

Abstract
The molecular underpinnings of the uncontrolled release of proinflammatory cytokines and chemokines (‘cytokine storm’), which can cause organ damage and even mortality, are not completely understood. Furthermore, targeted therapeutic options to dampen such hyperinflammation are scarce. Here, we highlight the ways in which technological advances have set the stage for a new age of synergy between experimental and computational researchers to guide the discovery of novel therapeutic targets for modulating hyperinflammation.
Hyperinflammation, or aberrant activation of inflammatory signaling pathways above the level needed to control disease, can be initiated by the innate immune response and inflammatory cell death in the context of infection or sterile inflammation. While innate immune activation and inflammatory signaling provide the first line of defense against infection and damage, overactivation of these mechanisms can be pathological. As a result, the uncontrolled production of proinflammatory cytokines and chemokines [collectively known as ‘cytokine storm’ (CS) or ‘cytokine release syndrome’] can cause tissue and organ damage, as well as a life-threatening pathological condition [1–3].
The term CS has been used for many years in a broad sense, without a specific definition based on cytokine thresholds or mechanistic characteristics, to describe situations where the presence of high concentrations of cytokines was associated with pathology. Due to this ambiguity, studies comparing cytokine amounts across diseases have introduced controversy over whether this term can be applied in certain contexts; for example, IL-6 concentrations were found to be lower in the serum of patients with coronavirus disease 2019 (COVID-19) than in those with sepsis in a meta-analysis [4], leading to debates on whether CSs occur in COVID-19. However, the CS was recently mechanistically defined as a life-threatening condition caused by excessive production of cytokines mediated by a form of inflammatory cell death (with key features of pyroptosis, apoptosis, and/or necroptosis, but not accounting for any of these pathways alone) called PANoptosis (regulated by the PANoptosome); this has been observed in murine models of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, as well as sepsis, hemophagocytic lympho-histiocytosis, and cytokine shock [3]. Overall, the CS is associated with a variety of conditions, including severe infections, autoimmune and inflammatory disorders (e.g., rheumatoid arthritis), medical interventions (transplantation), drug administration, and anaphylaxis [1,3]. Moreover, different pathologies can result in similar proinflammatory cytokine profiles while their etiologies dramatically differ. For example, influenza viruses (IAV), SARS-CoV(−2), and hantavirus infect different cell types in different parts of the mammalian respiratory system, but can result, in certain instances, in acute lung injury, sepsis, and, potentially, CSs [2]. Therefore, understanding the mechanistic underpinnings of the hyperinflammatory response is a crucial step towards identifying new and putative life-saving therapeutic strategies.
Moreover, paracrine signaling, involving immune and nonimmune cells, can also form positive feedback loops that amplify and maintain certain pathways of immune responses [5]. Cells participating in these feedback loops secrete extracellular ligands in response to the signals present in their environment, which in turn are sensed by other cells in these loops. For example, during IAV-induced inflammation in human lungs, macrophages release IL-1 and TNF in response to granulocyte-macrophage colony-stimulating factor from alveolar epithelial cells [2,6].
In the era of big data, the steadily increasing availability of different -omics data (genomics, transcriptomics, proteomics, etc.) allows the refined development and application of computational systems biology approaches to immunology (Figure 1). This large amount of data enables more sophisticated computational models generating more accurate predictions. For instance, using single-cell sequencing data (i.e., single-cell RNA-seq, single-cell assay for transposase-accessible chromatin sequencing) it is possible to identify novel cellular subpopulations and provide a better molecular characterization of these cells. Indeed, single-cell sequencing platforms, such as 10XGenomics [7], allow the profiling of thousands of cells. Moreover, with the current progress in methodology of spatially resolved transcriptomics [8], scientists can obtain location-specific gene expression data, enabling the development of computational models that consider the spatial information of different cell types. These models can be employed to identify key molecules in inflammatory processes and design novel and more efficient strategies for modulating hyperinflammation in the context of diverse disease pathologies, including infectious and inflammatory diseases. Hence, the time is ripe to foster the synergy between experimental and computational researchers to accelerate the discovery of new, promising therapeutic targets for clinical intervention. This synergy must entail a coordinated research effort from the onset, aiming to build an adequate and accurate computational model, define the necessary input data, and design follow-up experiments to fully validate the computational predictions. To this end, we foresee some rate-limiting steps to enact such a synergy, namely, technological limitations, such as current scale of single-cell phosphoproteomics [9] and resolution of tissue spatial information [8], shortage of good experimental models impeding the validation of computational predictions, and mutual unawareness of the advances in experimental and computational methods.
