

Abstract
African Americans (AA) have lower prevalence of alcohol dependence and higher subjective response to alcohol than European Americans. Genome-wide association studies (GWAS) have identified genes/variants associated with alcohol dependence specifically in AA; however, the sample sizes are still not large enough to detect variants with small effects. Admixture mapping is an alternative way to identify alcohol dependence genes/variants that may be unique to AA. In this study, we performed the first admixture mapping of DSM-IV alcohol dependence diagnosis, DSM-IV alcohol dependence criterion count, and two scores from the Self-Rating of Effects of Ethanol (SRE) as measures of response to alcohol: the first five times of using alcohol (SRE-5) and average of SRE across three times (SRE-T). Findings revealed a region on chromosome 4 that was genome-wide significant for SRE-5 (P-value=4.18E-05). Fine mapping did not identify a single causal variant to be associated with SRE-5; instead, conditional analysis concluded that multiple variants collectively explained the admixture mapping signal. PPARGC1A, a gene that has been linked to alcohol consumption in previous studies, is located in this region. Our finding suggests that admixture mapping is a useful tool to identify genes/variants that may have been missed by current GWAS approaches in admixed populations.
Keywords: Admixture mapping, African American, DSM-IV alcohol dependence, criterion count, response to ethanol
INTRODUCTION
Alcohol dependence is one of the most common and costly diseases worldwide. In the United States, 12.5% of the population meets criteria for alcohol dependence during their lifetime (Hasin & Grant, 2015), and about 88,000 deaths and 2.5 million years of potential life lost annually are attributable to excessive alcohol use (Stahre, Roeber, Kanny, Brewer, & Zhang, 2014). The estimated economic cost attributable to excessive drinking is almost $250 billion (Sacks, Gonzales, Bouchery, Tomedi, & Brewer, 2015).
It has been long observed that drinking behavior and drinking-related problems differ among ethnic groups (K. Chartier & Caetano, 2010; Delker, Brown, & Hasin, 2016; Gibbs et al., 2013; Hasin, Stinson, Ogburn, & Grant, 2007; Vaeth, Wang-Schweig, & Caetano, 2017; Witbrodt, Mulia, Zemore, & Kerr, 2014; Zapolski, Pedersen, McCarthy, & Smith, 2014). Compared to European Americans (EA), African Americans (AA) drink less frequently; are more likely to stop drinking; have fewer heavy drinking episodes, later onset of drinking, and slower progression to alcohol dependence (Alvanzo et al., 2011; Dawson, Goldstein, & Grant, 2013; Klima, Skinner, Haggerty, Crutchfield, & Catalano, 2014). As a result, AA have significantly lower prevalence of alcohol dependence (Hasin & Grant, 2015). Notably, this is despite the increased exposure to stressful experiences in AA, which is an important factor associated with progression to alcohol dependence (Gibbs et al., 2013; Ransome & Gilman, 2016). However, when alcohol dependence occurs, AA have higher rates of recurrent and persistent alcohol dependence than EA (Breslau, Kendler, Su, Gaxiola-Aguilar, & Kessler, 2005; K. Chartier & Caetano, 2010; Dawson et al., 2005). In addition, AA reported a sharper increase in stimulation in an alcohol administration study (Pedersen & McCarthy, 2013), and experienced different neuroendocrine and inflammation responses due to alcohol misuse (Ransome, Slopen, Karlsson, & Williams, 2017, 2018). Furthermore, rates of alcohol-related diseases, mortality, and consequences are higher in AA (K. Chartier & Caetano, 2010; Flores et al., 2008; Polednak, 2007; Russo, Purohit, Foudin, & Salin, 2004; Sempos, Rehm, Wu, Crespo, & Trevisan, 2003; Shield et al., 2013; Yang, Vadhavkar, Singh, & Omary, 2008).
The reasons for the disparities in drinking and alcohol-related problems between AA and EA are not fully understood (Hasin & Grant, 2015; Zapolski et al., 2014). Studies have suggested that both environmental and genetic factors contribute to these differences (K. G. Chartier et al., 2014). Relevant to the current study, there is evidence for differential heritability of problem drinking in EA and AA (Sartor et al., 2013). Genome-wide association studies (GWAS) of alcohol-related phenotypes have also identified variants that are only significant in AA or EA (Kranzler et al., 2019; Lai, Wetherill, Bertelsen, et al., 2019; Lai, Wetherill, Kapoor, et al., 2019; Walters et al., 2018). For example, different functional single nucleotide polymorphisms (SNP) in ADH1B, the gene encoding the main alcohol-metabolizing enzyme in liver, have been linked to alcohol dependence in various populations partially due to their population specific allele frequencies: rs2066702 in AA and rs1229984 in EA (Bierut et al., 2012; Edenberg & McClintick, 2018; Walters et al., 2018). Polimanti and colleagues studied functional variants in 24 genes related to alcohol dependence and found frequencies of these variants to vary between AA and EA (Polimanti, Yang, Zhao, & Gelernter, 2015).
There is a great need for identifying genes/variants specifically related to AA drinking behavior and problems (Zemore et al., 2018). The identification of population specific genes/variants can advance our knowledge of the etiology of alcohol dependence in AA and contribute to the development of novel prevention and therapeutic strategies. However, there are several methodological challenges specific to conducting genetic studies in AA. First, there are fewer and smaller studies of AA compared to EA. Of the 32 GWAS of alcohol dependence and related phenotypes in the NHGRI-EBI GWAS catalog (https://www.ebi.ac.uk/gwas/home), only 11 include AA and sample sizes are much smaller compared to other populations. Recently, in the largest GWAS of alcohol dependence, only 6,280 AA were included in an analysis of >52,000 individuals (Walters et al., 2018). In another recent GWAS using the Alcohol Use Disorders Identification Test (AUDIT) in the Million Veteran Project (MVP), there were about 57,000 AA samples, however, the EA population consisted of > 209,000 participants (Kranzler et al., 2019). Second, people of African ancestry have more genetic variants and a faster decay of linkage disequilibrium (LD) with an increase in physical distance (Altshuler et al., 2015). Therefore, more independent tagging variants are needed to fully cover the entire genome in AA as compared with other populations. As a result, the traditional genome-wide threshold, 5 x 10E-8, may not be appropriate. Third, the proportion of African and European ancestries differ among AA populations in the U.S. (Dick, Barr, Guy, Nasim, & Scott, 2017). In genetic studies, this admixture is usually modeled by including ancestral principal components (PCs) as covariates in analysis. However, these PCs are a genome-wide adjustment and may result in over- or under- adjustment in some chromosomal regions due to different proportions of local (i.e., region-specific) admixture.
Admixture mapping might provide novel insights into one potential source of the differential prevalence of alcohol dependence between EA and AA (Seldin, Pasaniuc, & Price, 2011). One study found that the degree of African admixture is correlated with alcohol dependence; and those with alcohol dependence have less African ancestry (Zuo et al., 2009). Since admixture in AA occurred relatively recently (usually <10 generations), only a small number of recombination events have likely occurred and the size of ancestry-specific regions is expected to be large. That is, the average size of an African ancestry block in AA is about 17 centimorgans (Patterson et al., 2004). Thus, a much smaller number of genetic markers would be needed to tag such regions than is required in a typical GWAS. Admixture mapping has been successfully applied to other traits, e.g. blood pressure, obstructive sleep apnea, systemic lupus erythematosus, etc., (Molineros et al., 2013; Sofer et al., 2017; Wang et al., 2019; Winkler, Nelson, & Smith, 2010); however, to our knowledge, it has not been applied to the study of alcohol dependence and related phenotypes in AA.
