

Abstract
Objective
Vasculitis is characterised by inflammation of the blood vessels. While all layers of the vessel can be affected, inflammation within the intimal layer can trigger thrombosis and arterial occlusion and is therefore of particular clinical concern. Given this pathological role, we have examined how intimal inflammation develops by exploring which (and how) macrophages come to populate this normally immune‐privileged site during vasculitis.
Methods
We have addressed this question for Kawasaki disease (KD), which is a type of vasculitis in children that typically involves the coronary arteries. We used confocal microscopy and flow cytometry to characterise the macrophages that populate the coronary artery intima in KD patient samples and in a mouse model of KD, and furthermore, have applied an adoptive transfer system to trace how these intimal macrophages develop.
Results
In KD patients, intimal hyperplasia coincided with marked macrophage infiltration of the coronary artery intima. Phenotypic analysis revealed that these ‘intimal macrophages’ did not express markers of resident cardiac macrophages, such as Lyve‐1, and instead, were uniformly positive for the chemokine receptor Ccr2, suggesting a monocytic lineage. In support of this origin, we show that circulating monocytes directly invade the intima via transluminal migration during established disease, coinciding with the activation of endothelial cells lining the coronary arteries.
Conclusions
During KD, intimal macrophages develop from circulating monocytes that infiltrate the inflamed coronary artery intima by transluminal migration.
Keywords: intimal hyperplasia, Kawasaki disease, macrophages, monocytes, vasculitis
During vasculitis, macrophages populate the intimal layer of the blood vessels driving pathogenic inflammation. Here, we describe the phenotype and origin of these intimal macrophages. We show that intimal macrophages develop from circulating monocytes that invade the intima through transluminal migration.

Introduction
Vasculitis encompasses a family of diseases characterised by inflammation of the blood vessels. 1 For many forms of vasculitis, vascular inflammation appears to arise when immune cells enter the vessel through activated vasa vasorum. 2 , 3 This results in inflammation within the adventitia, which unchecked, can spread to the medial and intimal layers. 4 , 5 Such transmural inflammation is a feature of severe vasculitis and can have significant pathological consequences. Inflammation of the media can cause aneurysmal formation, while intimal inflammation can precipitate thrombosis, and can subsequently lead to arterial stenosis and tissue ischaemia. 4 , 5 , 6
Thus, while all layers of the vessel can become inflamed in vasculitis, intimal inflammation is of particular clinical concern because of the risk of life‐threatening thrombosis and tissue ischaemia. As such, we have investigated how inflammation develops within this normally immune‐privileged site, focussing on the contribution by macrophages. Macrophages infiltrate the intimal layer in multiple forms of vasculitis 7 , 8 and are reported to produce factors that drive vascular remodelling and myofibroblast proliferation. 9 , 10 , 11 However, given the heterogeneity that exists within the macrophage pool (in terms of function and origin), which macrophages access the intima and how they do so is unclear. In this regard, it is possible that tissue‐resident macrophages infiltrate the intima by adventitial‐to‐intimal migration, or alternatively, that intimal macrophages develop from circulating precursors that directly invade the intima from the bloodstream.
We have addressed this question in the context of Kawasaki disease (KD), a vasculitis typically affecting the coronary arteries that occurs in children. 12 KD is thought to be triggered by an infection activating a systemic inflammatory response in genetically predisposed children. 12 While multiple organs are affected in KD patients, inflammation of the coronary arteries is the major cause of morbidity and mortality. In severe cases, KD patients can develop transmural inflammation of the coronary arteries, 6 , 13 which can lead to coronary aneurysms and intimal hyperplasia, with the latter precipitating thrombosis and/or occlusive stenosis. 5 , 14 , 15 , 16 Here, we have investigated how macrophages contribute to this pathological sequence by describing the phenotype and origin of the macrophages that invade the coronary artery intima in KD.
Results
CD68 + macrophages infiltrate the coronary artery intima during acute Kawasaki disease
To investigate the role of macrophages in driving intimal inflammation, we first determined whether macrophages infiltrate the coronary artery intima during KD. We analysed coronary artery sections from two infants who died from myocardial infarction in the acute stages of KD. Case 1 died on day 15 of disease. The day of death for KD case 2 was not determined, but extensive immune cell infiltrate indicates acute vasculitis (Figure 1). For comparison, we also analysed ‘normal’ coronary arteries obtained from three adult, cardiac disease‐free, organ donors. Sections were analysed by histology (H&E) and confocal microscopy, staining for CD68 (to identify monocyte and macrophages) and α‐SMA [smooth muscle actin (SMA), to identify smooth muscle cells (SMC) and myofibroblasts]. As seen in Figure 1a, all three normal coronary arteries showed minimal immune cell infiltrate, an intact media and a narrow intimal layer. Moreover, while CD68+ macrophages were present within the adventitia and surrounding epicardial adipose tissue, the coronary artery intima was largely devoid, or was sparsely populated, by macrophages (Figures 1a and 3a). By comparison, the coronary arteries from both KD patients showed marked immune cell infiltration (Figure 1b). Immunofluorescence analysis revealed abundant CD68+ monocytes/macrophages, which were distributed widely throughout the vessel. In both KD cases, CD68+ monocyte/macrophages were present within the adventitia and media, where they appeared to displace SMCs in the latter. In addition, CD68+ cells also populated the thickened coronary artery intima of KD patients, accounting for a large proportion of cells within this region. Thus, macrophages infiltrate the coronary artery intima during the acute phase of human KD.
