

Abstract
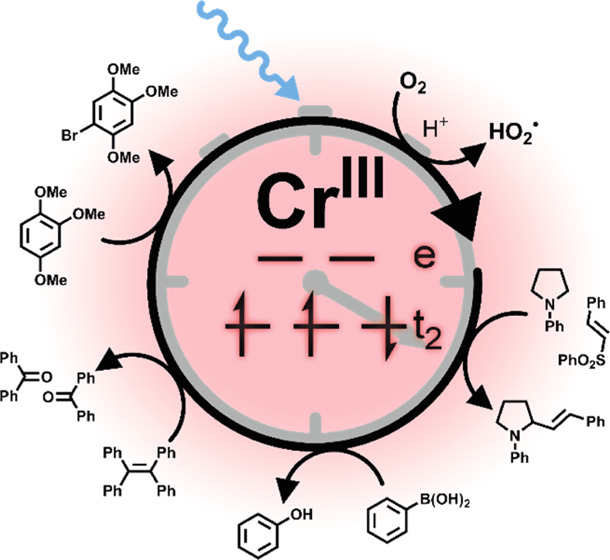
The photoredox activity of well-known RuII complexes stems from metal-to-ligand charge transfer (MLCT) excited states, in which a ligand-based electron can initiate chemical reductions and a metal-centered hole can trigger oxidations. CrIII polypyridines show similar photoredox properties, although they have fundamentally different electronic structures. Their photoactive excited state is of spin-flip nature, differing from the electronic ground state merely by a change of one electron spin, but with otherwise identical d-orbital occupancy. We find that the driving-force dependence for photoinduced electron transfer from 10 different donors to a spin-flip excited state of a CrIII complex is very similar to that for a RuII polypyridine, and thereby validate the concept of estimating the redox potential of d3 spin-flip excited states in analogous manner as for the MLCT states of d6 compounds. Building on this insight, we use our CrIII complex for photocatalytic reactions not previously explored with this compound class, including the aerobic bromination of methoxyaryls, oxygenation of 1,1,2,2-tetraphenylethylene, aerobic hydroxylation of arylboronic acids, and the vinylation of N-phenyl pyrrolidine. This work contributes to understanding the fundamental photochemical properties of first-row transition-metal complexes in comparison to well-explored precious-metal-based photocatalysts.
Introduction
Transition-metal complexes with the 4d6 and 5d6 valence electron configurations are widely employed in photophysics and photochemistry. Prominent representatives are RuII polypyridine and cyclometalated IrIII complexes, with applications in light-emitting devices,1−3 dye-sensitized solar cells,4 artificial photosynthesis,5−7 photocatalysis,8−11 and photodynamic therapy.12,13 The versatility of these d6 metal complexes is largely due to their electronic structure with metal-centered highest occupied molecular orbitals (HOMOs) and ligand-based lowest unoccupied molecular orbitals (LUMOs), leading to an energetically lowest excited state of 3MLCT type (Figure 1). In this photoactive state, the ligand-based excited electron can easily be transferred to suitable acceptors to accomplish photochemical reductions, or the metal-centered electron vacancy can induce photo-oxidations.14 The metal-to-ligand charge transfer (MLCT) character of the lowest excited state with an electron delocalized over one or several ligands and a hole centered on the metal is therefore often considered as the key to these photoredox properties.15 Similar reactivity is observable for CuI (3d10) complexes with photoactive MLCT excited states.16−19
Figure 1.

Energy-level diagrams and pertinent electron configurations of octahedrally coordinated low-spin d6 and d3 metal complexes (including metal-based t2 and e orbitals along with ligand-centered π* orbitals) in their respective electronic ground states (1A1, 4A2) and lowest electronically excited states (3MLCT, 2E). For low-spin d6 metal complexes, the 3MLCT–1A1 energy difference roughly equals the difference between the π* and the t2 orbitals. For d3 metal complexes, the 2E–4A2 energy difference reflects the change in electron exchange interactions. The structures of [Ru(bpz)3]2+ and [Cr(dqp)2]3+ are shown at the bottom.
CrIII polypyridine complexes look structurally similar to well-known RuII analogues at first glance but have fundamentally different electronic structures due to their 3d3 valence electron configuration. In strong ligand fields, the two lowest (usually thermally equilibrated) 2E and 2T1 states are so-called spin-flip excited states, which are metal-centered and do not involve the ligands. The 2E state differs from the electronic ground state merely by the change of one electron spin, but otherwise the d-orbital occupancy is identical (Figure 1).20 Evidently, there is no ligand-based electron and no metal-centered hole as in the case of 3MLCT-excited d6 metal complexes (Figure 1).
The reduction potential of any electronically excited state (E0(*A/ A•–)), regardless of its specific nature, is usually estimated by adding the energy difference between the ground state and the energetically lowest excited state (E00, in units of eV, divided by the elementary charge e) to the ground-state reduction potential (E0(A/A•–))
| 1 |
Equation 1 can be rationalized within the orbital picture of low-spin d6 metal complexes (Figure 1) because the MLCT excitation creates an electron vacancy in a metal-centered t2-orbital, energetically roughly by the value of E00 below the ligand-based π* orbital, in which electrochemical reduction in the electronic ground state occurs. Consequently, excited-state reduction occurs more easily by the value E00/e than ground-state reduction. The same logic can however not be applied to the 2E state of CrIII because in this case no electron vacancy in a metal-centered t2-orbital is newly created, merely one of the electron spins is flipped. In this case, E00 reflects essentially a change in electron exchange interactions.
Some CrIII complexes have long been known as strong photo-oxidants,21−25 whereas more recently developed CrIII compounds are yet underexplored in this capacity, presumably because the main focus until now has been on their photoluminescence behavior.26−41 So far, mostly photo-cycloadditions and direct reactions with oxygen have been investigated with CrIII photocatalysts,42−48 representing a much more restricted reactivity scope than that of RuII and IrIII compounds.15
In the first part of this study, we directly compare the photo-oxidation behavior of the recently developed [Cr(dqp)2]3+ complex (Figure 1, dqp = 2,6-bis(8′-quinolinyl)pyridine)28 with the well-known [Ru(bpz)3]2+ complex (bpz = 2,2′-bipyrazine). We find that the driving-force dependence for photo-oxidation of 10 different electron donors is very similar for both [Cr(dqp)2]3+ and [Ru(bpz)3]2+. Our analysis confirms that the redox potential of the lowest spin-flip excited state in CrIII complexes can be determined in the same manner as for 3MLCT states in RuII compounds, despite the very fundamental differences explained earlier (Figure 1). In the second part of this study, we have been able to increase the scope of photoredox catalytic applications of CrIII spin-flip excited states substantially beyond the current state-of-the-art.
Results and Discussion
Photoinduced Electron Transfer Reactivity
The UV–vis absorption spectrum of [Cr(dqp)2](PF6)3 in dry acetonitrile (Figure 2a, black trace) features the typical absorptions at 335 and 377 nm and a shoulder tailing to 450 nm. Upon excitation at 410 nm, two narrow emission bands at 724 and 747 nm are observable (Figure 2a, red trace), attributable to luminescence from the energetically lowest 2E state (747 nm) and the thermally populated 2T1 state (724 nm).28 Excitation spectra monitoring these two emission wavelengths match the ground state UV–vis absorption spectrum of [Cr(dqp)2]3+ well (Figure S6). With increasing temperature, the emission intensity at 724 nm increases relative to the emission intensity at 747 nm (Figure S10). At 77 K, only the emission at 747 nm remains observable (Figure 2a, red dotted trace). The 2E state is typically energetically below the 2T1 state for most CrIII complexes with pseudo-octahedral geometry,28,30,49 though there seem to be exceptions.27,38,39,50
Figure 2.
