Lai L, Riley NM, Richards AL, Vega RB, Leone TC, Pagliarini DJ, Muoio DM, Bedi KC Jr, et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight. 2016;2:e84897. doi: 10.1172/jci.insight.84897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lee CF, Chavez JD, Garcia‐Menendez L, Choi Y, Roe ND, Chiao YA, Edgar JS, Goo YA, Goodlett DR, Bruce JE, et al. Normalization of NAD+ redox balance as a therapy for heart failure. Circulation. 2016;134:883–894. doi: 10.1161/CIRCULATIONAHA.116.022495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Tong D, Schiattarella GG, Jiang N, Altamirano F, Szweda PA, Elnwasany A, Lee DI, Yoo H, Kass DA, Szweda LI, et al. NAD(+) repletion reverses heart failure with preserved ejection fraction. Circ Res. 2021;128:1629–1641. doi: 10.1161/CIRCRESAHA.120.317046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Tannous C, Booz GW, Altara R, Muhieddine DH, Mericskay M, Refaat MM, Zouein FA. Nicotinamide adenine dinucleotide: biosynthesis, consumption and therapeutic role in cardiac diseases. Acta Physiol (Oxf). 2021;231:e13551. doi: 10.1111/apha.13551 [DOI] [PubMed] [Google Scholar]
- 105. Belke DD, Larsen TS, Gibbs EM, Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab. 2000;279:E1104–E1113. doi: 10.1152/ajpendo.2000.279.5.E1104 [DOI] [PubMed] [Google Scholar]
- 106. Rosen P, Herberg L, Reinauer H. Different types of postinsulin receptor defects contribute to insulin resistance in hearts of obese Zucker rats. Endocrinology. 1986;119:1285–1291. doi: 10.1210/endo-119-3-1285 [DOI] [PubMed] [Google Scholar]
- 107. Christoffersen C, Bollano E, Lindegaard ML, Bartels ED, Goetze JP, Andersen CB, Nielsen LB. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology. 2003;144:3483–3490. doi: 10.1210/en.2003-0242 [DOI] [PubMed] [Google Scholar]
- 108. Papinska AM, Soto M, Meeks CJ, Rodgers KE. Long‐term administration of angiotensin (1‐7) prevents heart and lung dysfunction in a mouse model of type 2 diabetes (db/db) by reducing oxidative stress, inflammation and pathological remodeling. Pharmacol Res. 2016;107:372–380. doi: 10.1016/j.phrs.2016.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62:263–271. doi: 10.1016/j.jacc.2013.02.092 [DOI] [PubMed] [Google Scholar]
- 110. Collier P, Watson CJ, Voon V, Phelan D, Jan A, Mak G, Martos R, Baugh JA, Ledwidge MT, McDonald KM. Can emerging biomarkers of myocardial remodelling identify asymptomatic hypertensive patients at risk for diastolic dysfunction and diastolic heart failure? Eur J Heart Fail. 2011;13:1087–1095. doi: 10.1093/eurjhf/hfr079 [DOI] [PubMed] [Google Scholar]
- 111. Kalogeropoulos A, Georgiopoulou V, Psaty BM, Rodondi N, Smith AL, Harrison DG, Liu Y, Hoffmann U, Bauer DC, Newman AB, et al. Inflammatory markers and incident heart failure risk in older adults: the Health ABC (Health, Aging, and Body Composition) study. J Am Coll Cardiol. 2010;55:2129–2137. doi: 10.1016/j.jacc.2009.12.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sanders‐van Wijk S, van Empel V, Davarzani N, Maeder MT, Handschin R, Pfisterer ME, Brunner‐La Rocca HP; Investigators T‐C . Circulating biomarkers of distinct pathophysiological pathways in heart failure with preserved vs. reduced left ventricular ejection fraction. Eur J Heart Fail. 2015;17:1006–1014. doi: 10.1002/ejhf.414 [DOI] [PubMed] [Google Scholar]
- 113. van Heerebeek L, Borbely A, Niessen HW, Bronzwaer JG, van der Velden J, Stienen GJ, Linke WA, Laarman GJ, Paulus WJ. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation. 2006;113:1966–1973. doi: 10.1161/CIRCULATIONAHA.105.587519 [DOI] [PubMed] [Google Scholar]