Figure 1.
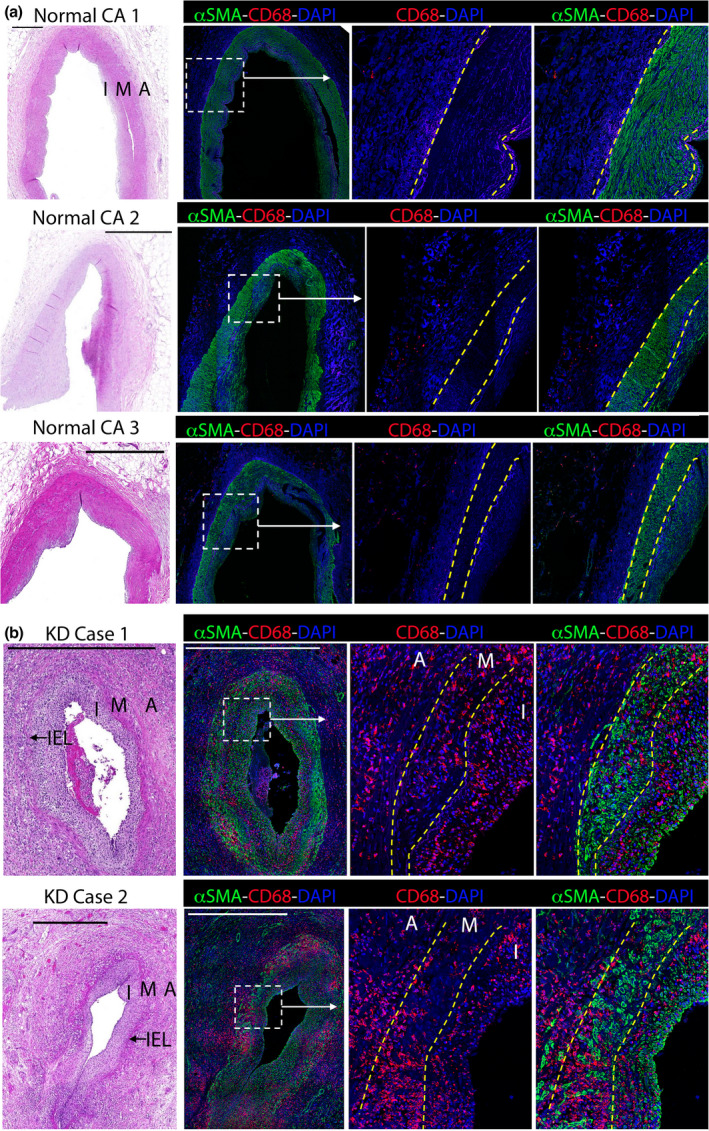
CD68+ monocyte/macrophages infiltrate the coronary artery intima during Kawasaki disease (KD). Coronary artery sections from three cardiac‐disease‐free, adult organ donors (a) or two infants who died from myocardial infarction following KD (b) were analysed by H&E staining and confocal microscopy (staining for α‐SMA, CD68 and DAPI). The intima (I), media (M), adventitia (a) and internal elastic lamina (IEL) are annotated and their borders are marked by dashed lines on inset images. Scale bars are 1000 μm.
Figure 3.
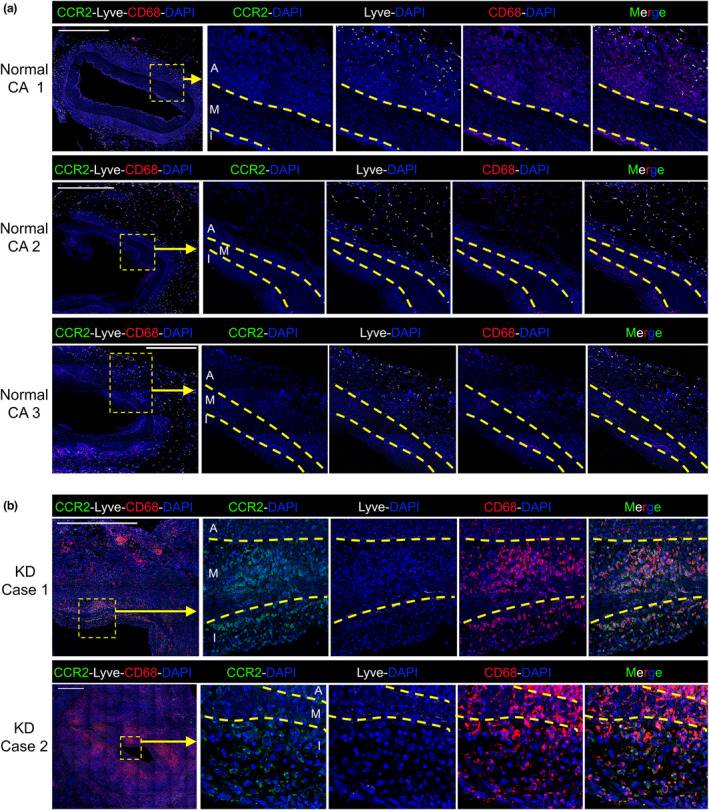
Ccr2+ macrophages infiltrate the coronary artery intima in human Kawasaki disease (KD): Coronary artery sections from three cardiac‐disease‐free adult organ donors (a) or two infants who died from myocardial infarction following KD (b) were analysed by confocal microscopy with staining for CD68, CCR2, Lyve and DAPI. The intima (I), media (M) and adventitia (A) are annotated and their borders marked by dashed lines. Inset shows magnified image and scale bars are 1000 μm.
