

Abstract
Genomic tools applied to non-model organisms are critical to design successful conservation strategies of particularly threatened groups. Freshwater mussels of the Unionida order are among the most vulnerable taxa and yet almost no genetic resources are available. Here, we present the gill transcriptomes of five European freshwater mussels with high conservation concern: Margaritifera margaritifera, Unio crassus, Unio pictorum, Unio mancus and Unio delphinus. The final assemblies, with N50 values ranging from 1069–1895 bp and total BUSCO scores above 90% (Eukaryote and Metazoan databases), were structurally and functionally annotated, and made available. The transcriptomes here produced represent a valuable resource for future studies on these species’ biology and ultimately guide their conservation.
Subject terms: Transcriptomics, Conservation biology
| Measurement(s) | transcriptomics |
| Technology Type(s) | Illumina sequencing |
| Sample Characteristic - Organism | Margaritifera margaritifera • Unio crassus • Unio delphinus • Unio mancus • Unio pictorum |
| Sample Characteristic - Location | Europe |
Background & Summary
Ever since genomics approaches have been applied to non-model organisms, they have been recognized as fundamental tools to study biodiversity and guide conservation actions, coining the term conservation genomics1–4. Genomic data provides a comprehensive and accurate framework enhancing the characterization of genetic variation, population structure and dynamics, selective pressures and adaptative traits that ultimately guide and prioritize applied conservation efforts1–4. Furthermore, genomic data are fundamental to construct predictive models to access the impact of human-mediated threats, such as biological invasions, resource depletion, and climate change1,3,5.
Freshwater mussels (Order Unionida) are molluscs extremely important to freshwater ecosystems where they play key ecological roles, such as nutrient and energy cycling and retention6–8. They also provide important direct (e.g., as food, pearls, and other raw materials) and indirect (e.g., water clearance, sediment mixing) services to humans6,7,9. These organisms are among the most threatened worldwide, with many species near extinction10–12. Of the thousand known species, only four whole genomes13–16 and less than 20 transcriptomes are available17–29. Of these, only one is from the European continent23. Here, we produce reference transcriptomes of five European species as baseline tools to support future studies. Genomic tools, such as transcriptomes, are key resources to study evolutionary and adaptive traits. Examples include, in the case of freshwater mussels, the unique obligatory parasitic interaction with a freshwater fish host (and occasionally other vertebrates), essential to disperse their larvae and complete the life cycle or the response to human-mediated threats, including climate change and habitat degradation8,10. Moreover, these species are ecological indicators, and the transcriptomes provide a catalogue of key genes and pathways, related to important stressors (e.g., temperature, oxygen availability), as well as basic mechanisms underlying freshwater mussel’s stress adaptation17,19,23,24,30,31.
We present the gill transcriptome of the most emblematic freshwater pearl mussel, Margaritifera margaritifera (Linnaeus, 1758). This species was famous as a source of pearls throughout the last two millennia13. Currently, is among the most threatened freshwater mussel species in Europe, with many populations suffering massive declines, with up to 90% of European populations depleted by the 90 s, which is reflected in the current scattered distribution32 (Fig. 1). Recently, a whole-genome assembly was published13, adding to unique transcriptomic dataset of a very specialized tissue (i.e., kidney23). The current species conservation status is Endangered by the IUCN and is also listed in the EC Habitats Directive33. The other four transcriptomes are from the Unio genus, the type genus of the order Unionida, i.e., Unio delphinus Spengler, 1793, Unio crassus Philipsson in Retzius, 1788, Unio pictorum (Linnaeus, 1758) and Unio mancus Lamarck, 1819, for which no genomic resources have been produced at all. Two of these species, i.e., U. crassus and U. pictorum, although widely distributed (Fig. 1), have also suffered recent declines, with U. crassus, once considered the most abundant unionid in Europe, now listed as Endangered by the IUCN and also listed in the EC Habitats Directive34. The other two species have much more restricted distributions (Fig. 1), both suffering strong population losses, with U. delphinus listed as Near Threatened and U. mancus as Endangered by the IUCN35,36. The depleted conservative state of Unionida mussels is a global concern, being the second group with the highest percentage of threatened species (43%) and the group with the highest number of wild extinct species (6.3%)37.
Fig. 1.

Maps of the five species’ potential distributions produced by overlapping points of recent presence records (obtained from Lopes-Lima et al.10) with the Hydrobasin level 5 polygons59. Overlapping distribution polygons between Unio mancus and Unio crassus are represented by a light purple shade, in the left panel. Overlapping distribution polygons between Unio pictorum and Margaritifera margaritifera are represented by an orange shade, in the right panel.
In this context, increasing the genomic resources available for freshwater mussels, particularly of European species, is vital. The transcriptomes produced here offer a unique opportunity to explore and decipher the capability of these species to cope with current and future threats and ultimately guide conservation genomic studies to protect this highly threatened group of organisms.
Methods
Animal sampling
One individual of M. margaritifera was collected from the Tuela River in Portugal, one U. crassus, and one U. pictorum from the Dobra River in Croatia, one U. mancus from the Taravu River in France and one U. delphinus from the Rabaçal River in Portugal (Table 1), all adult individuals. Differentiated tissues were promptly flash frozen and stored at −80 °C, at CIIMAR tissue and mussels’ collection, as well as their respective shells.
Table 1.