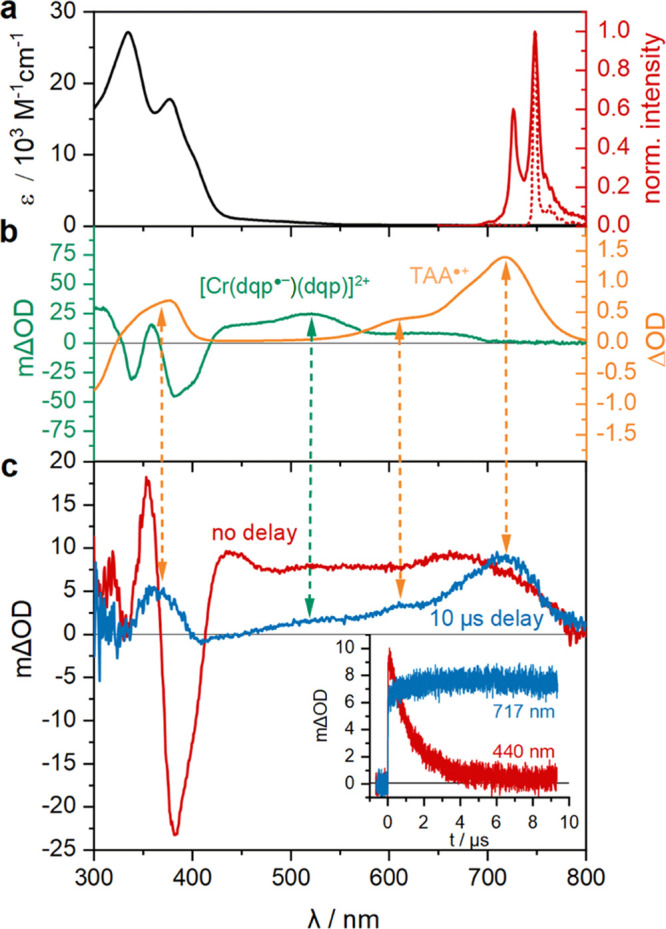
(a) UV–vis absorption (black) and emission spectra at 293 K in acetonitrile (solid red) and at 77 K in 1:1 (v/v) dimethyl sulfoxide (DMSO)/water (dotted red) of 35 μM [Cr(dqp)2](PF6)3. The emission spectra were recorded following excitation at 410 nm. (b) Spectro-electrochemical UV–vis difference spectrum of 350 μM [Cr(dqp)2](PF6)3 in acetonitrile containing 0.1 M tetra-n-butylammonium hexafluorophosphate (TBAPF6), obtained at an applied potential of −0.5 V vs SCE (green) along with the UV–vis difference spectrum of 400 μM TAA obtained in the same solvent by applying a potential of +0.8 V vs SCE (orange). The UV–vis spectra recorded prior to application of the potential served as a baseline in both cases. (c) Transient UV–vis absorption spectrum of 35 μM [Cr(dqp)2](PF6)3 and 100 μM TAA measured after excitation with 425 nm laser pulses of ∼10 ns duration directly after the laser pulse (red) and after a delay of 10 μs (blue). Inset: Transient absorption decay at 440 nm, corresponding to the disappearance of 2E/2T1-excited [Cr(dqp)2]3+ (red) and absorption growth at 717 nm, corresponding to TAA•+ formation (blue). All measurements were performed in dry and argon-saturated acetonitrile at 20 °C.
In cyclic voltammetry, the first reduction of [Cr(dqp)2]3+ is observed at −0.4 V vs SCE (Figure S1), in line with the literature.28 Consequently, a potential of −0.5 V vs SCE was applied for reductive UV–vis (Figure 2b, green trace) and NIR spectro-electrochemistry (Figure S3). The resulting difference spectra (for which the UV–vis and NIR absorption spectra of the [Cr(dqp)2]3+ complex prior to applying the potential served as baseline) are assigned to the CrIII complex with one dqp ligand reduced to its radical anion form. In particular, the presence of an intervalence charge-transfer band in the NIR region is in line with reports on CrIII complexes with other redox noninnocent polypyridyl ligands.51,52
We started our studies of photoinduced electron transfer with a solution containing 35 μM [Cr(dqp)2]3+ and 100 μM tris(p-anisyl)amine (TAA) in dry argon-saturated acetonitrile at 20 °C. [Cr(dqp)2]3+ was excited at 425 nm with ∼10 ns laser pulses and transient UV–vis absorption spectra were recorded after different delay times. Immediately after the laser pulses, the observable transient UV–vis absorption spectrum (Figure 2c, red trace) is identical to the transient UV–vis absorption spectrum of a neat solution of [Cr(dqp)2]3+ in dry argon-saturated acetonitrile recorded under identical conditions, but without any TAA additive present (Figure S7). This transient UV–vis absorption spectrum features a band maximum at 440 nm along with a prominent bleach at 380 nm, and these two signals are assigned to the 2E/2T1-excited [Cr(dqp)2]3+ complex because they show identical decay behavior (τ0 = 2.1 ms) as the photoluminescence signal at 747 nm (Figure S8). For the solution containing both [Cr(dqp)2]3+ and TAA, a fundamentally different transient UV–vis absorption spectrum becomes recordable after a delay of 10 μs (Figure 2c, blue trace). The initially present spectral features have disappeared, and a new absorption maximum at 717 nm is instead observable. This new band is due to the TAA radical cation, the spectrum of which was obtained in an independent spectro-electrochemical UV–vis absorption measurement, by applying a constant potential of +0.8 V vs SCE to a 400 μM solution of TAA in acetonitrile (Figure 2b, orange trace). The remaining absorption bands in that transient UV–vis absorption spectrum are caused by the one-electron reduced complex ([Cr(dqp)2]2+), in particular a weak absorption near 520 nm (dashed green double arrow in Figure 2). In other words, the TA spectrum recorded with a time delay of 10 μs (Figure 2c, blue trace) is essentially a linear combination of the spectro-electrochemical UV–vis difference spectra of one-electron reduced metal complex (Figure 2b, green trace) and one-electron oxidized TAA (Figure 2b, orange trace). The inset in Figure 2c shows the decay of the transient UV–vis absorption signal of 2E/2T1-excited [Cr(dqp)2]3+ at 440 nm and the growth of TAA•+ absorption at 717 nm (single-wavelength kinetics), both of which follow the same kinetics. Collectively, the data in Figure 2 clearly demonstrate that electron transfer occurs from TAA directly to photoexcited [Cr(dqp)2]3+, i.e., reductive excited-state quenching is taking place. For this specific experiment, a comparatively low concentration of electron donor (100 μM) was employed to facilitate the recording of spectra over the entire visible range and to decelerate the electron transfer event to a convenient timescale.
To explore the driving-force dependence of photoinduced electron transfer with [Cr(dqp)2]3+, a series of electron donors with oxidation potentials (E0(D•+/D)) in the range of 0.55–1.54 V vs SCE (Table S1) were chosen. Using time-resolved luminescence and UV–vis transient absorption spectroscopy, the reductive quenching of 2E/2T1-excited [Cr(dqp)2]3+ was studied in Stern–Volmer-type experiments. Full experimental details are reported in the Supporting Information, the individual Stern–Volmer plots are given in Figures S11–S20, and the obtained rate constants for bimolecular excited-state quenching (kq) are summarized in Table S1. The blue circles in Figure 3 illustrate how kq depends on ΔGET, the reaction free energy for electron transfer from the individual donors to 2E/2T1-excited [Cr(dqp)2]3+. Equation 2 was used to estimate ΔGET, where E0(D•+/D) is the donor oxidation potential and E0(*A/A•–) represents the excited-state acceptor reduction potential as defined by eq 1; work terms are neglected in eq 2.
| 2 |
The trend observable for the blue circles in Figure 3 fits qualitatively with the observations by Rehm and Weller in their studies of bimolecular quenching of excited organic acceptors with a similar set of electron donors.53,54 To compare the photoinduced electron transfer behavior of [Cr(dqp)2]3+ to a d6 metal complex, we identified [Ru(bpz)3]2+ as a suitable reference compound in combination with the same electron donor set. The respective luminescence quenching data and Stern–Volmer plots, from which the bimolecular excited-state quenching rate constants kq were obtained are displayed in Figures S21–S30, and the respective kq values are summarized in Table S1. Using eq 2 and the relevant donor oxidation (Table S1) and acceptor reduction potentials (Figure S5), ΔGET values between +0.09 and −0.9 eV are calculated, and the red squares in Figure 3 illustrate the dependence of kq on ΔGET for 3MLCT-excited [Ru(bpz)3]2+. For 2E/2T1-excited [Cr(dqp)2]3+, the ΔGET range is slightly different and ranges from +0.28 to −0.71 eV, due to the 0.19 V difference in excited-state reduction potentials estimated for [Cr(dqp)2]3+ and [Ru(bpz)3]2+ on the basis of eq 1. Evidently, the two data sets in Figure 3 are very similar despite the fundamental differences between 3MLCT states and 2E/2T1 spin-flip state outlined above.
Figure 3.

Rate constants for bimolecular electron transfer (kET) from selected electron donors (inset at the bottom left) to photoexcited [Cr(dqp)2]3+ (blue circles) and [Ru(bpz)3]2+ (red squares) as a function of reaction free energy (ΔGET), as estimated by eqs 1 and 2 (using reduction potentials and E00 values as indicated in Figures S4 and S5). Least-square fits using eq 3 are shown as solid blue and red lines. Best fits were obtained with kd values of (1.7 ± 0.2) × 1010 and (1.8 ± 0.2) × 1010 M–1 s–1, and ΔGET‡(0) values of 0.14 ± 0.01 and 0.16 ± 0.01 eV for [Cr(dqp)2]3+ and [Ru(bpz)3]2+ in acetonitrile, respectively.
Quantitatively, the dependence of kq on ΔGET can be described with the empirical Rehm–Weller equation
| 3 |
| 4 |
where kd is the rate constant for diffusional encounter, ΔGET‡ is the free energy of activation, ΔGET‡(0) is the activation free energy at ΔGET = 0, kB is the Boltzmann constant, and T is the temperature.53,54 Least-square fits for both data sets to eq 3 are shown as solid blue and red lines in Figure 3. For [Cr(dqp)2]3+, best fits yielded kd = (1.7 ± 0.2) × 1010 M–1 s–1 and ΔGET‡(0) = 0.14 ± 0.01 eV, whereas for [Ru(bpz)3]2+, kd = (1.8 ± 0.2) × 1010 M–1 s–1 and ΔGET‡(0) = 0.16 ± 0.01 eV were obtained. These values are similar to those previously reported in a comparative study of [Cr(bpy)3]3+ and [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) with similar electron donors.24 Furthermore, the parameters obtained for [Cr(dqp)2]3+ and [Ru(bpz)3]2+ are close to each other, reflecting the finding that the dependence of kq on ΔGET is very similar in these two cases. Since the ΔGET values in Figure 3 rely on the assumption that eq 1 is applicable to both metal complexes, the similarity of the two experimental data sets strongly suggests that this fundamental assumption is indeed valid also for the photoactive spin-flip excited state of CrIII.
