

Abstract
G protein-coupled receptors (GPCRs) are the largest group of receptors involved in cellular signaling across the plasma membrane and a major class of drug targets. The canonical model for GPCR signaling involves three components — the GPCR, a heterotrimeric G protein and a proximal plasma membrane effector — that have been generally thought to be freely mobile molecules able to interact by ‘collision coupling’. Here, we synthesize evidence that supports the existence of GPCR–effector macromolecular membrane assemblies (GEMMAs) comprised of specific GPCRs, G proteins, plasma membrane effector molecules and other associated transmembrane proteins that are pre-assembled prior to receptor activation by agonists, which then leads to subsequent rearrangement of the GEMMA components. The GEMMA concept offers an alternative and complementary model to the canonical collision-coupling model, allowing more efficient interactions between specific signaling components, as well as the integration of the concept of GPCR oligomerization as well as GPCR interactions with orphan receptors, truncated GPCRs and other membrane-localized GPCR-associated proteins. Collision-coupling and pre-assembled mechanisms are not exclusive and likely both operate in the cell, providing a spectrum of signaling modalities which explains the differential properties of multitude of GPCRs in their different cellular environments. Here, we explore the unique pharmacological characteristics of individual GEMMAs, which could provide new opportunities to therapeutically modulate GPCR signaling.
Keywords: G protein-coupled receptors, G protein subnits, plasma membrane effector, GPCR oligomerization, GPCR allosterism
1. Introduction: The GEMMA concept
G protein-coupled receptors (GPCRs) form the largest group of receptors involved in cellular signaling across the plasma membrane. Heterotrimeric (Gαβγ) guanine nucleotide-binding proteins (G proteins) are the initial transducers that convey information from agonist-occupied GPCRs to a variety of effector proteins, many of them localized to the plasma membrane (PM-effectors). The canonical model thus involves three components, the GPCR, a heterotrimeric G protein and a proximal PM-effector, thought initially to be freely mobile molecules able to interact by ‘collision coupling’.
In 1978, Tolkovsky and Levitzki articulated the notion of the collision coupling for GPCR signaling from studies on β-adrenergic receptor (βAR)-mediated adenylyl cyclase (AC) activation in turkey erythrocytes (Tolkovsky and Levitzki, 1978a). That same year, they reported that collision coupling could not explain results obtained using the same system on activation of AC by adenosine receptors. Instead, they observed a first-order process of AC activation, that could be better explained by the existence of pre-coupled GPCR-AC complexes (Tolkovsky and Levitzki, 1978b; Braun and Levitzki, 1979). Collision coupling was consistent with the ‘fluid mosaic’ model of the plasma membrane proposed by Singer and Nicholson in the early 1970s, which described the lipid bilayer as an isotropic milieu, allowing membrane-embedded proteins to diffuse and interact with each other by random collision (Singer and Nicholson, 1970). Initial support for collision coupling in GPCR signaling came from early studies on rhodopsin and its G protein partner transducin (Gt) in rod photoreceptor cells and on purified βAR, G proteins and AC reconstituted in phospholipid vesicles (Neubig, 1994; Levitzki and Klein, 2002).
However, the cellular models used in these influential studies were not generally representative of the common cellular environment of GPCRs and their interacting membrane signaling molecules. The outer segment disks of photoreceptors have a unique lipid composition that provides rhodopsin and Gt with much greater lateral mobility than that of most mammalian membrane proteins. Similarly, receptors and G proteins reconstituted in lipid vesicles are likely to be relatively mobile without the constraints on lateral motion imposed in cellular plasma membranes, such as interactions with the cytoskeleton and membrane microdomains with different protein and lipid compositions, such as ‘lipid rafts’ (see below). Furthermore, the collision coupling model is less compatible with the existence, in the same cell, of a large variety of different GPCRs, heterotrimeric G proteins and PM-effectors (Fig. 1). In this context, the binding of an agonist must promote a series of specific and sequential intermolecular interactions between the activated GPCR, one or more heterotrimeric G proteins comprised of particular Gα, Gβ and Gα subunit combinations and, in many cases, multiple PM-effectors, including AC and phospholipase C (PLC) subtypes and a number of GPCR-modulated ion channels (Cabrera-Vera et al., 2002).
Fig. 1. G protein-coupled receptor signaling.
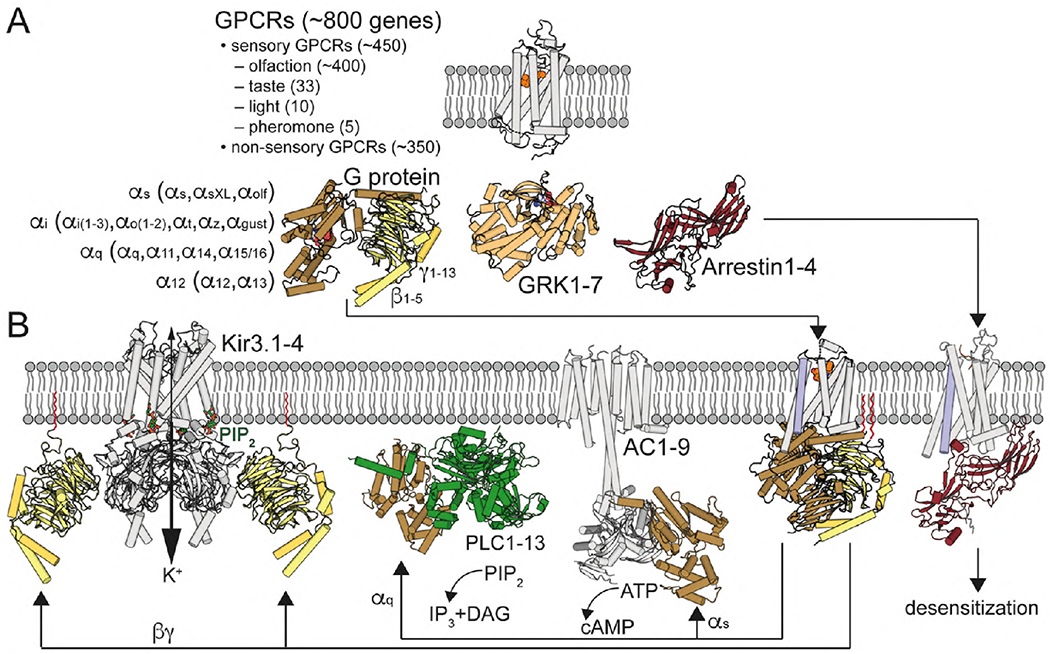
(A) GPCRs are illustrated with the structure of retinal-bound rhodopsin in white (Palczewski et al., 2000). In humans, there are 16 Gα subunits (classified as Gαs, Gαi, Gαq and Gα12) 5 Gβ and 12 Gγ (Gγ1-13, but no Gγ6) subunits. Active GPCRs interact with Gαβγ proteins, illustrated with the structure of GDP-bound Gαi1β1γ2 in brown/yellow (Wall et al., 1995), G protein-coupled receptor kinases (GRKs), with 7 mammalian isoforms, illustrated with the structure of ATP-bound GRK1 in light orange (Singh, Wang, Maeda et al., 2008), and arrestins, with 4 isoforms, illustrated with the structure of arrestin1 in red (Granzin et al., 1998). (B) Agonist binding triggers the movement of TM6 (in blue), opening an intracellular cavity for G protein arrangement, illustrated with the crystal structure of the agonist-β2AR-Gs complex (Rasmussen et al., 2011). Gαq activates PLC, with 13 mammalian isozymes, illustrated with the crystal structure of PLC-β3 bound to Gαq (Waldo et al., 2010), which catalyzes the hydrolysis of PIP2 to the Ca2+-mobilizing second messenger inositol 1,4,5-trisphosphate (IP3) and the PKC-activating second messenger diacylglycerol (DAG). Gαs stimulates and Gαi inhibits the activity of AC, with 9 mammalian isoforms, illustrated with the cryo-EM structure of AC9 bound to Gαs (Qi et al., 2019). Gβγ subunits increase the activity of Kir3 channels, illustrated with the crystal structure of the Kir3.2-Gβγ (Whorton and MacKinnon, 2011). Desensitization is mediated by GRK-mediated GPCR phosphorylation and subsequent binding of arrestin, illustrated with the cryo-EM structure of neurotensin NT1 receptor in complex with arrestin2 (Huang et al., 2020). Gγ isoprenylated group is shown in red.
Here, we establish the concept of a GPCR-effector macromolecular membrane assembly or GEMMA. A GEMMA is defined as a pre-assembled signaling complex composed of particular combinations of GPCRs, G proteins, effectors and other associated transmembrane (TM) proteins localized to the plasma membrane. Further, GEMMAs possess emergent functional and pharmacological characteristics making them potentially unique drug targets. Inherent in the definition of GEMMA is that the interactions between different components are membrane-delimited, which means that all the components are either intrinsic membrane or membrane-associated proteins, with no diffusible cytosolic intermediates between core components.
We must distinguish between the three primary components of membrane-delimited GPCR-mediated signaling pathways (GPCRs, G proteins and PM-effectors), from additional components that modulate or scaffold these core components. Among others, additional elements include G protein-coupled receptor kinases (GRKs) and arrestins, which are primarily recruited to the plasma membrane upon GPCR activation and may not directly interact with PM-effectors (Gurevich et al., 2012; Homan et al., 2013; Gurevich and Gurevich, 2019).
This notion revises the classical model of sequential ligand-induced association-dissociation of GPCR, G protein subunits and PM-effector, and posits that ligands can induce rearrangements within elements of these pre-assembled macromolecular complexes. Metaphorically, we can imagine that GPCRs, G proteins and PM-effectors act as ‘clock gears’, instead of ‘billiard balls’, as described by collision coupling. It can nevertheless be surmised that both alternative modes of interactions coexist in the same cell, particularly for GPCR-G protein interactions.
Within the GEMMA concept, it is necessary to address the impact of GPCR oligomerization. GEMMAs can include a number of identical or different GPCRs as well as orphan GPCRs, truncated GPCRs and other associated TM proteins, to generate unique macromolecular complexes with distinct functional and pharmacological properties. The homomeric nature of class C GPCRs is not a matter of dispute, Their obligate dimeric nature depends on an inter-protomer disulfide bridge formed via a conserved cysteine in their characteristic large binding domain (Ellaithy et al., 2020). The cryo-EM structures of several mGluR homodimers have been recently reported. These include the inactive and active conformations of mGlu5R and mGlu2R homodimers (Koehl et al., 2019; Du et al., 2021) (Fig. 2A), the active conformation of mGlu2R and mGlu4R homodimers coupled to a heterotrimeric Gi protein (Lin et al., 2021) and the inactive conformation of the mGlu7R homomer and the mGlu2R-mGlu7 heteromer (Du et al., 2021). Whether class A (rhodopsin-like family) and family B (secretin receptor family) GPCR dimers are also constitutively formed has been a more contentious issue, even though the crystal structures of several GPCRs revealed homo-oligomers, including the chemokine CXC4R (Wu et al., 2010), the μ-opioid receptor (MOR) (Manglik et al., 2012) and the β1-adrenoceptor (β1AR) (Huang et al., 2013) (Figs. 2B–2D).
Fig. 2. Molecular structures of GPCR homomers.
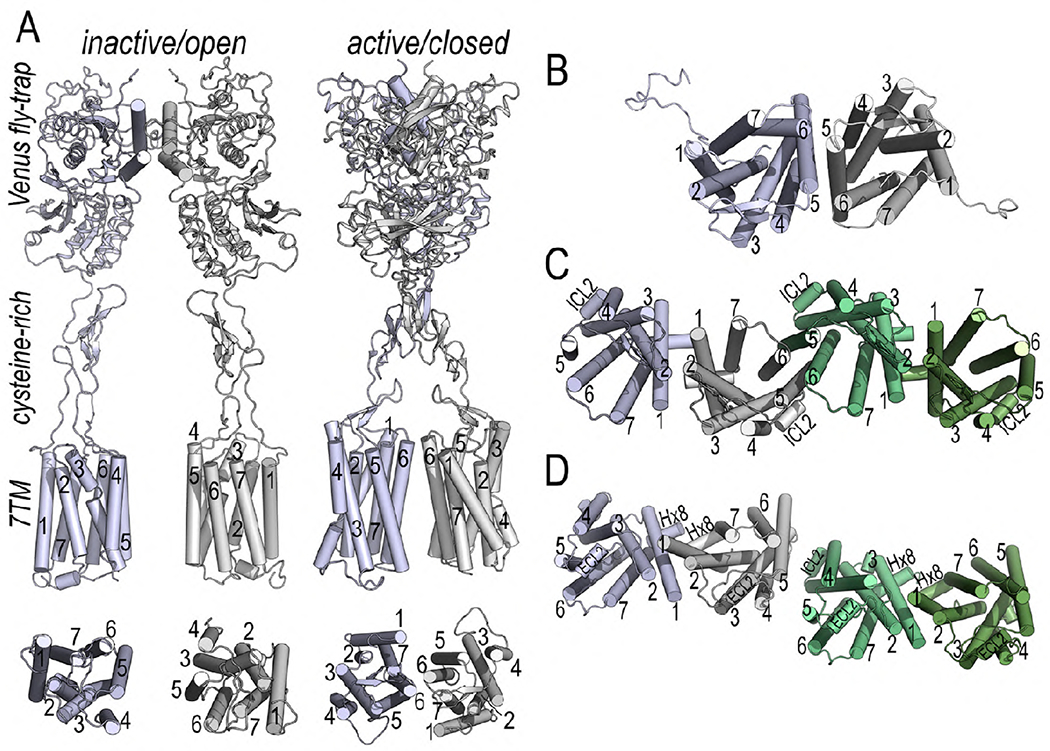
(A) Cryo-EM structures of the full length mGlu5R in the apo inactive/open and active/closed onformations (Koehl et al., 2019). Class C GPCRs are obligate cysteine-linked homomers and each protomer is formed by a large extracellular ‘Venus fly-trap’ domain that binds orthosteric ligands and promotes an inter-protomer disulfide bridge formed via a conserved cysteine, a cysteine rich domain that connects these two domains and a seven TM domain (7TM). The helix interface between protomers of the Venus fly-trap domains is represented by cylinders. Activation leads to compacting of the mGlu5R homomer, bringing the cysteine rich domains into proximity and enabling the 7TM domains to approximate to initiate signaling. The inactive state shows a TM5-TM5 orientation but with substantial separation between the 7TM domains. Activation leads to a 20° rotation of each 7TM domain, resulting in a close contact TM6-TM6 homodimeric interface. Both protomers are colored in light blue and gray. (B) CXC4R structure (Wu et al., 2010) reveals a homomer with an interface including TM5 and TM6. (C) MOR structure (Manglik et al., 2012) shows receptor protomers associated in pairs through two different interfaces. One interface is via TM1, TM2 and helix-8 (Hx8) (blue/white and light/dark green protomers) and the other interface comprises TM5 and TM6 (white/green protomers). (D) β1AR structure (Huang, Chen, Zhang, and Huang, 2013)15 also displays two homomeric interfaces. One interface also involves TM1, TM2 and Hx8 (blue/white and light/dark green protomers), as with MOR. In contrast, the other interface engages residues from TM4, TM5 and intracellular loop 2 (ICL2) white/green protomers).
Apart from the crystallographic evidence, during the last two decades, numerous studies using different technical approaches have provided strong support to the increasingly accepted notion that GPCR homomers and heteromers constitute primary functional GPCR signaling units (for reviews, see Ferré et al., 2014, 2020; Gomes et al., 2016; Sleno and Hébert, 2018; Gaitonde and González-Maeso, 2017; Bourque et al., 2020). GPCR oligomerization significantly broadens the allosteric mechanisms inherent in GPCRs, imposing conformational alterations and constraints of the individual GPCR units (Ferré et al., 2014, 2020). This gives the possibility of developing GEMMAs as targets for drug discovery, with the rationale of the existence of different GEMMAs with common components but with different oligomeric partners localized in different types of cells or cellular environments. Screening campaigns should look for molecules with the ability to promote distinct states of specific GEMMAs, using single agents or combinations of molecules with selectivity for their individual components.
2. Evidence for pre-assembly and ligand-induced rearrangement of specific GPCR, G proteins and effector complexes
2.1. Pre-assemblies of specific Gα, Gβ and Gγ subunits
Since Gilman’s classic conceptualization (Gilman, 1987) it was generally assumed that upon binding to the ligand (a GPCR agonist), GPCRs undergo conformational changes activating heterotrimeric G proteins by promoting GTP binding to Gα subunits and triggering the release of Gβγ subunits. When dissociated, both α and βγ subunits modulate the activities of effector molecules directly responsible for generating cellular responses (Gilman, 1987). However, as we discuss, an alternative model suggests that G protein heterotrimers can be found in more stable complexes that rearrange rather than dissociate upon receptor activation.
In many cases, coupling between receptors and G proteins is pleiotropic such that a single receptor can activate more than one G protein heterotrimer (Flock et al., 2017; Okashah et al., 2019, which allows initiation of multiple signal transduction pathways. However, signaling in vivo most often results in regulation of selective effectors with high fidelity, suggesting that the cell is endowed with different modalities to allow segregation of specific connections between different GPCRs and their effectors. In humans, there are at least 16 different isoforms of Gα subunits, grouped into families- Gαs (Gαs, Gαolf and GαsXL), Gαi (Gαi1, Gαi2, Gαi3, Gαo1, Gαo2, Gαz, Gαt and Gαgust), Gαq (Gαq, Gα11, Gα14 and α15/16) and α12 (α12 and α13); and 5 different isoforms of Gβ (β1-5) and 12 Gγ (Gγ1-13, but no Gγ6) subunits (Downes and Gautam, 1999). The combinatorial association of different G protein subunits potentially provides the bandwidth necessary to generate a broad range of signals independently transduced from different GPCRs to different effectors transmitted by G proteins.
Several seminal studies have provided evidence that specific combinations of Gα, Gβ and Gγ subunits are necessary to generate signal transduction from a given GPCR to a specific effector (Gudermann et al., 1996; Cabrera-Vera et al., 2002; Rebois and Hébert, 2003; Dupré et al., 2009). Studies in the early nineties using antisense oligonucleotides demonstrated that, even in the same cells, different combinations of specific Gα, Gβ and Gγ subunits allowed different receptors to regulate the same effector (Kleuss et al., 1994). Somatostatin, muscarinic M4 and galanin receptors inhibited voltage gated Ca2+ channels by specifically coupling to Gαo2β1γ3, Gαo1β3γ4 and Gαo1β2γ2, respectively (Kleuss et al., 1993; Kalkbrenner et al., 1995). The same studies showed that galanin receptors selectively used the same combination of G protein subunits, Gαo1β2γ2, to inhibit voltage gated Ca2+ channels in a different cell type (Kalkbrenner et al., 1995).
However, there is also experimental evidence indicating that one GPCR couples to different combinations of G protein subunits in different cells. Particularly significant is the example of the α2A-adrenoceptor (α2AAR) in the mouse brain, which, without considering the Gα subunit of the heterotrimer involved, was found to associate selectively to Gβ2, Gγ2 and Gγ3 in noradrenergic cells, but to Gβ4 and Gγ12 in non-noradrenergic cells (Yim et al., 2019). Another remarkable example for the need of specific Gαβγ subunit compositions comes from genetic studies, which demonstrate that, specifically in the striatum, the signaling of adenosine A2A receptors (A2AR) and dopamine D1 receptor (D1R) with AC, more specifically the AC5 isoform, is dependent on specific coupling to Gαolfβ2γ7 (Schwindinger et al., 2003, 2010; Hervé, 2011; Xie et al., 2015). The involvement of specific asemblies of G protein subunits from a potential large number of combinations as components of specific complexes of GPCRs, G proteins and PM-effectors do not seem to depend on a specific preferential affinity between those subunits, but on their synchronized ER synthesis, allowing pre-assembly and delivery to the plasma membrane.
