

SUMMARY
Nonalcoholic steatohepatitis (NASH) is becoming one of the leading causes of hepatocellular carcinoma (HCC). Sorafenib is the only first-line therapy for advanced HCC despite its serious adverse effects. Here, we report that at an equivalent of approximately one-tenth the clinical dose for HCC, sorafenib treatment effectively prevents the progression of NASH in both mice and monkeys without any observed significant adverse events. Mechanistically, sorafenib’s benefit in NASH is independent of its canonical kinase targets in HCC, but involves the induction of mild mitochondrial uncoupling and subsequent activation of AMP–activated protein kinase (AMPK). Collectively, our findings demonstrate a previously unappreciated therapeutic effect and signaling mechanism of low-dose sorafenib treatment in NASH. We envision that this new therapeutic strategy for NASH has the potential to translate into a beneficial anti-NASH therapy with fewer adverse events than is observed in the drug’s current use in HCC.
Graphical Abstract
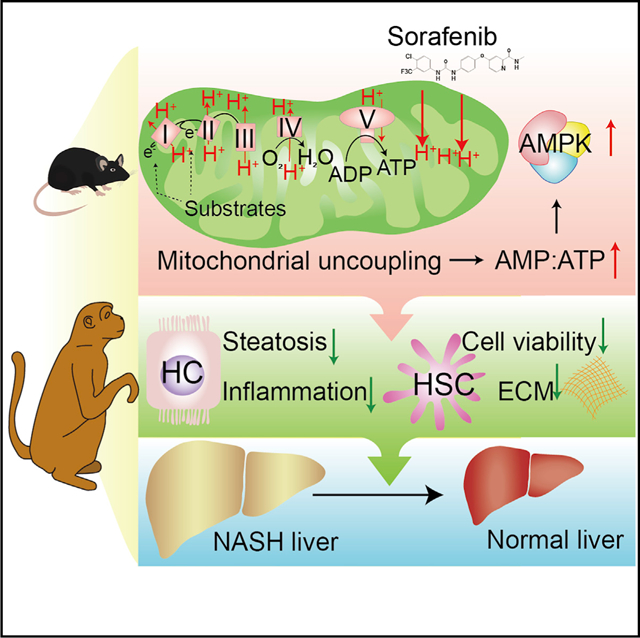
In Brief
Jian et al. show that low-dose sorafenib safely and effectively suppressed NASH progression in both mice and monkeys. Mechanistically, induction of mitochondrial uncoupling and subsequent AMPK activation primarily underlies the therapeutic effects of sorafenib in NASH.
INTRODUCTION
Sorafenib is currently the only US FDA-approved first-line therapy for advanced hepatocellular carcinoma (HCC) (Heimbach et al., 2018). Its potent inhibition of multiple kinases, in particular Raf-mitogen-activated extracellular-signal-regulated protein kinase (ERK) kinase (MEK)-ERK signaling, contributes to its antitumor effect by decreasing tumor cell proliferation, inducing apoptosis, and suppressing angiogenesis (Liu et al., 2006; Wan et al., 2004; Wilhelm et al., 2004). However, the efficacy of sorafenib in HCC therapy is far from satisfactory. An oral treatment of sorafenib at 400 mg twice daily (bid) only prolongs patient median overall survival by 2.8 months (Llovet et al., 2008). Moreover, at this dosage, sorafenib causes severe and extensive adverse events including fatigue, anorexia, diarrhea, rash and desquamation, hand-foot skin reaction, and hypertension. These adverse effects significantly limit the tolerance of the drug in the clinic and compromise its beneficial outcome (Minami et al., 2008; Strumberg et al., 2007).
The prevalence of nonalcoholic steatohepatitis (NASH) has increased sharply in recent decades and has become one of the leading pathogenic promoters of end-stage liver diseases, including cirrhosis, HCC, and liver failure (Cai et al., 2019a; Cohen et al., 2011; Zhang et al., 2018). It has been estimated that NASH will become the leading cause for liver transplantation by 2030 (Shaker et al., 2014; Wang et al., 2017). The pathogenic progression of NASH is a continuum, characterized by hepatic steatosis, hepatocyte ballooning, lobular inflammation, and fibrosis (Bai et al., 2019; Cai et al., 2019b; Ji et al., 2018; Zhao et al., 2017). Previous studies clearly demonstrated an anti-fibrotic function of sorafenib and uncovered the underlying mechanisms on multiple in vivo and in vitro models; e.g., toxin injection, bile duct ligation, and chronic hepatitis B (Su et al., 2015; Thabut et al., 2011; Wang et al., 2010). In a rat NASH model induced by choline-deficient high-fat diet and diethylnitrosamine (DEN), sorafenib treatment also effectively attenuated fibrosis formation (Stefano et al., 2015). However, whether sorafenib could render benefits on other pathological NASH features like steatosis and inflammation and the underlying mechanism(s) remains to be explored.
In this study, we tested the efficacy of a new treatment strategy of sorafenib in a mouse model of NASH-HCC by initiating low-dose sorafenib treatment at the early stage of HCC. Remarkably, we found that at an equivalent dose as low as one-tenth of the current clinical application sorafenib effectively blocked HCC occurrence in this model. More importantly, this low-dose treatment of sorafenib markedly ameliorated major hallmarks of NASH, including hepatic steatosis, inflammation, and fibrosis, without inducing any detectable adverse events. Encouragingly, the therapeutic benefit of low-dose sorafenib on NASH treatment was also observed in monkeys. Therefore, our findings suggest that sorafenib could be repurposed for the treatment of NASH in the clinic.
RESULTS AND DISCUSSION
Low-Dose Sorafenib Blocks Induction of NASH-HCC without Significant Adverse Effects
We tested the efficacy of sorafenib therapy in the pathological progression of NASH to HCC in mice treated with DEN and high-fat/high-cholesterol (HFHC) diet (Liu et al., 2018) by initiating sorafenib administration at 10 weeks after initiation of HFHC feeding, a time point when NASH is well established but HCC has yet to occur (Kishida et al., 2016). The dose gradient of sorafenib was selected as 10, 15, and 30 mg/kg/2 days, which are all lower than a previously reported dose of 30 mg/kg/day in prior mouse HCC models (Enomoto et al., 2017; Kuczynski et al., 2015), and tumor formation was assessed after 8 weeks of treatment (Figure 1A). The human equivalent doses (HEDs) of sorafenib used in mice (10, 15, and 30 mg/kg/2 days) were 45.0, 67.5, and 135 mg/2 days for a 60-kg person (Table S1), respectively, based on body surface area normalization (Cheung et al., 2009; FDA, 2005; Reagan-Shaw et al., 2008), which were largely lower than that applied in clinic for HCC therapy (800 mg/day).
Figure 1. Efficacy and Safety of Low-Dose Sorafenib in Treating NASH-HCC.
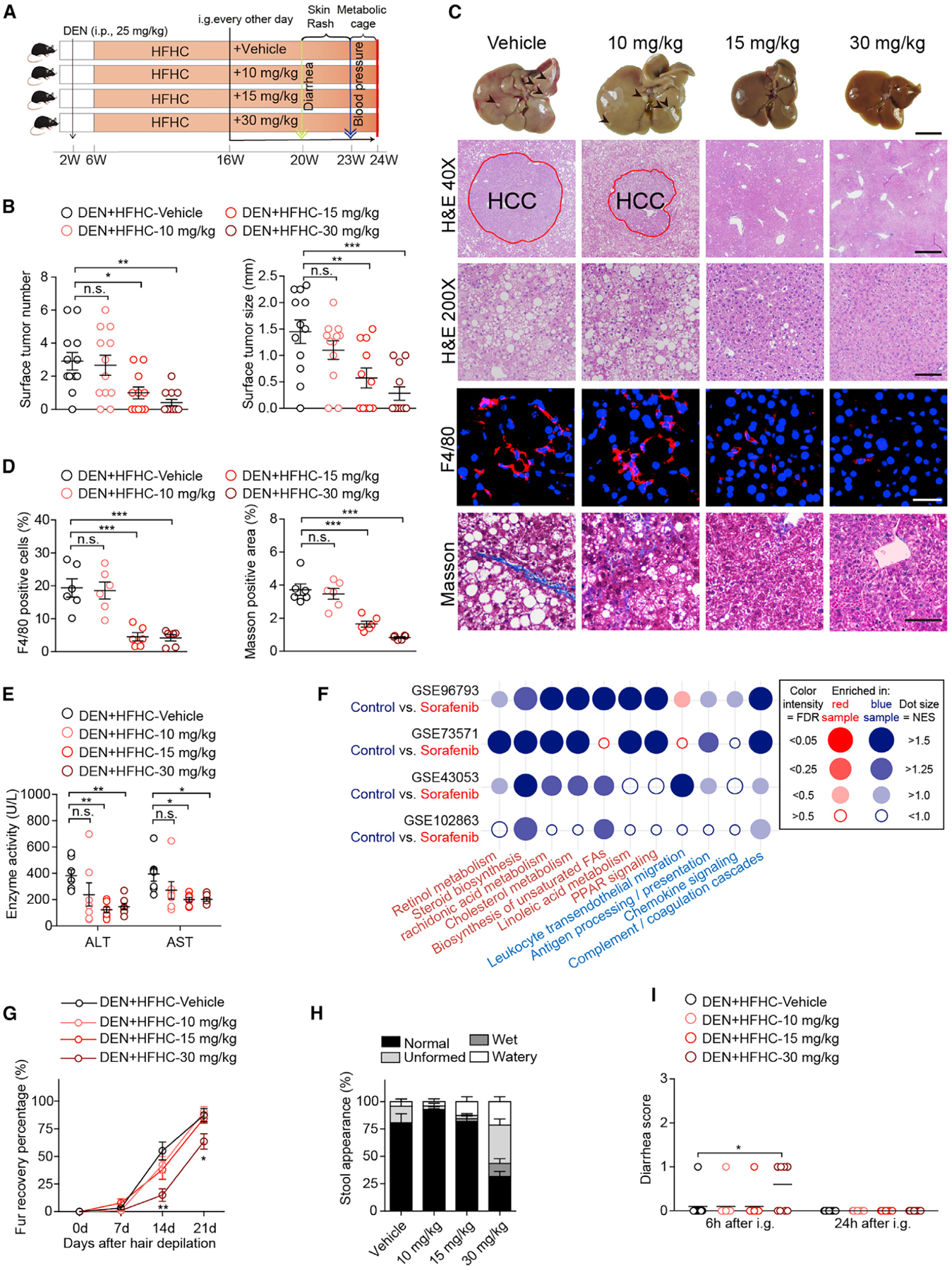
(A) Scheme for the experimental strategy on DEN-injected and HFHC-diet-fed mice treated with vehicle, 10, 15, or 30 mg/kg sorafenib every other day. i.p., intraperitoneal injection. i.g., intragastric administration.
(B) Surface tumor number and size in each indicated group. n = 11 mice for 15 mg/kg sorafenib group and n = 12 mice for other groups.
(C) Representative macroscopic and histological images of livers (scale bar, 1 cm) and liver sections. H&E (40×, scale bar 500 μm; 200×, scale bar 100 μm), F4/80 (scale bar, 20 μm), and Masson (scale bar, 50 μm) staining images of liver sections from mice treated with vehicle or indicated doses of sorafenib. Arrowheads and red circles indicate hepatocellular carcinomas. n = 6 mice per group.
(D) Quantitative results for F4/80 and Masson staining shown in (C). n = 6 mice per group.
(E) Serum ALT and AST levels of mice in the indicated groups. n = 7 mice per group.
(F) Dot plot representing pairwise gene set enrichment analysis (GSEA) comparison of publicly available transcriptomic data obtained in sorafenib-treated HCC cells in the GEO database. Dot color indicates that the gene signature set is enriched in the group with corresponding font color. NES, normalized enrichment score. FDR, false discovery rate.
(G) Fur recovery rate of mice in the indicated groups after wax depilation of the dorsal hair. n = 11 mice per group. **p < 0.01 versus vehicle group at 14 days; *p < 0.05 versus vehicle group at 21 days.
(H) Stool appearance of mice in the indicated groups. n = 10 mice per group.
(I) Diarrhea score of mice in the indicated groups. n = 10 mice per group.
The data in (B), (D), and (E) were presented as the means ± SEMs and analyzed by one-way ANOVA. A Kruskal-Wallis test was used for statistical analysis of the data in (G) and (I). *p < 0.05; **p < 0.01; ***p < 0.001; n.s., no significance, p > 0.05. See also Figure S1 and Tables S1 and S2.
We found that sorafenib at the doses of 15 and 30 mg/kg/2 days significantly reduced both tumor incidence and size, while the 10 mg/kg/2 days group showed negligible effect compared to the vehicle controls (Figure 1B). Notably, in addition to its expected inhibition of tumorigenesis, we observed that sorafenib at 15 and 30 mg/kg/2 days dramatically suppressed the pathological features of NASH, including hepatic steatosis, inflammation, and fibrosis, compared to the vehicle controls (Figures 1C and 1D). The improvement in histopathology also correlated with lower alanine transaminase (ALT) and aspartate aminotransferase (AST) levels in mice treated with sorafenib at 15 and 30 mg/kg/2 days than in vehicle-treated mice (Figure 1E). Importantly, based on the analysis of publicly available transcriptomic data in the GEO database, we also found that sorafenib treatment exerted a potent and broad-based impact on lipid metabolism and inflammatory pathways in hepatocarcinoma cells (Figure 1F). These data strongly suggested that low-dose sorafenib has a potent protective effect against NASH progression.
To determine the possibility of toxicity of low-dose sorafenib therapy, we included kidney function, physical activity, food intake, and blood pressure in our analysis. Sorafenib treatment at all three doses did not produce any significant impact on these parameters compared to the vehicle controls (Figures S1A–S1D). Except for the liver, no significant changes were observed in the indexes (ratio of organ weight to body weight) and histomorphology of other organs in any group (Table S2; Figure S1E). Skin rash was not presented in any group (Figure S1F), but blunted fur regeneration and mild diarrhea were detected in the 30 mg/kg/2 days cohort comparing to vehicle-treated controls (Figures 1G–1I and S1F). Thus, sorafenib at a low dose of 15 mg/kg/2 days is both effective and safe in resolving major NASH symptoms and inhibiting HCC occurrence in our mouse model.
Low-Dose Sorafenib Protects against Diet-Induced NASH in Mice
To directly evaluate the potential benefit of low-dose sorafenib treatment for NASH progression, we subjected mice to HFHC feeding for 10 weeks, followed by oral gavage of sorafenib at 15 mg/kg/2 days along with HFHC feeding for another 8 weeks (Figure 2A). Sorafenib treatment (15 mg/kg/2 days) completely reversed the increase in body weight, liver weight, and liver index in HFHC-fed mice, but had negligible impact on the normal chow (NC)-diet-fed mice treated by the same regiment (Figure 2B). In line with the above effect on HFHC feeding, diet-induced increases in serum ALT and AST levels were significantly lower in the HFHC-sorafenib group compared to the HFHC-vehicle group (Figure 2C). Moreover, hepatic lipid accumulation, inflammatory cell infiltration into the liver, circulating inflammatory response, and liver fibrosis were all markedly mitigated by sorafenib treatment compared to vehicle treatment in HFHC-fed mice (Figures 2D–2G). The comparable postprandial serum triglyceride (TG) level, intestinal lipid content, and fecal lipid content between vehicle- and sorafenib-treated mice (Figures S2A–S2C) suggests that sorafenib showed negligible influence on intestinal lipid absorption.
Figure 2. Sorafenib Protects against HFHC-Diet-Induced Hepatic Steatosis, Inflammation, and Fibrosis in Mice.
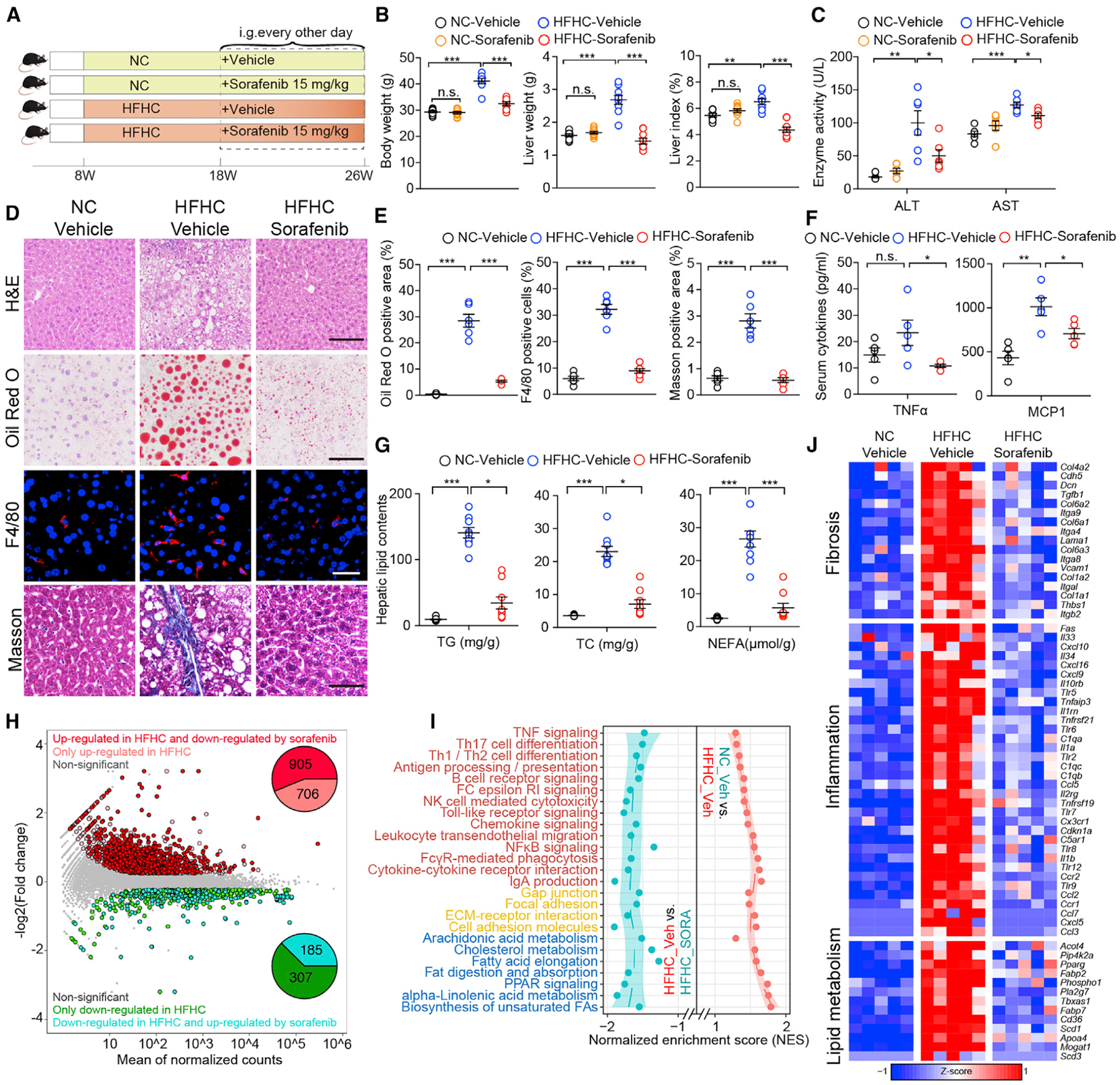
(A) Scheme for the experimental strategy on NC- or HFHC-diet-fed mice treated with vehicle or sorafenib (15mg/kg/2 days).