MixS descriptors for the five freshwater mussel species.
| Sample | Margaritifera margaritifera | Unio crassus | Unio pictorum | Unio mancus | Unio delphinus |
|---|---|---|---|---|---|
| Investigation_type | Eukaryote | Eukaryote | Eukaryote | Eukaryote | Eukaryote |
| Project_name | Gill transcriptome of five freshwater musssles’ european species | ||||
| Lat_lon | 41.862414; −6.931596 | 45.515500; 15.473240 | 45.515500; 15.473240 | 41.710606; 8.828512 | 41.564361; −7.258665 |
| Geo_loc_name | Portugal | Croatia | Croatia | France | North of Portugal |
| Collection_date | 7/6/2021 | 7/12/2019 | 7/12/2019 | 4/21/2021 | 3/20/2021 |
| Env_package | Water | Water | Water | Water | Water |
| Seq_meth | Illumina HiSeq 4000 | Illumina HiSeq 4000 | Illumina HiSeq 4000 | Illumina HiSeq 4000 | Illumina HiSeq 4000 |
| Assembly method | Trinity | Trinity | Trinity | Trinity | Trinity |
| Collector | Amilcar Teixeira | Manuel Lopes-Lima | Manuel Lopes-Lima | Vincent Prié | Amilcar Teixeira |
| Sex | Undetermined | Undetermined | Undetermined | Undetermined | Undetermined |
| Maturity | Mature | Mature | Mature | Mature | Mature |
RNA extraction, library construction, and sequencing
Total RNA of gills was extracted using the NZY Total RNA Isolation kit (NZYTech, Lda. - Genes and Enzymes), following the manufacturer’s instructions. RNA concentration (ng/μl) and quality measurement (OD260/280 ratio values) were obtained using a DS-11 Series Spectrophotometer/Fluorometer (M. margaritifera - 380.75 ng/μl, U. crassus – 478.290 ng/μl, U. pictorum - 375.461 ng/μl, U. mancus - 225.815 ng/μl, U. delphinus – 230.234 ng/μl). The extracted total RNA from the five samples was sent to Macrogen, Inc to build strand-specific libraries, with an insert size of 250–300 bp and sequenced using 150 bp paired-end reads on the Illumina HiSeq 4000 platform.
Pre-assembly processing
Raw reads datasets for each sample were first inspected with FastQC (version 0.11.8) software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Afterwards, reads were quality-filter and Illumina adaptors were removed using Trimmomatic (version 0.38)38, using the parameters LEADING:5 TRAILING:5 SLIDINGWINDOW:5:20 MINLEN:36 (Fig. 2). Trimmed reads were correct for random sequencing errors using a kmer-based error correction approach in Rcorrector (version 1.0.3)39 with default parameters and after imported to Centrifuge (version 1.0.3-beta)40 to taxonomically classify them using a pre-compiled nucleotide database from NCBI (ftp://ftp.ccb.jhu.edu/pub/infphilo/centrifuge/data/) (version nt_2018_3_3). All reads whose classification did not belong to the Mollusca superclass (Taxon Id: 6447) were removed (Fig. 2).
Fig. 2.
Bioinformatics pipeline applied for the transcriptome assembly and annotation. Auxiliary representative figures were created with BioRender.com.
De novo transcriptome assembly
The fully processed reads were used for the whole transcriptome de novo assembly for each sample, with Trinity (version 2.13.2)41,42 using the default parameters. To ensure the removal of contamination, the assembled transcripts were blasted against nucleotide database of NCBI (NCBI-nt; (Download; 24/08/2021)43) and Univec (Download; 02/04/2019) databases using Blast-n (version 2.11.0)44 (Fig. 2). Afterwards, transcripts that held a minimum alignment length of 100 bp, an e-value cut-off of 1e-5, identity score of 90%, and a match to Mollusca phylum (NCBI: taxid 6447) or without matches at all, were retained. On the other hand, transcripts matching other taxa in the NCBI-nt database or any match to the Univec database were considered contaminants and removed from the datasets.
Redundancy removal
Before proceeding to open reading frame (ORF) prediction, transcript redundancy was removed using a hierarchical contig clustering approach, implemented with Corset (version 1.0.9)45. For that, raw reads for each sample were mapped onto their respective transcriptome assemblies using Bowtie2 (version 2.3.5) (parameter:–no-mixed–no-discordant–end-to-end–all–score-min L,− 0.1,− 0.1). After Corset (version 1.0.9)45 was used to cluster contigs, filtered redundancies, and exclude any transcripts containing less than 10 mapped reads. The overall quality of the five transcriptomes (before and after redundancy removal) was assessed for completeness, using Benchmarking Universal Single-Copy Orthologs tool (BUSCO version 3.0.2) with the lineage-specific libraries for Eukaryota and Metazoa46 and for structural integrity using TransRate (version 1.0.3)47 (Fig. 2).
Open reading frame prediction and transcriptome annotation
The open reading frames (ORFs) for each non-redundant transcriptome, were produced using Transdecoder (version 5.3.0) (https://transdecoder.github.io/) (Fig. 2). During the ORF prediction process, the homology and protein searches were performed in UniProtKB/Swiss-Prot48 and PFAM databases49 using the Blast-p (version 2.12.0)44 and hmmscan of hmmer2 package (version 2.4i)50 software, respectively. Next, the Gtf/Gff Analysis Toolkit (AGAT) (version 0.8.0)51 was applied to produce the structural annotation file (in gff3 format) from the Transdecoder output file (.gff) and transcriptome assembly file (.fasta). In the end, the AGAT tool was used to extract the protein and transcript fasta files with the names properly uniformized and formatted per species. Afterwards, the functional annotation was performed with InterProScan tool (version 5.44.80) and Blast-n/p/x searches in several databases. While the proteins per species were queried against InterPro (Download; 30/03/2019) and protein databases of NCBI (NCBI-RefSeq – Reference Sequence Database (Download; 10/03/2022)52 NCBI-nr – non-redundant database of NCBI (Download; 15/12/2021)43 with the Blast-p/x tool of DIAMOND software (version version 2.0.13)53, the transcripts were searched by Blast-n/x in NCBI-nt and NCBI-nr databases, with Blast-n tool of NCBI and Blast-x tool of DIAMOND software. In the end, all blast (outfmt6 files) and InterProScan (tsv file) outputs were integrated into the gff3 annotation file with the AGAT tool. The putative gene name per sequence was assigned based on the best blast hit (Gene symbol – NCBI Accession Number) and following the ranking: 1- Blast-p Hit in RefSeq database; 2 - Blast-p Hit in NCBI-nr database; 3 - Blast-x Hit in NCBI-nr database; 4 - Blast-n Hit in NCBI-nt database.