Several studies indicate that Gαβγ heterotrimers pre-assemble before delivery to the plasma membrane, although there remains controversy over the secretory pathway involved. One study suggested that fully processed Gβγ subunits form heterotrimers with Gα on the cytosolic face of the Golgi apparatus (Michaelson et al., 2002). However, another study favored Golgi-independent trafficking of the Gαβγ heterotrimer, where formation was prevented by a dominant mutant of the small GTPase Sar1 (Takida and Wedegaertner, 2004), responsible for the assembly of coated vesicles at the endoplasmic reticulum (ER) membrane (Mizuno-Yamasaki, Rivera-Molina, and Novick, 2012). In any case, assembly of the heterotrimer precedes acylation of the Gα subunit, which is necessary for delivery of the heterotrimer to the plasma membrane (Marrari et al., 2007; Dupré et al., 2009). The evidence for pre-assembly of the full heterotrimeric G protein in its journey to the plasma membrane indicates that the different specific Gαβγ subunit combinations are not selected from a pool of subunits localized at the plasma membrane and sorted by random collision or another so far unidentified mechanism following receptor activation.
2.2. Stable interactions between specific GPCRs, G proteins and PM-effectors
Biophysical techniques have been widely used to evaluate the possible existence of intermolecular interactions between specific GPCRs, G proteins and PM-effectors in living cells (Bouvier, 2001; Milligan, 2001; Guo et al., 2017; El Khamlichi et al., 2019; Soave et al., (2020). In most of these techniques, two chromophores that can change their biophysical properties when they are in contact, or in very close proximity, are separately fused to the two putative interacting proteins and transfected to mammalian cells in culture. These techniques include bioluminescence and Förster or fluorescence resonance energy transfer (BRET and FRET), time-resolved FRET (TR-FRET), bimolecular bioluminescence or fluorescence complementation (BiBC or BiFC), and fluorescence recovery after photobleaching (FRAP). A series of studies using combinations of these biophysical techniques with biochemical techniques (co-immunoprecipitation and pull-down strategies) has provided strong support for the existence of stable interactions between GPCRs and G proteins (Galés et al., 2005, 2006; Nobles et al., 2005; Dupré et al., 2005; Ayoub et al., 2010; Qin et al., 2011; Damian et al., 2015; Andressen et al., 2018) and between G proteins and PM-effectors (Rebois et al., 2006; Riven et al., 2006; Sadana et al., 2009; Yuan et al., 2007). Further evidence has also accumulated for the existence of direct interactions between GPCRs and PM-effectors, including L-type and N-type voltage-dependent Ca2+ channels (Beedle et al., 2004; Rebois et al., 2006), inwardly rectifying potassium Kir3 channels (Lavine et al., 2002; David et al., 2006) and ACs (Lavine et al., 2002; Dupré et al., 2007; Navarro et al., 2018). These studies strongly support the existence of specific assemblies of GPCRs, G proteins and effectors localized in plasma membrane-delimited signaling complexes. Their specific make up raises the notion that components of such complexes could be pre-assembled before delivery to the plasma membrane. It could be demonstrated that dominant negative small GTPase isoforms promote retention of GPCR-G protein, G protein-PM-effector and GPCR-PM-effector assemblies. For instance, prevention of anterograde trafficking to the plasma membrane (using mutants of the coat-recruitment small GTPase Sar1 or the also small GTPase Rab1, which guides transport vesicles to the plasma membrane (Mizuno-Yamasaki et al., 2012), reduced β2AR plasma membrane localization but not interactions between β2AR and Gβ1/Gγ2 subunits, AC2 and β2AR or AC2 and β1 or Gγ2 fused to BRET chromophores (Dupré et al., 2006, 2007), indirectly indicating the pre-assembly of β2AR-β1γ2-AC2 complexes.
Several recent findings suggest plausible scenarios of GPCR-G protein interactions that are different from the fully engaged and agonist-promoted interactions in which helix 5 of the Ras-GTPase domain of the Gα protein interacts with the intracellular cavity of the GPCR formed by the agonist-promoted outward movement of TM6 (Weis and Kobilka, 2018). Whereas initial X-ray and cryo-EM structures of GPCR-G protein complexes were captured in their nucleotide-free state, recent structures based on time-resolved structural mass spectrometry techniques begin to observe alternative complexes and conformational transitions, all indicating the plasticity of interacting interfaces and dynamic nature of the GPCR-G protein complex (Du et al., 2019; Kato et al., 2019; Liu et al., 2019). Such techniques are expected to further reveal the molecular nature of GPCR-G protein pre-assemblies in the near future.
2.3. Association-dissociation of GPCRs and G proteins or ligand-induced rearrangement of GPCR-G protein complexes?
Biophysical techniques have also allowed the analysis of ligand-induced changes in the interactions between GEMMA components. The insertion of BRET chromophores in the α2AAR and in different positions of the Gαi1, β1 and γ2 subunits was used to establish that during the early stages of receptor activation, G protein activation is associated with a relative movement between the Gα and Gβγ subunits that does not involve their complete dissociation (Galés et al., 2006). More precisely, these BRET experiments showed a ligand-induced separation of the Gα subunit helical domain versus the Ras-GTPase domain coupled to the Gβγ subunit (Galés et al., 2006). The relatively large separation of the helical and Ras-GTPase domains of the Gα subunit, which tightly sandwich the nucleotide in all nucleotide-bound G protein crystal structures, was later on confirmed by double electron-electron resonance spectroscopy (Van Eps et al., 2011), a crystal structure of a GPCR-G protein complex (Rasmussen et al., 2011), and deuterium-exchange and electron microscopy data (Chung et al., 2011; Westfield et al., 2011).
Subsequent studies confirmed the ability of different GPCRs to transit from inactive to active conformations by a rearrangement of a pre-assembled receptor-G protein complex, such as a study on monomeric ghrelin GHS1a receptor (GHS1aR) reconstituted in nanodiscs, using TR-FRET and normal mode (NM) analysis (Damian et al., 2015). On the other hand, a more recent study using single-molecule tracking (SMT) with total internal reflection fluorescence (TIRF) obtained results indicating the existence of a predominant population of short-lived (1 second) complexes of α2AAR and Gαi1 labelled with two different organic fluorophores and expressed at low densities in mammalian transfected cells, in the absence of ligands (Sugkaworn et al., 2017). The authors proposed a model dependent on collision coupling, where agonists specifically regulate the kinetics of GPCR-G protein interactions by increasing their association rate (Sugkaworn et al., 2017).
Thus, the debate about a pre-assembly and rearrangement versus collision coupling and association-dissociation of GPCRs and G proteins remains open. Nevertheless, both mechanisms seem to operate and be dependent on protein density, the specific GPCR-G protein pair, and the cellular environment. With BRET and TR-FRET techniques, the GPCR protease-activated PAR1 receptor was shown to specifically form assemblies with Gαi1 but not with Gα12 (Ayoub et al., 2010). Nevertheless, Gα12 was slowly recruited upon receptor activation, indicating the possibility of the two modes of interaction of the same receptor, pre-assembly and collision coupling, depending on the G protein partner (Ayoub et al., 2010). Similar conclusions were obtained from a study using FRAP and BRET techniques examining pre-assembly and agonist-induced rearrangement of the M3R and Gαq (Qin et al., 2011). Moreover, the polybasic epitope was also necessary for an efficient M3R-mediated Gq activation, showing that GPCR-G protein pre-assembly significantly increased the rate of Gq-dependent signaling (Qin et al., 2011). Results obtained with FRAP and FRET techniques also showed a differential ability of 5-HT7R and 5-HT4R to preassemble with Gs proteins (Andressen et al., 2018). The results support a model where pre-associated 5-HT7R-Gαsβ1γ2 complexes undergo agonist-induced rearrangement involving a rapid movement of Gα relative to the receptor, followed by a slower dissociation of Gβγ subunits from both the receptor and the Gα subunit. In contrast, 5-HT4R displayed properties more consistent with collision coupling (Andressen et al., 2018). These studies (Ayoub et al., 2010; Qin et al., 2011; Andressen et al., 2018) therefore indicated the possibility of the coexistence of two alternative signaling modes in the same cell, with pre-assembly providing a means to regulate more specific signaling events as compared to the collision-coupling mode.
2.4. Association-dissociation of G proteins and PM-effectors or ligand-induced rearrangement of G protein-PM-effector assemblies?
A substantial number of studies provided evidence for pre-assembly (inactive state) and rearrangement (active state) of G proteins and PM-effectors, mainly dependent on interactions with Gβγ subunits. Gβγ subunits were initially thought to passively facilitate termination of intracellular information transfer by binding to the Gα subunit and preventing its further spontaneous activation in the absence of receptor activation. This dogma was challenged when Gβγ was shown to directly activate a Kir3 channel in cardiac atrial cells (Logothetis et al., 1987). Gβγ subunits are now known to modulate different isoforms of ACs, PLCs, K+ and Ca2+ channels and many other PM-effectors (Dupré et al., 2009; Khan et al., 2013, 2016; Smrcka and Fisher, 2019). The Gβγ structure includes the seven β-sheet WD40 repeat architecture of the Gβ subunit (the seven “blades” of the Gβ subunit “propeller”) and a “hotspot” surface where the turns between blades intersect. The Gβγ hotspot is required for interactions with numerous effectors and it is concealed in the inactive state by its binding to the Ras-GTPase domain of the Gα subunit. Upon ligand-induced activation, the hotspot is exposed and interacts with available effectors (Cabrera-Vera et al., 2002; Davis et al., 2005). Distinct but overlapping motifs of the hotspot area are involved in the interactions with the Gα subunit and the different effectors (Cabrera-Vera et al., 2002; Mirshahi et al., 2002; Davis et al., 2005; Smrcka and Fisher, 2019). The sites required for agonist-induced activation of effectors by the Gβγ (or by the Gα) subunits are likely to be inaccessible in the GDP-bound, inactive, Gαβγ assembly, thus necessitating certain degree of dissociation of the assembly for activation of the effectors.
In addition to their key modulatory role in effector function, cumulative evidence supports an additional key role of Gβγ subunits in the pre-assembly of G proteins to PM-effectors in their inactive state. This has been addressed in vitro by demonstrating direct interactions between Gβγ subunits with the PM-effector when forming part of the GDP-bound Gαβγ assembly or without involving the hotspot area. Initial evidence was obtained from pulldown experiments with purified bovine Gβγ subunits and the cytosolic NT and CT of the Kir3.1 subunit of Kir3 channels (Huang et al., 1995). Kir3 channels are prototypic PM-effectors of Gβγ subunits and are activated by direct interactions with Gβγ that follow GPCR-dependent activation of Gi/o proteins (Logothetis et al., 1987, 2015; Dascal and Kahanovitch, 2015). Kir3 channels are homo- or hetero-tetrameric and can be composed of four distinct subunits, Kir3.1-4. The basic architecture of the Kir3 channel subunits consists of two TM helices with the cytosolic NT and CT and an extracellular loop which folds back to form the pore-lining ion selectivity filter. Those initial pulldown experiments showed that Gβγ subunits bind to both cytosolic domains and that a GDP-bound Gαβγ assembly specifically binds to the NT of the Kir3.1 subunit, involving contacts mediated by both Gβγ and Gα subunits (Huang et al., 1995). The results also indicated that both cytoplasmic regions of the Kir3 channel subunit are involved in the activation of the Kir3 channel (Huang et al., 1995).
Subsequent studies, using different biochemical and biophysical techniques, provided additional evidence for the involvement of the Gβ subunit in the interaction with Kir3 channels, both in its active and inactive states. Key residues that constituted significant contact points with Kir3 channels and were not concealed by the non-activated Gα subunit were found to be involved in Gβγ-Kir3 channel interactions in the absence of agonist stimulation (Mirshahi et al., 2002). Experiments with internal reflection fluorescence (TIRF) microscopy combined with FRET in cells transfected with type 2 metabotropic glutamate receptor (mGlu2R), Kir3.1 or Kir3.4 fused at various positions of their cytosolic NT and CT with a FRET chromophore and Gβ1 fused to the other chromophore, indicated an interaction between the Gβγ subunits and the Kir3 channel in the resting state and a change in FRET in the presence of the endogenous ligand glutamate (Riven et al., 2006). The results suggested that receptor activation should promote an orientation switch of the Gβγ dimer, without affecting the position of Gα relative to the channel, which would allow the interaction of the Gβγ with the channel at a separate, independent site to promote opening (Riven et al., 2006).
Experiments with BRET in a cell line stably expressing β2AR allowed further analysis of interactions between Gβγ and Kir3 channels and another PM-effector, AC2, in the absence and presence of ligands, again indicating both pre-assembly and agonist-induced rearrangement (Rebois et al., 2006). The same study provided evidence for pre-assembly and ligand-induced rearrangement of G proteins and Kir3 channels before trafficking to the plasma membrane, by fusing Kir3.1 and Gγ2 to BRET chromophores and measuring BRET changes in response to membrane-permeable and non-membrane agonists in the presence and absence of Kir3.4. In the absence of Kir3.4, the β1γ2-Kir3.1 complex did not traffic to the plasma membrane and changes in BRET were only evident using membrane-permeable agonists (Rebois et al., 2006). These results therefore indicated an intracellular localization of functional β2AR as part of a GEMMA.
The subsequent publication of the crystal structure of the Kir3 channel with four Kir3.2 subunits in complex with Gβ1γ2 and PIP2 confirmed the inferences made from many of the previous biochemical and biophysical studies (Glaaser and Slesinger, 2015). The biologically relevant complex consists of one channel tetramer, four Gβγ subunits, four PIP2 molecules and four Na+ ions bound to regulatory sites (Whorton and MacKinnon, 2013). Gβγ subunits interact directly with the cytosolic CT and they are oriented such that the CT of the Gγ subunit, which contains a covalent geranylgeranyl group, points directly to the membrane layer as if to function as an anchor (Whorton and MacKinnon, 2013). Thus, the Kir3.2-Gβ1γ2 crystal structure is compatible with a physiological membrane-delimited Gβγ activation of Kir3. We are now waiting for structures of the Kir3 channel in complex with a GPCR and its heterotrimeric Gi protein in both inactive and active states. In the meantime, the studies described here provide evidence for pre-assembly and rearrangement of Gαβγ-Kir3 complexes in response to agonist. Both Gα and Gβγ subunits appear to be directly associated with Kir3 under both ligand-free inactive and agonist-induced active states. Agonist-induced rearrangement of the complex suggests separation of the Gβγ subunits from Gα subunit, but within a metastable complex, with Gβγ subunits moving away from the initial Kir3 NT- and CT-bound Gαβγ assembly.
The existence of G proteins and AC as a stable complex was first proposed by Levitzki based upon co-purification of AC and G proteins from turkey erythrocyte membranes independent of the activation state of the G proteins (Levitzki, 1988; Levitzki and Klein, 2002;Bar-Sinai et al., 1992). A role of Gβγ subunits in the pre-assembly and scaffolding of ACs has also been supported by in vitro and in cellulo experiments (see below). Further, we also know that Gβγ subunits can directly interact with AC facilitating or inhibiting its function depending on the AC isoform. This modulatory role of Gβγ is also conditional on Gαs-mediated activation. ACs are generally classified into four different categories based on their regulatory properties. All isoforms of TM ACs are stimulated by GTP-bound Gαs. Group I includes Gβγ-inhibited and Ca2+-stimulated AC1, AC3 and AC8; group II includes Gβγ-stimulated AC2, AC4 and AC7; group III includes Gαi and Ca2+-inhibited and Gβγ-stimulated AC5 and AC6; and group IV includes AC9, which is not regulated by Gαi or Ca2+ and was initially believed to be forskolin-insensitive (Dessauer et al., 2017; Baldwin et al., 2019; Qi et al., 2019). The topology of all ACs includes a long NT, two membrane-spanning domains, M1 and M2, each with six TM domains and two large cytoplasmic catalytic domains, C1 and C2. The C1 and C2 domains are homologous and dimerize to form the catalytic core at their interface, where ATP is converted to cAMP. When a GTP-bound Gαs subunit binds to C2, it increases the affinity between C1 and C2, promoting catalysis, while for group III, Gαi subunits bind to C1 and have the opposite effect (Sadana and Dessauer, 2009; Dessauer et al., 2017; Baldwin et al., 2019).
FRET experiments using truncated AC5 provided compelling evidence for a significant role of the NT of AC5 in scaffolding inactive, GDP-bound Gαβγ (Sadana et al., 2009). It was also shown that AC5 NT interacts with its catalytic domains to enhance Gαs- or forskolin-stimulated AC5 activity. These results support a model of Gαβγ-AC5 interactions where the AC5 NT brings the inactive heterotrimeric G protein and AC5 catalytic core in close proximity for efficient GPCR activation (Sadana et al., 2009). A subsequent study showed that Gαβγ binds to the NT of most AC isoforms (Brand et al., 2015). Mutational analysis indicated that the Gβγ hotspot does not interact with the AC5 NT scaffolding side, although it remains necessary for conditional Gβγ-mediated stimulation of AC5. On the other hand, the Gβγ hotspot was required for both stimulating AC6 and interacting with its NT domain, indicating that Gβγ regulation of AC involves multiple binding events, and that the AC NT plays unique isotype-specific roles (Brand et al., 2015).
The recently reported structure of the active state (GTPγS-bound) of AC9 in complex with Gαs, using cryo-electron microscopy (cryo-EM) and single-molecule analysis, revealed for the first time the twelve TM domains of a TM AC isoform, as well as a helical domain (HD) that spans between the membrane and the catalytic domains of AC9 (Qi et al., 2019) (Fig. 1B). The HD, in fact, corresponds to a 40-residue-long cytosolic extension of TM6 and TM12 (Qi et al., 2019). This implies the necessity of dissociation of the Gα subunit from the pre-assembled GPCR-Gαβγ complex allowing the concealed domain of the Ras-GTPase domain of the Gα subunit to interact with the corresponding AC catalytic domain. Similarly to Kir3 channel-Gαβγ assembly, this suggests that ligand-induced rearrangements of the components of such complexes are associated with a separation of the Gα subunit from the Gβγ subunits, but within the framework of the assembly, guided by the modifications in the interactions between the Gα and Gβγ subunits with the AC NT.
3. Compartmentalization of signaling molecules at the plasma membrane by membrane nanodomains and scaffold proteins
3.1. Membrane nanodomains
Apart from pre-assembly of receptor-based signaling complexes, to achieve fidelity and maintain the efficiency of signaling across the plasma membrane, the cell uses two additional complementary mechanisms that promote co-localization or association of specific components of distinct GPCR signaling complexes: i) membrane nanodomains, such as lipid rafts and ii) scaffold proteins. Lipid rafts are defined as heterogeneous, dynamic, cholesterol- and sphingolipid-enriched membrane nanodomains (10–200 nm), which have the potential to form microscopic domains (>300 nm) upon clustering induced by protein-protein and protein-lipid interactions (Pike, 2016). Enriched hydrophobic components and phospholipids that contain saturated fatty acyl chains increase lipid packing and order and subsequent decreased fluidity (Ahmed et al., 1997; Pike, 2003; Sezgin et al., 2017). Certain structural proteins enriched in lipid rafts serve as scaffolds or anchors for other proteins, including caveolins, glycosylphosphatidylinositol (GPI)-linked proteins and cortical actin, which also play an active part in the maintenance and remodeling of lipid rafts (Pike, 2003; Sezgin et al., 2017).
Since lipid rafts and other membrane nanodomains are not resolvable on a conventional optical microscope, super-resolution optical microscopy approaches such as photoactivated localization microscopy (PALM), stimulated emission depletion (STED) or stochastic optical reconstruction microscopy (STORM) have played a significant role in the visualization of lipid-mediated membrane protein clustering (for reviews, see Curthoys et al., 2015; Sezgin et al., 2017). For more dynamic measurements, the addition of SMT analysis has allowed to evaluate the diffusion of membrane molecules and relate it to models of heterogeneous organization of the membrane (Curthoys et al., 2015; Sezgin et al., 2017). Those include the membrane “hot spots” of interacting α2AAR and Gαi1 described by Sugkaworn et al. (2017).