This finding implies that relative to the electronic ground state, ligand-based reduction in the metal-centered 2E/2T1-excited state of [Cr(dqp)2]3+ is facilitated to the same extent as in the case of 3MLCT-excited d6 metal complexes. In the simplified orbital picture of Figure 1, this cannot be readily rationalized because it fails to capture differences in electron–electron interaction between the individual microstates. The key point is that in the 4A2 ground state of CrIII, three electron spins are aligned and thus experience exchange stabilization, whereas in the 2E excited state of CrIII that exchange stabilization is lowered due to the spin-flip. The energy difference between the 2E and 4A2 states (E00 in Figure 4a) then essentially corresponds to that change in exchange stabilization, rather than a difference in orbital energies, such as the case for the MLCT excited state of RuII.14,55Equation 1 can be seen as the adaptation of a thermodynamic cycle, in which every change to free energy of a state directly influences its thermodynamic properties including its redox potentials. Consequently, the nature of the excited state is in principle irrelevant to the applicability of eq 1, except if major structural changes have to be considered that influence the entropy of the system. In the MLCT excited states of d6 metal complexes with a formally oxidized metal center and a formally reduced ligand, such structural changes are present, as visualized by a horizontal shift along the nuclear coordinate axis of the potential well diagram in Figure 4b. In a spin-flip excited state, on the other hand, this is not the case, and consequently, the estimation of the excited state redox potential by eq 1 should be even more valid than for MLCT excited states.
Figure 4.

(a) Qualitative energy-level diagrams containing the pertinent electronically excited states of [Cr(dqp)2]3+ (left) and [Ru(bpz)3]2+ (right), along with relevant photophysical processes (ISC = intersystem crossing, IC = internal conversion) and selected microstates (gray). (b) Qualitative single-configurational coordinate diagrams for [Cr(dqp)2]3+ (left) and [Ru(bpz)3]2+ (right).
Cage Escape Yield and Photostability
To determine the cage escape yield of the photoproducts between 2E/2T1-excited [Cr(dqp)2]3+ and TAA, relative actinometry in combination with laser flash photolysis was performed. TAA was used as an electron donor because its radical cation has a characteristic absorption at 717 nm with a molar extinction coefficient (ε717) of 32 800 M–1 cm–1.56 In comparison, the one-electron reduced chromium complex features negligible extinction at that wavelength (Figure 2); hence, the TAA•+ photoproduct can be detected relatively cleanly at 717 nm. Solutions of [Cr(dqp)2]3+ in acetonitrile and [Ru(bpy)3]2+ in water with an identical optical density at 417 nm were prepared (Figure 5, inset). TAA was added to the [Cr(dqp)2]3+ sample to reach a concentration of 10 mM, and since TAA is transparent at 417 nm, this does not affect the optical density at that wavelength. The high TAA concentration of 10 mM ensures fast and efficient quenching of the 2E/2T1-excited CrIII complex with nearly instantaneous formation of electron transfer photoproducts. To quantify the number of photons absorbed by both solutions at 417 nm, the recovery of the MLCT ground-state bleach of [Ru(bpy)3]2+ at 455 nm was monitored (red trace in the main part of Figure 5). Based on the known change in molar extinction coefficient at that wavelength (Δε455= −10 100 M–1 cm–1),57 and based on the fact that intersystem crossing from the initially excited 1MLCT to the 3MLCT state for [Ru(bpy)3]2+ in water is quantitative,58 the concentration of 3MLCT-excited [Ru(bpy)3]2+ complexes can be determined from the experimentally observable change in optical density at t = 0 of the respective bleach recovery. In the representation chosen for Figure 5a, the respective concentration has been set to a normalized value of 1 (red trace). Since the [Cr(dqp)2]3+/TAA solution has the same optical density at the excitation wavelength of 417 nm, [Cr(dqp)2]3+ is expected to absorb an equal amount of photons as the [Ru(bpy)3]2+ solution. Based on the experimentally detectable change in optical density at 717 nm for the [Cr(dqp)2]3+/TAA solution (blue trace in the main part of Figure 5a) and the above-mentioned molar extinction coefficient (ε717 = 32 800 M–1 cm–1),56 the concentration of TAA•+ can be determined. Relative to the 3MLCT concentration after [Ru(bpy)3]2+ excitation, the TAA•+ concentration amounts to 13%, as emphasized by the horizontal black dotted line in Figure 5a. This photoproduct quantum yield of 13% is higher than the cage escape yields of 1–7% determined recently for an FeIII complex and various electron donors in polar solvents.59 This finding illustrates that the formation of electron transfer photoproducts from a spin-flip excited state can be similarly efficient as from the charge-transfer excited state of another first-row transition-metal complex.
Figure 5.
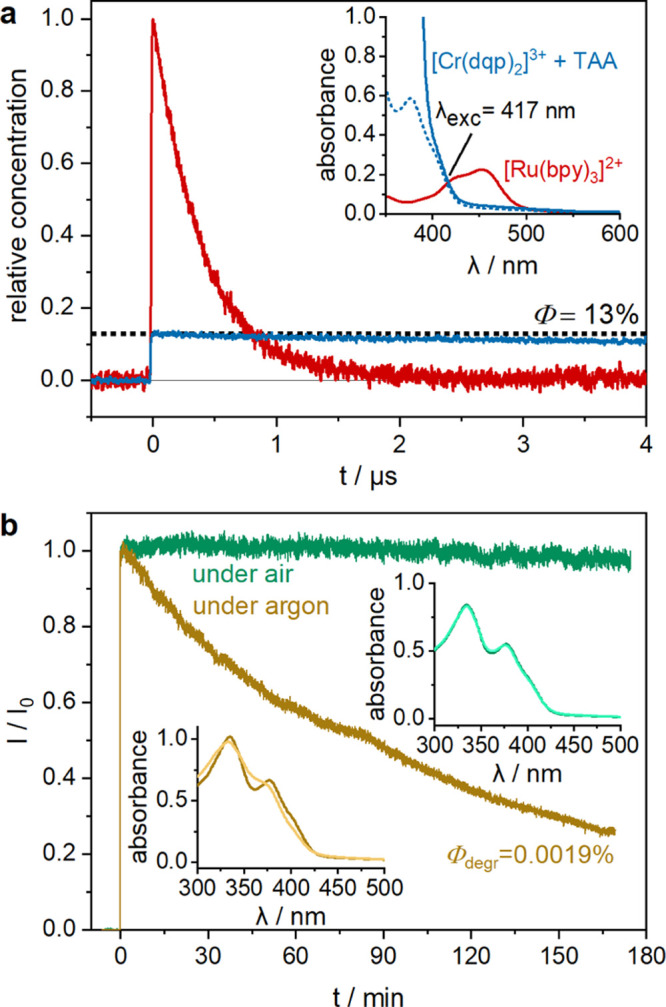
(a) TAA•+ cage escape yield after excitation of 35 μM [Cr(dqp)2](PF6)3 at 417 nm in the presence of 10 mM TAA in aerated dry acetonitrile at 20 °C. The concentration of photoproduced TAA•+ was determined using the molar extinction coefficient of TAA•+ at 717 nm (ε717 = 32 800 M–1 cm–1)56 and the experimentally determined ΔOD value. The amount of absorbed light at 417 nm was determined by measuring the concentration of 3MLCT excited [Ru(bpy)3]2+, using the known change in molar extinction coefficient related to the MLCT ground-state bleach at 455 nm (Δε455 = −10 100 M–1 cm–1)57 and the experimentally accessible ΔOD value. Inset: UV–vis absorption spectra of [Cr(dqp)2](PF6)3 (blue dotted line) and [Cr(dqp)2](PF6)3 + TAA (blue solid line) in acetonitrile and [Ru(bpy)3]Cl2 in water (red solid line) showing the same optical density at 417 nm (to ensure absorption of equal amounts of light by both solutions at that wavelength). (b) Photoluminescence intensity of [Cr(dqp)2](PF6)3 (detected at 747 nm) in argon-saturated (yellow) and air-saturated (green) acetonitrile at 20 °C over the course of continuous irradiation with a high-power (520 mW) blue laser (405 nm). The initial optical density at 405 nm was 0.32 for the argon-saturated solution and 0.25 for the air-saturated solution; the luminescence intensities were normalized to the initial luminescence intensity (I0). The insets contain the respective UV–vis absorption spectra before (dark colored) and after (light colored) irradiation for 170 min.