(B) Body weight, liver weight, and liver index of NC or HFHC-diet-fed mice treated with vehicle or sorafenib. n = 9 mice per group. A one-way ANOVA was used for statistical analysis.
(C) Serum ALT and AST levels of NC- or HFHC-diet-fed mice treated with vehicle or sorafenib. n = 6 mice per group. A Student’s t test was used for statistical analysis.
(D) Representative images of liver sections stained with H&E (scale bar, 100 μm), Oil Red O (scale bar, 100 μm), F4/80 (scale bar, 20 μm), and Masson (scale bar, 50 μm). n = 6 mice per group.
(E) Quantitative results for Oil Red O, F4/80, and Masson staining shown in (D). n = 6 mice per group. A one-way ANOVA was used for statistical analysis.
(F) Serum concentration of cytokines TNFα and MCP1 of NC or HFHC-diet-fed mice treated with vehicle or sorafenib. n = 5 mice per group. A Student’s t test was used for statistical analysis.
(G) Liver TG (triglyceride), TC (total cholesterol), and NEFA (non-esterified fatty acid) concentrations of NC- or HFHC-diet-fed mice treated with vehicle or sorafenib. n = 9 mice per group. For statistical analysis, one-way ANOVA was used for NEFA and Kruskal-Wallis test was used for TG and TC.
(H) The scatterplot showing the fold change of all genes in the RNA-seq dataset obtained from vehicle- or sorafenib-treated mice on NC or HFHC diet, with differentially expressed genes (DEGs) shown with corresponding font color. The pie charts indicate the number of DEGs regulated by sorafenib and HFHC feeding. n = 5 mice per group.
(I) KEGG pathway enrichment results showing the cellular pathways involved in inflammation, lipid metabolism, and fibrosis, which were upregulated by HFHC feeding but downregulated by sorafenib treatment. The font color of pathways related to inflammation, fibrosis, and lipid metabolism are in red, yellow, and blue, respectively. n = 5 mice per group.
(J) Heatmap showing the expression profile of genes related to inflammation, lipid metabolism, and fibrosis that were upregulated by HFHC diet feeding but reversed by sorafenib treatment based on the RNA-seq dataset. n = 5 mice per group.
The data in (B), (C), (E), (F), and (G) were presented as the means ± SEMs. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., no significance, p > 0.05. See also Figure S2.
To determine the specific impact of sorafenib at the molecular level, we performed transcriptomic analysis in liver tissue obtained from NC-vehicle, HFHC-vehicle, and HFHC-sorafenib groups. Unsupervised hierarchical clustering analysis showed a clear separation of NC-vehicle and HFHC-vehicle samples, indicating successful establishment of the HFHC diet model. The closer distance of the HFHC-sorafenib cluster to the NC-vehicle than to the HFHC-vehicle group indicates that sorafenib treatment significantly ameliorated the diet-induced detrimental gene expression profile (Figure S2D). Among the differentially expressed genes (DEGs) detected between the NASH (HFHC-vehicle) and the normal (NC-vehicle) liver, sorafenib treatment reversed 905 of 1611 upregulated genes and 185 of 492 downregulated genes induced by HFHC (Figure 2H). Gene set enrichment analysis revealed that the genes affected by sorafenib treatment were enriched for inflammation, lipid metabolism, and fibrosis (Figures 2I and 2J).
We further tested the effect of sorafenib in a high fat diet (HFD)-induced nonalcoholic fatty liver disease (NAFLD) mouse model (Figure S2E). Consistent with our observations with the HFHC-induced NASH model, sorafenib treatment effectively lowered liver weight, improved liver function, and dissipated lipid accumulation and inflammation as compared to the vehicle controls (Figures S2F–S2L). Furthermore, the RNA sequencing (RNA-seq)-based global gene expression profiling also demonstrated the potent impact of sorafenib on hepatic lipid metabolism and inflammatory response (Figures S2M–S2O).
Overall, our results demonstrate that low-dose sorafenib treatment is capable of resolving major pathological features of NASH, including attenuation of steatosis, inflammation, and fibrosis.
Sorafenib Ameliorates NASH Independent of Its Canonical Targets
In order to explore the molecular mechanism(s) underlying the anti-NASH effect of sorafenib, we investigated the effect of sorafenib treatment on free fatty acid (FFA) challenge in L02 hepatocytes. Sorafenib treatment dose-dependently reduced the level of lipid accumulation in L02 hepatocytes in response to high levels of palmitic acid and oleic acid (PAOA) based on Oil Red O staining and lipid content measurements (Figures S3A and S3B). Meanwhile, palmitic acid (PA)-stimulated expression of inflammatory cytokines such as interleukin-6 (IL-6) and interleukin-8 (IL-8) was significantly blunted by sorafenib treatment compared to DMSO treatment (Figure S3C). These data are consistent with the in vivo observations above, suggesting that sorafenib targets hepatocyte lipid metabolism and inflammation to exert its cytoprotective effect.
Sorafenib is known to inhibit tumor cell proliferation and angiogenesis by, respectively, targeting Raf kinases and receptor tyrosine kinases (RTKs; e.g., vascular endothelial growth factor receptor [VEGFR] 1–3 and platelet-derived growth factor receptor β [PDGFRβ]) (Wan et al., 2004; Wilhelm et al., 2004). To clarify whether these tyrosine kinases participate in the sorafenib-mediated anti-NASH effect, we first assessed the impact of sorafenib on the activities of these kinases and their downstream signaling cascades. Unexpectedly, we found that the activities of various kinases, including VEGFR2, PDGFRβ, and Raf-MEK-ERK cascades, were not influenced by sorafenib treatment in HFHC-fed mouse livers (Figures S3D and S3E). Further, genetic ablation of BRAF or CRAF in L02 hepatocytes neither protected hepatocytes from FFA-induced lipid accumulation and inflammation, nor impacted the protective effect of sorafenib (Figures 3A–3H). Our pharmacologic studies applying pazopanib (multi-kinase inhibitor targeting VEGFR1–3, PDGFR, c-KIT, and so on), ZM336372 (Raf inhibitor), SU1498 (VEGFR inhibitor), and Tyrphostin AG1296 (PDGFR inhibitor) further validated the negligible influence of those kinases on sorafenib function in hepatocyte lipid accumulation and the inflammatory response (Figure S3F–S3H). Therefore, the anti-NASH effect of sorafenib cannot be explained by inhibition of its canonical kinase targets in HCC.
Figure 3. Effects of Sorafenib on Lipid Accumulation and Inflammation Are Not Dependent on B-Raf or C-Raf.
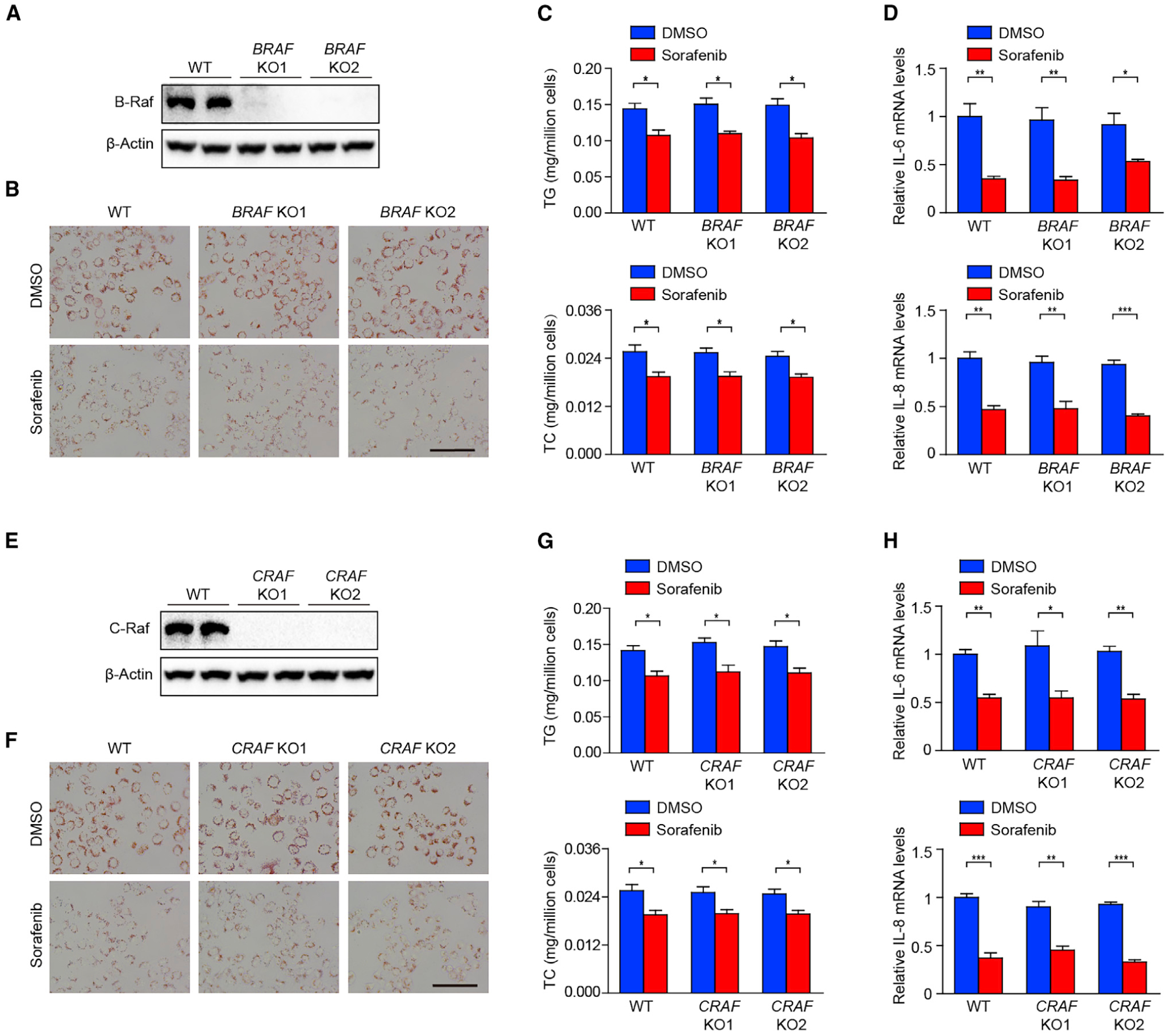
(A) Immunoblotting analysis of B-Raf in wild type (WT) and two strains of BRAF knockout (KO) L02 hepatocytes.
(B and C) Oil Red O staining (B), and TG and TC contents (C) of WT and two strains of BRAF KO L02 hepatocytes stimulated by palmitic acid and oleic acid (PAOA), together with DMSO or sorafenib. n = 3 replicates. Scale bar, 100 μm.
(D) qPCR analyses of pro-inflammatory factors IL-6 and IL-8 mRNA levels in WT and two strains of BRAF KO L02 hepatocytes stimulated by palmitic acid (PA) with DMSO or sorafenib. n = 3 replicates.
(E) Immunoblotting analysis of C-Raf in WT and two strains of CRAF KO L02 hepatocytes stimulated by PA and treated with DMSO or sorafenib.
(F and G) Oil Red O staining (F), and TG and TC contents (G) of WT and two strains of CRAF KO L02 hepatocytes stimulated by PAOA, together with DMSO or sorafenib. n = 3 replicates. Scale bar, 100 μm.
(H) qPCR analyses of IL-6 and IL-8 mRNA levels in WT and two strains of CRAF KO L02 hepatocytes stimulated by PA and treated with DMSO or sorafenib. n = 3 replicates.
The mRNA expression levels of target genes were normalized to that of ACTB (β-Actin). The data in (C), (D), (G), and (H) were presented as the means ± SEMs and analyzed by Student’s t test. *p < 0.05; **p < 0.01; ***p < 0.001. See also Figure S3.
Sorafenib Ameliorates NASH via AMPK Activation
To uncover the key downstream effector(s) that contribute to sorafenib-induced protection from NASH, we performed a label-free phospho-proteomic analysis on PA-treated hepatocytes in the presence or absence of sorafenib treatment. Based on the differentially phosphorylated peptides and their enrichment of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, we found that the differentially phosphorylated peptides were primarily enriched in mechanistic target of rapamycin (mTOR), AMP–activated protein kinase (AMPK), and Erb-B2 receptor tyrosine kinase (ErbB) signaling (Figure 4A). It’s well known that mTOR is a canonical negative downstream target of AMPK signaling (Gwinn et al., 2008; Inoki et al., 2003). Western blotting validated that the AMPK signaling was suppressed in HFHC-fed mouse livers compared to NC-fed controls (Figure S4A). Sorafenib markedly activated AMPK signaling in both PA-treated hepatocytes and HFHC-fed mouse livers (Figure 4B). However, ErbB signaling did not respond to sorafenib treatment (Figure S3E). By immunohistochemistry staining, we observed a periportal zonation of p-AMPKα (Thr172) and a perivenous zonation of p-mTOR (Ser2448) overlapped with lipid accumulation, which is consistent with a recent report (Adebayo Michael et al., 2019). Sorafenib treatment remarkably enhanced AMPK but reduced mTOR activity without altering their zonation (Figure 4C). Moreover, by integrating the results from the phospho-proteomic analysis and the transcriptomic characterization (Figure 2), we found a strong correlation between the differentially expressed genes and the differentially phosphorylated AMPK signaling molecules induced by the sorafenib treatment as illustrated by the STRING protein interaction network database and PubMed database correlation analysis (Figure 4D).
Figure 4. AMPK Is Required for the Effects of Sorafenib on Lipid Accumulation and Inflammation In Vitro.
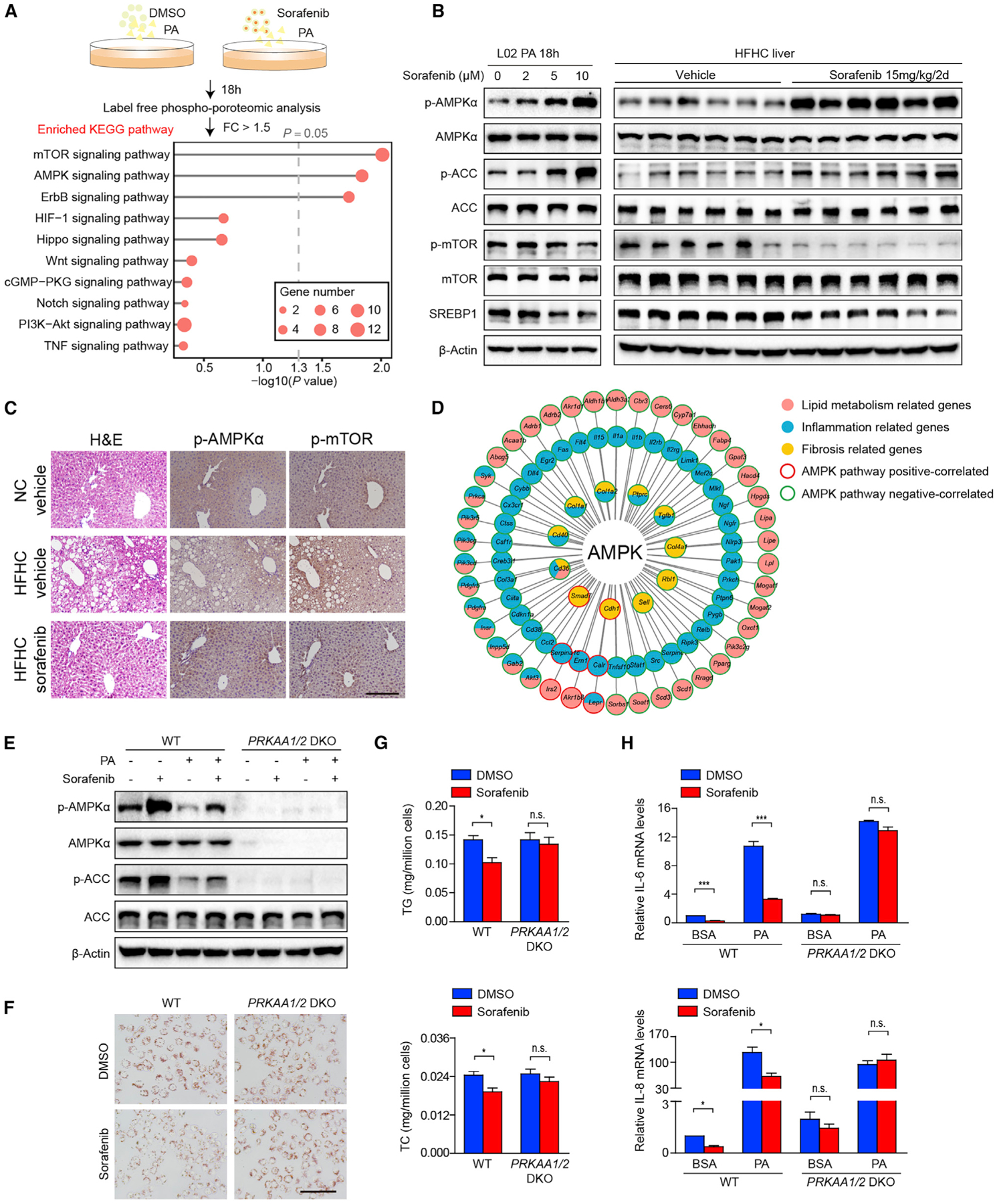
(A) Scheme (upper) showing the phospho-proteomic analysis of L02 hepatocytes stimulated by PA and treated with DMSO or sorafenib. The top ten enriched signaling pathways are shown (bottom). The gray lines represent −log10 (p value) for the enriched KEGG pathways. The dashed gray line indicates p value = 0.05, whereas the red circles represent the number of DEGs. FC, fold change.
(B) Immunoblotting analyses of total and phosphorylated AMPKα, ACC, mTOR, and total SREBP1 proteins in L02 hepatocytes stimulated by PA with indicated concentrations of sorafenib (left, n = 3 replicates) and in the liver tissues of HFHC-fed mice treated with vehicle or 15 mg/kg/2 days sorafenib (right, n = 6 mice per group).
(C) H&E and immunohistochemistry staining of serial liver sections from HFHC-fed mice in indicated groups show the inverse zonation of p-AMPKα (Thr172) and p-mTOR (Ser2448). n = 6 mice per group. Scale bar, 100 μm.
(D) STRING and PubMed database correlation analyses show that differentially phosphorylated AMPK signaling molecules are correlated to a variety of differentially expressed genes, which are related to inflammation, lipid metabolism, and fibrosis in RNA-seq data obtained on livers of HFHC-fed mice.
(E) Immunoblotting analyses of total and phosphorylated AMPKα and ACC proteins in WT and PRKAA1/2 (encoding AMPKα1/α2) double-knockout (DKO) L02 hepatocytes challenged by BSA vehicle (−) or PA (+), with or without sorafenib. n = 3 replicates.
(F and G) Oil Red O staining (F), and TG and TC contents (G) of WT and PRKAA1/2 DKO L02 hepatocytes challenged by PAOA with DMSO or sorafenib. n = 3 replicates. Scale bar, 100 μm.
(H) qPCR analyses of IL-6 and IL-8 mRNA levels in WT and PRKAA1/2 DKO L02 hepatocytes challenged by BSA or PA and treated with DMSO or sorafenib. The mRNA expression levels of target genes were normalized to that of ACTB (β-Actin). n = 3 replicates.
The data in (G) and (H) were presented as the means ± SEMs and analyzed by Student’s t test. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., no significance, p > 0.05. See also Figure S4.
