

Abstract
The trityl-substituted bisoxazoline (TrHBOX) was prepared as a chiral analogue to previously reported nickel dipyrrin system capable of ring-closing amination catalysis. Ligand metallation with divalent NiI2(py)4 followed by potassium graphite reduction afforded the monovalent (TrHBOX)Ni(py) (4). Slow addition of 1.4 equivalent of a benzene solution of 1-adamantylazide to 4 generated the tetrazido (TrHBOX)Ni(κ2-N4Ad2) (5) and terminal iminyl adduct (TrHBOX)Ni(NAd) (6). Investigation of 6 via single-crystal X-ray crystallography, NMR and EPR spectroscopies, and computations revealed a Ni(II)-iminyl radical formulation, similar to its dipyrrinato congener. Complex 4 exhibits enantioselective intramolecular C–H bond amination to afford N-heterocyclic products from 4-aryl-2-methyl-2-azidopentanes. Catalytic C–H amination occurs under mild conditions (5 mol% catalyst, 60 °C) and provides pyrrolidine products in decent yield (29%–87%) with moderate ee (up to 73%). Substrates with a 3,5-dialkyl substitution on the 4-aryl position maximized the observed enantioselectivity. Kinetic studies to probe the reaction mechanism were conducted using 1H and 19F NMR spectroscopies. A small, intermolecular kinetic isotope effect (1.35 ± 0.03) suggests an H-atom abstraction step with an asymmetric transition state while the reaction rate is measured to be first order in catalyst and zeroth order in substrate concentrations. Enantiospecific deuterium labelling studies show that the enantioselectivity is dictated by both the H-atom abstraction and radical recombination steps due to the comparable rate between radical rotation and C–N bond formation. Furthermore, the competing elements of the two-step reaction where H-removal from the pro-R configuration is preferred while the preferential radical capture occurs with the Si face of the carboradical likely lead to the diminished ee observed, as corroborated by theoretical calculations. Based on these enantio-determining steps, catalytic enantioselective synthesis of 2,5-bis-tertiary pyrrolidines is demonstrated with good yield (50–78%) and moderate ee (up to 79 %).
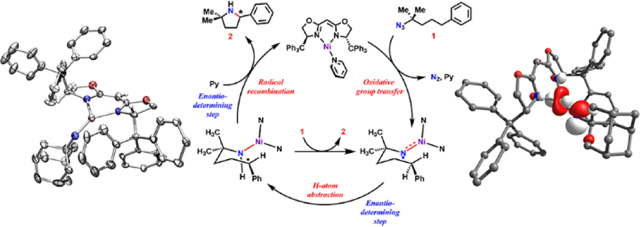
1. INTRODUCTION
Selective amination of C–H bonds remains one of the most challenging objectives in organic synthesis due to the ubiquity of saturated N-heterocycles in bioactive alkaloids,1 pharmaceutical agents,2–3 and as chiral elements in enantioselective catalysis.4 Asymmetric pyrrolidine motifs, in particular, are common building blocks for numerous bioactive materials.2, 5–6 Chiral amine heterocycles traditionally are accessed by four methods: (1) selective deprotonation facilitated by chiral auxiliaries-bound base, followed by transmetalation mediated C−C bond formation;7–8 (2) asymmetric addition to Ellman’s aldimines;9–10 (3) enantioselective catalytic hydroamination;11 and (4) enantioselective chemical or enzymatic imine hydrogenation.12–18 An alternative to this method could entail direct, enantioselective C−H bond amination cyclization. Transition metal mediated nitrene transfer methods have emerged as a viable path for amination catalysis, providing atom economical access to substituted pyrrolidine products from simple aliphatic azide substrates.19–21
The current state of the art method in pyrrolidine synthesis via C−H bond amination largely depends on late transition metal catalysts that template the C−H bond activation followed by a C−N bond forming step. Several groups have contributed to developing this method using different catalytic systems, including iron/cobalt porphyrins,22–24 cobalt corroles,25 iron bound within redox-active pincer ligands,26 and iron beta-diketiminate metal organic frameworks (Figure 1).27 Nevertheless, little progress has been made towards the enantioselective synthesis of these 5-membered N-heterocycles using such approaches. To the best of our knowledge, only two reported systems have demonstrated enantioselective pyrrolidine synthesis via nitrene insertion catalysis.24, 28 A chiral Co(II) porphyrin converts linear alkyl azides to pyrrolidines in low overall yields (22%) and moderate enantiomeric excess (ee, 46%),24 whereas a dual catalytic system comprising of a chiral-at-metal Ru complex and a phosphine achieves excellent enantioselectivity (up to 99% ee), albeit with relatively low product yields (15–57%).28 Considering the need for a precious metal in the latter system, as well as the requirement for product sequestration with tert-butyloxycarbonyl (Boc) to increase the turnover numbers for either catalyst, more sustainable and atom-economical alternatives are of interest. To overcome these limitations, it is thus critical to understand the nature and reactivity profile of key reactive intermediates implicated in transition metal mediated C–H amination processes.24, 29–40
Figure 1.

Pyrrolidine synthesis via cyclization C–H amination using base metal catalysts.
We recently reported a dipyrrin-supported Ni(I) system that catalyzes conversion of unactivated, alkyl azides into pyrrolidines in high yield, at low catalyst loadings, and without requiring product sequestration.41 Additionally, a Ni(II)-iminyl species (i.e., NiII(2NR)) was isolated and identified as the species responsible for the C–H activation event.33 To take advantage of the efficiency of this system, we explored ways to induce enantioselective pyrrolidine formation. To this end, it was reasoned that deprotonated bisoxazoline ligands (BOX) are structurally and electronically similar to the weak-field dipyrrin platform,42–43 yet more sterically enforcing and offer access to a chiral environment around the Ni center.44 Specifically, we sought to address the following questions: (1) can we access BOX-Ni nitrene moieties akin to those isolated with the dipyrrin platform? If so, (2) how is the electronic structure of such species impacted by the BOX ligand? (3) Can these BOX-Ni complexes aminate C–H bonds and impart enantioselectivity? And (4) what is the mechanism through which the asymmetrical formation of product is enforced?
Herein, we report the synthesis and characterization of a BOX-supported Ni(I) synthon, which can be readily converted to a Ni nitrenoid upon exposure to organic azides.33 The electronic structure of the resulting metal nitrenoid is best described as a Ni(II) iminyl radical akin to its dipyrrinato congeners. The Ni(I) complex is an effective catalyst for enantioselective C–H amination with decent yields (29–87%) and moderate ee (as high as 73%). A comprehensive mechanistic investigation corroborated by theoretical calculations indicates that both H-atom abstraction and radical recombination steps are enantiodetermining,45 providing the unique opportunity to synthesize asymmetric, fully substituted 2,5-bis-tertiary pyrrolidines that are otherwise difficult to access.
2. RESULTS AND DISCUSSION
2.1. Preparation of a Ni(I) synthon supported by a novel mono-anionic bisoxazoline ligand
Bisoxazoline ligands have been successfully employed in numerous enantioselective catalytic transformations, including hydrogenation, carbenoid cyclopropanation, nitrenoid aziridination, Michael addition, Nazarov cyclization, aldol-type, Mannich-type, Diels-Alder, and ene reactions.44, 46–48 In all instances, transition metals are supported by neutral BOX ligands. To simulate the electronic features of dipyrrinato ligands, however, we set out to prepare a mono-anionic BOX ligand instead.49 To the best of our knowledge, few studies on mono-anionic BOX scaffolds have been reported, most of which make use of a meso-cyano group to increase the ligand meso-H acidity and stabilize the monoanionic form.49–51 To expand upon this series, BOX ligands prepared52–53 from commercially available or synthetic54 enantiopure substituted 2-aminoethanols (1a-c) were deprotonated using potassium hydride49 to cleanly afford the corresponding mono-anionic BOX ligands (2a-c) at ambient temperature (25 °C, Scheme 1).
Scheme 1.

Following previously reported protocols for preparing the dipyrrinato-supported Ni(I) precatalyst ((AdFL)Ni(py); AdFL: 1,9-di(1–4 adamantyl)-5-perfluorophenyldipyrrin),33 dropwise addition of a THF-solution of variants of 2 to a frozen suspension of NiI2(py)4 in THF resulted in an instantaneous color change from light yellow to dark red. The 1H NMR spectra of the crude reaction mixtures showed total consumption of the deprotonated ligands 2 with clean conversion to new paramagnetic products. Crystals of each product were obtained by layering hexanes onto a concentrated solution of the material in benzene at room temperature. Single-crystal X-ray diffraction analysis revealed that bis-substitution (L2M) is thermodynamically favored for ligands with phenyl or tert-butyl substituents (2b-c), preventing access to the Ni complex of interest. However, the enhanced steric protection of trityl substituents (TrHBOX–,2a) inhibits bis-substitution and enables isolation of a dark-red, pseudo tetrahedral pyridine-bound complex (TrHBOX)NiI(py) (3, Scheme 2, Figure 2a). Complex 3 displays a paramagnetically shifted 1H NMR spectrum, indicative of a triplet (S = 1) ground state.33
Scheme 2.