For photocatalytic applications, high quantum yields
for photoproduct
formation are desirable because low yields imply that more excitation
events are necessary until full substrate conversion is reached, whereby
the risk of photodegradation increases. To probe the photostability
of [Cr(dqp)2]3+, an argon-saturated acetonitrile
solution with an optical density of 0.32 at 405 nm was irradiated
by a 520 mW continuous-wave laser at that wavelength, and the emission
intensity at 747 nm was monitored as a function of irradiation time
(yellow trace in Figure 5b). After 170 min of continuous high-power illumination, roughly
75% of the initially present [Cr(dqp)2]3+ have
degraded, presumably because the very long 2E/2T1-excited state lifetime of 2.1 ms (Figure S8) gives ample opportunity for decomposition. A photodegradation
quantum yield 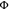 degr, defined as the number of
decomposed photosensitizer molecules divided by the number of absorbed
photons (see Supporting Information for
details), of 0.0019% was determined. This value compares favorably
to the photodegradation quantum yield determined for [Ru(bpy)3]2+ (
degr, defined as the number of
decomposed photosensitizer molecules divided by the number of absorbed
photons (see Supporting Information for
details), of 0.0019% was determined. This value compares favorably
to the photodegradation quantum yield determined for [Ru(bpy)3]2+ (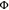 degr = 0.028%) and fac-[Ir(ppy)3] (ppy = 2-phenylpyridine) under similar conditions
(
degr = 0.028%) and fac-[Ir(ppy)3] (ppy = 2-phenylpyridine) under similar conditions
(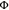 degr = 0.0018%).60 Remarkably, in an air-saturated solution, essentially no
photodegradation is detectable after 170 minutes of irradiation with
the same experimental setup, even though under these conditions 1O2 is likely formed, thereby shortening the 2E/2T1 lifetime to 31 μs (Figure S9). The importance of oxygen to enhance
the stability of CrIII catalysts has been noted already
in previous studies of catalytic Diels–Alder cycloaddition
reactions,43 and a similar improvement
in photostability in the presence of a quencher has been observed
recently for an IrIII photocatalyst.61 Though the experiments in Figure 5b involved very low [Cr(dqp)2]3+ concentrations in closed cuvettes, their outcome suggests
that aerobic conditions can be used for photoredox reactions requiring
higher concentrations and an extended amount of irradiation time.
degr = 0.0018%).60 Remarkably, in an air-saturated solution, essentially no
photodegradation is detectable after 170 minutes of irradiation with
the same experimental setup, even though under these conditions 1O2 is likely formed, thereby shortening the 2E/2T1 lifetime to 31 μs (Figure S9). The importance of oxygen to enhance
the stability of CrIII catalysts has been noted already
in previous studies of catalytic Diels–Alder cycloaddition
reactions,43 and a similar improvement
in photostability in the presence of a quencher has been observed
recently for an IrIII photocatalyst.61 Though the experiments in Figure 5b involved very low [Cr(dqp)2]3+ concentrations in closed cuvettes, their outcome suggests
that aerobic conditions can be used for photoredox reactions requiring
higher concentrations and an extended amount of irradiation time.
Photocatalytic Applications
According to the driving-force dependence in Figure 3, an estimate of 1.26 V vs SCE based on eq 1 is adequate for 2E/2T1-excited [Cr(dqp)2]3+ (Figure S4). This opens the possibility for photocatalytic applications that have classically been performed with RuII or IrIII complexes or organic photo-oxidants.15,63 So far, mostly photo-cycloaddition reactions have been investigated with CrIII complexes based on polypyridine ligands, and important insights concerning the role of oxygen as a redox shuttle in the catalytic cycle of CrIII have been gained.42−45,48 A more recently developed CrIII complex with a much more negative and metal-centered reduction was used to generate 1O2, which subsequently triggered C–H activation of aliphatic amines followed by trapping with nitriles.47 However, 1O2 generation can be accomplished with many different photosensitizers,64−66 and there seems to be no obvious benefit from using CrIII complexes for that purpose. However, compared to most RuII polypyridine and many cyclometalated IrIII complexes,67 [Cr(dqp)2]3+ and its CrIII congeners should be relatively strong photo-oxidants,46 and therefore we explored several new examples of photocatalytic applications that specifically exploit the strong oxidative power of 2E/2T1-excited [Cr(dqp)2]3+.
Acridinium cations have evolved into increasingly popular photo-oxidants over the past decade,68,69 and the prototypical 9-mesityl-10-methylacridinium ion (Acr+-Mes) with an excited-state reduction potential of 1.88 V vs SCE70 has been used for diverse photocatalytic applications including a photocatalytic oxidative bromination method.62 Although 2E/2T1-excited [Cr(dqp)2]3+ is a weaker oxidant than Acr+-Mes, we anticipated that this CrIII complex could catalyze the bromination of methoxyaryls because the Stern–Volmer experiments summarized in Figure 3 indicate that this compound class is readily photo-oxidized. Using 1,2,4-trimethoxybenzene (1,2,4-TMB) as substrate, the singly brominated product is obtained with 100% conversion and 91% yield after 120 min of CrIII irradiation at 415 nm (Figures 6a and S34). The selective bromination at the unsubstituted 5-position was followed as a function of irradiation time using 1H NMR spectroscopy (Figure S35). In a control experiment, in which 5 mol % of neat ligand instead of [Cr(dqp)2](PF6)3 was added, no reactivity was observed, and the employed substrates do not absorb at the output of the 415 nm light-emitting diode (LED) (Figures S33 and S41). When using tetra-n-butylammonium bromide instead of HBr, no reaction occurs, indicating that acidic conditions are crucial for this reaction to proceed. To assess whether a radical chain mechanism could be operative, an irradiation pause of 30 min was made after 30 min of irradiation (leading to 31% conversion). The 1H NMR spectra before and after the pause were identical and showed no further reaction progress, and additional photo-irradiation was needed to drive the reaction to completion (Figure S36).
Figure 6.
(a) Photocatalytic oxidative bromination of methoxyarenes with HBr. Irradiation with a 415 nm LED (7.0 W output) with 400 nm long pass filter, 4 mM substrate, 1.0 equiv of 1,2,4,5-tetrachlorobenzene as internal 1H NMR standard, 5.0 equiv of HBr, and 1 mol % of catalyst in 0.6 mL of CD3CN in closed NMR tubes under air at up to 35 °C (temperature fluctuations caused by the LED radiation). Yields are referenced to the internal standard. aIsolated yield with 17.5 mM substrate in 20 mL of acetonitrile. (b) Plausible reaction mechanism with 1,2,4-trimethoxybenzene as substrate, based on a previous study with 9-mesityl-10-methylacridinium ion (Acr+-Mes) as a photocatalyst.62
Photocatalytic bromination of 1,3,5-trimethoxybenzene (1,3,5-TMB) led to a maximum yield for the mono-brominated product of 78% after 30 min (Figure 6a), along with minor formation (8%) of the dibrominated product and a 96% conversion of the starting material. After 210 min of irradiation, full conversion of the starting material and of the singly brominated product was achieved, to yield 91% of doubly brominated product (Figure S37). The reaction progress as a function of irradiation time was followed by 1H NMR spectroscopy and revealed almost textbook-quality A → B → C reaction kinetics (Figure S39).
Bromination of 1-methoxynaphthalene (1-MN) was performed on a larger scale with 17.5 mM substrate in 20 mL of acetonitrile, and the product 1-bromo-4-methoxynaphthalene was isolated with 91% yield (Figures 6a and S40). In principle, the reductive excited-state quenching of the photocatalyst by 1-MN, 1,3,5-TMB and the mono-brominated form of the latter (the redox potentials of 1,3,5-TMB and its mono-brominated form are similar, Figure S38 and Table S1) are endergonic by up to 0.3 eV (Figure 3), yet the respective photoreactions readily proceed with excellent yields. This is likely due to the very long lifetime of 2E/2T1-excited [Cr(dqp)2]3+ even in aerated acetonitrile (τair = 31 μs, Figure S9), giving ample opportunity for collisional encounters with substrate molecules and thereby increasing the probability of productive electron transfer. We term this behavior “high kinetic reactivity”,71 which compensates for the lower thermodynamic reactivity compared to some acridinium ions.