Figure 1. Synergy between computational and experimental efforts to tackle hyperinflammation-associated morbidity and mortality.
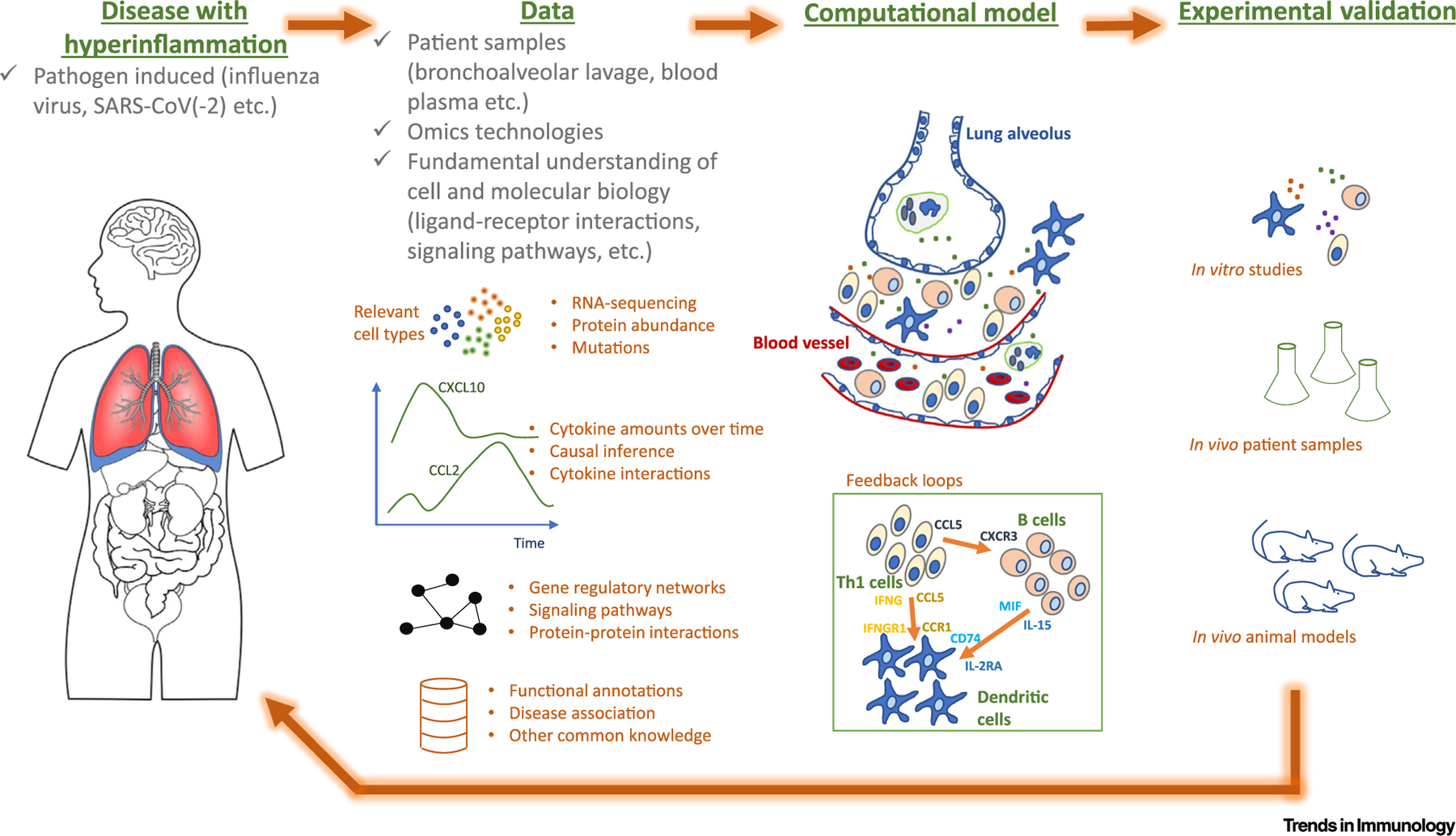
Patient samples are processed with the most advanced -omics technologies and are partnered with a fundamental understanding of cell and molecular biology and immunology to yield accurate biological predictions together with computational modeling (e.g., reconstruction of the feedback loops during cytokine storms). Experimentalists coordinate building adequate and accurate computational models and validate the predictions (using in vitro/vivo experimental models). Experimental trials generate new hypotheses and renew the modeling paradigms.