Figure 2.

Solid-state molecular structure for (a) (TrHBOX)NiI(py) (3) and (b) (TrHBOX)Ni(py) (4) with thermal ellipsoids at 50% probability level. Color scheme: Ni, pink; N, blue; C, gray; O, red; I, purple. H atoms (except for the ones on chiral centers and meso carbon) and solvent molecules omitted for clarity. (c) Frozen solution EPR spectrum of (TrHBOX)Ni(py) (4) collected at 77 K in toluene (Red). Blue line represents a fit of the data using the program EasySpin.55 Fitting parameters: S = ½, g1 = 2.43, g2 = 2.13, g3 = 2.07.
Chemical reduction of 3 with KC8 in a frozen-thawing THF solution cleanly generates the Ni(I) pyridine adduct (TrHBOX)Ni(py) (4, Scheme 3, Figure 2b) as a dark yellow solid. The EPR spectrum of 4 collected at 77 K in toluene indicates a doublet spin ground state (S = ½) in a rhombic environment (g1 = 2.43, g2 = 2.13, g3 = 2.07; Figure 2c).56 Layering hexanes on a concentrated benzene solution of 4 at room temperature overnight afforded needle-shaped yellow crystals suitable for single-crystal X-ray diffraction analysis. The solid-state structure of 4 (Figure 2b) reveals an unusual pyramidally-distorted, T-shape geometry around the Ni(I) center with the NL–Ni–Npy angle of 146.4(2)°, akin to the previously reported three-coordinated Ni(I) dipyrrinato and β-diketiminate complexes.33, 57 Complex 4 decomposes instantaneously upon exposure to air; however, it is stable at room temperature in the solid state for a month under a nitrogen atmosphere, showing only trace amount of decomposition as indicated by NMR and EPR spectroscopies.
Scheme 3.

2.2. Isolation and structural characterization of a BOX supported terminal iminyl complex.
With a well-characterized Ni(I) synthon in hand, we next sought to probe the general reactivity of 4 with azides and compare the electronic nature of the resulting Ni-nitrenoid species with its dipyrrinato congeners.33 Treatment of 4 with 1.4 equiv of AdN3 in benzene resulted in a slow color change from dark yellow to dark orange along with effervescence over 12 h. The EPR spectrum of the resulting mixture revealed disappearance of 4 along with formation of two new S = ½ paramagnetic species (Scheme 4). Fortunately, one of the species is only marginally soluble in diethyl ether, enabling the two species to be separated. Indeed, the orange residue following diethyl ether extraction can be recrystallized by layering hexanes on a concentrated THF solution. After standing at room temperature overnight, long needle-shaped dark orange crystals suitable for single-crystal X-ray diffraction were obtained to reveal a tetrazido complex (TrHBOX)Ni(κ2-N4Ad2) (5, Scheme 4, Figure 3a). The solid-state molecular structure of 5 (Figure 3a) resembles those of previously published tetrazido complexes supported by dipyrrinato ligands.37, 39 The N1–N2, N2–N3 and N3–N4 bond lengths of 1.300(4) Å, 1.348(4) Å and 1.302(4) Å, respectively, (Figure 3a) fall between typical N–N single and double bonds, suggesting a mono-anionic tetrazido radical formulation.37, 39 The EPR spectrum of 5 collected at 77 K in toluene indicates an S = ½ spin state with the unpaired electron occupying a rhombic environment (g1 = 2.33, g2 = 2.17, g3 = 2.01; Figure 3b).
Scheme 4.

Figure 3.

Solid-state molecular structure for (a) (TrHBOX)Ni(κ2-N4Ad2) (5) and (c) (TrHBOX)Ni(NAd) (6) with thermal ellipsoids at 50% probability level. Color scheme: Ni, pink; N, blue; C, gray; O, red. H atoms (except for the ones on chiral centers and meso carbon) omitted for clarity. (b) Frozen solution EPR spectrum of (TrHBOX)Ni(κ2-N4Ad2) (5) collected at 77 K in toluene (Red). Blue line represents a fit of the data using the program EasySpin.55 Fitting parameters: S = ½, g1 = 2.33, g2 = 2.17, g3 = 2.01. (d) Frozen solution EPR spectrum of (TrHBOX)Ni(NAd) (6) collected at 77 K in toluene (Red). Blue line represents a fit of the data using the program EasySpin.55 Fitting parameters: S = ½, g1 = 2.19, g2 = 2.06, g3 = 1.93 with hyperfine splitting constant, A = 20.9 G (14N, I = 1).
The second species formed upon addition of AdN3 to 4 can be recrystallized from a concentrated THF solution slowly through vapor-diffusion with hexanes at −35 °C to afford dark green plate-shaped crystals suitable for single-crystal X-ray diffraction. The solid-state structure of the product reveals a terminal iminyl adduct, (TrHBOX)Ni(NAd) (6, Scheme 4, Figure 3c) with a Ni–Nim bond length of 1.680(8) Å, similar to those reported for both dipyrrinato- and β-diketiminate-supported nickel nitrenoid adducts.32–33, 57 Two different molecules of 6 are found in the asymmetric unit and display significantly different Ni–Nim–C bond angles [139.5(7)°, 152.8(8)°], potentially due to crystal packing effects. The local geometry at nickel is close to planar [∑ N–Ni–N (avg) = 355.6°], consistent with previously reported three-coordinate Ni nitrenoid species.33, 57–61
2.3. Electronic structure considerations of ancillary ligands
With the isolation of terminal iminyl 6, we sought to compare the electronic effects of BOX ligand 2a to dipyrrin (AdFL) on the bound Ni center. In our previous studies with Ni-dipyrrinato system, we discovered that the amount of radical density on the nitrenoid nitrogen atom was critical to effecting H−atom abstraction (HAA).34–35, 41, 57 From a molecular orbital perspective, the unpaired electron resides in a singly occupied molecular orbital (SOMO) with contributions from both the metal 3dxz and nitrogen 2px orbitals (Figure 4a); therefore, a SOMO with lower energy would delocalize more unpaired electron density onto the nitrenoid nitrogen due to smaller energy gap with N-valence shell. The σ-donation ability of the bidentate ancillary ligand impacts the metal dyz orbital, whereas the dxz orbital is of appropriate symmetry to participate in π-interactions with the ligand and raises energy of the SOMO. The most notable difference between the two ligand platforms arises from the greater π-donor ability of BOX that is attenuated in the dipyrrin (AdFL) owing to its extended π-delocalization.34, 42 Due to the structural similarities between terminal iminyls supported by these ligand platforms (6, (AdFL)Ni(NAd)), the amount of radical density delocalized onto the nitrenoid nitrogen can be crudely correlated with the observed 14N hyperfine splitting constants. The EPR spectrum of 6 collected at 77 K in toluene revealed an S = ½ spin ground state with a rhombic environment for the unpaired electron (g1 = 2.19, g2 = 2.06 g3 = 1.93; Figure 3d) and an 14N hyperfine splitting constant of 20.9 G (14N, I = 1), similar to those reported for (AdFL)Ni(NAd) (AN = 21.3 G).33 Previous N K-edge X-ray absorption studies for (AdFL)Ni(NAd) identified a low-energy, pre-edge absorption, suggesting a NiII(2NAd−) formulation (i.e., iminyl radical anion as opposed to imido, NiIII(NAd2−)).33 Although the corresponding data is not available for 6, a similar electronic structure is proposed herein in light of the similarity between their structural and electronic properties observed spectroscopically. To further probe the frontier molecular orbital structure of these iminyl complexes, geometry optimizations of 6 and (AdFL)Ni(NAd) were conducted to produce coordinates from which to carry out electronic structure calculations. The complete structures for both complexes were optimized using the unrestricted Kohn-Sham methods, B3LYP functional62–65 and Ahlrich’s def2-SVP (C, H, F) and def2-TZVP (Ni, N, O) basis sets.66–67 Indeed, the SOMO of 6 has only marginally smaller contribution from the nitrogen 2px orbital as compared to (AdFL)Ni(NAd) (Figure 4b–c), 52.0 vs 55.3%, revealing minimal impact of the π-donating ability of BOX ligand on the spin distribution and energy of SOMOs.33
Figure 4.