Data Records
The raw reads for each sample were deposited at the NCBI Sequence Read Archive with the accessions numbers: SRR19261768 (MM), SRR19261764 (UD), SRR19261767 (UP), SRR19261765 (UM), SRR19261766 (UC)54; the BioSample accessions numbers: SAMN28495338 (MM), SAMN28495283 (UD), SAMN28495235 (UP), SAMN28495263 (UM), SAMN28495214 (UC) and under BioProject PRJNA83906255. The remaining information was uploaded to figshare56. In detailed, the files uploaded to figshare include, the filtered trinity redundant assemblies (_trinity_filtered.fasta), the non-redundant transcriptomes (_transcriptome.fa), transcripts files (_genes.fa), messenger RNA file (_mrna.fa), open reading frames predictions (_cds.fa), open reading frames proteins predictions (_proteins.fa) as well as the annotation files (_annotation_sorted.gff3.gz).
Technical Validation
Raw datasets and pre-assembly processing quality control
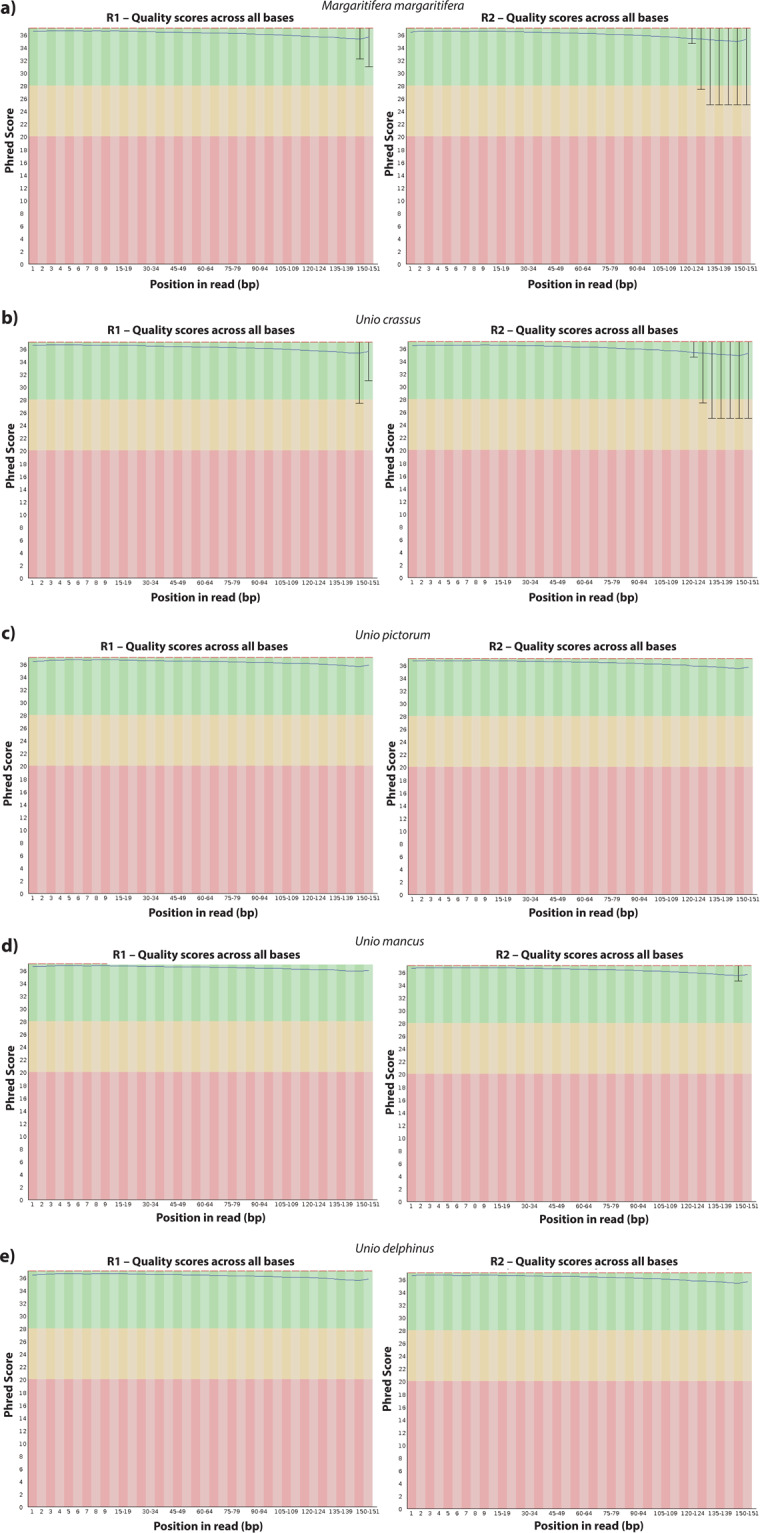
The raw sequencing outputs resulted in a total of 131051306 million reads (M) for M. margaritifera, 132002266 M for U. crassus, 104108396 M for U. pictorum, 100704688 M for U. mancus, and 112439686 M for U. delphinus. Although the initial overall quality of raw data was considerably good (Fig. 3), the datasets were further improved by quality trimming (Trimmomatic), error-correction (Rcorrector), and decontaminated (Centrifuge) (Fig. 3). The number of reads removed during the pre-assembly processing represented less than 3% of each dataset (Table 2) and the overall Phred scores were all above 25 (Fig. 3a–e).