A clear example of how experimental and computational approaches support each other includes the discovery of modulators of hyperinflammation and was recently shown by two individual studies [10,11]. Independently, these studies predicted Toll-like receptor (TLR) 2 as a putative immunomodulator of COVID-19-induced lung hyperinflammation: one report was solely based on a computational systems biology model that relied on scRNAseq data [10], while the other used in vitro and in vivo mouse screening of key innate pattern recognition receptors activating inflammatory cytokine production [11]. In the computational study, a holistic model of intercellular ligand–receptor interactions, intracellular signaling pathways, and transcriptional regulation allowed the reconstruction of the positive feedback loops maintaining and amplifying the hyperinflammatory immune response in critically ill patients with COVID-19 [10].
Computational simulation of the perturbation of the feedback loops allowed the prioritization of inflammatory regulators based on their capacity to disrupt pathological yet preserve physiological loops. Thus, TLR2 was identified as one of the target molecules implicated in modulating the uncontrolled immune response in severe COVID-19 patients [10]. In an independent study [11], increased expression of TLR2 in the peripheral blood of human patients correlated with disease severity during COVID-19, with TLR2 expression increasing from healthy patients to those with moderate, severe, or critical COVID-19. Experimental evaluation of the specific role of TLR2 in disease showed that TLR2 responded to the SARS-CoV-2 E protein to activate the production of proinflammatory cytokines; indeed, inhibition or deletion of TLR2 in human or murine macrophages, respectively, reduced cytokine release compared with controls. These cytokines included TNF-α and IFN-γ [11,12], known to be key mediators of the CS via PANoptosis (see earlier) [3,11,]). Murine macrophages treated with TNF-α and IFN-γ activated biochemical markers of PANoptosis and treatment of mice with this combination or with SARS-CoV-2 infection led to mortality that was characterized by key features of the CS and COVID-19, including lymphopenia, thrombocytopenia, and increased numbers of caspase-3- and TUNEL-positive cells in the tissue [12]. Treatment of K18-hACE2 transgenic mice with a TLR2 inhibitor, oxPAPC, protected the animals against SARS-CoV-2-mediated inflammatory cytokine production and mortality [11]. This further suggested that TLR2 might act as a key modulator of COVID-19-induced hyperinflammation, providing a proof-of-concept for the utility of targeting TLR2 to modulate inflammation [11].
In another COVID-19-related study [13], sepsis and severe COVID-19 manifestations were associated with increased number of suppressive myeloid cells relative to controls. Using computational tools and scRNA-seq data, the authors identified an expanded CD14+ monocyte state: a gene expression program that correlated with sepsis severity and was activated in monocytes from severe COVID-19 patients [13]. They further showed that plasma from such patients induced myelopoiesis in healthy human bone marrow stem and progenitor cells and the expression program in monocytes and neutrophils induced differentiation from these progenitors [13].
Additional studies have also shown the utility of combining computational and experimental approaches to understand inflammatory processes. In the context of hepatic ischemia-reperfusion (IR), time-dependent changes in cytokine concentrations were correlated to computationally predict interactions between inflammatory mediators [14]. These interactions grew in number and complexity following IR in the liver. This computational analysis implicated IL-17A as an early central driver of inflammation and inducer of a self-propagating CS. The subsequent in vivo validation in mice confirmed that IL-17A promoted liver injury after reperfusion by sustaining neutrophil recruitment to areas of inflammation, identifying IL-17A as a potential therapeutic target for liver damage after hepatic IR [14]. In a recent study, a network-based model enabled the inference of a novel control network of chemokine interactions, which was used to identify crucial mediators of acute systemic inflammation in post-traumatic injury [15]. Namely, this model predicted that CCL2, CXCL9, and CXCL10 formed a regulatory core upstream of IL-6. These cytokine concentrations correlated with the severity of injury and the magnitude of systemic inflammation. Thus, the model accurately predicted hospitalization time of blunt trauma survivors based on molecular patterns [15] and suggested potential treatment strategies.