CAWS injection elicits macrophage infiltration of the coronary artery intima in mice
We next explored how intimal macrophages develop using a well‐described mouse model of KD, where cardiac vasculitis is induced by the injection of the water‐soluble, carbohydrate fraction of Candida albicans (referred to as Candida albicans water‐soluble complex; CAWS). 17 , 18 , 19 To determine whether CAWS injection induced intimal macrophage formation, mice were injected with CAWS, and 4–5 weeks later, cardiac sections were analysed by histology and confocal microscopy (staining for CD68 to identify monocyte/macrophages, CD31 for endothelial cells and using autofluorescence to identify the elastic fibres of the media). As seen in Figure 2, while CD68+ macrophages populated the adventitia (and epicardial adipose tissue) of the coronary arteries of naïve mice, they did not infiltrate the media or intima. By comparison, we observed profound infiltration of CD68+ macrophages within the intimal layer of the coronary arteries of CAWS‐injected mice, which coincided with marked intimal thickening (Figure 2b and c).
Figure 2.
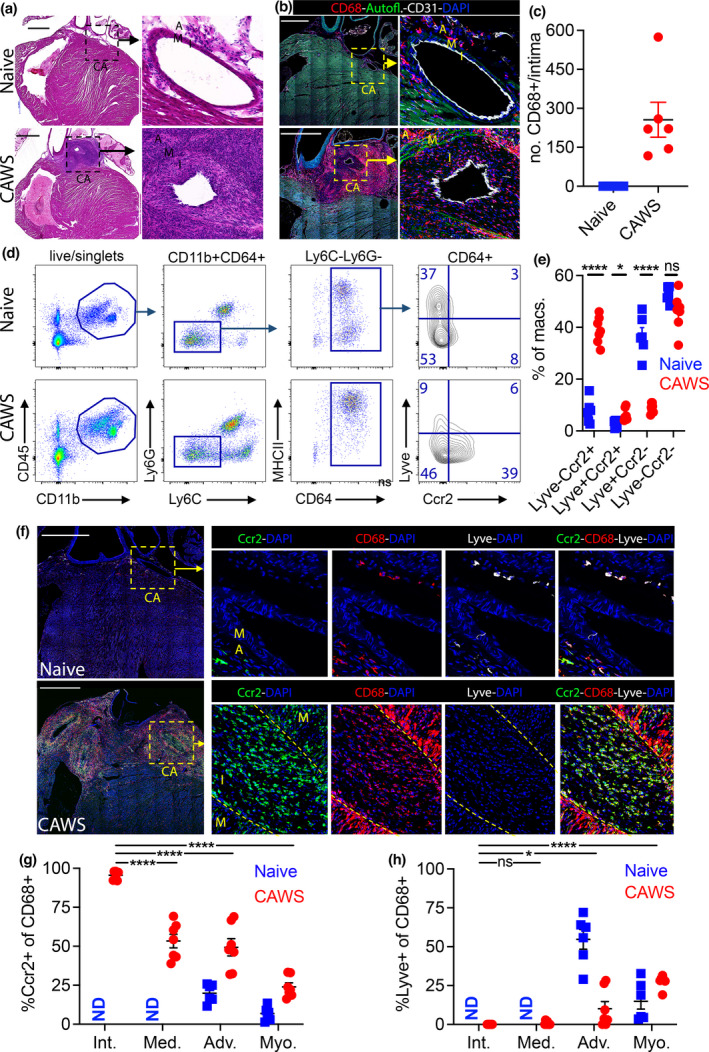
Ccr2+ macrophages dominate the coronary artery intima in the CAWS model of Kawasaki disease. (a–c) Cardiac sections from naïve and CAWS‐injected (4–5 weeks postinjection) mice were analysed by H&E staining (a) and confocal microscopy (b); staining for CD68, CD31, DAPI and using autofluorescence to identify the elastic fibres of the media. Inset shows the coronary artery (CA) with the intima (I), media (M) and adventitia (A) annotated. Scale bars are 1000 μm and (c) shows the number of intimal CD68+ cells/section for individual mice (with mean ± SEM) pooled from three experiments. (d, e) Flow cytometric analysis of hearts from naïve and CAWS‐injected (4–5 weeks postinjection) mice. Doublets and dead (PI+) cells were excluded and the gating strategy is depicted by representative plots. (e) Graphs show the percentage for each macrophage subset for individual mice (n = 6 or 7 mice/group with mean ± SEM) pooled from three experiments. (f–h) Cardiac sections from naïve and CAWS‐injected (4–5 weeks postinjection) Ccr2.CFP mice stained for CD68, Lyve, GFP (to identify Ccr2+ cells) and DAPI, then analysed by confocal microscopy. Inset shows the coronary artery (CA) with the intima (I), media (M) and adventitia (A) annotated and their borders are marked by a dashed line. Scale bars are 1000 μm. (g, h) Percentages of CD68+ macrophages that are Ccr2+ (g) or Lyve+ (h) within the intima, media, adventitia and myocardium are graphed, showing individual mice (n = 6–9 mice/group, with mean ± SEM) pooled from three experiments. Macrophages were not detected (ND) in the intima or the media in naïve mice. Statistical significance levels are expressed as * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001.