The presence within lipid rafts of a variety of membrane proteins involved in cell signaling led to the consensus that membrane nanodomains play an important role in facilitating rapid and specific signal transduction events (Smart et al., 1999; Simons and Toomre, 2000). Many GPCRs and their cognate signaling partners, including G protein subunits and AC isoforms, are enriched in lipid rafts (Ostrom et al., 2000). In the simplest scenario, lipid rafts could serve to co-localize specific GPCRs, G proteins and PM-effectors. Alternatively, rafts could contain a much larger array of signaling molecules activated when a receptor or other required molecule is recruited into the raft (Pike, 2003). There is also evidence that the targeting of GPCRs and their proximal signaling partners to lipid rafts can be cell specific (Ostrom et al., 2000, 2002). Finally, there is evidence indicating that not all lipid rafts are equivalent and may have distinctly different protein and/or lipid components that coexist in cells (Pike, 2003). However, there is no evidence for mechanisms to isolate specific GPCRs, G protein subunits and PM-effectors by segregating them into distinct lipid rafts or other membrane nanodomains and, given the large number of different signaling molecules that they can harbor, membrane nanodomains cannot be the primary means for generating signaling specificity.
3.2. Plasma membrane scaffold proteins and A kinase anchoring proteins
The most extensively studied plasma membrane scaffold proteins are those localized in and around the postsynaptic density of the glutamatergic synapse. PSD-95/Discs large/Zona occludens-1 (PDZ) domain-containing proteins (PDZ proteins) are the most abundant and PDZ domains represent the most common protein-protein interaction domain (Dunn and Ferguson, 2015). PDZ proteins directly bind to the PDZ-binding ligand motifs found in the CT region of many adhesion molecules and receptors and often contain additional modular interacting domains, which can bind to each other and to various signaling proteins encompassing both sides of the synapse (Sheng and Sala, 2001; Dunn and Ferguson, 2015; Funke et al., 2005; Zheng et al., 2011). PSD-95 family PDZ domains bind to the CT of several GPCRs, including β1AR and β2AR (Hu et al., 2000; Xiang et al., 2002; He et al., 2006; Joiner et al., 2010; Valentine and Haggie, 2011; Li et al., 2013; Dunn and Ferguson, 2015) and serotonin 5-HT2A and 5-HT2C receptors (Xia et al., 2004). The PDZ domain of another PDZ protein family, Shank, interacts indirectly with GPCRs, including group I metabotropic glutamate receptors through the additional scaffold protein Homer, which binds to the Shank proline-rich domain (Fig. 3A) (Dunn and Ferguson, 2015; Zheng et al., 2011; Bécamel et al., 2004). At the postsynaptic density, a key scaffolding link is determined by indirect interactions between PSD-95 and Shank proteins established by guanylate kinase-associated protein (GKAP), which refers to a family of four scaffold proteins which connect the guanylate kinase domain of PSD-95 with Shank PDZ domain (Shin et al., 2012) (Fig. 3A). Apart from specific interactions with GPCRs, direct interactions have been reported between PDZ proteins and the G protein subunit Gγ13, particularly expressed in taste and olfactory cells (Li et al., 2006; Liu et al., 2012) and between Shank and the CT of the central pore subunit of the L-type voltage-dependent Cav1.3 Ca2+ channels (α1.3) (Olson et al., 2005). However, to our knowledge, there is no evidence to date for other direct interactions between PSD-95 and Shank families of PDZ proteins and G protein subunits or other PM-effectors.
Fig. 3. Scaffolding of GPCRs, ligand-gated ion channels and PM-effectors mediated by PDZ proteins and AKAPs.
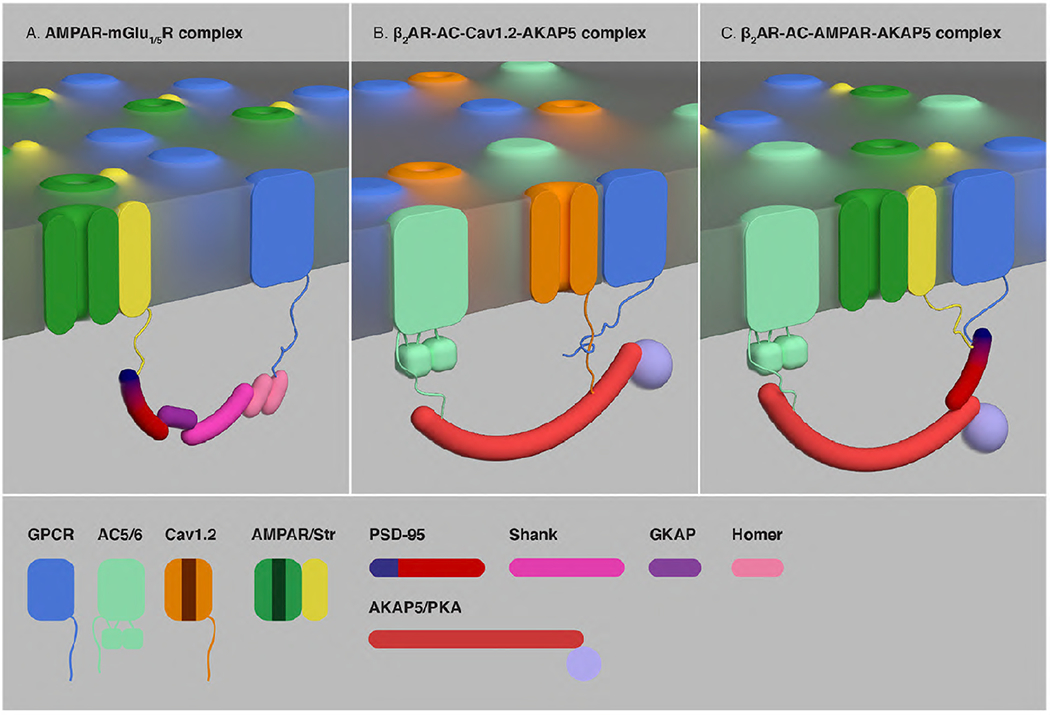
(A) Complexes of ionotropic glutamate AMPAR group I metabotropic receptors (mGlu1R or mGlu5R) established by a chain of interactions of the scaffold proteins PSD-95, GKAP, Shank and Homer. Homer directly interacts with the CT of mGlu1R or mGl5R, while AMPAR indirectly binds to PSD-95 by oligomerization with the TM AMPAR regulatory protein stargazing (Str). (B) Complexes of β2AR, AC and L-type voltage-dependent Cav1.2 Ca2+ channels mediated by AKAP5. (C) Complexes of β2AR, AC and AMPAR mediated by AKAP5 and PSD-95. By bringing together β2AR, AC and PKA, AKAP5-mediated complexes provide the frame for an efficient β2AR-dependent, PKA-mediated phosphorylation of the α1.2 subunit of Cav1.2 channels (B) and the GluA1 subunit of AMPAR (C).
A-kinase anchoring proteins (AKAPs) are a family of structural diverse proteins which share a common motif that binds to the regulatory subunits of the cyclic AMP-dependent protein kinase A (PKA) (Wong and Scott, 2004). More than 50 AKAPs have been identified which can be found in most cellular organelles, as well as the plasma membrane. The scaffolding of unique signaling elements by different types of AKAPs provides a framework for integration and modulation of distinct AC-cAMP-PKA-mediated cell signals (Dessauer, 2009). For instance, the plasma membrane AKAP5 (also known as AKAP79/150) targets not only PKA, but also PKC and protein phosphatase 2B (PP2B), to the inner face of the plasma membrane (Scott et al., 2013; Patriarchi et al., 2018). In addition, AKAP5 binds to the intracellular domains of several membrane-bound signaling molecules, including β1AR and β2AR (Fraser et al., 2000; Gardner et al., 2006; Valentine and Haggie, 2011; Li et al., 2013) and the PM-effectors AC5 and AC6 (Bauman et al., 2006; Efendiev et al., 2010) and L-type voltage-dependent Cav1.2 Ca2+ channels (Hall et al., 2007; Oliveria et al., 2007; Patriarchi et al., 2018). In addition, PSD-95 family proteins can interact through their SH3 and GK domains with a broad surface in the central region of AKAP5. PDZ proteins and AKAPs can therefore combine their interactions with different molecules to scaffold distinct macromolecular signaling complexes that include GPCRs. In this way, AKAP5 can promote the interaction of PKA with its plasma membrane-localized phosphorylation targets, either directly, such as for β1AR and β2AR (Fraser et al., 2000; Valentine and Haggie, 2011; Li et al., 2013), AC5 and AC6 (Bauan et al., 2006; Zhang et al., 2013) and the α1.2 subunit of Cav1.2 channels (Oliveria et al., 2007), or via binding to PSD-95 family proteins, such as for the GluA1 subunit of ionotropic glutamate AMPA receptors (AMPARs) (Colledge at al., 2000; Joiner et al., 2010; Diering et al., 2014).
Interestingly, the β2AR has been shown to be involved in AKAP5-dependent PKA-mediated phosphorylation of both the α1.2 subunit of Cav1.2 channels (Patriarchi et al., 2018) and the GluA1 subunit of AMPAR (Joiner et al., 2010); both events occur in dendritic spines of excitatory synapses (Patriarchi et al., 2018). Thus, at the postsynaptic density, there is evidence for the existence of different AKAP5-scaffolded macromolecular β2AR-AC-AKAP5 complexes that include either Cav1.2 channels or AMPAR (Figs. 3B and 3C). In both complexes, the β2AR does not seem to be directly connected to AKAP5. In the β2AR-AC-Cav1.2-AKAP5 complex, β2AR oligomerizes with Cav1.2 (Davare et al., 2001), which directly binds to AKAP5 (Hall et al., 2007; Oliveria et al., 2007) (Fig. 3B). In the β2AR-AC-AMPAR-AKAP5 complex, β2AR binds directly and GluA1 binds indirectly to PSD-95, through its oligomerization with the TM AMPAR regulatory protein stargazin; PSD-95 then couples to AKAP5 bringing AC5 and PKA to form the larger complex (Joiner et al., 2010; Zhang et al., 2013) (Fig. 3C). The AMPA-stargazin complex can also indirectly interact with group I mGluRs by means of a PSD-95-GKAP-Shank-Homer link (Tu et al., 1999; Zheng et al., 2011; Dunn and Ferguson, 2015) (Fig. 3A). In summary, there is evidence for specificity in the interactions of structurally similar GPCRs for some PDZ domains (He et al., 2006; Valentine and Haggie, 2011; Li et al., 2013) and different AKAPs (Fan et al., 2001; Gardner et al., 2006; Valentine and Haggie, 2011). However, those involve relatively few GPCRs, which likely does not account for the multiplicity of specific combinations of GPCRs, G protein subunits and PM-effectors expressed in different cells or even the same cell. In summary, although complementary, neither membrane nanodomains nor scaffold proteins and AKAPs can generate the specificity of combinations of GPCRs, G protein subunits and PM-effectors provided by GEMMAs.
4. GPCR oligomerization
4.1. Homomers and heteromers as common functional GPCR units
Initial evidence for GPCR homomerization came from radioligand binding studies, with the demonstration of ligand binding cooperativity. This phenomenon has been recognized for many years and reproduced in many experimental preparations, including experiments with membrane extracts from multiple native tissues, mammalian transfected cells and other artificial systems, including GPCRs reconstituted in detergent micelles, liposomes (phospholipid vesicles with or without different proportions of cholesterol and integral membrane proteins) or nanodiscs (phospholipid bilayer preparations held together by membrane scaffold proteins) (reviewed in Ferré et al., 2014, 2020). Additional evidence for GPCR homomerization came with pioneering studies with BRET and FRET techniques (reviewed in Bouvier, 2001; Milligan, 2001; Guo et al., 2017; El Khamlichi et al., 2019; Soave et al., 2020). This was followed by a large number of studies using these and other biophysical techniques in living cells, such as FRAP (Dorsch et al., 2009), TR-FRET (Cottet et al., 2012) and with single molecule-based methods, with the analysis of single fluorescence-labeled receptor molecules by fluorescence autocorrelation and cross-correlation spectroscopy (FCS and FCCS; Herrick-Davis et al., 2013; Teichmann et al., 2014) and SMT analysis by TIRF (Calebiro et al. 2013; Scarselli et al., 2016). Single molecule-based methods in transfected cells also provided evidence for a differential degree of oligomerization and stoichiometry of GPCR oligomers versus monomers, depending on the type and density of GPCR (Calebiro et al., 2013; Teichmann et al., 2014; Scarselli et al., 2016). GPCR type-dependent extent of oligomerization was also recently reproduced using fluorescence-microscopy-based techniques in liposome preparations (Walsh et al., 2018).
Biophysical techniques have also been fundamental in the study of GPCR heteromers (Ferré et al., 2014, 2020; Gomes et al., 2016; Sleno and Hébert, 2018; Gaitonde and González-Maeso, 2017; Bourque et al., 2020). Furthermore, several studies also using biophysical techniques, dominant negative mutant GTPases and other complementary strategies, strongly supported the pre-assembly of not only family C (Margeta-Mitrovic et al., 2000), but also family A GPCR homomers and heteromers in the ER (Salahpour et al., 2004; Dupré et al., 2006; Herrick-Davis et al., 2006; Décaillot, et al., 2008).
The application of techniques that selectively disrupt specific oligomeric interfaces have not only provided the means to establish the quaternary structure of GPCR oligomers, but also their emergent functional and pharmacological properties, as compared with parent monomers. Those techniques include mutating key residues, using chimeras, cysteine crosslinking techniques, or using synthetic peptides with the amino acid sequence of putative interacting domains, for instance of specific TM domains (TM peptides) (Ferré et al., 2014). Disrupting TM peptides were initially introduced to investigate the TM interface of the β2AR homodimer in co-immunoprecipitation experiments, suggesting the involvement of TM6 (Hébert et al., 1996). More recently, the TM-peptide strategy has been applied to BiFC experiments to identify the interfaces of several GPCR homodimers and heteromers (Guitart et al., 2014, 2019; Navarro et al., 2018; Rivera-Oliver et al., 2019; Köfalvi et al., 2020). With this approach, TM6 was also found to be involved in the interface of adenosine A2A receptor (A2AR) and dopamine D2R (D2R) homodimers (Navarro et al., 2018). The involvement of TM6 in D2R homomerization was further substantiated by selective ability of peptides derived from TM6 of D2R to completely counteract the decrease in binding of a bivalent ligand (see below) with selective picomolar affinity for the D2R homodimer (Pulido et al., 2018). TM6 has also been shown to be part of the homomeric interface of high-resolution crystallographic structures of several GPCRs, including CXC4R and MOR (Wu et al., 2010; Manglik et al., 2012) (Figs. 2B and 2C). In addition, using the TM-peptide-BiFC approach, TM6 was found to be involved in several interfaces of GPCR heteromers, including adenosine A1R-dopamine D1R, dopamine D1R-D3R and adenosine A2AR-cannabinoid CB1R heteromers (Guitart et al., 2014, 2019; Rivera-Oliver et al., 2019; Köfalvi et al., 2020).
The fact that rearrangement of TM6 constitutes a critical ligand-induced conformational change that determines G protein activation and modulation of ligand affinity (Dupré et al., 2007), provides a framework for understanding allosteric communication through protomers of GPCR homomers or heteromers with a TM6 interface (Navarro et al., 2018; Ferré et al., 2020). Nevertheless, as reviewed elsewhere (Ferré et al., 2014), TM6 is not always involved in GPCR homomer or heteromer interfaces (see for instance the alternative interfaces obtained with the β1AR and MOR crystal structures, shown in Figs. 2C and 2D). In some cases, the homomeric interface can change in the presence of an agonist, as shown for mGluRs. Recent results obtained with the cryo-EM structures of the full-length mGlu5R shows a TM5-TM5 orientation in its inactive state, but with substantial separation between the seven TM domains. Activation of the receptor leads to a 20° rotation of each seven TM domain which results in a close contact TM6-TM6 homodimeric interface (Fig. 2A) (Koehl et al., 2019). Nevertheless, the disulfide bridge is not the only determinant of homodimerization in the inactive state of mGlu2R, which shows extensive interactions along the whole length of TM4 (Du et al., 2021). Agonist binding to the mGlu2R is then associated with a TM interface change mainly contributed by TM6 (Du et al., 2021). Finally, the same GPCR can utilize different TMs in their heteromeric interactions with different GPCRs and it can also display different homomeric interfaces when forming heteromers with different GPCRs (heteromers of homodimers; see below and Köfalvi et al., 2020).
4.2. Allosteric interactions of GPCR heteromers
There is now a long list of putative GPCR receptor heteromers (Gomes et al., 2016; Gaitonde and González-Maeso, 2017), discussion of which is beyond the scope of this review. Here, we will only summarize findings for the A2AR-D2R heteromer, a paradigmatic example that demonstrates the different types of allosteric interactions in the context of a GPCR heteromer which constitutes the core of a prototype GEMMA. Allosteric interactions constitute a common property of GPCR heteromers (Ferré et al., 2009, 2014, 2020). Importantly, these allosteric properties provide a framework for translational significance, for their potential role as therapeutic targets.
Allosterism is currently defined as “the process by which the interaction of a chemical or protein at one location on a protein or macromolecular complex (the allosteric site) influences the binding or function of the same or another chemical or protein at a topographically distinct site” (Smith and Milligan, 2010). Here we suggest three types of allosterism that can be identified in GPCR heteromers: type I allosterism, or allosterism between ligands, which entails the ability of an orthosteric ligand of one protomer in the GPCR heteromer to modify the affinity or efficacy of an orthosteric ligand of the other molecularly distinct protomer (Fig. 4A); type II allosterism, or ligand-independent allosterism, where heteromerization per se modifies the properties of a specific orthosteric ligand for one of the protomers of the GPCR heteromer, independent of ligand binding to the other molecularly distinct protomer (Fig. 4B); and type III allosterism, or allosterism through the effector, where the effector (PM-effector of a GEMMA) acts as an interface for the interactions between orthosteric ligands of the molecularly distinct protomers of a GPCR heteromer (Fig. 4C).
Fig. 4. Allosterism in GPCR heteromers.
