

Abstract
Despite their clinical success, chimeric antigen receptor (CAR)-T cell therapies for B-cell malignancies are limited by lengthy, costly and labor-intensive ex vivo manufacturing procedures that may lead to cell products with heterogeneous composition. Here we describe an implantable, multifunctional alginate scaffold for T cell engineering and release (MASTER) that streamlines in vivo CAR-T cell manufacturing and reduces processing time to a single day. When seeded with human peripheral blood mononuclear cells and CD19-encoding retroviral particles, MASTER provides the appropriate interface for viral vector-mediated gene transfer and, following subcutaneous implantation, mediates the release of functional CAR-T cells in mice. We further demonstrate that in vivo-generated CAR-T cells enter the bloodstream, and control distal tumor growth in a mouse xenograft model of lymphoma, showing greater persistence than conventional CAR-T cells. MASTER promises to transform CAR-T cell therapy by fast-tracking manufacture and potentially reducing the complexity and resources needed for provision of this type of therapy.
Keywords: Adoptive cellular therapy, Viral transduction, CAR-T cell manufacturing, Implantable scaffold, Immunotherapy
Introduction:
CAR T cell therapy has demonstrated unprecedented success against CD19-expressing B cell malignancies1–4, resulting in two FDA approvals5,6 and inspiring hundreds of ongoing clinical trials7. Despite the revolutionary potential of CAR-T cell therapies to treat human malignancies, the complex procedures and expense required to produce clinical grade CAR-T cells is a major obstacle to widespread clinical use8–11. Therapeutic CAR-T cell manufacturing requires an extensive array of steps, including: (i) T cell collection via leukapheresis from the patient and shipment to the manufacturing center; (ii) labor-intensive procedures under good manufacturing practice (GMP) conditions to activate, expand and engineer the T cells using viral vectors; (iii) quality control of the produced cells; and (iv) shipment of the final CAR-T cell product to the hospital and re-infusion into the patient12. The complete manufacturing process can cost up to half a million dollars and can take several weeks. This delay is problematic because the aggressiveness of many cancers may not allow sufficient time to complete the production13–15. In addition, extensive ex vivo culture is linked to T cell differentiation, which impairs the potency of CAR-T cells by compromising their engraftment and persistence in vivo16–18.
The costs and challenges associated with producing CAR-T cells have driven research into improved manufacturing methods. One approach aims for closed and automatic manufacturing to reduce the labor and time needed to make CAR-T cells19. Closed-loop production of autologous CAR-T cells maintains the sequential application of three procedures (activation, transduction, expansion) ex vivo, and is limited by a theoretical lower bound of 6–8 days20. A highly attractive alternative uses allogeneic “off-the-shelf” CAR-T cells depleted of TCR expression, overcoming challenges stemming from prior patient chemotherapeutic treatments including low T cell counts, poor quality, and reduced functional capacity21. Additional engineering is required to prevent allogeneic CAR-T cells from creating life-threatening graft-versus-host disease21,22 as well as reducing alloreactivity against the CAR-T cells by the host’s immune system21,22. Finally, fully in vivo CAR-T generation is being actively explored with systemic administration of CAR-encoding nanocarriers23 and viral constructs24. In vivo generation of CAR-T cells eliminates extensive ex vivo culture and could prevent the terminal differentiation of CAR-T cells due to ex vivo procedures altogether. Several challenges remain to this approach, including the short plasma half-life of systemically administered carriers25 and possible nonspecific targeting of carriers to off-target cells26. Due to these limitations, in situ generation of CAR-T cells with minimal ex vivo manipulation remains highly attractive. Such an approach would eliminate the need for extensive ex vivo culture, prevent the terminal differentiation of CAR-T cells prior to administration, and facilitate use of autologous T cells as compared to allogeneic CAR-T cell products.
Herein we describe an all-in-one Macroporous Alginate Scaffold for T cell Engineering and Release (MASTER) that provides for in vivo CAR-T cell generation with minimal extracorporeal manipulation. MASTER can be directly loaded with patient-derived T cells and viral particles encoding the CAR and implanted on the same day to generate CAR-T cells in vivo (Scheme 1). MASTER is designed to: (i) host T cells and viral particles; (ii) stimulate T cell activation and proliferation, iii) promote T cell transduction; and (iv) locally expand CAR-T cells and (v) sustainably release fully functional CAR-T cells to control tumor growth.
Bioinstructive Scaffolds Efficiently Generate CAR-T Cells In Vitro.
Alginate was selected as the basis for these multifunctional scaffolds (MASTER) due to its biocompatibility, biodegradability, mild gelation requirements27 and extensive application as a 3D structure for cell culture28,29. Macroporosity was desired in the scaffold initially to provide an interface for efficient contact between the cells and retrovirus30 and subsequently for mass transfer of nutrients to the proliferating CAR-T cells30. Macroporosity was achieved in MASTER through mild cryogelation (Fig. 1A)31,32. SEM and X-ray CT analysis of MASTER revealed well connected, 100–200 µm pores throughout the scaffold (Fig. 1B, Extended Fig. 1A–D), with a calculated porosity of 75.8%. Most pores exhibited an oblong shape and were connected with at least three other pores (Fig. 1 C–E, Extended Fig. 1E). Cell distribution within the scaffold was assessed by 3D confocal microscopy of GFP-expressing T cells seeded on to MASTER scaffolds, revealing homogeneously distributed T cells (~12 µm diameter) throughout the pores of the scaffold (Fig. 1F, Extended Fig 2).
Figure 1:
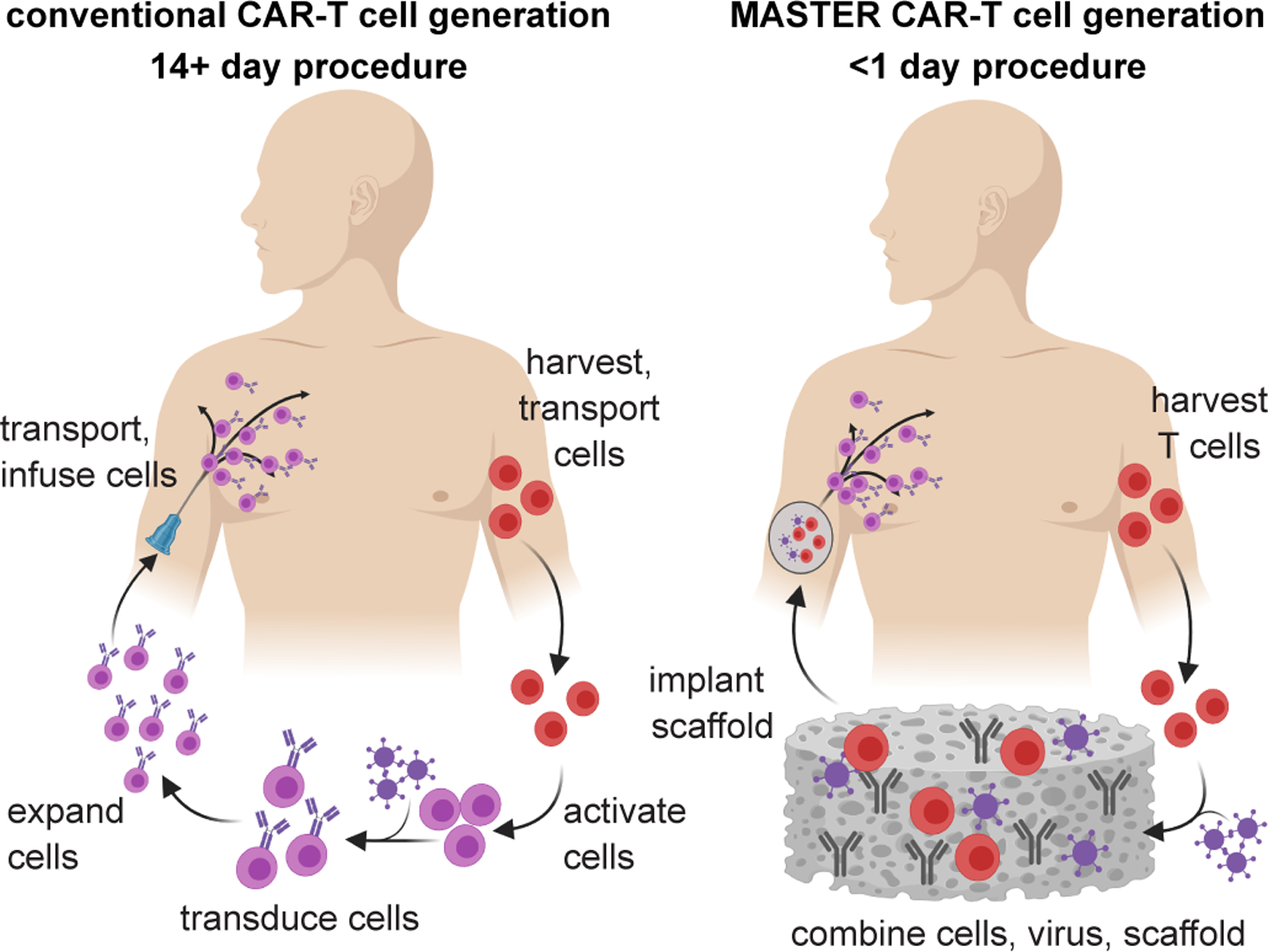
Schematic showing conventional CAR-T cell therapy (left, 2–4 weeks process) compared to rapid MASTER-mediated CAR T cell generation and therapy (right, <1 day process).
Efficient T cells activation and transduction is crucial for CAR-T cell manufacturing. To promote T cell activation within the scaffold cyclooctyne-conjugated anti-CD3 and anti-CD28 antibodies were immobilized on azide-modified alginate33–36 through mild and efficient copper-free click chemistry (Supplementary Fig. 1). To ensure that T cells loaded in MASTER receive the appropriate activation cues necessary for retroviral-mediated gene transfer, we tested escalating doses of agonistic anti-CD3 and anti-CD28 antibodies (0, 0.5, 1, 2, or 4 μg at 1:1 w/w per mg alginate) conjugated to MASTER. Human peripheral blood mononuclear cells (PBMCs) from three different donors were seeded in MASTER and analyzed after 18 hours for the expression of the early T-cell activation marker CD69. As shown in Fig. 1G, cells seeded on MASTER demonstrated a ~10-fold increase in CD69 expression as compared to cells seeded on alginate scaffolds without antibodies. Highest CD69 expression was achieved at an antibody concentration of 1 µg/mg alginate, and this density was used for further studies. Although αCD3 and αCD28 are expected to be randomly conjugated to the scaffolds, T cell activation as measured by CD69 expression was broadly similar after incubation with soluble antibodies, cyclooctyne-conjugated antibodies, or MASTER, suggesting the functionality of antibodies remains unaltered after conjugation with scaffold (Supplementary Fig. 2). To test MASTER-mediated simultaneous activation and transduction of non-activated T cells, naive PBMCs and GFP-encoding retrovirus were added to MASTER (Fig. 1H) and to scaffold controls lacking antibodies and the resulting CAR-T cell populations compared to those produced using conventional spinoculation (centrifugation onto retronectin-coated plates)37,38. After 72 hours of incubation, GFP expression was barely detected when non-activated PBMCs and virus were incubated on blank scaffold or spinoculated (<0.4%). In contrast, incubation of non-activated PBMCs on MASTER resulted in 20% transduction of the T cells (Fig. 1I), indicating that MASTER functions as a platform for both T-cell activation and static transduction via retroviral gene transfer.
To activate and expand T cells within the scaffold, the cytokine IL-2 was physically encapsulated in the scaffold34,39. The physical encapsulation enabled sustained release of IL-2 over five days while maintaining bioactivity of released IL-2 as assessed by proliferation of IL-2-dependent human T cells (Extended Fig. 3). Although IL-2 plays a crucial role in the proliferation of lymphocytes, transduction was found to be independent of the presence of IL-2. (Extended Fig. 4).
Next, we assessed the functionality of CAR-T cells generated within MASTER using retrovirus encoding a CD19-specific CAR (CD19.CAR)40. PBMCs and CD19.CAR gamma retroviral particles were loaded on MASTER and incubated for 3 or 10 days. As shown in Fig. 2A, CAR expression was detected in 22% ± 1% T cells at day 3 and remained stable at day 10 (Supplementary Fig. 3). Transduction occurred predominantly in T cells and the final composition of cells was predominantly T cell enriched (Fig. 2B). This selectivity likely arises because retroviruses transduce actively proliferating cells and only T cells upon activation meet this criterion. Since T cell phenotype is an important determinant for successful engraftment and persistence of CAR-T cells41, MASTER-generated CAR-T cells were analyzed for phenotypic composition and compared to CAR-T cells generated using the conventional clinical procedure of spinoculation of activated T cells and retrovirus on retronectin-coated plates42,43. Both MASTER generated and spinoculation-generated CAR-T cells contained similar amounts of CD4+ and CD8+ T cells (Fig. 2C). However, MASTER-generated CAR-T cells contained a higher percentage of CCR7+CD45RA+ central memory T cells (14.8% vs 2.57%), CCR7+CD45RA+ stem cell-like T cells (21.5% vs 9.8%) and CCR7+CD62L+ T cells with lymphoid homing capacity (13.1% vs 2.98%) than spinoculation-generated CAR-T cells (Fig. 2D, Supplementary Fig. 4). Characterization for expression of exhaustion markers, PD-1 and LAG3 on day 12 post-transduction showed less than 1% of CAR-T cells generated by either method were PD-1+LAG3+ (Fig. 2E). In addition, all three cell types (non-transduced, MASTER, and conventionally produced CAR-T cells) showed comparable proliferative capacity in response to CD19+ tumor cells (Fig. 2F).