Fig. 3.

FastQC quality report of the trimmed and decontaminated RNA-seq reads (after Centrifuge for each species. (a) Margaritifera margaritifera; (b) Unio crassus; (c) Unio pictorum; (d) Unio mancus; and (e) Unio delphinus.
Table 2.
Basic statistics of raw sequencing datasets and percentages of removed reads at each step of the preassembly processing strategy.
| Basic Statistics | Total Transcriptome | Non redundant Transcriptome | Total Transcriptome | Non redundant Transcriptome | Total Transcriptome | Non redundant Transcriptome | Total Transcriptome | Non redundant Transcriptome | Total Transcriptome | Non redundant Transcriptome | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Margaritifera margaritifera | Margaritifera margaritifera | Unio crassus | Unio crassus | Unio pictorum | Unio pictorum | Unio mancus | Unio mancus | Unio delphinus | Unio delphinus | ||
| Number of transcripts | 1694677 | 470852 | 1304611 | 169668 | 232124 | 68670 | 234695 | 65620 | 280001 | 82542 | |
| n bases | 1052464277 | 442302372 | 1002862692 | 262637793 | 189129150 | 83762650 | 198791465 | 89666570 | 224567067 | 103248722 | |
| Mean transcript lenght (bp) | 621.02389 | 939.36603 | 768.67926 | 1547.94894 | 814.75652 | 1219.7852 | 847.00815 | 1366.44881 | 802.01073 | 1250.86286 | |
| Number of transcripts over 1 K nt | 214128 | 134690 | 235872 | 104192 | 53293 | 28701 | 54754 | 31276 | 62078 | 35904 | 27.78417362 |
| Number of transcripts over 10 K | 1189 | 261 | 1905 | 453 | 7 | 5 | 33 | 15 | 24 | 12 | |
| N90 trancript lenght (bp) | 284 | 499 | 313 | 816 | 314 | 582 | 322 | 659 | 309 | 612 | |
| N70 trancript lenght (bp) | 462 | 759 | 589 | 1324 | 697 | 1037 | 732 | 1168 | 677 | 1047 | |
| N50 trancript lenght (bp) | 773 | 1069 | 1187 | 1889 | 1447 | 1688 | 1569 | 1895 | 1400 | 1669 | |
| N30 trancript lenght (bp) | 1475 | 1619 | 2409 | 2864 | 2438 | 2589 | 2635 | 2870 | 2426 | 2600 | |
| N10 trancript lenght (bp) | 3783 | 3281 | 5504 | 5458 | 4073 | 4174 | 4427 | 4592 | 4108 | 4252 | |
| Percentage of GC (%) | 0.36365 | 0.35712 | 0.35352 | 0.34896 | 0.35511 | 0.35179 | 0.35899 | 0.35468 | 0.36814 | 0.36893 | |
| Busco analysis (%) | |||||||||||
| BUSCO Complete (Single + Duplicated) | 93.7/94.5 | 85.8/89.4 | 97.1/98.1 | 92.1/93.1 | 87.5/83.1 | 83.8/79.7 | 89.8/88.2 | 85.2/83.9 | 92.1/88.3 | 89.1/84.8 | |
| BUSCO Single* | 45.5/47.4 | 83.8/85.8 | 44.6/43.6 | 90.8/90.5 | 58.1/57.8 | 80.5/77.8 | 62.7/64.6 | 82.2/82.7 | 62.7/64.0 | 81.2/80.8 | |
| BUSCO Duplicated* | 48.2/47.1 | 2.0/3.6 | 52.5/54.5 | 1.3/2.6 | 29.4/25.3 | 3.3/1.9 | 27.1/23.6 | 3.0/1.2 | 29.4/24.3 | 7.9/4.0 | |
| BUSCO Fragmented* | 4.0/4.5 | 8.3/6.1 | 2.3/1.6 | 3.6/3.9 | 7.9/10.2 | 6.9/7.4 | 6.6/8.0 | 7.6/6.4 | 5.6/7.8 | 5.0/6.1 | |
| BUSCO Missing* | 2.3/1.0 | 5.9/4.5 | 0.6/0.3 | 4.3/3.0 | 4.6/6.7 | 9.3/12.9 | 3.6/3.8 | 7.2/9.7 | 2.3/3.9 | 5.9/9.1 | |
| Total Buscos Found* | 97.7/99.0 | 94.1/95.5 | 99.4/99.7 | 95.7/97.0 | 95.4/93.3 | 90.7/87.1 | 96.4/96.8 | 92.8/90.3 | 97.7/96.1 | 94.1/90.4 | |
Transcriptome assembly metrics
The de novo transcriptome assemblies were performed using Trinity, with default paraments, which has been successfully applied for other Unionida transcriptome assembly projects17,20–23. Furthermore, the overall completeness of the transcriptome assemblies was evaluated using Benchmarking Universal Single-Copy Orthologs (BUSCO), by searching the Eukaryota (n:303) and Metazoa (n:978) near-universal single-copy orthologs databases, for all species. The overall metrics for each transcriptome de novo assembly, as well as their corresponding BUSCO scores, are presented in Table 3. The general assembly metrics of U. pictorum, U. mancus, and U. delphinus are very similar, both in the number of transcripts (~250,000) and N50 values (>1400 bp) (Table 3). On the other hand, M. margaritifera and U. crassus transcriptomes, have a much higher number of assembled transcripts (>1,000,000) and, consequently lower N50 lengths (Table 3). However, all these values are within the reported for other Unionida transcriptomes assembly projects17–21,23,25–27,29. Furthermore, M. margaritifera and U. crassus transcriptome assemblies also have a considerably high level of duplicated BUSCO scores, i.e., around 50%, compared with the remaining species which presented values around 30% (Table 3). The percentage of total genes found (complete + fragmented) in all BUSCO analyses, for all species, was above 95%, except for the U. pictorum transcriptome in the Metazoan lineage-specific profile library, which had a total of 93.3%. These results reveal that despite being produced from a single tissue the initial assemblies were highly efficient in capturing conserved and widely express genes, thus providing a highly complete gill transcriptomic repertoire.