In this study, we performed admixture mapping using AA individuals from the Collaborative study on the Genetics of Alcoholism (COGA) (Reich et al., 1998), Study of Addiction: Genetics and Environment (SAGE) (Bierut et al., 2010), Alcohol Dependence GWAS in European and African Americans (Yale-Penn) (Gelernter et al., 2014), and an African American cohort from the National Institute on Alcohol Abuse and Alcoholism (NIAAA). Duplicate individuals among those studies were removed. We focused on four phenotypes: DSM-IV (American Psychiatric Association, 1994) alcohol dependence diagnosis; DSM-IV alcohol dependence criterion count as a measure of alcohol dependence severity (Lai, Wetherill, Bertelsen, et al., 2019), and two scores from the Self-Rating of Effects of Ethanol (SRE) questionnaire (Schuckit, Smith, & Tipp, 1997) as measures of response to alcohol. In genome-wide significant (GWS) regions, we conducted fine mapping using genotyped and imputed data to identify potentially causal variants. Lastly, we performed conditional analyses to test whether the variants identified during fine mapping could explain the admixture mapping association signal.
MATERIALS AND METHODS
Samples:
COGA recruited alcohol dependent probands and their family members from inpatient and outpatient AD treatment facilities in seven sites, and community comparison families were also recruited from a variety of sources in the same areas (Nurnberger et al., 2004; Reich et al., 1998). Institutional review boards from all sites approved the study and every participant provided informed consent or assent. The Semi-Structured Assessment for the Genetics of Alcoholism (SSAGA) and the child version of the SSAGA (Bucholz et al., 1994; Hesselbrock, Easton, Bucholz, Schuckit, & Hesselbrock, 1999) were administered to individuals age 18 or over and younger than 18, respectively. SAGE (phs000092.v1.p1, https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000092.v1.p1) and Yale-Penn (phs000425.v1.p1, https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000425.v1.p1) were downloaded from the database of Genotypes and Phenotypes (dbGaP). For the NIAAA cohort, participants were recruited using the NIH Institutional Review Board-approved screening and assessment protocols conducted at the National Institutes of Health Clinical Center (Bethesda, Maryland, USA) from 2005 to 2015. All participants provided written informed consent.
Phenotypes:
Individuals who endorsed three or more of the seven DSM-IV criteria occurring within a 12-month period were diagnosed with DSM-IV alcohol dependence. Affected individuals were age 15 or older and met criteria for DSM-IV alcohol dependence. Unaffected individuals were defined as those who had consumed at least one full drink of alcohol, were ≥ 21 years old, endorsed < 2 criteria for DSM-IV dependence, and did not meet criteria for abuse of alcohol, cocaine, opioids, marijuana, sedatives, and stimulants (Lai, Wetherill, Bertelsen, et al., 2019). For SAGE and Yale-Penn datasets, DSM-IV alcohol dependence diagnoses were downloaded from dbGaP; and unaffected individuals with alcohol abuse, or other substance dependence were excluded.
The seven DSM-IV alcohol dependence criteria were summed to create a criterion count. Individuals with comorbid use and misuse of other drugs were not excluded.
The Self-Rating of Effects of Ethanol (SRE) questionnaire is a retrospective, self-report instrument to measure the numbers of standard drinks required to produce four effects of ethanol (Schuckit, Tipp, Smith, Wiesbeck, & Kalmijn, 1997): a) “how many (standard) drinks did it take for you to begin to feel an effect?”; b) “how many drinks did it take for you to feel a bit dizzy or begin to slur your speech?”; c) “how many drinks did it take you to begin to stumble or walk in an uncoordinated manner?”; d) “how many drinks did it take you to pass out or fall asleep when you did not want to?”. The SRE queries drinking at three time points: the first five times using alcohol (SRE-5); the period of heaviest drinking; and the most recent 3 months of consumption (Schuckit, Tipp, et al., 1997). In this study, we used SRE-5 as well as the average scores across the three time points (SRE-T). Individuals who drank >=2 drinks on one occasion were included in the analysis with extreme observations winsorized at the mean plus 2 standard deviations. The natural logarithm of SRE-5 and square root of SRE-T were used in analyses based on their distributions (Lai, Wetherill, Kapoor, et al., 2019).
Genotyping, Ancestry and Imputation:
Detailed information about data processing and QC applied in each study was reported previously (Lai, Wetherill, Bertelsen, et al., 2019; Lai, Wetherill, Kapoor, et al., 2019). To identify duplicate samples among studies, confirm the reported pedigree structure, and calculate PCs representing population stratification, all available data from COGA, SAGE, Yale-Penn, and NIAAA were combined. Then, variants meeting the following criteria: common (defined as minor allele frequency (MAF) >10% in the combined sample), independent (defined as r2 <0.5), high quality (missing rate <2% and Hardy-Weinberg Equilibrium (HWE) P-values >0.001), were used to identify duplicate samples and confirm the reported pedigree structure using PLINK (Chang et al., 2015; Purcell et al., 2007). To remove the same individual included in multiple datasets, (i.e., between COGA and SAGE), duplicate samples were removed from the study with less phenotypic information and fewer family members (e.g. SAGE). Family structures were updated, as needed. Using genetically-confirmed pedigrees, Pedcheck (O’Connell & Weeks, 1998) was used to identify Mendelian errors and inconsistencies were removed. The same set of variants were used to estimate PCs using Eigenstrat (Price et al., 2006) with 1000 Genomes data serving as the reference (Phase 3, version 5). Only AA samples, designated based on the first two PCs (COGA N=2,939; SAGE N=959; Yale-Penn N=2,044; NIAAA N=169), were included in analyses (Lai, Wetherill, Bertelsen, et al., 2019; Lai, Wetherill, Kapoor, et al., 2019). For the purposes of fine mapping, all samples were imputed to 1000 Genomes (Phase 3, version 5, hg19) separately by cohort using SHAPEIT2 (Delaneau, Howie, Cox, Zagury, & Marchini, 2013) followed by Minimac3 (Das et al., 2016). Only variants with non A/T or C/G alleles, missing rates <5%, MAF >3%, and HWE P-values >0.0001 were used for imputation. Imputed variants with imputation quality score R2 <0.30 were excluded.
Inference of African ancestry
Due to differences in genotyping arrays, RFMix (V1.5.4) (Maples, Gravel, Kenny, & Bustamante, 2013) was used to estimate local African ancestry in each cohort separately. RFMix is a discriminative modeling approach that uses random forests trained on reference samples. Ninety-nine CEU and 99 YRI samples from 1000 Genomes Phase 3 were used as European and African reference samples, respectively, as recommended by RFMix. Only genotyped, high quality variants (defined as missing rate <0.05, Hardy-Weinberg P-values >0.001, and MAF >3%) were included to improve inference accuracy. SHAPEIT2 (Delaneau et al., 2013) was used for haplotype phasing, then RFMix was used to estimate the number of African alleles at each locus (i.e., 0, 1 or 2 copies of African alleles, referred as local African ancestry). For each individual, global African ancestry was also calculated as the average percentage of African ancestry across the entire genome.
Admixture mapping:
We used RVTESTS (Zhan, Hu, Li, Abecasis, & Liu, 2016) to perform admixture mapping within each dataset. For each phenotype, the association with the number of African alleles at each locus was tested after adjusting for study specific covariates and a kinship matrix estimated by RVTESTS. For COGA and SAGE, sex and birth cohort were significantly associated with alcohol dependence, and were therefore used as covariates (Lai, Wetherill, Bertelsen, et al., 2019; Lai, Wetherill, Kapoor, et al., 2019). For Yale-Penn and NIAAA, birth cohort was not available, therefore, sex and age were included, as in previous studies (Lai, Wetherill, Bertelsen, et al., 2019; Lai, Wetherill, Kapoor, et al., 2019). Global African ancestry was included as a covariate in all tests, as suggested (Molineros et al., 2013; Parra et al., 2017). Results from each dataset were meta-analyzed with the effect size weighted by the inverse of the estimated standard error using METAL (Willer, Li, & Abecasis, 2010). Since the block sizes were different for each cohort, only the overlapping part of the blocks from each cohort were included in meta-analysis.