(a) Molecular orbital picture of (LX)Ni(NR); SOMO plot for (b) (TrHBOX)Ni(NAd) and (c) (AdFL)Ni(NAd) from geometry optimized DFT calculations. Nitrogen 2px and nickel 3dxz contributions toward SOMO are listed. uB3LYP/def2-TZVP(Ni, N, O)+def2-SVP(C, H).
2.4. Enantioselective ring-closing amination catalysis using 4
Having established the similar ligand strength of dipyrrinato and anionic BOX platforms, we next sought to examine the catalytic efficacy of 4 for pyrrolidine formation via C–H amination. As the 1H NMR spectra of both 5 and 6 show very few, indiscernible broad features, a pentafluorophenyl group was introduced at the meso position of the ligand to monitor the identity of metal containing species during catalysis via 19F NMR spectroscopy.33, 41–42 Treatment of mono-anionic 2a with five equivalence of hexafluorobenzene in THF and heating at 95 °C for 12 h afforded the perfluorophenyl-substituted BOX ligand with concomitant production of a stoichiometric equivalent of KF as evidenced by 1H and 19F NMR spectroscopies (TrFBOX–, 2d; Scheme 5). The corresponding Ni precatalyst 4F was prepared using the same procedure as outlined for 4 (Scheme 5). The 19F NMR spectrum of 4F displays three features (Figure S8), indicating rapid rotation of the perfluorophenyl group on the NMR timescale.33, 41–42 We note that the reactivity results in the following discussion were the same regardless of catalyst (i.e., 4 or 4F).
Scheme 5.

As an initial test, 10 mol% of 4F was subjected to (4-azido-4-methylpentyl)fluorobenzene (7) in C6D6 (Table 1, entry 1). Tertiary azides were used to inhibit the unproductive ɑ-H abstraction previously observed with Ni-iminyl species.33, 41 Although no reaction was observed at room temperature (25 °C), complete substrate consumption occurred within 12 h upon heating to 60 °C, leading to isolation of the corresponding pyrrolidine product 8 in 85% yield. We note that the electron-rich Ni center permits catalysis without product pyrrolidine sequestration akin to the previously reported dipyrrin-Ni catalyst system.41 Conversion of the catalyst to tetrazido species along with other unidentifiable decomposition products was noted toward the end of the reaction as evidenced by EPR and 19F NMR spectroscopies. Lowering the catalyst loading to 5 mol% (entry 2) produces the same outcome, albeit the reaction time is doubled. A maximum turn-over number of 36 is achieved when 1 mol% catalyst loading (entry 3) is used, and no further conversion is observed after heating the reaction for one week at 60 °C. The overall catalyst performance is lower than that of the dipyrrin system, for which the optimized conditions only require 1 mol% catalyst loading at 25 °C for full conversion of 7 to 8 in 2 h (94% yield) with full regeneration of catalyst at the end.41 We hypothesize that the comparably lower catalyst activity of 4F is a result of the formation of tetrazene species.37–39 We propose that the two vacant quadrants around the Ni center promote rapid reaction of Ni iminyl with another azide equivalent, whereas the more directional steric profile of the dipyrrin is sufficient to block this pathway.
Table 1.
Optimization of Amination Conditionsa
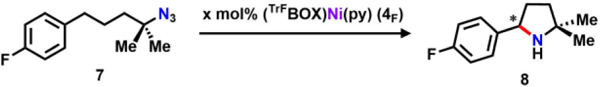
With the identified optimized reaction conditions, we next sought to examine the enantio-inducing effect of the chiral BOX ligand. A 28% ee was determined by condensing 8 with (S)-(+)-α-methoxy-α-(trifluoromethyl)phenylacetyl chloride (S-Mosher acyl chloride) and integration of representative peaks in both 1H and 19F NMR spectra.41, 68–71 Lowering the reaction temperature to 40 °C (entry 4) increases the ee for 8 to 33%, albeit at longer reaction times (72 h) and lower yield (22%) due to catalyst decomposition. The catalysis can also be carried out in either toluene or THF (Table 1, entries 6 and 7) with comparable results, but diethyl ether or hexanes are not suitable solvents due to low catalyst solubility. Additionally, catalyst 4F reacts with DCM to generate (TrFBOX)NiCl(py) through halogen atom abstraction, akin to the Ni-dipyrrin system.33, 41
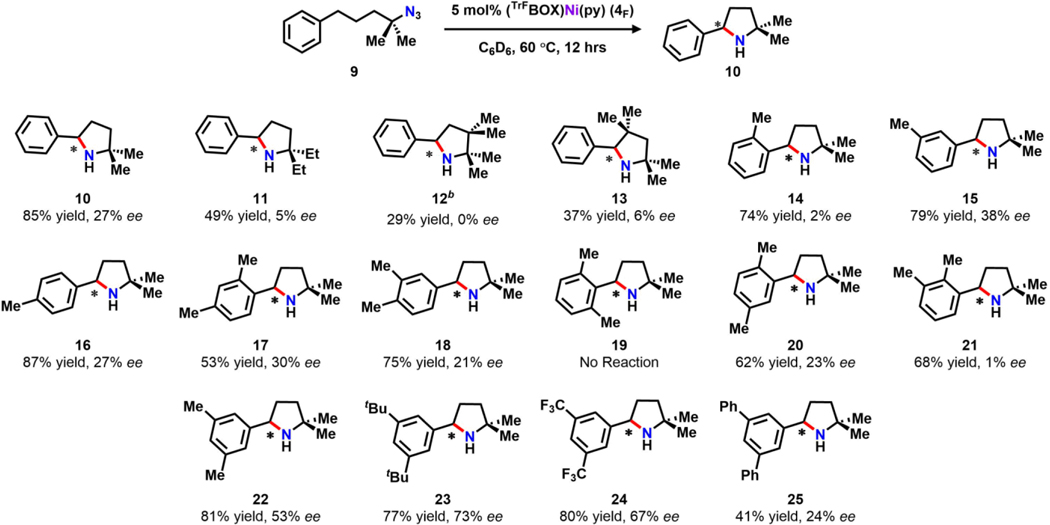
To systematically probe the effect of the steric profile on enantioselectivity, we surveyed a series of organic azides featuring different methyl substitution patterns on both the aliphatic chain and the aryl moiety in 9 (Table 2). Increasing the steric profile on the aliphatic chain proved to have an adverse effect (11–13), resulting in lower enantioselectivity (5–12% ee) for the respective products. Substitutions on the aryl moiety, however, produced significant results (14–25). For substrates with mono-methyl substitutions (14–16) on the phenyl group, the highest ee was observed with meta substitution (15, 38%). Likewise, introducing two methyl groups at the meta positions of the aromatic group (22) induces the highest enantioselectivity comparing to other substitution patterns (17–21), achieving a moderate ee of 53% (22). Interestingly, when one meta position is occupied by a methyl group, substitution at para (18) or ortho (20–21) sites results in a diminished ee as compared to 15. Given the significant improvement on ee upon bis-meta substitution, different functional groups were introduced at the meta positions in an attempt to increase the enantioselectivity (23–25). Despite significantly improved results obtained with both sterically bulky electron-donating (23, tBu–, 73% ee) and withdrawing (24, CF3–, 67% ee) substituents, phenyl substitution resulted in an attenuated ee of 24% (25).
Table 2.
Substrate scopea
|
Isolation yield
50 mol% of catalyst was used due to the side product generated by carbocation rearrangement during the substrate preparation through SN1 mechanism (see SI).
2.5. Mechanistic evaluation of ring-closing amination catalysis
To understand what factors are important for enantioselectivity, we set out to probe the ring-closing amination mechanism using kinetic analysis. Due to observed catalyst transformation into tetrazido complexes at late stage of the catalysis (80% conversion, Figure S9), initial rate measurements were conducted to determine the reaction rate dependence on catalyst and substrate concentrations during the initial phase of the reaction.39 Concentrations of azide 7 and product 8 during the catalytic reaction at 60 °C were monitored as a function of time using 19F NMR spectroscopy. While holding the initial concentration of azide 7 constant and varying 4F catalyst loadings, a plot of slopes obtained from the initial 10% conversion of each run versus catalyst loadings revealed a linear relationship, indicating a first order rate dependence of the reaction on catalyst 4F concentration (Figure 5a). Similar experiments were carried out holding the catalyst 4F concentration constant while changing the initial concentration of azide 7. Analogous initial rate analysis reveals a zeroth order dependence on the substrate (Figure 5b). Of note, the same rate law was obtained in our previous nickel dipyrrin study.41 Monitoring the catalysis via 19F NMR spectroscopy, three similar features to those of 4F are detected prior to 30% conversion of substrate, and no significant amount of catalyst decomposition or conversion into the tetrazido species is noted (Figure S8). The Ni-species observed during catalysis can be a combination of Ni(I) L-type donor coordination compounds such as pyridine (4F), organoazide (ɑ- and/or γ-binding) as well as (R)- and (S)-pyrrolidine adducts.
Figure 5.