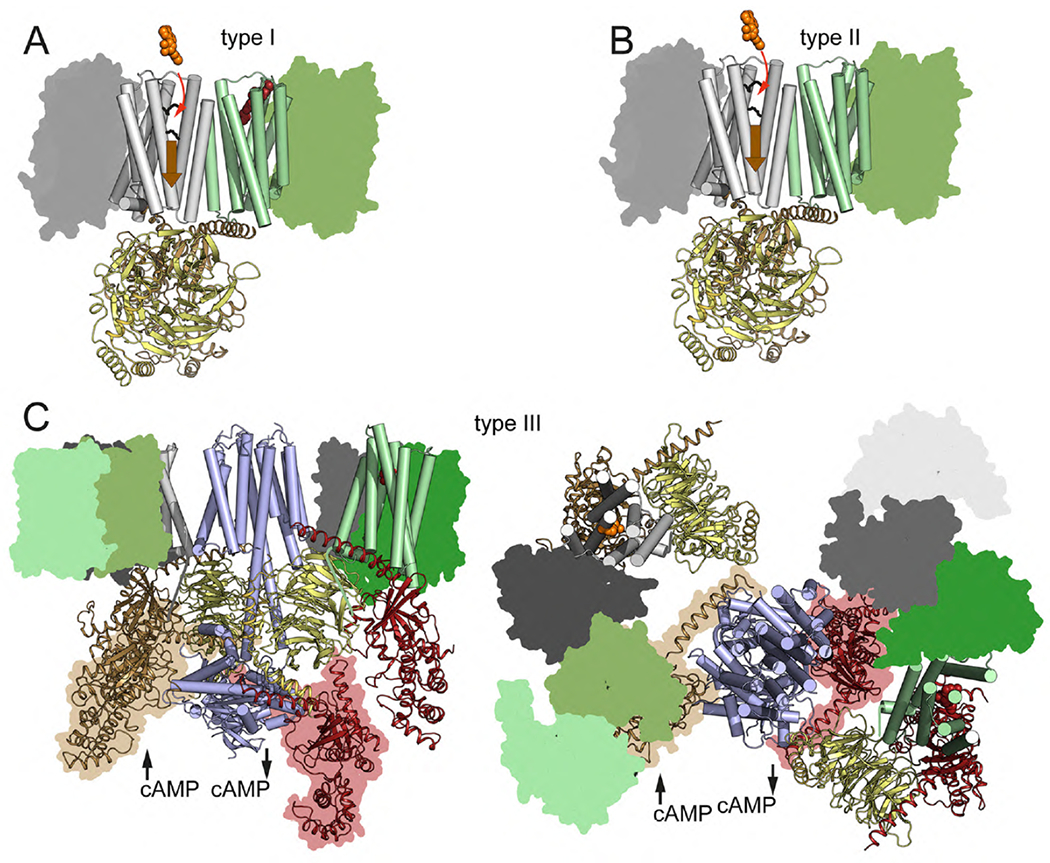
(A) Type I allosterism, in which a ligand (red spheres) binding to one protomer (green cylinders) in the GPCR heteromer can modify the affinity (ligand binding depicted by the red arrow) or the efficacy (receptor activation depicted by the wide orange arrow) of another ligand (orange spheres) binding to the partner receptor (gray cylinders) via their TM helices (see Fig. 5). (B) Type II allosterism, or ligand-independent allosterism, in which the affinity or efficacy of a ligand (orange spheres) for a GPCR (gray cylinders) can be modified just by heteromerization with a molecularly different GPCR (green cylinders). (C) Lateral view (left) and extracellular view (right) of a computer model of a GEMMA including two heterotetramers composed of two different GPCR homomers (one represented by white cylinders and grey surfaces and the other represented by green cylinders and surfaces), AC (light blue cylinders; based on the cryo-EM structure of AC9; Qi et al, 2019), Gs (brown for Gαs and yellow for Gβγ) and Gi (red for Gαi and yellow for Gβγ) (based on Navarro et al., 2018). This type of GEMMA can explain the ability of a Gi-coupled GPCR to counteract AC activation mediated by a Gs-coupled GPCR (type III allosterism). The proposed contact between the CT domain of the receptor and the Gβ subunit is shown by a color line (Tsai et al. 2019). To facilitate visualization of all protomers of the GPCR oligomers, one of the protomers is represented by cylinders and the other protomers are represented as color surfaces (with different shades of the same color for the same GPCR). Both positions of the Gαs and Gαi subunits, in their Gβγ-associated and receptor-bound and state (without surface) and in their Gβγ-dissociated and AC-bound state (with surface), are shown. The agonist-induced dissociation of Gαs and Gαi from their respective Gβγ takes place within the framework of the GEMMA..
The most described type I allosteric interaction in GPCR heteromers involves an agonist or an antagonist of one of the protomers that inhibit the affinity or efficacy of an agonist of the other molecularly distinct protomer. Such interactions are usually referred as ‘negative crosstalk’ or ‘cross-antagonism’, respectively, and their molecular mechanisms are beginning to be understood (Ferré et al., 2014, 2020) (Fig. 5). Since the first report in 1991 (Ferré et al., 1991), A2AR ligands have been repeatedly reported to antagonistically modulate the binding properties of D2R ligands in membrane preparations from transfected mammalian cells and native tissues (reviewed in Ferré et al., 2018). Demonstration of the dependence of these ligand interactions on the integrity of the A2AR-D2R heteromer (with disruptive synthetic peptides or mutations of key residues of the heteromeric interface) (Borroto-Escuela et al., 2010a,b; Bonaventura et al., 2015) implied a type I allosterism in the A2AR-D2R heteromer. This allosterism determines the therapeutic effects of A2AR antagonists in Parkinson’s disease, which target A2AR-D2R heteromers localized in striatopallidal neurons (for recent review, see Ferré et al., 2018). Significantly, the selective A2AR antagonist istradefylline, in combination with L-DOPA, is the first non-dopaminergic drug approved by FDA for the treatment of Parkinson’s Disease (Chen and Cunha, 2020).
Fig. 5. Mechanisms of type I allosterism in GPCR oligomers heteromers.
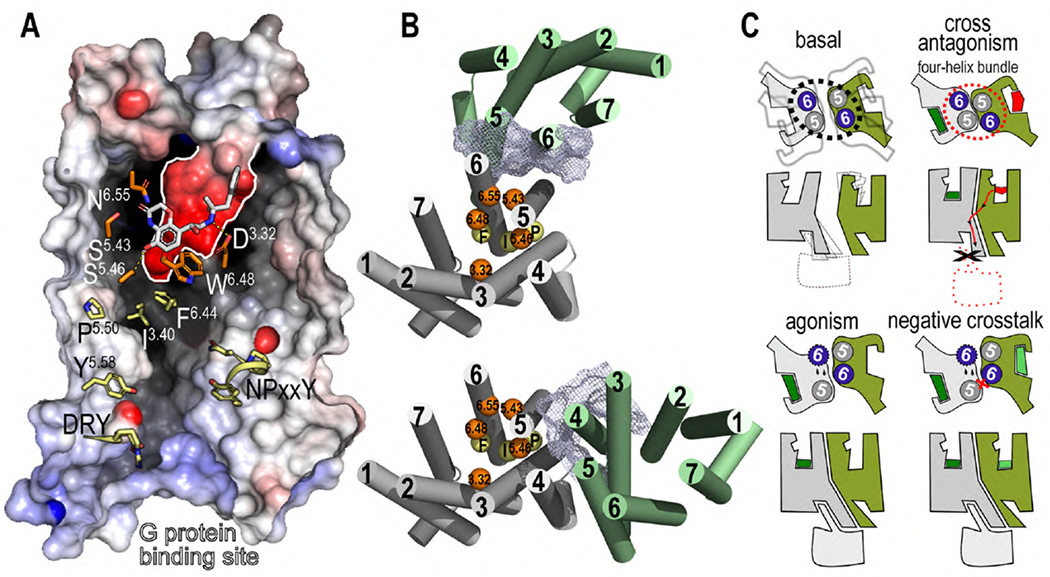
(A) Cross-section through a prototypical class A GPCR, highlighting the agonist (white sticks), the amino acids of the orthosteric site (delineated by a white line) involved in ligand binding (orange), the conserved PIF motif (yellow), and the NPxxY and DRY motifs and Y, (yellow) that transmit the signal from the PIF motif (transmission switch) to the G protein site (Weis and Kobilka, 2018). (B) Position of these amino acids involved in ligand binding (orange spheres) and signal transmission (yellow) in models of GPCR dimers via TM5 and TM6 (top) and TM4 and TM5 and intracellular loop 2 (bottom). Clearly, a ligand-bound (type I allosterism) or ligand-free (type II allosterism) or orphan GPCR (green cylinders) can modify the affinity (orange spheres) or efficacy (yellow spheres) of the partner receptor (gray) via the protein-protein interface (blue mesh). (C) GPCRs are dynamic proteins that adopt, in a ligand-free form, a number of conformations (shades in gray) that not only involve the extracellular and intracellular sites (Weis and Kobilka, 2018), but also a potential TM oligomeric interface. An inverse agonist (red polygon) binding to one of the protomers of a GPCR heteromer triggers a high surface complementarity between TM5 and TM6 of the two molecularly different protomers, via the four-helix bundle (dashed red circle) (Manglik et al., 2012), which blocks the opening of the intracellular cavity for G protein binding at the other protomer (cross-antagonism) (Viñals et al., 2015). An agonist (green polygon) binding to one of the protomers triggers the outward movement of TM 6 (see arrows), opening the intracellular cavity for G protein binding (Rasmussen et al., 2011), whereas the TM5 and TM6 heteromeric interface impedes simultaneous agonist-induced movement of both TM6 (negative crosstalk) due to a steric clash (red cross) (Guinart et al., 2020).
Type II allosterism has been described in the A2AR-D2R heteromer involving a specific A2AR antagonist (Fig. 4B). When comparing the binding properties of several selective orthosteric A2AR antagonists (including istradefylline) in mammalian transfected cells, only one compound, SCH442416, showed a selective low affinity for A2AR when co-expressed with D2R, as compared with A2AR when expressed alone or co-expressed with A1R (Orrú et al., 2011). The specific behavior of SCH442416 in the A2AR-D2R heteromer could also be demonstrated in the mouse striatum, but not in mice with conditional genetic deletion of striatal D2R (Ferré et al., 2018). These results represented proof of concept for the possibility of using GPCR heteromers as targets for specific ligands. Thus, it could be demonstrated that SCH442416 is a preferential presynaptic striatal A2AR antagonist, because of the preferential pre-synaptic localization of A1R-A2AR heteromers in striatal glutamatergic terminals, as compared with the post-synaptic striatal localization of A2AR-D2R heteromers in striatopallidal neurons (Ferré et al., 2018). The preferential striatal presynaptic effect of SCH442416 was suggested to determine its ability to reduce cannabinoid self-administration in monkeys and its possible application to cannabinoid use disorder (Justinová et al., 2014).
Apart from the ability of A2AR ligands to allosterically modulate D2R ligands, a reciprocal canonical Gs-Gi protein-dependent antagonistic interaction was discovered early on, where D2R agonists, by activating Gi-coupled D2R, could oppose Gs-coupled, A2AR-mediated activation of AC-cAMP-PKA signaling (Kull et al., 1999). The balance between antagonistic allosteric and Gs-Gi-AC interactions determines the final output of A2AR-D2R heteromer-dependent signaling in the striatopallidal neuron (Ferré et al., 2018). Importantly, the Gs-Gi-AC interaction depends on the integrity of the A2AR-D2R heteromer, since the Gi-dependent regulation of Gs-mediated AC activation could also be disrupted by TM peptides that disrupt the heteromer (Navarro et al., 2018). This discovery represents a significant shift in our understanding of crosstalk between GPCR signaling pathways, since such interactions at the effector level were previously understood as related to independent changes in second messenger levels (Zoli et al., 1993). GPCR heteromer-dependent Gs-Gi-AC interactions have been demonstrated for several other GPCR heteromers, including A1R-D1R, D1R-D3R and A2AR-CB1R heteromers (Guitart et al., 2019; Rivera-Oliver et al., 2019; Köfalvi et al., 2020). In addition to GPCR heteromerization, the Gs-Gi-AC interaction suggests simultaneous respective interaction of Gαs and Gαi subunits with the C2 and C1 catalytic domains of the same molecule of AC, more specifically with group III ACs, the AC5 and AC6 isoforms. This depends on a pseudo-symmetrical arrangement of the C1 and C2 domains (Dessauer et al., 1988). Another mechanism for antagonistic interactions between Gs- and Gi-coupled receptors can also occur with group I ACs, AC1, AC3 and AC8, involving interactions with Gβγ rather than the Gαi subunits of an activated Gi protein (Taussig et al., 1993; Steiner et al., 2006).
The need for heteromerization of a Gs-coupled and a Gi-coupled GPCR, for oligomerization of those GPCRs with AC, and for the simultaneous binding of Gs and Gi to the C2 and C1 catalytic domains of AC, strongly suggests that a canonical antagonistic Gs-Gi-AC interaction should most commonly occur within GEMMAs that include a GPCR heteromer, Gs and Gi proteins and the PM-effector AC. Following the definition of allosterism, we can consider the antagonistic Gs-Gi-AC interaction as an allosteric interaction of a GPCR heteromer, as part of a macromolecular complex that includes a PM-effector, which we labeled type III allosterism (Fig. 4C).
4.3. GPCR heterotetramers
The relatively large size of the heterotrimeric G protein makes simultaneous binding of more than one G protein to a GPCR unlikely. Therefore, the pentameric structure consisting of a GPCR homodimer and one heterotrimeric G protein is likely the primary functional unit, as initially supported by studies with the detergent-solubilized leukotriene B4 receptor BLT1 (BLT1R) (Banéres and Parello, 2003). The minimal functional quaternary structure of a GPCR heteromer able to sustain a Gs-Gi-AC interaction would then be tetrameric, including Gs- and Gi-coupled homomers, or a Gs-Gi-coupled heterotetramer, as previously hypothesized (Ferré, 2015). With computational analysis of information provided from TM-peptide-BiFC experiments and several reported crystallographic structures of GPCRs alone or bound to G protein partners, it was possible to infer an optimal quaternary structure of the A2AR-D2R heterotetramer: a linear structure with the two internal protomers involved in the heteromer interaction (via symmetrical TM4-TM5/TM5-TM4 interfaces) and the two external protomers of each homodimer (with a TM6/TM6 interface) coupled to their respective G proteins (Navarro et al., 2018) (Fig. 6A). Since AC5 and Golf are the major striatal AC and Gs family isoforms (Schwindinger et al., 2003, 2010; Hervé, 2011; Xie et al., 2015), evidence for Gαβγ-AC5 pre-assembly (see above) suggests that Gα(olf)βγ can indirectly pre-assemble AC5 with the A2AR-D2R heterotetramer, forming a A2AR-D2R-AC5 GEMMA, providing an example of an allosteric machine that integrates types I, II and III allosteric mechanisms (Fig. 6A).
Fig. 6. The A2AR-D2R heterotetramer-AC5 and mGlu2R-5-HT2AR-PLC-Kir3 GEMMAs.

(A) The A2AR-D2R heterotetramer-AC5 GEMMA, constituted by A2AR homomers coupled to Gs, D2R homomers coupled to Gi and AC5, can be considered as a prototype of GEMMA that integrates canonical antagonistic Gs-Gi-AC interactions. (B) The mGlu2R-5-HT2AR-PLC-Kir3 GEMMA, constituted by mGlu2R homomers coupled to Gi, 5-HT2AR homomers coupled to Gq, PLC and Kir3, can be considered as a prototype of GEMMA that integrates canonical antagonistic Gi-Gq-Kir3 interactions.
Following the definition of GEMMA, the same study (Navarro et al., 2018) also provides, to our knowledge, the first non-equivocal demonstration of a ligand-dependent rearrangement of a complex including a GPCR or a GPCR oligomer and PM-effector. Using TM-peptide-BiFC experiments it could be demonstrated that specific TM domains of the A2AR and the D2R directly interact with the TM domains of AC5, and that those interfaces changed upon the binding of agonists (Navarro et al., 2018). It could also be shown that disruption of the GPCRs-AC5 interfaces also disrupted the Gs-Gi-AC type III allosteric interaction within the A2AR-D2R-AC5 GEMMA (Navarro et al., 2018). The quaternary structure of the A2AR-D2R-AC5 GEMMA obtained by computer modeling suggested that two molecules of AC5 would be needed to interact simultaneously with A2AR and D2R protomers in a single heterotetramer, while two heterotetramers are needed to allow the simultaneous interaction of A2AR and D2R with a single AC5 molecule, to allow full Gs-Gi-AC interactions (Fig. 6A). According to this model, the A2AR-D2R heterotetramer is the more rigid part of the complex and the model also predicts the possible extension of this basic unit into a higher-order GEMMA (Navarro et al., 2018) (Fig. 6A).
The mGlu2R-5-HT2AR heteromer is another example of a well-studied GPCR heteromer with evidence of both type I and II allosteric mechanisms. It is localized in cortical glutamatergic pyramidal neurons and represents an important target for the hallucinogenic effects of 5-HT2AR agonists and the antipsychotic effect of both 5-HT2AR antagonists/inverse agonists and mGlu2R agonists (González-Maeso et al., 2008; Fribourg et al., 2011; Moreno et al., 2012, 2016; Baki et al., 2016). Its quaternary structure also seems to be tetrameric, constituted of mGlu2R and 5-HT2AR homodimers respectively coupled to Gi and Gq, with a clear involvement of TM4 in the heteromeric interface (Moreno et al., 2016; Shah et al., 2020). In transfected mammalian cells, Kir3 channels were identified as a common PM-effector for both Gi and Gq protein-mediated signaling of the mGlu2R-5-HT2AR heteromer (Baki et al., 2016). As mentioned above, Kir3 channels are activated by direct interactions with Gβγ subunits following activation of Gi-coupled receptors (Logothetis et al., 1987; Riven et al., 2006; Logothetis et al., 2015; Dascal and Kahanovitch, 2015). However, stimulation of Kir3 channels is critically dependent on phosphatidylinositol 4,5-bisphosphate (PIP2) (Huang et al., 1998; Sui et al., 1998). A canonical effect of Gq activation is PLC activation, which leads to PIP2 hydrolysis and opposes Gβγ-mediated activation (Logothetis et al., 2015). When added to evidence for pre-assembly of Gαβγ with Kir3 (see above) and with PLC (Yuan et al., 2007; Smrcka, and Fisher, 2019), we suggest the existence of a mGlu2R-5-HT2AR-PLC-Kir3 GEMMA (Fig. 6B). This GEMMA would represent an allosteric machine that controls neuronal excitability by integrating antagonistic effects of serotonin and glutamate on Kir3 activity. As with the A2AR-D2R-AC5 GEMMA, it might be predicted that the mGlu2R-5-HT2AR-PLC-Kir3 GEMMA could be the basic unit of a higher-order GEMMA (Fig. 6B).
4.4. Oligomerization of GPCRs with orphan GPCRs, truncated GPCRs and other TM proteins
From the 800 GPCR genes identified, more than 100 GPCRs remain orphans, including 86 family A GPCRs (Davenport et al., 2013; Ngo et al., 2016). There is evidence indicating that some GPCRs will not be deorphanized because they may not have a natural ligand. Rather, they can oligomerize with other GPCRs and change their functional and pharmacological properties via allosteric interactions. This concept was originally suggested from studies with melatonin receptors and the orphan receptor GPR50 (Levoye et al., 2006a). The melatonin receptor subfamily consists of MT1R and MT2R, with melatonin as their natural ligand, and the orphan receptor GPR50, which does not bind to melatonin or any other known endogenous ligand to date (Cecon et al., 2018). MT1R and MT2R signal preferentially through Gi/o protein coupling, while GPR50 does not couple to G proteins and has a characteristic very long CT (~300 amino acids) (Oishi et al., 2018). Several studies support the ability of the melatonin receptor family to oligomerize, including heteromerization of GPR50 with MT1R and MT2R (Levoye et al., 2006b; Oishi et al., 2018) (Fig. 7A). Experiments using BRET in transfected mammalian cells suggested that MT1R and MT2R form constitutive homomers as well as MT1R-MT2R heteromers (Ayoub et al., 2002). In transfected cells the propensity for homomer and heteromer formation differs between both receptor subtypes, with a lower propensity of MT2R to form homomers as compared with the MT1R and a higher propensity of forming MT1R-MT2R heteromers, suggesting that the MT2R preferentially exists as a heteromeric complex with MT1R (Ayoub et al., 2004). In fact, by inducing expression of tagged MT1R and MT2R in genetically modified mice, it was possible to demonstrate the existence of MT1R-MT2R complexes in the retina, where they mediate the effect of melatonin on light sensitivity of rod photoreceptors (Baba et al., 2013). Significantly, in transfected mammalian cells, MT1R-MT2R heteromers seem to preferentially couple to Gq and promote Gq-PLC-PKC signaling, a primary signaling pathway involved in the functional effects of melatonin in rod photoreceptors (Baba et al., 2013) (Fig. 7A). GPR50 promotes a decreased ability of melatonin to bind and signal though the MT1R, which depends on the ability of the long GPR50 CT to alter the pre-coupling or rearrangement of G proteins to MT1R in the MT1R-GPR50 heteromer (Levoye et al., 2006b; Oishi, et al., 2018) (Fig. 7A).
Fig, 7. Oligomerization of GPCRs with orphan GPCRs, truncated GPCRs and RAMPs.