Ccr2+ macrophages dominate the coronary artery intima in CAWS‐injected mice
We next used the CAWS model to characterise the phenotype of the intimal macrophages that develop during vasculitis, asking whether they comprise tissue‐resident or monocyte‐derived macrophages (or both). For this analysis, we analysed the expression of lymphatic vessel endothelial hyaluronan receptor (Lyve), which is widely expressed by tissue‐resident macrophages, 20 , 21 and the chemokine receptor Ccr2, which is expressed by short‐lived monocyte‐derived macrophages. 20 , 21 Flow cytometry revealed that the cardiac macrophage compartment (defined as CD45+CD11b+Ly6G−Ly6C−CD64+ cells) of naïve mice contained a large proportion of Lyve+ macrophages and relatively few Ccr2+ cells (Figure 2d and e). By comparison, CAWS‐injected mice had a marked increase in Ccr2+ macrophages and a concomitant loss of the Lyve+/Ccr2− subset (Figure 2d and e). Thus, Ccr2+ macrophages increase within the heart during coronary vasculitis.
We next used microscopy to analyse Lyve and Ccr2 expressions by intimal macrophages. To measure Ccr2 by microscopy, we utilised the Ccr2.DTR.CFP mouse line (referred to as Ccr2‐CFP). This mouse has a diphtheria toxin receptor/cyan fluorescent fusion protein (DTR‐CFP) expressed as a transgene under the Ccr2 promoter, 22 allowing Ccr2‐expressing cells to be identified by CFP/GFP antibody staining. We stained cardiac sections from Ccr2‐CFP mice for CD68, Lyve‐1 and GFP (to identify Ccr2‐expressing cells) with analysis by confocal microscopy. As seen in Figure 2f, Lyve+ macrophages were the major population in the coronary arteries in naive mice and were positioned in the adventitia/adipose tissue (although a small number of Ccr2+ macrophages were routinely detected). In contrast, there was a profound reduction in Lyve+ macrophages around the coronary arteries of CAWS‐injected mice (Figure 2h), coincident with the emergence of CD68+ cells within the intima. Phenotypic analysis revealed that these intimal macrophages were positive for Ccr2 but did not express Lyve‐1 (Figure 2f). Indeed, quantification revealed that nearly all (~96%) macrophages that populated the coronary artery intima in CAWS‐injected mice expressed Ccr2 (Figure 2g), while about 50% of the macrophages within the media or adventitia were Ccr2+. Thus, while the adventitia and media are populated by a diverse pool of Ccr2− and Ccr2+ macrophages, the intima is infiltrated by a homogeneous population of Ccr2+ macrophages.
Ccr2+ macrophages populate the coronary artery intima in human KD
We next determined whether CCR2+ macrophages also populated the intima in human KD. To address this question, coronary artery sections from the three organ donors and two KD fatalities described in Figure 1 were stained for CD68, Lyve‐1 and CCR2 for analysis by confocal microscopy. In keeping with our observations in mice, this analysis revealed that the normal human coronary arteries are populated predominantly by Lyve+ macrophages, with only a small number of CCR2+ macrophages detected (Figure 3a). Thus, Lyve+ macrophages are the dominant subset surrounding normal human coronary arteries. By comparison, there was a marked reduction in Lyve‐1+ macrophages in both KD cases and an increase in CCR2+ macrophages. Moreover, the vast majority of CD68+ macrophages within the intimal layer of KD cases expressed CCR2, but not Lyve (Figure 3b). Thus, the intimal macrophages that appear during KD are characterised by the expression CCR2 and the absence of Lyve‐1.
Circulating Ccr2+ monocytes infiltrate the coronary artery intima by transluminal migration
The expression of Ccr2 indicates that intimal macrophages have a monocytic origin. However, whether monocytes access the intima from the tissue (via adventitial‐to‐intimal migration) or from the bloodstream (via transluminal migration) is unclear. To answer this question, we developed an adoptive transfer system. Wild‐type mice were injected with CAWS and rested for 4–5 weeks to allow intimal inflammation to develop. At this point, CFP+ monocytes (enriched from Ccr2.CFP donor mice) were intravenously injected into recipient CAWS mice, and their migration into the coronary arteries was tracked at 0.5, 1 or 24 h later by microscopy (Figure 4a). As seen in Figure 4, at 0.5 h after transfer, donor CFP+ monocytes were found tethered to the luminal side of the coronary artery and adventitial capillaries but had not yet infiltrated the tissue (Figure 4b). However, at 1‐h post‐transfer, donor monocytes had completed tissue extravasation and were detected within the coronary artery intima and migrating through smaller capillaries in the outer adventitia. However, donor monocytes had not reached the media at 1 h after transfer and only infiltrated this layer at 24 h (Figure 4b–d). This sequence suggests that in established disease, monocytes migrate directly into both the adventitial and the intimal layers of the coronary artery before moving towards the media. Therefore, in regard to the origin of intimal macrophages, these findings suggest that this population develop from monocytes directly invading the intimal layer from the circulation (via transluminal migration), rather than migrating from the tissue (via adventitial‐to‐intimal migration).