(a) Initial rate for conversion of 7 into 8 over the first 10% as a function of catalyst loadings while holding initial azide 7 concentration constant. Every measurement is done in triplicate, and error bars represent standard deviations; (b) Initial rate for conversion of 7 into 8 over the first 10% as a function of initial azide 7 concentration while holding catalyst concentration constant. Every measurement is done in triplicate, and error bars represent standard deviations; (c) Plotting the first 10% conversion of 9H2 (blue) and 9D2 (red) into 10H and 10D over time, respectively. Intermolecular KIE is derived by taking the ratio of the slopes of the linear fits [KIE = 1.35 ± 0.03 (60 °C)]. Every measurement is done in triplicate, and error bars represent standard deviations.
Due to the observation of a tetrazyl species by EPR spectroscopy at late stage of the transformation (Figure S9), we set out to probe the viability of Ni tetrazyl complexes to release azide and reenter the catalytic cycle. Despite our reported catalytically inactive dipyrrinato cobalt tetrazyl complexes,38–39 precedent from the Jenkins’ lab establishes iron tetrazido complexes can release organic azide and the corresponding imide which can aziridinate olefins. 72–73 To test the reversibility of tetrazyl formation in this system, we heated an equimolar C6D6 solution of 5F and substrate 9 to 80 °C for 20 h while monitoring the reaction using 1H and 19F NMR spectroscopies. If the formation of Ni tetrazyl is reversible, the cyclized product 10 along with liberated AdN3 should be observed via sequential azide metathesis reactions (Scheme 6). Indeed, the 1H NMR spectrum of the final reaction mixture shows consumption of substrate 9 as well as generation of 10 and AdN3, suggesting that the tetrazyl complex 5F is capable of releasing the iminyl intermediate to reenter the catalytic cycle (Figure S10). However, due to the zeroth order rate dependence on azide during our initial rate study, this pathway likely only contributes during the final stages of catalysis following sufficient build-up of the tetrazyl complex and lower remaining azide substrate concentration.
Scheme 6.

To further probe the reaction mechanism, kinetic isotope effect (KIE) was evaluated by comparing the initial rates of treating 4F with bis-deutero- (4-azido-4-methylpentyl-1,1-d2)benzene (9D2) and corresponding proteo-substrate (9H2, Figure 5c), and an intermolecular KIE of 1.35 ± 0.03 (60 °C) was determined.75–76 Relative to the large intermolecular KIE of 31.9 ± 1.0 (23 °C) observed with the dipyrrinato nickel system,41 the small value suggests that the H-atom abstraction step likely involves a linear yet asymmetric transition state. Nevertheless, a concerted C–H insertion cannot be excluded.39, 77–78
The two possible mechanisms above for C–H activation (radical-mediated vs. concerted) suggest different possibilities for the enantio-determining step. If the C–H activation is radical-mediated, depending on the relative rate between radical rotation (Re vs Si) and radical recombination, either the HAA or the radical recombination step is enantio-determining.79 However, if the C–H functionalization follows a concerted mechanism, the enantioselectivity is directly determined by the relative barrier of activating different C–H bonds (pro-R vs pro-S).
To distinguish between these possibilities and elucidate the enantio-determining step, a pair of enantiopure mono deuterium-labeled substrates (S-9HD and R-9HD) was prepared according to modified literature procedures41, 74 and subjected to standard catalytic conditions, leading to four potential products (S-10H, R-10H, S-10D and R-10D; Figure 6). The two pairs of enantiomers (S-10H, R-10H; S-10D, R-10D) were resolved using Mosher analysis,70–71 and the product distributions were obtained from integration of 1H NMR spectra of the final mixture. We note that the integration values derived from S- and R-10H using S-9HD are relatively small comparing to other enantiomer pairs. In the event of a concerted C–H activation, the only products observable from S-9HD (or R-9HD) should be R-10D and S-10H (or S-10D and R-10H) due to stereospecificity of the mechanism. However, all four products were found in both cases, suggesting a radical mechanism for C–H activation.
Figure 6.

Product distribution using enantiopure mono D-labeled substrates S-9HD and R-9HD.41, 74 The two pairs of enantiomers (S-10H and R-10H; S-10D and R-10D) are resolved using Mosher analysis and the ratios are obtained using 1H NMR spectroscopy.70–71 All reactions were carried out using standard catalytic conditions at 60 °C. Figure here showcases the product generation using S-9HD as an example. See SI for detailed analysis for both S-9HD and R-9HD.41
The HAA step (H-atom vs. D-atom abstraction) of the reaction can be analyzed using the level of deuterium incorporation in the final product (R-10D + S-10D vs. R-10H + S-10H; Figure 6). Two factors contribute to this distribution: (1) chiral ligand preference for pro-R or pro-S hydrogen; (2) intramolecular kinetic isotope effects (HAA favored over DAA). Upon isolation of the product, the ratio between D-labeled (R-10D + S-10D) and H-labeled (R-10H + S-10H) product was determined to be 18:1 and 1.9:1 starting from S-9HD and R-9HD (Figure 6) as a result of a match or mismatch between the two effects, respectively. The higher ratio obtained from S-9HD suggests a lower barrier for pro-R hydrogen abstraction enforced by the chiral BOX ligand.
Once the H/D-atom is abstracted, two competing processes occur: (1) rotation of the planar, carbon-based benzyl radical to expose either the Re or Si face near the iminyl N (Figure 6), and (2) radical rebound to form the C–N bond. If the radical trapping process is faster than carboradical rotation (stereo-retentive radical recombination),79 the final product distribution is solely determined by the HAA step (enantio-determining step), and again the only products observable from S-9HD (or R-9HD) should be R-10D and S-10H (or S-10D and R-10H). Otherwise, if the radical rebound process is sufficiently slow such that carboradical rotation reaches thermo-equilibrium first, the final product distribution is determined by the relative energy of the transition states for radical recombination with the Re or Si face (Curtin-Hammett control).75, 79 Hence, similar ratios between S-10H and R-10H (or S-10D and R-10D) should be observed regardless of starting material, and the radical recombination becomes the enantio-determining step.
Mosher analysis revealed that the ratios of S-10H to R-10H derived from S-9HD and R-9HD are both 2:1, suggesting that the radical recombination is under Curtin-Hammett control. However, the ratios of S-10D to R-10D derived from S-9HD and R-9HD are different (1.8:1 and 3.3:1, respectively; Figure 6), suggesting the rate of radical recombination is comparable to the rate for the D-bound carboradical to reach thermo-equilibrium. The racemization of H-bound carboradical should be faster than its D-bound counterpart due to 2° KIE; however, since such 2° KIEs are normally close to unity,75 the rate for radical recombination is likely very similar to that of the rotation of a carbon-based radical, indicating that both the HAA and radical recombination steps can potentially contribute to the enantioselectivity.79 Interestingly, the preferred prochirality of the carboradical (Si) is the opposite to that during the HAA step (pro-R), likely resulting in the low overall enantioselectivity observed. The similar strategy to probe the rate of radical rebound has been used to examine the mechanism of C−H hydroxylation using cytochrome P450,80 intramolecular C−H insertion using iron-carbene complexes,81 allylic C−H abstraction by singlet oxygen,82 and more recently the enantioselective carbene C−H insertion catalyzed by chiral cobalt porphyrin complexes79 as well as our recently reported dipyrrinato nickel system for catalytic C−H amination.41
With detailed understanding of the enantio-determining processes in the stepwise mechanism, the increase in ee from meta substitution on the aryl group favoring S-pyrrolidine product can be the result from either less preferential pro-R H-atom removal or more relatively favored radical recombination with Si compared to Re face of the carboradical. However, due to the similar ee increase with meta substitution observed in the amination of tertiary benzylic C–H bond (vide infra), the higher ee observed is likely caused by a larger discrepancy between kinetic barriers toward C–N bond formation with the Si and Re face of the carboradical either by steric repulsion to disfavor the R-pyrrolidine formation or attractive dispersion interaction between the meta-alkyl and ligand-based trityl groups to favor the S-pyrrolidine synthesis.83
Taken together, we propose the following catalytic cycle similar to that for the nickel dipyrrin system (Figure 7).41 The pyridine adduct 4 first undergoes ligand exchange with substrate 9 to generate an azide adduct. Dinitrogen extrusion from the azide adduct generates a nickel iminyl species.41 Intramolecular HAA by the iminyl followed by radical recombination between the Ni-amide and the carboradical intermediate generated in the previous step furnishes the final pyrrolidine product 10, which is displaced upon ligand substitution with pyridine or substrate 9. The iminyl species can also react with another equivalent of substrate 9 to generate the tetrazyl complex,35, 39 which can release organoazide and the corresponding Ni-iminyl to reenter the catalytic cycle.72–73 Even though we do not observe accumulation of the iminyl during catalysis, we note that the HAA step does contribute to the observed kinetics with the non-unity KIE in addition to a higher barrier (pro-R: 21.3 kcal/mol; pro-S: 23.5 kcal/mol) in comparison to the N2 loss step (9.7 kcal/mol) derived from DFT calculation (vide infra). Furthermore, our studies reveal that the enantio-determining step is impacted by the relative rates of radical recombination and rotation.
Figure 7.