The following procedure was used to determine the GWS threshold. First, matSpD was used to account for correlations across the four phenotypes by spectral decomposition of the correlation matrix (Li & Ji, 2005; Nyholt, 2004), resulting in 2.52 effective independent tests. Second, using the autocorrelation function of the R package CODA (Plummer, Best, Cowles, & Vines, 2005), 273.82 effective African ancestry blocks were estimated across the entire genome using a combined sample from all cohorts. Therefore, the GWS threshold was determined as 0.05/(2.52*273.82)=7.25E-05.
Fine mapping and conditional analysis:
For GWS regions that were identified, all genotyped and imputed variants within that region that had a missing rate <0.05, Hardy-Weinberg P-values >0.0001, and MAF >3% were tested using the same model as in the admixture mapping, except the global African ancestry index was replaced by PCs (the first four PCs in COGA and the first three PCs in other samples as in the original studies (Lai, Wetherill, Bertelsen, et al., 2019; Lai, Wetherill, Kapoor, et al., 2019)) to adjust for population stratification among AA. RVTESTS (Zhan et al., 2016) was used to perform association tests within each cohort separately, then results from each cohort were meta-analyzed using METAL (Willer et al., 2010). The significance threshold was determined by the estimated effective number of tests using the R package matSpDlite (Li & Ji, 2005; Nyholt, 2004), which decomposes the LD between variants to arrive at the approximate number of independent variants.
Conditional analysis was performed by including variants identified in fine mapping with the lowest P-values as covariates in another round of admixture mapping in GWS regions. A conditional analysis P-value >0.01 indicated that the variants included as covariates in admixture mapping were the driving factors of an admixture mapping association signal. We first tested each variant individually. If single variants did not explain the admixture mapping signal, then we tested multiple variants following the framework proposed by Molineros et al (Molineros et al., 2013). Starting with the variant with the lowest P-value, we added the variant having the next lowest P-value and not in LD (defined as r2<0.5) with variants that were already in the model, one at a time, until the conditional P-value was greater than 0.01.
RESULTS
Table 1 summarizes the study samples. COGA, SAGE, and Yale-Penn were used in the analysis of DSM-IV alcohol dependence diagnosis and DSM-IV alcohol dependence criterion count. In total, there were 2,872 alcohol dependence cases and 1,672 controls. A total of 5,942 individuals had data on DSM-IV criterion count. COGA and NIAAA datasets were used for SRE analysis; SRE-5 and SRE-T analyses included data on 1,546 individuals in total. In terms of variants, 632,882, 601,545, 637,753, and 580,705 SNPs were included in the COGA, SAGE, Yale-Penn, and NIAAA data respectively (Table 1). RFMix estimated 14,376, 14,958, 15,336, and 14,118 African ancestry blocks for COGA, SAGE, Yale-Penn, and NIAAA respectively (Table 1). Using the R package CODA (Plummer et al., 2005), 273.82 effective African ancestry blocks were estimated across these four cohorts.
Table 1:
Summary of study samples
| Cohort | DSM-IV alcohol dependence (#cs/#ctl) | # DSM-IV alcohol dependence criterion count | # SRE-5 | # SRE-T | # variants | # African ancestry blocks |
|---|---|---|---|---|---|---|
| COGA | 875/840 | 2,939 | 1,377 | 1,377 | 632,882 | 14,376 |
| SAGE | 400/341 | 959 | NA | NA | 601,545 | 14,958 |
| Yale-Penn | 1552/491 | 2,044 | NA | NA | 637,753 | 15,336 |
| NIAAA | NA | NA | 169 | 169 | 580,705 | 14,118 |
| Total | 2,827/1,672 | 5,942 | 1,546 | 1,546 |
One region on chromosome 4 reached genome-wide significance for SRE-5 (P-value=4.18E-05) (Table 2; Figure 1). The most significant blocks in this region were between 24,377,777 bp and 24,512,590 bp and were supported by both the COGA and NIAAA cohorts. The estimate of effect size is larger in NIAAA than in COGA due to the proportion of study participants with higher SRE scores and larger variation in the NIAAA sample. Individuals carrying African ancestry blocks in this region had higher SRE scores (BETA=0.21, SE=0.05), i.e., lower the response to alcohol. No other region reached genome-wide significance for other phenotypes (Supplement Figures 1A, 1B, 1C).
Table 2:
SRE-5 admixture mapping results of the chromosome 4 region
| chr | Start | end | Ancestry tested | Meta-analysis | COGA | NIAAA | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BETA | SE | P-value | African ancestry Frequency | BETA | SE | P-value | African ancestry Frequency | BETA | SE | P-value | ||||
| 4 | 24,225,918 | 24,377,777 | AFR | 0.20 | 0.05 | 7.91E-05 | 0.78 | 0.17 | 0.06 | 2.08E-03 | 0.77 | 0.35 | 0.12 | 5.18E-03 |
| 4 | 24,377,777 | 24,439,865 | AFR | 0.20 | 0.05 | 7.15E-05 | 0.78 | 0.17 | 0.06 | 1.88E-03 | 0.77 | 0.34 | 0.12 | 5.64E-03 |
| 4 | 24,439,865 | 24,512,590 | AFR | 0.21 | 0.05 | 4.18E-05 | 0.78 | 0.18 | 0.06 | 9.02E-04 | 0.78 | 0.32 | 0.12 | 9.15E-03 |
Note: Genome-wide significant blocks are in bold. AFR: African ancestry allele.
Figure 1.