Figure 2. MASTER promotes activation and retrovirus-mediated transduction of primary human T cells.
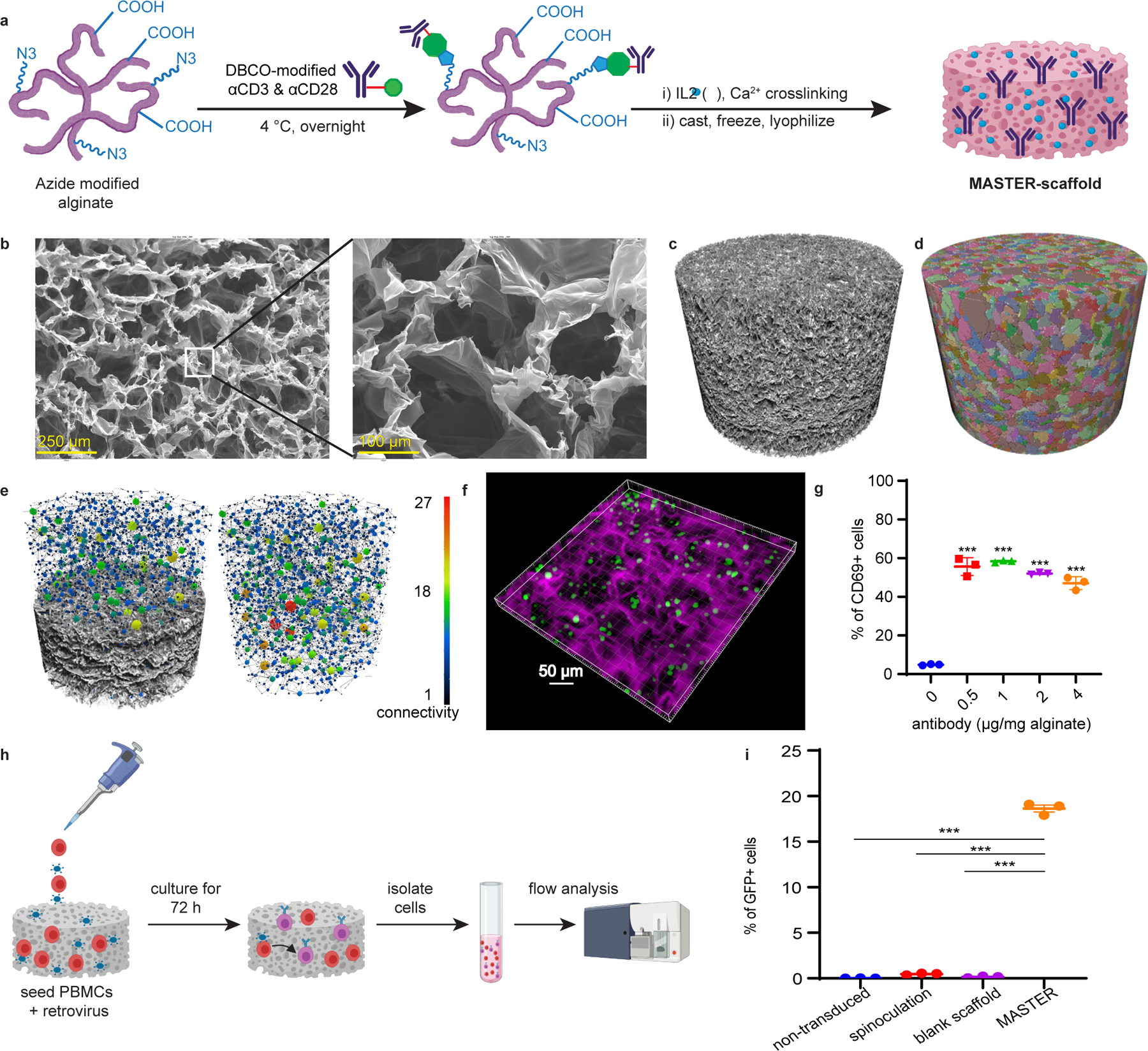
A) Schematic for synthesis of MASTER. B) SEM image of MASTER showing homogenous macroporous structure throughout the scaffold. C) Xray-CT volume of MASTER. D) Individual pores were isolated and color labeled so that similar pore volumes have similar colors. E) Isolated pores can be analyzed individually to calculate porosity (75.8%) as well as to analyze connectivity between the pores. Connectivity maps display individual pore as spheres with lines signifying connections to neighbors. Sphere size and coloring signify connectivity. A partial cut into sample showing the connectivity (left) and full the connectivity in sample (right). F) 3D confocal fluorescence micrograph of GFP+ T cells in AlexaFluor-647 labeled MASTER, depicting cell distribution along the pores. G) Quantification of T cells expressing CD69, an early T cell activation marker. MASTER with increasing amounts of anti-CD3 and anti-CD28 antibodies were seeded with PBMCs and analyzed for CD69+ cells after 24 hours by flow cytometry. (***p<0.001, one-way ANOVA with Tukey’s correction). Data represent mean ± SD of n=3 biologically independent samples H) Overview of the process of MASTER-mediated T cell transduction in vitro. PBMCs and gamma retroviral particles are seeded on MASTER and incubated at 37°C. After seventy-two hours, cells are collected from the scaffold and analyzed by flow cytometry. I) FACS quantification of GFP+ cells (***p<0.001, one-way ANOVA with Tukey’s correction). Data represent mean ± SEM of n=3 biologically independent samples.
To evaluate anti-tumor effects in vitro, control non-transduced T cells and CD19.CAR-T cells generated either by MASTER or by spinoculation were co-cultured with CD19+ target cells (Daudi) and CD19− target cells (U937) at 1:5 effector-to-target ratio (E:T). While non-transduced cells did not eliminate either of the tumor cells, both MASTER and spinoculation-generated CD19.CAR-T cells eliminated CD19+ cells, but not CD19− cells (Fig. 2G and Supplementary Fig. 5). In these co-culture experiments, CD19.CAR-T cells released IL-2 and interferon IFN-γ in response to CD19+ cells (Fig. 2H, I). Taken together, these results demonstrate that MASTER generates highly functional CAR-T cells with improved differentiation phenotypes in comparison to conventionally produced CAR-T cells.
We next evaluated whether MASTER releases resident T cells. MASTER with or without encapsulated IL-2 was seeded with PBMCs (1 x 106) and placed into transwell inserts (pore size 40 µm). At defined time points, each scaffold was transferred to a new well and the cells released into the bottom chamber were counted. MASTER efficiently released cells over five days (Extended Fig. 5) likely owing to inherent migratory properties of activated T cells and the well-connected macroporous structure of the scaffold.
Bioinstructive Scaffolds Serve as Biocompatible CAR-T Cell Factories In Vivo.
We evaluated the biocompatibility of MASTER in immunocompetent mice at four weeks following implantation. Histopathological evaluation of major organs showed no deleterious impact (Extended Fig. 6). At the implant site, only a thin layer of fibrous connective tissue and few inflammatory cells were observed around the scaffold. No evidence of toxicity was observed in skin-associated skeletal muscle, adnexa or epidermis or in any of the other tissues examined. To further confirm biocompatibility, we did a thorough blood biochemical analyses which showed no significant changes were induced by scaffold implantation (Supplementary Fig. 6).
To test whether surrounding host cells might be transduced upon implantation, we designed an in vitro transwell experiment wherein MASTER is placed at the top of the transwell and fibroblast cells are seeded in the bottom well. When MASTER was seeded with virus alone, ~30% GFP expression was observed in the fibroblasts, indicating virus leakage from the scaffold in the absence of co-seeded PBMCs. In contrast, when MASTER was co-seeded with both PBMCs and virus, no transduction was observed in the fibroblast cells, suggesting virus encounters and transduces the cells at its closest proximity (Extended Fig. 7) and providing indirect proof that MASTER will not transduce host cells upon implantation. To evaluate whether host cells could infiltrate the scaffold and be transduced within the scaffold, MASTER scaffolds carrying GFP-encoding retrovirus were implanted into NSG mice engrafted with human PBMCs (Extended Fig. 8). MASTER was predominantly infiltrated by mouse myeloid lineage cells, with small percentages of human T cells (<5%) and NK cells (<2%) and mouse fibroblast cells (<1%) (Extended Fig. 8D–F). Importantly, we observed no transduced cells among those infiltrating the scaffold, circulating in blood, or in the skin surrounding the scaffold (Extended Fig. 8G), suggesting that host cells do not appreciably get transduced by resident virus, likely due to the virus’ short stability half-life of 4 to 6 hours48. Although infiltrating cells are not transduced by resident virus, the possibility of infiltrating CD4+ T cells converting into Tregs44 due to constant stimulation remains and may require further exploration.
Motivated by the in vivo biocompatibility of MASTER and its ability to function as an all-in-one platform for T-cell activation, transduction, expansion, and release, we tested the antitumor activity of MASTER-produced CAR-T cells in an in vivo xenograft tumor model. MASTER was seeded with PBMCs and CD19.CAR-encoding retroviral particles, incubated for one hour, and subcutaneously implanted in NSG mice engrafted with Ffluc-labeled Daudi cells. Tumor-bearing mice i.v. infused with conventionally generated CAR-T cells at a clinically relevant dose45 served as positive controls (Fig. 3A). Mice subcutaneously implanted with MASTER seeded with only PBMCs or infused i.v. with non-transduced cells served as negative controls. Tumors grew rapidly in mice treated with control non-transduced cells, while MASTER-produced CAR-T cells and conventional i.v. infused CAR-T cells equally controlled tumor progression up to day 45, without significant changes in body weight (Fig. 3B–D, Supplementary Fig. 7). At 100 days, i.v infusion of 4 million conventionally produced CAR-T cells led to 16.6% tumor-free survival. In contrast, implantation of MASTER with 2 million PBMCs increased tumor-free survival to 50% (p=0.15, Fig. 3E).
Figure 3. MASTER-mediated gene transfer generates highly functional CAR-T cells.
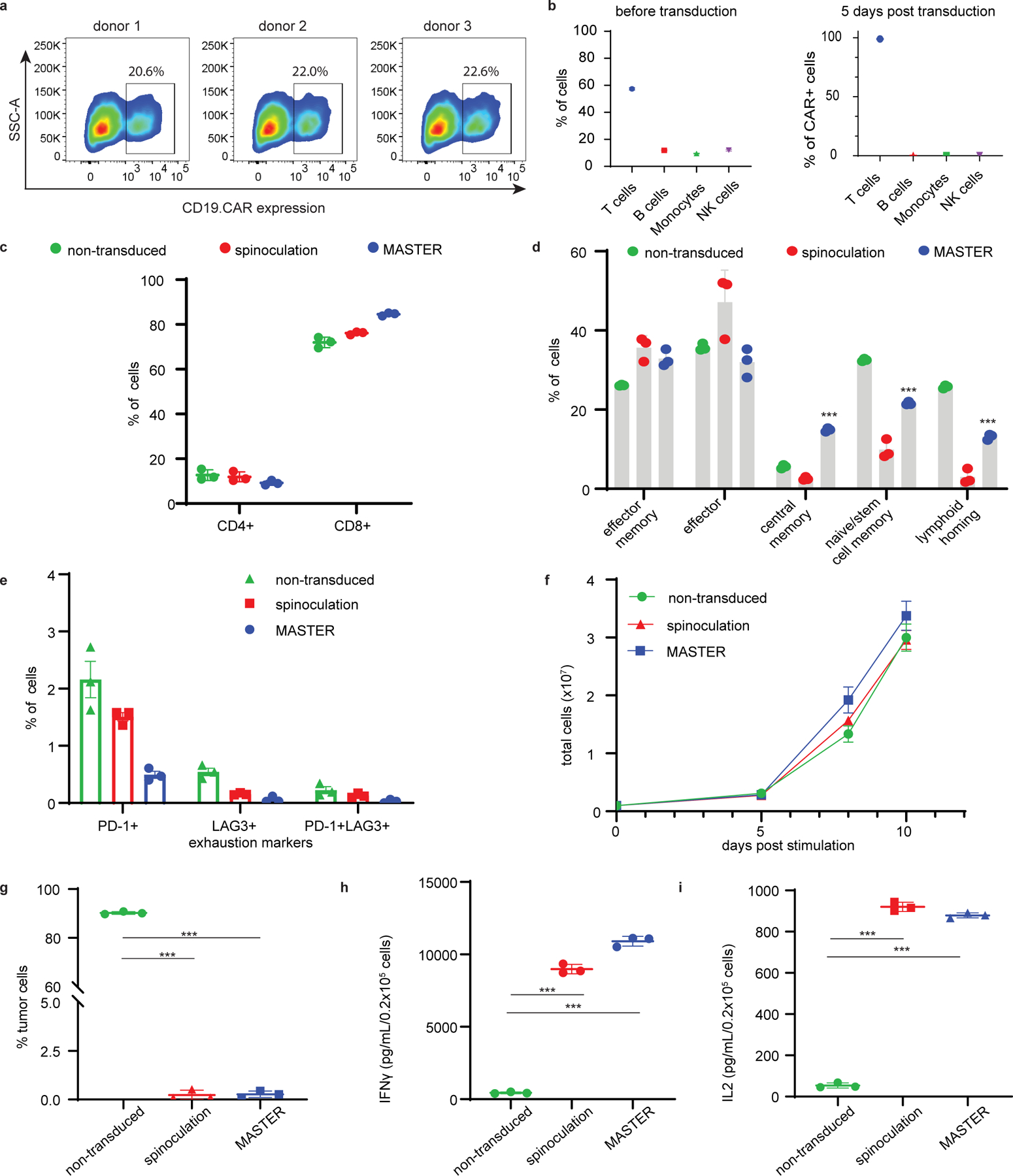
A) Flow cytometric plots depicting CD19.CAR expression in T cells transduced on MASTER. B) MASTER mediated retroviral transduction of PBMCs results in T cell enriched cell population. PBMC fractions before transduction (left) and PBMC fraction 5 days post transduction (right). C,D) Immunophenotypic composition of CAR-T cells obtained via MASTER-mediated transduction of PBMCs or by conventional spinoculation of activated T cells at day 12 of culture. Non-transduced cells were used as control. Analysis was performed gating on CAR-expressing T cells except for non-transduced cells. E) FACS quantification of CAR+ cells expressing exhaustion markers PD-1 and/or LAG-3. F) Ex vivo expansion of non-transduced T cells or T cells transduced on MASTER or by spinoculation. G) Percentage of viable CD19+ Daudi cells when co-cultured with non-transduced cells, CAR-T cells obtained by MASTER or by spinoculation. Tumor cells and CAR-T cells were plated at 1:5 effector to target ratio. T cells and tumor cells were quantified by flow cytometry at day 5 of co-culture. H, I) IFN-γ and IL-2 released into the co-culture supernatant by MASTER-generated, spinoculation-generated CAR-T cells and non-transduced cells after 24 hours as assessed by ELISA. ***P < 0.001; one-way ANOVA with Tukey correction. All data represented as the mean ± SD from three experiments, each derived from a different PBMC donor.