Proposed mechanism for C–H amination.
2.6. DFT evaluation of the amination mechanism
Density functional theory (DFT) was employed to provide support for the proposed mechanism in Figure 7 and to gain insight into geometric factors that may influence enantioselectivity during catalysis. Calculations of the reaction trajectory and relevant intermediates (Figure 8) were performed with the B3LYP functional62–65 and 6–31+G(d) basis sets84–87 on an untruncated system. The full computational methodology is described in detail in the Supporting Information.
Figure 8.

DFT calculated [B3LYP/6–31+G(d)] free energy diagram (in kcal/mol) for the proposed C–H amination mechanism for the cyclization of 9 into 2-phenyl-5,5-dimethylpyrrolidine (10) mediated by (TrHBOX)Ni(py) (4) with energies corresponding to the S enantiomer in red and the R enantiomer in blue.
The nitrenoid structure (A) was found to favor a doublet spin state with a T-shaped geometry about the nickel. A quartet excited state was close in energy, being only 2.9 kcal/mol higher in free energy. Spin densities of 0.40 e- and 0.54 e- were found for nickel and iminyl nitrogen atoms, respectively, supporting the experimentally proposed iminyl character.41 However, the spin density on the iminyl nitrogen being < 1 e- implies an admixture of iminyl and imido character.
The amide intermediate (C) generated by intramolecular HAA from the iminyl was identified with a similar T-shaped geometry to A. Spin densities of 1.45 e- for nickel and 0.39 e- for the amide nitrogen were calculated, suggesting an amide description with some aminyl character. The amine product of RR was determined to favor the S enantiomer (E1) with a calculated free energy relative to the R enantiomer (E2) of −4.6 kcal/mol, both featuring a doublet spin state. The spin densities of nickel and nitrogen were determined to be 0.99 and −0.01 e-, respectively, indicating a NiI-amine formulation as expected for a d9 complex.
The reaction from iminyl to amide is exergonic and has a ΔG of −4.5 kcal/mol with a reaction barrier of 23.5 (B1, pro-S) and 21.3 kcal/mol (B2, pro-R) for the two isomeric HAA transition states, favoring abstraction of the pro-R hydrogen atom. The formation of the S enantiomer via radical recombination (D1) is also exergonic (ΔG = −13.1 kcal/mol), with a barrier of 9.1 kcal/mol relative to the amide C. The TS from C to the R enantiomer of the amine (D2) possesses a higher energy barrier of 11.1 kcal/mol (ΔΔG‡ = 2.0 kcal/mol) and is also a less exergonic (ΔΔG = 4.6 kcal/mol) reaction with a ΔG of −8.5 kcal/mol for the radical rebound step. These results suggest that the S enantiomer is favored upon radical recombination, consistent with isotope labelling experiments. The greater stability of the S-amine product can be attributed to the improved steric profile resulting from positioning the phenyl group at a greater distance from the bulky CPh3 substituents on the ligand (Figure S27). The calculated result reproduced the switch in preferred prochirality during the HAA (pro-R) and RR (Si) transition states, further supporting the mechanistic proposal in Figure 7.
The barrier toward radical capture with the Si (9.1 kcal/mol) and Re (11.1 kcal/mol) faces of the radical is close to the upper limit of a C–C bond rotational barrier for saturated alkanes (3–10 kcal/mol).88 The actual rotational barrier around PhHC•–CH2 bond in the amide intermediate (C) is likely higher than that of a typical C(sp3)–C(sp3) bond due to two factors: (1) the •C–CH2 bond is shorter than the other C(sp3)–C(sp3) bonds present in the optimized structure of C likely due to hyperconjugation between the radical p-orbital and neighboring C(sp3)–H σ-bond (Figure S28), resulting in heightened rotation barrier; and (2) the radical rotation must occur in a sterically restricted pocket created by the TrBOX ligand, incurring an additional energetic penalty for C−C rotation. Despite our best attempts at locating the C–C rotational transition states for the C-based radical, satisfactory result was not obtained due to a large ensemble of torsional modes plausible for the overall radical rotation.
2.7. Catalytic enantioselective synthesis of 2,5-bis-tertiary pyrrolidine
With a comprehensive understanding for enantio-determining steps, the proposed mechanism provides the unique opportunity for enantioselective synthesis of 2,5-bis-tertiary pyrrolidines. These products are important precursors for a range of pharmaceutical agents.89–92 A common pathway to synthesize chiral pyrrolidines is asymmetric hydrogenation of cyclic imines via either organometallic or enzymatic catalysis.12–18 However, this strategy necessarily introduces a H-atom on the ɑ-position to the amino group in the product, rendering 2,5-bis-tertiary pyrrolidines inaccessible.
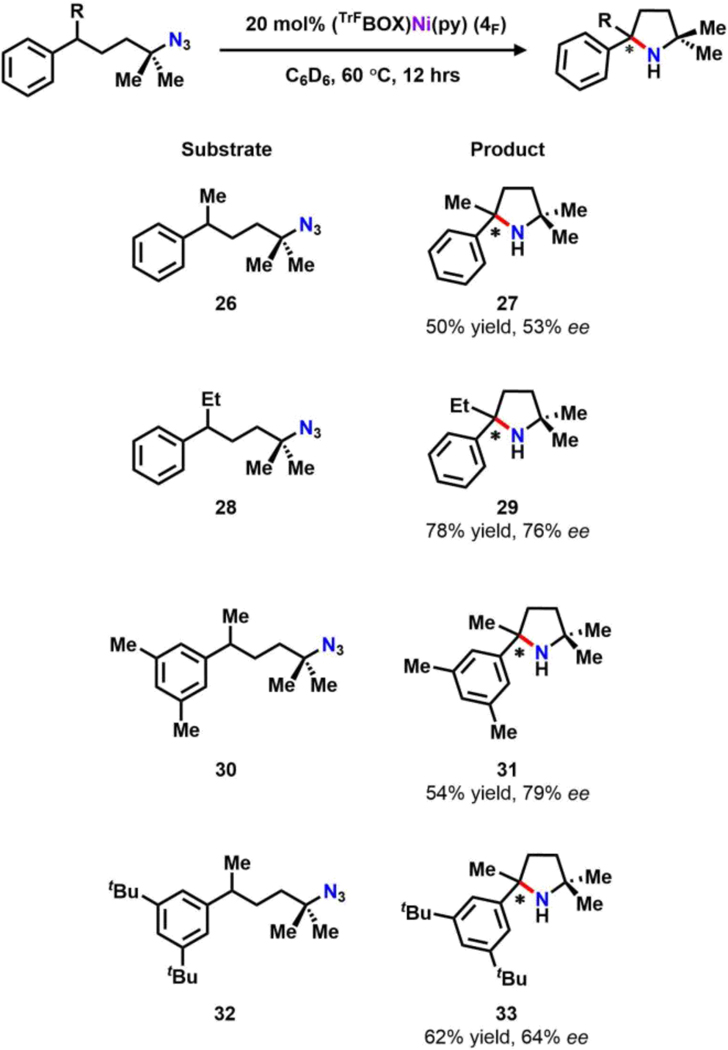
As an initial test, racemic (5-azido-5-methylhexan-2-yl)benzene (26) was synthesized and subjected to catalytic conditions. Gratifyingly, upon full conversion the corresponding product, 2,2,5-trimethyl-5-phenylpyrrolidine (27) can be isolated in moderate yield (50%) and much improved ee (53%) as compared to 10. Additionally, replacing the methyl with an ethyl group at the benzylic position (28) or elaborating the aryl group with 1,3-bis-methyl (30) or bis-tert butyl substituents (32) afforded the corresponding products in good yield (54–78%) and with enhanced ee (64–79%).
The result suggests that the chiral environment enforced by the ligand better differentiates the steric of 2,2,5,5-tetra-substituted pyrrolidines comparing to their 2,2,5-tri-substituted congeners. Detailed mechanistic and computational investigations for further improved ligand system are currently under study.
3. CONCLUSIONS
In conclusion, we have developed a nickel catalyst supported by a trityl-substituted, mono-anionic BOX ligand capable of enantioselective C–H bond amination. The BOX supported Ni-imide adopts a similar Ni iminyl electronic structure formulation as its dipyrrinato congener despite stronger π-donating ability of the BOX ligand.33 The (TrBOX)Ni system operates under mild reaction conditions to afford pyrrolidines in good yield (29%–87%) and with moderate ee (as high as 73%). A detailed mechanistic study was carried out leading to our proposal that a similar catalytic cycle to the previously reported dipyrrinato nickel complex was operative. Inter- and intramolecular kinetic isotope labelling experiments point to a radical-based C–H activation step. Furthermore, due to the similar rate between competing radical rotation and C–N bond formation, both the HAA and radical recombination steps contribute to enantioselectivity of the reaction. However, while HAA favors the pro-R hydrogen removal, the formation of S-pyrrolidine is slightly favored for the RR step leading to the diminished overall enantiomeric excess observed. Despite that switch in configurational preference, increasing the steric bulk at the meta positions on the