The development of strategies aiming to mitigate the CS is essential for reducing the mortality associated with the severe form of various infectious and inflammatory diseases. Thus, computational modeling can certainly inform traditional experimental approaches to identify targets that may reduce the hyperinflammatory immune response. Indeed, in a growing number of studies, systems immunology approaches are being successfully employed to help predict novel therapeutic targets for modulating uncontrolled immune responses. Nevertheless, the full potential of computational modeling to systematically assist experimental endeavors has not been exploited.
We envision new challenges when building computational models in the context of hyperinflammation. These include access to tissue-specific data to model local inflammatory signatures, which cannot always be inferred from blood samples, integration of time-series data to determine predictors and effectors of hyperinflammation, and timing of intervention strategies to modulate hyperinflammation while maintaining the beneficial immune response.
Currently, advances in systems biology and the increasing generation of large amounts of big -omics data are contributing to the development of more sophisticated computational models. We posit that the key to designing novel and more efficient therapeutic intervention strategies for reducing hyperinflammation is to synergize experimental and computational efforts from the onset. We argue that computational approaches and experimental validation should represent a key partnership in biomedical research, needed for identifying fundamental therapeutic targets.
Acknowledgments
Research in the Kanneganti lab is supported by grants from the US National Institutes of Health (AI101935, AI124346, AI160179, AR056296, and CA253095) and the American Lebanese Syrian Associated Charities to T.D-K. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. I.P. and A.d.S. were supported by the Fonds National de la Recherche (FNR), Luxembourg (references: SysBioCOVID19 & 11662681/InTRinSIC).
Footnotes
Declaration of interests
No interests are declared.
References
- 1.Lukan N (2020) “Cytokine storm”, not only in COVID-19 patients. Mini-review. Immunol. Lett 228, 38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tisoncik JR et al. (2012) Into the eye of the cytokine storm. Am. Soc. Microb 76, 16–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karki R and Kanneganti T-D (2021) The ‘cytokine storm’: molecular mechanisms and therapeutic prospects. Trends Immunol 42, 681–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leisman DE et al. (2020) Cytokine elevation in severe and critical COVID-19: a rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med 8, 1233–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramilowski J et al. (2015) A draft network of ligand–receptor-mediated multicellular signalling in human. Nat. Commun 6, 7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ito Y et al. (2015) Influenza induces IL-8 and GM-CSF secretion by human alveolar epithelial cells through HGF/c-Met and TGF-α/EGFR signaling. Am. J. Physiol. Lung Cell Mol. Physiol 308, L1178–L1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zheng G et al. (2017) Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marx V (2021) Method of the year: spatially resolved transcriptomics. Nat. Methods 18, 9–14 [DOI] [PubMed] [Google Scholar]
- 9.Qin X et al. (2020) Cell-type-specific signaling networks in heterocellular organoids. Nat. Methods 17, 335–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jung S et al. (2021) Leveraging systems biology for predicting modulators of inflammation in patients with COVID-19. Sci. Adv 7, eabe5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng M et al. (2021) TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol 22, 829–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karki R et al. (2021) Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 184, 149–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reyes M et al. (2021) Plasma from patients with bacterial sepsis or severe COVID-19 induces suppressive myeloid cell production from hematopoietic progenitors in vitro. Sci. Transl. Med 13, eabe9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tohme S et al. (2019) Computational analysis supports IL-17A as a central driver of neutrophil extracellular trap-mediated injury in liver ischemia reperfusion. J. Immunol 202, 268–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Azhar N et al. (2021) A putative “chemokine switch” that regulates systemic acute inflammation in humans. Sci. Rep 11, 9703. [DOI] [PMC free article] [PubMed] [Google Scholar]