Given the well-established role of AMPK in the pathogenesis of NASH (Smith et al., 2016b), we speculated that the targeted AMPK activation by sorafenib is the key downstream mechanism of its protective function against NASH. Notably, PRKAA1 and PRKAA2 (encoding AMPKα1 and α2) double-knockout (DKO) L02 hepatocytes showed a complete abolishment of sorafenib-mediated inhibition on lipid accumulation and inflammatory activation (Figures 4E–4H). Furthermore, co-treatment of an AMPK inhibitor, Compound C (CC), also largely eliminated the inhibitory effects of sorafenib on lipid accumulation and inflammation in hepatocytes (Figures S4B–S4E). In line with these observations in hepatocytes, sorafenib treatment also significantly inhibited the proliferative and migratory activities of hepatic stellate cells (HSCs) (Figures S4F and S4G). Again, sorafenib treatment markedly activated AMPK signaling in the HSCs but had no effect on Raf signaling (Figure S4H).
Finally, pre-treatment of mice with CC one week prior to sorafenib administration (Figure 5A) effectively blocked AMPK activity in liver (Figure 5B) and significantly abrogated the protective effect of sorafenib against the development of hepatic steatosis, inflammation, and fibrosis in HFHC-fed mice (Figures 5C–5H). Furthermore, RNA-seq data also illustrated a global reversal of gene expression in lipid metabolism, inflammation, and fibrosis-related pathways following CC treatment (Figure 5I). Collectively, pharmacological and genetic evidence strongly implicate AMPK activation as a necessary downstream signaling effector for sorafenib-mediated protection against NASH.
Figure 5. AMPK Activation Is Required for the Effects of Sorafenib on HFHC-Induced NASH in Mice.
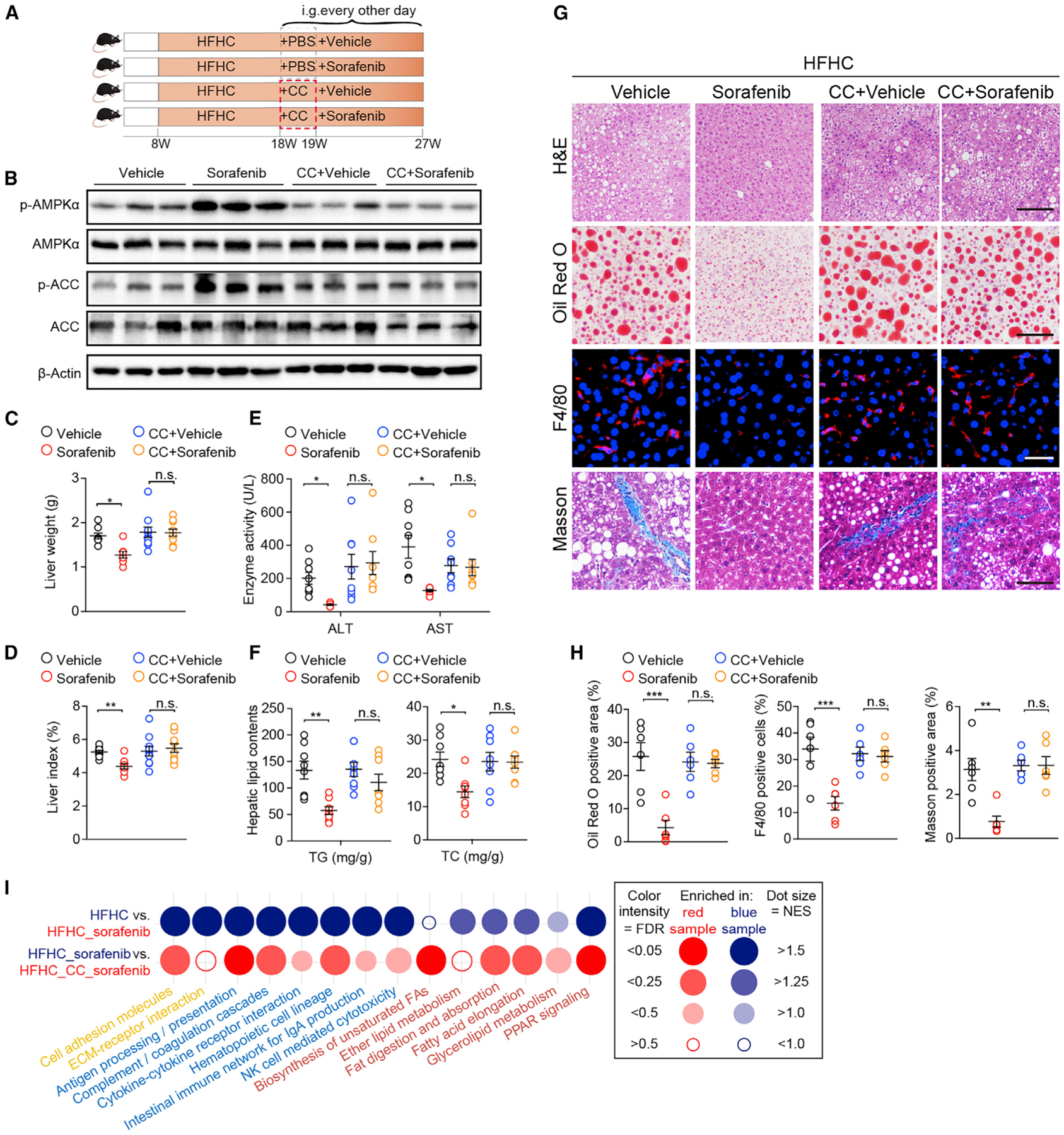
(A) Scheme showing the experimental procedure on mice fed a HFHC diet and treated with vehicle or sorafenib (15 mg/kg/2 days) in the absence or presence of Compound C (CC, 10 mg/kg/2 days).
(B) Immunoblotting analyses of total and phosphorylated AMPKα and ACC in livers from mice in the indicated groups. n = 3 mice per group.
(C and D) Liver weight (C) and liver index (D) of mice in indicated groups. n = 8 mice for vehicle and sorafenib group while n = 10 mice for CC-vehicle and CC-sorafenib group.
(E) Enzyme ALT and AST levels of mice in the indicated groups. n = 8 mice per group.
(F) Liver TG and TC concentrations of mice in the indicated groups. n = 8 mice per group.
(G) Staining of liver sections for H&E (scale bar, 100 μm), Oil Red O (scale bar, 100 μm), F4/80 (scale bar, 20 μm), and Masson (scale bar, 50 μm). n = 6 mice per group.
(H) Quantitative results for Oil Red O, F4/80, and Masson staining shown in (G). n = 6 mice per group.
(I) Dot plot representing pairwise GSEA comparison of transcriptomic data obtained in mouse livers from HFHC-vehicle, HFHC-sorafenib, and HFHC-CC-sorafenib groups. Dot color indicates that the gene signature set is enriched in the group with corresponding font color. NES, normalized enrichment score. FDR, false discovery rate. n = 5 mice per group.
The data in (C), (D), (E), (F), and (H) were presented as the means ± SEMs and analyzed by one-way ANOVA. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., no significance, p > 0.05.
Sorafenib Activates AMPK by Inducing Mitochondrial Uncoupling
While we found that sorafenib potently activated AMPK activity, none of the known upstream activators were affected, including liver kinase B1 (LKB1), calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2), and transforming growth factor-beta-activated kinase 1 (TAK1), nor the negative regulators such as Protein Phosphatase 2A (PP2A) and PP2C (Figure 6A; Inoki et al., 2012). However, sorafenib treatment did result in a higher AMP/ATP ratio in primary mouse hepatocytes and mouse livers compared to the corresponding vehicle treatment (Figure 6B).
Figure 6. Sorafenib Activates AMPK by Inducing Mitochondrial Uncoupling.
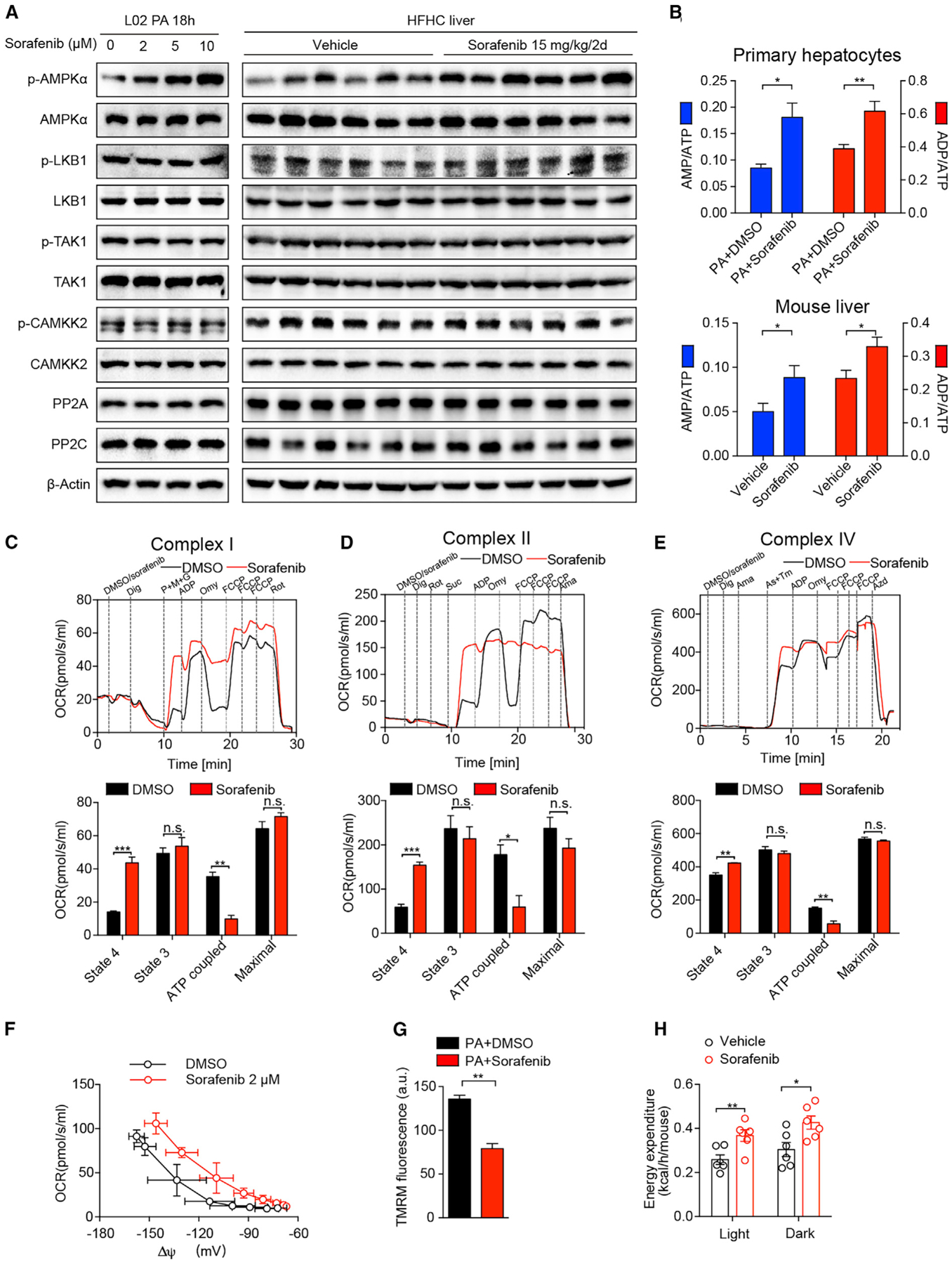
(A) Immunoblotting analyses of PP2A, PP2C, and total and phosphorylated AMPKα, LKB1, TAK1, and CAMKK2 proteins in L02 hepatocytes (left, n = 3 replicates) stimulated by PA with indicated concentrations of sorafenib, and in the liver tissues (right) of vehicle- or sorafenib-treated mice on HFHC diet (n = 6 mice per group).
(B) AMP/ATP and ADP/ATP ratios in mouse primary hepatocytes (upper) stimulated by PA with DMSO or sorafenib (n = 6 mice per group) and in mouse liver tissues treated with vehicle or sorafenib (bottom) (n = 7 mice per group).
(C) Representative (upper) and statistical (bottom) results of complex-I-related respiration on digitonin (Dig)-permeabilized primary mouse hepatocytes treated with DMSO or sorafenib. OCR, oxygen consumption rate. P+M+G, complex I substrates pyruvate, malate, and glutamate. Omy, F1Fo ATP synthase inhibitor oligomycin. FCCP, mitochondrial uncoupler. Rot, complex I inhibitor rotenone. n = 3 replicates.
(D) Representative (upper) and statistical (bottom) results of complex-II-related respiration on digitonin-permeabilized primary mouse hepatocytes treated with DMSO or 10 μM sorafenib. Rotenone (Rot) is used to pre-inhibit complex I. Suc, complex II substrates succinate. Ama, complex III inhibitor antimycin A. n = 3 replicates.
(E) Representative (upper) and statistical (bottom) results of complex-IV-related respiration measured from digitonin-permeabilized primary mouse hepatocytes treated with DMSO or 10 μM sorafenib. As+Tm, ascorbate (As) and N,N,N’,N’-Tetramethyl-p-phenylenediamine dihydrochloride (TMPD). Sodium azide (Azd) is a complex IV inhibitor. n = 3 replicates.
(F) Dependence of proton leak rate (measured as the respiration in the presence of oligomycin) on membrane potential in the presence or absence of sorafenib. n = 3 replicates.
(G) Statistical results of mitochondrial membrane potential indicator TMRM fluorescence on PA-stimulated primary mouse hepatocytes with or without sorafenib treatment. n = 3 replicates. a.u., arbitrary unit.
(H) Energy expenditure of HFHC-fed mice treated with vehicle or sorafenib (15 mg/kg/2 days). n = 6 mice per group.
The data in (B), (C), (D), (E), (G), and (H) were presented as the means ± SEMs and analyzed by Student’s t test. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., no significance, p > 0.05. See also Figure S5.
To determine the mechanism by which sorafenib increases the AMP/ATP ratio, we assessed the respiratory capacity of electron transfer chain complexes on permeabilized primary mouse hepatocytes. Interestingly, compared with DMSO, sorafenib significantly elevated the state 4 respiration induced by complex I substrates pyruvate, malate, and glutamate (P+M+G) in the absence of ADP (Figure 6C), indicating induction of a proton leak. In contrast, the state 3 respiration stimulated by ADP and the maximal respiration induced by carbonyl cyanide p-trifluoro-methoxyphenyl hydrazone (FCCP) were comparable between the sorafenib-treated and control samples (Figure 6C). Therefore, the loss of ATP-production-related respiration was not due to a direct inhibition of complex I respiratory capacity per se, but by inducing mitochondrial uncoupling. Consistent with this notion, sorafenib-treated cells exhibited higher state 4 respiration and lower ATP production-related respiration compared to DMSO-treated controls when complex II- or IV-specific respiration was assessed. In both cases, ADP-stimulated respiration and maximal respiratory capacity were not significantly affected by sorafenib as compared with DMSO-treated controls (Figures 6D and 6E). Thus, sorafenib treatment induces proton leak in mitochondria without significant impact on the intrinsic capacity of electron transfer chain. Consistent with the elevation in the AMP/ATP ratio and the reduction of ATP-coupled respiration, sorafenib treatment significantly reduced ATP content in PA-treated cultured primary mouse hepatocytes compared to DMSO treatment (Figure S5A).
To gain direct evidence of mitochondrial uncoupling, we performed a mitochondrial proton conductance assay. In the presence of sorafenib, proton leak coupled respiration was elevated at a given membrane potential, indicating a higher proton conductance (Figure 6F). In line with the concept of mitochondrial uncoupling, sorafenib increased fatty-acid-induced respiration, especially the fatty-acid-induced uncoupled respiration that cannot be inhibited by the F1Fo ATP synthase (complex V) inhibitor oligomycin (Figure S5B). Moreover, sorafenib decreased mitochondrial membrane potential as revealed by tetramethylrhodamine methyl ester (TMRM) staining (Figure 6G). As a result of membrane depolarization, mitophagy was remarkably activated by sorafenib treatment (Figure S5C). However, sorafenib had negligible impact on mitochondrial morphology, as the percentages of cells with tubular, tubular and fragmented, and fragmented mitochondria were comparable between sorafenib-treated and vehicle-treated cells under both control and PA-stimulated conditions (Figure S5D). For in vivo evidence of mitochondrial uncoupling, we found that HFHC-fed mice receiving sorafenib had a higher energy expenditure rate than the vehicle-treated controls, in both the light and dark cycle (Figure 6H).
Low-Dose Sorafenib Is Efficacious and Safe in Monkeys with NASH
To demonstrate the potential clinical translation of our findings, we evaluated the impact of a low-dose sorafenib therapy on the development of NASH in a nonhuman primate model. Twelve cynomolgus monkeys (Macaca fascicularis) with spontaneously developed and biopsy-proven NASH features were chosen and randomly divided into two groups and treated with either vehicle or sorafenib at 1 mg/kg by intravenous injection every 3 days. The selected dose was extrapolated from that of mice (15 mg/kg/2 days) using the body surface area normalization method with consideration of sorafenib’s oral bioactivity (38%–49%). The HED of sorafenib dose for monkey was 56.4–72 mg/3 days for a 60-kg person (Table S1), which is less than one-tenth of the dose (800 mg/day) used for HCC therapy in clinic. Monkeys were fed with a HFHC diet to promote further NASH progression during the process of treatment (Figure 7A).
Figure 7. Efficacy of Sorafenib in Monkeys with NASH.
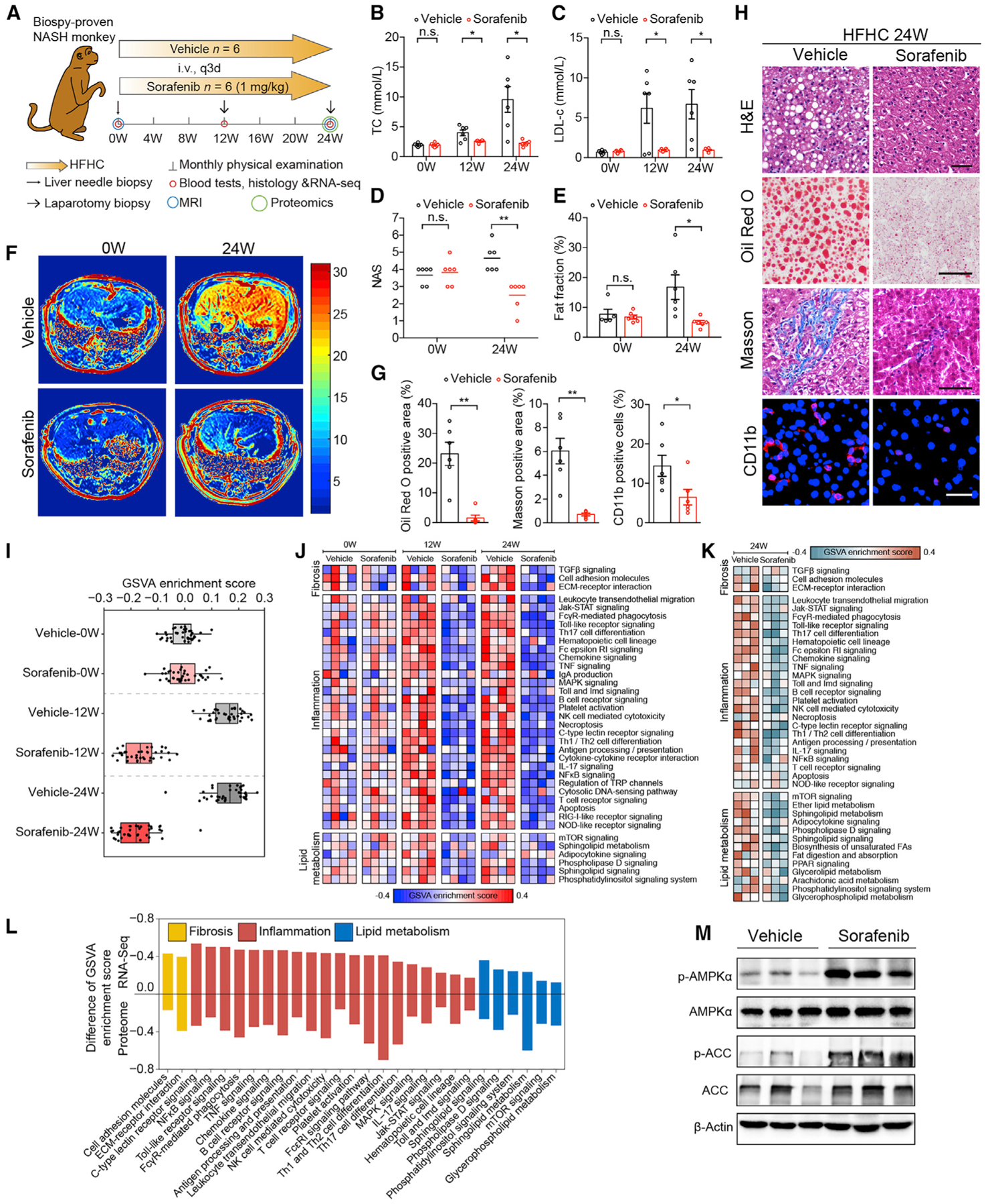
(A) Schematic diagram of experimental strategy on cynomolgus monkey treated with vehicle or sorafenib (1mg/kg/3 days). i.v., intravenous injection. q3d, every three days. MRI, magnetic resonance imaging.