aryl substituents leads to a higher observed product ee. We propose that the increased meta-position bulk either suppresses capture of re face of the carboradical by steric repulsion or favors the S-configuration by attractive dispersion interaction during the radical recombination step. Theoretical studies corroborate the proposed mechanistic cycle and the energetically favored RR transition states leading to S-pyrrolidines. Following this comprehensive mechanistic investigation, we conclude that future catalyst design that operate under a similar stepwise mechanism aiming for better stereo-control need to address: (a) the trajectory of radical capture for rate control with respect to carboradical rotation to establish the enantio-determining step; and (b), conform to the geometry of the corresponding HAA or RR transition state to improve the overall enantioselectivity. We note that the previously mentioned Ru system showcasing excellent stereo-control is proposed to go through a concerted pathway where the C−H bond activation step solely determines the enantiomeric outcome. The C−H bond functionalization that operates via a similar two-step mechanism as the one observed herein can also achieve higher enantioselectivity. For example, excellent enantioselectivity from Co-porphyrin catalyzed carbene insertion is reported whose mechanism is described as occurring via two steps where the HAA is enantio-determining as the rapid radical recombination step is stereo-retentive.79 Based on our proposed mechanism and enantio-determining processes, catalytic synthesis of 2,5-bis-tertiary pyrrolidines inaccessible from the well-established asymmetric imine hydrogenation is demonstrated in decent yield (50–78%) and with moderate ee (53–79%).
Supplementary Material
Table 3.
Substrate scope for catalytic enantioselective 2,5-bis-tertiary pyrrolidine synthesisa
|
Isolation yield.
ACKNOWLEDGMENT
This work was generously supported by a grant from the NIH (GM-115815), the Dreyfus Foundation (Teacher-Scholar Award to T.A.B.), and Harvard University. R.M.C. gratefully acknowledges an NSERC Postdoctoral Fellowship. C. J. L. and T. R. C. acknowledge support from the U.S. Department of Energy, Office of Science, Basic Energy Sciences (DE-FG02-03ER15387), and thank the National Science Foundation for their generous support of the UNT CASCaM HPC cluster via grant CHE-1531468.
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website.
General experimental considerations and procedures, multinuclear NMR data, solid state molecular structures (PDF)
Crystallography data (CIF)
The authors declare no competing financial interest.
REFERENCE
- 1.Fattorusso E; Taglialatela-Scafati O, Modern Alkaloids: Structure, Isolation, Synthesis, and Biology. Wiley: 2008. [Google Scholar]
- 2.Vitaku E; Smith DT; Njardarson JT, Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257. [DOI] [PubMed] [Google Scholar]
- 3.Taylor RD; MacCoss M; Lawson ADG, Rings in Drugs. J. Med. Chem. 2014, 57, 5845. [DOI] [PubMed] [Google Scholar]
- 4.Banik SM; Levina A; Hyde AM; Jacobsen EN, Lewis acid enhancement by hydrogen-bond donors for asymmetric catalysis. Science 2017, 358, 761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuhnert M; Blum A; Steuber H; Diederich WE, Privileged Structures Meet Human T-Cell Leukemia Virus-1 (HTLV-1): C2-Symmetric 3,4-Disubstituted Pyrrolidines as Nonpeptidic HTLV-1 Protease Inhibitors. J. Med. Chem. 2015, 58, 4845. [DOI] [PubMed] [Google Scholar]
- 6.Adrio J; Carretero JC, Stereochemical diversity in pyrrolidine synthesis by catalytic asymmetric 1,3-dipolar cycloaddition of azomethine ylides. Chem. Comm. 2019, 55, 11979. [DOI] [PubMed] [Google Scholar]
- 7.Campos KR; Klapars A; Waldman JH; Dormer PG; Chen C. y., Enantioselective, Palladium-Catalyzed α-Arylation of N-Boc-pyrrolidine. J. Am. Chem. Soc. 2006, 128, 3538. [DOI] [PubMed] [Google Scholar]
- 8.Stead D; O’Brien P; Sanderson A, A New Sparteine Surrogate for Asymmetric Deprotonation of N-Boc Pyrrolidine. Org. Lett. 2008, 10, 1409. [DOI] [PubMed] [Google Scholar]
- 9.Reddy LR; Prashad M, Asymmetric synthesis of 2-substituted pyrrolidines by addition of Grignard reagents to γ-chlorinated N-tert-butanesulfinyl imine. Chem. Comm. 2010, 46, 222. [DOI] [PubMed] [Google Scholar]
- 10.Ellman JA; Owens TD; Tang TP, N-tert-Butanesulfinyl Imines: Versatile Intermediates for the Asymmetric Synthesis of Amines. Acc. Chem. Res. 2002, 35, 984. [DOI] [PubMed] [Google Scholar]
- 11.Dai X-J; Engl OD; León T; Buchwald SL, Catalytic Asymmetric Synthesis of α-Arylpyrrolidines and Benzo-fused Nitrogen Heterocycles. Angew. Chem. Int. Ed. 2019, 58, 3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aleku GA; Man H; France SP; Leipold F; Hussain S; Toca-Gonzalez L; Marchington R; Hart S; Turkenburg JP; Grogan G; Turner NJ, Stereoselectivity and Structural Characterization of an Imine Reductase (IRED) from Amycolatopsis orientalis. ACS Catalysis 2016, 6, 3880. [Google Scholar]
- 13.Li H; Zhang G-X; Li L-M; Ou Y-S; Wang M-Y; Li C-X; Zheng G-W; Xu J-H, A Novel (R)-Imine Reductase from Paenibacillus lactis for Asymmetric Reduction of 3 H-Indoles. ChemCatChem 2016, 8, 724. [Google Scholar]
- 14.Velikogne S; Resch V; Dertnig C; Schrittwieser JH; Kroutil W, Sequence-Based In-silico Discovery, Characterisation, and Biocatalytic Application of a Set of Imine Reductases. ChemCatChem 2018, 10, 3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verdaguer X; Lange UEW; Reding MT; Buchwald SL, Highly Enantioselective Imine Hydrosilylation Using (S,S)-Ethylenebis(η5-tetrahydroindenyl)titanium Difluoride. J. Am. Chem. Soc. 1996, 118, 6784. [Google Scholar]
- 16.Aleku GA; France SP; Man H; Mangas-Sanchez J; Montgomery SL; Sharma M; Leipold F; Hussain S; Grogan G; Turner NJ, A reductive aminase from Aspergillus oryzae. Nat. Chem. 2017, 9, 961. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y; Kong D; Wang R; Hou G, Synthesis of chiral cyclic amines via Ir-catalyzed enantioselective hydrogenation of cyclic imines. Org. Biomol. Chem. 2017, 15, 3006. [DOI] [PubMed] [Google Scholar]
- 18.Takei I; Nishibayashi Y; Arikawa Y; Uemura S; Hidai M, Iridium-Catalyzed Asymmetric Hydrosilylation of Imines Using Chiral Oxazolinyl-Phosphine Ligands. Organometallics 1999, 18, 2271. [Google Scholar]
- 19.Nakamura I; Yamamoto Y, Transition-Metal-Catalyzed Reactions in Heterocyclic Synthesis. Chem. Rev. 2004, 104, 2127. [DOI] [PubMed] [Google Scholar]
- 20.Gandeepan P; Müller T; Zell D; Cera G; Warratz S; Ackermann L, 3d Transition Metals for C–H Activation. Chem. Rev. 2019, 119, 2192. [DOI] [PubMed] [Google Scholar]
- 21.Park Y; Kim Y; Chang S, Transition Metal-Catalyzed C–H Amination: Scope, Mechanism, and Applications. Chem. Rev. 2017, 117, 9247. [DOI] [PubMed] [Google Scholar]
- 22.Du Y-D; Xu Z-J; Zhou C-Y; Che C-M, An Effective [FeIII(TF4DMAP)Cl] Catalyst for C–H Bond Amination with Aryl and Alkyl Azides. Org. Lett. 2019, 21, 895. [DOI] [PubMed] [Google Scholar]