A plausible mechanism for the reactions from Figure 6a, based on a previously reported mechanism for acridinium photocatalysis,62 is shown in Figure 6b. After irradiation of [Cr(dqp)2]3+ with visible light, the substrate reductively quenches the 2E/2T1 excited states and forms a methoxyaryl radical cation, that is subsequently trapped by bromide. Oxygen recovers the photocatalyst and is itself protonated to give the hydroperoxyl radical, which then acts as an oxidant for the brominated radical intermediate of the substrate. In this reaction, 1 equiv of product is formed together with hydrogen peroxide, and the latter can act as an oxidant in combination with HBr to form a second equivalent of product.62
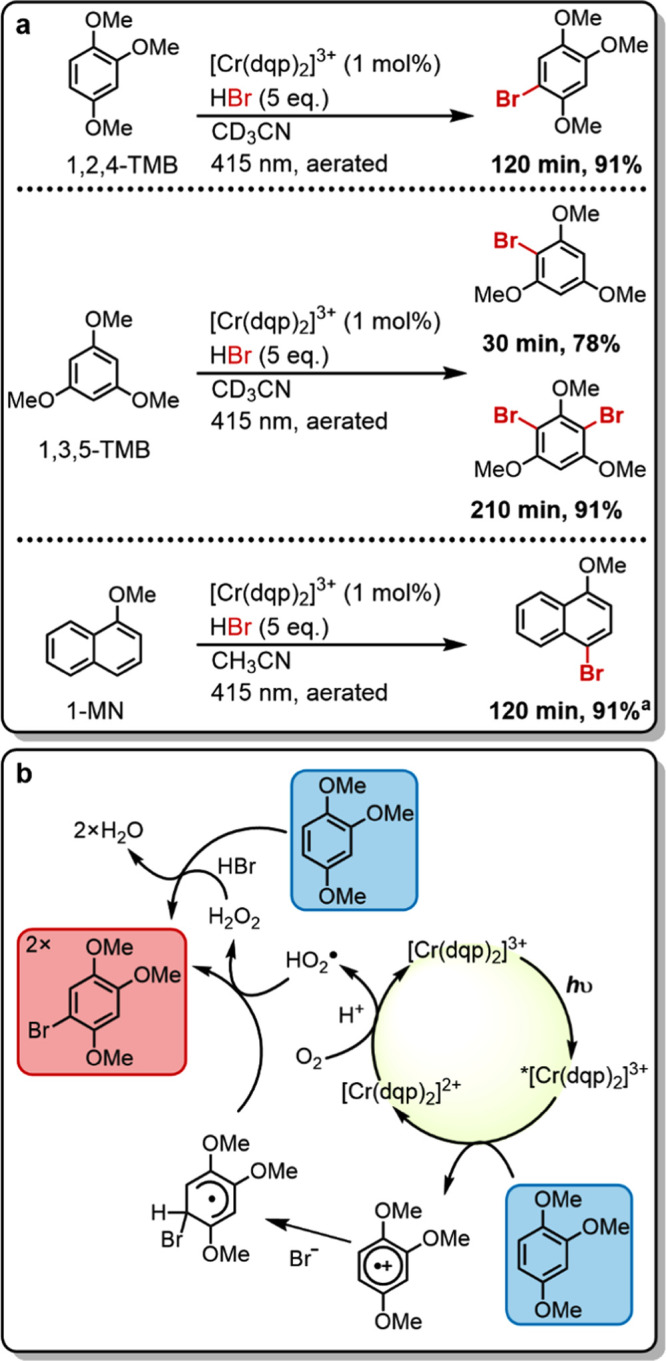
Next, we explored the photocatalytic oxygenation of 1,1,2,2-tetraphenylethene (TPE) using [Cr(dqp)2](PF6)3 (Figure 7a). The reaction progress was followed by 1H NMR spectroscopy and yields were referenced to an internal standard. Product formation was confirmed by comparison with a representative 1H NMR spectrum of benzophenone. The insolubility of [Cr(dqp)2](PF6)3 in apolar solvents and the low solubility of TPE in polar solvents necessitated the use of a mixture of deuterated acetonitrile and chloroform (1/1, v/v). An aerated sample reached full conversion after 6 h of irradiation at 415 nm with 43% formation of the desired product along with a known peroxide intermediate formed in the overall reaction to the desired product (marked purple in Figure 7b, see mechanistic discussion below).72 After an additional hour of irradiation, only minor conversion of the respective peroxide intermediate to the final product was observed. The reaction was repeated under an oxygen atmosphere (instead of air), but full conversion was again reached after 6 h with a product yield of only 36%, and further irradiation gave no improvement. In the photocatalytic bromination experiments from Figure 6a, acidic conditions had a beneficial effect on product formation, and consequently, the reaction from Figure 7a was repeated under air in the presence of 20 equiv of trifluoroacetic acid (TFA). Under these conditions, full conversion of the starting material was already reached after 4 h of irradiation with a product yield of 61%. After two additional hours of irradiation, the product yield further increased to 64%, and after a total of 24 h of irradiation, a yield of 75% was reached, with no detectable NMR resonances attributable to the above-mentioned peroxide intermediate left.
Figure 7.
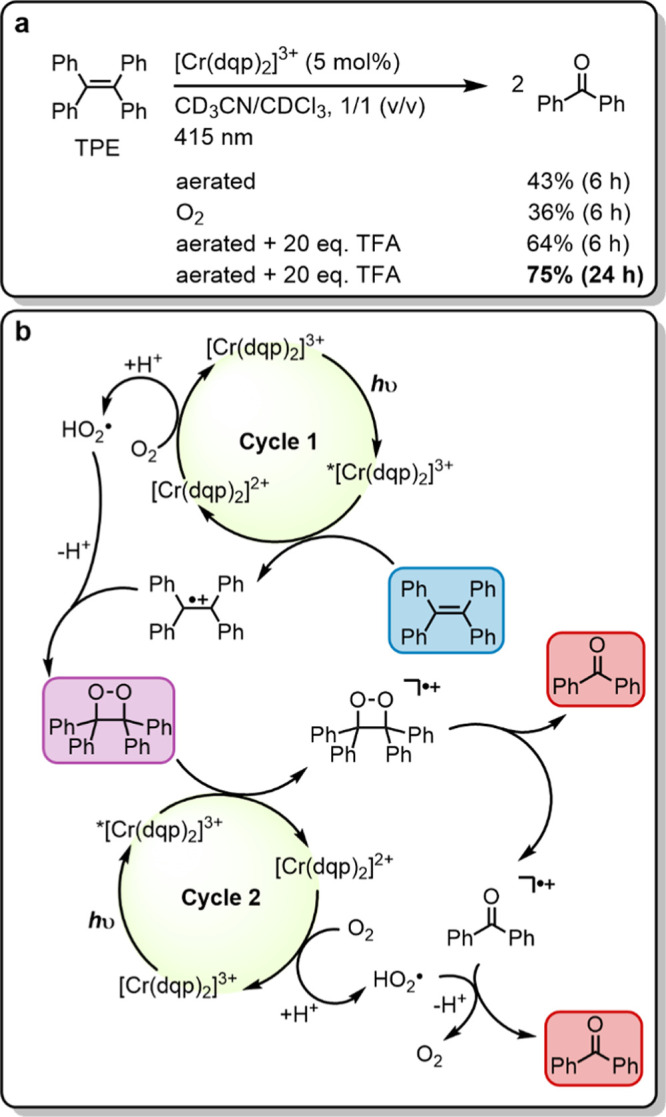
(a) Photocatalytic oxygenation of 1,1,2,2-tetraphenylethene (TPE) with oxygen followed by C–C bond cleavage to two benzophenone units. Irradiation with a 415 nm LED (7.0 W output) with 400 nm long pass filter, 15 mM substrate, 2.0 equiv of 1,4-dichlorobenzene as internal 1H NMR standard, 20 equiv of trifluoroacetic acid (TFA), and 5 mol % of catalyst in 0.6 mL of CD3CN/CDCl3 (1/1, v/v) in closed NMR tubes under air at up to 35 °C (temperature fluctuations caused by the LED radiation). Yields are referenced to the internal standard. (b) Plausible mechanism based on a previous report with 9-mesityl-10-methylacridinium ion (Acr+-Mes) as a photocatalyst.72
Based on these observations, the previously reported mechanistic proposal for the oxygenation of TPE by an acridinium photocatalyst is adapted as illustrated in Figure 7b.72 In the first catalytic cycle, TPE reductively quenches 2E/2T1-excited [Cr(dqp)2]3+ and forms the radical cation of TPE. Oxygen recovers the photocatalyst under protonation to the hydroperoxyl radical, which subsequently reacts with TPE•+ to form the above-mentioned peroxide intermediate (TPE-O2, marked in purple in Figure 7b). In the second catalytic cycle, TPE-O2 reductively quenches 2E/2T1-excited [Cr(dqp)2]3+ and forms TPE-O2•+, and the latter decomposes into one benzophenone molecule and one benzophenone radical cation. Oxygen likely mediates both the oxidation of [Cr(dqp)2]2+ back to [Cr(dqp)2]3+, as well as the reduction of the benzophenone radical cation to a second equivalent of product. This mechanistic proposal seems in line with the observation of improved product yields under acidic conditions. Although cycle 1 seemed fully functional in the absence of TFA, as is evident from the observable formation of the peroxide intermediate, the functioning of cycle 2 seems to be enhanced by acid. A similar electron shuttle role of oxygen was observed previously in a photo-cycloaddition experiment with [(Cr(Ph2phen)3)]3+ (Ph2phen = 4,7-diphenyl-1,10-phenanthroline) as photocatalyst.43 The reduced yield when using an oxygen atmosphere (instead of simply aerated samples) can be understood on the basis of more efficient 1O2 formation, which causes side product formation.
The reductive 2E/2T1 excited-state quenching of [Cr(dqp)2]3+ by TPE is endergonic by 0.32 eV (E0= 1.58 V vs SCE)72 and therefore evidently profits from the long excited state lifetime of [Cr(dqp)2]3+ under standard atmospheric oxygen concentrations (τair = 31 μs, Figure S9). The lower oxygen concentration under atmospheric conditions compared to neat O2 atmosphere can be compensated by the addition of a strong Brønsted acid, similar to that found for Brønsted acid-promoted CuI to CuII oxidation by oxygen in a [Cu(dap)2]+ (dap = 2,9-bis(4-methoxyphenyl)-1,10-phenanthroline)-catalyzed C–H bond functionalization.73 In principle, a doublet–triplet energy transfer reaction pathway74 is also conceivable, but TPE has a triplet energy above 2.2 eV, which makes this reaction endergonic by at least 0.5 eV.75 Against this background, photoinduced electron transfer seems more plausible than doublet–triplet energy transfer.