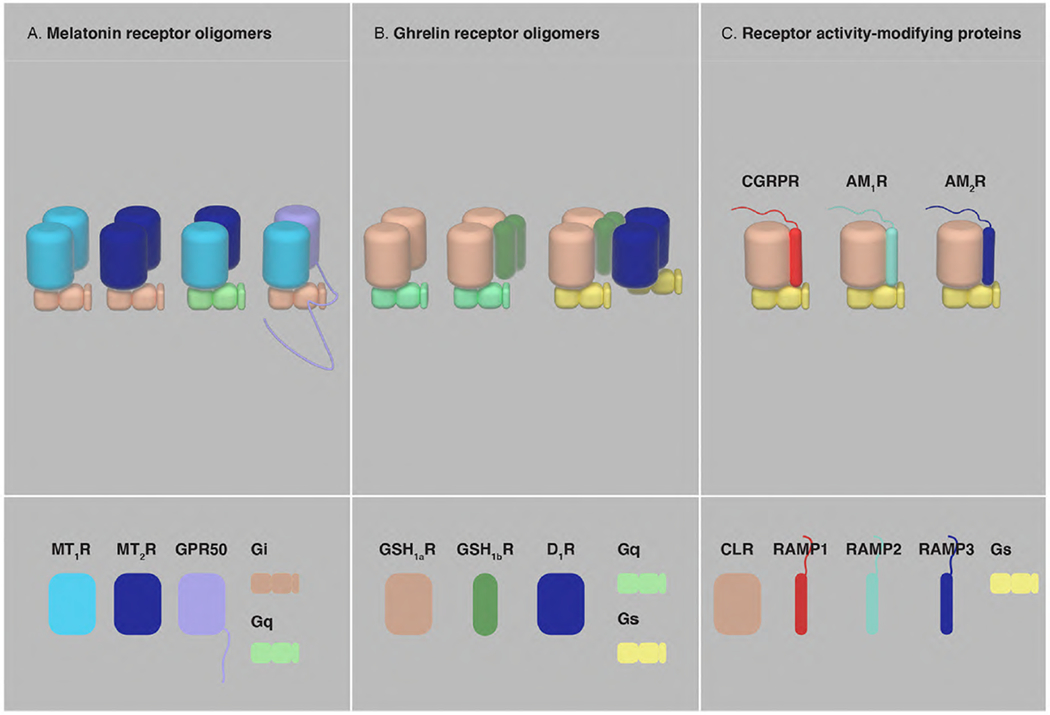
(A) Homomers and heteromers of MT1R, MT2R and GPR50. MT2R preferentially exists as a heteromeric complex with MT1R and MT1R-MT2R heteromerization drives a change in the preference of G protein subtype coupling, from Gi to Gq. The long CT of GPR50 significantly alters MT1R-Gi protein signaling in the MT1R-GPR50 heteromer (see text). (B) GHS1aR forms homomers and oligomerizes with its non-functional truncated isoform GHS1bR to form homomers and heteromers. One of the properties of the GHS1aR-GHS1bR oligomers is the facilitation of an additional interaction with the D1R, leading to a change in the preference of G protein subtype coupling of GHS1aR, from Gq to Gs. (C) The single TM domain proteins RAMP1, RAMP2 and RAMP3 associate with CLR and form the Gs protein-coupled receptors CGRP, AM1R and AM2R, respectively, determining the ligands potentially binding to CLR.
Some GPCRs exist in different splice variants, which can be expressed by the same cell and, therefore potentially oligomerize leading to distinct functional outcomes. Some of these variants represent truncated versions of receptors, missing several TM domains, often resulting in their inability to bind to endogenous ligands per se (Wise, 2012). Truncated GPCR isoforms may be expressed at the plasma membrane and constitute components of GEMMAs. Those include D3nf and GHS1b, truncated splice variants of dopamine D3 receptor (D3R) and ghrelin GHS1a receptor GHS1aR, respectively. Both isoforms lack TM6 and TM7 domains, do not bind ligands or promote signaling themselves, but oligomerize with their respective functional isoforms (Elmhurst et al., 2000; Karpa et al., 2000; Leung et al., 2007; Chow et al., 2012; Mary et al., 2013; Navarro et al., 2016). Although under some experimental conditions both isoforms seem able to promote intracellular localization of their functional GPCR partners, there is evidence for the ability of D3R-D3nf and GHS1aR-GHS1b oligomers to localize to the plasma membrane, where the truncated partner exerts ligand-independent negative allosteric modulation (type II allosterism) of D3R and GHS1aR ligands (Elmhurst et al., 2000; Mary et al., 2013; Navarro et al., 2016). In addition, BRET and signaling experiments in transfected mammalian cells indicate that GHS1aR-GHS1bR can be part of a GEMMA that includes D1R, in which ghrelin, instead of promoting its preferred GHS1aR-dependent Gq-PLC signaling, activates a Gs-AC signaling, which is opposed by D1R antagonists (Navarro et al., 2016b) (Fig. 7B).
Receptor activity-modifying proteins (RAMPs) are an example of membrane-spanning accessory proteins that can alter the function of GPCRs. A small family of three proteins (RAMP1, RAMP2 and RAMP3) has substantial capacity for introducing functional diversity by directly interacting with GPCRs (Hay and Pioszak, 2016; Serafin et al., 2020). Structurally, RAMPs comprise a single TM domain with a long extracellular NT (90–100 amino acids) and a short intracellular CT (9 amino acids). The interactions of RAMPs with the family B GPCR calcitonin-like receptor (CLR) and the calcitonin receptor (CTR) are the most extensively studied and provide a picture of the broad influence that a single TM-domain protein can have when interacting with a GPCR (Fig. 7C). RAMPs are required to chaperone CLR to the cell surface, where RAMP-CLR complexes act as receptors for the peptide hormones calcitonin gene-related peptide (CGRP), adrenomedullin (AM) or adrenomedullin 2/intermedin (AM2), depending on the subtype of RAMP co-expressed (McLatchie et al., 1998; Serafin et al., 2020). Thus, the RAMP subtype determines which endogenous ligands preferentially bind to CLR and CLR-RAMP1, CLR-RAMP2 and CLR-RAMP3 constitute CGRP, AM and AM2 receptors, respectively (McLatchie et al., 1998; Poyner et al., 2002; Serafin et al., 2020) (Fig. 7C). CTR is the closest relative to CLR and it can reach the cell surface in the absence of RAMP, but CTR can drive RAMP translocation to the cell surface and CTR pharmacology is altered in the presence of RAMP, which increases the affinity of the endocrine hormone amylin (AMY), such that three amylin receptor subtypes (AMY1–3 receptors) result from each respective RAMP-CTR complex (Poyner et al., 2002).
5. Targeting specific GEMMAs
5.1. Specific GPCR ligands for individual GEMMAs
The main properties of GPCR oligomers (allosterism), orphan receptors, truncated GPCRs and other membrane-localized GPCR-associated proteins (such as RAMPs) can potentially provide significant functional and pharmacological properties to the GEMMAs that include them. The remarkable varied influence of TM RAMPs in modifying the properties of ligand binding to CLR and CTR, provides a clear demonstration for exploiting GEMMAs as targets for drug discovery. Namely, that different components of GEMMAs directly interacting with a particular GPCR can potentially modify the effects of specific ligands for this GPCR. Another reason is that localization of the same GPCR with additional components in different cells likely creates a new and unique GEMMA that can be targeted in a way distinct from the same receptor in a different complex. This raises the possibility of obtaining ligands with selectivity for the GPCRs in distinct GEMMAs which could mediate desired therapeutic effects of the putative ligand, while avoiding targeting GEMMAs that mediate the adverse side effects.
Type II allosterism of GPCR heteromers and the changes it brings to pharmacological properties is noted with the example of A2AR heteromers discussed above. A significant clinically translational type II allosterism of GPCR heteromers for substance use disorders has been generated by the study of MOR heteromers. Since the pioneering studies from Lakshmi Devi’s research group, MOR oligomerization has been the focus of a significant amount of experimental work, providing substantial evidence for MOR-δ opioid receptor (DOR) heteromerization and their role in the analgesic effects of opioids (Fujita, Gomes and Devi, 2015). Although the initially proposed localization of MOR-DOR heteromers in the nociceptive sensory neurons of the dorsal root ganglia and dorsal horn of spinal cord was challenged (Scherrer et al., 2009), subsequent studies provided strong confirmatory evidence (Wang et al., 2010; Yekkirala et al., 2012; Tiwari, et al., 2020). More recently, a major population of MOR localized in the mesencephalon, in the ventral tegmental area, has been shown to form heteromers with galanin Gal1 receptors (Gal1R) and to mediate the ability of opioids to activate the dopaminergic system and therefore abuse liability (Moreno et al., 2017; Cai et al., 2019). These studies support the rationale of selectively targeting MOR-DOR heteromers in the search for new effective analgesic drugs devoid of addictive properties.
Different approaches have been shown to be effective pre-clinically, including high-throughput screening of small-molecule libraries in cells expressing MOR-DOR heteromers, which led to the discovery of CYM51010, which showed a significant increase in its potency and efficacy to promote G protein activation mediated by the MOR in heteromers with DOR (Gomes et al., 2013). CYM51010 exhibited a potent antinociceptive activity with reduced tolerance potential compared to morphine in mice (Gomes et al., 2013) and, more recently, CYM51010 was also found effective in a rat model of neuropathic pain (Tiwari et al., 2020). Because of structural similarities of CYM51010 with carfentanyl, structure-activity relationship (SAR) analysis was used to develop several carfentanyl derivatives (Faouzi et al., 2020). One of the derivatives, MP135, showed even higher relative efficacy at MOR-DOR heteromer versus MOR alone, as compared with CYM51010 (Faouzi et al., 2020). As expected, in rodents, it produced pronounced analgesic effects, but still showed rewarding properties and was readily self-administered (Faouzi et al., 2020). Although not tested yet, MP135 might also exhibit significant potency and efficacy at the MOR-Gal1R heteromer, which determines the dopaminergic/euphoric effects of opioids (Cai et al., 2019).
The effect of morphine, methadone and fentanyl, representatives of the three principal opioid structures, has been separately analyzed for both MOR-DOR and MOR-Gal1R heteromers (Yekkirala et al., 2010, 2012; Cai et al., 2019). The research group of Philip Portoghese found evidence for a significantly higher efficacy of the three opioids for MOR-DOR versus MOR alone (Yekkirala et al., 2010, 2012). In a more recent study, for the first time, differential pharmacodynamic effects were observed for methadone versus morphine or fentanyl, a significant decrease in potency which depended on heteromerization of MOR with Gal1R (Cai et al., 2019). As predicted from the predominant role of MOR-Gal1R heteromers in the modulation of the dopaminergic system, methadone was a much weaker activator of the dopaminergic system as compared with morphine and fentanyl and, it could be demonstrated clinically that it was also much weaker at producing euphoric effects (Cai et al., 2019). Elucidation of the molecular mechanisms of this type II allosterism in the MOR-Gal1R heteromer should facilitate discovery of more methadone-like compounds, with strong analgesic and reduced addictive properties. An additional challenge is to determine specific components of the MOR-containing GEMMAs responsible for the other major unwanted effects of opioids, such as respiratory depression and constipation.
5.2. Simultaneous targeting of different components of specific GEMMAs
Another approach to obtain opioid analgesia with low abuse liability would be using effective MOR-DOR ligands while selectively blocking the unwanted MOR signaling via the MOR-Gal1R heteromer. This could be achieved by administering Gal1R ligands, based on recently described type I allosterism in the MOR-Gal1R heteromer, by which those ligands significantly reduce the affinity and efficacy of MOR agonists (Cai et al., 2019). Gal1R are also localized in the spinal cord, where previous studies indicate their activation produces analgesic effects synergistic to those of opioids (Hua et al., 2004). Since the only available selective and potent Gal1R ligands are galanin-derived peptides (Freimann et al., 2015), the discovery of small molecules with significant brain penetrability targeting Gal1R could provide an important new approach for the treatment of pain. Gal1R agonists could be co-administered with lower therapeutic doses of MOR agonists, promoting analgesia and significantly reducing side effects, particularly the dopaminergic effects mediated by MOR-Gal1R heteromers.
Type I allosterism in GPCR heteromers could then be used as a therapeutic strategy to target specific GEMMAs, either by specifically increasing the therapeutic effects or decreasing the unwanted effects of GPCR ligands. This is illustrated by the increase in the therapeutic index of L-DOPA by A2AR antagonists in Parkinson’s Disease (see above), which decreases the effective dose of L-DOPA required and, therefore, both short- and long-term side effects (Chen and Cunha, 2020).
A further step in drug discovery would be to obtain bivalent compounds that can simultaneously bind both protomers in the GPCR heteromer, defined as single chemical entities composed of two pharmacophore units covalently linked by an appropriate spacer. These ligands are designed to interact simultaneously within a GPCR homo- or heterooligomer, to enhance affinity and subtype selectivity and, in case of a GPCR heteromer, promote type I allosteric modulation (Daniels et al., 2005; Soriano et al., 2009; Pulido et al., 2018). Although it could be argued that the pharmacologic effects of bivalent ligands are related to the separate binding of each pharmacophore to two different GPCR oligomers, true simultaneous bivalent binding to the same GPCR oligomer can be demonstrated by dependence on the integrity of the GPCR oligomer. For instance, in a recent study, the dramatic increase of affinity of a D2R ligand (from nanomolar to picomolar affinity) obtained when two molecules of the same ligand were attached using an appropriate spacer length was reversed upon disruption of the D2R homomeric interface (see above) (Pulido et al., 2018).
Portoghese’s group pioneered studies with bivalent ligands and opioid receptor heteromers. Their studies revealed additional implications with potential therapeutic importance. Thus, MOR-DOR bivalent ligands with specific length spacers and a MOR agonist and a DOR antagonist as pharmacophores produced analgesia without the development of tolerance (Daniels et al., 2005). This effect was attributed to the bivalent ligand-induced stabilization (“bridging”) of the MOR-DOR heteromer with concomitant blockade of DOR signaling (Daniels et al., 2005), which facilitates internalization of the MOR-DOR heteromer and, therefore, subsequent development of tolerance to MOR agonist-mediate analgesia (Gomes et al., 2013).
Finally, apart from simultaneously targeting two different GPCRs, there are also possibilities of simultaneously targeting a GPCR protomer and another component of a specific GEMMA. This is best exemplified by the recent discovery of the specific activator of Kir3.1-Kir3.2 channels, GAT1508 (Xu et al., 2020). Kir3 channels are widely expressed in the brain and in the heart, although the localization of the Kir3.1-Kir3.4 channel is predominantly in the heart, whereas Kir3.1-Kir3.2 channels are more restricted to the central nervous system (CNS) (Dascal and Kahanovitch, 2015). GAT1508 represents the first specific activator of Kir3.1-Kir3.2 channels, making them CNS-selective. The mechanism of GAT1508-induced channel activation is related to strengthening channel-PIP2 interactions (Xu et al., 2020). Importantly, at lower concentrations that are insufficient to produce significant activation of Kir3.1-Kir3.2 channels, GAT1508 significantly potentiated the ligand-induced activating effect of an associated GPCR (such as baclofen-induced activation of the GABAB receptor), suggesting synergism in the allosteric effects of GAT1508 and Gβγ subunits in fostering channel-PIP2 interactions (Xu et al., 2020). This provides further selectivity of the effects of GAT1508 in the brain and evidence was provided by a specific effect of the baclofen-GAT1508 in the basolateral amygdala, with possible translational implications for posttraumatic stress disorder (Xu et al., 2020).
5.3. G protein subtype-dependent functional selectivity
The properties of a GPCR ligand can also depend on specific pre-assembled G protein subtypes, as recently shown in BRET-based experiments. This also extends the notion of functional selectivity, where a selective ligand regulates a subset G protein-dependent or independent signaling (Violin and Lefkowitz, 2007), to biased G protein subtype-dependent signaling. For instance, the effect of several D1R ligands were compared in their ability to modify BRET in cells transfected with D1R and Gαs or Gαolf fused to BRET chromophores. The D1R agonist dihydrexidine (DHX) was found to be a full D1R agonist with Gαs but a weak partial agonist with Gαolf (Yano et al., 2018). It was then expected that DHX would be more effective in the cerebral cortex, where Gαs is the predominant isoform, than in the striatum, where Gαolf predominates. In fact, with electrophysiological and behavioral experiments, it was demonstrated that DHX had greater efficacy in cortical versus striatal neurons. It was then suggested that DHX or other functionally Gαs subtype-functionally selective D1R agonists could be used as pro-cognitive drugs with reduced extrapyramidal side effects (Yano et al., 2018).
The same phenomenon had been previously observed for other GPCRs and G protein subtypes, although it was not conceptualized as functional selectivity. For instance, the selective CB1R agonist WIN55,212-2 facilitated Gq-mediated signaling, while other classes of CB1R agonists (including Δ9-tetrahydrocannabinol and the endocannabinoids 2-arachidonoylglycerol and methanandamide) preferentially signaled through Gi/o proteins (Lauckner et al., 2005). Another example is the selective ability of the A1R agonist 5’-N-cyclopentyl-carboxyamidoadenosine, as compared with a series of adenosine analogues, to facilitate Gs-mediated signaling, although A1R preferentially signals through Gi/o proteins (Cordeaux et al., 2004). In those studies, the results were interpreted as dependent on a ligand-induced stabilization of a GPCR conformation that favored preferential coupling with a specific G protein subtype. This interpretation would be in line with collision-coupling. The GEMMAs concept provides another interpretation, the ability of ligands to differentially activate GPCRs pre-assembled with specific G protein subtypes.
6. Concluding remarks and future directions
The idea that GPCRs can serve as signaling platforms for assembly of macromolecular complexes of G proteins and other related signaling proteins to provide highly efficient and spatially restricted signaling events, with no requirement for G protein subunit dissociation and lateral diffusion within the membrane, has been recurrently considered over the years (Neubig, 1994; Levitzki and Klein, 2002; Rebois and Hébert, 2003; Dupré et al., 2006, 2009; Hepler, 2014; Navarro et al., 2018; Sleno and Hébert, 2019). The GEMMA concept restricts the components of these macromolecular assemblies to GPCRs, G proteins, PM-effectors and other associated transmembrane proteins putatively pre-assembled prior to receptor activation by agonists, although we cannot ignore the idea that these core components of transmembrane signaling complexes will interact with additional cytosolic elements that will be recruited by the assembly upon GPCR activation, including GRKs and arrestins.
The GEMMA concept represents a divergent mechanism from collision coupling and both signaling modes are likely used by the cell, depending on the GPCRs and effectors involved and their need for providing more efficient and restricted signaling (GEMMA) or less efficient but more amplified signaling (collision-coupling mode). There is also evidence suggesting that these two poles are part of a spectrum of signaling mechanisms. Mixed models have been proposed where some of the components of the macromolecular membrane complex would still dissociate (Levitzki, 1986; Neubig, 1994). Such models can still explain the catalytic behavior of GPCRs observed in some cell systems, such as the amplification of signaling seen when one receptor activates numerous AC molecules, a phenomenon that, in fact, led to the formulation of the collision-coupling model (Tolkovsky and Levitzki, 1978a). On the other hand, amplification of signaling may not be expected with a ligand-induced rearrangement without dissociation of the GEMMA components. Nevertheless, as discussed, agonist-induced rearrangement of the G protein subunits of GEMMAs including the PM-effectors Kir3 or AC might necessarily imply limited dissociation between G protein subunits within the framework of the macromolecular assembly.