- 23.Shing K-P; Liu Y; Cao B; Chang X-Y; You T; Che C-M, N-Heterocyclic Carbene Iron(III) Porphyrin-Catalyzed Intramolecular C(sp3)–H Amination of Alkyl Azides. Angew. Chem. Int. Ed. 2018, 57, 11947. [DOI] [PubMed] [Google Scholar]
- 24.Kuijpers PF; Tiekink MJ; Breukelaar WB; Broere DLJ; van Leest NP; van der Vlugt JI; Reek JNH; de Bruin B, Cobalt-Porphyrin-Catalysed Intramolecular Ring-Closing C−H Amination of Aliphatic Azides: A Nitrene-Radical Approach to Saturated Heterocycles. Chem. Eur. J. 2017, 23, 7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goswami M; Geuijen P; Reek JNH; de Bruin B, Application of [Co(Corrole)]– Complexes in Ring-Closing C–H Amination of Aliphatic Azides via Nitrene Radical Intermediates. Eur. J. Inorg. Chem. 2018, 2018, 617. [Google Scholar]
- 26.Bagh B; Broere DLJ; Sinha V; Kuijpers PF; van Leest NP; de Bruin B; Demeshko S; Siegler MA; van der Vlugt JI, Catalytic Synthesis of N-Heterocycles via Direct C(sp3)–H Amination Using an Air-Stable Iron(III) Species with a Redox-Active Ligand. J. Am. Chem. Soc. 2017, 139, 5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thacker NC; Lin Z; Zhang T; Gilhula JC; Abney CW; Lin W, Robust and Porous β-Diketiminate-Functionalized Metal–Organic Frameworks for Earth-Abundant-Metal-Catalyzed C–H Amination and Hydrogenation. J. Am. Chem. Soc. 2016, 138, 3501. [DOI] [PubMed] [Google Scholar]
- 28.Qin J; Zhou Z; Cui T; Hemming M; Meggers E, Enantioselective intramolecular C–H amination of aliphatic azides by dual ruthenium and phosphine catalysis. Chem. Sci. 2019, 10, 3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lyaskovskyy V; Suarez AIO; Lu H; Jiang H; Zhang XP; de Bruin B, Mechanism of Cobalt(II) Porphyrin-Catalyzed C–H Amination with Organic Azides: Radical Nature and H-Atom Abstraction Ability of the Key Cobalt(III)–Nitrene Intermediates. J. Am. Chem. Soc. 2011, 133, 12264. [DOI] [PubMed] [Google Scholar]
- 30.Goswami M; Lyaskovskyy V; Domingos SR; Buma WJ; Woutersen S; Troeppner O; Ivanović-Burmazović I; Lu H; Cui X; Zhang XP; Reijerse EJ; DeBeer S; van Schooneveld MM; Pfaff FF; Ray K; de Bruin B, Characterization of Porphyrin-Co(III)-’Nitrene Radical’ Species Relevant in Catalytic Nitrene Transfer Reactions. J. Am. Chem. Soc. 2015, 137, 5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuijpers PF; van der Vlugt JI; Schneider S; de Bruin B, Nitrene Radical Intermediates in Catalytic Synthesis. Chem. Eur. J. 2017, 23, 13819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wiese S; McAfee JL; Pahls DR; McMullin CL; Cundari TR; Warren TH, C–H Functionalization Reactivity of a Nickel–Imide. J. Am. Chem. Soc. 2012, 134, 10114. [DOI] [PubMed] [Google Scholar]
- 33.Dong Y; Lukens JT; Clarke RM; Zheng S-L; Lancaster KM; Betley TA, Synthesis, characterization and C–H amination reactivity of nickel iminyl complexes. Chem. Sci. 2020, 11, 1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carsch KM; DiMucci IM; Iovan DA; Li A; Zheng S-L; Titus CJ; Lee SJ; Irwin KD; Nordlund D; Lancaster KM; Betley TA, Synthesis of a copper-supported triplet nitrene complex pertinent to copper-catalyzed amination. Science 2019, 365, 1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilding MJT; Iovan DA; Wrobel AT; Lukens JT; MacMillan SN; Lancaster KM; Betley TA, Direct Comparison of C–H Bond Amination Efficacy through Manipulation of Nitrogen-Valence Centered Redox: Imido versus Iminyl. J. Am. Chem. Soc. 2017, 139, 14757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iovan DA; Betley TA, Characterization of Iron-Imido Species Relevant for N-Group Transfer Chemistry. J. Am. Chem. Soc. 2016, 138, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilding MJT; Iovan DA; Betley TA, High-Spin Iron Imido Complexes Competent for C–H Bond Amination. J. Am. Chem. Soc. 2017, 139, 12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baek Y; Hennessy ET; Betley TA, Direct Manipulation of Metal Imido Geometry: Key Principles to Enhance C–H Amination Efficacy. J. Am. Chem. Soc. 2019, 141, 16944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baek Y; Betley TA, Catalytic C–H Amination Mediated by Dipyrrin Cobalt Imidos. J. Am. Chem. Soc. 2019, 141, 7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hennessy ET; Betley TA, Complex N-Heterocycle Synthesis via Iron-Catalyzed, Direct C–H Bond Amination. Science 2013, 340, 591. [DOI] [PubMed] [Google Scholar]
- 41.Dong Y; Clarke RM; Porter GJ; Betley TA, Efficient C–H Amination Catalysis Using Nickel-Dipyrrin Complexes. J. Am. Chem. Soc. 2020, 142, 10996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dong Y; Clarke RM; Zheng S-L; Betley TA, Synthesis and electronic structure studies of a Cr-imido redox series. Chem. Comm. 2020, 56, 3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.King ER; Hennessy ET; Betley TA, Catalytic C−H Bond Amination from High-Spin Iron Imido Complexes. J. Am. Chem. Soc. 2011, 133, 4917. [DOI] [PubMed] [Google Scholar]
- 44.Desimoni G; Faita G; Jørgensen KA, C2-Symmetric Chiral Bis(Oxazoline) Ligands in Asymmetric Catalysis. Chem. Rev. 2006, 106, 3561. [DOI] [PubMed] [Google Scholar]
- 45.Gridnev ID; Dub PA, Enantioselection in Asymmetric Catalysis. CRC Press: 2016. [Google Scholar]
- 46.Ollevier T, Iron bis(oxazoline) complexes in asymmetric catalysis. Catal. Sci. 2016, 6, 41. [Google Scholar]
- 47.Evans DA; Woerpel KA; Hinman MM; Faul MM, Bis(oxazolines) as chiral ligands in metal-catalyzed asymmetric reactions. Catalytic, asymmetric cyclopropanation of olefins. J. Am. Chem. Soc. 1991, 113, 726. [Google Scholar]
- 48.Ghosh AK; Mathivanan P; Cappiello J, C2-Symmetric chiral bis(oxazoline)–metal complexes in catalytic asymmetric synthesis1Dedicated to Professor E. J. Corey with deep respect and sincere appreciation.1. Tetrahedron: Asymmetry 1998, 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsutsumi K; Itagaki K; Nomura K, Synthesis and Structural Analysis of Palladium(II) Complexes Containing Neutral or Anionic C2-Symmetric Bis(oxazoline) Ligands: Effects of Substituents in the 5-Position. ACS Omega 2017, 2, 3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Corey EJ; Zhe W, Enantioselective conversion of aldehydes to cyanohydrins by a catalytic system with separate chiral binding sites for aldehyde and cyanide components. Tetrahedron Lett. 1993, 34, 4001. [Google Scholar]
- 51.Walli A; Dechert S; Meyer F, Tautomerism in Bis(oxazoline)s. Eur. J. Org. Chem. 2013, 2013, 7044. [Google Scholar]
- 52.McElvain SM; Schroeder JP, Orthoesters and Related Compounds from Malono- and Succinonitriles. J. Am. Chem. Soc. 1949, 71, 40. [Google Scholar]
- 53.Denmark SE; Stiff CM, Effect of Ligand Structure in the Bisoxazoline Mediated Asymmetric Addition of Methyllithium to Imines. J. Org. Chem. 2000, 65, 5875. [DOI] [PubMed] [Google Scholar]
- 54.Denmark SE; Nakajima N; Nicaise OJC; Faucher A-M; Edwards JP, Preparation of Chiral Bisoxazolines: Observations on the Effect of Substituents. J. Org. Chem. 1995, 60, 4884. [Google Scholar]