- 114. Hamdani N, Bishu KG, von Frieling‐Salewsky M, Redfield MM, Linke WA. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc Res. 2013;97:464–471. doi: 10.1093/cvr/cvs353 [DOI] [PubMed] [Google Scholar]
- 115. Linke WA, Hamdani N. Gigantic business: titin properties and function through thick and thin. Circ Res. 2014;114:1052–1068. doi: 10.1161/CIRCRESAHA.114.301286 [DOI] [PubMed] [Google Scholar]
- 116. Ali MA, Cho WJ, Hudson B, Kassiri Z, Granzier H, Schulz R. Titin is a target of matrix metalloproteinase‐2: implications in myocardial ischemia/reperfusion injury. Circulation. 2010;122:2039–2047. doi: 10.1161/CIRCULATIONAHA.109.930222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Paulus WJ, Dal Canto E. Distinct myocardial targets for diabetes therapy in heart failure with preserved or reduced ejection fraction. JACC Heart Fail. 2018;6:1–7. doi: 10.1016/j.jchf.2017.07.012 [DOI] [PubMed] [Google Scholar]
- 118. Raya TE, Gay RG, Lancaster L, Aguirre M, Moffett C, Goldman S. Serial changes in left ventricular relaxation and chamber stiffness after large myocardial infarction in rats. Circulation. 1988;77:1424–1431. doi: 10.1161/01.cir.77.6.1424 [DOI] [PubMed] [Google Scholar]
- 119. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. 2018;122:624–638. doi: 10.1161/CIRCRESAHA.117.311586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. McCandless MG, Altara R, Booz GW, Kurdi M. What role do mitochondria have in diastolic dysfunction? Implications for diabetic cardiomyopathy and heart failure with preserved ejection function. J Cardiovasc Pharmacol. 2022;79:399–406. doi: 10.1097/FJC.0000000000001228 [DOI] [PubMed] [Google Scholar]
- 121. Tofovic SP, Kusaka H, Jackson EK, Bastacky SI. Renal and metabolic effects of caffeine in obese (fa/fa(cp)), diabetic, hypertensive ZSF1 rats. Ren Fail. 2001;23:159–173. doi: 10.1081/jdi-100103488 [DOI] [PubMed] [Google Scholar]
- 122. Park SH, Farooq MA, Gaertner S, Bruckert C, Qureshi AW, Lee HH, Benrahla D, Pollet B, Stephan D, Ohlmann P, et al. Empagliflozin improved systolic blood pressure, endothelial dysfunction and heart remodeling in the metabolic syndrome ZSF1 rat. Cardiovasc Diabetol. 2020;19:19. doi: 10.1186/s12933-020-00997-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Cuijpers I, Papageorgiou AP, Carai P, Herwig M, Mugge A, Klein T, Hamdani N, Jones EAV, Heymans S. Linagliptin prevents left ventricular stiffening by reducing titin cleavage and hypophosphorylation. J Cell Mol Med. 2021;25:729–741. doi: 10.1111/jcmm.16122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Schauer A, Adams V, Augstein A, Jannasch A, Draskowski R, Kirchhoff V, Goto K, Mittag J, Galli R, Mannel A, et al. Sacubitril/valsartan improves diastolic function but not skeletal muscle function in a rat model of HFpEF. Int J Mol Sci. 2021;22:3570. doi: 10.3390/ijms22073570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Leite S, Moreira‐Costa L, Cerqueira R, Sousa‐Mendes C, Angelico‐Goncalves A, Fontoura D, Vasques‐Novoa F, Leite‐Moreira AF, Lourenco AP. Chronic sildenafil therapy in the ZSF1 obese rat model of metabolic syndrome and heart failure with preserved ejection fraction. J Cardiovasc Pharmacol Ther. 2021;26:690–701. doi: 10.1177/10742484211034253 [DOI] [PubMed] [Google Scholar]
- 126. Franssen C, Chen S, Unger A, Korkmaz HI, De Keulenaer GW, Tschope C, Leite‐Moreira AF, Musters R, Niessen HW, Linke WA, et al. Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail. 2016;4:312–324. doi: 10.1016/j.jchf.2015.10.007 [DOI] [PubMed] [Google Scholar]