Many questions about GEMMAs remain to be answered, such as whether they can form higher-order macromolecular structures, as here suggested for the A2AR-D2R-AC5 GEMMA and the mGlu2R-5-HT2AR-PLC-Kir3 GEMMA. In that case, it will be important to resolve which basic units are pre-assembled in the ER and what determines and modulates the assembly and disassembly of the final macromolecular complex. It will also be important to establish the fate of GEMMAs and whether they can internalize as intact signaling complexes. In fact, internalization of GEMMAs would provide a very plausible framework for the now well established endosomal signaling mediated by internalized GPCRs (Ferrandon et al., 2009; Calebiro et al., 2010; von Zastrow and Sorkin, 2021), although current evidence suggests that β2AR and AC may traffic independently and potentially reform in endosomes (Lazar et al., 2020). Furthermore, it will be important to determine what is the role of arrestins and other cytosolic proteins that interact with GPCRs, G proteins and PM-effectors in the modulation of internalization and both G protein-dependent and independent signaling of GEMMAs.
Apart from their relevance as functional signaling units, the GEMMA concept will be important for further exploration of GPCR pharmacology. The localization in specific cellular environments, together with the unique properties of each GEMMA, determined by the large variety of potential components, should promote new directions in the search for new therapeutic agents. Screens should focus on molecules with the ability to promote changes in the function of a specific GEMMA, using single agents or combination of molecules with selectivity for the different components in particular GEMMAs.
Our primary recommendation is the further discovery and characterization of GEMMAs in their native environments with the identification of their primary components. This can only be achieved by combining the same approaches used for identification of GPCR oligomers with proteomic, biochemical or antibody-based techniques, such as proximity ligation or AlphaLISA assays (see, for instance, Moreno-Delgado et al., 2020; Valle-León et al., 2021). Such studies should be performed in parallel with reconstitution of the putative GEMMA in an artificial system, which should allow determination of interaction sites and interfaces between its putative components, using, for instance, the TM-peptide strategy. This should allow the establishment of unique pharmacological properties of the GEMMA that could be used for its identification in native tissue and potentially targeted for drug discovery.
We should also aim at elucidating the precise molecular structure of GEMMAs in the presence and absence of ligands that stabilize active and inactive states, which should already be possible with cryo-EM techniques. Dynamic transitions between active and inactive states could be pursued by molecular dynamics simulations. The elucidation of the molecular structures of GEMMAs and their dynamic interrelationships should provide a deeper understanding of the intricate mechanisms of membrane-delimited GPCR-mediated cell signaling and an evaluation of their potential as therapeutic targets.
Acknowledgements
This article was written as the result of a roundtable meeting of the coauthors at the University of Barcelona in February 2020, organized by Esteve Foundation and the Institute of Neurosciences of the University of Barcelona. S.F. is supported with the intramural funds of the National Institute on Drug Abuse. C.W.D. is supported by the National Institute of General Medicine (GM60419). J.G.-M. is supported by the National Institute of Mental Health (R01MH084894 and R01MH111940). D.E.L. is supported by the National Institute of Heart, Lung and Blood Institute (HL09549-24). T.E.H. holds the Canadian Pacific Chair in Biotechnology. F.C. and L.P. are supported by the Spanish Ministry of Economy and Innovation with FEDER funds (SAF2017-87349-R and PID2019-109240RB-I00). R.J. is supported by ”Agence Nationale de la Recherche” (ANR-19-CE16-0025-01), ”Fondation de la Recherche Médicale” (FRM DEQ20130326503), ”Institut National de la Santé et de la Recherche Médicale and ”Centre National de la Recherche Scientifique”.
Abbreviations:
GPCR nomenclature used in the current review follows the guidelines of the International Union of Basic and Clinical Pharmacology Committee on Receptor Nomenclature and Drug Classification (https://www.guidetopharmacology.org/nomenclature.jsp) and adds an ‘R’ at the end as an abbreviation of ‘receptor’
- AC
adenylate cyclase
- AKAP
A-kinase anchoring protein
- AMPAR
ionotropic glutamate AMPA receptors
- BiBC or BiFC
bimolecular bioluminescence or fluorescence complementation
- BRET or FRET
bioluminescence or fluorescence resonance energy transfer
- CT or NT
carboxy terminus or amino terminus
- ER
endoplasmic reticulum
- FRAP
fluorescence recovery after photobleaching
- GEMMA
GPCR-effector macromolecular membrane assembly
- GPCR
G protein-coupled receptor
- Kir
inwardly rectifying potassium channel
- PDZ
PSD-95/Discs large/Zona occludens-1
- PKA
protein kinase A
- PM-effector
plasma membrane effector
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PLC
phospholipase C
- RAMP
Receptor activity-modifying protein
- Rluc
Renilla luciferase
- TM
transmembrane
- YFP
yellow fluorescence protein
Footnotes
Conflict of interest statement
J.G.-M. has a sponsored research contract with Neuristic Pharma. The remaining authors declare that there are no conflicts of interest.
References
- Ahmed SN, Brown DA, & London E (1997). On the origin of sphingolipid/cholesterol-rich detergent-insoluble cell membranes: physiological concentrations of cholesterol and sphingolipid induce formation of a detergent-insoluble, liquid-ordered lipid phase in model membranes. Biochemistry, 36(36), 10944–10953. [DOI] [PubMed] [Google Scholar]
- Andressen KW, Ulsund AH, Krobert KA, Lohse MJ, Bünemann M, & Levy FO (2018). Related GPCRs couple differently to Gs: preassociation between G protein and 5-HT7 serotonin receptor reveals movement of Gαs upon receptor activation. FASEB Journal, 32(2), 1059–1069. [DOI] [PubMed] [Google Scholar]
- Ayoub MA, Couturier C, Lucas-Meunier E, Angers S, Fossier P, Bouvier M, et al. (2002). Monitoring of ligand-independent dimerization and ligand-induced conformational changes of melatonin receptors in living cells by bioluminescence resonance energy transfer. The Journal of Biological Chemistry, 277(24), 21522–21528. [DOI] [PubMed] [Google Scholar]
- Ayoub MA, Levoye A, Delagrange P, & Jockers R (2004). Preferential formation of MT1/MT2 melatonin receptor heterodimers with distinct ligand interaction properties compared with MT2 homodimers. Molecular Pharmacology, 66(2), 312–321. [DOI] [PubMed] [Google Scholar]
- Ayoub MA, Trinquet E, Pfleger KD, & Pin JP (2010). Differential association modes of the thrombin receptor PAR1 with Galphai1, Galpha12, and beta-arrestin 1. FASEB Journal, 24(9), 3522–3535. [DOI] [PubMed] [Google Scholar]
- Baba K, Benleulmi-Chaachoua A, Journé AS, Kamal M, Guillaume JL, Dussaud S, et al. (2013). Heteromeric MT1/MT2 melatonin receptors modulate photoreceptor function. Science Signaling, 6(296), ra89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baki L, Fribourg M, Younkin J, Eltit JM, Moreno JL, Park G, et al. (2016). Cross-signaling in metabotropic glutamate 2 and serotonin 2A receptor heteromers in mammalian cells. Pflugers Archiv, 468(5), 775–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin TA, Li Y, Brand CS, Watts VJ, & Dessauer CW (2019). Insights into the Regulatory Properties of Human Adenylyl Cyclase Type 9. Molecular Pharmacology, 95(4), 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banères JL, & Parello J (2003). Structure-based analysis of GPCR function: evidence for a novel pentameric assembly between the dimeric leukotriene B4 receptor BLT1 and the G-protein. Journal of Molecular Biology, 329(4), 815–829. [DOI] [PubMed] [Google Scholar]
- Bar-Sinai A, Marbach I, Shorr RG, & Levitzki A (1992). The GppNHp-activated adenylyl cyclase complex from turkey erythrocyte membranes can be isolated with its beta gamma subunits. European Journal of Biochemistry, 207(2), 703–708. [DOI] [PubMed] [Google Scholar]
- Bauman AL, Soughayer J, Nguyen BT, Willoughby D, Carnegie GK, Wong W, et al. (2006). Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Molecular Cell, 23(6), 925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bécamel C, Gavarini S, Chanrion B, Alonso G, Galéotti N, Dumuis A, et al. (2004). The serotonin 5-HT2A and 5-HT2C receptors interact with specific sets of PDZ proteins. The Journal of Biological Chemistry, 279(19), 20257–20266. [DOI] [PubMed] [Google Scholar]
- Beedle AM, McRory JE, Poirot O, Doering CJ, Altier C, Barrere C et al. (2004). Agonist-independent modulation of N-type calcium channels by ORL1 receptors. Nature Neuroscience, 7(2), 118–125. [DOI] [PubMed] [Google Scholar]
- Bonaventura J, Navarro G, Casadó-Anguera V, Azdad K, Rea W, Moreno E, et al. (2015). Allosteric interactions between agonists and antagonists within the adenosine A2A receptor-dopamine D2 receptor heterotetramer. Proceedings of the National Academy of Sciences of the United States of America, 112(27), E3609–E3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borroto-Escuela DO, Romero-Fernandez W, Tarakanov AO, Gómez-Soler M, Corrales F, Marcellino, et al. (2010a). Characterization of the A2AR-D2R interface: focus on the role of the C-terminal tail and the transmembrane helices. Biochemical and Biophysical Research Communications, 402(4), 801–807. [DOI] [PubMed] [Google Scholar]
- Borroto-Escuela DO, Marcellino D, Narvaez M, Flajolet M, Heintz N, Agnati, et al. (2010b). A serine point mutation in the adenosine A2AR C-terminal tail reduces receptor heteromerization and allosteric modulation of the dopamine D2R. Biochemical and Biophysical Research Communications, 394(1), 222–227. [DOI] [PubMed] [Google Scholar]
- Bourque K, Jones-Tabah J, Devost D, Clarke P, & Hébert TE (2020). Exploring functional consequences of GPCR oligomerization requires a different lens. Progress in Molecular Biology and Translational Science, 169, 181–211. [DOI] [PubMed] [Google Scholar]
- Bouvier Μ (2001). Oligomerization of G-protein-coupled transmitter receptors. Nature reviews. Neuroscience, 2(4), 274–286. [DOI] [PubMed] [Google Scholar]
- Brand CS, Sadana R, Malik S, Smrcka AV, & Dessauer CW (2015). Adenylyl Cyclase 5 Regulation by Gβγ Involves Isoform-Specific Use of Multiple Interaction Sites. Molecular Pharmacology, 88(4), 758–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun S, & Levitzki A (1979). Adenosine receptor permanently coupled to turkey erythrocyte adenylate cyclase. Biochemistry, 18(10), 2134–2138. [DOI] [PubMed] [Google Scholar]
- Cabrera-Vera TM, Thomas TO, Vanhauwe J, Depree KM, Graber SG, & Hamm HE (2002). Dissecting receptor-G protein specificity using G alpha chimeras. Methods in Enzymology, 344, 69–81. [DOI] [PubMed] [Google Scholar]
- Cai NS, Quiroz C, Bonaventura J, Bonifazi A, Cole TO, Purks J, et al. (2019). Opioid-galanin receptor heteromers mediate the dopaminergic effects of opioids. The Journal of Clinical Investigation, 129(7), 2730–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calebiro D, Nikolaev VO, Persani L, & Lohse MJ (2010). Signaling by internalized G-protein-coupled receptors. Trends in Pharmacological Sciences, 31(5), 221–228. [DOI] [PubMed] [Google Scholar]
- Calebiro D, Rieken F, Wagner J, Sungkaworn T, Zabel U, Borzi A, et al. (2013). Single-molecule analysis of fluorescently labeled G-protein-coupled receptors reveals complexes with distinct dynamics and organization. Proceedings of the National Academy of Sciences of the United States of America, 110(2), 743–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecon E, Oishi A, & Jockers R (2018). Melatonin receptors: molecular pharmacology and signalling in the context of system bias. British Journal of Pharmacology, 175(16), 3263–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, & Cunha RA (2020). The belated US FDA approval of the adenosine A2A receptor antagonist istradefylline for treatment of Parkinson’s disease. Purinergic Signalling, 16(2), 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow KB, Sun J, Chu KM, Tai Cheung W, Cheng CH, et al. (2012). The truncated ghrelin receptor polypeptide (GHS-R1b) is localized in the endoplasmic reticulum where it forms heterodimers with ghrelin receptors (GHS-R1a) to attenuate their cell surface expression. Molecular and Cellular Endocrinology, 348(1), 247–254. [DOI] [PubMed] [Google Scholar]
- Chung KY, Rasmussen SG, Liu T, Li S, DeVree BT, Chae PS, et al. (2011). Conformational changes in the G protein Gs induced by the β2 adrenergic receptor. Nature, 477(7366), 611–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, & Scott JD (2000). Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron, 27(1), 107–119. [DOI] [PubMed] [Google Scholar]
- Cordeaux Y, Ijzerman AP, & Hill SJ (2004). Coupling of the human A1 adenosine receptor to different heterotrimeric G proteins: evidence for agonist-specific G protein activation. British Journal of Pharmacology, 143(6), 705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottet M, Faklaris O, Maurel D, Scholler P, Doumazane E, Trinquet E, et al. (2012). BRET and Time-resolved FRET strategy to study GPCR oligomerization: from cell lines toward native tissues. Frontiers in Endocrinology, 3, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curthoys NM, Parent M, Mlodzianoski M, Nelson AJ, Lilieholm J, Butler MB, et al. (2015). Dances with Membranes: Breakthroughs from Super-resolution Imaging. Current Topics in Membranes, 75, 59–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damian M, Mary S, Maingot M, M’Kadmi C, Gagne D, Leyris JP, et al. (2015). Ghrelin receptor conformational dynamics regulate the transition from a preassembled to an active receptor:Gq complex. Proceedings of the National Academy of Sciences of the United States of America, 112(5), 1601–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels DJ, Lenard NR, Etienne CL, Law PY, Roerig SC, & Portoghese PS (2005). Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proceedings of the National Academy of Sciences of the United States of America, 102(52), 19208–19213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dascal N, & Kahanovitch U (2015). The Roles of Gβγ and Gα in Gating and Regulation of GIRK Channels. International Review of Neurobiology, 123, 27–85. [DOI] [PubMed] [Google Scholar]
- Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, et al. (2001). A beta2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science, 293(5527), 98–101. [DOI] [PubMed] [Google Scholar]
- Davenport AP, Alexander SP, Sharman JL, Pawson AJ, Benson HE, Monaghan AE, et al. (2013). International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list: recommendations for new pairings with cognate ligands. Pharmacological Reviews, 65(3), 967–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David M, Richer M, Mamarbachi AM, Villeneuve LR, Dupré DJ, & Hébert TE (2006). Interactions between GABA-B1 receptors and Kir 3 inwardly rectifying potassium channels. Cellular Signalling, 18(12), 2172–2181. [DOI] [PubMed] [Google Scholar]
- Davis TL, Bonacci TM, Sprang SR, & Smrcka AV (2005). Structural and molecular characterization of a preferred protein interaction surface on G protein beta gamma subunits. Biochemistry, 44(31), 10593–10604. [DOI] [PubMed] [Google Scholar]
- Décaillot FM, Rozenfeld R, Gupta A, & Devi LA (2008). Cell surface targeting of mu-delta opioid receptor heterodimers by RTP4. Proceedings of the National Academy of Sciences of the United States of America, 105(41), 16045–16050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessauer CW (2009). Adenylyl cyclase--A-kinase anchoring protein complexes: the next dimension in cAMP signaling. Molecular Pharmacology, 76(5), 935–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessauer CW, Tesmer JJ, Sprang SR, & Gilman AG (1998). Identification of a Gialpha binding site on type V adenylyl cyclase. The Journal of Biological Chemistry, 273(40), 25831–25839. [DOI] [PubMed] [Google Scholar]
- Dessauer CW, Watts VJ, Ostrom RS, Conti M, Dove S, & Seifert R (2017). International Union of Basic and Clinical Pharmacology. CI. Structures and Small Molecule Modulators of Mammalian Adenylyl Cyclases. Pharmacological Reviews, 69(2), 93–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diering GH, Gustina AS, & Huganir RL (2014). PKA-GluA1 coupling via AKAP5 controls AMPA receptor phosphorylation and cell-surface targeting during bidirectional homeostatic plasticity. Neuron, 84(4), 790–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsch S, Klotz KN, Engelhardt S, Lohse MJ, & Bünemann M (2009). Analysis of receptor oligomerization by FRAP microscopy. Nature Methods, 6(3), 225–230. [DOI] [PubMed] [Google Scholar]
- Downes GB, & Gautam N (1999). The G protein subunit gene families. Genomics, 62(3), 544–552. [DOI] [PubMed] [Google Scholar]
- Du Y, Duc NM, Rasmussen S, Hilger D, Kubiak X, Wang L, et al. (2019). Assembly of a GPCR-G Protein Complex. Cell, 177(5), 1232–1242.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Wang D, Fan H, Xu C, Tai L, Lin S, et al. (2021). Structures of human mGlu2 and mGlu7 homo- and heterodimers. Nature, 594(7864), 589–593. [DOI] [PubMed] [Google Scholar]
- Dunn HA, & Ferguson SS (2015). PDZ Protein Regulation of G Protein-Coupled Receptor Trafficking and Signaling Pathways. Molecular Pharmacology, 88(4), 624–639. [DOI] [PubMed] [Google Scholar]
- Dupré DJ, Robitaille M, Ethier N, Villeneuve LR, Mamarbachi AM, & Hébert TE (2006). Seven transmembrane receptor core signaling complexes are assembled prior to plasma membrane trafficking. The Journal of Biological Chemistry, 281(45), 34561–34573. [DOI] [PubMed] [Google Scholar]
- Dupré DJ, Baragli A, Rebois RV, Ethier N, & Hébert TE (2007). Signalling complexes associated with adenylyl cyclase II are assembled during their biosynthesis. Cellular Signalling, 19(3), 481–489. [DOI] [PubMed] [Google Scholar]
- Dupré DJ, Robitaille M, Rebois RV, & Hébert TE (2009). The role of Gbetagamma subunits in the organization, assembly, and function of GPCR signaling complexes. Annual Review of Pharmacology and Toxicology, 49, 31–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efendiev R, Samelson BK, Nguyen BT, Phatarpekar PV, Baameur F, Scott JD, et al. (2010). AKAP79 interacts with multiple adenylyl cyclase (AC) isoforms and scaffolds AC5 and -6 to alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptors. The Journal of Biological Chemistry, 285(19), 14450–14458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khamlichi C, Reverchon-Assadi F, Hervouet-Coste N, Blot L, Reiter E, & Morisset-Lopez S (2019). Bioluminescence Resonance Energy Transfer as a Method to Study Protein-Protein Interactions: Application to G Protein Coupled Receptor Biology. Molecules, 24(3), 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellaithy A, Gonzalez-Maeso J, Logothetis DA, & Levitz J (2020). Structural and Biophysical Mechanisms of Class C G Protein-Coupled Receptor Function. Trends in biochemical sciences, 45(12), 1049–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmhurst JL, Xie Z, O’Dowd BF, & George SR (2000). The splice variant D3nf reduces ligand binding to the D3 dopamine receptor: evidence for heterooligomerization. Brain research. Molecular Brain Research, 80(1), 63–74. [DOI] [PubMed] [Google Scholar]
- Fan G, Shumay E, Wang H, & Malbon CC (2001). The scaffold protein gravin (cAMP-dependent protein kinase-anchoring protein 250) binds the beta 2-adrenergic receptor via the receptor cytoplasmic Arg-329 to Leu-413 domain and provides a mobile scaffold during desensitization. The Journal of Biological Chemistry, 276(26), 24005–24014. [DOI] [PubMed] [Google Scholar]