We also examined the in vivo persistence of CAR-T cells. Blood from mice treated with therapeutic doses of CAR T cells or MASTER (Fig. 3) was sampled on day 14 and 22. In addition, when control mice reached the humane endpoint (day 32), 3 mice from each of the CAR-T treated groups were euthanized, and blood, bone marrow (BM) and spleen were collected. Cell suspensions were analyzed for human CD45+CD3+CAR+ cells (Supplementary Fig. 8). Remarkably, MASTER-generated CAR-T cells had nearly 30-fold higher absolute counts in the peripheral blood at 22 days (Fig. 3F) and significantly increased counts in the bone marrow and spleen at 32 days compared to conventional CAR-T cells (Fig. 3G&H, Supplementary Fig. 9). CAR+ cells isolated from blood and bone marrow were further analyzed for the expression of memory and exhaustion markers. MASTER-generated and conventional CAR-T cells were phenotypically similar; with MASTER exhibiting significantly higher number of effector memory T cells (TEM: CCR7−CD45RA−) in both blood and bone marrow as compared to conventional CAR T cells (Fig. 3I, Supplementary Fig. 10). In both blood and bone marrow, the absolute number of PD-1+LAG3+CAR+ cells were similar between the two groups (Fig. 3I, Extended Fig. 9). These data indicate that MASTER generated CAR-T cells have increased population of effector memory cells and persist better after tumor clearance.
To increase our ability to detect small differences in efficacy between these two systems, we performed a stress test46 in which T cell dose is purposefully lowered to levels where CAR T cell therapy begins to fail. Under stressed dose conditions MASTER was more efficacious in controlling tumor growth and significantly increased survivability compared to i.v. infusion of CAR T cells (Fig. 4).
Figure 4: Subcutaneously implanted MASTER generates and release fully functional CAR-T cells in a xenograft model of lymphoma.
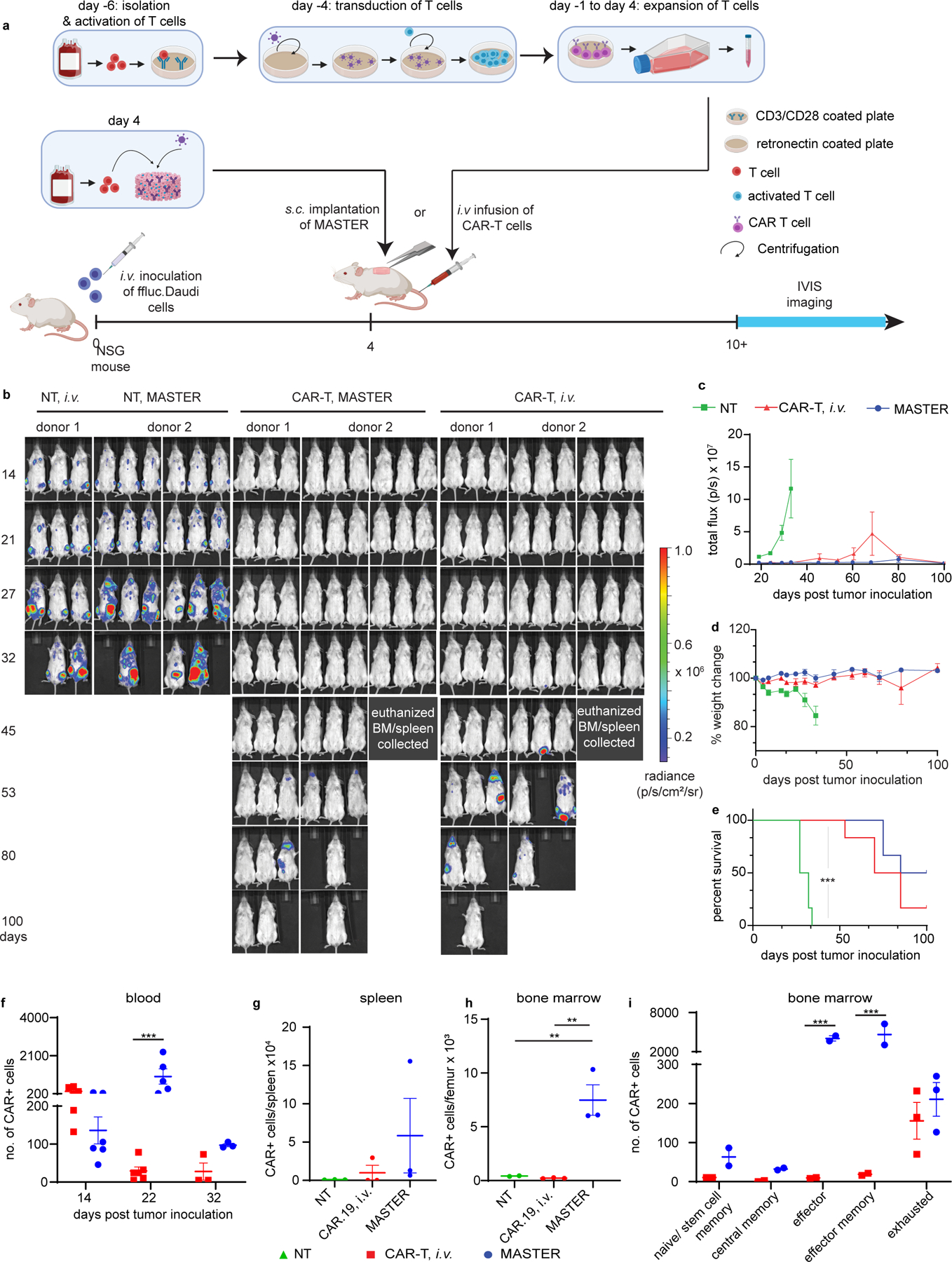
A) Experimental timeline of the lymphoma xenograft model in NSG mice engrafted with FFLuc- labeled CD19+ human Daudi tumor cells. B) In vivo tumor bioluminescence imaging (BLI) of NSG mice treated with MASTER, conventional CAR-T cells or control non-transduced (NT) cells. C) Kinetics of tumor growth measured by quantification of BLI. D) Percentage change in body weight (BW) of treated mice. Data in c-d represent mean ± SEM of nine biologically independent animals examined over two independent experiments. E) Survival of mice shown as Kaplan- Meier curves. Six mice per treatment group are shown. ***p<0.001, Log-rank (Mantel-Cox) test, Gehan-Breslow-Wilcoxon test. F) Flow cytometric quantification of CD3+CAR+ cells in peripheral blood of mice described in (A). ***p<0.001, two-way ANOVA with Sidak’s multiple comparison test G, H) Quantification of CD3+CAR+ cells in bone marrow and spleen of mice euthanized on day 32 in the model described in (A) determined by flow cytometry **p<0.01, one-way ANOVA with Tukey’s correction Data in f-h represent mean ± SEM of three biologically independent samples I) Analysis of memory and exhaustion markers in CAR T cells isolated from the bone marrow of mice treated as described in (A). Data in i represent mean ± SEM of two biologically independent samples.
The better expansion and persistence of MASTER-generated CAR-T cells were even more prominent in tumor rechallenge study when mice received an additional dose of tumor cells (Fig. 5A) at 29 days following initial tumor inoculation. MASTER-treated mice were tumor free up to 40 days post rechallenge, while tumor growth was evident within 2 weeks after the rechallenge in mice i.v infused with conventional CAR T cells (Fig. 5A–C). The enhanced persistence of MASTER generated CAR T cells could be attributed to two factors. First, MASTER provides a “depot” for cell generation, wherein the scaffold continually releases cells into rather than providing a bolus administration as with intravascular infusion. Second, since MASTER-generated CAR T cells bypass ex vivo expansion, the resultant cell population has significantly more memory T cells16. The enhanced generation of these self-renewing T cell populations could further contribute to the improved persistence of cells47. Although MASTER generated CAR T cells have higher persistence and superior antitumor activity, further development of innovative strategies to overcome resistance mechanisms to CAR T cell therapy such as antigen loss48,49 will be necessary to expand the clinical use of CAR-T cell therapies.
Figure 5: Subcutaneously implanted MASTER outperforms i.v. administered CAR-T cells in a rechallenge model of lymphoma.
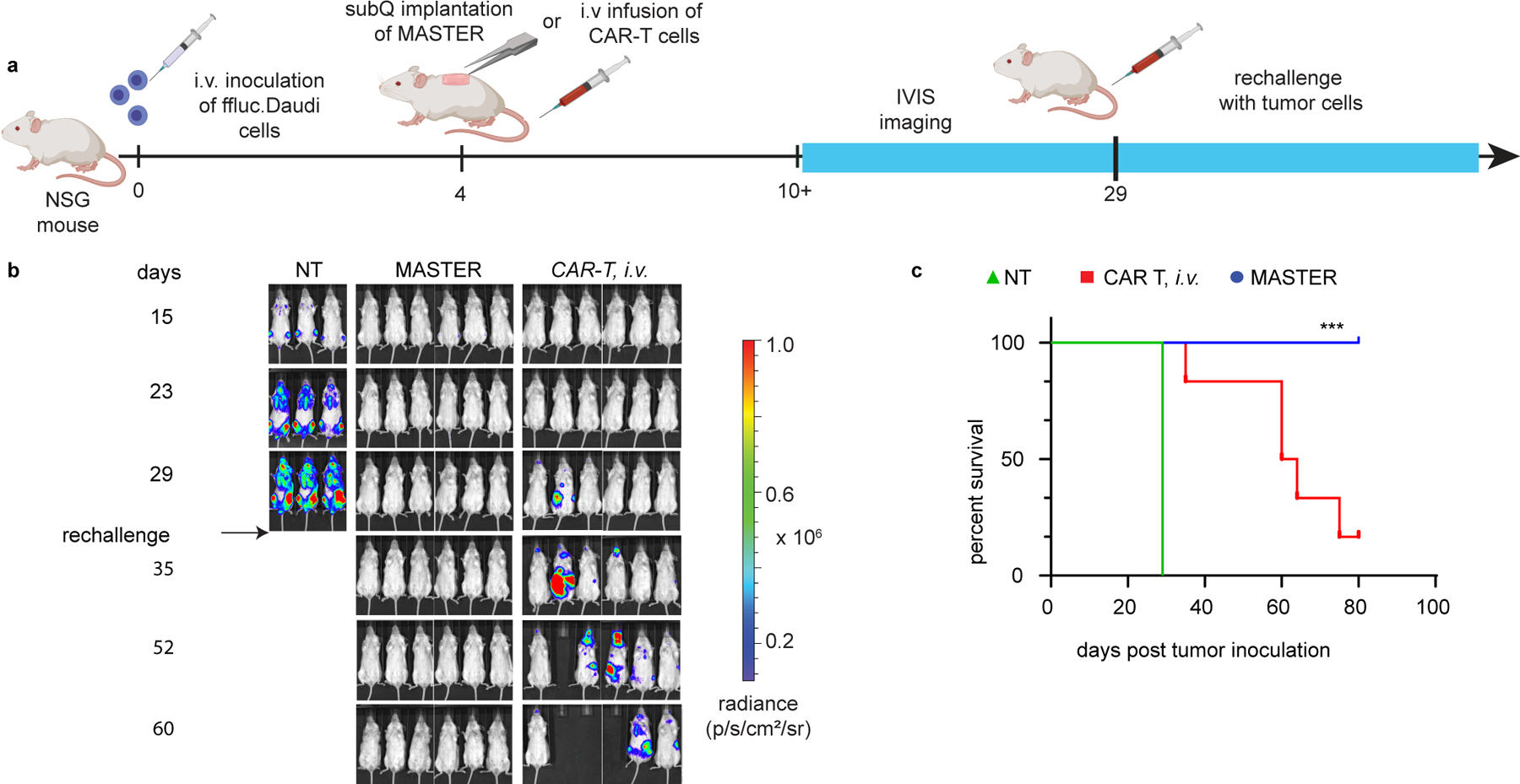
A) Experimental timeline of the lymphoma xenograft model in NSG mice engrafted and rechallenged with FFLuc− labeled CD19+ human Daudi tumor cells. B) In vivo tumor bioluminescence imaging of NSG mice treated with MASTER, conventional CAR-T cells or control non-transduced (NT) cells. C) Survival of mice shown as Kaplan-Meier curves. Six mice per treatment group are shown. ***p<0.001 w.r.t CAR T, MASTER, Log-rank (Mantel-Cox) test, Gehan-Breslow-Wilcoxon test.