Table 3.
Transrate and Busco scores of redundant and non-redundant gill transcriptome assemblies for each species.
| Raw Reads | Margaritifera margaritifera | Unio crassus | Unio pictorum | Unio mancus | Unio delphinus |
|---|---|---|---|---|---|
| Raw sequencing reads | 131051306 | 132002266 | 104108396 | 100704688 | 112439686 |
| Trimmomatic reads removed | 1524256 (1.16%) | 1761532 (1.33%) | 937250 (0.90%) | 714904 (0.71%) | 1074338 (0.96%) |
| Centrifuge reads removed | 157718 (0.12%) | 118410 (0.090%) | 101442 (0.097%) | 145422 (0.14%) | 250936 (0.22%) |
| Reads used in assembly | 129369332 (98.72%) | 130122324 (98.56%) | 103069704 (99.00%) | 99844362 (99.15%) | 111114412 (98.82%) |
*euk/met. Euk: Dataset with 303 genes of Eukaryota library profile. Met: Dataset with 978 genes of Metazoa library profile.
Post-assembly processing and annotation verification
The newly assembled transcriptomes were after subject to a decontamination process by Blast-n search against NCBI-nt and Univec databases. The Blast-n hits against NCBI-nt, were manually validated based on the reads with a minimum alignment length of 100 bp, an e-value of 1e-5, an identity score of 90% and a match to Mollusca phylum (NCBI: taxid 6447) or without matches at all, were retained. On the other hand, all Blast-n hits against Univec database were considered exogenous and removed. This decontamination approach has been routinely and successfully used by the team (e.g.57,58) and focuses the analyses on the identification, by homology, of putative contaminations and only excluded them if they are well supported and thus avoiding the exclusion of unambiguous matches.
Subsequently, before proceeding to the annotation, the decontaminated transcriptomes were subjected to redundancy removal using Corset. This software relies on hierarchical clustering of contigs that share read alignments and thus allows an unbiased removal of redundancy without discarding non-coding transcripts from the process45. The general transcriptome metrics after redundancy removal are presented in Table 3. Corser was extremely efficient in removing the redundancy from the filtered assemblies (Table 3). In fact, over 70% of the initial transcripts were removed during the process, suggesting that although Trinity was effective in producing a complete transcriptome assembly, it as has also generated several duplicated transcripts as well as many transcripts with low read support (Table 3). These results highlight the importance of using read clustering approach to remove redundancy, rather than simply relying on coding transcripts and selection of the largest isoform. The efficiency of the redundancy removal is also supported by the BUSCO analyses, where duplicated scores were on average 3.5% for Eukaryota (n:303) and 2.66% for Metazoa (n:978) after Corset, in opposition to an average 37.32% for Eukaryota (n:303) and 34.96% for Metazoa (n:978) before redundancy removal (Table 3). Furthermore, redundancy removal did not impact the overall completeness of the transcriptome assemblies, which still maintained the total BUSCO scores of over 90% (Table 3). In the end, the final gill transcriptomes were significantly reduced, fairly complete and cleared of putative errors introduced during the assembly, thus properly adjusted for annotation.
TransDecoder prediction of transcripts with an assigned ORF, resulted in a total of 56,730 for M. margaritifera, 35,069 for U. crassus, 19,830 for U. pictorum, 19,881 for U. mancus, and 28,216 for U. delphinus (Table 4). These predictions were performed in the non-redundant transcriptomes and were deposited in FigShare56. Finally, the results of the functional annotation are presented in Table 4, where a thorough listing of hits counts from distinct databases used in the functional annotation processes is presented. The number of transcripts functionally annotated was InterProScan:25,267; Blast:71,046 for M. margaritifera, InterProScan:20,432; Blast:51,937 for U. crassus, InterProScan:14,723; Blast:24,194 for U. pictorum, InterProScan:14,971; Blast:24,775 for U. mancus and InterProScan:20,637; Blast:32,688 for U. delphinus (Table 4). These values are within the observed values for other Unionida genomics projects, both in transcriptomes17,19–21,23,25,26 and genome14–16,19. Particularly for M. margaritifera, the number of genes functionally annotated, is very similar to the values obtained for the annotated genome assembly available for the species, i.e., 26,836 transcripts13.
Table 4.