Photocatalytic aerobic hydroxylation of arylboronic acids and their pinacol esters to phenols has been explored previously with [Ru(bpy)3]2+ as a photocatalyst, using aliphatic amines as sacrificial electron donors in dimethylformamide (DMF) under an oxygen atmosphere.76 Later, a metal-free alternative with methylene blue in a mixture of acetonitrile and water was developed.77 The same photoreaction was furthermore accomplished in neat water with water-soluble N-substituted 3(10H)-acridone.78 The same reactivity turns out to be reproducible with [Cr(dqp)2](PF6)3 for different substrates and N,N-diisopropylethylamine (DIPEA) as a sacrificial electron donor in a 4/1 (v/v) acetonitrile–water mixture under an oxygen atmosphere. Boronic acid and 4-bromo and 4-methoxyboronic acid reacted with full conversion and high yields after 4–6.5 h of irradiation at 415 nm (Figures 8a and S45–S47). 4-Cyanophenylboronic acid pinacol ester was studied on a larger scale with 83.3 mM substrate in 3 mL of acetonitrile/water (4/1, v/v), and the desired product was isolated in 91% yield (see the Supporting Information and Figure S49 for more details).
Figure 8.
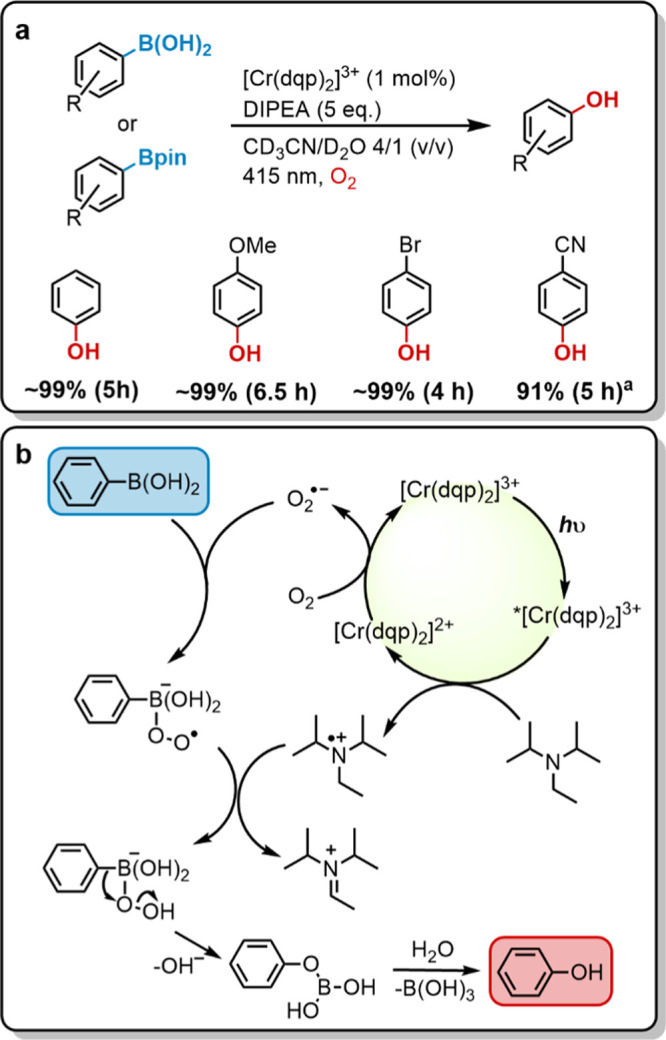
(a) Photocatalytic aerobic oxidative hydroxylation of arylboronic acids. Irradiation with a 415 nm LED (7.0 W output) with 400 nm long pass filter, 50 mM substrate, 1.0 equiv of phenyl trimethylsilane as internal 1H NMR standard, 5 equiv of N,N-diisopropylethylamine (DIPEA), and 1 mol % catalyst in 0.6 mL of CD3CN/D2O (4/1, v/v) in closed NMR tubes under an oxygen atmosphere up to 35 °C (temperature fluctuations caused by the LED radiation). Yields are referenced to the internal standard. aReaction was performed with 83.3 mM 4-cyanophenylboronic acid pinacol ester in 3 mL of acetonitrile/water (4/1, v/v). The yield was determined by isolating the product. (b) Plausible reaction mechanism as suggested previously for different photocatalysts.76−78
The reactions were conducted under an oxygen atmosphere because reactions under air resulted in a maximum of 50% conversion after 20 h of irradiation. At high oxygen concentrations, energy transfer to 3O2 could potentially become a viable excited-state quenching pathway that competes with the reductive quenching by DIPEA. The latter is oxidized at a potential of +0.72 V vs SCE,79,80 and therefore the reductive quenching pathway is exergonic by 0.54 eV. Based on the Rehm–Weller relationship from Figure 3, a kq value close to the diffusion limit can be expected for the reaction of DIPEA with 2E/2T1-excited [Cr(dqp)2]3+. On the other hand, given the shortening of the 2E/2T1 lifetime between deaerated acetonitrile (τ0 = 2.1 ms, Figure S8) and air-saturated acetonitrile (τair = 31 μs, Figure S9), a rate constant of ∼1.3 × 107 M–1 s–1 can be estimated for energy transfer from [Cr(dqp)2]3+ to 3O2 (see Figure S9 for details). Given a DIPEA concentration of 250 mM and an estimated oxygen concentration of 8.1 mM (based on an oxygen-saturated solution at ambient pressure),81 reductive excited-state quenching is by far the dominant reaction pathway, and consequently, the mechanism proposed earlier for other photocatalysts (Figure 8b) seems also plausible for [Cr(dqp)2]3+.76
The photocatalytic reactions accomplished with [Cr(dqp)2]3+ discussed above all represent overall photo-oxidation reactions, in which oxygen acts as an electron acceptor. The vinylation reaction in Figure 9 is an overall redox-neutral reaction, which however relies on an initial photoredox step. This reaction was originally described with a well-known IrIII-based photo-oxidant,82 which is reductively quenched by an N-aryl pyrrolidine substrate, resulting in an α-amino alkyl radical. The latter reacts with phenyl trans-styryl sulfone to a β-sulfonyl radical, which eliminates a sulfonyl radical to form the final vinylated product (marked in red). The sulfonyl radical is sufficiently oxidizing (+0.5 V vs SCE) to recycle the photocatalyst (Figure 9b).82
Figure 9.
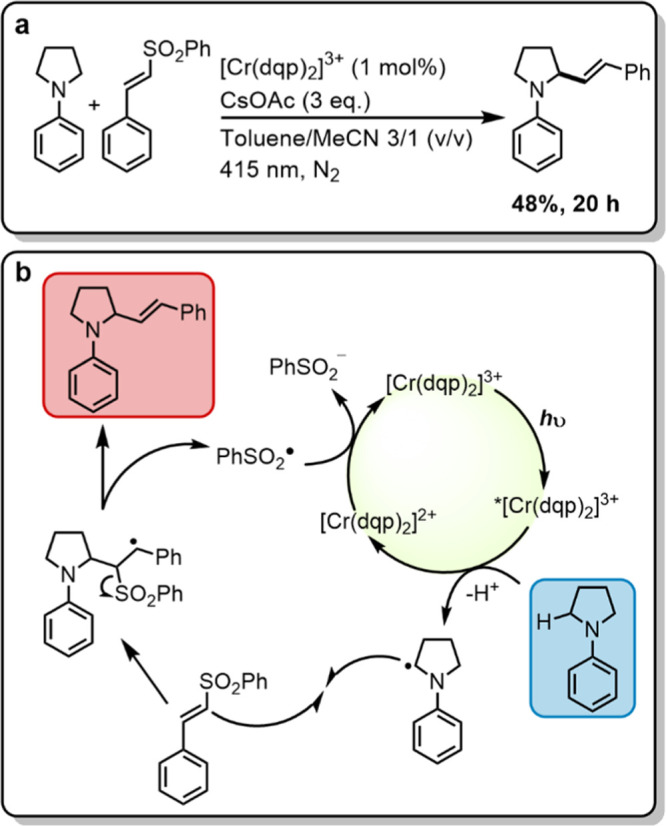
(a) Photocatalytic vinylation of an N-aryl amine. Irradiation with a 415 nm LED (7.0 W output) with a 400 nm long pass filter, 100 mM phenyl trans-styryl sulfone, 2.5 equiv N-phenyl pyrrolidine (NPP), 3.0 equiv cesium acetate, 1 mol % catalyst in 2 mL of deaerated toluene/acetonitrile (3/1 v/v) in a Schlenk tube under nitrogen at up to 35 °C (temperature fluctuations caused the LED radiation). The yield was determined by isolating the product. (b) Plausible reaction mechanism similar to that postulated for an IrIII-based photocatalyst.82
The vinylation reaction in Figure 9 is known to proceed with good yields in apolar solvents such as toluene and 1,2-dichloroethane (DCE),82 but the hexafluorophosphate salt of [Cr(dqp)2]3+ is only soluble in more polar solvents. Consequently, the reaction was initially conducted in dry nitrogen-saturated acetonitrile, using [Cr(dqp)2]3+ (1 mol %) as photocatalyst with 2.5 equiv of N-phenyl pyrrolidine (NPP), 200 μmol phenyl trans-styryl sulfone as substrate, and 3.0 equiv of cesium acetate as a base. After 20 h of irradiation at 415 nm, the product yield was only 36%. This behavior is similar to the previously reported reaction with an IrIII catalyst in polar N,N′-dimethylacetamide, which gave 28% product yield.82 When instead performing the photoreaction with [Cr(dqp)2]3+ in a nitrogen-saturated dry 3/1 (v/v) toluene/acetonitrile mixture, the isolated yield improved to 48% after 20 h of irradiation. The NPP substrate absorbs weakly in the spectral range, in which the used 415 nm LED emits (Figure S52), yet the absorption by the CrIII photocatalyst is clearly dominant, and no product formation was observable in the absence of [Cr(dqp)2]3+ under otherwise identical conditions.