Finally, we pushed the limit of MASTER and investigated its efficacy in a more established tumor model of lymphoma50. NSG mice were injected with Ffluc-labeled Daudi cells. On day 9, when tumor engraftment was visible by IVIS imaging, mice were either implanted with MASTER or i.v. infused with conventional CAR T cells. Under these conditions, both MASTER and conventional CAR T cells promoted equal tumor control demonstrating MASTER is also effective in controlling high tumor burden (Fig. 6). Taken together, these results demonstrate that CAR-T cells produced using MASTER were equally functional to conventional CAR-T cells in controlling tumor growth, but had better expansion and persistence. Importantly, MASTER drastically reduced the time, complexity, and cost of CAR-T cell production.
Figure 6: MASTER and conventional CAR T cells exhibit equal anti-tumor efficacy against established tumor in vivo.
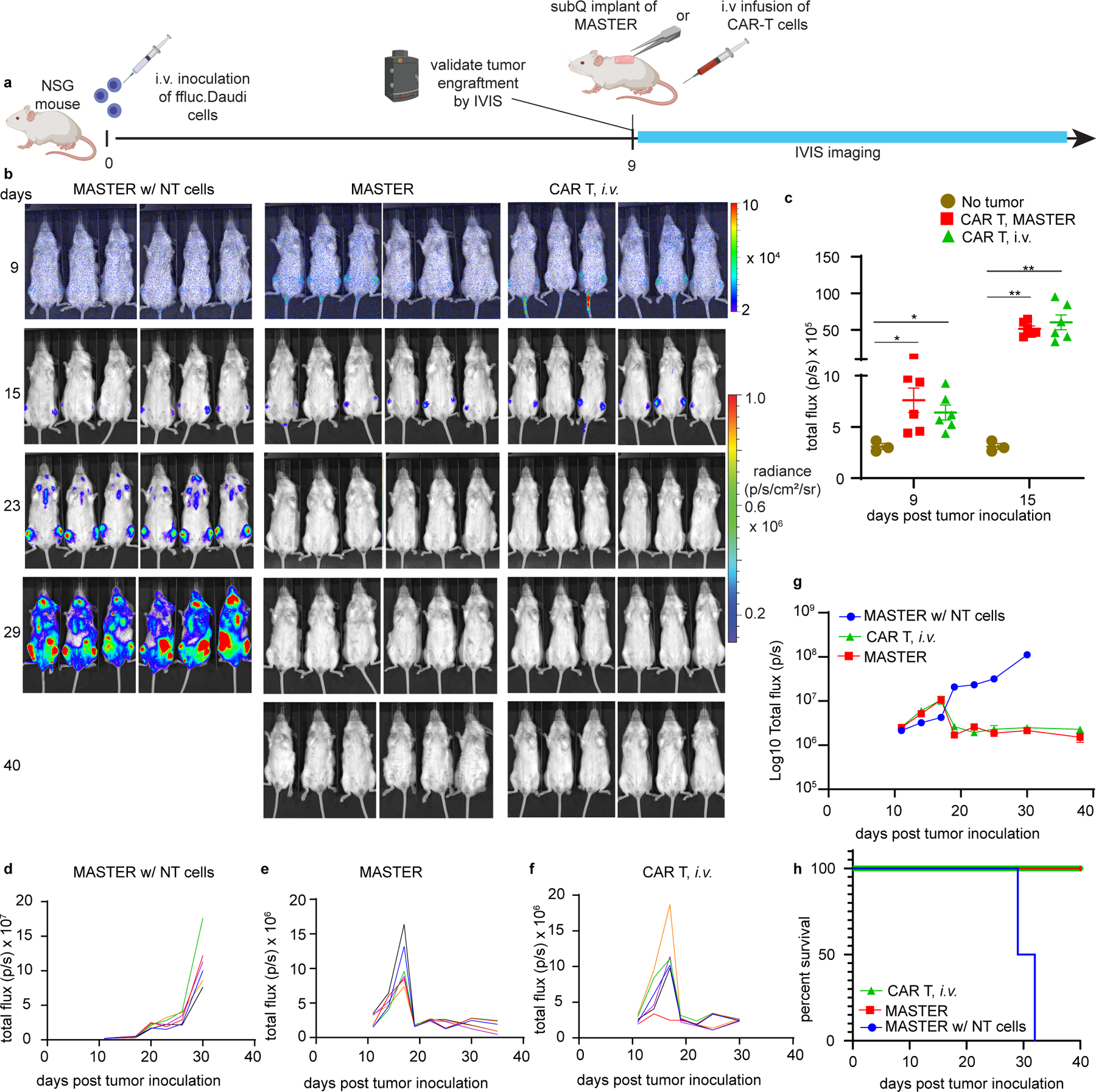
A) Experimental timeline of the lymphoma xenograft model in NSG mice engrafted with FFLuc-labeled CD19+ human Daudi tumor cells. B) In vivo tumor bioluminescence imaging of NSG mice treated with MASTER, conventional CAR-T cells or control non-transduced (NT) cells. Cells were normalized to transduction efficiency and mice were treated with equivalent number of CAR T cells (2 x 106 CAR T cells/mouse). C) Engraftment of the established tumor as confirmed on day 9 and day 15 by comparing luminescent intensity of healthy mice (background, n = 3 biologically independent animals) and tumor bearing mice (n = 6 biologically independent animals). (*p<0.05, **p<0.01, two-tailed unpaired t test). Data represent mean ± SEM D-F) Kinetics of tumor growth measured by quantification of luminescence. G) Combine tumor growth kinetics shown on logarithmic plot (n = 6 biologically independent animals). Data represent mean ± SEM H) Survival of mice shown as Kaplan- Meier curves. Six mice per treatment group are shown.
Conclusion
In summary, we generated a multifunctional scaffold that brings together the key aspects of CAR-T cell manufacturing and delivery under one platform, reducing the entire process of CAR-T cell manufacturing to a single day. The scaffold, which uses FDA-approved, nonimmunogenic materials, creates a local, nurturing niche for αCD3 and αCD28-mediated cell activation and interleukin-mediated proliferation. Scaffold macroporosity facilitates homogeneous cell distribution and creates an interface for interaction between viral particles and T cells, preventing viral shedding and off-target gene transfer that may result from systemic approaches23. The scaffolds then enable in vivo release of fully functional reprogrammed CAR-T cells. MASTER provides a modular platform technology that can be adapted to reprogram other immune cells or to deliver immunomodulatory factors to support cell function synergistically. Beyond its potential for cancer therapy, the MASTER technology may inspire new treatments harnessing the capacity for reprogramming and release of therapeutic cells.
Methods
DBCO-modification of antibodies:
A 10-fold molar excess of Dibenzocyclooctyne-PEG4-N-hydroxysuccinimidyl ester (NHS-PEG4-DBCO, A134–10, Click Chemistry Tools, USA) was added to anti-CD3 (Bio X Cell BE0001–2, clone OKT-3, 2 μg/μL) or anti-CD28 antibodies (Bio X Cell BE0291, clone CD28.2, 2 μg/μL) and incubated at room temperature for 1 h. Then the solution was purified by Amicon centrifugation (MWCO 10 kDa), 10000 g, 10 mins until the flow through was free from NHS-PEG4-DBCO (measured by characteristic absorbance of DBCO moiety at 309 nm using Nanodrop). The degree of DBCO incorporation (i.e. the number of DBCO per antibody) was determined from the absorbance scan of the purified conjugate (235‐400 nm) using the following equation.
(Molarity of DBCO) / (Molarity of antibody) = (A309 DBCO x ε280 Ab) / (ε309 DBCO x A280c Ab)
A309 DBCO = DBCO-Ab conjugate’s absorbance at 309 nm
ε280 Ab = 210,000M−1cm−1
A280C Ab = conjugate’s corrected absorbance at 280 nm = A280 - (A309 x CF DBCO)
ε309 DBCO = 12000M-1cm-1
A280Ab = DBCO-Ab conjugate’s absorbance at 280 nm
CF DBCO = DBCO correction factor at 280 nm = 1.089
Preparation and fabrication of MASTER:
Azide-modified alginate was prepared as previously reported1,2. To quantify the number of azides per alginate strand, azide alginate was incubated with excess DBCO-amine. The decrease in characteristic absorbance of DBCO at 309 nm was used to calculate the number of azides per alginate strand2. To prepare MASTER, a 2% (w/v) solution of azide alginate in molecular biology grade water was incubated with DBCO-modified anti-CD3 and anti-CD28 antibody (1 µg Ab/mg alginate, and 0.2 µmol DBCO per mol of azide) at 4°C overnight. Recombinant human IL2 (PeproTech), at a concentration of 0.2 µg/mg alginate was next added, and the solution was stirred for 15 mins. Finally, the resulting solution was vigorously stirred with equal volume of 0.4% calcium gluconate for 15 mins, cast in 24 well plates for in vitro studies (1 mL/well) or 48 well plates for in vivo studies (300 µL/well), frozen at −20 °C overnight and lyophilized. MASTER was stored at 4 °C before use for in vitro or in vivo study.
Scanning electron microscopy (SEM):
Dry macroporous scaffold was cut with a sharp razor blade, coated with 70 nm AuPd (Au: 60%, Pd: 40%) for 10 minutes at 7 nm/min and analyzed on Hitachi S-3200N variable pressure SEM using Oxford Aztec software.
X-ray CT:
The X-ray computed tomography (CT) scans were performed in a Xradia Versa 510 using Zeiss Scout and Scan v13 with the acquisition parameters below.
| Scanning parameters | |
|---|---|
| Projections | 1600 |
| Filter | None |
| Voltage | 40 kV |
| Current | 74 µA |
| Pixel size | 2.6 µm |
| Exposure | 8 seconds |
|
| |
| Optical magnification | 4X |
A cylindrical volume of 2.50x2mm was scanned in each sample. These volumes were used to calculate the porosity of the samples and the dimensions of the pores. A smaller sub-volume of 10x1mm was extracted to calculate and show connectivity between the cells. This was done to reduce the size of calculation and enhance the visualization of the results.
The CT data was analyzed using the software Dragonfly [Dragonfly 2020.1 [Computer software]. Object Research Systems (ORS) Inc, Montreal, Canada, 2018; software available at http://www.theobjects.com/dragonfly.]. To segment the samples a training dataset was created manually for each sample using histogram thresholding and masking techniques. Once the training data was created it was used to train a deep learning image segmentation model called U-net. The resulting model was then used to segment the full samples into scaffolds and porosity. To calculate the connectivity between the pores an open-source package (openPNM3 2.8) was used.
Confocal Microscopy:
AF647 DBCO (click chemistry tools) was click conjugated to azide modified alginate. Excess unbound dye was removed by dialysis against water for 1 day. AF647-labeled scaffolds were fabricated as described above. 1 x 106 GFP+ T cells were seeded on these scaffolds, incubated overnight at 37 °C and imaged using Zeiss LSM 880 confocal microscope using Zeiss Zen Black 2.3 software.
Cell lines:
Daudi cells and U937 cells were purchased from ATCC. Daudi cells were transduced with a retroviral vector encoding the Firefly-Luciferase gene (FFLuc)4,5. After transduction, cells were selected in puromycin (Sigma, St Louis, MO). All cells were maintained in RPMI 1640 (Gibco) supplemented with 10% FBS (Gibco), 2 mmol/L GlutaMax (Gibco) and penicillin (100 units/mL) and streptomycin (100 mg/mL; Gibco). All cells were maintained at 37 °C with 5% CO2.
Conventional CAR-T cell generation:
T cells expressing CAR were generated in accordance with standard operating procedures currently used to manufacture CAR-T cells for clinical use6 and for clinical trials at the University of North Carolina - Chapel Hill7,8. Peripheral blood mononuclear cells were isolated from human buffy coat fractions (Gulf Coast Regional Blood Center) using Lymphoprep medium (Accurate Chemical and Scientific Corporation) and activated on plates coated with 1 µg/mL anti-CD3 (Miltenyi Biotec 130–093-387, clone OKT-3) and anti-CD28 (BD Biosciences 555725, clone CD28.2) monoclonal antibodies. Retroviral supernatants used for the transduction were prepared as previously described4. To transduce human T cells, retrovirus with RD114 envelope was used. For murine cells transduction, retrovirus was generated using packaging vector encoding Eco envelope protein. Activated T cells were transduced with retroviral supernatants on retronectin-coated 24-well plates (Takara Bio, Inc.) 2 days after activation5. Medium containing retrovirus was spinoculated on retronectin coated plates for 90 mins at 1500 g, viral supernatant was removed and then activated T cells were spinoculated on retrovirus and retronectin coated plates for 10 mins at 1000 g. 72 h post transduction, cells were transferred and expanded in tissue culture flasks. Transduced T cells were expanded in Click’s Medium (Irvine Scientific) and RPMI-1640 (1:1 v/v) supplemented with 10% HyClone FBS (GE Healthcare), 2 mmol/L GlutaMax (Gibco) and penicillin (100 units/mL) and streptomycin (100 mg/mL; Gibco) with 10 ng/mL IL7 and 5 ng/mL of IL15 (PeproTech) for 10 to 14 days of culture before being used for in vitro or in vivo experiments.