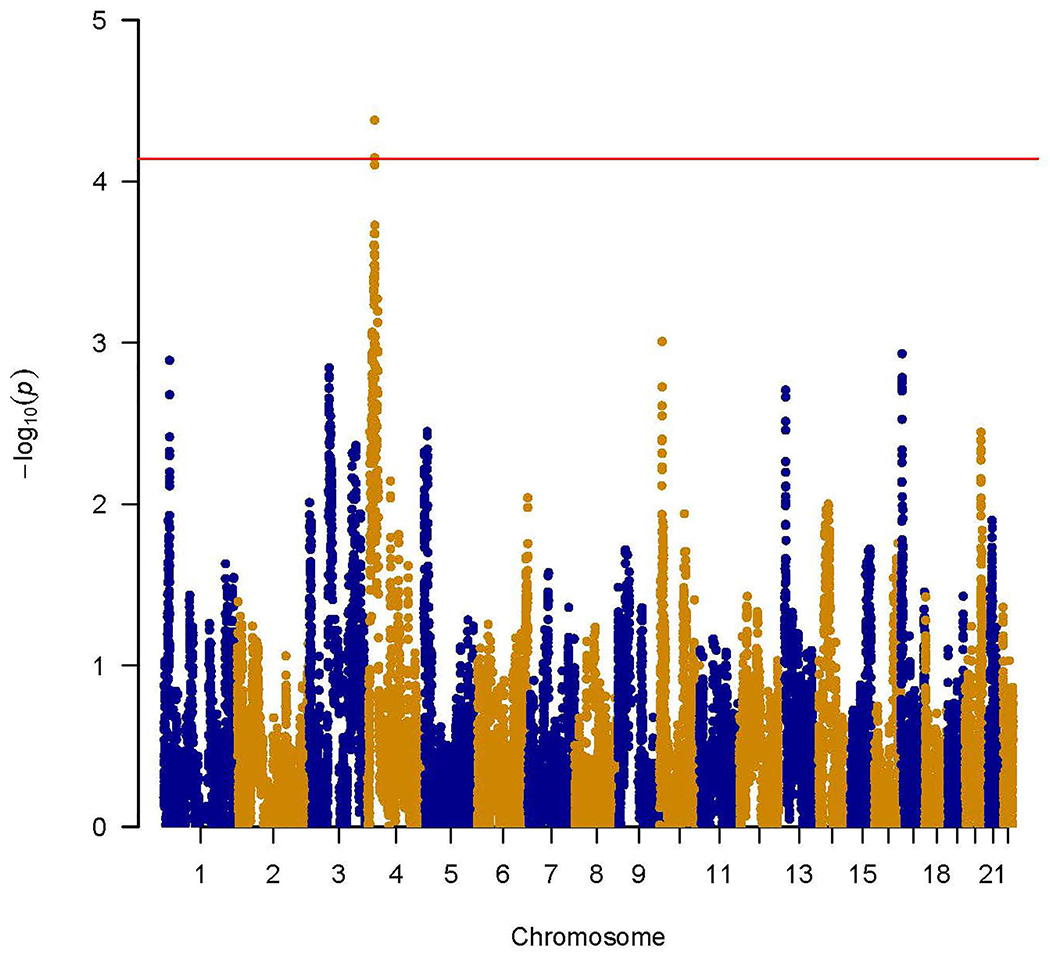
Genome-wide admixture mapping of SRE-5. Y-axis is the –log(P-value) for associations. X-axis is physical position of African ancestry blocks across the genome. The red horizontal line indicates genome-wide significance.
There were 298 genotyped and imputed variants located in the SRE-5 chromosome 4 GWS region. Using matSpDlite (Li & Ji, 2005; Nyholt, 2004), the estimated number of effective tests was 132; therefore, the significance threshold for the fine mapping analysis was determined as P-value <3.79E-04. None of the variants tested individually reached this significance threshold. Table 3 lists all variants with P-values <0.01 in fine mapping; some, for instance, had P-values <0.01 in COGA but P-values >0.05 in NIAAA, possibly due to the much smaller sample size of the NIAAA cohort. These variants from both COGA and NIAAA had similar allele frequencies as the African sample in the genome aggregation database (genomAD, http://gnomad.broadinstitute.org/). However, they had dramatically different allele frequencies from the gnomAD non-Finnish European sample, indicating that they were ancestry informative, as expected. Carrying an effective allele increased SRE-5 scores (Table 3) and the effective allele frequencies were higher in Africans than in non-Finnish Europeans for all of these variants, except rs79462764. This was consistent with the results of admixture mapping. All variants in Table 3 individually had conditional P-values <0.01, indicating that they did not individually explain the admixture mapping association signal. When conditioned collectively on rs76004436, rs3966916, rs11931595, and rs10018808 (four independent variants with the lowest P-values), the conditional analysis had P-value >0.01, demonstrating that these four variants (or variants in LD with them) were driving the admixture mapping association signal. Supplemental Figure 2 shows the regional association plots that those four variants were index variants. Variants that were in LD with those index variants were all located in a small region between 24.37 Mbp and 24.52 Mbp. Supplemental Figure 3 depicts the proportion of African ancestry on chromosome 4 for all cohorts. As can be seen, the proportion of African ancestry differed dramatically at different locations for all four cohorts.
Table 3:
Variants having P-value < 0.01 in fine mapping of chromosome 4 region for SRE-5.
| gnomAD | Meta-analysis | COGA | NIAAA | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| chr | bp | rs | Effective Allele | Other Allele | AFR EAF | NFE EAF | BETA | SE | P-value | EAF | BETA | SE | P-value | EAF | BETA | SE | P-value |
| 4 | 24,509,859 | rs76004436 | T | C | 0.05 | 1.00E-04 | 0.20 | 0.06 | 1.04E-03 | 0.04 | 0.21 | 0.06 | 9.20E-04 | 0.04 | 0.05 | 3.92 | 0.86 |
| 4 | 24,506,961 | rs376595619 | T | C | 0.05 | 6.67E-05 | 0.20 | 0.06 | 1.13E-03 | 0.04 | 0.20 | 0.06 | 1.02E-03 | 0.04 | 0.06 | 3.92 | 0.85 |
| 4 | 24,377,799 | rs3966916 | G | A | 0.09 | 9.00E-04 | 0.13 | 0.04 | 1.56E-03 | 0.08 | 0.14 | 0.04 | 1.15E-03 | 0.07 | −0.04 | 2.89 | 0.86 |
| 4 | 24,463,807 | rs11931595 | C | G | 0.60 | 0.17 | 0.07 | 0.02 | 1.85E-03 | 0.55 | 0.07 | 0.02 | 1.38E-03 | 0.56 | −0.01 | 1.41 | 0.95 |
| 4 | 24,496,851 | rs115697448 | A | G | 0.07 | 1.00E-04 | 0.16 | 0.05 | 2.34E-03 | 0.05 | 0.17 | 0.05 | 1.95E-03 | 0.05 | 0.01 | 3.61 | 0.98 |
| 4 | 24,451,810 | rs10018808 | A | G | 0.28 | 0.05 | 0.08 | 0.03 | 2.60E-03 | 0.25 | 0.08 | 0.03 | 3.76E-03 | 0.27 | 0.11 | 1.67 | 0.39 |
| 4 | 24,454,550 | rs10025734 | G | C | 0.37 | 0.12 | 0.07 | 0.02 | 2.81E-03 | 0.33 | 0.07 | 0.02 | 2.83E-03 | 0.35 | 0.04 | 1.59 | 0.76 |
| 4 | 24,450,286 | rs10034872 | G | A | 0.65 | 0.47 | 0.07 | 0.02 | 4.26E-03 | 0.62 | 0.07 | 0.02 | 2.37E-03 | 0.64 | −0.06 | 1.49 | 0.58 |
| 4 | 24,413,183 | rs79462764 | C | T | 0.02 | 0.11 | 0.31 | 0.11 | 4.75E-03 | 0.01 | 0.36 | 0.11 | 1.48E-03 | 0.02 | −0.39 | 5.43 | 0.35 |
| 4 | 24,456,232 | rs9992361 | G | A | 0.63 | 0.47 | 0.06 | 0.02 | 5.21E-03 | 0.60 | 0.07 | 0.02 | 2.96E-03 | 0.64 | −0.07 | 1.48 | 0.55 |
| 4 | 24,455,932 | rs10026526 | C | T | 0.64 | 0.47 | 0.06 | 0.02 | 6.07E-03 | 0.61 | 0.07 | 0.02 | 3.56E-03 | 0.64 | −0.06 | 1.49 | 0.57 |
| 4 | 24,454,338 | rs10025483 | A | C | 0.53 | 0.18 | 0.06 | 0.02 | 6.56E-03 | 0.49 | 0.06 | 0.02 | 5.55E-03 | 0.50 | 0.00 | 1.45 | 1.00 |
| 4 | 24,456,195 | rs35429805 | C | CA | 0.62 | 0.47 | 0.06 | 0.02 | 6.58E-03 | 0.60 | 0.07 | 0.02 | 3.82E-03 | 0.63 | −0.07 | 1.48 | 0.56 |
| 4 | 24,466,090 | rs6847029 | A | G | 0.48 | 0.15 | 0.06 | 0.02 | 7.91E-03 | 0.45 | 0.06 | 0.02 | 8.79E-03 | 0.49 | 0.05 | 1.49 | 0.66 |
| 4 | 24,412,938 | rs12331764 | A | G | 0.14 | 1.30E-03 | 0.09 | 0.03 | 9.28E-03 | 0.13 | 0.08 | 0.03 | 1.83E-02 | 0.08 | 0.40 | 2.90 | 0.08 |
Note: gnomAD: the genome aggregation database; AFR: African; NFE: non-Finnish European; EAF: Effective allele frequency. Variants that are in bold were independent of each other and used in multi-variants conditional analysis.