Figure 4.
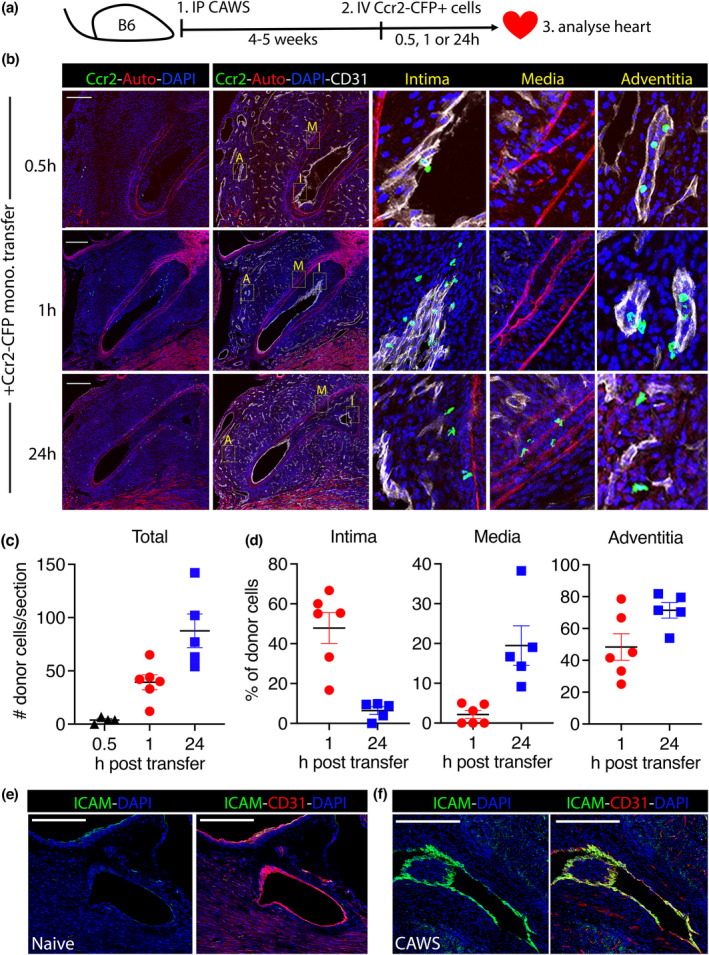
Ccr2+ monocytes infiltrate the coronary artery intima via transluminal migration during vasculitis. (a) Experimental design of the adoptive transfer study. (b) Representative confocal microscopy images of the coronary arteries from CAWS mice at 0.5, 1 or 24 h after the transfer of Ccr2−CFP+ donor monocytes (scale bars are 250 μm). Inset images show magnified regions of the intima (I), media (M) and adventitia (A). (c, d) The total number of donor monocytes within the heart (c) and the percentage of donor cells within each layer of the vessel (d) is graphed. Points depict individual mice (with mean ± SEM) pooled from three independent experiments. (e) Images show coronary arteries of naive and (f) CAWS‐injected mice (4–5 weeks postinjection) stained for ICAM, CD31 and DAPI. A representative image from two experiments (n = 4 mice/group) is shown.
In support of the direct recruitment of circulating monocytes, we found that the CD31+ endothelial layer of the coronary arteries upregulated intercellular adhesion molecule‐1 (ICAM) expression following CAWS injection (Figure 4e). Thus, the endothelium of the coronary arteries becomes activated during vasculitis, acquiring the ability to directly recruit circulating monocytes into the underlying intima. Collectively, these findings reveal that intimal macrophages develop from circulating monocytes via transluminal migration.
Discussion
Through our analysis of human arterial sections from fatal KD cases, we demonstrate that CD68+ monocyte/macrophages infiltrate the coronary artery intima during acute disease. These findings implicate macrophages in the initiation and/or exacerbation of intimal hyperplasia. Moreover, we reveal that these ‘intimal macrophages’ comprise a uniform population, characterised by the expression of CCR2 and an absence of Lyve‐1. Thus, the intimal macrophages that develop in human KD are phenotypically distinct from the Lyve+ tissue‐resident macrophages that normally dominate in nondiseased coronary arteries, and instead, resemble a monocyte‐derived population.