(B and C) Serum TC (B) and LDL-c concentrations (C) of monkeys treated with vehicle or sorafenib (1mg/kg/3 days) at 0, 12, and 24 weeks. n = 6 monkeys per group.
(D) NASs in vehicle- and sorafenib-treated monkeys at 0 and 24 weeks. n = 6 monkeys per group.
(E) Monkey liver was scanned and reconstructed by MRI and the fat fraction was calculated. n = 6 monkeys per group.
(F) Representative images of liver MRI from monkey before and after 24-week treatment with vehicle or sorafenib. n = 6 monkeys per group.
(G) Quantitative results for Oil Red O, Masson, and CD11b staining of liver sections from monkeys treated with vehicle or sorafenib for 24 weeks. n = 6 monkeys per group.
(H) Representative histological and immunofluorescence images of monkey liver sections stained by H&E (scale bar, 50 μm), Oil Red O (scale bar, 50 μm), Masson (scale bar, 50 μm), and CD11b (scale bar, 20 μm). n = 6 monkeys per group.
(I) Gene set variation analysis (GSVA) score of inflammation, lipid metabolism, and fibrosis-related pathways from pairs comparisons of monkeys with same weeks of vehicle or sorafenib treatment. n = 4 monkeys per group.
(J) Heatmap showing the GSVA-enriched pathways related to fibrosis, inflammation, and lipid metabolism based on RNA-seq dataset obtained as in (I).
(K) Heatmap showing the GSVA-enriched pathways related to fibrosis, inflammation, and lipid metabolism based on proteomic dataset obtained on livers from monkeys from vehicle or sorafenib groups at 24 weeks. n = 3 monkeys per group.
(L) Histogram showing inhibition degree of inflammation, lipid metabolism, and fibrosis based on GSVA enrichment score difference of each related pathway in RNA-seq and proteomic dataset at 24 weeks. Pathways with mean GSVA score difference ≤ −0.1 were shown.
(M) Immunoblotting analyses of total and phosphorylated AMPKα and ACC in livers from monkeys treated with vehicle or sorafenib for 24 weeks. n = 3 monkeys per group.
The data in (B), (C), (E), and (G) were presented as the means ± SEMs and analyzed by Student’s t test. A Mann-Whitney U test was used for statistical analysis of the data in (D). *p < 0.05; **p < 0.01; n.s., no significance, p > 0.05. See also Figure S6 and Table S1.
Following 24 weeks of HFHC feeding, monkeys in the vehicle-treated groups developed increased plasma total cholesterol (TC) and low-density lipoprotein cholesterol (LDL-c) contents and NAFLD activity score (NAS). In contrast, these pathological features were ameliorated in the sorafenib-treated monkeys (Figures 7B–7D). Magnetic resonance imaging data demonstrated that sorafenib treatment markedly blocked the deterioration of hepatic lipid accumulation induced by HFHC diet (Figures 7E and 7F). Histological analysis showed significant improvement in hepatic ballooning, hepatic steatosis, fibrosis, and inflammation in the sorafenib-treated monkeys compared to the vehicle-treated controls (Figures 7G and 7H).
Furthermore, transcriptomic analyses of liver samples collected at 0, 12, and 24 weeks unveiled a progressive and extensive reprogramming of gene expression profiles between the vehicle- and sorafenib-treated monkeys (Figure 7I). Notably, the cellular pathways significantly affected by sorafenib treatment also include lipid accumulation, inflammation, and fibrosis (Figure 7J). The broad effects of sorafenib on NASH-related pathological features were further validated by proteomic analysis using liver samples collected at 24 weeks from the sorafenib-treated and vehicle-treated monkeys (Figure 7K). Combined analysis of RNA-seq and proteomic data revealed the consistency of sorafenib-affected pathways involved in inflammation, lipid metabolism, and fibrosis (Figure 7L). In line with the observations in hepatocytes and mice, AMPK activity was remarkably increased in the liver of the sorafenib-treated monkeys compared to the vehicle-treated controls (Figure 7M).
During the treatment period of sorafenib, we did not observe any significant impact on body weight, BMI, waist hip rate (WHR), blood pressure, or plasma TG and HDL-c levels (Figures S6A–S6E). The general organ function, as reflected by plasma levels of urea and creatinine for renal function; ALT, AST, and alkaline phosphatase (ALP) for liver function; and creatine kinase (CK), creatine kinase-MB (CKMB), and lactate dehydrogenase (LDH) for muscle and heart injury, was comparable between sorafenib-treated and vehicle-treated monkeys (Figures S6F–S6H). In addition, we did not observe adverse events as noted in the clinic, including diarrhea and skin rashes in either group.
As aforementioned, the HEDs of the doses we used in mice and monkeys for NASH treatment were relatively low compared to the clinical dose for HCC treatment. Consistently, our pharmacokinetic data showed that the area under the curve yield by 1 mg/kg of sorafenib in monkeys (4.73 ± 0.43 mg×h/L) was about 10% of the clinical range (43 ~ 76.5 mg×h/L) (Strumberg et al., 2007). Safety data for sorafenib from phase I clinical trials indicate that no toxicities were presented at doses lower than 100 mg bid (Awada et al., 2005; Strumberg et al., 2005). Therefore, both clinical safety data and our results support that this low dose of sorafenib might be an effective therapy for NASH patients with minimal adverse risk.
As a multi-kinase inhibitor, sorafenib has been reported to affect numerous processes that are important in HCC and liver injury (Shi et al., 2011; Su et al., 2015; Wan et al., 2004; Wilhelm et al., 2004). Our data clarified that independent from its canonical kinase targets, AMPK signaling activation is important for sorafenib function in NASH. The benefits of AMPK activation for NASH therapy could be achieved by the regulation of fatty acid metabolism homeostasis, which contributes to the relief of inflammatory responses (Jeon, 2016). AMPK activation also directly phosphorylates caspase-6 to inhibit lipotoxicity-induced hepatocellular apoptosis in NASH (Zhao et al., 2020). Moreover, resolution of fibrosis (Jiang et al., 2017) through the AMPK-mTOR pathway (Jeon, 2016) might also be a therapeutic action of AMPK in hepatic fibrosis. However, the benefit of mTOR inhibition for liver injury is controversial. Our observation that sorafenib inhibited mTOR activity (Figure 4D) was in line with previous reports that mTOR inhibition can revert metabolic disorders (Cornu et al., 2013). However, persistent mTORC1 inhibition by liver-specific Raptor ablation results in worsening of liver damage and tumorigenesis (Umemura et al., 2014). This paradox indicates that the homeostasis of mTOR activity is critical for liver pathogenesis. We speculate that proper timing and dosing of sorafenib for moderate mTOR inhibition and/or mTOR-independent signaling events downstream of AMPK activation might explain the beneficial effects of sorafenib in NASH.
To our knowledge, this is the first time that sorafenib has been shown to be a mitochondrial uncoupler, though impairment of mitochondrial function has been implicated in its cancer-killing ability (Chiou et al., 2009; Fiume et al., 2011). Here, our data suggest that, under the condition of nutrient overload, mild mitochondrial uncoupling induced by low-dose sorafenib “burns” out excess nutrients, activates AMPK, and affords protection against metabolic disease, notably NASH. Mitochondrial uncoupling, which is defined as a futile proton influx without ATP generation, has long been viewed as a therapeutic target for metabolic diseases. For instance, the mitochondrial uncoupler 2,4-dinitrophenol (DNP) was approved for obesity in the 1930s, but was withdrawn from the market due to risk of fatal hyperthermia. Recently, Gerald Shulman’s group has developed several liver-targeted derivatives of DNP, which produce mild hepatic mitochondrial uncoupling and safely reverse hepatic steatosis, hepatic inflammation, and liver fibrosis in rodent and non-human primate models (Goedeke et al., 2019; Perry et al., 2013; Perry et al., 2015). Moreover, several chemicals, such as salicylate (Smith et al., 2016a) and niclosamide ethanolamine (Tao et al., 2014), have been shown to improve hepatic steatosis and diabetes by inducing mitochondrial uncoupling. Collectively, these studies support our findings that sorafenib ameliorates NASH by acting as a mitochondrial uncoupler while providing a compelling argument that low-dose sorafenib treatment may offer therapeutic benefit for other metabolic disorders.
Limitations of Study
Although we have provided both in vitro and in vivo evidence demonstrating the role of sorafenib on mitochondrial uncoupling, the direct target and actions of sorafenib on the mitochondrion remains unknown. Further, we have not directly benchmarked low-dose sorafenib to other mild mitochondrial uncouplers or other anti-NASH therapeutics in these studies to determine relative efficacies or safety profiles, but it should be noted that unlike other such agents reported to date, sorafenib is an approved drug, whereas currently there are no approved medications for the treatment of NASH. Finally, while we suggest the HED for NASH therapy can be as low as 10% of current clinical dosage based on the effective doses used in mice and monkeys, it is important to note that the HED calculated by the body surface area normalization method cannot be directly applied in patients with NASH. The effectiveness, dosage, and safety profile of sorafenib for NASH therapy in patients remains to be determined in future clinical trials.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Hongliang Li (lihl@whu.edu.cn).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
All RNA sequencing data reported in this study have been deposited to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database. The accession numbers for the data obtained on HFHC-fed mice, HFD-fed mice, HFHC-fed mice treated with compound C and sorafenib in combination and NASH monkeys are: SRA: PRJNA613854, PRJNA613938, PRJNA613940, PRJNA614574, respectively.
The phospho-proteomic data obtained on PA-treated L02 hepatocytes has been deposited to the ProteomeXchange Consortium via the proteomics identification (PRIDE) partner repository and the accession number is: PRIDE: PXD018214. The proteomic data obtained on NASH monkeys has also been deposited to the ProteomeXchange Consortium via the PRIDE partner repository and the accession number is: PRIDE: PXD018256.
The publicly available datasets from sorafenib-treated HCC cells can be found at GEO: GSE96793, GSE73571, GSE43053, GSE102863. All major software and code used to analyze these datasets are referenced the KEY RESOURCES TABLE.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-β-Actin, dil: 1/1000 | Abcam | Cat#ab8226; RRID: AB_306371 |
| Anti-phospho-AMPKα(Thr172), dil: 1/1000 for WB, dil: 1/50 for IHC | Cell Signaling Technology | Cat#2535; RRID:AB_331250 |
| Anti-AMPKα, dil: 1/1000 | Cell Signaling Technology | Cat#2603; RRID:AB_490795 |
| Anti-phospho-Acetyl-CoA Carboxylase (Ser79), dil: 1/1000 | Cell Signaling Technology | Cat#3661; RRID:AB_330337 |
| Anti-Acetyl-CoA Carboxylase, dil: 1/1000 | Cell Signaling Technology | Cat#3676; RRID:AB_2219397 |
| Anti-phospho-mTOR (Ser2448), dil: 1/1000 for WB | Cell Signaling Technology | Cat#2971; RRID:AB_330970 |
| Anti-phospho-mTOR-S2448 Rabbit mAb, dil: 1/50 for IHC | Abclonal | Cat#AP0115; RRID:AB_2832985 |
| Anti-mTOR, dil: 1/1000 | Cell Signaling Technology | Cat#2983; RRID:AB_2105622 |
| Anti-phospho-B-Raf (Ser445), dil: 1/1000 | Cell Signaling Technology | Cat#2696; RRID:AB_390721 |
| Anti-B-Raf, dil: 1/1000 | Cell Signaling Technology | Cat#9433; RRID:AB_2259354 |
| Anti-Phospho-c-Raf (Ser338), dil: 1/1000 | Cell Signaling Technology | Cat#9427; RRID:AB_2067317 |
| Anti-C-Raf, dil: 1/1000 | Cell Signaling Technology | Cat#12552; RRID:AB_2728706 |
| Anti-phospho-MEK1/2 (Ser217/221), dil: 1/1000 | Cell Signaling Technology | Cat#9154; RRID:AB_2138017 |
| Anti-MEK1/2, dil: 1/1000 | Cell Signaling Technology | Cat#9122; RRID:AB_823567 |
| Anti-phospho-Erk1/2 (Thr202/Tyr204), dil: 1/1000 | Cell Signaling Technology | Cat#4370; RRID:AB_2315112 |
| Anti-p44/42 MAPK (Erk1/2), dil: 1/1000 | Cell Signaling Technology | Cat#4695; RRID:AB_390779 |
| Anti-phospho-TAK1 (Thr184/187), dil: 1/1000 | Cell Signaling Technology | Cat#4508; RRID:AB_561317 |
| Anti-TAK1, dil: 1/1000 | Cell Signaling Technology | Cat#4505; RRID: AB_490858 |
| Anti-phospho-LKB1 (Ser334), dil: 1/1000 | Cell Signaling Technology | Cat#3055; RRID:AB_330071 |
| Anti-LKB1, dil: 1/1000 | Cell Signaling Technology | Cat#3050; RRID:AB_823559 |
| Anti-phospho-CaMKK2 (Ser511), dil: 1/1000 | Cell Signaling Technology | Cat#12818; RRID:AB_2798034 |
| Anti-CaMKK2, dil: 1/1000 | Cell Signaling Technology | Cat#16810; RRID:AB_2798771 |
| Anti-PP2A C Subunit, dil: 1/1000 | Cell Signaling Technology | Cat#2038; RRID:AB_2169495 |
| Anti-PP2C-α, dil: 1/1000 | Cell Signaling Technology | Cat#3549; RRID:AB_2169764 |
| Anti-EGFR, dil: 1/1000 | Cell Signaling Technology | Cat#2232; RRID:AB_331707 |
| Anti-Phospho-EGFR (Tyr1068), dil: 1/1000 | Cell Signaling Technology | Cat#3777; RRID:AB_2096270 |
| Anti-Beclin-1, dil: 1/1000 | Cell Signaling Technology | Cat#3738; RRID:AB_490837 |
| Anti-LC3B, dil: 1/1000 | Cell Signaling Technology | Cat#3868; RRID:AB_2137707 |
| Anti-Phospho-PDGFR beta-Y751, dil: 1/1000 | Abclonal | Cat#AP0493; RRID:AB_2771408 |
| Anti-PDGF Receptor beta, dil: 1/1000 | Abclonal | Cat#A19531; RRID:AB_2832987 |
| Anti-Phospho-VEGFR2-Y1175, dil: 1/1000 | Abclonal | Cat#AP0382; RRID:AB_2771247 |
| Anti-VEGFR2, dil: 1/1000, dil: 1/1000 | Abclonal | Cat#A5609; RRID:AB_2766373 |
| Anti- ErbB2, dil: 1/1000 | Abclonal | Cat#A2071; RRID:AB_2832988 |
| Anti-phospho-ERBB2-Y1221/1222, dil: 1/1000 | Abclonal | Cat#AP0527; RRID:AB_2771088 |
| Anti-Pink1, dil: 1/1000 | Abclonal | Cat#A11435; RRID:AB_2758558 |
| Anti-Parkin, dil: 1/1000 | Abclonal | Cat#A11172; RRID:AB_2758446 |
| Anti-BNIP3, dil: 1/1000 | Abclonal | Cat#A19593; RRID:AB_2832989 |
| Anti-BNIP3L, dil: 1/1000 | Abclonal | Cat#A6283; RRID:AB_2766888 |
| Anti-SREBP1, dil: 1/1000 | Abcam | Cat#ab28481; RRID:AB_778069 |
| Anti-αSMA, dil: 1/1000 | Abcam | Cat#ab7817; RRID:AB_262054 |
| Anti-CTGF(L-20), dil: 1/200 | Santa Cruz | Cat#sc-14939; RRID:AB_638805 |
| Goat anti-mouse IgG-HRP, dil:1/5000 | Jackson | Cat#115-035-003; RRID:AB_10015289 |
| Goat anti-rabbit IgG-HRP, dil:1/5000 | Jackson | Cat#111-035-003; RRID:AB_2313567 |
| Anti-F4/80, dil: 1/50 | Bio-Rad | Cat#MCA497; RRID:AB_2098196 |
| Anti-CD11b, dil: 1/100 | Boster Biological Technology | Cat#BM3925; RRID:AB_2832991 |
| Alexa Fluor® 555 Conjugate Anti-rat IgG (H+L), dil: 1/200 | Cell Signaling Technology | Cat#4417; RRID:AB_10696896 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Pentobarbital sodium | Sigma-Aldrich | Cat#P3761 |
| DMSO | Sigma-Aldrich | Cat#D2650 |
| Sorafenib | Selleck | Cat#S7397 |
| Dorsomorphin (Compound C) 2HCl | Selleck | Cat#S7306 |
| DEN(Diethylnitrosamine) | Sigma-Aldrich | Cat#N0756 |
| β-Cyclodextrin | RHAWN | Cat#R010666 |
| Zoletil | Virbac | Cat#83888102 |
| Xylazine hydrochloride | Jilin Huamu | Cat#180606 |
| PhosStop phosphatase inhibitor | Roche | Cat#4906837001 |
| cOmplete Protease Inhibitor Cocktail | Roche | Cat#04693132001 |
| Clarity™ Western ECL Substrate | Bio-Rad | Cat#1705061 |
| TRIzol | Sigma-Aldrich | Cat#T9424 |
| SYBR Green | Roche | Cat#04887352001 |
| Dulbecco’s modified Eagle’s medium (DMEM) | GIBCO | Cat# C11995500BT |
| fetal bovine serum (FBS) | Tico Europe | Cat#FBSEU500 |
| Polybrene | Sigma | Cat#H9268 |
| Puromycin | GIBCO | Cat#A1113803 |
| Trypsin | GIBCO | Cat#27250-018 |
| 1% penicillin/streptomycin | GIBCO | Cat#15140-122 |
| Sudan III | Solarbio | Cat#S8460 |
| Hematoxylin | Google biology | Cat#G1004 |
| Eosin | BASO | Cat#BA-4024 |
| Masson | BASO | Cat#BA-4079B |
| Tissue-Tek O.C.T. Compound | Servicebio | Cat#4583 |
| Oil Red O | Sigma-Aldrich | Cat# 625 |
| TMRM | AAT Bioquest | Cat# 22221 |
| MitoTracker® Red CMXRos | YEASEN | Cat# 40741ES50 |
| Safranin O | Sigma-Aldrich | Cat#S2255 |
| Rotenone | Dalian Meilun | Cat#MB7162 |
| Digitonin | Sigma-Aldrich | Cat# D5628 |
| Succinate | Shanghai Maclin | Cat# D822253 |
| Pyruvate | Sigma-Aldrich | Cat# P2256 |
| Glutamate | Shanghai Maclin | Cat#L810495 |
| Malate | Shanghai Maclin | Cat#L813179 |
| Ascorbate | Sigma-Aldrich | Cat#A7631 |
| TMPD | Shanghai Maclin | Cat#T819055 |
| ADP | Shanghai Maclin | Cat#A844435 |
| AMP | Sigma-Aldrich | Cat#A2252 |
| ATP | Shanghai Maclin | Cat#A800906 |
| FCCP | Dalian Meilun | Cat#MB3642 |
| Malonic acid | Shanghai Maclin | Cat#M813040 |
| Antimycin A | Dalian Meilun | Cat# MB1384 |
| Sodium azide | Sigma-Aldrich | Cat#S8032 |
| Oligomycin | Sigma-Aldrich | Cat#O4876 |
| Lactobionic acid | Shanghai Maclin | Cat#L812453 |
| Taurine | Sigma-Aldrich | Cat#T0625 |
| Valinomycin | Shanghai Maclin | Cat#V820378 |
| Nigericin | MedChemExpress | Cat# HY-100381 |
| BSA, essentially fatty acid free | Equitech Bio | Cat# BAH66-0100 |
| Sodium palmitate | Sigma-Aldrich | Cat# P9767 |
| Oleic acid | Sigma-Aldrich | Cat#O1008 |