DISCUSSION
In this study, we performed admixture mapping of DSM-IV alcohol dependence diagnosis, DSM-IV alcohol dependence criterion count, and two SRE scores in four cohorts of AA. To our knowledge, this is the first genome-wide admixture mapping analysis of any of those four alcohol-related phenotypes in AA. One region on chromosome 4 was genome-wide significant (i.e., P-value <7.25E-05) for SRE-5.
For the chromosome 4 locus, carrying an African ancestry allele in this region increased the SRE-5 score, indicating a lower response to alcohol during the first five times the individual used alcohol. This might initially appear counterintuitive, because AA typically report faster rates of stimulation in response to alcohol compared to EA (Pedersen & McCarthy, 2013). Recent studies suggested that findings from admixture mapping need not conform to expectations regarding the direction of disease prevalence; i.e., even though a disorder is more common in one ancestral group, admixture mapping may result in identification of variants of protective effect (Molineros et al., 2013; Sofer et al., 2017; Wang et al., 2019). As long as the disease-causing variants have different allele frequencies between different ancestries, these variants will be detected by admixture mapping (Patterson et al., 2004), regardless of their directions of effect on the phenotype. Other variants that cannot be detected in our current analysis might be responsible for the population-specific effect.
Multiple genes are located in the chromosome 4 GWS region. One of them is PPARGC1A (PPARG coactivator 1 alpha). Among the 15 variants that have P-values < 0.01 in fine mapping (Table 3), 13 are in introns of PPARGC1A. This gene is broadly expressed in multiple tissues including liver and brain. The protein product of this gene interacts with cAMP response element binding protein (CREB) and nuclear respiratory factors (NRFs). Studies have found that the expression of the protein product of PPARGC1A was altered in post-mortem brain tissue from alcohol dependent individuals (Blednov et al., 2015; Ponomarev, Wang, Zhang, Harris, & Mayfield, 2012). Chronic alcohol exposure has also been shown to dramatically reduce cellular cAMP levels via a pathway involving PPARGC1A (Avila et al., 2016). In cultured neuronal cells, Liu et al (2014) found that ethanol suppressed PPARGC1A expression, causing impaired mitochondrial functioning and increased cellular toxicity; while over-expression of PPARGC1A alleviated the alcohol-induced cellular toxicity (Liu et al., 2014). In a Spanish Mediterranean sample, PPARGC1A was found to be associated with alcohol consumption (Frances et al., 2008). Animal studies have reported that chronic alcohol treatment increased liver PPARGC1A expression levels in rats and this was reversed with an anti-oxidant N-acetylcysteine (Caro et al., 2014). In addition, PPARGC1A was related to reduced alcohol intake in mice (Blednov et al., 2015). Finally, while not studying SRE-5 as an outcome, one association study found that interactions between variants in PPARGC1A and alcohol consumption were significantly associated with obesity in AA but not EA (Edwards et al., 2012). Thus, there is persuasive evidence for a potential ancestry specific effect of this gene on alcohol-related response. Other genes such as DHX15 (DEATH-box helicase 15), SOD3 (superoxide dismutase 3), CCDC149 (coiled-coil domain containing 149), and LGI2 (leucine rich repeat LDI family member 2) are also located within or near the chromosome 4 GWS region; however, none of them have previously been associated with alcohol dependence or related phenotypes.
The samples included in this study for SRE scores were also utilized in a previous GWAS (Lai, Wetherill, Kapoor, et al., 2019). In that study, no variant was genome-wide significant in meta-analysis of COGA and NIAAA cohorts, and that null finding was attributed to the relatively small sample size (N=1,546)) (Lai, Wetherill, Kapoor, et al., 2019). In contrast, the current admixture mapping approach successfully identified a region on chromosome 4. Consistent with other studies, these findings corroborate the importance of applying admixture mapping for variant discovery in recently admixed samples, including those that might have been underpowered for detection using standard approaches. In that previous GWAS study of SRE, one variant (rs4770359, P-value=2.92E-08, Beta=0.16; SE=0.03; effective allele: A) on chromosome 13 was genome-wide significant for SRE-5 in COGA only but not replicated in the NIAAA cohort (P-value=0.82), and meta-analysis has a P-value of 6.33E-08 (Lai, Wetherill, Kapoor, et al., 2019). In the current admixture mapping analysis, this region was marginally associated with SRE-5 (P-value=0.08), with African ancestry increasing SRE-5 scores, which is in agreement with the prior GWAS result. This much higher P-value could be due to the large size of the inferred African ancestry block: it is about twice the size of the identified region in previous GWAS, and included only one association signal; therefore, the regional effect size might have been too small to be detected by admixture mapping.
For DSM-IV alcohol dependence diagnosis and DSM-IV alcohol dependence criterion count, we did not find any genome-wide significant blocks. Most samples in this study were included in a GWAS meta-analysis of alcohol dependence in AA and the significant association of rs2066702 in ADH1B was identified (Walters et al., 2018). The protective role of rs2066702 was confirmed in a GWAS of alcohol dependence from the Million Veteran project (Kranzler et al., 2019). However, other variants in this gene, e.g., rs1229984, also have protective effects in other ancestries, including EA. In the current admixture mapping, the inferred African ancestry block around this gene was large and included all known variants with protective effects; therefore, admixture mapping doesn’t have sufficiently high resolution to detect association. We also examined the other AA-only finding from the Million Veteran Project (rs72900220) (Kranzler et al., 2019) and found no evidence of association using the current admixture mapping approach. One possible explanation could be the low MAF of this variant (3.9%); therefore, even with admixture mapping, a much larger sample size may be required to detect the association with this variant.
Although admixture mapping can detect genes/variants that may not have been identified by current GWAS, it has several limitations. First, as shown in the ADH1B gene, multiple ancestry-specific disease-causing variants could be located in the same ancestry block, which limits the ability of admixture mapping to detect them. Second, ancestry blocks are determined by relatively common variants. If disease-causing variants have low MAF, e.g., rs72900220 identified by Kranzler et al (2019) (Kranzler et al., 2019), then larger sample sizes and much smaller blocks are needed to detect them in admixture mapping. Third, we performed a power analysis using QuantoV1.2.4 (Gauderman, 2002), assuming a MAF of 30% and the same sample size as in this study. We estimate 80% power to detect an odds ratio > 1.3 and change of score > 0.4 for binary and continuous traits, respectively. To detect variants with smaller effect, a larger sample size would be needed. Fourth, due to the design of admixture mapping and the complex LD patterns in admixed populations, no gene- or set-based tests could be performed. While large-scale GWAS will still be the major tool for genes/variants discovery, admixture mapping is a great complement to these mainstay methods. As shown in our chromosome 4 GWS region, all single variants had P-values >0.001. In GWAS, these variants would likely have been discounted; however, collectively these variants were associated with SRE-5 and explained the admixture mapping signal. Continued recruitment of participants from underrepresented and admixed populations are essential (Peterson et al., 2019) and we suggest that admixture mapping should also be performed to detect ancestry-specific disease genes/variants that may be missed by GWAS.