- Faouzi A, Uprety R, Gomes I, Massaly N, Keresztes AI, Le Rouzic V, et al. (2020). Synthesis and Pharmacology of a Novel μ-δ Opioid Receptor Heteromer-Selective Agonist Based on the Carfentanyl Template. Journal of Medicinal Chemistry, 63(22), 13618–13637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrandon S, Feinstein TN, Castro M, Wang B, Bouley R, Potts JT, et al. (2009). Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nature Chemical Biology, 5(10), 734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S (2015). The GPCR heterotetramer: challenging classical pharmacology. Trends in Pharmacological Sciences, 36(3), 145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, von Euler G, Johansson B, Fredholm BB, & Fuxe K (1991). Stimulation of high-affinity adenosine A2 receptors decreases the affinity of dopamine D2 receptors in rat striatal membranes. Proceedings of the National Academy of Sciences of the United States of America, 88(16), 7238–7241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Baler R, Bouvier M, Caron MG, Devi LA, Durroux T, et al. (2009). Building a new conceptual framework for receptor heteromers. Nature Chemical Biology, 5(3), 131–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Casadó V, Devi LA, Filizola M, Jockers R, Lohse MJ, et al. (2014). G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacological Reviews, 66(2), 413–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Bonaventura J, Zhu W, Hatcher-Solis C, Taura J, Quiroz C, et al. (2018). Essential Control of the Function of the Striatopallidal Neuron by Pre-coupled Complexes of Adenosine A2A-Dopamine D2 Receptor Heterotetramers and Adenylyl Cyclase. Frontiers in Pharmacology, 9, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Ciruela F, Casadó V, & Pardo L (2020). Oligomerization of G protein-coupled receptors: Still doubted?. Progress in Molecular Biology and Translational Science, 169, 297–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flock T, Hauser AS, Lund N, Gloriam DE, Balaji S, & Babu MM (2017). Selectivity determinants of GPCR-G-protein binding. Nature, 545(7654), 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser ID, Cong M, Kim J, Rollins EN, Daaka Y, Lefkowitz RJ, et al. (2000). Assembly of an A kinase-anchoring protein-beta(2)-adrenergic receptor complex facilitates receptor phosphorylation and signaling. Current Biology : CB, 10(7), 409–412. [DOI] [PubMed] [Google Scholar]
- Freimann K, Kurrikoff K, & Langel Ü (2015). Galanin receptors as a potential target for neurological disease. Expert Opinion on Therapeutic Targets, 19(12), 1665–1676. [DOI] [PubMed] [Google Scholar]
- Fribourg M, Moreno JL, Holloway T, Provasi D, Baki L, Mahajan R, et al. (2011). Decoding the signaling of a GPCR heteromeric complex reveals a unifying mechanism of action of antipsychotic drugs. Cell, 147(5), 1011–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita W, Gomes I, & Devi LA (2015). Heteromers of μ-δ opioid receptors: new pharmacology and novel therapeutic possibilities. British Journal of Pharmacology, 172(2), 375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funke L, Dakoji S, & Bredt DS (2005). Membrane-associated guanylate kinases regulate adhesion and plasticity at cell junctions. Annual Review of Biochemistry, 74, 219–245. [DOI] [PubMed] [Google Scholar]
- Gaitonde SA, & González-Maeso J (2017). Contribution of heteromerization to G protein-coupled receptor function. Current Opinion in Pharmacology, 32, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galés C, Rebois RV, Hogue M, Trieu P, Breit A, Hébert TE, et al. (2005). Real-time monitoring of receptor and G-protein interactions in living cells. Nature Methods, 2(3), 177–184. [DOI] [PubMed] [Google Scholar]
- Galés C, Van Durm JJ, Schaak S, Pontier S, Percherancier Y, Audet M, et al. (2006). Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nature Structural & Molecular Biology, 13(9), 778–786. [DOI] [PubMed] [Google Scholar]
- Gardner LA, Tavalin SJ, Goehring AS, Scott JD, & Bahouth SW (2006). AKAP79-mediated targeting of the cyclic AMP-dependent protein kinase to the beta1-adrenergic receptor promotes recycling and functional resensitization of the receptor. The Journal of Biological Chemistry, 281(44), 33537–33553. [DOI] [PubMed] [Google Scholar]
- Gilman AG (1987). G proteins: transducers of receptor-generated signals. Annual Review of Biochemistry, 56, 615–649. [DOI] [PubMed] [Google Scholar]
- Glaaser IW, & Slesinger PA (2015). Structural Insights into GIRK Channel Function. International Review of Neurobiology, 123, 117–160. [DOI] [PubMed] [Google Scholar]
- Gomes I, Fujita W, Gupta A, Saldanha SA, Negri A, Pinello CE, et al. (2013). Identification of a μ-δ opioid receptor heteromer-biased agonist with antinociceptive activity. Proceedings of the National Academy of Sciences of the United States of America, 110(29), 12072–12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Ayoub MA, Fujita W, Jaeger WC, Pfleger KD, & Devi LA (2016). G Protein-Coupled Receptor Heteromers. Annual Review of Pharmacology and Toxicology, 56, 403–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Maeso J, Ang RL, Yuen T, Chan P, Weisstaub NV, López-Giménez JF, et al. (2008). Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature, 452(7183), 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzin J, Wilden U, Choe HW, Labahn J, Krafft B, & Büldt G (1998). X-ray crystal structure of arrestin from bovine rod outer segments. Nature, 391(6670), 918–921. [DOI] [PubMed] [Google Scholar]
- Guinart D, Moreno E, Galindo L, Cuenca-Royo A, Barrera-Conde M, Pérez EJ, et al. (2020). Altered Signaling in CB1R-5-HT2AR Heteromers in Olfactory Neuroepithelium Cells of Schizophrenia Patients is Modulated by Cannabis Use. Schizophrenia Bulletin, 46(6), 1547–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guitart X, Navarro G, Moreno E, Yano H, Cai NS, Sánchez-Soto M, et al. (2014). Functional selectivity of allosteric interactions within G protein-coupled receptor oligomers: the dopamine D1-D3 receptor heterotetramer. Molecular Pharmacology, 86(4), 417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guitart X, Moreno E, Rea W, Sánchez-Soto M, Cai NS, Quiroz C, et al. (2019). Biased G Protein-Independent Signaling of Dopamine D1-D3 Receptor Heteromers in the Nucleus Accumbens. Molecular Neurobiology, 56(10), 6756–6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudermann T, Kalkbrenner F, & Schultz G (1996). Diversity and selectivity of receptor-G protein interaction. Annual Review of Pharmacology and Toxicology, 36, 429–459. [DOI] [PubMed] [Google Scholar]
- Guo H, An S, Ward R, Yang Y, Liu Y, Guo XX, et al. (2017). Methods used to study the oligomeric structure of G-protein-coupled receptors. Bioscience Reports, 37(2), BSR20160547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, & Gurevich EV (2019). The structural basis of the arrestin binding to GPCRs. Molecular and Cellular Endocrinology, 484, 34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich EV, Tesmer JJ, Mushegian A, & Gurevich VV (2012). G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacology & Therapeutics, 133(1), 40–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DD, Davare MA, Shi M, Allen ML, Weisenhaus M, McKnight GS, et al. (2007). Critical role of cAMP-dependent protein kinase anchoring to the L-type calcium channel Cav1.2 via A-kinase anchor protein 150 in neurons. Biochemistry, 46(6), 1635–1646. [DOI] [PubMed] [Google Scholar]
- Hay DL, & Pioszak AA (2016). Receptor Activity-Modifying Proteins (RAMPs): New Insights and Roles. Annual Review of Pharmacology and Toxicology, 56, 469–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Bellini M, Inuzuka H, Xu J, Xiong Y, Yang X, et al. (2006). Proteomic analysis of beta1-adrenergic receptor interactions with PDZ scaffold proteins. The Journal of Biological Chemistry, 281(5), 2820–2827. [DOI] [PubMed] [Google Scholar]
- Hébert TE, Moffett S, Morello JP, Loisel TP, Bichet DG, Barret C, et al. (1996). A peptide derived from a beta2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. The Journal of Biological Chemistry, 271(27), 16384–16392. [DOI] [PubMed] [Google Scholar]
- Hepler JR (2014). G protein coupled receptor signaling complexes in live cells. Cellular Logistics, 4, e29392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick-Davis K, Weaver BA, Grinde E, & Mazurkiewicz JE (2006). Serotonin 5-HT2C receptor homodimer biogenesis in the endoplasmic reticulum: real-time visualization with confocal fluorescence resonance energy transfer. The Journal of Biological Chemistry, 281(37), 27109–27116. [DOI] [PubMed] [Google Scholar]
- Herrick-Davis K, Grinde E, Cowan A, & Mazurkiewicz JE (2013). Fluorescence correlation spectroscopy analysis of serotonin, adrenergic, muscarinic, and dopamine receptor dimerization: the oligomer number puzzle. Molecular Pharmacology, 84(4), 630–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervé D (2011). Identification of a specific assembly of the g protein golf as a critical and regulated module of dopamine and adenosine-activated cAMP pathways in the striatum. Frontiers in Neuroanatomy, 5, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homan KT, Glukhova A, & Tesmer JJ (2013). Regulation of G protein-coupled receptor kinases by phospholipids. Current Medicinal Chemistry, 20(1), 39–46. [PubMed] [Google Scholar]
- Hu LA, Tang Y, Miller WE, Cong M, Lau AG, Lefkowitz RJ, et al. (2000). beta 1-adrenergic receptor association with PSD-95. Inhibition of receptor internalization and facilitation of beta 1-adrenergic receptor interaction with N-methyl-D-aspartate receptors. The Journal of Biological Chemistry, 275(49), 38659–38666. [DOI] [PubMed] [Google Scholar]
- Hua XY, Hayes CS, Hofer A, Fitzsimmons B, Kilk K, Langel U, et al. (2004). Galanin acts at GalR1 receptors in spinal antinociception: synergy with morphine and AP-5. The Journal of Pharmacology and Experimental Therapeutics, 308(2), 574–582. [DOI] [PubMed] [Google Scholar]
- Huang CL, Slesinger PA, Casey PJ, Jan YN, & Jan LY (1995). Evidence that direct binding of G beta gamma to the GIRK1 G protein-gated inwardly rectifying K+ channel is important for channel activation. Neuron, 15(5), 1133–1143. [DOI] [PubMed] [Google Scholar]
- Huang CL, Feng S, & Hilgemann DW (1998). Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature, 391(6669), 803–806. [DOI] [PubMed] [Google Scholar]
- Huang J, Chen S, Zhang JJ, & Huang XY (2013). Crystal structure of oligomeric β1-adrenergic G protein-coupled receptors in ligand-free basal state. Nature Structural & Molecular Biology, 20(4), 419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Masureel M, Qu Q, Janetzko J, Inoue A, Kato HE, et al. (2020). Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature, 579(7798), 303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joiner ML, Lisé MF, Yuen EY, Kam AY, Zhang M, Hall DD, et al. (2010). Assembly of a beta2-adrenergic receptor--GluR1 signalling complex for localized cAMP signalling. The EMBO Journal, 29(2), 482–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justinová Z, Redhi GH, Goldberg SR, & Ferré S (2014). Differential effects of presynaptic versus postsynaptic adenosine A2A receptor blockade on Δ9-tetrahydrocannabinol (THC) self-administration in squirrel monkeys. The Journal of Neuroscience, 34(19), 6480–6484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkbrenner F, Degtiar VE, Schenker M, Brendel S, Zobel A, Heschler J, et al. (1995). Subunit composition of G(o) proteins functionally coupling galanin receptors to voltage-gated calcium channels. The EMBO Journal, 14(19), 4728–4737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpa KD, Lin R, Kabbani N, & Levenson R (2000). The dopamine D3 receptor interacts with itself and the truncated D3 splice variant d3nf: D3-D3nf interaction causes mislocalization of D3 receptors. Molecular Pharmacology, 58(4), 677–683. [DOI] [PubMed] [Google Scholar]
- Kato HE, Zhang Y, Hu H, Suomivuori CM, Kadji F, Aoki J, et al. (2019). Conformational transitions of a neurotensin receptor 1-Gi1 complex. Nature, 572(7767), 80–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SM, Sleno R, Gora S, Zylbergold P, Laverdure JP, Labbé JC, et al. (2013). The expanding roles of Gβγ subunits in G protein-coupled receptor signaling and drug action. Pharmacological Reviews, 65(2), 545–577. [DOI] [PubMed] [Google Scholar]
- Khan SM, Sung JY, & Hébert TE (2016). Gβγ subunits-Different spaces, different faces. Pharmacological Research, 111, 434–441. [DOI] [PubMed] [Google Scholar]
- Kleuss C, Scherübl H, Hescheler J, Schultz G, & Wittig B (1993). Selectivity in signal transduction determined by gamma subunits of heterotrimeric G proteins. Science, 259(5096), 832–834. [DOI] [PubMed] [Google Scholar]
- Kleuss C, Schultz G, & Wittig B (1994). Microinjection of antisense oligonucleotides to assess G-protein subunit function. Methods in Enzymology, 237, 345–355. [DOI] [PubMed] [Google Scholar]
- Koehl A, Hu H, Feng D, Sun B, Zhang Y, Robertson MJ, et al. (2019). Structural insights into the activation of metabotropic glutamate receptors. Nature, 566(7742), 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kӧfalvi A, Moreno E, Cordomí A, Cai NS, Fernández-Dueñas V, Ferreira SG, et al. (2020). Control of glutamate release by complexes of adenosine and cannabinoid receptors. BMC Biology, 18(1), 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kull B, Ferré S, Arslan G, Svenningsson P, Fuxe K, Owman C, et al. (1999). Reciprocal interactions between adenosine A2A and dopamine D2 receptors in Chinese hamster ovary cells co-transfected with the two receptors. Biochemical Pharmacology, 58(6), 1035–1045. [DOI] [PubMed] [Google Scholar]
- Lauckner JE, Hille B, & Mackie K (2005). The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proceedings of the National Academy of Sciences of the United States of America, 102(52), 19144–19149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavine N, Ethier N, Oak JN, Pei L, Liu F, Trieu P, et al. (2002). G protein-coupled receptors form stable complexes with inwardly rectifying potassium channels and adenylyl cyclase. The Journal of Biological Chemistry, 277(48), 46010–46019. [DOI] [PubMed] [Google Scholar]
- Lazar AM, Irannejad R, Baldwin TA, Sundaram AB, Gutkind JS, Inoue A, et al. (2020). G protein-regulated endocytic trafficking of adenylyl cyclase type 9. eLife, 9, e58039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung PK, Chow KB, Lau PN, Chu KM, Chan CB, Cheng CH, et al. (2007). The truncated ghrelin receptor polypeptide (GHS-R1b) acts as a dominant-negative mutant of the ghrelin receptor. Cellular Signalling, 19(5), 1011–1022. [DOI] [PubMed] [Google Scholar]
- Levitzki A (1986). Beta-adrenergic receptors and their mode of coupling to adenylate cyclase. Physiological Reviews, 66(3), 819–854. [DOI] [PubMed] [Google Scholar]
- Levitzki A (1988). From epinephrine to cyclic AMP. Science (New York, N.Y.), 241(4867), 800–806. [DOI] [PubMed] [Google Scholar]
- Levitzki A, & Klein S (2002). G-protein subunit dissociation is not an integral part of G-protein action. Chembiochem, 3(9), 815–818. [DOI] [PubMed] [Google Scholar]
- Levoye A, Dam J, Ayoub MA, Guillaume JL, & Jockers R (2006a). Do orphan G-protein-coupled receptors have ligand-independent functions? New insights from receptor heterodimers. EMBO Reports, 7(11), 1094–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levoye A, Dam J, Ayoub MA, Guillaume JL, Couturier C, Delagrange P, et al. (2006b). The orphan GPR50 receptor specifically inhibits MT1 melatonin receptor function through heterodimerization. The EMBO Journal, 25(13), 3012–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Benard O, & Margolskee RF (2006). Ggamma13 interacts with PDZ domain-containing proteins. The Journal of Biological Chemistry, 281(16), 11066–11073. [DOI] [PubMed] [Google Scholar]
- Li X, Nooh MM, & Bahouth SW (2013). Role of AKAP79/150 protein in β1-adrenergic receptor trafficking and signaling in mammalian cells. The Journal of Biological Chemistry, 288(47), 33797–33812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Han S, Cai X, Tan Q, Zhou K, Wang D, et al. (2021). Structures of Gi-bound metabotropic glutamate receptors mGlu2 and mGlu4. Nature, 594(7864), 583–588. [DOI] [PubMed] [Google Scholar]
- Liu Z, Fenech C, Cadiou H, Grall S, Tili E, Laugerette F, et al. (2012). Identification of new binding partners of the chemosensory signaling protein Gγ13 expressed in taste and olfactory sensory cells. Frontiers in Cellular Neuroscience, 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Xu X, Hilger D, Aschauer P, Tiemann J, Du Y, et al. (2019). Structural Insights into the Process of GPCR-G Protein Complex Formation. Cell, 177(5), 1243–1251.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logothetis DE, Kurachi Y, Galper J, Neer EJ, & Clapham DE (1987). The beta gamma subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature, 325(6102), 321–326. [DOI] [PubMed] [Google Scholar]
- Logothetis DE, Petrou VI, Zhang M, Mahajan R, Meng XY, Adney SK, et al. (2015). Phosphoinositide control of membrane protein function: a frontier led by studies on ion channels. Annual Review of Physiology, 77, 81–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, et al. (2012). Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature, 485(7398), 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margeta-Mitrovic M, Jan YN, & Jan LY (2000). A trafficking checkpoint controls GABA(B) receptor heterodimerization. Neuron, 27(1), 97–106. [DOI] [PubMed] [Google Scholar]
- Marrari Y, Crouthamel M, Irannejad R, & Wedegaertner PB (2007). Assembly and trafficking of heterotrimeric G proteins. Biochemistry, 46(26), 7665–7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mary S, Fehrentz JA, Damian M, Gaibelet G, Orcel H, Verdié P, et al. (2013). Heterodimerization with Its splice variant blocks the ghrelin receptor 1a in a non-signaling conformation: a study with a purified heterodimer assembled into lipid discs. The Journal of Biological Chemistry, 288(34), 24656–24665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, et al. (1998). RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. Nature, 393(6683), 333–339. [DOI] [PubMed] [Google Scholar]
- Michaelson D, Ahearn I, Bergo M, Young S, & Philips M (2002). Membrane trafficking of heterotrimeric G proteins via the endoplasmic reticulum and Golgi. Molecular Biology of the Cell, 13(9), 3294–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G (2001). Oligomerisation of G-protein-coupled receptors. Journal of Cell Science, 114(Pt 7), 1265–1271. [DOI] [PubMed] [Google Scholar]
- Mirshahi T, Robillard L, Zhang H, Hébert TE, & Logothetis DE (2002). Gbeta residues that do not interact with Galpha underlie agonist-independent activity of K+ channels. The Journal of Biological Chemistry, 277(9), 7348–7355. [DOI] [PubMed] [Google Scholar]
- Mizuno-Yamasaki E, Rivera-Molina F, & Novick P (2012). GTPase networks in membrane traffic. Annual Review of Biochemistry, 81, 637–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JL, Muguruza C, Umali A, Mortillo S, Holloway T, Pilar-Cuéllar F, et al. (2012). Identification of three residues essential for 5-hydroxytryptamine 2A-metabotropic glutamate 2 (5-HT2ȦmGlu2) receptor heteromerization and its psychoactive behavioral function. The Journal of Biological Chemistry, 287(53), 44301–44319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JL, Miranda-Azpiazu P, García-Bea A, Younkin J, Cui M, Kozlenkov A, et al. (2016). Allosteric signaling through an mGlu2 and 5-HT2A heteromeric receptor complex and its potential contribution to schizophrenia. Science Signaling, 9(410), ra5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno E, Quiroz C, Rea W, Cai NS, Mallol J, Cortés A, et al. (2017). Functional μ-Opioid-Galanin Receptor Heteromers in the Ventral Tegmental Area. The Journal of Neuroscience, 37(5), 1176–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Delgado D, Puigdellívol M, Moreno E, Rodríguez-Ruiz M, Botta J, Gasperini P, et al. (2020). Modulation of dopamine D1 receptors via histamine H3 receptors is a novel therapeutic target for Huntington’s disease. eLife, 9, e51093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro G, Aguinaga D, Angelats E, Medrano M, Moreno E, Mallol J, et al. (2016). A Significant Role of the Truncated Ghrelin Receptor GHS-R1b in Ghrelin-induced Signaling in Neurons. The Journal of Biological Chemistry, 291(25), 13048–13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro G, Cordomí A, Casadó-Anguera V, Moreno E, Cai NS, Cortés A, et al. (2018). Evidence for functional pre-coupled complexes of receptor heteromers and adenylyl cyclase. Nature Communications, 9(1), 1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubig RR (1994). Membrane organization in G-protein mechanisms. FASEB Journal, 8(12), 939–946. [DOI] [PubMed] [Google Scholar]