In line with these observations from patient samples, we found that the intimal macrophages that emerge in a mouse model of vasculitis also have a monocytic origin. Specifically, CAWS‐induced vasculitis triggered the appearance of Ccr2+/Lyve−/CD68+ macrophages within the intimal layer of the coronary arteries, similar to that which occurs in human KD. Furthermore, using an adoptive transfer strategy, we revealed that circulating monocytes directly infiltrate the intima of CAWS mice, demonstrating that intimal macrophages develop from circulating monocytes undergoing extravasation through the coronary artery endothelium. In support of this conclusion, the coronary artery endothelium of CAWS‐injected mice was found to be ‘activated’ (as evidenced by increased ICAM expression), acquiring the ability to support monocyte extravasation during vasculitis. These findings not only resolve the direction of monocyte recruitment into the inflamed vascular intima, but also identify potential therapeutic targets. Antagonising adhesion molecules, selectins and/or chemokines that facilitate monocyte‐endothelial interactions may prevent monocytes entering the intima, and thereby reduce inflammation at what is normally an immune‐privileged site.
Furthermore, our findings offer insight into how pathology develops in the intima. We and others have shown that Ccr2+ monocyte/macrophages are essential for cardiac inflammation in the CAWS model. 21 , 23 , 24 Mechanistically, monocytes and monocyte‐derived macrophage are potent producers of factors that could cause intimal pathology. These include IL‐1, 25 which is an essential cytokine for vasculitis, 21 , 24 , 26 , 27 and tissue‐factor, 28 , 29 , 30 which can initiate a coagulation cascade. Thus, by directly invading the intima, infiltrating monocytes may localise inflammation and thrombosis (via the localised production of pathogenic mediators) into the intimal layer.
It is important to note a number of limitations of this study. First, in our human analysis, there is a substantial age discrepancy between normal controls (aged between 40 and 70) and KD cases (which are infants). Thus, while we believe that macrophage infiltration of the intima is driven by vasculitis (a conclusion supported by our studies in aged‐matched mice), we cannot exclude the possibility that the age difference between control and KD cases may influence our observations. Ideally, this conclusion should be confirmed by analysis of coronary arteries from children without vasculitis, although in practice obtaining such samples is difficult. Second, we have used a murine model for mechanistic studies of KD. While the vascular pathology observed in CAWS mice recapitulates many features of human KD (most notably the emergence of Ccr2+ intimal macrophages), it is possible that there are important species differences. As always, some caution is required when relying on animal models of human disease.
In summary, our findings reveal that in coronary vasculitis, intimal macrophages develop from circulating monocytes that access the intima via transluminal migration. These findings provide insight into the pathogenesis of a key pathological event – namely vascular intimal hyperplasia and identify CCR2 and/or adhesion molecules that facilitate monocyte extravasation (such as ICAM) as potential therapeutic targets in vasculitis.
Methods
Mice and the CAWS model of Kawasaki disease
C57BL/6 and Ccr2.DTR.CFP 22 mice were bred at WEHI (Parkville, Australia) under specific pathogen‐free conditions. The CAWS complex was prepared as previously described. 17 , 18 , 19 To induce Kawasaki‐like disease, adult mice received a single intraperitoneal injection of 4 mg of CAWS. All procedures were performed at the WEHI and approved by the WEHI Animal Ethics Committee.
Microscopy of cardiac tissue
Coronary artery sections from fatal cases of KD were obtained from the Victorian Institute of Forensic Medicine (VIFM). Normal cardiac tissue from three adult donors (in the fourth to seventh decades of life) was obtained through the Australian Donor Tissue Biobank (ADTB). Paraffin sections (7 μm) were dewaxed, subjected to citrate antigen retrieval, treated with 0.1% triton‐X and then non‐specific staining blocked with a sequence of donkey serum, TrueBlack Lipofuscin Autofluorescence Quencher (Biotium, CA, USA) and avidin‐biotin‐protein block (Dako, CA, USA). Sections were then stained with antibodies against human‐CD68 (KP1; Novus Biologicals, CO, USA), α‐SMA (1A4; R&D systems, MN, USA), CCR2 (7A7; abcam, UK) and Lyve‐1 (goat polyclonal; R&D systems AF2089) before detection with fluorochrome‐conjugated secondary antibodies or streptavidin (Invitrogen, MA, USA or Abcam). Slides were counterstained with DAPI, imaged on a Zeiss LSM‐880 Confocal Microscope and analysed with ImageJ software. All procedures were approved by the VIFM, ADTB and WEHI Ethics Committees.
For imaging of mouse tissue, mice were perfused with PBS and the hearts fixed in 2% paraformaldehyde (4 h on ice), dehydrated in 30% sucrose (12–18 h at 4°C) and embedded in OCT (Tissue‐Tek, USA). Hearts were sectioned (10 μm) in the coronal plane and analysed by histopathology or immunofluorescence microscopy. For immunostaining, cardiac sections were stained with primary antibodies against GFP (ab290; Abcam), CD31(MEC13.3; BD, USA), CD68 (FA11; BD) or Lyve‐1 (ALY7; BD) before analysis as described above.