Structural and functional annotation statistics for the final gill transcriptome assemblies for each species.
| Structural annotation | Margaritifera margaritifera | Unio crassus | Unio pictorum | Unio mancus | Unio delphinus |
|---|---|---|---|---|---|
| Number of transcripts | 470852 | 169668 | 68670 | 65620 | 82542 |
| Number of cdss | 56730 | 35069 | 19830 | 19881 | 28216 |
| Number of exons | 56730 | 35069 | 19830 | 19881 | 28216 |
| Total gene length | 442302372 | 262637793 | 83762650 | 89666570 | 103248722 |
| Total cds length | 41461605 | 34346592 | 17039142 | 18840849 | 22564185 |
| Total exon length | 95381543 | 85666986 | 36059402 | 41076667 | 48847415 |
| mean gene length | 939 | 1547 | 1219 | 1366 | 1250 |
| mean cds length | 730 | 979 | 859 | 947 | 799 |
| mean exon length | 1681 | 2442 | 1818 | 2066 | 1731 |
| Functional annotation Blast | Margaritifera margaritifera | Unio crassus | Unio pictorum | Unio mancus | Unio delphinus |
| Blast-p/x/n hits (NCBI-RefSeq; NCBI-nr; NCBI-nt) | 71046 | 51937 | 24194 | 24775 | 32688 |
| Functional annotation InterPro | Margaritifera margaritifera | Unio crassus | Unio pictorum | Unio mancus | Unio delphinus |
| CDD | 6295 | 6475 | 4357 | 4693 | 5542 |
| Coils | 4943 | 4558 | 2815 | 2930 | 3821 |
| GO | 10784 | 9966 | 7243 | 7701 | 10272 |
| Gene3D | 15077 | 13342 | 9681 | 9975 | 13499 |
| Hamap | 270 | 266 | 221 | 229 | 254 |
| InterPro | 19126 | 16611 | 12116 | 12524 | 16717 |
| KEGG | 909 | 874 | 575 | 625 | 802 |
| MetaCyc | 835 | 781 | 581 | 574 | 777 |
| MobiDBLite | 10629 | 8238 | 5225 | 5737 | 6786 |
| PIRSF | 628 | 687 | 484 | 556 | 582 |
| PRINTS | 2609 | 2645 | 1961 | 2232 | 2589 |
| Pfam | 15788 | 14394 | 10591 | 11116 | 14428 |
| ProSitePatterns | 3585 | 3546 | 2445 | 2708 | 3346 |
| ProSiteProfiles | 9079 | 8323 | 5716 | 6034 | 7612 |
| Reactome | 3717 | 3515 | 2580 | 2732 | 3564 |
| SFLD | 69 | 72 | 54 | 60 | 67 |
| SMART | 7138 | 6869 | 4534 | 4958 | 6036 |
| SUPERFAMILY | 15070 | 13240 | 9376 | 9729 | 13190 |
| TIGRFAM | 757 | 751 | 552 | 617 | 815 |
| Total | 25267 | 20432 | 14723 | 14971 | 20637 |
Overall, these results provide evidence of the quality and completeness of the five gill transcriptome assemblies, which represent timely needed genomic resources for this highly threatened group of organisms. Although future studies should also aim to obtain transcriptomic information from other tissues/development stages, these five annotated gill transcriptomes represent a valuable baseline tool to study these organisms and can ultimately help and guide future conservation actions.
Acknowledgements
AGS was funded by the Portuguese Foundation for Science and Technology (FCT) under the grant SFRH/BD/137935/2018, that also supported MLL (2020.03608.CEECIND) and EF (CEECIND/00627/2017). This research was developed under the project EdgeOmics - Freshwater Bivalves at the edge: Adaptation genomics under climate-change scenarios (PTDC/CTA-AMB/3065/2020) funded by FCT through national funds. Additional strategic funding was provided by FCT UIDB/04423/2020 and UIDP/04423/2020.
Author contributions
E.F., M.L.L., L.F.C.C. designed and conceived this work. M.L.L., V.P. and A.T. collected the samples. A.G.S. and A.M.M. carry on all the analysis. A.G.S., E.F and M.L.L. wrote the first version of the manuscript. All authors read, revised, and approved the final manuscript.
Code availability
All software with respective versions and parameters used for producing the resources here presented (i.e., transcriptome assembly, pre and post-assembly processing stages, and transcriptome annotation) are listed in the methods section. Software programs with no parameters associated were used with the default settings.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
André Gomes-dos-Santos, Email: andrepousa64@gmail.com.
Elsa Froufe, Email: elsafroufe@gmail.com.
References
- 1.Allendorf FW, Hohenlohe PA, Luikart G. Genomics and the future of conservation genetics. Nature Reviews Genetics 2010 11:10. 2010;11:697–709. doi: 10.1038/nrg2844. [DOI] [PubMed] [Google Scholar]
- 2.Formenti G, et al. The era of reference genomes in conservation genomics. Trends in Ecology and Evolution. 2022;37:197–202. doi: 10.1016/j.tree.2021.11.008. [DOI] [PubMed] [Google Scholar]
- 3.Hohenlohe PA, Funk WC, Rajora OP. Population genomics for wildlife conservation and management. Molecular Ecology. 2021;30:62–82. doi: 10.1111/mec.15720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meek MH, Larson WA. The future is now: Amplicon sequencing and sequence capture usher in the conservation genomics era. Molecular Ecology Resources. 2019;19:795–803. doi: 10.1111/1755-0998.12998. [DOI] [PubMed] [Google Scholar]
- 5.McCartney, M. A. et al. The genome of the zebra mussel, Dreissena polymorpha: a resource for comparative genomics, invasion genetics, and biocontrol. G3 Genes|Genomes|Genetics12 (2022). [DOI] [PMC free article] [PubMed]
- 6.Vaughn, C. C., Nichols, S. J. & Spooner, D. E. Community and foodweb ecology of freshwater mussels. 27, 409–423, 10.1899/07-058.1 (2015).