- Ngo T, Kufareva I, Coleman J, Graham RM, Abagyan R, & Smith NJ (2016). Identifying ligands at orphan GPCRs: current status using structure-based approaches. British Journal of Pharmacology, 173(20), 2934–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobles M, Benians A, & Tinker A (2005). Heterotrimeric G proteins precouple with G protein-coupled receptors in living cells. Proceedings of the National Academy of Sciences of the United States of America, 102(51), 18706–18711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi A, Cecon E, & Jockers R (2018). Melatonin Receptor Signaling: Impact of Receptor Oligomerization on Receptor Function. International Review of Cell and Molecular Biology, 338, 59–77. [DOI] [PubMed] [Google Scholar]
- Okashah N, Wan Q, Ghosh S, Sandhu M, Inoue A, Vaidehi N, et al. (2019). Variable G protein determinants of GPCR coupling selectivity. Proceedings of the National Academy of Sciences of the United States of America, 116(24), 12054–12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveria SF, Dell’Acqua ML, & Sather WA (2007). AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron, 55(2), 261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson PA, Tkatch T, Hernandez-Lopez S, Ulrich S, Ilijic E, Mugnaini E, et al. (2005). G-protein-coupled receptor modulation of striatal CaV1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. The Journal of Neuroscience, 25(5), 1050–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrú M, Bakešová J, Brugarolas M, Quiroz C, Beaumont V, Goldberg SR, et al. (2011). Striatal pre- and postsynaptic profile of adenosine A(2A) receptor antagonists. PloS One, 6(1), e16088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrom RS, Violin JD, Coleman S, & Insel PA (2000). Selective enhancement of beta-adrenergic receptor signaling by overexpression of adenylyl cyclase type 6: colocalization of receptor and adenylyl cyclase in caveolae of cardiac myocytes. Molecular Pharmacology, 57(5), 1075–1079. [PubMed] [Google Scholar]
- Ostrom RS, Liu X, Head BP, Gregorian C, Seasholtz TM, & Insel PA (2002). Localization of adenylyl cyclase isoforms and G protein-coupled receptors in vascular smooth muscle cells: expression in caveolin-rich and noncaveolin domains. Molecular Pharmacology, 62(5), 983–992. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, et al. (2000). Crystal structure of rhodopsin: A G protein-coupled receptor. Science, 289(5480), 739–745. [DOI] [PubMed] [Google Scholar]
- Patriarchi T, Buonarati OR, & Hell JW (2018). Postsynaptic localization and regulation of AMPA receptors and Cav1.2 by β2 adrenergic receptor/PKA and Ca2+/CaMKII signaling. The EMBO Journal, 37(20), e99771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike LJ (2003). Lipid rafts: bringing order to chaos. Journal of Lipid Research, 44(4), 655–667. [DOI] [PubMed] [Google Scholar]
- Pike LJ (2006). Rafts defined: a report on the Keystone Symposium on Lipid Rafts and Cell Function. Journal of Lipid Research, 47(7), 1597–1598. [DOI] [PubMed] [Google Scholar]
- Poyner DR, Sexton PM, Marshall I, Smith DM, Quirion R, Born W, et al. (2002). International Union of Pharmacology. XXXII. The mammalian calcitonin gene-related peptides, adrenomedullin, amylin, and calcitonin receptors. Pharmacological Reviews, 54(2), 233–246. [DOI] [PubMed] [Google Scholar]
- Pulido D, Casadó-Anguera V, Pérez-Benito L, Moreno E, Cordomí A, López L, et al. (2018). Design of a True Bivalent Ligand with Picomolar Binding Affinity for a G Protein-Coupled Receptor Homodimer. Journal of Medicinal Chemistry, 61(20), 9335–9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi C, Sorrentino S, Medalia O, & Korkhov VM (2019). The structure of a membrane adenylyl cyclase bound to an activated stimulatory G protein. Science, 364(6438), 389–394. [DOI] [PubMed] [Google Scholar]
- Qin K, Dong C, Wu G, & Lambert NA (2011). Inactive-state preassembly of G(q)-coupled receptors and G(q) heterotrimers. Nature Chemical Biology, 7(10), 740–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, et al. (2011). Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature, 477(7366), 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebois RV, & Hébert TE (2003). Protein complexes involved in heptahelical receptor-mediated signal transduction. Receptors & Channels, 9(3), 169–194. [PubMed] [Google Scholar]
- Rebois RV, Robitaille M, Galés C, Dupré DJ, Baragli A, Trieu P, et al. (2006). Heterotrimeric G proteins form stable complexes with adenylyl cyclase and Kir3.1 channels in living cells. Journal of Cell Science, 119(Pt 13), 2807–2818. [DOI] [PubMed] [Google Scholar]
- Rivera-Oliver M, Moreno E, Álvarez-Bagnarol Y, Ayala-Santiago C, Cruz-Reyes N, Molina-Castro GC, et al. (2019). Adenosine A1-Dopamine D1 Receptor Heteromers Control the Excitability of the Spinal Motoneuron. Molecular Neurobiology, 56(2), 797–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riven I, Iwanir S, & Reuveny E (2006). GIRK channel activation involves a local rearrangement of a preformed G protein channel complex. Neuron, 51(5), 561–573. [DOI] [PubMed] [Google Scholar]
- Robishaw JD, & Berlot CH (2004). Translating G protein subunit diversity into functional specificity. Current Opinion in Cell Biology, 16(2), 206–209. [DOI] [PubMed] [Google Scholar]
- Sadana R, & Dessauer CW (2009). Physiological roles for G protein-regulated adenylyl cyclase isoforms: insights from knockout and overexpression studies. NeuroSignals, 17(1), 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadana R, Dascal N, & Dessauer CW (2009). N terminus of type 5 adenylyl cyclase scaffolds Gs heterotrimer. Molecular Pharmacology, 76(6), 1256–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salahpour A, Angers S, Mercier JF, Lagacé M, Marullo S, & Bouvier M (2004). Homodimerization of the beta2-adrenergic receptor as a prerequisite for cell surface targeting. The Journal of Biological Chemistry, 279(32), 33390–33397. [DOI] [PubMed] [Google Scholar]
- Scarselli M, Annibale P, McCormick PJ, Kolachalam S, Aringhieri S, Radenovic A, et al. (2016). Revealing G-protein-coupled receptor oligomerization at the single-molecule level through a nanoscopic lens: methods, dynamics and biological function. The FEBS Journal, 283(7), 1197–1217. [DOI] [PubMed] [Google Scholar]
- Scherrer G, Imamachi N, Cao YQ, Contet C, Mennicken F, O’Donnell D, et al. (2009). Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell, 137(6), 1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwindinger WF, Betz KS, Giger KE, Sabol A, Bronson SK, & Robishaw JD (2003). Loss of G protein gamma 7 alters behavior and reduces striatal alpha(olf) level and cAMP production. The Journal of Biological Chemistry, 278(8), 6575–6579. [DOI] [PubMed] [Google Scholar]
- Scott JD, Dessauer CW, & Taskén K (2013). Creating order from chaos: cellular regulation by kinase anchoring. Annual Review of Pharmacology and Toxicology, 53, 187–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafin DS, Harris NR, Nielsen NR, Mackie DI, & Caron KM (2020). Dawn of a New RAMPage. Trends in Pharmacological Sciences, 41(4), 249–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sezgin E, Levental I, Mayor S, & Eggeling C (2017). The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nature reviews. Molecular Cell Biology, 18(6), 361–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah UH, Toneatti R, Gaitonde SA, Shin JM, & González-Maeso J (2020). Site-Specific Incorporation of Genetically Encoded Photo-Crosslinkers Locates the Heteromeric Interface of a GPCR Complex in Living Cells. Cell Chemical Biology, 27(10), 1308–1317.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, & Sala C (2001). PDZ domains and the organization of supramolecular complexes. Annual Review of Neuroscience, 24, 1–29. [DOI] [PubMed] [Google Scholar]
- Shin SM, Zhang N, Hansen J, Gerges NZ, Pak DT, Sheng M, et al. (2012). GKAP orchestrates activity-dependent postsynaptic protein remodeling and homeostatic scaling. Nature Neuroscience, 15(12), 1655–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, & Toomre D (2000). Lipid rafts and signal transduction. Nature Reviews. Molecular Cell Biology, 1(1), 31–39. [DOI] [PubMed] [Google Scholar]
- Singer SJ, & Nicolson GL (1972). The fluid mosaic model of the structure of cell membranes. Science, 175(4023), 720–731. [DOI] [PubMed] [Google Scholar]
- Singh P, Wang B, Maeda T, Palczewski K, & Tesmer JJ (2008). Structures of rhodopsin kinase in different ligand states reveal key elements involved in G protein-coupled receptor kinase activation. The Journal of Biological Chemistry, 283(20), 14053–14062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleno R, & Hébert TE (2018). The Dynamics of GPCR Oligomerization and Their Functional Consequences. International Review of Cell and Molecular Biology, 338, 141–171. [DOI] [PubMed] [Google Scholar]
- Smart EJ, Graf GA, McNiven MA, Sessa WC, Engelman JA, Scherer PE, et al. Caveolins, liquid-ordered domains, and signal transduction. Molecular and Cellular Biology, 19(11), 7289–7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NJ, & Milligan G (2010). Allostery at G protein-coupled receptor homo- and heteromers: uncharted pharmacological landscapes. Pharmacological Reviews, 62(4), 701–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smrcka AV, & Fisher I (2019). G-protein βγ subunits as multi-functional scaffolds and transducers in G-protein-coupled receptor signaling. Cellular and Molecular Life Sciences, 76(22), 4447–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soave M, Briddon SJ, Hill SJ, & Stoddart LA (2020). Fluorescent ligands: Bringing light to emerging GPCR paradigms. British Journal of Pharmacology, 177(5), 978–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano A, Ventura R, Molero A, Hoen R, Casadó V, Cortés A, et al. (2009). Adenosine A2A receptor-antagonist/dopamine D2 receptor-agonist bivalent ligands as pharmacological tools to detect A2A-D2 receptor heteromers. Journal of Medicinal Chemistry, 52(18), 5590–5602. [DOI] [PubMed] [Google Scholar]
- Steiner D, Saya D, Schallmach E, Simonds WF, & Vogel Z (2006). Adenylyl cyclase type-VIII activity is regulated by G(betagamma) subunits. Cellular Signalling, 18(1), 62–68. [DOI] [PubMed] [Google Scholar]
- Sungkaworn T, Jobin ML, Burnecki K, Weron A, Lohse MJ, & Calebiro D (2017). Single-molecule imaging reveals receptor-G protein interactions at cell surface hot spots. Nature, 550(7677), 543–547. [DOI] [PubMed] [Google Scholar]
- Sui JL, Petit-Jacques J, & Logothetis DE (1998). Activation of the atrial KACh channel by the betagamma subunits of G proteins or intracellular Na+ ions depends on the presence of phosphatidylinositol phosphates. Proceedings of the National Academy of Sciences of the United States of America, 95(3), 1307–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takida S, & Wedegaertner PB (2004). Exocytic pathway-independent plasma membrane targeting of heterotrimeric G proteins. FEBS Letters, 567(2-3), 209–213. [DOI] [PubMed] [Google Scholar]
- Taussig R, Quarmby LM, & Gilman AG (1993). Regulation of purified type I and type II adenylylcyclases by G protein beta gamma subunits. The Journal of Biological Chemistry, 268(1), 9–12. [PubMed] [Google Scholar]
- Teichmann A, Gibert A, Lampe A, Grzesik P, Rutz C, Furkert J, et al. (2014). The specific monomer/dimer equilibrium of the corticotropin-releasing factor receptor type 1 is established in the endoplasmic reticulum. The Journal of Biological Chemistry, 289(35), 24250–24262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari V, He SQ, Huang Q, Liang L, Yang F, Chen Z, et al. (2020). Activation of μ-δ opioid receptor heteromers inhibits neuropathic pain behavior in rodents. Pain, 161(4), 842–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolkovsky AM, & Levitzki A (1978a). Mode of coupling between the beta-adrenergic receptor and adenylate cyclase in turkey erythrocytes. Biochemistry, 17(18), 3795. [DOI] [PubMed] [Google Scholar]
- Tolkovsky AM, & Levitzki A (1978b). Coupling of a single adenylate cyclase to two receptors: adenosine and catecholamine. Biochemistry, 17(18), 3811–3817. [DOI] [PubMed] [Google Scholar]
- Tsai CJ, Marino J, Adaixo R, Pamula F, Muehle J, Maeda S, et al. (2019). Cryo-EM structure of the rhodopsin-Gαi-βγ complex reveals binding of the rhodopsin C-terminal tail to the gβ subunit. eLife, 8, e46041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, et al. (1999). Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron, 23(3), 583–592. [DOI] [PubMed] [Google Scholar]
- Valentine CD, & Haggie PM (2011). Confinement of β(1)- and β(2)-adrenergic receptors in the plasma membrane of cardiomyocyte-like H9c2 cells is mediated by selective interactions with PDZ domain and A-kinase anchoring proteins but not caveolae. Molecular Biology of the Cell, 22(16), 2970–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle-León M, Callado LF, Aso E, Cajiao-Manrique MM, Sahlholm K, López-Cano M, et al. (2021). Decreased striatal adenosine A2A-dopamine D2 receptor heteromerization in schizophrenia. Neuropsychopharmacology, 46(3), 665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eps N, Preininger AM, Alexander N, Kaya AI, Meier S, Meiler J, et al. (2011). Interaction of a G protein with an activated receptor opens the interdomain interface in the alpha subunit. Proceedings of the National Academy of Sciences of the United States of America, 108(23), 9420–9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viñals X, Moreno E, Lanfumey L, Cordomí A, Pastor A, de La Torre R, et al. (2015). Cognitive Impairment Induced by Delta9-tetrahydrocannabinol Occurs through Heteromers between Cannabinoid CB1 and Serotonin 5-HT2A Receptors. PLoS Biology, 13(7), e1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violin JD, & Lefkowitz RJ (2007). Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends in Pharmacological Sciences, 28(8), 416–422. [DOI] [PubMed] [Google Scholar]
- von Zastrow M, & Sorkin A (2021). Mechanisms for Regulating and Organizing Receptor Signaling by Endocytosis. Annual Review of Biochemistry, 10.1146/annurev-biochem-081820-092427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldo GL, Ricks TK, Hicks SN, Cheever ML, Kawano T, Tsuboi K, et al. (2010). Kinetic scaffolding mediated by a phospholipase C-beta and Gq signaling complex. Science, 330(6006), 974–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh SM, Mathiasen S, Christensen SM, Fay JF, King C, Provasi D, et al. (2018). Single Proteoliposome High-Content Analysis Reveals Differences in the Homo-Oligomerization of GPCRs. Biophysical Journal, 115(2), 300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall MA, Coleman DE, Lee E, Iñiguez-Lluhi JA, Posner BA, Gilman AG, et al. (1995). The structure of the G protein heterotrimer Gi alpha 1 beta 1 gamma 2. Cell, 83(6), 1047–1058. [DOI] [PubMed] [Google Scholar]
- Wang HB, Zhao B, Zhong YQ, Li KC, Li ZY, Wang Q, et al. (2010). Coexpression of delta- and mu-opioid receptors in nociceptive sensory neurons. Proceedings of the National Academy of Sciences of the United States of America, 107(29), 13117–13122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis WI, & Kobilka BK (2018). The Molecular Basis of G Protein-Coupled Receptor Activation. Annual Review of Biochemistry, 87, 897–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westfield GH, Rasmussen SG, Su M, Dutta S, DeVree BT, Chung KY, et al. (2011). Structural flexibility of the G alpha s alpha-helical domain in the beta2-adrenoceptor Gs complex. Proceedings of the National Academy of Sciences of the United States of America, 108(38), 16086–16091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR, & MacKinnon R (2013). X-ray structure of the mammalian GIRK2-βγ G-protein complex. Nature, 498(7453), 190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise H (2012). The roles played by highly truncated splice variants of G protein-coupled receptors. Journal of Molecular Signaling, 7(1), 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong W, & Scott JD (2004). AKAP signalling complexes: focal points in space and time. Nature reviews. Molecular Cell Biology, 5(12), 959–970. [DOI] [PubMed] [Google Scholar]
- Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, et al. (2010). Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 330(6007), 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Gray JA, Compton-Toth BA, & Roth BL (2003). A direct interaction of PSD-95 with 5-HT2A serotonin receptors regulates receptor trafficking and signal transduction. The Journal of Biological Chemistry, 278(24), 21901–21908. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Devic E, & Kobilka B (2002). The PDZ binding motif of the beta 1 adrenergic receptor modulates receptor trafficking and signaling in cardiac myocytes. The Journal of Biological Chemistry, 277(37), 33783–33790. [DOI] [PubMed] [Google Scholar]
- Xie K, Masuho I, Shih CC, Cao Y, Sasaki K, Lai CW, et al. (2015). Stable G protein-effector complexes in striatal neurons: mechanism of assembly and role in neurotransmitter signaling. eLife, 4, e10451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Cantwell L, Molosh AI, Plant LD, Gazgalis D, Fitz SD, et al. (2020). The small molecule GAT1508 activates brain-specific GIRK1/2 channel heteromers and facilitates conditioned fear extinction in rodents. The Journal of Biological Chemistry, 295(11), 3614–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano H, Cai NS, Xu M, Verma RK, Rea W, Hoffman AF, et al. (2018). Gs- versus Golf-dependent functional selectivity mediated by the dopamine D1 receptor. Nature Communications, 9(1), 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekkirala AS, Kalyuzhny AE, & Portoghese PS (2010). Standard opioid agonists activate heteromeric opioid receptors: evidence for morphine and [d-Ala(2)-MePhe(4)-Glyol(5)]enkephalin as selective μ-δ agonists. ACS Chemical Neuroscience, 1(2), 146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekkirala AS, Banks ML, Lunzer MM, Negus SS, Rice KC, & Portoghese PS (2012). Clinically employed opioid analgesics produce antinociception via μ-δ opioid receptor heteromers in Rhesus monkeys. ACS Chemical Neuroscience, 3(9), 720–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim YY, Betke KM, McDonald WH, Gilsbach R, Chen Y, Hyde K, et al. (2019). The in vivo specificity of synaptic Gβ and Gγ subunits to the α2a adrenergic receptor at CNS synapses. Scientific Reports, 9(1), 1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan C, Sato M, Lanier SM, & Smrcka AV (2007). Signaling by a non-dissociated complex of G protein βγ and α subunits stimulated by a receptor-independent activator of G protein signaling, AGS8. The Journal of Biological Chemistry, 282(27), 19938–19947. [DOI] [PubMed] [Google Scholar]
- Zhang M, Patriarchi T, Stein IS, Qian H, Matt L, Nguyen M, et al. (2013). Adenylyl cyclase anchoring by a kinase anchor protein AKAP5 (AKAP79/150) is important for postsynaptic β-adrenergic signaling. The Journal of Biological Chemistry, 288(24), 17918–17931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng CY, Seabold GK, Horak M, & Petralia RS (2011). MAGUKs, synaptic development, and synaptic plasticity. The Neuroscientist, 17(5), 493–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoli M, Agnati LF, Hedlund PB, Li XM, Ferré S, & Fuxe K (1993). Receptor-receptor interactions as an integrative mechanism in nerve cells. Molecular Neurobiology, 7(3-4), 293–334. [DOI] [PubMed] [Google Scholar]