| DAPI | Invitrogen | Cat#S36939 |
| Pazopanib | Selleck | Cat#S3012 |
| ZM336372 | Selleck | Cat#S2720 |
| SU1498 | Selleck | Cat#S6535 |
| Tyrphostin AG 1296 | Selleck | Cat#S8024 |
| Liver Perfusion Medium | Thermo Fisher Scientific | Cat#17701-038 |
| Liver Digestion Medium | Thermo Fisher Scientific | Cat#17701-034 |
| Pronase E | Roche | Cat#10165921001 |
| Collagenase IV | GIBCO | Cat#17104019 |
| DNase I | Roche | Cat#10104159001 |
| Crystal violet | Sinopharm Chemical Reagent | Cat#71012314 |
| Critical Commercial Assays | ||
| BCA Protein assay kit | Thermo Fisher Scientific | Cat#23225 |
| Transcriptor first-strand cDNA synthesis kit | Roche | Cat#04896866001 |
| Triglyceride (TG) assay kit | Wako | Cat#290-63701 |
| Cholesterol assay kit | Wako | Cat#294-65801 |
| NEFA assay kit | Wako | Cat#294-63601 |
| Triglyceride (TG) assay kit | Solarbio | Cat#BC0625 |
| Mouse TNF ELISA kit | Cloud-Clone Co. | Cat#SEA133Mu |
| Mouse MCP1(CCL2) ELISA kit | Cloud-Clone Co. | Cat#SEA087Mu |
| Cell Counting Kit-8 assay kit | Bimake | Cat#B34304 |
| Enhanced enzyme-labeled goat anti-rabbit IgG kit | Beijing ZSGB-Bio | Cat#PV-9001 |
| 3,3′-Diaminobenzidine (DAB) substrate kit | Beijing ZSGB-Bio | Cat#ZLI-9018 |
| RNA 6000 Nano kit | Agilent | Cat#5067-1511 |
| MGIEasy RNA Library Prep Kit | MGI Tech Co. | Cat#1000006384 |
| Deposited Data | ||
| RNA-seq data of HFHC-fed mice | This paper, NCBI SRA | PRJNA613854 |
| RNA-seq data of HFD-fed mice | This paper, NCBI SRA | PRJNA613938 |
| RNA-seq data of HFHC-fed mice treated with CC and sorafenib | This paper, NCBI SRA | PRJNA613940 |
| RNA-seq data of NASH monkeys | This paper, NCBI SRA | PRJNA614574 |
| Proteomic data of NASH monkeys | This paper, PRIDE | PXD018256 |
| Phospho-proteomic data of PA-treated L02 | This paper, PRIDE | PXD018214 |
| Publicly available transcriptomic datasets from sorafenib treated HCC cell lines | GEO database | GSE43053, GSE73571, GSE96793, GSE102863 |
| Experimental Models: Cell Lines | ||
| Human: L02 (HL-7702) | China Center for Type Culture Collection, Wuhan, China. | N/A |
| Human: HEK293T | Cell Bank of Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China. |
Cat#GNHu17 |
| LX-2 | Xu et al., 2005 | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J mice | Vital River | N/A |
| Cynomolgus monkey | Topgene Biotechnology | N/A |
| Oligonucleotides | ||
| sgRNA target sequences to generate gene knockout cell lines, see Table S3 | This paper | N/A |
| Primers used to genotype knockout cell lines, see Table S3 | This paper | N/A |
| Primers used for qPCR, see Table S4 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: lentiCRISPRv2 puro | Addgene | Cat#98290; RRID:Addgene_98290 |
| Plasmid: pMD2.G | Addgene | Cat#12259; RRID:Addgene_12259 |
| Plasmid: psPAX2 | Addgene | Cat#12260; RRID:Addgene_12260 |
| Software and Algorithms | ||
| Las X (v3.4.2) | Leica Microsystems | N/A |
| Graphpad Prism 8 | Graphpad | https://www.graphpad.com/ |
| SPSS statistics 19.0 | IBM Corporation | http://www.spss.com.hk/software/statistics/ |
| Image-Pro plus 6.0 | Media Cybernetics | http://www.mediacy.com/imageproplus |
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| Phoenix WinNonlin 6.3 | Pharsight | https://lp.certara.com/Download-Phoenix-8.2.html |
| HISAT2 2.1.0 | Kim et al., 2015 | http://ccb.jhu.edu/software/hisat2/index.shtml |
| SAMtools 1.4 | Li et al., 2009 | http://samtools.sourceforge.net |
| StringTie 1.3.3b | Pertea et al., 2015 | http://ccb.jhu.edu/software/stringtie/index.shtml |
| DESeq2 1.24.0 | Love et al., 2014 | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| GSEA 3.0 | Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/index.jsp |
| GSVA 1.32.0 | Hänzelmann et al., 2013 | https://bioconductor.org/packages/release/bioc/html/GSVA.html |
| MaxQuant 1.3.0.5 | Cox and Mann, 2008 | https://www.maxquant.org/ |
| Perseus 1.3.0.4 | Tyanova et al., 2016 | https://maxquant.net/perseus/ |
| oligo 1.46.0 | Carvalho and Irizarry, 2010 | https://www.bioconductor.org/packages/release/bioc/html/oligo.html |
| limma 3.38.3 | Ritchie et al., 2015 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| ggplot2 3.1.1 | Wickham, 2016 | https://cran.r-project.org/web/packages/ggplot2/index.html |
| Others | ||
| Mouse high fat diet | Huafukang Bioscience | Cat#D12492 |
| Mouse normal chow diet | Huafukang Bioscience | Cat#D12450B |
| Mouse high fat high cholesterol diet | TrophicDiet | Cat#TP26304 |
| Monkey high fat high cholesterol diet | Beijing Keao Xieli Feed Co. | N/A |
| PVDF membranes | Millipore | Cat#IPVH00010 |
| Transwell plate | Corning | Cat#3428 |
| HyperCarb HPLC Columns,150 × 2.1 mm, 3 mm | Thermo Fisher Scientific | Cat#35003-152130 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Line
The human hepatocyte L02 cell line was purchased from the China Center for Type Culture Collection, Wuhan, China. HEK293T cell line was purchased from the Cell Bank of the Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China. L02 and HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. The human hepatic stellate cell (HSC) line LX-2 (Xu et al., 2005) was kindly provided by Dr. Xinbing Yu (Sun Yat-Sen University, Guangzhou, China) and cultured in DMEM supplemented with 2% FBS and 1% penicillin/streptomycin. The cell lines were assessed for mycoplasma contamination, and the test results were negative.
To establish a cell model of lipid accumulation, L02 cells were challenged by 0.5 mM palmitic acid (PA) and 1 mM oleic acid (OA) (dissolved in 0.5% fatty acid–free BSA) and treated with DMSO vehicle or sorafenib (10 μM) for 18 h. To establish a cell model of hepatic inflammation, L02 cells were challenged by 0.5 mM PA and treated with DMSO or sorafenib (10 μM) for 18 h. Fatty acid–free BSA (0.5%) alone was used as a vehicle control. To inhibit the AMPK pathway, 4 μM compound C was used.
Cell lines deficient for specific genes were generated using the CRISPR/Cas9 system. sgRNAs targeting the human BRAF, CRAF, PRKAA1, PRKAA2 genes were designed and cloned into the lentiCRISPRv2 plasmid (#98290, Addgene). The sgRNA-expressing plasmid, together with lentivirus packaging plasmids pMD2.G (#12259, Addgene) and psPAX2 (#12260, Addgene) were transfected into HEK293T cells at a ratio of 2:1:1. Supernatants were collected and filtered through a 0.22 μm-filter 48 h after transfection. Then, the L02 cells were transduced with lentiviral supernatants in the presence of polybrene (2 μg/mL). To generate AMPKα1/α2 double knockout cells, lentiviruses targeting PRKAA1 and PRKAA2 were combined and added into a single dish of L02 cells. Puromycin (2 μg/mL) was added to select positive candidates. Single cells were placed in 96-well plates to facilitate the growth of cell clones. Then, the positive clones were screened by western blotting using the indicated antibodies and further confirmed by sequencing. The sgRNA target sequences and genotyping primers are listed in Table S3.
Primary Cells
Mouse primary hepatocytes were isolated from 8-week-old C57BL/6J male mice by a two-step collagenase perfusion process, as previously described (Tong et al., 2019). Briefly, after anesthetized with 3% pentobarbital sodium (90 mg/kg, #P3761, Sigma-Aldrich, St. Louis, Missouri, USA), mice were perfused through the portal vein with Liver Perfusion Medium (#17701038, Thermo Fisher Scientfic, Waltham, MA, USA) followed by Liver Digestion Medium (#17701034, Thermo Fisher Scientfic). After digestion, the liver was excised, minced, filtered through a 100 μm steel mesh. Hepatocytes were separated after two centrifugations at 50g for 1 min. The isolated hepatocytes were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin for other tests.
Rat primary HSCs were isolated from 12-week-old male Sprague–Dawley rats as described previously (Maschmeyer et al., 2011) with some modifications. Briefly, rat liver was perfused and digested with 0.04% pronase (#10165921001, Roche, Basel, Switzerland) and 0.05% collagenase IV (#17104019, GIBCO) for 15 min each. The liver is then excised, minced and suspended in DMEM containing 0.1% DNase (#10104159001, Roche). The cell suspension was filtered through sterile gauze and centrifuged at 50g for 2 min to remove hepatocytes. The remaining cell suspension was centrifuged at 580g for 10 min. The cell pellet is re-suspended and subjected to density gradient centrifugation. Before experiments, the HSCs were spontaneously differentiated in DEME supplemented with 10% FBS and 1% penicillin/streptomycin for 8 days.
Animals
The animal protocols for mouse studies were approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University. The animal protocol for monkey study was approved Institutional Animal Care and Use Committee of the Institute of Model Animals of Wuhan University. The animals received humane care according to the Guide for the Care and Use of Laboratory Animals published by the National Academy of Sciences and the National Institutes of Health. Besides obesity, animals were in generally good health.
Male C57BL/6J mice were housed in a temperature-controlled environment (23 ± 02°C) under a 12 h light/dark cycle with free access to food and water. To establish a NASH-HCC model, C57BL/6J mice were injected with a single dose of DEN (25 mg/kg) at the age of 2 weeks and fed a HFHC diet beginning at the age of 6 weeks. After 10 weeks of HFHC diet feeding, the mice were divided randomly into 4 groups, each of which received vehicle (1% DMSO and 15% β-cyclodextrin in saline) or 10, 15 or 30 mg/kg sorafenib (dissolved in 1% DMSO and 15% β-cyclodextrin in saline) by gavage every other day. At the age of 24 weeks, the mice were sacrificed, and their livers were analyzed. The adverse effects of sorafenib, including diarrhea, skin rash, blood pressure effects and fatigue (measured as the physical activity in a metabolic cage) were assessed.
To establish a fatty liver model, 8-week-old male C57BL/6J mice were fed a HFD (protein, 20%; fat, 60%; carbohydrates, 20%; #D12492, Huafukang Bioscience, Beijing, China) for 24 weeks. A NASH mouse model was established by feeding the C57BL/6J mice a HFHC diet (protein, 14%; fat, 42%; carbohydrates, 44%; cholesterol, 0.2%; #TP26304, TrophicDiet, Nantong, China) for 18 weeks. Mice that received an NC diet (protein, 20.6%; fat, 12%; carbohydrates, 67.4%; #D12450B, Huafukang) served as controls. Sorafenib (15 mg/kg) or vehicle was administered for 8 weeks by gavage to both mouse models. To test the in vivo requirement of AMPK activation in therapeutic effects of sorafenib on NASH, 8-week-old male C57BL/6J mice were fed a HFHC diet for 19 weeks. After 10 weeks’ of HFHC-feeding, mice were randomly divided into two groups and treated with AMPK inhibitor CC (10 mg/kg/2 days) or vehicle for 1 week. Then mice were further divided into four groups and treated with vehicle, sorafenib (15mg/kg/2 days), CC (10 mg/kg/2 days) or sorafenib (15mg/kg/2 days) in combination with CC (10 mg/kg/2 days) for 8 weeks.
For the monkey experiments, 12 male cynomolgus monkeys (Macaca fascicularis) aged 8–12 years (6–12 kg) with naturally developed NASH symptoms were purchased from Topgene Biotechnology (Guangzhou, China). The purchase procedures adhered to the legal and regulatory requirements of the People’s Republic of China and were approved by the Department of Forestry of Hubei Province and the Department of Forestry of Guangdong Province. All monkeys included in this study passed the physical examination and met the standards of local quarantine inspection. After transportation to the animal facility at the Institute of Model Animal of Wuhan University, monkeys were individually housed and acclimated for 1 week before conducting the formal experiments. The initially selected monkeys were screened by liver biopsy, and the NAFLD activity score (NAS) was independently evaluated by two pathologists according to the NASH-CRN scoring system (Kleiner et al., 2005). Monkeys with NAS at 3~4 plus liver fibrosis were chosen. Monkeys were randomly divided into vehicle and sorafenib groups with 6 monkeys per group. To induce more severe NASH symptoms, these monkeys were fed a HFHC diet (containing lard, 10%; sucrose, 15%; cholesterol, 1%; corn flour, 14.5%; wheat flour, 14.5%; Beijing Keao Xieli Feed Co., Ltd., Beijing, China) during the treatment process. Sorafenib (1 mg/kg, dissolved in 1% DMSO and 15% β-Cyclodextrin in saline) or vehicle (with parallel volume to sorafenib group) was administrated by intravenous injection every three days. Physical examinations were performed every 4 weeks, and liver biopsy and MRI were performed at 0, 12, and 24 weeks. Monkeys were fasted overnight and anesthetized with Zoletil and Xylazine (5:2, v/v; 0.03 ml/kg) before liver needle biopsy or MRI. A Bard Monopty biopsy gun (Bard Biopsy Systems, Tempe, AZ, USA) loaded with a 16-gauge biopsy needle was used for liver needle biopsy under the guidance of an ultrasound system as previously described (Wang et al., 2017). For laparotomy biopsy, after intra-muscular injection of atropine (0.04 mg/kg) and fensulfamide (0.1 ml/kg), monkeys were anesthetized with Zoletil and Xylazine (5:4, v/v; 0.05 ml/kg). Monkeys were intra-muscularly injected with analgesic torpidine (0.15 ml/kg) before the surgery to relieve the pain. Median laparotomy was then performed to expose the liver. Liver tissues were collected at a distance of about 1 cm from the edge of the liver. The electrocautery was used to stop bleeding, and then the abdomen was closed. After the surgery, monkeys were intra-muscularly injected with torpidine (0.15 ml/kg/2 days) and penicillin (150,000 IU/kg/day) for 3 days. The abdominal wounds were kept dry and treated with iodophor twice a day.
METHOD DETAILS
Western Blotting
Briefly, protein from cells or mouse liver tissues was extracted with RIPA lysis buffer (65 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate and 0.1% SDS) together with protease inhibitor cocktail tablets (#04693132001, Roche) and quantified with a BCA Protein Assay Kit (#23225, Thermo Fisher Scientific). The protein samples were separated using 10% SDS-PAGE gels and then transferred to polyvinylidene difluoride (PVDF) membranes. After blocking with 5% skim milk, the membranes were incubated with the indicated primary antibodies overnight at 4°C and then with secondary horseradish peroxidase (HRP)-conjugated antibodies for 1 h at room temperature. The protein expression signals were detected on a ChemiDoc MP Imaging System (Bio-Rad, Hercules, CA, USA). β-Actin was used as loading control.
Real-Time qPCR
Total RNA was extracted with Trizol (#T9424, Sigma-Aldrich) and then reverse-transcribed into cDNA by using a Transcriptor First-Strand cDNA Synthesis Kit (Roche) according to the manufacturer’s instructions. SYBR Green (#04896866001, Roche) was used to quantify the PCR-amplification products. The mRNA expression levels of the target genes were normalized to ACTB. Primers for qPCR are listed in Table S4.
Transwell Migration Assay
The transwell migration of LX-2 cells were performed as previously described (Wang et al., 2020). Briefly, LX-2 cells were starved for 24 h in serum-free DMEM before seeding in the upper chambers of transwell plate (8 μm pore size, #3428, Corning, NY, USA). The bottom chambers were filled with DMEM medium with or without 4 μM sorafenib as indicated. After incubation for 48 h, the unmigrated LX-2 cells were removed and the migrated cells were fixed in 4% paraformaldehyde followed by crystal violet (#71012314, Sinopharm Chemical Reagent, Beijing, China) staining.
Cell Viability
Cell viability was tested with a Cell Counting Kit-8 assay kit according to the manufacturer’s instructions. Briefly, LX-2 cells were seeded in 96-well plates with 5000 cells per well. After incubation with 5 μM or 10 μM sorafenib for the indicated times, 10 μL of Cell Counting Kit-8 reagent (#B34304, Bimake, TX, USA) was added into each well, and the cells were incubated for 3 h at 37°C. The absorbance of each well at 450 nm was measured to estimate the number of viable cells.
Measurement of AMP, ADP and ATP
For analysis of AMP, ADP and ATP in cells, 1 × 106 primary mouse hepatocytes were treated with PA for 18 h and then incubated with DMSO or sorafenib (10 μM) for 1 h before extraction. For analysis of AMP, ADP and ATP in liver tissues, mice were anesthetized 4 h after sorafenib (15 mg/kg) administration, and their livers were immediately freeze-clamped. Approximately 50 mg of liver tissue was analyzed for each mouse. The extraction of AMP, ADP and ATP were carried out as previously described (Zhang et al., 2017). Briefly, cells were rinsed with 5% mannitol solution (dissolved in water) and instantly frozen in liquid nitrogen. Cells were then scrapped off from the dish with 1 mL of methanol. The lysate was then mixed with 1 mL of chloroform and 400 μL of ultra-pure water. After centrifugation at 15,000 g for 5 min at 4°C, the aqueous phase was collected, diluted 20 times with 20% methanol solution (dissolved in water) and then detected by liquid chromatography-mass spectrometry (LC-MS).