MASTER-mediated generation of CAR-T cells:
Medium containing GFP encoded or human CD19.CAR encoded gamma retrovirus (RV) was concentrated 10-fold by Amicon centrifugation (MWCO 100 kDa, Millipore), 2500 g, 15–20 mins. Dry lyophilized MASTER scaffolds were transferred to non-tissue culture coated 24 well plates (Falcon), and 1 x 106 PBMCs isolated from human buffy coat fractions and concentrated RV at MOI 2 was pipetted onto each scaffold (total volume of virus and PBMC = 300 µl). Control scaffolds were seeded with 1 x 106 PBMCs suspended in cell culture medium. For in vitro studies only, the seeded scaffolds were incubated without any additional medium in the 5% CO2 incubator at 37°C for 1 h, then 1 ml of complete medium was added. Cells were isolated from scaffolds after 72 h by digesting with 0.25 M EDTA, washed twice with excess PBS and analyzed for GFP expression or CD19.CAR expression by flow cytometry. For in vivo studies, MASTER was seeded with PBMCs and CD19.CAR encoding retrovirus, incubated for ~1 h at 37°C until the media was completely absorbed by the scaffolds and implanted on the same day in the subcutaneous space of NSG mice.
Flow cytometry and antibodies
Monoclonal antibodies specific for human CD3 (APC-Cy7, 557832, SK7), CD4 (APC-Cy7, 561839, RPA-T4), CD8 (PerCP-Cy5.5, 565310, SK1), CD20 (FITC, 555622, 2H7), CD45RA (PE, 555489, HI100), CD62L (BV421, 563861, DREG-56), LAG3 (PE, 565617, T47–530), PD-1 (FITC, 561035, MIH4), TIM3 (BV421, 565563, 7D3), CD14 (PerCP, 340585, MφP9), CD56 (PE-Cy7, 557747, NCAM16.2), CD19 (APC, 555415, HIB19), and goat anti-mouse lg (APC, 550826, Poly1270) were purchased from BD Biosciences and used according to manufacturer’s instruction. Monoclonal antibodies specific for human CCR7 (FITC, FAB197F-100, 150503) was purchased from R&D Systems and used according to manufacturer’s instruction. Monoclonal antibodies specific for mouse CD45 (BV510, 103137, 30-F11), CD11b (PerCP-Cy5.5, 101228, M1/70), Ly6C (PE-Cy7, 128018, HK1.4) and CD140a (APC, 135907, APA5) were purchased from Biolegend and used according to manufacturer’s instruction. An anti-idiotype scFv monoclonal antibody was used to detect the expression of the CD19.CAR as previously described5. All samples were acquired on a BD LSRII using BD FACSDiva v8 software, and a minimum of 10,000 events were acquired per sample. Count bright absolute counting beads (C36950, thermo fisher) were used for calculating absolute number of cells. Samples were analyzed on FlowJo 10 (FlowJo LLC).
Cytokine production by CAR-T cells
CAR-T cells were cocultured with Daudi tumor cells at a 1:5 effector to target [E:T] ratio for 24 hours, and the culture supernatant was collected. IL-2 and IFN-gamma was quantified by ELISA as per manufacturer’s protocol (R&D Systems) and imaged on the Biotek Cytation 5 plate reader with Gen5 3.0 software.
In vitro cytotoxicity
Tumor cells (Daudi) were seeded at 1x105 cells per well in 24-well plates. CAR-T cells normalized for transduction efficiency were added at a 1:5 Effector : Target (E:T) ratio. On day 5 of coculture, cells were collected, and the frequency of T cells (CD3+) and residual tumors (CD20+) cells was measured by flow cytometry.
In vitro T cell release assay:
Scaffolds were seeded with 1x 106 PBMCs and placed on 40 µm cell mesh (Corning). The mesh was used as a transwell insert and placed in 6 well plate. The bottom of the well contained complete T cell medium. At definite time points, cells in the bottom chamber were counted and the scaffold with the insert was moved to a new well containing fresh media.
In vivo studies:
All procedures involving animals were done in compliance with North Carolina State University’s Institutional Animal Care and Use Committee and approved by North Carolina State University Institutional Animal Care and Use Committee. Ten- to twelve-week-old, female, immune-compromised NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) were infused with 1 x 106 FFluc expressing Daudi cells intravenously. For initial tumor regression and rechallenge study (Figure 3), two scaffolds —each loaded with 1 x 106 PBMCs and CD19.CAR encoded gamma retrovirus that transduces ~20% of the PBMCs and results into ~0.2 x 106 CAR T cells equivalent— was implanted in subcutaneous space of the tumor engrafted mice. Mice intravenously injected with either 2 x 106 non-transduced (NT) T cells or implanted with MASTER seeded with 1 x 106 PBMCs only (no retrovirus) were used as negative control. Mice infused with 2x106 CD19 CAR T cells (clinically relevant dose) generated by conventional method as described above were used as positive control. For stress test and established tumor model, cells were normalized to transduction efficiency and equivalent number of CAR T cells were compared between all groups. Treatment was started on day 4 post tumor cell infusion except for established tumor model where treatment started on day 9. Tumor burden was monitored using the Xenogen-IVIS Imaging System using Living Image 4.2 software. Mice were monitored for signs of discomfort and euthanized upon losing more than 15% of initial body weight, the development of hind-limb paresis, or reaching humane endpoints based on tumor burden. All procedures involving animals were done in compliance with the University’s Institutional Animal Care and Use Committee
To analyze CAR+ cells in blood, bone marrow or spleen, single cell suspensions from these organs were stained with anti-human CD45, anti-human CD3, and CAR.19 antibody and analyzed by flow cytometry. Single-cell suspensions of spleen were obtained by mincing the organ through a 70 µm cell mesh. Red blood cells were lysed by resuspending splenocytes in ACK lysis buffer (Lonza). Bone marrow single-cell suspensions were obtained by flushing the bones with a syringe containing PBS. The cell suspension was passed through a 70 µm cell mesh and red blood cells were lysed by resuspending cells in lysis buffer (BD Pharm Lyse). Similarly, blood collected via cheek bleed was incubated with lysis buffer (BD Pharm Lyse), washed with PBS and passed through 70 µm cell mesh before staining with antibodies. Failure to cheek bleed on day 22 resulted in exclusion of one mouse treated with MASTER scaffolds.
To analyze host cell infiltration into implanted scaffold, NSG mice were i.v inoculated with 10 x 106 hu-PBMCs via tail vein injection. Successful engraftment was validated by the presence of >10% human-CD45+CD3+ cells in blood. MASTER seeded with GFP encoded retrovirus was implanted in the subcutaneous space of space. Since the retrovirus used for all studies is pseudotyped with the Galv envelope, which predominantly transduces human cells, an additional group was implanted with ecotropic GFP encoded retrovirus that transduces mouse cells. Four days post implantation, scaffold was explanted, digested with 0.25M EDTA and isolated cells were analyzed by flow cytometry. Mice were also bled and skin surrounding the scaffold was isolated. Skin was digested with collagenase and cells were analyzed for GFP expression by flow cytometry.
Histological analysis:
12-week-old C57BL/6J mice (Jackson laboratory) were implanted with MASTER, MASTER seeded with mouse PBMCs (isolated from mouse spleen) and MASTER seeded with mouse PBMCs + GFP encoding ecotropic retrovirus (n=3). These mice were sacrificed four weeks after subcutaneous implantation, and the scaffold, surrounding skin and main organs (heart, liver, kidney, lung, and spleen) were excised, embedded in paraffin, sectioned and stained with hematoxylin and eosin (H&E) by UNC-CH histology core to evaluate inflammatory host response to the MASTER and its components. The sections were sent to a certified, veterinary pathologist who was blinded to the groups for evaluations
Statistical and Reproducibility.
All statistical analysis was done using two-tailed Student’s t-test, one way ANOVA or two-way ANOVA with Tukey post hoc analysis using GraphPad prism and noted in figures as *=p<0.05, **=p<0.01, ***=p<0.001. In vitro experiments were conducted at least two times and were reliably reproduced with similar effect sizes. In vivo studies in Fig 2 were performed with cells from multiple donors with similar effects. In vivo experiments other than those reported in figure 3 were not independently replicated.
Extended Data
Extended Data Fig. 1. Quantitative characterization of MASTER scaffold structure.
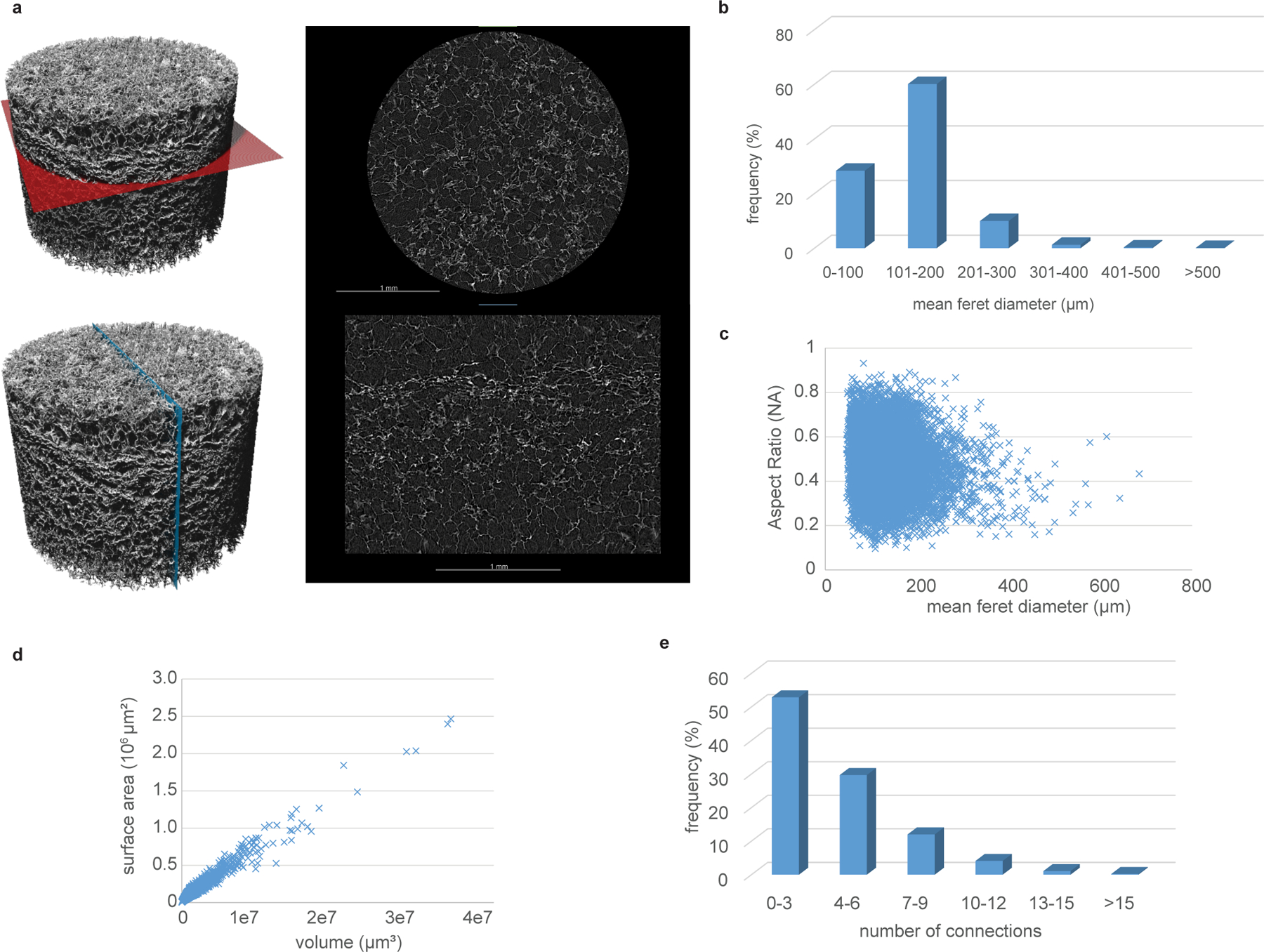
A) Scanned volume of MASTER (left) with a colored plane indicating the cross-section seen on the right. In the cross sections a brighter value indicates a higher density (scaffolds) and a darker value a lower density (air porosity). B) Relative frequency of pores of different dimensions. C) The aspect ratio of the pores showing most of the pores has an oblong shape. An aspect ratio of 1 corresponds to a sphere and close to 0 corresponds to a flat plane or stick. D) The surface area as a function of volume plotted. The total surface area inside of MASTER is roughly 810 mm2 E) Connectivity of sample showing most of the pores are connected to 0–3 other pores with a very few pores (around 6%) have more connections than 9.
Extended Data Fig. 2. Confocal images of CAR-T cells within MASTER scaffold.