- 7.Vaughn CC. Ecosystem services provided by freshwater mussels. Hydrobiologia 2017 810:1. 2017;810:15–27. [Google Scholar]
- 8.Lopes-Lima M, et al. Biology and conservation of freshwater bivalves: Past, present and future perspectives. Hydrobiologia. 2014;735:1–13. doi: 10.1007/s10750-014-1902-9. [DOI] [Google Scholar]
- 9.Haag, W. R. North American Freshwater Mussels: Natural History, Ecology, and Conservation. (Cambridge University Press, 2012).
- 10.Lopes-Lima M, et al. Conservation status of freshwater mussels in Europe: state of the art and future challenges. Biological Reviews. 2017;92:572–607. doi: 10.1111/brv.12244. [DOI] [PubMed] [Google Scholar]
- 11.Cuttelod, A., Seddon, M. & Neubert, E. European red list of non-marine molluscs. (Publications Office of the European Union Luxembourg, 2011).
- 12.Lopes-Lima M, et al. Conservation of freshwater bivalves at the global scale: diversity, threats and research needs. Hydrobiologia. 2018;810:1–14. doi: 10.1007/s10750-017-3486-7. [DOI] [Google Scholar]
- 13.Gomes-dos-Santos, A. et al. The Crown Pearl: a draft genome assembly of the European freshwater pearl mussel Margaritifera margaritifera (Linnaeus, 1758). DNA Research, 10.1093/dnares/dsab002 (2021). [DOI] [PMC free article] [PubMed]
- 14.Smith, C. H. A High-Quality Reference Genome for a Parasitic Bivalve with Doubly Uniparental Inheritance (Bivalvia: Unionida). Genome Biology and Evolution13 (2021). [DOI] [PMC free article] [PubMed]
- 15.Rogers RL, et al. Gene family amplification facilitates adaptation in freshwater unionid bivalve Megalonaias nervosa. Molecular Ecology. 2021;30:1155–1173. doi: 10.1111/mec.15786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Renaut S, et al. Genome Survey of the Freshwater Mussel Venustaconcha ellipsiformis (Bivalvia: Unionida) Using a Hybrid De Novo Assembly Approach. Genome Biology and Evolution. 2018;10:1637–1646. doi: 10.1093/gbe/evy117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roznere I, Sinn BT, Watters GT. The Amblema plicata Transcriptome as a Resource to Assess Environmental Impacts on Freshwater Mussels. Freshwater Mollusk Biology and Conservation. 2018;21:57–64. [Google Scholar]
- 18.Wang, R. et al. Rapid development of molecular resources for a freshwater mussel, Villosa lienosa (Bivalvia:Unionidae), using an RNA-seq-based approach. 31, 695–708, 10.1899/11-149.1 (2015).
- 19.Luo Y, et al. Transcriptomic Profiling of Differential Responses to Drought in Two Freshwater Mussel Species, the Giant Floater Pyganodon grandis and the Pondhorn Uniomerus tetralasmus. PLOS ONE. 2014;9:e89481. doi: 10.1371/journal.pone.0089481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patnaik BB, et al. Sequencing, De Novo Assembly, and Annotation of the Transcriptome of the Endangered Freshwater Pearl Bivalve, Cristaria plicata, Provides Novel Insights into Functional Genes and Marker Discovery. PLOS ONE. 2016;11:e0148622. doi: 10.1371/journal.pone.0148622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Liu Z, Wu W. Transcriptome analysis of the freshwater pearl mussel (Cristaria plicata) mantle unravels genes involved in the formation of shell and pearl. Molecular Genetics and Genomics. 2017;292:343–352. doi: 10.1007/s00438-016-1278-9. [DOI] [PubMed] [Google Scholar]
- 22.Yang Q, et al. Histopathology, antioxidant responses, transcriptome and gene expression analysis in triangle sail mussel Hyriopsis cumingii after bacterial infection. Developmental & Comparative Immunology. 2021;124:104175. doi: 10.1016/j.dci.2021.104175. [DOI] [PubMed] [Google Scholar]
- 23.Bertucci A, et al. Transcriptomic responses of the endangered freshwater mussel Margaritifera margaritifera to trace metal contamination in the Dronne River, France. Environmental Science and Pollution Research. 2017;24:27145–27159. doi: 10.1007/s11356-017-0294-6. [DOI] [PubMed] [Google Scholar]
- 24.Robertson LS, Galbraith HS, Iwanowicz D, Blakeslee CJ, Cornman RS. RNA sequencing analysis of transcriptional change in the freshwater mussel Elliptio complanata after environmentally relevant sodium chloride exposure. Environmental Toxicology and Chemistry. 2017;36:2352–2366. doi: 10.1002/etc.3774. [DOI] [PubMed] [Google Scholar]
- 25.Capt C, et al. Deciphering the Link between Doubly Uniparental Inheritance of mtDNA and Sex Determination in Bivalves: Clues from Comparative Transcriptomics. Genome Biology and Evolution. 2018;10:577–590. doi: 10.1093/gbe/evy019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang D, Shen J, Li J, Bai Z. Integrated transcriptome analysis of immunological responses in the pearl sac of the triangle sail mussel (Hyriopsis cumingii) after mantle implantation. Fish & Shellfish Immunology. 2019;90:385–394. doi: 10.1016/j.fsi.2019.05.012. [DOI] [PubMed] [Google Scholar]
- 27.Capt C, Renaut S, Stewart DT, Johnson NA, Breton S. Putative Mitochondrial Sex Determination in the Bivalvia: Insights From a Hybrid Transcriptome Assembly in Freshwater Mussels. Frontiers in Genetics. 2019;10:840. doi: 10.3389/fgene.2019.00840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen X, Bai Z, Li J. The Mantle Exosome and MicroRNAs of Hyriopsis cumingii Involved in Nacre Color Formation. Marine Biotechnology. 2019;21:634–642. doi: 10.1007/s10126-019-09908-8. [DOI] [PubMed] [Google Scholar]