Conclusions
With tridendate polypyridyl ligands ensuring nearly perfect octahedral and rigid coordination geometries, CrIII complexes are currently undergoing a renaissance, especially with regard to their photoluminescence properties.20,26−41,83 By contrast, their photoredox behavior has received comparatively little attention, and studies so far seem to be mostly limited to photocatalytic cycloadditions and singlet oxygen formation.24,42−48,84 In this work, the application scope of CrIII complexes in photoredox catalysis has been extended to three different types of aerobic photo-oxidation reactions and to an overall redox-neutral vinylation reaction (Figures 6–9). The similarity of the driving-force dependence for photoinduced electron transfer from 10 different donors to 2E/2T1-excited [Cr(dqp)2]3+ and 3MLCT-excited [Ru(bpz)3]2+ (Figure 3) demonstrates that the excited state oxidation potential of [Cr(dqp)2]3+ approaches that of [Ru(bpz)3]2+. Furthermore, and perhaps more importantly, it verifies that the oxidation power of d3 spin-flip excited states can indeed be estimated in an analogous manner as in the case of d6 3MLCT excited states (using eq 1), despite the fact the lowest spin-flip excitation does not change the d-orbital occupancy (Figure 1), in line with early studies.23 This contrasts with FeII polypyridine complexes that exhibit photoreactivity from distorted metal-centered (MC) states, and where the estimation of the pertinent excited-state redox potential is more cumbersome because E00 is tricky to obtain.85 The results in Figure 3 imply that relative to the electronic ground state, ligand-based reduction in the metal-centered 2E/2T1-excited state of [Cr(dqp)2]3+ is facilitated to the same extent as in the case of 3MLCT excited d6 metal complexes, despite the fact that in the chromium(III) complex no electron is promoted from a metal- to a ligand-based orbital and merely an electron spin is flipped instead. However, the spin-flip implies that the exchange stabilization is lowered, which is perhaps not readily evident from the simplistic orbital picture in Figure 1. Photoexcitation to the 2E state creates an α-spin hole (easier to reduce) and a β-spin electron, which is easier to remove due to lack of exchange stabilization. In principle, the terms “α-spin hole” and “β-spin electron” are synonyms, and these terms are merely used here in an attempt of illustrating some analogy between the CrIII spin-flip excited state and the RuII MLCT excited state (Figure 1). In one case it is primarily a change in electron–electron interaction (CrIII), whereas in the other (RuII), it is largely a difference in orbital energies that governs the redox properties in the excited state relative to the ground state. Some complications of this simple picture emerge from the fact that the primary reduction of [Cr(dqp)2]3+ is ligand-based (not metal-centered), yet the photoredox behavior of this complex can be rationalized within this framework by formally viewing its overall one-electron reduction as an initial metal-centered process, followed by intramolecular electron transfer to the ligand. Ultimately, eq 1 is best considered as the outcome of a thermodynamic cycle, in which any change in the state energy of the system caused by photon absorption directly influences the thermodynamic properties, including the redox potentials. In this treatment, the nature of the electronically excited state becomes irrelevant for the estimation of redox potentials. On a more general level, this discussion illustrates the important difference between simplistic orbital pictures (Figure 1), which do not necessarily provide a good visualization of electron–electron interactions, and electronic state pictures (Figure 4). Recent work on an isoelectronic V2+ complex presented a new computational procedure that enables the visualization of electron–electron interactions.86
The cage escape yield of 13% determined herein for the photoinduced electron transfer (PET) reaction between tris(p-anisyl)amine (TAA) and the spin-flip excited state of [Cr(dqp)2]3+ compares to values between 5 and 60% for 3MLCT-excited RuII polypyridine complexes.87−94 Unwanted in-cage charge recombination between geminate radical pairs follows spin selection rules,95,96 and hence the doublet nature of the lowest spin-flip excited state of CrIII polypyridyls could in principle lead to different behavior of spin-correlated radical pairs than in the case of 3MLCT excited states in RuII analogues. In the above-mentioned PET between TAA and 2E/2T1-excited [Cr(dqp)2]3+ complex, a singlet (S = 0) and a doublet (S = 1/2) species react with each other. The expectable primary photoproducts are a doublet species (TAA•+, S = 1/2) and the [Cr(dqp•–)(dqp)]2+ complex with different possible total spins (S = 0, 1, 2; Figure S53). In-cage charge recombination then is expected to yield TAA in its singlet ground state (S = 0) and [Cr(dqp)2]3+ in its quartet ground state (S = 3/2). This situation is fundamentally different from PET between TAA and RuII polypyridines, which leads from a singlet (S = 0) and a 3MLCT state (S = 1) to two doublets as primary photoproducts (S = 1/2 for both TAA•+ and [Ru(bpy)3]+; Figure S54), and where in-cage charge recombination leads to two singlets (S = 0). Thus, it seems that spin effects might deserve further attention in future photoredox studies with CrIII polypyridines because this could potentially provide further insight into spin control of photoinduced electron transfer and radical pair reactions.97−100
The strong photo-oxidizing properties of CrIII polypyridines compared to classical RuII complexes such as [Ru(bpy)3]2+ are attributable mostly to the higher metal oxidation state (+III vs +II), which in turn makes the ligand-centered reduction easier. This effect is somewhat counteracted by the lower 2E/2T1 energy (roughly 1.7 eV) compared to the 3MLCT energy (usually around 2.1 eV) of RuII polypyridines. CrIII polypyridines typically need to be excited in the blue spectral range (due to lack of sizeable extinction at longer wavelengths), and then intersystem crossing from the initially excited 4T (or charge-transfer) states to the 2E/2T1 states is associated with substantially greater energy loss than in common RuII complexes (Figure 4).101−103 In this sense, the “photon economy” is worse for CrIII polypyridines, and even though this may seem less important than “atom economy” in chemical reactions,104−106 the dissipation of excess excitation energy is often associated with detrimental photophysical and photochemical processes.14,107 Interestingly, [Cr(dqp)2]3+ exhibits a remarkable photostability in coordinating acetonitrile upon continuous high-power photo-irradiation at 405 nm (Figure 5b), far better than [Ru(bpy)3]2+ under comparable conditions.60 This is likely related to the relatively undistorted nature of the 2E/2T1 spin-flip excited states, in which structural changes such as Cr–N bond elongations are comparatively minor,108 but then it becomes important that reverse intersystem crossing from 2E/2T1 to 4T is minimized.109 This is the case in the near-perfectly octahedrally coordinated complexes obtained with dqp and other newly developed ligands, highlighting the importance of these recently discovered molecular rubies.26−41,47,74,110−112 Tridentate ligands such as dqp furthermore offer the possibility for “self-healing” during photo-irradiation, whereby a temporarily de-coordinated pyridine subunit can re-coordinate over time because the two remaining pyridine subunits still hold the overall ligand attached to the metal center.113
Compared to organic photo-oxidants with short-lived electronically excited states such as acridinium dyes,68−70 CrIII polypyridines seem to benefit from a substantially increased kinetic reactivity. Acridinium dyes typically react from singlet excited states with lifetimes on the order of a few nanoseconds, whereas [Cr(dqp)2]3+ exhibits a 2E/2T1 excited state lifetime on the millisecond timescale in deaerated solution and 31 μs in air-saturated acetonitrile at 20 °C. Remarkably, the above-mentioned photostability is maintained in aerated solution (Figure 5b), which opens the door to photoredox reactions under aerobic conditions, whereby oxygen typically acts to recover the initial catalyst state following its photo-reduction by a substrate.
New ligand designs for CrIII continue to be published and a recently disclosed complex surpassed the excited-state properties of the prototypical d6-metal-based [Ru(bpy)3]2+ in several respects.39 However, until now, the photoredox properties of CrIII might have been underappreciated, and we hope that our study contributes to the general understanding of photocatalysis based on first-row transition-metal compounds. Now, as an increasing number of complexes based on different Earth-abundant transition metals with photoactive excited states in solution are discovered,114−118 it seems possible that some of them will be amenable to unusual photochemical applications that are not necessarily viable with traditional precious-metal-based compounds.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c04465.
Equipment and methods; synthetic protocols and characterization data; electrochemical characterization; spectroscopic data; 1H NMR data; details of Stern–Volmer experiments; photostability; and microstates in photoinduced electron transfer (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was funded by the Swiss National Science Foundation through grant number 200021_178760.
The authors declare no competing financial interest.