- 127. Bode D, Semmler L, Wakula P, Hegemann N, Primessnig U, Beindorff N, Powell D, Dahmen R, Ruetten H, Oeing C, et al. Dual SGLT‐1 and SGLT‐2 inhibition improves left atrial dysfunction in HFpEF. Cardiovasc Diabetol. 2021;20:7. doi: 10.1186/s12933-020-01208-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Miranda‐Silva D, Wust RCI, Conceicao G, Goncalves‐Rodrigues P, Goncalves N, Goncalves A, Kuster DWD, Leite‐Moreira AF, van der Velden J, de Sousa Beleza JM, et al. Disturbed cardiac mitochondrial and cytosolic calcium handling in a metabolic risk‐related rat model of heart failure with preserved ejection fraction. Acta Physiol (Oxf). 2020;228:e13378. doi: 10.1111/apha.13378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hamdani N, Franssen C, Lourenco A, Falcao‐Pires I, Fontoura D, Leite S, Plettig L, Lopez B, Ottenheijm CA, Becher PM, et al. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circ Heart Fail. 2013;6:1239–1249. doi: 10.1161/CIRCHEARTFAILURE.113.000539 [DOI] [PubMed] [Google Scholar]
- 130. Nguyen ITN, Brandt MM, van de Wouw J, van Drie RWA, Wesseling M, Cramer MJ, de Jager SCA, Merkus D, Duncker DJ, Cheng C, et al. Both male and female obese ZSF1 rats develop cardiac dysfunction in obesity‐induced heart failure with preserved ejection fraction. PLoS One. 2020;15:e0232399. doi: 10.1371/journal.pone.0232399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Stolina M, Luo X, Dwyer D, Han CY, Chen R, Zhang Y, Xiong Y, Chen Y, Yin J, Shkumatov A, et al. The evolving systemic biomarker milieu in obese ZSF1 rat model of human cardiometabolic syndrome: characterization of the model and cardioprotective effect of GDF15. PLoS One. 2020;15:e0231234. doi: 10.1371/journal.pone.0231234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Summer G, Kuhn AR, Munts C, Miranda‐Silva D, Leite‐Moreira AF, Lourenco AP, Heymans S, Falcao‐Pires I, van Bilsen M. A directed network analysis of the cardiome identifies molecular pathways contributing to the development of HFpEF. J Mol Cell Cardiol. 2020;144:66–75. doi: 10.1016/j.yjmcc.2020.05.008 [DOI] [PubMed] [Google Scholar]
- 133. Frisk M, Le C, Shen X, Roe AT, Hou Y, Manfra O, Silva GJJ, van Hout I, Norden ES, Aronsen JM, et al. Etiology‐dependent impairment of diastolic cardiomyocyte calcium homeostasis in heart failure with preserved ejection fraction. J Am Coll Cardiol. 2021;77:405–419. doi: 10.1016/j.jacc.2020.11.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Tanwar J, Singh JB, Motiani RK. Molecular machinery regulating mitochondrial calcium levels: the nuts and bolts of mitochondrial calcium dynamics. Mitochondrion. 2021;57:9–22. doi: 10.1016/j.mito.2020.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Buttner P, Werner S, Baskal S, Tsikas D, Adams V, Lurz P, Besler C, Knauth S, Bahls M, Schwedhelm E, et al. Arginine metabolism and nitric oxide turnover in the ZSF1 animal model for heart failure with preserved ejection fraction. Sci Rep. 2021;11:20684. doi: 10.1038/s41598-021-00216-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ghimire K, Altmann HM, Straub AC, Isenberg JS. Nitric oxide: what's new to NO? Am J Physiol Cell Physiol. 2017;312:C254–C262. doi: 10.1152/ajpcell.00315.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Durante W, Johnson FK, Johnson RA. Arginase: a critical regulator of nitric oxide synthesis and vascular function. Clin Exp Pharmacol Physiol. 2007;34:906–911. doi: 10.1111/j.1440-1681.2007.04638.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Paolocci N, Biondi R, Bettini M, Lee CI, Berlowitz CO, Rossi R, Xia Y, Ambrosio G, L'Abbate A, Kass DA, et al. Oxygen radical‐mediated reduction in basal and agonist‐evoked NO release in isolated rat heart. J Mol Cell Cardiol. 2001;33:671–679. doi: 10.1006/jmcc.2000.1334 [DOI] [PubMed] [Google Scholar]