Flow cytometry of cardiac tissue
For flow cytometric analysis, murine hearts were digested in type I collagenase (1 mg mL−1; Worthington, NJ, USA) with DNase I (5 μg mL−1; Sigma). Single‐cell suspensions were stained with anti‐mouse CD11b (M1/70), CD45.2 (104), Ly6G (1A8), Ly6C (HK1.4), CD64 (X54.5/7.1), MHC‐II (M5/114), Ccr2 (465301) and Lyve‐1 (ALY7) directly conjugated mAbs (BD Bioscience, R&D Systems or Biolegend, CA, USA). Propidium iodide (100 ng mL−1) was added immediately prior to acquisition on a Fortessa (BD) FACS machine and analysed using Flowjo software.
Adoptive transfer of murine Ccr2+ monocytes
To purify donor monocytes, bone marrow cells and splenocytes were pooled from Ccr2.CFP donor mice and coated with a cocktail of anti‐Ter119 (TER119), B220 (RA3‐6B2), CD3 (145‐2C11) and Ly6G (1A8) biotinylated antibodies (Biolegend). Coated cells were then mixed with streptavidin MACs beads and purified on LS columns (Miltenyi, Germany). The negative, monocyte‐enriched fraction was collected and ~1 × 106 CFP+/Ly6C+ monocytes were injected intravenously into sex‐matched adult mice that had received CAWS 4–5 weeks earlier. At 0.5, 1 or 24 h after transfer, hearts were harvested and analysed by confocal microscopy as described above.
Statistical analysis
Throughout this study, 4–7 mice were analysed for each group in 2 or 3 independent experiments (N for each experiment is provided in the Figure captions). Statistical analysis was performed with Prism 6.0 (GraphPad Software) using unpaired, two‐tailed Student's t‐tests. Statistical significance levels are expressed as *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
AUTHOR CONTRIBUTIONS
Angus T Stock: Conceptualization; data curation; formal analysis; writing – original draft; writing – review and editing. Sarah Parsons: Formal analysis; resources; writing – review and editing. Varun J Sharma: Resources. Fiona James: Resources. Graham Starkey: Resources. Rohit D'Costa: Resources. Claire L Gordon: Resources. Ian P Wicks: Funding acquisition; writing – original draft; writing – review and editing.
Conflict of interest
IPW has received funding from CSL and Med‐Immune for research on cytokine antagonists. The remaining authors declare no conflict of interest.
Acknowledgments
We thank Tom Kitson, Lauren Wilkins, Jacinta Hansen, Huon Wong and Damian D'Silva (WEHI, Melbourne, Australia) for outstanding technical assistance and Drs Andrew Lew and Yifan Zhan (WEHI, Melbourne, Australia) for the provision of mice. This research has been conducted using samples and data from the Australian Donation and Transplantation Biobank (ADTB). We acknowledge the following ADTB collaborators: Lisa Davies, Effie Mouhtouris, Robert Jones, Bao Zhong Wang, M. Lindsay Grayson, Helen Opdam, Jai Raman, Angela Vago and The Austin Transplant Perfusionist Group. We gratefully acknowledge the generosity of the organ donor families for providing invaluable tissue samples. This work was supported by the Australian National Health and Medical Research Council (NHMRC) Program Grant 1113577 and Clinical Practitioner Fellowship 1154235 (to IPW), the Reid Charitable Trusts and NHMRC Early Career Fellowship GNT 1160963 (CLG). This study was made possible through Victorian State Government Operational Infrastructure Support and the Australian Government National Health and Medical Research Council Independent Research Institute Infrastructure Support scheme.
Contributor Information
Angus T Stock, Email: stock.a@wehi.edu.au.
Ian P Wicks, Email: wicks@wehi.edu.au.
References
- 1. Jennette JC, Falk RJ, Bacon PA et al. 2012 Revised international Chapel Hill consensus conference nomenclature of vasculitides. Arthritis Rheum 2013; 65: 1–11. [DOI] [PubMed] [Google Scholar]
- 2. Akiyama M, Ohtsuki S, Berry GJ, Liang DH, Goronzy JJ, Weyand CM. Innate and adaptive immunity in giant cell arteritis. Front Immunol 2020; 11: 621098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hotchi M. Pathological studies on Takayasu arteritis. Heart Vessels Suppl 1992; 7: 11–17. [DOI] [PubMed] [Google Scholar]
- 4. Weyand CM, Goronzy JJ. Immune mechanisms in medium and large‐vessel vasculitis. Nat Rev Rheumatol 2013; 9: 731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Orenstein JM, Shulman ST, Fox LM et al. Three linked vasculopathic processes characterize Kawasaki disease: a light and transmission electron microscopic study. PLoS One 2012; 7: e38998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Friedman KG, Gauvreau K, Hamaoka‐Okamoto A et al. Coronary artery aneurysms in Kawasaki disease: risk factors for progressive disease and adverse cardiac events in the US population. J Am Heart Assoc 2016; 5: e003289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kobayashi M, Ito M, Nakagawa A, Nishikimi N, Nimura Y. Immunohistochemical analysis of arterial wall cellular infiltration in Buerger's disease (endarteritis obliterans). J Vasc Surg 1999; 29: 451–458. [DOI] [PubMed] [Google Scholar]