The AMP, ADP and ATP levels were measured using an ultrahigh-performance liquid chromatography (UHPLC) system (LC-30 AD, Shimadzu, Kyoto, Japan) equipped with a triple-quadrupole mass spectrometer (Triple Quad 4500, AB Sciex, Boston, MA, USA) at Wuhan Biobank Co., Ltd. (Wuhan, China). The compounds were separated on a porous graphitic carbon column (HyperCarb 150 × 2.1 mm, 3 μm, #35003-152130, Thermo Fisher Scientific) under an optimized column temperature of 25°C with an injection volume of 2 μl. The low rate was set at 0.4 mL/min, and chromatographic separation was achieved with the following gradient program: 2 min, 10% B; 4 min, 95% B; 7 min, 95% B; 8 min, 10% B; 10 min, stop. The mobile phase buffer A consisted of 0.3% (v/v) formic acid adjusted to pH 9 with ammonia. The mobile phase buffer B was 90% (v/v) acetonitrile. The samples were kept at 4°C during the whole process. Three transitions were used to monitor each of the three compounds: ATP (506 > 159, 506 > 408 and 506 > 273), ADP (426 > 159, 426 > 328 and 426 > 408) and AMP (346 > 78.8, 346 > 134 and 346 > 210.9).
Pharmacokinetic Study
Pharmacokinetic analyses of sorafenib in monkeys were performed using methods described previously (Kim et al., 2012). After intravenous injection of 1 mg/kg sorafenib in monkeys, blood was collected in heparinized tubes at 0, 5, 10, 15 and 30 min and at 1, 3, 6, 12, 24, 48, and 72 h. The plasma was separated by centrifugation and frozen at −80°C until analysis. Sorafenib was extracted by protein precipitation through addition of 0.5 mL of acetonitrile to 50 μL of plasma. The samples were centrifuged at 10,000 rpm for 10 min, and the supernatant was collected for analysis by HPLC/MS/MS system at Wuhan Biobank Co., Ltd. The samples were separated on a Waters Acquity UPLC BEH C18 (2.1 × 50 mm, 1.7 μm) at a flow rate of 0.4 mL/min using the following gradient program: 1 min, 5% B; 5 min, 95% B; 6.5 min, 95% B; 7 min, 5% B; 8 min, stop. The mobile phase buffer A consisted of 0.1% (v/v) formic acid, and the mobile phase buffer B was acetonitrile. Three transitions were used to monitor sorafenib (465 > 252, 465 > 406 and 465 > 447). The pharmacokinetic parameters were calculated through non-compartmental pharmacokinetic analysis using Phoenix WinNonlin software (version 6.3, Pharsight, CA, USA) with the linear/log trapezoidal rule.
Mouse Metabolic Cage Studies
The activity and energy expenditure of mice were measured in metabolic cages (Promethion, Sable Systems International Inc., NV, USA). After vehicle or sorafenib gavage, mice were put into metabolic cages under a 12 h light/dark cycle with free access to water and HFHC diet. Data were collected from each mouse every 5 min, and collection continued for more than 24 h (including a 12 h light phase and a 12 h dark cycle). The results of activity and energy expenditure were calculated using the ExpDate software provided by the manufacturer.
Mitochondrial Respiration
Mitochondrial respiration was measured by high-resolution respirometry (Oxygraph-2k, Oroboros, Innsbruck, Austria) at 37°C. A total of 2 × 105 freely isolated primary mouse hepatocytes were suspended in 2 mL of MiR05 respiration buffer (20 mM Taurine, 0.5 mM EGTA, 3 mM MgCl2·6H2O, 60 mM K-lactobionate, 10 mM KH2PO4, 20 mM HEPES, 110 mM D-sucrose, and 1 g/L fatty acid–free BSA). Digitonin (0.5 μg/mL) was added to the chambers to permeabilize the cells. Assessment of complex I–related respiration was performed through sequential additions of complex I substrates pyruvate, malate and glutamate (P+M+G, 5 mM, 2 mM, 10 mM, respectively), ADP (2.5 mM), F1Fo ATP synthase inhibitor oligomycin (2 μg/mL), mitochondrial uncoupler FCCP (0.5 μM/step untill maximal OCR reached), and complex I inhibitor rotenone (0.5 μM). Assessment of complex II–related respiration was performed by sequential additions of rotenone (0.5 μM), complex II substrate succinate (5 mM), ADP (2.5 mM), oligomycin (2 μg/mL), FCCP (0.5 μM/step untill maximal OCR reached) and complex III inhibitor antimycin A (2.5 μM). The complex IV-related respiration was assessed by sequential additions of elector donor ascorbate and TMPD (As+Tm, 2 mM and 0.5 mM, respectively), ADP (2.5 mM), oligomycin (2 μg/mL), FCCP (0.5 μM/step untill maximal OCR reached) and complex IV inhibitor Sodium azide (100 mM). State 4 respiration refers to the oxygen consumption rate (OCR) in the absence of ADP (before ADP/after oligomycin addition). State 3 respiration refers to OCR in the presence of ADP. ATP coupled respiration was calculated by by subtracting the state 4 OCR from state 3 OCR. Maximal respiration refers to the OCR in the presence of FCCP.
The PA-stimulated respiration was used to assess the effect of sorafenib on fatty acid oxidation on intact primary mouse hepatocytes. PA was conjugated to BSA at a molecular ratio of 6:1 to create an aqueous-soluble respiratory substrate. A total of 2 × 105 freely isolated primary mouse hepatocytes were suspended in 2 mL of KHB buffer (111 mM NaCl, 4.7 mM KCl, 2 mM MgSO4, 1.2 mM Na2HPO4, 2.5 mM glucose, 0.5 mM carnitine, pH 7.4) with DMSO or sorafenib (5 μM). The oxygen consumption rate (OCR) was monitored following sequential additions of PA (100 μM), oligomycin (2.5 μM), FCCP (0.5 μM), rotenone (0.5 μM) and antimycin A (2.5 μM). FAO OCR refers to PA stimulated respiration. FAO-Leak related OCR was calculated by substrating the OCR in the presence rotenone and antimycin A from those in the presence of oligomycin. Maximal OCR was calculated by subtracting the OCR in the presence of rotenone and antimycin A from those in the presence of FCCP.
Confocal Microscopy
For measurement of mitochondrial membrane potential and morphological changes, primary mouse hepatocytes were stained with tetramethyl rhodamine methyl ester (TMRM, 50 nM) and MitoTracker® Red CMXRos (100nM), respectively, and imaged on an inverted confocal microscope (TCS SP8, Leica, Wetzler, Germany). TMRM and mitoTracker fluorescence was taken by excitation at 561nm and emission collection at 570~650nm. Before dye loading, the cells had been treated with BSA vehicle or PA (0.5 mM) in the presence or absence of sorafenib (10 μM) for 18 h.
Mitochondrial Proton Conductance
Proton conductance was measured as previously described (Brand et al., 2005). OCR and ΔΨm (indicated by Safranin O fluorescence) were simultaneously measured on the Oroboros system at 37°C. Permeabilized primary mouse hepatocytes (2 × 105) were suspended in MiR05 solution containing oligomycin (2.5 μM, to inhibit ATP synthesis), nigericin (100 ng/mL, to dissipate the pH gradient), and rotenone (2.5 μM, to inhibit complex I). Safranin O (#S2255, Sigma-Aldrich) was calibrated through sequential titration up to 10 μM, and then succinate (a complex II substrate) was added to initiate respiration. ΔΨm and OCR were progressively inhibited through successive addition of the complex II inhibitor malonate up to 3 mM. A standard cure for conversion of safranin fluorescence to absolute values of ΔΨm was established as described previously (Vercesi et al., 1998). Briefly, digitonin-permeabilized primary mouse hepatocytes (2 × 105) were added to a potassium-free medium containing 200 mM sucrose, 10 mM Na-HEPES buffer (pH 7.2), 0.25 mM EGTA, 2 mM MgCl2, 2 mM sodium phosphate, 5 mM succinate, 5 μg/mL digitonin, 10 μM safranine and 5 μM valinomycin. Titration of ΔΨm was performed through sequential addition of KCl to give final concentrations of 0.6, 1.2, 2.2, 3.2, 5.2, and 10.2 mM. The ΔΨm values after each KCl addition were determined by the Nernst equation assuming [K+]in = 120 mM.
Lipid Analyses
Commercial kits were used to measure the triglyceride (TG), total cholesterol (TC) and non-esterified fatty acid (NEFA) contents in the liver (#290-63701 for TG, #294-65801 for TC, #294-63601 for NEFA; Wako, Tokyo, Japan), according to the manufacturer’s instructions..
Serum Assays
Cytokine concentrations in mouse serum were examined via ELISA (#SEA133Mu for TNF and #SEA087Mu for MCP1, Cloud-Clone Corp., Wuhan, China). Serum TG, TC, ALT, AST, ALP, LDL-c, HDL-c, LDH, CK, CKMB, UREA and CRE levels were measured by using an ADVIA 2400 Chemistry System analyzer (Siemens, Tarrytown, NY, USA) according to the manufacturer’s instructions.
Immunofluorescence Staining
Tissues were fixed in 10% neutral buffered formalin, embedded in paraffin and then stained with anti-F4/80 (1:50, #MCA497, AbD Serotec, Kidlington, UK) or anti-CD11b (1:100, #BM3925, Boster, Wuhan, China) polyclonal antibodies overnight. The samples were then incubated with fluorophore-conjugated secondary antibodies for 1 h. Images were captured by a fluorescence microscope with DP2-BSW software (version 2.2, Olympus, Tokyo, Japan).
Histopathologic Analysis
For histopathologic analysis, H&E staining was performed on paraffin-embedded tissues. Lipid droplets were visualized by Oil Red O (Sigma-Aldrich) staining of Tissue-Tek O.C.T. Compound (Servicebio)–embedded frozen liver tissues. Liver fibrosis was assessed via Masson (BASO) staining. Histopathological images were captured under a light microscope (Olympus). Images were quantified using Image-Pro plus 6.0 and SPSS 19.0 software.
Immunohistochemistry
Immunohistochemistry of AMPKα and mTOR activities were performed on paraffin embedded liver sections (5 μm) using p-AMP-Kα-T172 (1:50, Cell Signaling Technology) and p-mTOR-S2448 (1:50, Abclonal) antibodies. For antigen retrieval, samples were heated in a pressure cocker for 20 min in pH9.0 EDTA buffer. After cooling, samples were placed in 3% H2O2 for 20 min to quench endogenous peroxide activity. After washing with PBS, slides were blocked with 10% BSA for 10 min. Sections were incubated with the primary antibodies overnight at 4°C, washed with PBS buffer for 3 times (3min/wash) and then incubated with enhanced enzyme-labeled goat anti-rabbit IgG (Beijing ZSGB Biotech) for 1 h at room temperature. Immunohistochemical staining was visualized using 3,3′-diaminobenzidine (DAB) substrate kit (Beijing ZSGB Biotech) and were counterstained with hematoxylin. Images were captured on a light microscope (Olympus).
Diarrhea Assessment
To assess diarrhea severity, stools were classified into four forms according to their consistency: normal, wet, unformed and watery, as previously described (Yeung et al., 2015). Each mouse was placed individually into a cage lined with filter paper, and the numbers of feces of various forms were counted 6 h after gavage. The diarrhea score was recorded 6 h and 24 h after sorafenib treatment, as previously described (Wardill et al., 2016). Briefly, there were four grades: 0, no diarrhea; 1, mild diarrhea (slightly wet and soft stool); 2, moderate diarrhea (with staining of the tops of the legs and the lower abdomen); and 3, severe diarrhea (with continual anal leakage that stained the legs and upper abdomen).
Skin Rash and Fur Recovery Assessment
Wax depilation was performed on the dorsal surfaces of anesthetized mice (Zimmerman et al., 2016) to assess the skin toxicity of sorafenib at the age of 20 weeks. Skin rashes were monitored daily and were graded as “rash,” “no rash” or “resolved rash.” Pictures of the dorsal hair of mice were taken weekly for 4 weeks and the fur recovery rate was recorded by measuring the percentage of recovery area.
RNA-sequencing
Total RNA was extracted from liver tissues using TRIzol reagent (#T9424, Sigma-Aldrich) and the quality of the extracted total RNA samples was checked with RNA 6000 Nano kit (#5067-1511, Agilent, CA, USA). The MGIEasy RNA Library Prep Kit (#1000006384, MGI Tech Co., Ltd, Shenzhen, China) was used for library preparation with an according to the manufacturer’s instructions. Briefly, after mRNA enrichment, the samples were incubated with fragmentation buffer at 94°C for 8 min to obtain a target insert fragment size of ~150 bp. Then, the products were reverse-transcribed into cDNA. After repair and A-tailing, the double-stranded cDNA products were ligated with an adaptor and subjected to PCR amplification following a 14-cycle procedure (95°C for 30 s, 56°C for 30 s, and 72°C for 60 s; 14 cycles). The PCR products were cleaned using DNA Clean Beads. Then quality control of the purified PCR products was carried out with an Agilent DNA 1000 Kit. PCR products at ~230 bp in size were subjected to multiple-sample pooling, amplification, and digestion to obtain the resulting libraries. Gene expression analysis was performed by mRNA sequencing on a BGISEQ-500 (MGI Tech Co.) with a single-end 50 bp module.
Phospho-proteomic Analysis
L02 cells (1 × 107) were treated with 0.5 mM PA and DMSO or sorafenib (10 μM) for 18 h for label-free phospho-proteomic analysis. The cells were lysed with SDT buffer (4% SDS, 100 mM dithiothreitol (DTT), and 150 mM Tris-HCl, pH 8.0). Then, 400 μg of protein was reduced with 10 mM DTT at 37°C for 2.5 h. Subsequently, the samples were alkylated with iodoacetamide (50 mM) for 30 min in the dark. The proteins were then digested in 1.5 M urea buffer containing trypsin (1 μg/100 μg protein) for 18 h at 37°C. The digests were desalted using Sep-Pak C18 cartridges (#WAT051910, Waters, MA, USA), dried in vacuo, and stored at −80°C for further use. The phosphopeptides were enriched using TiO2 beads as previously described (de Graaf et al., 2014).
The phosphopeptides were subjected to LC-MS/MS analysis using a Proxeon EASY-nLC 1000 (Thermo Scientific; in Shanghai Applied Protein Technology Co., Ltd., Shanghai, China). The peptides were first trapped (Thermo EASY column SC001 traps, 150 μm × 20 mm [RP-C18], Thermo Fisher Scientific) at a maximum pressure of 800 bar with 100% solvent A (0.1% formic acid and 2% acetonitrile in water) before being separated on the analytical column (EASY column SC200, 150 μm × 100 mm [RP-C18], Thermo Fisher Scientific). The peptides were chromatographically separated with the following 120 min gradient program at a flow rate of 300 nL/min: 0–110 min, 0%–55% solvent B (0.1% formic acid and 84% acetonitrile in water); 110–115 min, 55%–100% solvent B; and 115–120 min, 100% solvent B. The eluents were analyzed on a Q Exactive mass spectrometer (Thermo Finnigan, Odense, Denmark) with scanning from m/z 300 to m/z 1800 at a resolution of 70,000 at m/z 400.
Proteomic Analysis
Proteomic analysis were performed by data-dependent acquisition (DDA) mass spectrometry assay. Sample preparation and fractionation were performed as previously described (Wiśniewski et al., 2009). Equal aliquots of the samples in this experiment were pooled into one sample for DDA library generation and quality control. All fractions for DDA library generation were injected into a Thermo Scientific Q Exactive HF mass spectrometer connected to an Easy-nLC 1200 chromatography system (Thermo Scientific). Each sample peptides were analyzed in a data-independent acquisition (DIA) mode. The DIA cycle contained a full MS–selected ion monitoring (SIM) scan, and 30 DIA scans were performed covering a mass range of 350–1650 m/z with the following settings: SIM full scan resolution, 60,000 at 200 m/z; automatic gain control (AGC), 3e6; maximum ion trap (IT) time, 50 ms; profile mode; DIA scan resolution, 30,000; AGC target, 3e6; maximum IT, auto; and normalized collision energy, 30 eV. The runtime was 120 min with a linear gradient of buffer B (80% acetonitrile and 0.1% formic acid) at a flow rate of 250 nL/min. Quality control samples (pools of equal aliquots of all the samples in the experiment) were injected in DIA mode at the beginning of the MS study and after every 5 injections throughout the experiment and were used to monitor MS performance.
Monkey Liver Magnetic Resonance Imaging
The lipid contents in monkey livers were examined by magnetic resonance imaging (MRI) (MAGNETOM Prisma 3.0T, Siemens Medical Systems, Forchheim, Germany). The animals were anesthetized by intramuscular injection of Zoletil and Xylazine (5:2, v/v; 0.03 mL/kg). MRI images were acquired using T1WI sequences with two echo time in a respiratory triggering mode. For TIWI MRI imaging, the field of view was 224 mm, the slice thickness was 3 mm, the number of slices was 25, the repetition time was 1590.0 ms, the flip angle was 20 degrees, and the ISO voxel size was 0.5 × 0.5 × 3.0 mm. The echo time was 2.66 ms for in-phase T1 imaging, and 3.67 ms for opp-phase MRI. Liver fat fractions were measured by region of interest (ROI)-based methods in a region of the monkey liver using a formula of (SIP-SOP)/(2 × SIP) × 100%, where SIP is the value of in-phase signal and SOP is the value of opp-phase signal. Blood vessels and bile ducts were avoided. A pseudocolor liver map was drawn using MATLAB.
Dose conversions
We converted doses between species using the body surface area normalization method as described previously (Cheung et al., 2009; FDA, 2005; Reagan-Shaw et al., 2008). Briefly, the equations used for calculation of body surface area, Km factor and equivalent doses are as follows:
where A is body surface area, K is constant, W is body weight in gram (g), Km is factor for converting mg/kg dose to mg/m2 dose. The parameters for calculation and the calculated results are listed in Table S1.
Assessment of Intestinal Lipid Absorption.
After one week on HFHC diet with vehicle/sorafenib administration (15mg/kg, i.g., every two days), mice were fasted for 16 h, received vehicle or sorafenib by gavage, individually housed and then refed HFHC diet. The serum was collected before and 1 h after refeeding for TG analysis using commercial kit (#BC0625, Solarbio, Beijing, China), and feces were collected 5 h after refeeding for lipid staining by Sudan III (#S8460, Solarbio). Then mice were euthanized and proximal jejunum was immediately removed and immersed with PBS, fixed in 4% paraformaldehyde for 2 h, incubated in 20% sucrose over night and embedded in Tissue-Tek O.C.T. Compound. Intestinal lipid droplets were visualized by Oil Red O stainings of jejunum sections (10 μm).
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantitative Phospho-proteomics Data Analysis
The raw MS files were analyzed with MaxQuant (Cox and Mann, 2008) and searched against an integrated human proteome in the UniProt database. The resulting files obtained by MaxQuant were analyzed using Perseus (Tyanova et al., 2016). To compare the abundances of phosphopeptides between the control and treatment samples, label-free quantification was performed with a minimum fold change of 1.5 to determine the differentially expressed phosphopeptides. In addition, Student’s t test was employed to identify significant differences between the control and treatment samples. P values < 0.05 were considered to indicate significance.