Supplementary Material
ACKNOWLEDGMENT
COGA: The Collaborative Study on the Genetics of Alcoholism (COGA), Principal Investigators B. Porjesz, V. Hesselbrock, T. Foroud; Scientific Director, A. Agrawal; Translational Director, D. Dick, includes eleven different centers: University of Connecticut (V. Hesselbrock); Indiana University (H.J. Edenberg, T. Foroud, J. Nurnberger Jr., Y. Liu); University of Iowa (S. Kuperman, J. Kramer); SUNY Downstate (B. Porjesz, J. Meyers, C. Kamarajan, A. Pandey); Washington University in St. Louis (L. Bierut, J. Rice, K. Bucholz, A. Agrawal); University of California at San Diego (M. Schuckit); Rutgers University (J. Tischfield, A. Brooks, R. Hart); The Children’s Hospital of Philadelphia, University of Pennsylvania (L. Almasy); Virginia Commonwealth University (D. Dick, J. Salvatore); Icahn School of Medicine at Mount Sinai (A. Goate, M. Kapoor, P. Slesinger); and Howard University (D. Scott). Other COGA collaborators include: L. Bauer (University of Connecticut); L. Wetherill, X. Xuei, D. Lai, S. O’Connor, M. Plawecki, S. Lourens (Indiana University); L. Acion (University of Iowa); G. Chan (University of Iowa; University of Connecticut); D.B. Chorlian, J. Zhang, S. Kinreich, G. Pandey (SUNY Downstate); M. Chao (Icahn School of Medicine at Mount Sinai); A. Anokhin, V. McCutcheon, S. Saccone (Washington University); F. Aliev, P. Barr (Virginia Commonwealth University); H. Chin and A. Parsian are the NIAAA Staff Collaborators. We continue to be inspired by our memories of Henri Begleiter and Theodore Reich, founding PI and Co-PI of COGA, and also owe a debt of gratitude to other past organizers of COGA, including Ting-Kai Li, P. Michael Conneally, Raymond Crowe, Wendy Reich, and Robert E. Taylor, for their critical contributions. This national collaborative study is supported by NIH Grant U10AA008401 from the National Institute on Alcohol Abuse and Alcoholism (NIAAA) and the National Institute on Drug Abuse (NIDA).
The authors acknowledge the Indiana University Pervasive Technology Institute for providing [HPC (Big Red II, Karst, Carbonate), visualization, database, storage, or consulting] resources that have contributed to the research results reported within this paper.
Funding information
COGA (the Collaborative study on the Genetics of Alcoholism) is supported by NIH Grant U10AA008401 from the National Institute on Alcohol Abuse and Alcoholism (NIAAA) and the National Institute on Drug Abuse (NIDA).
A. Agrawal receives additional funding support from NIDA (DA032573).
Genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number HHSN268201200008I.
D. Goldman, M.L. Schwandt and V.A. Ramchandani are supported by the NIAAA Division of Intramural Clinical and Biological Research.
Footnotes
CONFLICT OF INTEREST
Alison Goate is listed as an inventor on Issued U.S. Patent 8080,371, “Markers for Addiction” covering the use of certain variants in determining the diagnosis, prognosis, and treatment of addiction.
All other authors have no potential conflicts of interest.
REFERENCES
- Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, … Consortium, G. P. (2015). A global reference for human genetic variation. Nature, 526(7571), 68-+. doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvanzo AAH, Storr CL, La Flair L, Green KM, Wagner FA, & Crum RM (2011). Race/ethnicity and sex differences in progression from drinking initiation to the development of alcohol dependence. Drug and Alcohol Dependence, 118(2-3), 375–382. doi: 10.1016/j.drugalcdep.2011.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association. (1994). Diagnostic and Statistical Manual of Mental Disorders (4th edition, Revised ed.). Washington, DC: American Psychiatric Association. [Google Scholar]
- Avila DV, Barker DF, Zhang J, McClain CJ, Barve S, & Gobejishvili L (2016). Dysregulation of hepatic cAMP levels via altered Pde4b expression plays a critical role in alcohol-induced steatosis. J Pathol, 240(1), 96–107. doi: 10.1002/path.4760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierut LJ, Agrawal A, Bucholz KK, Doheny KF, Laurie C, Pugh E, … GENEVA, G. E. A. S. (2010). A genome-wide association study of alcohol dependence. Proceedings of the National Academy of Sciences of the United States of America, 107(11), 5082–5087. doi: 10.1073/pnas.0911109107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierut LJ, Goate AM, Breslau N, Johnson EO, Bertelsen S, Fox L, … Edenberg HJ (2012). ADH1B is associated with alcohol dependence and alcohol consumption in populations of European and African ancestry. Molecular Psychiatry, 17(4), 445–450. doi: 10.1038/mp.2011.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Benavidez JM, Black M, Ferguson LB, Schoenhard GL, Goate AM, … Harris RA (2015). Peroxisome proliferator-activated receptors alpha and gamma are linked with alcohol consumption in mice and withdrawal and dependence in humans. Alcohol Clin Exp Res, 39(1), 136–145. doi: 10.1111/acer.12610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslau J, Kendler KS, Su M, Gaxiola-Aguilar S, & Kessler RC (2005). Lifetime risk and persistence of psychiatric disorders across ethnic groups in the United States. Psychol Med, 35(3), 317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucholz KK, Cadoret R, Cloninger CR, Dinwiddie SH, Hesselbrock VM, Nurnberger JI Jr., … Schuckit MA (1994). A new, semi-structured psychiatric interview for use in genetic linkage studies: a report on the reliability of the SSAGA. J Stud Alcohol, 55(2), 149–158. [DOI] [PubMed] [Google Scholar]
- Caro AA, Bell M, Ejiofor S, Zurcher G, Petersen DR, & Ronis MJ (2014). N-acetylcysteine inhibits the up-regulation of mitochondrial biogenesis genes in livers from rats fed ethanol chronically. Alcohol Clin Exp Res, 38(12), 2896–2906. doi: 10.1111/acer.12576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, & Lee JJ (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience, 4, 7. doi: 10.1186/s13742-015-0047-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier K, & Caetano R (2010). Ethnicity and health disparities in alcohol research. Alcohol Res Health, 33(1-2), 152–160. [PMC free article] [PubMed] [Google Scholar]
- Chartier KG, Scott DM, Wall TL, Covault J, Karriker-Jaffe KJ, Mills BA, … Arroyo JA (2014). Framing ethnic variations in alcohol outcomes from biological pathways to neighborhood context. Alcohol Clin Exp Res, 38(3), 611–618. doi: 10.1111/acer.12304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A, … Fuchsberger C (2016). Next-generation genotype imputation service and methods. Nat Genet, 48(10), 1284–1287. doi: 10.1038/ng.3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson DA, Goldstein RB, & Grant BF (2013). Prospective correlates of drinking cessation: variation across the life-course. Addiction, 108(4), 712–722. doi: 10.1111/add.12079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson DA, Grant BF, Stinson FS, Chou PS, Huang B, & Ruan WJ (2005). Recovery from DSM-IV alcohol dependence: United States, 2001-2002. Addiction, 100(3), 281–292. doi: 10.1111/j.1360-0443.2004.00964.x [DOI] [PubMed] [Google Scholar]
- Delaneau O, Howie B, Cox AJ, Zagury JF, & Marchini J (2013). Haplotype estimation using sequencing reads. Am J Hum Genet, 93(4), 687–696. doi: 10.1016/j.ajhg.2013.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delker E, Brown Q, & Hasin DS (2016). Alcohol Consumption in Demographic Subpopulations An Epidemiologic Overview. Alcohol Research-Current Reviews, 38(1), 7–15. [PMC free article] [PubMed] [Google Scholar]