- 55.Stoll S; Schweiger A, EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42. [DOI] [PubMed] [Google Scholar]
- 56.Drago RS, Physical Methods for Chemists. Saunders College Pub.: 1992. [Google Scholar]
- 57.Kogut E; Wiencko HL; Zhang L; Cordeau DE; Warren TH, A Terminal Ni(III)−Imide with Diverse Reactivity Pathways. J. Am. Chem. Soc. 2005, 127, 11248. [DOI] [PubMed] [Google Scholar]
- 58.Waterman R; Hillhouse GL, η2-Organoazide Complexes of Nickel and Their Conversion to Terminal Imido Complexes via Dinitrogen Extrusion. J. Am. Chem. Soc. 2008, 130, 12628. [DOI] [PubMed] [Google Scholar]
- 59.Harrold ND; Hillhouse GL, Strongly bent nickel imides supported by a chelating bis(N-heterocyclic carbene) ligand. Chem. Sci. 2013, 4, 4011. [Google Scholar]
- 60.Mindiola DJ; Hillhouse GL, Terminal amido and imido complexes of three-coordinate nickel. J. Am. Chem. Soc. 2001, 123, 4623. [DOI] [PubMed] [Google Scholar]
- 61.Iluc VM; Miller AJM; Anderson JS; Monreal MJ; Mehn MP; Hillhouse GL, Synthesis and Characterization of Three-Coordinate Ni(III)-Imide Complexes. J. Am. Chem. Soc. 2011, 133, 13055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Becke AD, Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar]
- 63.Lee C; Yang W; Parr RG, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter 1988, 37, 785. [DOI] [PubMed] [Google Scholar]
- 64.Vosko SH; Wilk L; Nusair M, Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 1980, 58, 1200. [Google Scholar]
- 65.Stephens PJ; Devlin FJ; Chabalowski CF; Frisch MJ, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623. [Google Scholar]
- 66.Schäfer A; Huber C; Ahlrichs R, Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829. [Google Scholar]
- 67.Schäfer A; Horn H; Ahlrichs R, Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571. [Google Scholar]
- 68.Allen DA; Tomaso AE; Priest OP; Hindson DF; Hurlburt JL, Mosher Amides: Determining the Absolute Stereochemistry of Optically-Active Amines. J. Chem. Educ. 2008, 85, 698. [Google Scholar]
- 69.Ichikawa A; Ono H; Mikata Y, Characteristic Conformation of Mosher’s Amide Elucidated Using the Cambridge Structural Database. Molecules 2015, 20, 12880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dale JA; Mosher HS, Nuclear magnetic resonance enantiomer regents. Configurational correlations via nuclear magnetic resonance chemical shifts of diastereomeric mandelate, O-methylmandelate, and .alpha.-methoxy-.alpha.-trifluoromethylphenylacetate (MTPA) esters. J. Am. Chem. Soc. 1973, 95, 512. [Google Scholar]
- 71.Dale JA; Dull DL; Mosher HS, .alpha.-Methoxy-.alpha.-trifluoromethylphenylacetic acid, a versatile reagent for the determination of enantiomeric composition of alcohols and amines. J. Org. Chem. 1969, 34, 2543. [Google Scholar]
- 72.Cramer SA; Hernández Sánchez R; Brakhage DF; Jenkins DM, Probing the role of an FeIV tetrazene in catalytic aziridination. Chem. Comm. 2014, 50, 13967. [DOI] [PubMed] [Google Scholar]
- 73.Cramer SA; Jenkins DM, Synthesis of Aziridines from Alkenes and Aryl Azides with a Reusable Macrocyclic Tetracarbene Iron Catalyst. J. Am. Chem. Soc. 2011, 133, 19342. [DOI] [PubMed] [Google Scholar]
- 74.Elsenbaumer RL; Mosher HS, Enantiomerically pure (R)-(+)-2-phenylethanol-2-d and −1,1,2-d3, and (S)-(+)-1-phenylethane-1-d, −1,2,-d2, −1,2,2-d3, and −1,2,2,2-d4. J. Org. Chem. 1979, 44, 600. [Google Scholar]
- 75.Anslyn EV; Dougherty DA; Dougherty EV; Books US, Modern Physical Organic Chemistry. University Science Books: 2006. [Google Scholar]
- 76.Simmons EM; Hartwig JF, On the Interpretation of Deuterium Kinetic Isotope Effects in C–H Bond Functionalizations by Transition-Metal Complexes. Angew. Chem. Int. Ed. 2012, 51, 3066. [DOI] [PubMed] [Google Scholar]
- 77.Ogliaro F; Harris N; Cohen S; Filatov M; de Visser SP; Shaik S, A Model “Rebound” Mechanism of Hydroxylation by Cytochrome P450: Stepwise and Effectively Concerted Pathways, and Their Reactivity Patterns. J. Am. Chem. Soc. 2000, 122, 8977. [Google Scholar]
- 78.Varela-Álvarez A; Yang T; Jennings H; Kornecki KP; Macmillan SN; Lancaster KM; Mack JBC; Du Bois J; Berry JF; Musaev DG, Rh2(II,III) Catalysts with Chelating Carboxylate and Carboxamidate Supports: Electronic Structure and Nitrene Transfer Reactivity. J. Am. Chem. Soc. 2016, 138, 2327. [DOI] [PubMed] [Google Scholar]
- 79.Lang K; Torker S; Wojtas L; Zhang XP, Asymmetric Induction and Enantiodivergence in Catalytic Radical C–H Amination via Enantiodifferentiative H-Atom Abstraction and Stereoretentive Radical Substitution. J. Am. Chem. Soc. 2019, 141, 12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.White RE; Miller JP; Favreau LV; Bhattacharyya A, Stereochemical dynamics of aliphatic hydroxylation by cytochrome P-450. J. Am. Chem. Soc. 1986, 108, 6024. [DOI] [PubMed] [Google Scholar]
- 81.Ishii S; Zhao S; Helquist P, Stereochemical Probes of Intramolecular C−H Insertion Reactions of Iron-Carbene Complexes. J. Am. Chem. Soc. 2000, 122, 5897. [Google Scholar]
- 82.Alberti MN; Vassilikogiannakis G; Orfanopoulos M, Stereochemistry of the Singlet Oxygenation of Simple Alkenes: A Stereospecific Transformation. Org. Lett. 2008, 10, 3997. [DOI] [PubMed] [Google Scholar]
- 83.Lu G; Liu RY; Yang Y; Fang C; Lambrecht DS; Buchwald SL; Liu P, Ligand–Substrate Dispersion Facilitates the Copper-Catalyzed Hydroamination of Unactivated Olefins. J. Am. Chem. Soc. 2017, 139, 16548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rassolov VA; Pople JA; Ratner MA; Windus TL, 6–31G* basis set for atoms K through Zn. J. Chem. Phys. 1998, 109, 1223. [Google Scholar]
- 85.Francl MM; Pietro WJ; Hehre WJ; Binkley JS; Gordon MS; DeFrees DJ; Pople JA, Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654. [Google Scholar]
- 86.Petersson GA; Bennett A; Tensfeldt TG; Al-Laham MA; Shirley WA; Mantzaris J, A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J. Chem. Phys. 1988, 89, 2193. [Google Scholar]
- 87.Petersson GA; Al-Laham MA, A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J. Chem. Phys. 1991, 94, 6081. [Google Scholar]
- 88.Mo Y, Rotational barriers in alkanes. WIREs Comput Mol Sci 2011, 1, 164. [Google Scholar]
- 89.Shen TYH, W D; Huang D; Gonzalez J Preparation of 7-azabicyclo[2.2.1]heptane and heptene derivatives as cholinergic receptor ligands. US 5817679, Oct 6, 1998. [Google Scholar]
- 90.Khan MA Preparation of spiro-lactam and bis-spiro-lactam NMDA receptor modulators and uses thereof. WO 2019152681, Aug 8, 2019. [Google Scholar]
- 91.Giblin GM; Witty DR; Macpherson DT; Stanway SJ; Wilson A; Gleave L Preparation of spiro derivatives as voltage-gated sodium channel modulators. WO 2013179049, Dec, 5, 2013. [Google Scholar]
- 92.Shi F; Tao Z-L; Luo S-W; Tu S-J; Gong L-Z, Scaffold-Inspired Enantioselective Synthesis of Biologically Important Spiro[pyrrolidin-3,2′-oxindoles] with Structural Diversity through Catalytic Isatin-Derived 1,3-Dipolar Cycloadditions. Chem. Eur. J. 2012, 18, 6885. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.