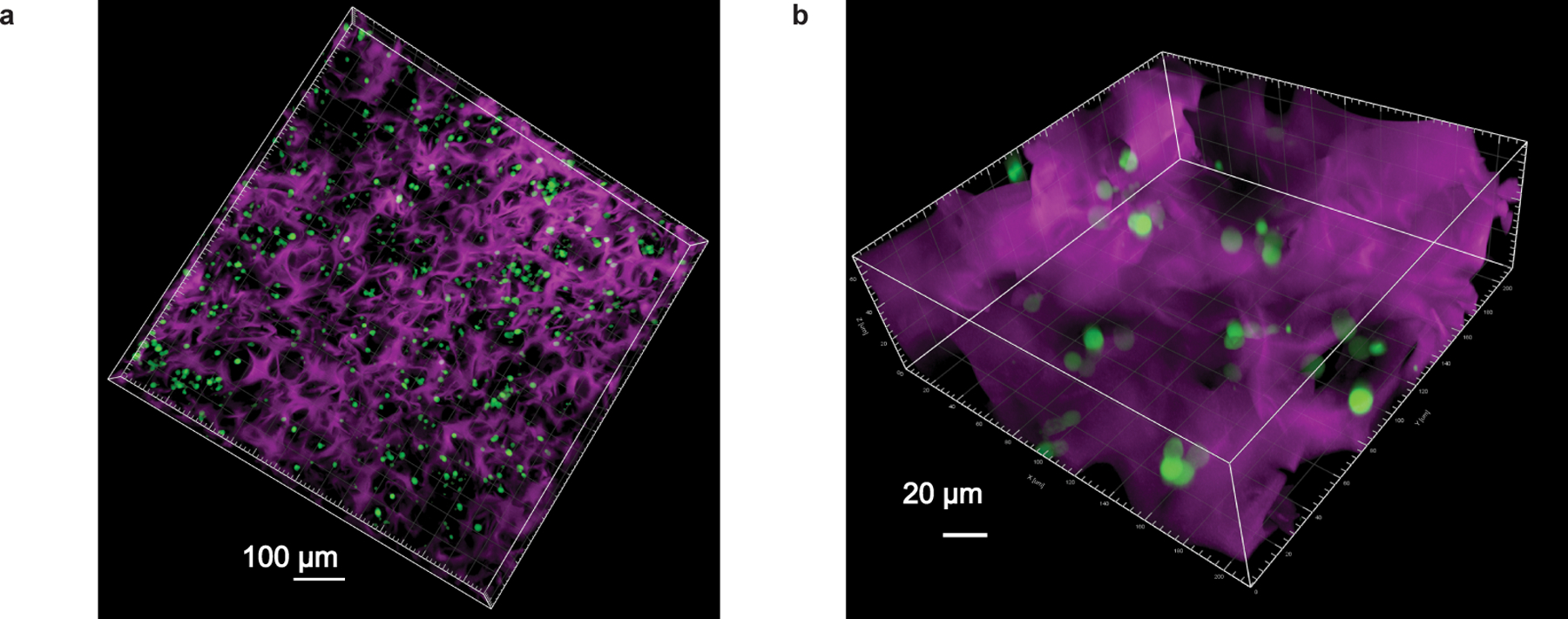
3D confocal micrograph showing distribution of GFP+ T cells in AF647 labeled MASTER at 10X (A) and 40X (B) magnification. This experiment was repeated twice independently with similar results.
Extended Data Fig. 3. IL-2 loaded onto MASTER released in a sustained manner over five days in vitro and retained its bioactivity.
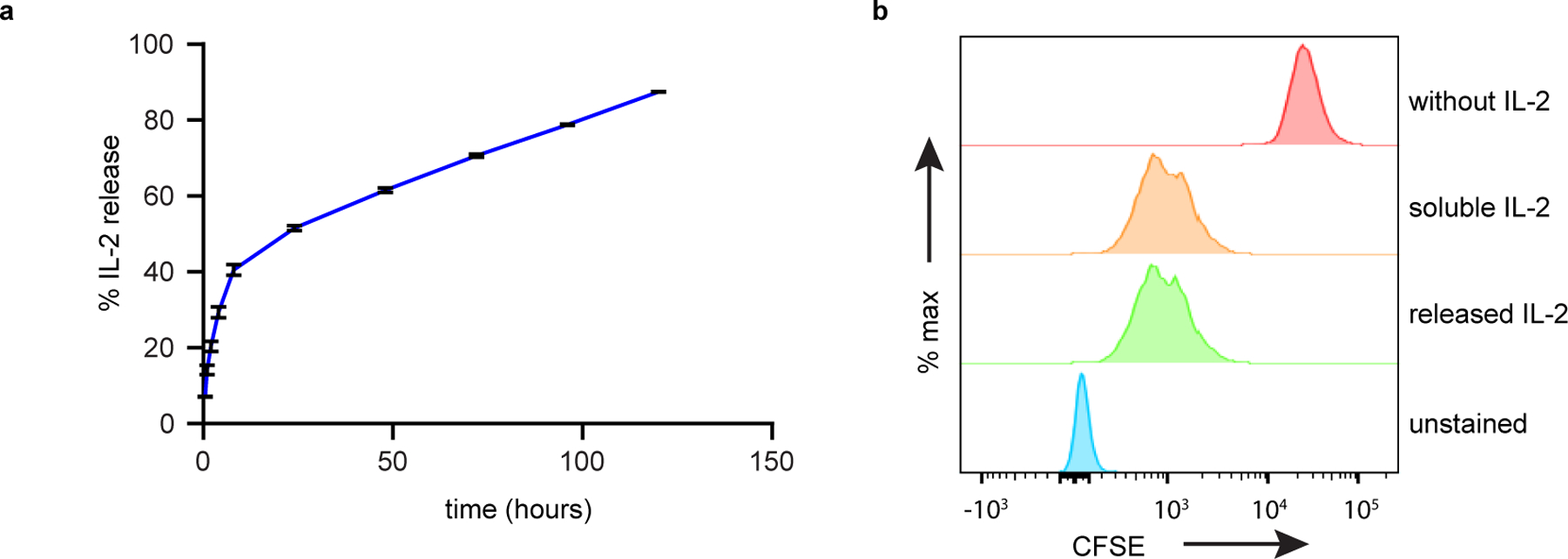
(A) Cumulative release of IL-2 from MASTER as quantified by ELISA assay. Data represent mean ± SD of three independent samples (B) Bioactivity of IL2 released at 24 hours as assessed by proliferation of CFSE stained T cells.
Extended Data Fig. 4. IL-2 promotes lymphocyte proliferation, but not transduction efficiency of MASTER.
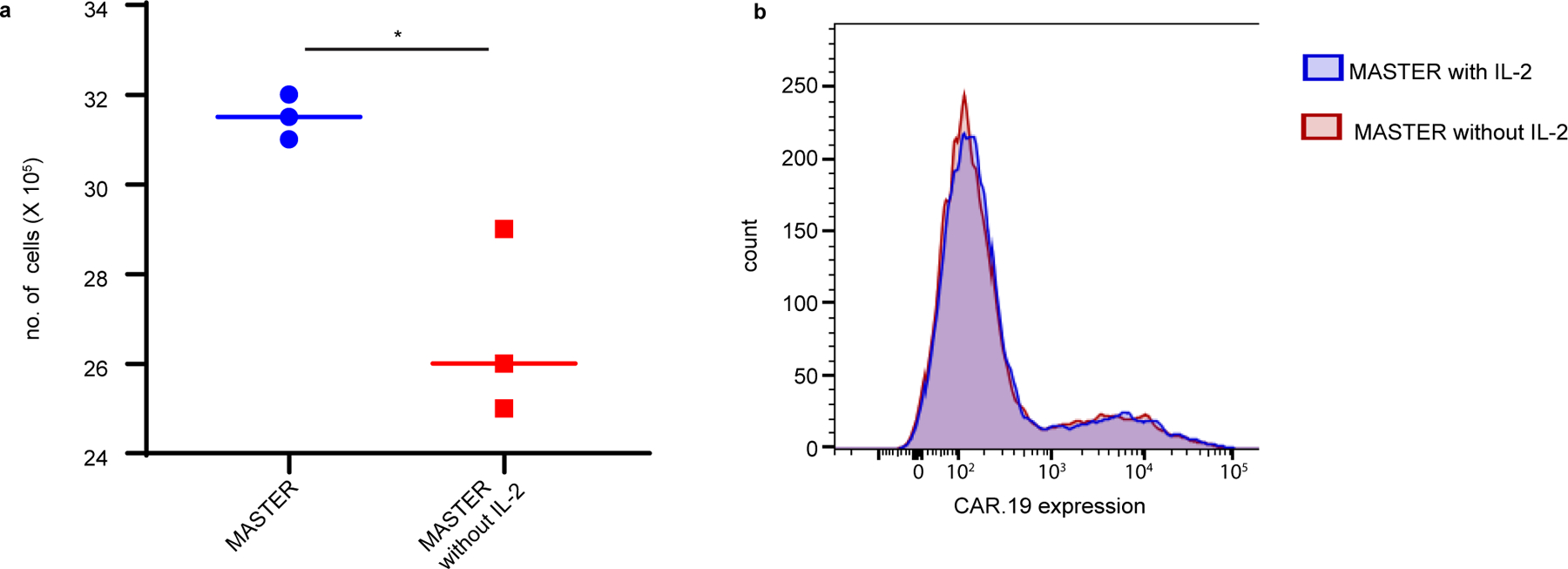
(A) MASTER and MASTER without IL-2 was seeded with PBMCs and virus and number of cells were counted 5 days post transduction. *p<0.05, two tailed unpaired t test, (B) CAR.19 expression in T cells 72 h post transduction. *p<0.05, two-tailed unpaired t test. Data represent median of n=3 biologically independent samples.
Extended Data Fig. 5. MASTER functions as an efficient T cell-release system.
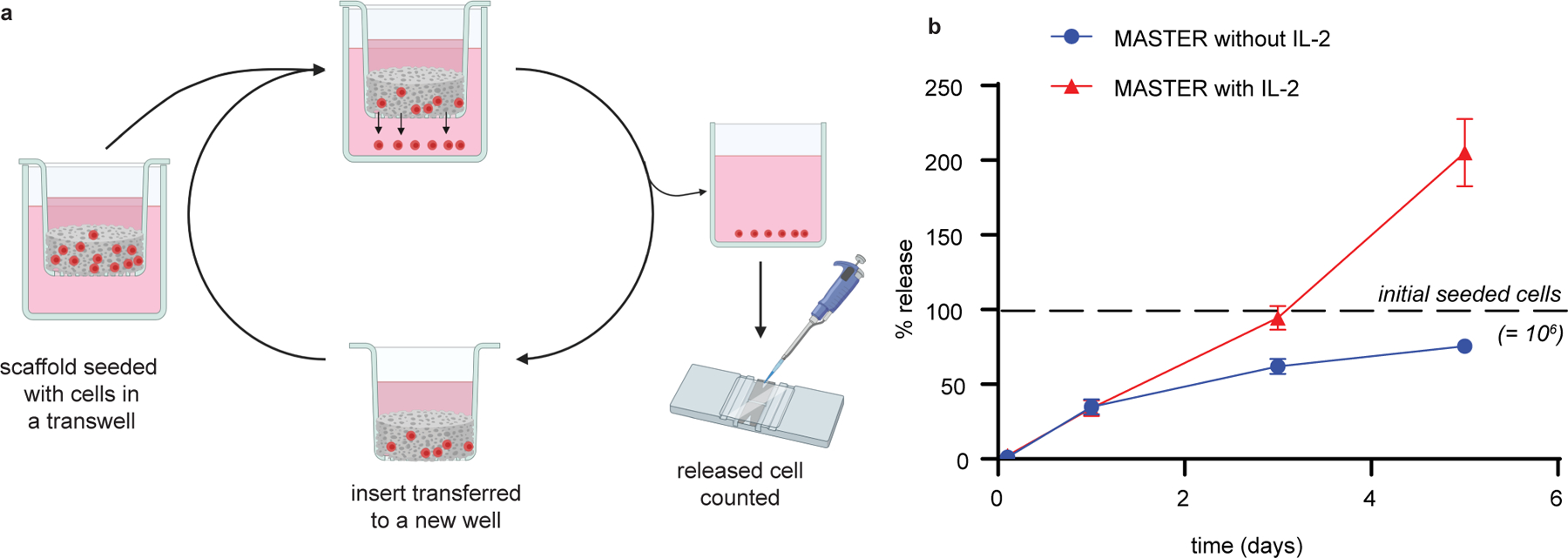
A) Schematic of in vitro release study. B) Percent of cells released from scaffold. Data in represent mean ± SD of n=3 independent samples.
Extended Data Fig. 6. Biocompatibility of MASTER and its components.