- Dick DM, Barr P, Guy M, Nasim A, & Scott D (2017). Review: Genetic research on alcohol use outcomes in African American populations: A review of the literature, associated challenges, and implications. American Journal on Addictions, 26(5), 486–493. doi: 10.1111/ajad.12495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenberg HJ, & McClintick JN (2018). Alcohol Dehydrogenases, Aldehyde Dehydrogenases, and Alcohol Use Disorders: A Critical Review. Alcoholism-Clinical and Experimental Research, 42(12), 2281–2297. doi: 10.1111/acer.13904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards TL, Velez Edwards DR, Villegas R, Cohen SS, Buchowski MS, Fowke JH, … Matthews CE (2012). HTR1B, ADIPOR1, PPARGC1A, and CYP19A1 and obesity in a cohort of Caucasians and African Americans: an evaluation of gene-environment interactions and candidate genes. Am J Epidemiol, 175(1), 11–21. doi: 10.1093/aje/kwr272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores YN, Yee HF Jr., Leng M, Escarce JJ, Bastani R, Salmeron J, & Morales LS (2008). Risk factors for chronic liver disease in Blacks, Mexican Americans, and Whites in the United States: results from NHANES IV, 1999-2004. Am J Gastroenterol, 103(9), 2231–2238. doi: 10.1111/j.1572-0241.2008.02022.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frances F, Verdu F, Portoles O, Castello A, Sorli JV, Guillen M, & Corella D (2008). PPAR-alpha L162V and PGC-1 G482S gene polymorphisms, but not PPAR-gamma P12A, are associated with alcohol consumption in a Spanish Mediterranean population. Clin Chim Acta, 398(1-2), 70–74. doi: 10.1016/j.cca.2008.08.011 [DOI] [PubMed] [Google Scholar]
- Gauderman WJ (2002). Sample size requirements for matched case-control studies of gene-environment interaction. Statistics in Medicine, 21(1), 35–50. doi:DOI 10.1002/sim.973 [DOI] [PubMed] [Google Scholar]
- Gelernter J, Kranzler HR, Sherva R, Almasy L, Koesterer R, Smith AH, … Farrer LA (2014). Genome-wide association study of alcohol dependence: significant findings in African-and European-Americans including novel risk loci. Molecular Psychiatry, 19(1), 41–49. doi: 10.1038/mp.2013.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs TA, Okuda M, Oquendo MA, Lawson WB, Wang S, Thomas YF, & Blanco C (2013). Mental health of African Americans and Caribbean blacks in the United States: results from the National Epidemiological Survey on Alcohol and Related Conditions. Am J Public Health, 103(2), 330–338. doi: 10.2105/AJPH.2012.300891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasin DS, & Grant BF (2015). The National Epidemiologic Survey on Alcohol and Related Conditions (NESARC) Waves 1 and 2: review and summary of findings. Soc Psychiatry Psychiatr Epidemiol, 50(11), 1609–1640. doi: 10.1007/s00127-015-1088-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasin DS, Stinson FS, Ogburn E, & Grant BF (2007). Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry, 64(7), 830–842. doi: 10.1001/archpsyc.64.7.830 [DOI] [PubMed] [Google Scholar]
- Hesselbrock M, Easton C, Bucholz KK, Schuckit M, & Hesselbrock V (1999). A validity study of the SSAGA--a comparison with the SCAN. Addiction, 94(9), 1361–1370. [DOI] [PubMed] [Google Scholar]
- Klima T, Skinner ML, Haggerty KP, Crutchfield RD, & Catalano RF (2014). Exploring Heavy Drinking Patterns Among Black and White Young Adults. Journal of Studies on Alcohol and Drugs, 75(5), 839–849. doi:DOI 10.15288/jsad.2014.75.839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzler HR, Zhou H, Kember RL, Vickers Smith R, Justice AC, Damrauer S, … Gelernter J (2019). Genome-wide association study of alcohol consumption and use disorder in 274,424 individuals from multiple populations. Nature Communications, 10(1), 1499. doi: 10.1038/s41467-019-09480-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai D, Wetherill L, Bertelsen S, Carey CE, Kamarajan C, Kapoor M, … Foroud T (2019). Genome-wide association studies of alcohol dependence, DSM-IV criterion count and individual criteria. Genes Brain Behav, 18(6), e12579. doi: 10.1111/gbb.12579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai D, Wetherill L, Kapoor M, Johnson EC, Schwandt M, Ramchandani VA, … Schuckit M (2019). Genome-wide association studies of the self-rating of effects of ethanol (SRE). Addict Biol, e12800. doi: 10.1111/adb.12800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, & Ji L (2005). Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity (Edinb), 95(3), 221–227. doi: 10.1038/sj.hdy.6800717 [DOI] [PubMed] [Google Scholar]
- Liu Z, Liu Y, Gao R, Li H, Dunn T, Wu P, … Fang X (2014). Ethanol suppresses PGC-1alpha expression by interfering with the cAMP-CREB pathway in neuronal cells. PLoS One, 9(8), e104247. doi: 10.1371/journal.pone.0104247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maples BK, Gravel S, Kenny EE, & Bustamante CD (2013). RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. Am J Hum Genet, 93(2), 278–288. doi: 10.1016/j.ajhg.2013.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molineros JE, Maiti AK, Sun C, Looger LL, Han S, Kim-Howard X, … Nath SK (2013). Admixture mapping in lupus identifies multiple functional variants within IFIH1 associated with apoptosis, inflammation, and autoantibody production. PLoS Genet, 9(2), e1003222. doi: 10.1371/journal.pgen.1003222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurnberger JI Jr., Wiegand R, Bucholz K, O’Connor S, Meyer ET, Reich T, … Porjesz B (2004). A family study of alcohol dependence: coaggregation of multiple disorders in relatives of alcohol-dependent probands. Arch Gen Psychiatry, 61(12), 1246–1256. doi: 10.1001/archpsyc.61.12.1246 [DOI] [PubMed] [Google Scholar]
- Nyholt DR (2004). A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am J Hum Genet, 74(4), 765–769. doi: 10.1086/383251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell JR, & Weeks DE (1998). PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet, 63(1), 259–266. doi: 10.1086/301904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra EJ, Mazurek A, Gignoux CR, Sockell A, Agostino M, Morris AP, … Cruz M (2017). Admixture mapping in two Mexican samples identifies significant associations of locus ancestry with triglyceride levels in the BUD13/ZNF259/APOA5 region and fine mapping points to rs964184 as the main driver of the association signal. PLoS One, 12(2), e0172880. doi: 10.1371/journal.pone.0172880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Hattangadi N, Lane B, Lohmueller KE, Hafler DA, Oksenberg JR, … Reich D (2004). Methods for high-density admixture mapping of disease genes. American Journal of Human Genetics, 74(5), 979–1000. doi:Doi 10.1086/420871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen SL, & McCarthy DM (2013). Differences in acute response to alcohol between African Americans and European Americans. Alcohol Clin Exp Res, 37(6), 1056–1063. doi: 10.1111/acer.12068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson RE, Kuchenbaecker K, Walters RK, Chen CY, Popejoy AB, Periyasamy S, … Duncan LE (2019). Genome-wide Association Studies in Ancestrally Diverse Populations: Opportunities, Methods, Pitfalls, and Recommendations. Cell, 179(3), 589–603. doi: 10.1016/j.cell.2019.08.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer M, Best N, Cowles K, & Vines K (2005). CODA: Convergence diagnosis and output analysis for MCMC. R News, 6. [Google Scholar]