Digital Gene Expression
Single-end libraries were sequenced using a BGISEQ-500 with a read length of 50 bp. The clean reads were aligned using HISAT2 software version 2.1.0 (Kim et al., 2015) with Ensembl reference genomes, including mouse (mm10/GRCm38), human (hg38/GRCh38), Crab-eating macaque (macFas5/Macaca_fascicularis_5.0) genomes. The mapped fragments were stored and converted to the BAM format by SAMtools (Li et al., 2009). StringTie version v1.3.3b (Pertea et al., 2015) was applied to quantify the number of reads associated with each gene. The read counts were used as input for DEG analysis in DESeq2 (Love et al., 2014). In pairwise comparisons, genes with fold changes greater than 1.5 and adjusted P values less than 0.05 were defined as DEGs.
KEGG Pathway Enrichment Analysis
KEGG pathway enrichment analysis was performed using Fisher’s exact test with our in-house R script, and the KEGG pathway annotations of all genes in the selected genome were downloaded from the KEGG database. Only pathways with a corresponding P value less than 0.05 were considered significantly enriched.
Gene Set Enrichment and Gene Set Variation Analysis
Each known biological pathways from KEGG database and involved genes were defined as gene sets, and gene set enrichment analysis (GSEA) was implemented on the Java GSEA version 3.0 (Subramanian et al., 2005) platform with the ‘Signal2Noise’ metric to generate a ranked list and a ‘gene set’ permutation type. Gene sets with nominal P values less than 0.05 and FDR values less than 0.25 were considered statistically significant.
Gene set variation analysis (GSVA) was carried out to estimate sample-wise KEGG pathway activity variation using the GSVA R package version 1.32.0 (Hänzelmann et al., 2013).
Clustering Analysis
A hierarchical clustering analysis was performed to construct a phylogenetic tree of samples using an unweighted average distance (unweighted pair group method with arithmetic mean, UPGMA) algorithm. Principal component analysis was applied to compare the difference between groups using R function “fast.prcomp” and visualized by R package “ggplot” (Wickham, 2016).
Identify AMPK-related Proteins
The proteins that were phosphorylated in the AMPK pathway were selected after sorafenib treatment. Proteins related to these phosphorylated proteins reported in the Pubmed or STRING database are considered to be AMPK-associated proteins.
HCC RNA Array Datasets Analysis
RNA array datasets, including GSE96793, GSE73571, GSE43053, GSE102863, from the GEO database (https://www.ncbi.nlm.nih.gov/gds/) were used to investigate the role of sorafenib in the treatment of HCC. The downloaded raw data were preprocessed using the oligo package (Carvalho and Irizarry, 2010) for Affymetrix array or limma package (Ritchie et al., 2015) for Illumina expression beadchip, including background correction, expression calculation and normalization.
Statistical Analysis
All data were analyzed in SPSS software and are expressed as the means ± SEMs. Student’s t test was employed to analyze differences between two groups. One-way ANOVA was applied for multiple comparisons and was followed by Bonferroni’s post hoc test (for data showing homogeneity of variance) or Tamhane’s T2 (M) post hoc test (for data showing heteroscedasticity). For datasets with a skewed distribution, nonparametric statistical analyses were performed using the Mann-Whitney U test for two groups and the Kruskal-Wallis test for multiple comparisons. Statistical differences with P values less than 0.05 were considered significant. The statistical methods of each experiment are indicated in the figure legends. For the sample size of the animals used in this study, it was fixed in a prospective manner; no statistical method was used to predetermine sample size; it was determined based on previous studies from our group and others (Ji et al., 2018; Kraus et al., 2014; Oh et al., 2014; Wang et al., 2017). Animals with similar baseline values were randomly assigned to groups using a simple random-sampling approach by a random-number method. Data from animal studies were collected in a blinded fashion. All in vitro experiments were replicated at least three independent times. All observations were included in the final statistical analysis and all analyzed variables were fixed using a pre-specification approach.
Supplementary Material
Highlights.
Low-dose sorafenib safely suppresses NASH progression in mice
Low-dose sorafenib resolves NASH in monkeys without detectable toxicities
AMPK activation is required for the therapeutic effects of sorafenib in NASH
Sorafenib activates AMPK by acting as a mitochondrial uncoupler
Context and Significance.
The prevalence of nonalcoholic steatohepatitis (NASH) is increasing sharply and has become the leading cause of end-stage liver disease, including cirrhosis, hepatocellular carcinoma (HCC) and liver failure. Sorafenib remains the only approved first-line therapy for HCC despite serious adverse effects at current clinical dosage. Here, Jian et al. report that treatment with sorafenib in both mouse and monkey models of NASH at the equivalent of one-tenth the current clinical dose resolved major hallmarks of the disease, including hepatic steatosis, inflammation, and fibrosis, without detectable toxicities. These data suggest a potential clinical translation of low-dose sorafenib as a NASH therapy with less risk of adverse events than is currently observed for its use in HCC.
ACKNOWLEDGMENTS
This work was supported by grants from the National Key R&D Program of China (2016YFF0101504 to Z.-G.S.), the Youth Program of National Natural Science Foundation of China (81800322 to C.J. and 81700356 to Y.-X.J.), the General Program of National Natural Science Foundation of China (81970070 to X.-J.Z.), the National Science Foundation of China (81630011 to H.L.), the Major Research Plan of the National Natural Science Foundation of China (91639304 and 91729303 to H.L.), the National Science Fund for Distinguished Young Scholars (81425005 to H.L.), the Creative Groups Project of Hubei Province (2016CFA010 to H.L.), the Hubei Science and Technology Support Project (2018BEC473 to H.L.), the Hubei Construction Project of Science and Technology Condition Platform in Province (2017BEC001 to H.L.), the Scientific Research Project of Hubei Health Commission (WJ2019Q046 to Y.-X.J.), and the Fundamental Research Funds for the Central Universities (2042018kf0073 to C.J., 2042018kf0063 to J.F., and 2042018kf0233 to Y.-X.J.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cmet.2020.04.011.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adebayo Michael AO, Ko S, Tao J, Moghe A, Yang H, Xu M, Russell JO, Pradhan-Sundd T, Liu S, Singh S, et al. (2019). Inhibiting Glutamine-Dependent mTORC1 Activation Ameliorates Liver Cancers Driven by beta-Catenin Mutations. Cell Metab. 29, 1135–1150.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awada A, Hendlisz A, Gil T, Bartholomeus S, Mano M, de Valeriola D, Strumberg D, Brendel E, Haase CG, Schwartz B, and Piccart M (2005). Phase I safety and pharmacokinetics of BAY 43-9006 administered for 21 days on/7 days off in patients with advanced, refractory solid tumours. Br. J. Cancer 92, 1855–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai L, Chen MM, Chen ZD, Zhang P, Tian S, Zhang Y, Zhu XY, Liu Y, She ZG, Ji YX, and Li H (2019). F-box/WD Repeat-Containing Protein 5 Mediates the Ubiquitination of Apoptosis Signal-Regulating Kinase 1 and Exacerbates Nonalcoholic Steatohepatitis in Mice. Hepatology 70, 1942–1957. [DOI] [PubMed] [Google Scholar]
- Brand MD, Pakay JL, Ocloo A, Kokoszka J, Wallace DC, Brookes PS, and Cornwall EJ (2005). The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem. J 392, 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Xu M, Zhang X, and Li H (2019a). Innate Immune Signaling in Nonalcoholic Fatty Liver Disease and Cardiovascular Diseases. Annu. Rev. Pathol 14, 153–184. [DOI] [PubMed] [Google Scholar]
- Cai J, Zhang XJ, and Li H (2019b). The Role of Innate Immune Cells in Nonalcoholic Steatohepatitis. Hepatology 70, 1026–1037. [DOI] [PubMed] [Google Scholar]
- Carvalho BS, and Irizarry RA (2010). A framework for oligonucleotide microarray preprocessing. Bioinformatics 26, 2363–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung MC, Spalding PB, Gutierrez JC, Balkan W, Namias N, Koniaris LG, and Zimmers TA (2009). Body surface area prediction in normal, hypermuscular, and obese mice. J. Surg. Res 153, 326–331. [DOI] [PubMed] [Google Scholar]
- Chiou JF, Tai CJ, Wang YH, Liu TZ, Jen YM, and Shiau CY (2009). Sorafenib induces preferential apoptotic killing of a drug- and radio-resistant Hep G2 cells through a mitochondria-dependent oxidative stress mechanism. Cancer Biol. Ther 8, 1904–1913. [DOI] [PubMed] [Google Scholar]
- Cohen JC, Horton JD, and Hobbs HH (2011). Human fatty liver disease: old questions and new insights. Science 332, 1519–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornu M, Albert V, and Hall MN (2013). mTOR in aging, metabolism, and cancer. Curr. Opin. Genet. Dev 23, 53–62. [DOI] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- de Graaf EL, Giansanti P, Altelaar AF, and Heck AJ (2014). Single-step enrichment by Ti4+-IMAC and label-free quantitation enables in-depth monitoring of phosphorylation dynamics with high reproducibility and temporal resolution. Mol. Cell. Proteomics 13, 2426–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto H, Tao L, Eguchi R, Sato A, Honda M, Kaneko S, Iwata Y, Nishikawa H, Imanishi H, Iijima H, et al. (2017). The in vivo antitumor effects of type I-interferon against hepatocellular carcinoma: the suppression of tumor cell growth and angiogenesis. Sci. Rep 7, 12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA (2005). Guidance for Industry. Estimating the safe starting dose in clinical trials for therapeutics in adult healthy volunteers. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/estimating-maximum-safe-starting-dose-initial-clinical-trials-therapeutics-adult-healthy-volunteers.
- Fiume L, Manerba M, Vettraino M, and Di Stefano G (2011). Effect of sorafenib on the energy metabolism of hepatocellular carcinoma cells. Eur. J. Pharmacol 670, 39–43. [DOI] [PubMed] [Google Scholar]
- Goedeke L, Peng L, Montalvo-Romeral V, Butrico GM, Dufour S, Zhang XM, Perry RJ, Cline GW, Kievit P, Chng K, et al. (2019). Controlled-release mitochondrial protonophore (CRMP) reverses dyslipidemia and hepatic steatosis in dysmetabolic nonhuman primates. Sci. Transl. Med 11, 10.1126/scitranslmed.aay0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, and Shaw RJ (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimbach JK, Kulik LM, Finn RS, Sirlin CB, Abecassis MM, Roberts LR, Zhu AX, Murad MH, and Marrero JA (2018). AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 67, 358–380. [DOI] [PubMed] [Google Scholar]
- Inoki K, Zhu T, and Guan KL (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. [DOI] [PubMed] [Google Scholar]
- Inoki K, Kim J, and Guan KL (2012). AMPK and mTOR in cellular energy homeostasis and drug targets. Annu. Rev. Pharmacol. Toxicol 52, 381–400. [DOI] [PubMed] [Google Scholar]
- Jeon SM (2016). Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med 48, e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji YX, Huang Z, Yang X, Wang X, Zhao LP, Wang PX, Zhang XJ, Alves-Bezerra M, Cai L, Zhang P, et al. (2018). The deubiquitinating enzyme cylindromatosis mitigates nonalcoholic steatohepatitis. Nat. Med 24, 213–223. [DOI] [PubMed] [Google Scholar]
- Kim A, McCully C, Cruz R, Cole DE, Fox E, Balis FM, and Widemann BC (2012). The plasma and cerebrospinal fluid pharmacokinetics of sorafenib after intravenous administration in non-human primates. Invest. New Drugs 30, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Li T, Yang Z, Yi W, Di S, Sun Y, Wang D, and Yang Y (2017). AMPK orchestrates an elaborate cascade protecting tissue from fibrosis and aging. Ageing Res. Rev 38, 18–27. [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida N, Matsuda S, Itano O, Shinoda M, Kitago M, Yagi H, Abe Y, Hibi T, Masugi Y, Aiura K, et al. (2016). Development of a novel mouse model of hepatocellular carcinoma with nonalcoholic steatohepatitis using a high-fat, choline-deficient diet and intraperitoneal injection of diethylnitrosamine. BMC Gastroenterol. 16, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, et al. ; Nonalcoholic Steatohepatitis Clinical Research Network (2005). Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321. [DOI] [PubMed] [Google Scholar]
- Kraus D, Yang Q, Kong D, Banks AS, Zhang L, Rodgers JT, Pirinen E, Pulinilkunnil TC, Gong F, Wang YC, et al. (2014). Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 508, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczynski EA, Lee CR, Man S, Chen E, and Kerbel RS (2015). Effects of Sorafenib Dose on Acquired Reversible Resistance and Toxicity in Hepatocellular Carcinoma. Cancer Res. 75, 2510–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, and Carter C (2006). Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 66, 11851–11858. [DOI] [PubMed] [Google Scholar]
- Liu D, Wong CC, Fu L, Chen H, Zhao L, Li C, Zhou Y, Zhang Y, Xu W, Yang Y, et al. (2018). Squalene epoxidase drives NAFLD-induced hepatocellular carcinoma and is a pharmaceutical target. Sci. Transl. Med 10, 10.1126/scitranslmed.aap9840. [DOI] [PubMed] [Google Scholar]
- Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. ; SHARP Investigators Study Group (2008). Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med 359, 378–390. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maschmeyer P, Flach M, and Winau F (2011). Seven steps to stellate cells. J. Vis. Exp 10, 2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami H, Kawada K, Ebi H, Kitagawa K, Kim YI, Araki K, Mukai H, Tahara M, Nakajima H, and Nakajima K (2008). Phase I and pharmacokinetic study of sorafenib, an oral multikinase inhibitor, in Japanese patients with advanced refractory solid tumors. Cancer Sci. 99, 1492–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DY, Walenta E, Akiyama TE, Lagakos WS, Lackey D, Pessentheiner AR, Sasik R, Hah N, Chi TJ, Cox JM, et al. (2014). A Gpr120-selective agonist improves insulin resistance and chronic inflammation in obese mice. Nat. Med 20, 942–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Kim T, Zhang XM, Lee HY, Pesta D, Popov VB, Zhang D, Rahimi Y, Jurczak MJ, Cline GW, et al. (2013). Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metab. 18, 740–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Zhang D, Zhang XM, Boyer JL, and Shulman GI (2015). Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 347, 1253–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, and Salzberg SL (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, and Ahmad N (2008). Dose translation from animal to human studies revisited. FASEB J. 22, 659–661. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaker M, Tabbaa A, Albeldawi M, and Alkhouri N (2014). Liver transplantation for nonalcoholic fatty liver disease: new challenges and new opportunities. World J. Gastroenterol 20, 5320–5330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi YH, Ding ZB, Zhou J, Hui B, Shi GM, Ke AW, Wang XY, Dai Z, Peng YF, Gu CY, et al. (2011). Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 7, 1159–1172. [DOI] [PubMed] [Google Scholar]
- Smith BK, Ford RJ, Desjardins EM, Green AE, Hughes MC, Houde VP, Day EA, Marcinko K, Crane JD, Mottillo EP, et al. (2016a). Salsalate (Salicylate) Uncouples Mitochondria, Improves Glucose Homeostasis, and Reduces Liver Lipids Independent of AMPK-β1. Diabetes 65, 3352–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BK, Marcinko K, Desjardins EM, Lally JS, Ford RJ, and Steinberg GR (2016b). Treatment of nonalcoholic fatty liver disease: role of AMPK. Am. J. Physiol. Endocrinol. Metab 311, E730–E740. [DOI] [PubMed] [Google Scholar]
- Stefano JT, Pereira IV, Torres MM, Bida PM, Coelho AM, Xerfan MP, Cogliati B, Barbeiro DF, Mazo DF, Kubrusly MS, et al. (2015). Sorafenib prevents liver fibrosis in a non-alcoholic steatohepatitis (NASH) rodent model. Braz. J. Med. Biol. Res 48, 408–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strumberg D, Richly H, Hilger RA, Schleucher N, Korfee S, Tewes M, Faghih M, Brendel E, Voliotis D, Haase CG, et al. (2005). Phase I clinical and pharmacokinetic study of the Novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43–9006 in patients with advanced refractory solid tumors. J. Clin. Oncol 23, 965–972. [DOI] [PubMed] [Google Scholar]
- Strumberg D, Clark JW, Awada A, Moore MJ, Richly H, Hendlisz A, Hirte HW, Eder JP, Lenz HJ, and Schwartz B (2007). Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: a review of four phase I trials in patients with advanced refractory solid tumors. Oncologist 12, 426–437. [DOI] [PubMed] [Google Scholar]
- Su TH, Shiau CW, Jao P, Liu CH, Liu CJ, Tai WT, Jeng YM, Yang HC, Tseng TC, Huang HP, et al. (2015). Sorafenib and its derivative SC-1 exhibit antifibrotic effects through signal transducer and activator of transcription 3 inhibition. Proc. Natl. Acad. Sci. USA 112, 7243–7248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao H, Zhang Y, Zeng X, Shulman GI, and Jin S (2014). Niclosamide ethanolamine-induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nat. Med 20, 1263–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thabut D, Routray C, Lomberk G, Shergill U, Glaser K, Huebert R, Patel L, Masyuk T, Blechacz B, Vercnocke A, et al. (2011). Complementary vascular and matrix regulatory pathways underlie the beneficial mechanism of action of sorafenib in liver fibrosis. Hepatology 54, 573–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J, Han CJ, Zhang JZ, He WZ, Zhao GJ, Cheng X, Zhang L, Deng KQ, Liu Y, Fan HF, et al. (2019). Hepatic Interferon Regulatory Factor 6 Alleviates Liver Steatosis and Metabolic Disorder by Transcriptionally Suppressing Peroxisome Proliferator-Activated Receptor γ in Mice. Hepatology 69, 2471–2488. [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. [DOI] [PubMed] [Google Scholar]
- Umemura A, Park EJ, Taniguchi K, Lee JH, Shalapour S, Valasek MA, Aghajan M, Nakagawa H, Seki E, Hall MN, and Karin M (2014). Liver damage, inflammation, and enhanced tumorigenesis after persistent mTORC1 inhibition. Cell Metab. 20, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercesi AE, Rodrigues CO, Uyemura SA, Zhong L, and Moreno SN (1998). Respiration and oxidative phosphorylation in the apicomplexan parasite Toxoplasma gondii. J. Biol. Chem 273, 31040–31047. [DOI] [PubMed] [Google Scholar]
- Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, and Marais R; Cancer Genome Project (2004). Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116, 855–867. [DOI] [PubMed] [Google Scholar]
- Wang Y, Gao J, Zhang D, Zhang J, Ma J, and Jiang H (2010). New insights into the antifibrotic effects of sorafenib on hepatic stellate cells and liver fibrosis. J. Hepatol 53, 132–144. [DOI] [PubMed] [Google Scholar]
- Wang PX, Ji YX, Zhang XJ, Zhao LP, Yan ZZ, Zhang P, Shen LJ, Yang X, Fang J, Tian S, et al. (2017). Targeting CASP8 and FADD-like apoptosis regulator ameliorates nonalcoholic steatohepatitis in mice and nonhuman primates. Nat. Med 23, 439–449. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wen H, Fu J, Cai L, Li PL, Zhao CL, Dong ZF, Ma JP, Wang X, Tian H, et al. (2020). Hepatocyte TNF Receptor-Associated Factor 6 Aggravates Hepatic Inflammation and Fibrosis by Promoting Lysine 6-Linked Polyubiquitination of Apoptosis Signal-Regulating Kinase 1. Hepatology 71, 93–111. [DOI] [PubMed] [Google Scholar]
- Wardill HR, Gibson RJ, Van Sebille YZ, Secombe KR, Coller JK, White IA, Manavis J, Hutchinson MR, Staikopoulos V, Logan RM, and Bowen JM (2016). Irinotecan-Induced Gastrointestinal Dysfunction and Pain Are Mediated by Common TLR4-Dependent Mechanisms. Mol. Cancer Ther 15, 1376–1386. [DOI] [PubMed] [Google Scholar]
- Wickham H (2016). Ggplot2: Elegant Graphics for Data Analysis. (Springer-Verlag New York; ). [Google Scholar]
- Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al. (2004). BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 64, 7099–7109. [DOI] [PubMed] [Google Scholar]
- Wiśniewski JR, Zougman A, Nagaraj N, and Mann M (2009). Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362. [DOI] [PubMed] [Google Scholar]
- Xu L, Hui AY, Albanis E, Arthur MJ, O’Byrne SM, Blaner WS, Mukherjee P, Friedman SL, and Eng FJ (2005). Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut 54, 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung CY, Chan WT, Jiang CB, Cheng ML, Liu CY, Chang SW, Chiang Chiau JS, and Lee HC (2015). Amelioration of Chemotherapy-Induced Intestinal Mucositis by Orally Administered Probiotics in a Mouse Model. PLoS ONE 10, e0138746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CS, Hawley SA, Zong Y, Li M, Wang Z, Gray A, Ma T, Cui J, Feng JW, Zhu M, et al. (2017). Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 548, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Wang PX, Zhao LP, Zhang X, Ji YX, Zhang XJ, Fang C, Lu YX, Yang X, Gao MM, et al. (2018). The deubiquitinating enzyme TNFAIP3 mediates inactivation of hepatic ASK1 and ameliorates nonalcoholic steatohepatitis. Nat. Med 24, 84–94. [DOI] [PubMed] [Google Scholar]
- Zhao GN, Zhang P, Gong J, Zhang XJ, Wang PX, Yin M, Jiang Z, Shen LJ, Ji YX, Tong J, et al. (2017). Tmbim1 is a multivesicular body regulator that protects against non-alcoholic fatty liver disease in mice and monkeys by targeting the lysosomal degradation of Tlr4. Nat. Med 23, 742–752. [DOI] [PubMed] [Google Scholar]
- Zhao P, Sun X, Chaggan C, Liao Z, In Wong K, He F, Singh S, Loomba R, Karin M, Witztum JL, and Saltiel AR (2020). An AMPK-caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science 367, 652–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman EI, Gibson AA, Hu S, Vasilyeva A, Orwick SJ, Du G, Mascara GP, Ong SS, Chen T, Vogel P, et al. (2016). Multikinase Inhibitors Induce Cutaneous Toxicity through OAT6-Mediated Uptake and MAP3K7-Driven Cell Death. Cancer Res. 76, 117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All RNA sequencing data reported in this study have been deposited to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database. The accession numbers for the data obtained on HFHC-fed mice, HFD-fed mice, HFHC-fed mice treated with compound C and sorafenib in combination and NASH monkeys are: SRA: PRJNA613854, PRJNA613938, PRJNA613940, PRJNA614574, respectively.