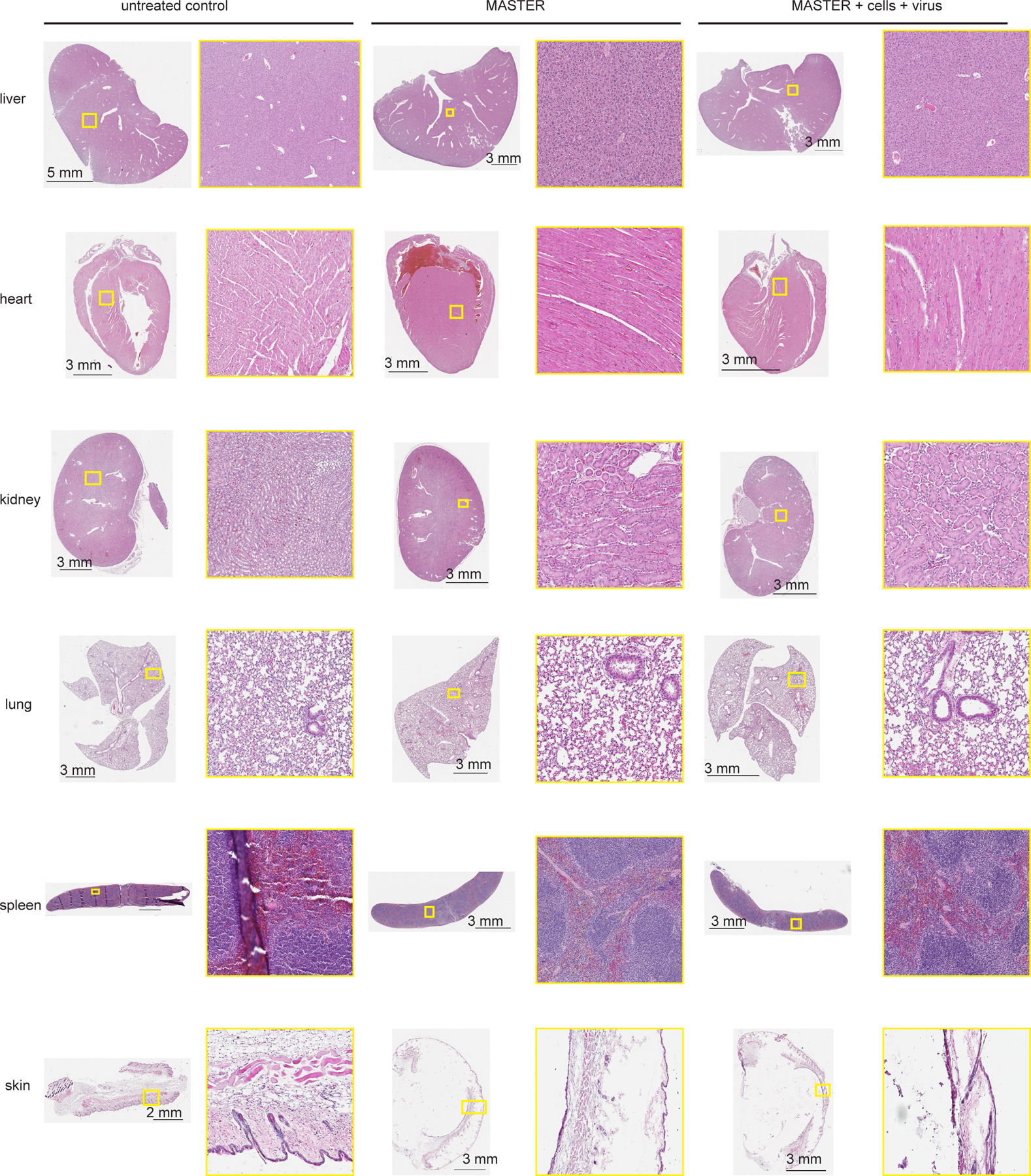
Representative images of H&E-stained sections of five major organs and implantation site four weeks after subcutaneous implant of MASTER, MASTER + mouse PBMCs + GFP-encoding gamma retrovirus and untreated controls in C57Bl6/J immunocompetent mice. Data is representative of three biologically independent animals.
Extended Data Fig. 7. MASTER loaded with PBMCs and retrovirus does not transduce host cells.
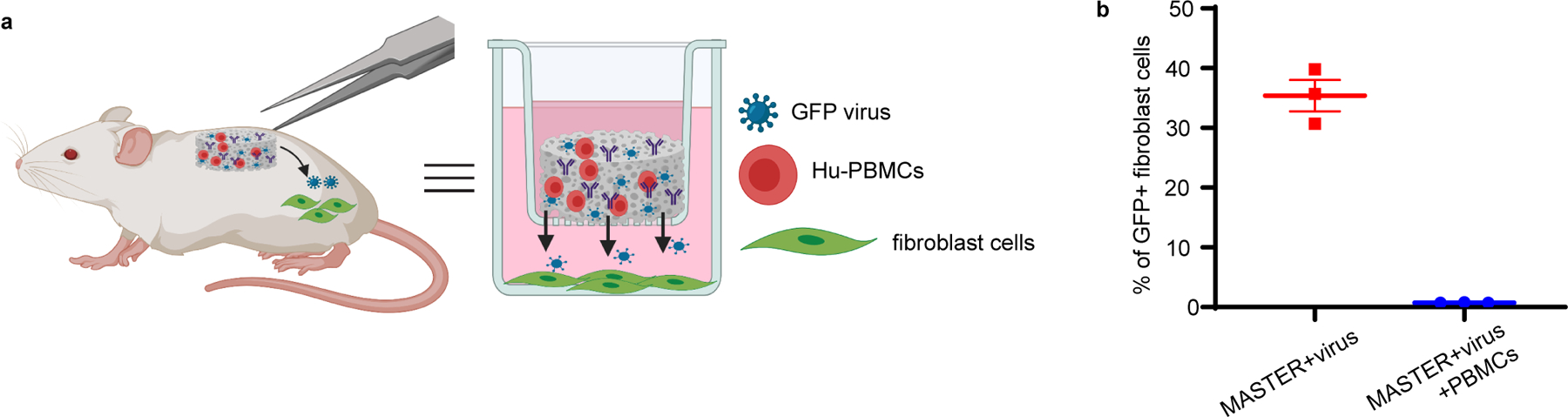
A) In vitro transwell model mimicking the in vivo system. B) GFP expression in fibroblast cells seeded on the bottom of transwell plate. Data represent mean ± SEM of n=3 biologically independent samples.
Extended Data Fig. 8. Characterization of host cells infiltrating MASTER.
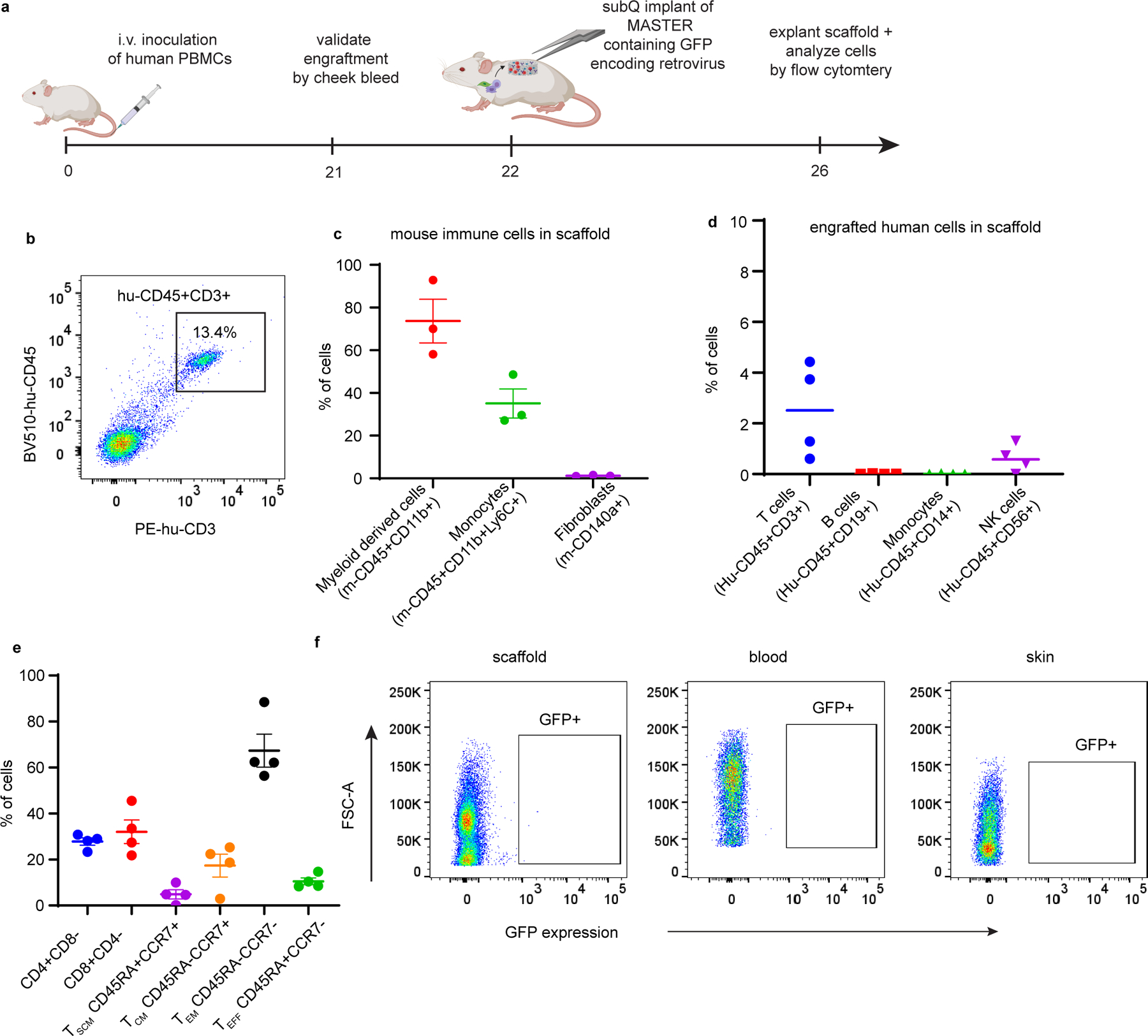
Characterization of host cells infiltrating MASTER. A) Schematic of host cells migrating into scaffold and timeline of experiment B) Representative FACS plot showing efficient engraftment of human PBMCs (Hu-CD45+CD3+) in blood. C-D) Different subsets of mouse and human cells that infiltrated MASTER. Cells were gated on live cells. Data in d-e represent mean ± SEM and median of three biologically independent samples. E) Phenotype of the engrafted human T cells that infiltrated into the scaffold. Cells were gated on human-CD45+CD3+ cells. Data represent mean ± SEM of four biologically independent samples F) FACS plot showing no GFP expression in cells infiltrating MASTER, in blood and in the skin surrounding the scaffold.
Extended Data Fig. 9. Similar numbers of exhausted cells in blood of mice with conventional and MASTER-generated CAR-T cells.
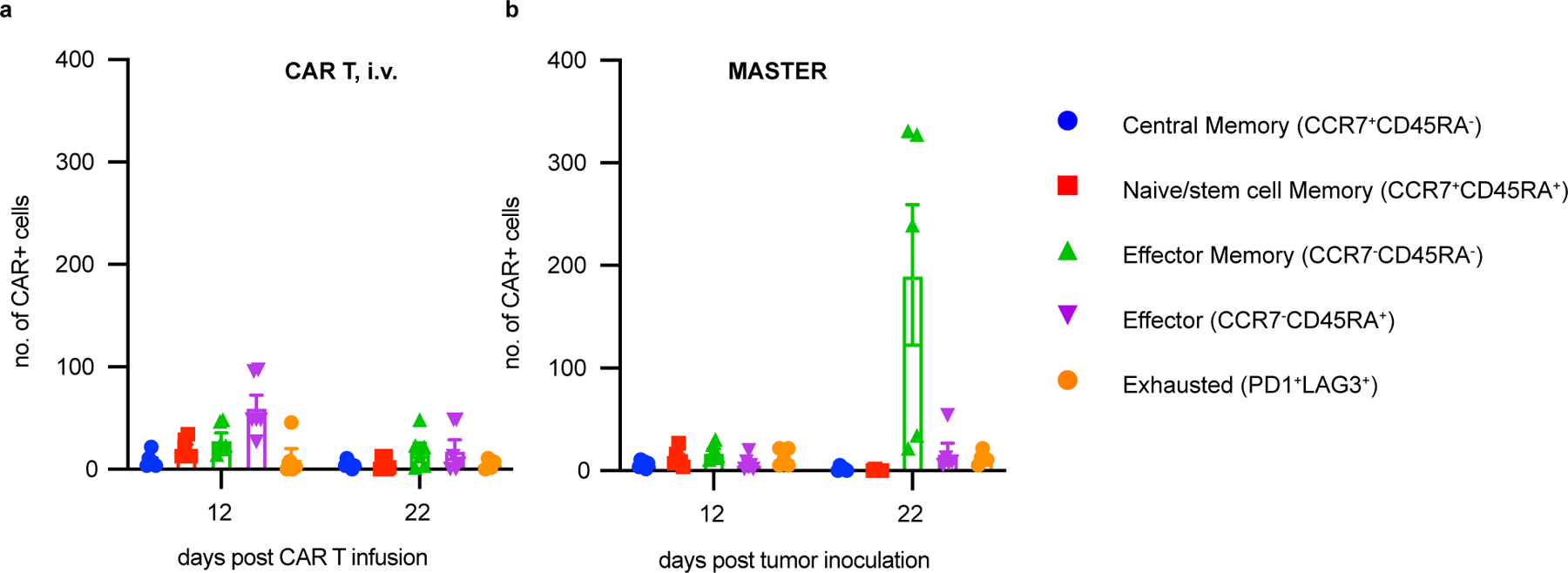
Immunophenotypic composition of CAR-T cells in blood of mice treated with conventionally expanded CAR-T cells i.v. (A) or MASTER (B) at day 12 and 22 post tumor inoculation. Data in (A) represents mean ± SD of six experimental replicates. Data in (B) represent mean ± SEM of five experimental replicates.
Extended Data Fig. 10. Subcutaneously implanted MASTER exhibited better control of tumor growth and ex-tended survival compared under stressed dose conditions.