- Polednak AP (2007). Secular trend in U.S. black-white disparities in selected alcohol-related cancer incidence rates. Alcohol Alcohol, 42(2), 125–130. doi: 10.1093/alcalc/agl121 [DOI] [PubMed] [Google Scholar]
- Polimanti R, Yang C, Zhao HY, & Gelernter J (2015). Dissecting ancestry genomic background in substance dependence genome-wide association studies. Pharmacogenomics, 16(13), 1487–1498. doi: 10.2217/Pgs.15.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev I, Wang S, Zhang LL, Harris RA, & Mayfield RD (2012). Gene Coexpression Networks in Human Brain Identify Epigenetic Modifications in Alcohol Dependence. Journal of Neuroscience, 32(5), 1884–1897. doi: 10.1523/Jneurosci.3136-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, & Reich D (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet, 38(8), 904–909. doi:ng1847 [pii] 10.1038/ng1847 [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, … Sham PC (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet, 81(3), 559–575. doi:AJHG44631 [pii] 10.1086/519795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransome Y, & Gilman SE (2016). The Role of Religious Involvement in Black-White Differences in Alcohol Use Disorders. J Stud Alcohol Drugs, 77(5), 792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransome Y, Slopen N, Karlsson O, & Williams DR (2017). The association between alcohol abuse and neuroendocrine system dysregulation: Race differences in a National sample. Brain Behavior and Immunity, 66, 313–321. doi: 10.1016/j.bbi.2017.07.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransome Y, Slopen N, Karlsson O, & Williams DR (2018). Elevated inflammation in association with alcohol abuse among Blacks but not Whites: results from the MIDUS biomarker study. Journal of Behavioral Medicine, 41(3), 374–384. doi: 10.1007/s10865-017-9905-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich T, Edenberg HJ, Goate A, Williams JT, Rice JP, Van Eerdewegh P, … Begleiter H (1998). Genome-wide search for genes affecting the risk for alcohol dependence. American Journal of Medical Genetics, 81(3), 207–215. doi:Doi [DOI] [PubMed] [Google Scholar]
- Russo D, Purohit V, Foudin L, & Salin M (2004). Workshop on alcohol use and health disparities 2002: a call to arms. Alcohol, 32(1), 37–43. doi: 10.1016/j.alcohol.2004.01.003 [DOI] [PubMed] [Google Scholar]
- Sacks JJ, Gonzales KR, Bouchery EE, Tomedi LE, & Brewer RD (2015). 2010 National and State Costs of Excessive Alcohol Consumption. Am J Prev Med, 49(5), e73–e79. doi: 10.1016/j.amepre.2015.05.031 [DOI] [PubMed] [Google Scholar]
- Sartor CE, Nelson EC, Lynskey MT, Madden PAF, Heath AC, & Bucholz KK (2013). Are There Differences Between Young African-American and European-American Women in the Relative Influences of Genetics Versus Environment on Age at First Drink and Problem Alcohol Use? Alcoholism-Clinical and Experimental Research, 37(11), 1939–1946. doi: 10.1111/acer.12185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuckit MA, Smith TL, & Tipp JE (1997). The Self-Rating of the Effects of alcohol (SRE) form as a retrospective measure of the risk for alcoholism. Addiction, 92(8), 979–988. [PubMed] [Google Scholar]
- Schuckit MA, Tipp JE, Smith TL, Wiesbeck GA, & Kalmijn J (1997). The relationship between Self-Rating of the Effects of alcohol and alcohol challenge results in ninety-eight young men. J Stud Alcohol, 58(4), 397–404. [DOI] [PubMed] [Google Scholar]
- Seldin MF, Pasaniuc B, & Price AL (2011). New approaches to disease mapping in admixed populations. Nat Rev Genet, 12(8), 523–528. doi: 10.1038/nrg3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sempos CT, Rehm A, Wu TJ, Crespo CJ, & Trevisan M (2003). Average volume of alcohol consumption and all-cause mortality in African Americans: The NHEFS cohort. Alcoholism-Clinical and Experimental Research, 27(1), 88–92. doi: 10.1097/01.Alc.0000046597.92232.73 [DOI] [PubMed] [Google Scholar]
- Shield KD, Gmel G, Kehoe-Chan T, Dawson DA, Grant BF, & Rehm J (2013). Mortality and Potential Years of Life Lost Attributable to Alcohol Consumption by Race and Sex in the United States in 2005. PLoS One, 8(1). doi:ARTN e51923 10.1371/journal.pone.0051923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofer T, Baier LJ, Browning SR, Thornton TA, Talavera GA, Wassertheil-Smoller S, … Franceschini N (2017). Admixture mapping in the Hispanic Community Health Study/Study of Latinos reveals regions of genetic associations with blood pressure traits. PLoS One, 12(11), e0188400. doi: 10.1371/journal.pone.0188400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahre M, Roeber J, Kanny D, Brewer RD, & Zhang X (2014). Contribution of excessive alcohol consumption to deaths and years of potential life lost in the United States. Prev Chronic Dis, 11, E109. doi: 10.5888/pcd11.130293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaeth PA, Wang-Schweig M, & Caetano R (2017). Drinking, Alcohol Use Disorder, and Treatment Access and Utilization Among U.S. Racial/Ethnic Groups. Alcohol Clin Exp Res, 41(1), 6–19. doi: 10.1111/acer.13285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters RK, Polimanti R, Johnson EC, McClintick JN, Adams MJ, Adkins AE, … Agrawal A (2018). Transancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nat Neurosci, 21(12), 1656–1669. doi: 10.1038/s41593-018-0275-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Cade BE, Sofer T, Sands SA, Chen H, Browning SR, … Zhu X (2019). Admixture mapping identifies novel loci for obstructive sleep apnea in Hispanic/Latino Americans. Hum Mol Genet, 28(4), 675–687. doi: 10.1093/hmg/ddy387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer CJ, Li Y, & Abecasis GR (2010). METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics, 26(17), 2190–2191. doi: 10.1093/bioinformatics/btq340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler CA, Nelson GW, & Smith MW (2010). Admixture Mapping Comes of Age. Annual Review of Genomics and Human Genetics, Vol 11, 11, 65–89. doi: 10.1146/annurev-genom-082509-141523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witbrodt J, Mulia N, Zemore SE, & Kerr WC (2014). Racial/Ethnic Disparities in Alcohol-Related Problems: Differences by Gender and Level of Heavy Drinking. Alcoholism-Clinical and Experimental Research, 38(6), 1662–1670. doi: 10.1111/acer.12398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AL, Vadhavkar S, Singh G, & Omary MB (2008). Epidemiology of alcohol-related liver and pancreatic disease in the United States. Arch Intern Med, 168(6), 649–656. doi: 10.1001/archinte.168.6.649 [DOI] [PubMed] [Google Scholar]
- Zapolski TCB, Pedersen SL, McCarthy DM, & Smith GT (2014). Less Drinking, Yet More Problems: Understanding African American Drinking and Related Problems. Psychological Bulletin, 140(1), 188–223. doi: 10.1037/a0032113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemore SE, Karriker-Jaffe KJ, Mulia N, Kerr WC, Ehlers CL, Cook WK, … Greenfield TK (2018). The Future of Research on Alcohol-Related Disparities Across U.S. Racial/Ethnic Groups: A Plan of Attack. J Stud Alcohol Drugs, 79(1), 7–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Hu Y, Li B, Abecasis GR, & Liu DJ (2016). RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics, 32(9), 1423–1426. doi: 10.1093/bioinformatics/btw079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo LJ, Luo XG, Listman JB, Kranzler HR, Wang S, Anton RF, … Gelernter J (2009). Population admixture modulates risk for alcohol dependence. Human Genetics, 125(5-6), 605–613. doi: 10.1007/s00439-009-0647-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.