- 29.Cornman RS, Robertson LS, Galbraith H, Blakeslee C. Transcriptomic Analysis of the Mussel Elliptio complanata Identifies Candidate Stress-Response Genes and an Abundance of Novel or Noncoding Transcripts. PLOS ONE. 2014;9:e112420. doi: 10.1371/journal.pone.0112420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ganser AM, Newton TJ, Haro RJ. Effects of elevated water temperature on physiological responses in adult freshwater mussels. Freshwater Biology. 2015;60:1705–1716. doi: 10.1111/fwb.12603. [DOI] [Google Scholar]
- 31.Haney A, Abdelrahman H, Stoeckel JA. Effects of thermal and hypoxic stress on respiratory patterns of three unionid species: implications for management and conservation. Hydrobiologia. 2020;847:787–802. doi: 10.1007/s10750-019-04138-4. [DOI] [Google Scholar]
- 32.Geist J. Strategies for the conservation of endangered freshwater pearl mussels (Margaritifera margaritifera L.): a synthesis of Conservation Genetics and Ecology. Hydrobiologia. 2010;644:69–88. doi: 10.1007/s10750-010-0190-2. [DOI] [Google Scholar]
- 33.Moorkens, E., Cordeiro, J., Seddon, M. B. & von Proschwitz, T. Woolnough, D. Margaritifera margaritifera (Freshwater Pearl Mussel). The IUCN Red List of Threatened Specieshttps://www.iucnredlist.org/species/12799/128686456 (2017).
- 34.Lopes-Lima, M., Kebapçı, U. & van Damme, D. Unio crassus (Thick Shelled River Mussel). The IUCN Red List of Threatened Specieshttps://www.iucnredlist.org/species/22736/42465628 (2014).
- 35.Lopes-Lima, M. & Seddon, M. B. Unio mancus. The IUCN Red List of Threatened Specieshttps://www.iucnredlist.org/species/22737/42466471 (2014).
- 36.Araujo, R. Unio delphinus. The IUCN Red List of Threatened Specieshttps://www.iucnredlist.org/species/195510/8975648 (2011).
- 37.Díaz, S. et al. IPBES, 2019: Summary for policymakers of the global assessment report on biodiversity and ecosystem services of the Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services. Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services (2019).
- 38.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song L, Florea L. Rcorrector: Efficient and accurate error correction for Illumina RNA-seq reads. Gigascience. 2015;4:48. doi: 10.1186/s13742-015-0089-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim D, Song L, Breitwieser FP, Salzberg SL. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Research. 2016;26:1721–1729. doi: 10.1101/gr.210641.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology 2011 29:7. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haas BJ, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Agarwala R, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Research. 2016;44:D7–D19. doi: 10.1093/nar/gkv1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Camacho C, et al. BLAST+: Architecture and applications. BMC Bioinformatics. 2009;10:1–9. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davidson NM, Oshlack A. Corset: Enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biology. 2014;15:1–14. doi: 10.1186/gb-2014-15-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 47.Lang-Unnasch N. Purification and properties of Plasmodium falciparum malate dehydrogenase. Molecular and Biochemical Parasitology. 1992;50:17–25. doi: 10.1016/0166-6851(92)90240-K. [DOI] [PubMed] [Google Scholar]
- 48.Bateman A, et al. UniProt: the universal protein knowledgebase. Nucleic Acids Research. 2017;45:D158–D169. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Punta M, et al. The Pfam protein families database. Nucleic Acids Research. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Research. 2011;39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dainat, J., Hereñú, D. & Pucholt, P. AGAT: Another Gff Analysis Toolkit to handle annotations in any GTF/GFF format, 10.5281/zenodo.4205393 (2020).
- 52.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Research. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nature Methods. 2015;12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 54.2022. NCBI Sequence Read Archive. SRP375793
- 55.2022. NCBI BioProject. PRJNA839062
- 56.Gomes-dos-Santos A. 2022. The gill transcriptome of threatened European freshwater mussels. figshare. [DOI] [PMC free article] [PubMed]
- 57.Machado AM, et al. The male and female gonad transcriptome of the edible sea urchin, Paracentrotus lividus: Identification of sex-related and lipid biosynthesis genes. Aquaculture Reports. 2022;22:100936. doi: 10.1016/j.aqrep.2021.100936. [DOI] [Google Scholar]
- 58.Machado AM, et al. Liver transcriptome resources of four commercially exploited teleost species. Scientific Data. 2020;7:1–9. doi: 10.1038/s41597-020-0565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lehner B, Grill G. Global river hydrography and network routing: Baseline data and new approaches to study the world’s large river systems. Hydrological Processes. 2013;27:2171–2186. doi: 10.1002/hyp.9740. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- 2022. NCBI Sequence Read Archive. SRP375793
- 2022. NCBI BioProject. PRJNA839062
- Gomes-dos-Santos A. 2022. The gill transcriptome of threatened European freshwater mussels. figshare. [DOI] [PMC free article] [PubMed]
Data Availability Statement
All software with respective versions and parameters used for producing the resources here presented (i.e., transcriptome assembly, pre and post-assembly processing stages, and transcriptome annotation) are listed in the methods section. Software programs with no parameters associated were used with the default settings.