- 8. Jiemy WF, van Sleen Y, van der Geest KS et al. Distinct macrophage phenotypes skewed by local granulocyte macrophage colony‐stimulating factor (GM‐CSF) and macrophage colony‐stimulating factor (M‐CSF) are associated with tissue destruction and intimal hyperplasia in giant cell arteritis. Clin Transl Immunology 2020; 9: e1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Watanabe R, Maeda T, Zhang H et al. MMP (matrix metalloprotease)‐9‐producing monocytes enable T cells to invade the vessel wall and cause vasculitis. Circ Res 2018; 123: 700–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kaiser M, Weyand CM, Bjornsson J, Goronzy JJ. Platelet‐derived growth factor, intimal hyperplasia, and ischemic complications in giant cell arteritis. Arthritis Rheum 1998; 41: 623–633. [DOI] [PubMed] [Google Scholar]
- 11. Weyand CM, Watanabe R, Zhang H, Akiyama M, Berry GJ, Goronzy JJ. Cytokines, growth factors and proteases in medium and large vessel vasculitis. Clin Immunol 2019; 206: 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Burns JC, Glode MP. Kawasaki syndrome. Lancet 2004; 364: 533–544. [DOI] [PubMed] [Google Scholar]
- 13. Kato H, Sugimura T, Akagi T et al. Long‐term consequences of Kawasaki disease. A 10‐ to 21‐year follow‐up study of 594 patients. Circulation 1996; 94: 1379–1385. [DOI] [PubMed] [Google Scholar]
- 14. Dietz SM, van Stijn D, Burgner D et al. Dissecting Kawasaki disease: a state‐of‐the‐art review. Eur J Pediatr 2017; 176: 995–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Patel AS, Bruce M, Harrington W, Portman MA. Coronary artery stenosis risk and time course in Kawasaki disease patients: experience at a US tertiary pediatric centre. Open Heart 2015; 2: e000206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tsuda E, Kamiya T, Ono Y, Kimura K, Kurosaki K, Echigo S. Incidence of stenotic lesions predicted by acute phase changes in coronary arterial diameter during Kawasaki disease. Pediatr Cardiol 2005; 26: 73–79. [DOI] [PubMed] [Google Scholar]
- 17. Nagi‐Miura N, Harada T, Shinohara H et al. Lethal and severe coronary arteritis in DBA/2 mice induced by fungal pathogen, CAWS, Candida albicans water‐soluble fraction. Atherosclerosis 2006; 186: 310–320. [DOI] [PubMed] [Google Scholar]
- 18. Tada R, Nagi‐Miura N, Adachi Y, Ohno N. The influence of culture conditions on vasculitis and anaphylactoid shock induced by fungal pathogen Candida albicans cell wall extract in mice. Microb Pathog 2008; 44: 379–388. [DOI] [PubMed] [Google Scholar]
- 19. Stock AT, Hansen JA, Sleeman MA, McKenzie BS, Wicks IP. GM‐CSF primes cardiac inflammation in a mouse model of Kawasaki disease. J Exp Med 2016; 213: 1983–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dick SA, Wong A, Hamidzada H et al. Three tissue resident macrophage subsets coexist across organs with conserved origins and life cycles. Sci Immunol 2022; 7: eabf7777. [DOI] [PubMed] [Google Scholar]
- 21. Stock AT, Collins N, Smyth GK et al. The selective expansion and targeted accumulation of bone marrow‐derived macrophages drive cardiac vasculitis. J Immunol 2019; 202: 3282–3296. [DOI] [PubMed] [Google Scholar]
- 22. Hohl TM, Rivera A, Lipuma L et al. Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe 2009; 6: 470–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martinez HG, Quinones MP, Jimenez F et al. Important role of CCR2 in a murine model of coronary vasculitis. BMC Immunol 2012; 13: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miyabe C, Miyabe Y, Bricio‐Moreno L et al. Dectin‐2‐induced CCL2 production in tissue‐resident macrophages ignites cardiac arteritis. J Clin Invest 2019; 129: 3610–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Porritt RA, Zemmour D, Abe M et al. NLRP3 inflammasome mediates immune‐stromal interactions in vasculitis. Circ Res 2021; 129: e183–e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stock AT, Jama HA, Hansen JA, Wicks IP. TNF and IL‐1 play essential but temporally distinct roles in driving cardiac inflammation in a murine model of Kawasaki disease. J Immunol 2019; 202: 3151–3160. [DOI] [PubMed] [Google Scholar]
- 27. Wakita D, Kurashima Y, Crother TR et al. Role of interleukin‐1 signaling in a mouse model of Kawasaki disease‐associated abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol 2016; 36: 886–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Egorina EM, Sovershaev MA, Bjorkoy G et al. Intracellular and surface distribution of monocyte tissue factor: application to intersubject variability. Arterioscler Thromb Vasc Biol 2005; 25: 1493–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Osterud B. Tissue factor expression by monocytes: regulation and pathophysiological roles. Blood Coagul Fibrinolysis 1998; 9(Suppl 1): S9–S14. [PubMed] [Google Scholar]
- 30. Hottz ED, Azevedo‐Quintanilha IG, Palhinha L et al. Platelet activation and platelet‐monocyte aggregate formation trigger tissue factor expression in patients with severe COVID‐19. Blood 2020; 136: 1330–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]