- 139. Altara R, Zouein FA, Brandao RD, Bajestani SN, Cataliotti A, Booz GW. In silico analysis of differential gene expression in three common rat models of diastolic dysfunction. Front Cardiovasc Med. 2018;5:11. doi: 10.3389/fcvm.2018.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Liao S, Tang Y, Yue X, Gao R, Yao W, Zhou Y, Zhang H. beta‐Hydroxybutyrate mitigated heart failure with preserved ejection fraction by increasing treg cells via Nox2/GSK‐3beta. J Inflamm Res. 2021;14:4697–4706. doi: 10.2147/JIR.S331320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Kitakata H, Endo J, Hashimoto S, Mizuno E, Moriyama H, Shirakawa K, Goto S, Katsumata Y, Fukuda K, Sano M. Imeglimin prevents heart failure with preserved ejection fraction by recovering the impaired unfolded protein response in mice subjected to cardiometabolic stress. Biochem Biophys Res Commun. 2021;572:185–190. doi: 10.1016/j.bbrc.2021.07.090 [DOI] [PubMed] [Google Scholar]
- 142. Schiattarella GG, Altamirano F, Kim SY, Tong D, Ferdous A, Piristine H, Dasgupta S, Wang X, French KM, Villalobos E, et al. Xbp1s‐FoxO1 axis governs lipid accumulation and contractile performance in heart failure with preserved ejection fraction. Nat Commun. 2021;12:1684. doi: 10.1038/s41467-021-21931-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Gonzalez‐Lopez E, Gallego‐Delgado M, Guzzo‐Merello G, de Haro‐Del Moral FJ, Cobo‐Marcos M, Robles C, Bornstein B, Salas C, Lara‐Pezzi E, Alonso‐Pulpon L, et al. Wild‐type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36:2585–2594. doi: 10.1093/eurheartj/ehv338 [DOI] [PubMed] [Google Scholar]
- 144. Metcalf MG, Higuchi‐Sanabria R, Garcia G, Tsui CK, Dillin A. Beyond the cell factory: homeostatic regulation of and by the UPR(ER). Sci Adv. 2020;6:eabb9614. doi: 10.1126/sciadv.abb9614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Tong D, Schiattarella GG, Jiang N, May HI, Lavandero S, Gillette TG, Hill JA. Female sex is protective in a preclinical model of heart failure with preserved ejection fraction. Circulation. 2019;140:1769–1771. doi: 10.1161/CIRCULATIONAHA.119.042267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Wallner M, Eaton DM, Berretta RM, Liesinger L, Schittmayer M, Gindlhuber J, Wu J, Jeong MY, Lin YH, Borghetti G, et al. HDAC inhibition improves cardiopulmonary function in a feline model of diastolic dysfunction. Sci Transl Med. 2020;12:eaay7205. doi: 10.1126/scitranslmed.aay7205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Gibb AA, Murray EK, Eaton DM, Huynh AT, Tomar D, Garbincius JF, Kolmetzky DW, Berretta RM, Wallner M, Houser SR, et al. Molecular signature of HFpEF: systems biology in a cardiac‐centric large animal model. JACC Basic Transl Sci. 2021;6:650–672. doi: 10.1016/j.jacbts.2021.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Chandramouli C, Ting TW, Tromp J, Agarwal A, Svedlund S, Saraste A, Hage C, Tan RS, Beussink‐Nelson L, Fermer ML, et al. Sex differences in proteomic correlates of coronary microvascular dysfunction among patients with heart failure and preserved ejection fraction. Eur J Heart Fail. 2022;24:681–684. doi: 10.1002/ejhf.2435 [DOI] [PMC free article] [PubMed] [Google Scholar]