The phospho-proteomic data obtained on PA-treated L02 hepatocytes has been deposited to the ProteomeXchange Consortium via the proteomics identification (PRIDE) partner repository and the accession number is: PRIDE: PXD018214. The proteomic data obtained on NASH monkeys has also been deposited to the ProteomeXchange Consortium via the PRIDE partner repository and the accession number is: PRIDE: PXD018256.
The publicly available datasets from sorafenib-treated HCC cells can be found at GEO: GSE96793, GSE73571, GSE43053, GSE102863. All major software and code used to analyze these datasets are referenced the KEY RESOURCES TABLE.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-β-Actin, dil: 1/1000 | Abcam | Cat#ab8226; RRID: AB_306371 |
| Anti-phospho-AMPKα(Thr172), dil: 1/1000 for WB, dil: 1/50 for IHC | Cell Signaling Technology | Cat#2535; RRID:AB_331250 |
| Anti-AMPKα, dil: 1/1000 | Cell Signaling Technology | Cat#2603; RRID:AB_490795 |
| Anti-phospho-Acetyl-CoA Carboxylase (Ser79), dil: 1/1000 | Cell Signaling Technology | Cat#3661; RRID:AB_330337 |
| Anti-Acetyl-CoA Carboxylase, dil: 1/1000 | Cell Signaling Technology | Cat#3676; RRID:AB_2219397 |
| Anti-phospho-mTOR (Ser2448), dil: 1/1000 for WB | Cell Signaling Technology | Cat#2971; RRID:AB_330970 |
| Anti-phospho-mTOR-S2448 Rabbit mAb, dil: 1/50 for IHC | Abclonal | Cat#AP0115; RRID:AB_2832985 |
| Anti-mTOR, dil: 1/1000 | Cell Signaling Technology | Cat#2983; RRID:AB_2105622 |
| Anti-phospho-B-Raf (Ser445), dil: 1/1000 | Cell Signaling Technology | Cat#2696; RRID:AB_390721 |
| Anti-B-Raf, dil: 1/1000 | Cell Signaling Technology | Cat#9433; RRID:AB_2259354 |
| Anti-Phospho-c-Raf (Ser338), dil: 1/1000 | Cell Signaling Technology | Cat#9427; RRID:AB_2067317 |
| Anti-C-Raf, dil: 1/1000 | Cell Signaling Technology | Cat#12552; RRID:AB_2728706 |
| Anti-phospho-MEK1/2 (Ser217/221), dil: 1/1000 | Cell Signaling Technology | Cat#9154; RRID:AB_2138017 |
| Anti-MEK1/2, dil: 1/1000 | Cell Signaling Technology | Cat#9122; RRID:AB_823567 |
| Anti-phospho-Erk1/2 (Thr202/Tyr204), dil: 1/1000 | Cell Signaling Technology | Cat#4370; RRID:AB_2315112 |
| Anti-p44/42 MAPK (Erk1/2), dil: 1/1000 | Cell Signaling Technology | Cat#4695; RRID:AB_390779 |
| Anti-phospho-TAK1 (Thr184/187), dil: 1/1000 | Cell Signaling Technology | Cat#4508; RRID:AB_561317 |
| Anti-TAK1, dil: 1/1000 | Cell Signaling Technology | Cat#4505; RRID: AB_490858 |
| Anti-phospho-LKB1 (Ser334), dil: 1/1000 | Cell Signaling Technology | Cat#3055; RRID:AB_330071 |
| Anti-LKB1, dil: 1/1000 | Cell Signaling Technology | Cat#3050; RRID:AB_823559 |
| Anti-phospho-CaMKK2 (Ser511), dil: 1/1000 | Cell Signaling Technology | Cat#12818; RRID:AB_2798034 |
| Anti-CaMKK2, dil: 1/1000 | Cell Signaling Technology | Cat#16810; RRID:AB_2798771 |
| Anti-PP2A C Subunit, dil: 1/1000 | Cell Signaling Technology | Cat#2038; RRID:AB_2169495 |
| Anti-PP2C-α, dil: 1/1000 | Cell Signaling Technology | Cat#3549; RRID:AB_2169764 |
| Anti-EGFR, dil: 1/1000 | Cell Signaling Technology | Cat#2232; RRID:AB_331707 |
| Anti-Phospho-EGFR (Tyr1068), dil: 1/1000 | Cell Signaling Technology | Cat#3777; RRID:AB_2096270 |
| Anti-Beclin-1, dil: 1/1000 | Cell Signaling Technology | Cat#3738; RRID:AB_490837 |
| Anti-LC3B, dil: 1/1000 | Cell Signaling Technology | Cat#3868; RRID:AB_2137707 |
| Anti-Phospho-PDGFR beta-Y751, dil: 1/1000 | Abclonal | Cat#AP0493; RRID:AB_2771408 |
| Anti-PDGF Receptor beta, dil: 1/1000 | Abclonal | Cat#A19531; RRID:AB_2832987 |
| Anti-Phospho-VEGFR2-Y1175, dil: 1/1000 | Abclonal | Cat#AP0382; RRID:AB_2771247 |
| Anti-VEGFR2, dil: 1/1000, dil: 1/1000 | Abclonal | Cat#A5609; RRID:AB_2766373 |
| Anti- ErbB2, dil: 1/1000 | Abclonal | Cat#A2071; RRID:AB_2832988 |
| Anti-phospho-ERBB2-Y1221/1222, dil: 1/1000 | Abclonal | Cat#AP0527; RRID:AB_2771088 |
| Anti-Pink1, dil: 1/1000 | Abclonal | Cat#A11435; RRID:AB_2758558 |
| Anti-Parkin, dil: 1/1000 | Abclonal | Cat#A11172; RRID:AB_2758446 |
| Anti-BNIP3, dil: 1/1000 | Abclonal | Cat#A19593; RRID:AB_2832989 |
| Anti-BNIP3L, dil: 1/1000 | Abclonal | Cat#A6283; RRID:AB_2766888 |
| Anti-SREBP1, dil: 1/1000 | Abcam | Cat#ab28481; RRID:AB_778069 |
| Anti-αSMA, dil: 1/1000 | Abcam | Cat#ab7817; RRID:AB_262054 |
| Anti-CTGF(L-20), dil: 1/200 | Santa Cruz | Cat#sc-14939; RRID:AB_638805 |
| Goat anti-mouse IgG-HRP, dil:1/5000 | Jackson | Cat#115-035-003; RRID:AB_10015289 |
| Goat anti-rabbit IgG-HRP, dil:1/5000 | Jackson | Cat#111-035-003; RRID:AB_2313567 |
| Anti-F4/80, dil: 1/50 | Bio-Rad | Cat#MCA497; RRID:AB_2098196 |
| Anti-CD11b, dil: 1/100 | Boster Biological Technology | Cat#BM3925; RRID:AB_2832991 |
| Alexa Fluor® 555 Conjugate Anti-rat IgG (H+L), dil: 1/200 | Cell Signaling Technology | Cat#4417; RRID:AB_10696896 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Pentobarbital sodium | Sigma-Aldrich | Cat#P3761 |
| DMSO | Sigma-Aldrich | Cat#D2650 |
| Sorafenib | Selleck | Cat#S7397 |
| Dorsomorphin (Compound C) 2HCl | Selleck | Cat#S7306 |
| DEN(Diethylnitrosamine) | Sigma-Aldrich | Cat#N0756 |
| β-Cyclodextrin | RHAWN | Cat#R010666 |
| Zoletil | Virbac | Cat#83888102 |
| Xylazine hydrochloride | Jilin Huamu | Cat#180606 |
| PhosStop phosphatase inhibitor | Roche | Cat#4906837001 |
| cOmplete Protease Inhibitor Cocktail | Roche | Cat#04693132001 |
| Clarity™ Western ECL Substrate | Bio-Rad | Cat#1705061 |
| TRIzol | Sigma-Aldrich | Cat#T9424 |
| SYBR Green | Roche | Cat#04887352001 |
| Dulbecco’s modified Eagle’s medium (DMEM) | GIBCO | Cat# C11995500BT |
| fetal bovine serum (FBS) | Tico Europe | Cat#FBSEU500 |
| Polybrene | Sigma | Cat#H9268 |
| Puromycin | GIBCO | Cat#A1113803 |
| Trypsin | GIBCO | Cat#27250-018 |
| 1% penicillin/streptomycin | GIBCO | Cat#15140-122 |
| Sudan III | Solarbio | Cat#S8460 |
| Hematoxylin | Google biology | Cat#G1004 |
| Eosin | BASO | Cat#BA-4024 |
| Masson | BASO | Cat#BA-4079B |
| Tissue-Tek O.C.T. Compound | Servicebio | Cat#4583 |
| Oil Red O | Sigma-Aldrich | Cat# 625 |
| TMRM | AAT Bioquest | Cat# 22221 |
| MitoTracker® Red CMXRos | YEASEN | Cat# 40741ES50 |
| Safranin O | Sigma-Aldrich | Cat#S2255 |
| Rotenone | Dalian Meilun | Cat#MB7162 |
| Digitonin | Sigma-Aldrich | Cat# D5628 |
| Succinate | Shanghai Maclin | Cat# D822253 |
| Pyruvate | Sigma-Aldrich | Cat# P2256 |
| Glutamate | Shanghai Maclin | Cat#L810495 |
| Malate | Shanghai Maclin | Cat#L813179 |
| Ascorbate | Sigma-Aldrich | Cat#A7631 |
| TMPD | Shanghai Maclin | Cat#T819055 |
| ADP | Shanghai Maclin | Cat#A844435 |
| AMP | Sigma-Aldrich | Cat#A2252 |
| ATP | Shanghai Maclin | Cat#A800906 |
| FCCP | Dalian Meilun | Cat#MB3642 |
| Malonic acid | Shanghai Maclin | Cat#M813040 |
| Antimycin A | Dalian Meilun | Cat# MB1384 |
| Sodium azide | Sigma-Aldrich | Cat#S8032 |
| Oligomycin | Sigma-Aldrich | Cat#O4876 |
| Lactobionic acid | Shanghai Maclin | Cat#L812453 |
| Taurine | Sigma-Aldrich | Cat#T0625 |
| Valinomycin | Shanghai Maclin | Cat#V820378 |
| Nigericin | MedChemExpress | Cat# HY-100381 |
| BSA, essentially fatty acid free | Equitech Bio | Cat# BAH66-0100 |
| Sodium palmitate | Sigma-Aldrich | Cat# P9767 |
| Oleic acid | Sigma-Aldrich | Cat#O1008 |
| DAPI | Invitrogen | Cat#S36939 |
| Pazopanib | Selleck | Cat#S3012 |
| ZM336372 | Selleck | Cat#S2720 |
| SU1498 | Selleck | Cat#S6535 |
| Tyrphostin AG 1296 | Selleck | Cat#S8024 |
| Liver Perfusion Medium | Thermo Fisher Scientific | Cat#17701-038 |
| Liver Digestion Medium | Thermo Fisher Scientific | Cat#17701-034 |
| Pronase E | Roche | Cat#10165921001 |
| Collagenase IV | GIBCO | Cat#17104019 |
| DNase I | Roche | Cat#10104159001 |
| Crystal violet | Sinopharm Chemical Reagent | Cat#71012314 |
| Critical Commercial Assays | ||
| BCA Protein assay kit | Thermo Fisher Scientific | Cat#23225 |
| Transcriptor first-strand cDNA synthesis kit | Roche | Cat#04896866001 |
| Triglyceride (TG) assay kit | Wako | Cat#290-63701 |
| Cholesterol assay kit | Wako | Cat#294-65801 |
| NEFA assay kit | Wako | Cat#294-63601 |
| Triglyceride (TG) assay kit | Solarbio | Cat#BC0625 |
| Mouse TNF ELISA kit | Cloud-Clone Co. | Cat#SEA133Mu |
| Mouse MCP1(CCL2) ELISA kit | Cloud-Clone Co. | Cat#SEA087Mu |
| Cell Counting Kit-8 assay kit | Bimake | Cat#B34304 |
| Enhanced enzyme-labeled goat anti-rabbit IgG kit | Beijing ZSGB-Bio | Cat#PV-9001 |
| 3,3′-Diaminobenzidine (DAB) substrate kit | Beijing ZSGB-Bio | Cat#ZLI-9018 |
| RNA 6000 Nano kit | Agilent | Cat#5067-1511 |
| MGIEasy RNA Library Prep Kit | MGI Tech Co. | Cat#1000006384 |
| Deposited Data | ||
| RNA-seq data of HFHC-fed mice | This paper, NCBI SRA | PRJNA613854 |
| RNA-seq data of HFD-fed mice | This paper, NCBI SRA | PRJNA613938 |
| RNA-seq data of HFHC-fed mice treated with CC and sorafenib | This paper, NCBI SRA | PRJNA613940 |
| RNA-seq data of NASH monkeys | This paper, NCBI SRA | PRJNA614574 |
| Proteomic data of NASH monkeys | This paper, PRIDE | PXD018256 |
| Phospho-proteomic data of PA-treated L02 | This paper, PRIDE | PXD018214 |
| Publicly available transcriptomic datasets from sorafenib treated HCC cell lines | GEO database | GSE43053, GSE73571, GSE96793, GSE102863 |
| Experimental Models: Cell Lines | ||
| Human: L02 (HL-7702) | China Center for Type Culture Collection, Wuhan, China. | N/A |
| Human: HEK293T | Cell Bank of Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China. |
Cat#GNHu17 |
| LX-2 | Xu et al., 2005 | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J mice | Vital River | N/A |
| Cynomolgus monkey | Topgene Biotechnology | N/A |
| Oligonucleotides | ||
| sgRNA target sequences to generate gene knockout cell lines, see Table S3 | This paper | N/A |
| Primers used to genotype knockout cell lines, see Table S3 | This paper | N/A |
| Primers used for qPCR, see Table S4 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: lentiCRISPRv2 puro | Addgene | Cat#98290; RRID:Addgene_98290 |
| Plasmid: pMD2.G | Addgene | Cat#12259; RRID:Addgene_12259 |
| Plasmid: psPAX2 | Addgene | Cat#12260; RRID:Addgene_12260 |
| Software and Algorithms | ||
| Las X (v3.4.2) | Leica Microsystems | N/A |
| Graphpad Prism 8 | Graphpad | https://www.graphpad.com/ |
| SPSS statistics 19.0 | IBM Corporation | http://www.spss.com.hk/software/statistics/ |
| Image-Pro plus 6.0 | Media Cybernetics | http://www.mediacy.com/imageproplus |
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| Phoenix WinNonlin 6.3 | Pharsight | https://lp.certara.com/Download-Phoenix-8.2.html |
| HISAT2 2.1.0 | Kim et al., 2015 | http://ccb.jhu.edu/software/hisat2/index.shtml |
| SAMtools 1.4 | Li et al., 2009 | http://samtools.sourceforge.net |
| StringTie 1.3.3b | Pertea et al., 2015 | http://ccb.jhu.edu/software/stringtie/index.shtml |
| DESeq2 1.24.0 | Love et al., 2014 | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| GSEA 3.0 | Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/index.jsp |
| GSVA 1.32.0 | Hänzelmann et al., 2013 | https://bioconductor.org/packages/release/bioc/html/GSVA.html |
| MaxQuant 1.3.0.5 | Cox and Mann, 2008 | https://www.maxquant.org/ |
| Perseus 1.3.0.4 | Tyanova et al., 2016 | https://maxquant.net/perseus/ |
| oligo 1.46.0 | Carvalho and Irizarry, 2010 | https://www.bioconductor.org/packages/release/bioc/html/oligo.html |
| limma 3.38.3 | Ritchie et al., 2015 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| ggplot2 3.1.1 | Wickham, 2016 | https://cran.r-project.org/web/packages/ggplot2/index.html |
| Others | ||
| Mouse high fat diet | Huafukang Bioscience | Cat#D12492 |
| Mouse normal chow diet | Huafukang Bioscience | Cat#D12450B |
| Mouse high fat high cholesterol diet | TrophicDiet | Cat#TP26304 |
| Monkey high fat high cholesterol diet | Beijing Keao Xieli Feed Co. | N/A |
| PVDF membranes | Millipore | Cat#IPVH00010 |
| Transwell plate | Corning | Cat#3428 |
| HyperCarb HPLC Columns,150 × 2.1 mm, 3 mm | Thermo Fisher Scientific | Cat#35003-152130 |