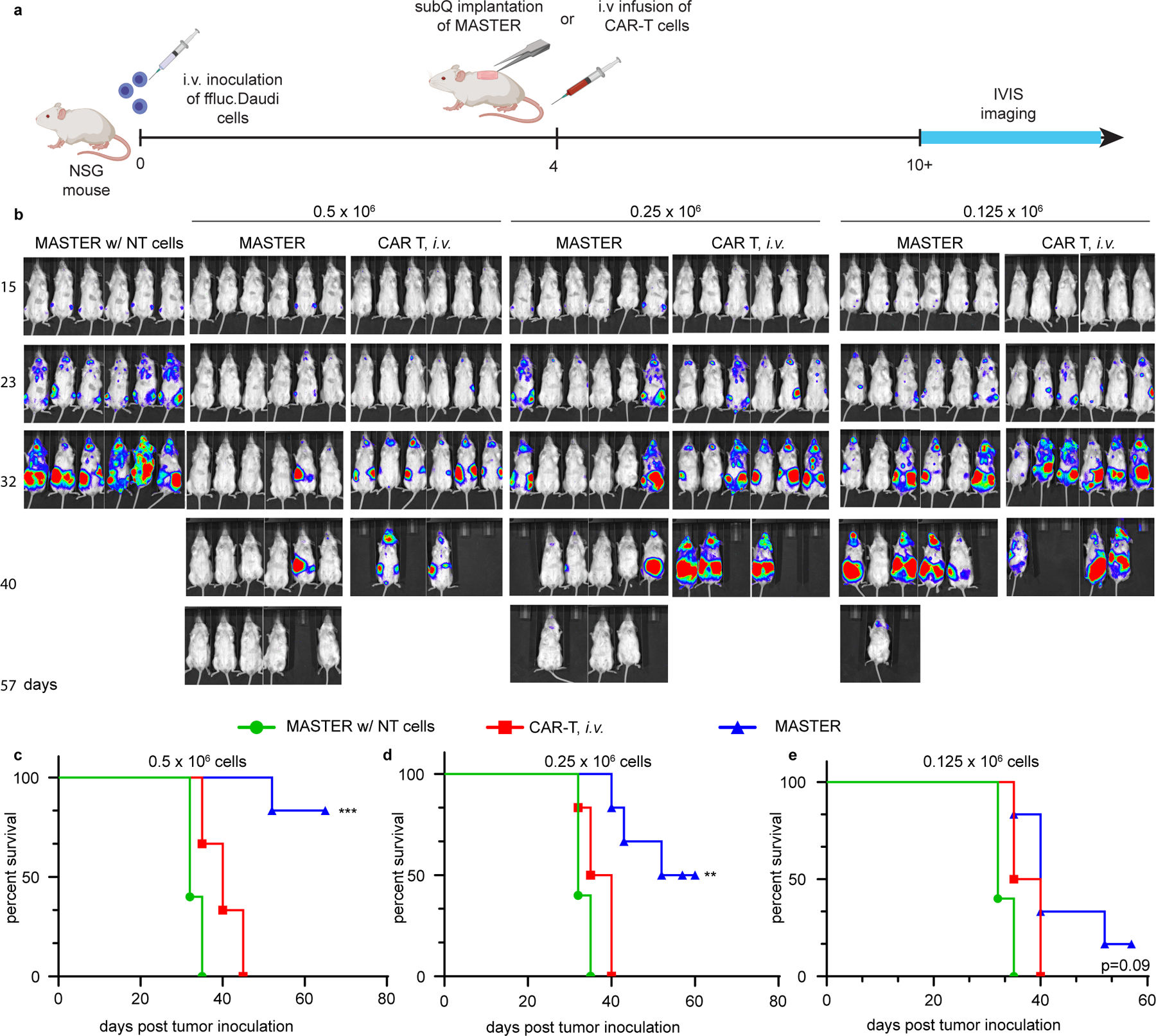
A) Timeline of the study B) In vivo tumor biolumines-cence imaging (BLI) of NSG mice (n=6) treated with MASTER, conventional CAR-T cells, or control non-transduced (NT) cells. Mice were treated with 0.5 x 106 (left), 0.25 x 106 (middle), or 0.125 x 106 (right) CAR T cells. PBMCs seeded onto MASTER were normalized to transduction efficiency and both groups (MASTER and CAR T, i.v) were treated with equivalent number of CAR T cells. C-E) Survival of mice shown as Kaplan- Meier curves. **p < 0.01; ***p < 0.001 Log-rank (Mantel-Cox) test, Gehan-Breslow-Wilcoxon test.
Supplementary Material
Acknowledgments.
This work was supported by the North Carolina Biotechnology Center Flash Grant 2019-FLG-3812, by the National Center for Advancing Translational Sciences (NCATS), and by the National Institutes of Health through Grant Award Numbers R37-CA260223, UL1-TR002489, R01-CA193140, R21-CA229938, T32-CA196589 and R25-NS094093 and by Start-Up funds provided by the University of North Carolina at Chapel Hill and North Carolina State University at Raleigh, the Lineberger Cancer Center, and the Ross M. Lampe endowed chair. We thank the NCSU CVM staff for proper care of animals used in experiments and valuable resources on training. We also thank the NCSU flow cytometry core and Javid Mohammed for training and guidance on flow cytometry analysis. We are also grateful to Dr Tan Xianming for assistance with power analysis and Dr. Charles Clifford for evaluating histology samples. SEM and X-ray CT images were taken at the Analytical Instrumentation Facility (AIF) at North Carolina State University, which is supported by the State of North Carolina and the National Science Foundation (award number ECCS-1542015). The AIF is a member of the North Carolina Research Triangle Nanotechnology Network (RTNN), a site in the National Nanotechnology Coordinated Infrastructure (NNCI). The authors acknowledge the use of the Cellular and Molecular Imaging Facility (CMIF) at North Carolina State University, which is supported by the State of North Carolina and the National Science Foundation. Schematics were created with Biorender.com
Footnotes
Competing Interest Statement. P.A., G.D, and Y.B. are inventors on patents related to the use of biomaterials for generation of CAR-T cell therapeutics. Y.B. receives an industry-sponsored research grant related to CAR-T cell therapeutic technology (unrelated to this work). GD is a paid consultant for Bellicum Pharmaceuticals, Tessa Therapeutics and Catamaran. The other authors declare no competing interests.
Data Availability Statement.
The authors declare that all data supporting the findings of this study are available within the paper and its extended data and supplementary information files. Source data are provided with this paper
References:
- 1.Lee DW et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385, 517–528 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude SL et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med 371, 1507–1517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Enblad G et al. A Phase I/IIa Trial Using CD19-Targeted Third-Generation CAR T Cells for Lymphoma and Leukemia. Clin. Cancer Res 24, 6185–6194 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Ramos CA, Savoldo B & Dotti G CD19-CAR trials. Cancer J 20, 112–118 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prasad V Immunotherapy: Tisagenlecleucel - the first approved CAR-T-cell therapy: implications for payers and policy makers. Nat. Rev. Clin. Oncol 15, 11–12 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Sharma P, King GT, Shinde SS, Purev E & Jimeno A Axicabtagene ciloleucel for the treatment of relapsed/refractory B-cell non-Hodgkin’s lymphomas. Drugs Today 54, 187–198 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Tang J, Hubbard-Lucey VM, Pearce L, O’Donnell-Tormey J & Shalabi A The global landscape of cancer cell therapy. Nat. Rev. Drug Discov 17, 465–466 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Bach PB National Coverage Analysis of CAR-T Therapies — Policy, Evidence, and Payment. New England Journal of Medicine vol. 379 1396–1398 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Hernandez I Analysis determines true cost for CAR T-cell therapy. Helio-In the Journals Plus Accessed Dec 3, (2018). [Google Scholar]
- 10.Leyfman Y Chimeric antigen receptors: unleashing a new age of anti-cancer therapy. Cancer Cell Int 18, 182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caffrey M With approval of CAR T-cell therapy comes the next challenge: payer coverage. Am. J. Manag. Care 24, SP35–SP36 (2018). [Google Scholar]
- 12.Jackson HJ, Rafiq S & Brentjens RJ Driving CAR T-cells forward. Nat. Rev. Clin. Oncol 13, 370–383 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shah NN & Fry TJ Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol 16, 372–385 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park JH et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med 378, 449–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maude SL et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med 378, 439–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghassemi S et al. Reducing Ex Vivo Culture Improves the Antileukemic Activity of Chimeric Antigen Receptor (CAR) T Cells. Cancer Immunol Res 6, 1100–1109 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Y & Dotti G Selection bias: maintaining less-differentiated T cells for adoptive immunotherapy. The Journal of clinical investigation vol. 126 35–37 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gattinoni L, Klebanoff CA & Restifo NP Paths to stemness: building the ultimate antitumour T cell. Nat. Rev. Cancer 12, 671–684 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mock U et al. Automated manufacturing of chimeric antigen receptor T cells for adoptive immunotherapy using CliniMACS prodigy. Cytotherapy 18, 1002–1011 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Lu TL et al. A rapid cell expansion process for production of engineered autologous CAR-T cell therapies. Hum. Gene Ther. Methods 27, 209–218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Depil S, Duchateau P, Grupp SA, Mufti G & Poirot L ‘Off-the-shelf’allogeneic CAR T cells: development and challenges. Nat. Rev. Drug Discov 1–15 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Kim DW & Cho J-Y Recent Advances in Allogeneic CAR-T Cells. Biomolecules 10, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith TT et al. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol 12, 813–820 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agarwal S, Weidner T, Thalheimer FB & Buchholz CJ In vivo generated human CAR T cells eradicate tumor cells. Oncoimmunology 8, e1671761 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dautzenberg IJC, Rabelink MJWE & Hoeben RC The stability of envelope-pseudotyped lentiviral vectors. Gene Ther 28, 89–104 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenblum D, Joshi N, Tao W, Karp JM & Peer D Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun 9, 1410 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee KY & Mooney DJ Alginate: properties and biomedical applications. Prog. Polym. Sci 37, 106–126 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersen T, Auk-Emblem P & Dornish M 3D Cell Culture in Alginate Hydrogels. Microarrays (Basel) 4, 133–161 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hwang CM et al. Fabrication of three-dimensional porous cell-laden hydrogel for tissue engineering. Biofabrication 2, 035003 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Agarwalla P et al. Scaffold‐Mediated Static Transduction of T Cells for CAR‐T Cell Therapy. Adv. Healthc. Mater 2000275 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Savina IN, Ingavle GC, Cundy AB & Mikhalovsky SV A simple method for the production of large volume 3D macroporous hydrogels for advanced biotechnological, medical and environmental applications. Sci. Rep 6, 21154 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shapiro L & Cohen S Novel alginate sponges for cell culture and transplantation. Biomaterials 18, 583–590 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Brudno Y et al. In vivo targeting through click chemistry. ChemMedChem 10, 617–620 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Cheung AS, Zhang DKY, Koshy ST & Mooney DJ Scaffolds that mimic antigen-presenting cells enable ex vivo expansion of primary T cells. Nat. Biotechnol 36, 160–169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stephan SB et al. Biopolymer implants enhance the efficacy of adoptive T-cell therapy. Nat. Biotechnol 33, 97–101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moody CT, Palvai S & Brudno Y Click cross-linking improves retention and targeting of refillable alginate depots. Acta Biomater 112, 112–121 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Savoldo B et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor–modified T cells in lymphoma patients. J. Clin. Invest 121, 1822–1826 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stock S et al. Influence of Retronectin-Mediated T-Cell Activation on Expansion and Phenotype of CD19-Specific Chimeric Antigen Receptor T Cells. Hum. Gene Ther 29, 1167–1182 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Hori Y, Winans AM & Irvine DJ Modular injectable matrices based on alginate solution/microsphere mixtures that gel in situ and co-deliver immunomodulatory factors. Acta Biomater 5, 969–982 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diaconu I et al. Inducible Caspase-9 Selectively Modulates the Toxicities of CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Mol. Ther 25, 580–592 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu Y et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 123, 3750–3759 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramos CA et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor–redirected lymphocytes. J. Clin. Invest 127, 3462–3471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramos CA et al. Clinical responses with T lymphocytes targeting malignancy-associated κ light chains. J. Clin. Invest 126, 2588–2596 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tay RE, Richardson EK & Toh HC Revisiting the role of CD4+ T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther 28, 5–17 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wen H et al. Preclinical safety evaluation of chimeric antigen receptor-modified T cells against CD19 in NSG mice. Ann Transl Med 7, 735 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao Z et al. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 28, 415–428 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McLellan AD & Ali Hosseini Rad SM Chimeric antigen receptor T cell persistence and memory cell formation. Immunol. Cell Biol 97, 664–674 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Sterner RC & Sterner RM CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J 11, 69 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Majzner RG & Mackall CL Tumor antigen escape from CAR T-cell therapy. Cancer Discov 8, 1219–1226 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Liu L et al. Enhanced CAR-T activity against established tumors by polarizing human T cells to secrete interleukin-9. Nat. Commun 11, 5902 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
References:
- 1.Brudno Y et al. In vivo targeting through click chemistry. ChemMedChem 10, 617–620 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Moody CT, Palvai S & Brudno Y Click cross-linking improves retention and targeting of refillable alginate depots. Acta Biomater (2020) doi: 10.1016/j.actbio.2020.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gostick J et al. OpenPNM: A pore network modeling package. Comput. Sci. Eng 18, 60–74 (2016). [Google Scholar]
- 4.Vera J et al. T lymphocytes redirected against the kappa light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood 108, 3890–3897 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diaconu I et al. Inducible Caspase-9 Selectively Modulates the Toxicities of CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Mol. Ther 25, 580–592 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu Y et al. Glycolysis determines dichotomous regulation of T cell subsets in hypoxia. J. Clin. Invest 126, 2678–2688 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramos CA et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor–redirected lymphocytes. J. Clin. Invest 127, 3462–3471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramos CA et al. Anti-CD30 CAR-T cell therapy in relapsed and refractory Hodgkin lymphoma. J. Clin. Oncol 38, 3794–3804 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the paper and its extended data and supplementary information files. Source data are provided with this paper