Supplementary Material
References
- Henwood A. F.; Zysman-Colman E. Lessons Learned in Tuning the Optoelectronic Properties of Phosphorescent Iridium(III) Complexes. Chem. Commun. 2017, 53, 807–826. 10.1039/C6CC06729H. [DOI] [PubMed] [Google Scholar]
- Mills I. N.; Porras J. A.; Bernhard S. Judicious Design of Cationic, Cyclometalated Ir(III) Complexes for Photochemical Energy Conversion and Optoelectronics. Acc. Chem. Res. 2018, 51, 352–364. 10.1021/acs.accounts.7b00375. [DOI] [PubMed] [Google Scholar]
- Kerr E.; Hayne D. J.; Soulsby L. C.; Bawden J. C.; Blom S. J.; Doeven E. H.; Henderson L. C.; Hogan C. F.; Francis P. S. A Redox-Mediator Pathway for Enhanced Multi-Colour Electrochemiluminescence in Aqueous Solution. Chem. Sci. 2022, 13, 469–477. 10.1039/D1SC05609C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardo S.; Meyer G. J. Photodriven Heterogeneous Charge Transfer with Transition-Metal Compounds Anchored to TiO2 Semiconductor Surfaces. Chem. Soc. Rev. 2009, 38, 115–164. 10.1039/B804321N. [DOI] [PubMed] [Google Scholar]
- Magnuson A.; Anderlund M.; Johansson O.; Lindblad P.; Lomoth R.; Polivka T.; Ott S.; Stensjö K.; Styring S.; Sundström V.; Hammarström L. Biomimetic and Microbial Approaches to Solar Fuel Generation. Acc. Chem. Res. 2009, 42, 1899–1909. 10.1021/ar900127h. [DOI] [PubMed] [Google Scholar]
- Troian-Gautier L.; Turlington M. D.; Wehlin S. A. M.; Maurer A. B.; Brady M. D.; Swords W. B.; Meyer G. J. Halide Photoredox Chemistry. Chem. Rev. 2019, 119, 4628–4683. 10.1021/acs.chemrev.8b00732. [DOI] [PubMed] [Google Scholar]
- Diluzio S.; Connell T. U.; Mdluli V.; Kowalewski J. F.; Bernhard S. Understanding Ir(III) Photocatalyst Structure-Activity Relationships: A Highly Parallelized Study of Light-Driven Metal Reduction Processes. J. Am. Chem. Soc. 2022, 144, 1431–1444. 10.1021/jacs.1c12059. [DOI] [PubMed] [Google Scholar]
- Sayre H.; Ripberger H. H.; Odella E.; Zieleniewska A.; Heredia D. A.; Rumbles G.; Scholes G. D.; Moore T. A.; Moore A. L.; Knowles R. R. PCET-Based Ligand Limits Charge Recombination with an Ir(III) Photoredox Catalyst. J. Am. Chem. Soc. 2021, 143, 13034–13043. 10.1021/jacs.1c01701. [DOI] [PubMed] [Google Scholar]
- Shon J. H.; Kim D.; Rathnayake M. D.; Sittel S.; Weaver J.; Teets T. S. Photoredox Catalysis on Unactivated Substrates with Strongly Reducing Iridium Photosensitizers. Chem. Sci. 2021, 12, 4069–4078. 10.1039/D0SC06306A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strieth-Kalthoff F.; James M. J.; Teders M.; Pitzer L.; Glorius F. Energy Transfer Catalysis Mediated by Visible Light: Principles, Applications, Directions. Chem. Soc. Rev. 2018, 47, 7190–7202. 10.1039/C8CS00054A. [DOI] [PubMed] [Google Scholar]
- Bevernaegie R.; Wehlin S. A. M.; Elias B.; Troian-Gautier L. A Roadmap Towards Visible Light Mediated Electron Transfer Chemistry with Iridium(III) Complexes. ChemPhotoChem 2021, 5, 217–234. 10.1002/cptc.202000255. [DOI] [Google Scholar]
- Mari C.; Pierroz V.; Ferrari S.; Gasser G. Combination of Ru(II) Complexes and Light: New Frontiers in Cancer Therapy. Chem. Sci. 2015, 6, 2660–2686. 10.1039/C4SC03759F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A.; Turro C.; Kodanko J. J. Ru(II) Polypyridyl Complexes Derived from Tetradentate Ancillary Ligands for Effective Photocaging. Acc. Chem. Res. 2018, 51, 1415–1421. 10.1021/acs.accounts.8b00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias-Rotondo D. M.; McCusker J. K. The Photophysics of Photoredox Catalysis: A Roadmap for Catalyst Design. Chem. Soc. Rev. 2016, 45, 5803–5820. 10.1039/C6CS00526H. [DOI] [PubMed] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls T. P.; Bissember A. C. Developments in Visible-Light-Mediated Copper Photocatalysis. Tetrahedron Lett. 2019, 60, 150883 10.1016/j.tetlet.2019.06.042. [DOI] [Google Scholar]
- Hernandez-Perez A. C.; Collins S. K. Heteroleptic Cu-Based Sensitizers in Photoredox Catalysis. Acc. Chem. Res. 2016, 49, 1557–1565. 10.1021/acs.accounts.6b00250. [DOI] [PubMed] [Google Scholar]
- Hossain A.; Bhattacharyya A.; Reiser O. Copper’s Rapid Ascent in Visible-Light Photoredox Catalysis. Science 2019, 364, eaav9713 10.1126/science.aav9713. [DOI] [PubMed] [Google Scholar]
- Glaser F.; Wenger O. S. Red Light-Based Dual Photoredox Strategy Resembling the Z-Scheme of Natural Photosynthesis. JACS Au 2022, 2, 1488–1503. 10.1021/jacsau.2c00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzmann W. R.; Moll J.; Heinze K. Spin-Flip Luminescence. Photochem. Photobiol. Sci. 2022, 10.1007/s43630-022-00186-3. [DOI] [PubMed] [Google Scholar]
- Bolletta F.; Maestri M.; Moggi L.; Balzani V. Bimolecular Electron Transfer Reactions of Electronically Excited States of Co-ordination Compounds. J. Chem. Soc., Chem. Commun. 1975, 901–902. 10.1039/c39750000901. [DOI] [Google Scholar]
- Serpone N.; Jamieson M. A.; Henry M. S.; Hoffman M. Z.; Bolletta F.; Maestri M. Excited-State Behavior of Polypyridyl Complexes of Chromium(III). J. Am. Chem. Soc. 1979, 101, 2907–2916. 10.1021/ja00505a019. [DOI] [Google Scholar]
- Ballardini R.; Varani G.; Scandola F.; Balzani V. Bimolecular Electron Transfer Processes of Electronically Excited Tris(2,2′-bipyridine)chromium(III). J. Am. Chem. Soc. 1976, 98, 7432–7433. 10.1021/ja00439a059. [DOI] [Google Scholar]
- Ballardini R.; Varani G.; Indelli M. T.; Scandola F.; Balzani V. Free Energy Correlation of Rate Constants for Electron Transfer Quenching of Excited Transition Metal Complexes. J. Am. Chem. Soc. 1978, 100, 7219–7223. 10.1021/ja00491a017. [DOI] [Google Scholar]
- Farran R.; Le-Quang L.; Mouesca J. M.; Maurel V.; Jouvenot D.; Loiseau F.; Deronzier A.; Chauvin J. [Cr(ttpy)2]3+ as a Multi-Electron Reservoir for Photoinduced Charge Accumulation. Dalton Trans. 2019, 48, 6800–6811. 10.1039/C9DT00848A. [DOI] [PubMed] [Google Scholar]
- Förster C.; Dorn M.; Reuter T.; Otto S.; Davarci G.; Reich T.; Carrella L.; Rentschler E.; Heinze K. Ddpd as Expanded Terpyridine: Dramatic Effects of Symmetry and Electronic Properties in First Row Transition Metal Complexes. Inorganics 2018, 6, 86. 10.3390/inorganics6030086. [DOI] [Google Scholar]
- Otto S.; Harris J. P.; Heinze K.; Reber C. Molecular Ruby under Pressure. Angew. Chem., Int. Ed. 2018, 57, 11069–11073. 10.1002/anie.201806755. [DOI] [PubMed] [Google Scholar]
- Jiménez J.-R.; Doistau B.; Cruz C. M.; Besnard C.; Cuerva J. M.; Campaña A. G.; Piguet C. Chiral Molecular Ruby [Cr(dqp)2]3+ with Long-Lived Circularly Polarized Luminescence. J. Am. Chem. Soc. 2019, 141, 13244–13252. 10.1021/jacs.9b06524. [DOI] [PubMed] [Google Scholar]
- Otto S.; Förster C.; Wang C.; Resch-Genger U.; Heinze K. A Strongly Luminescent Chromium(III) Complex Acid. Chem. - Eur. J. 2018, 24, 12555–12563. 10.1002/chem.201802797. [DOI] [PubMed] [Google Scholar]
- Jiménez J.-R.; Poncet M.; Doistau B.; Besnard C.; Piguet C. Luminescent Polypyridyl Heteroleptic CrIII Complexes with High Quantum Yields and Long Excited State Lifetimes. Dalton Trans. 2020, 49, 13528–13532. 10.1039/D0DT02872J. [DOI] [PubMed] [Google Scholar]
- Wang C.; Kitzmann W. R.; Weigert F.; Förster C.; Wang X.; Heinze K.; Resch-Genger U. Matrix Effects on Photoluminescence and Oxygen Sensitivity of a Molecular Ruby. ChemPhotoChem 2022, 6, e202100296 10.1002/cptc.202100296. [DOI] [Google Scholar]
- Boden P.; Di Martino-Fumo P.; Niedner-Schatteburg G.; Seidel W.; Heinze K.; Gerhards M. Transient FTIR Spectroscopy after One- And Two-Colour Excitation on a Highly Luminescent Chromium(III) Complex. Phys. Chem. Chem. Phys. 2021, 23, 13808–13818. 10.1039/D1CP01077H. [DOI] [PubMed] [Google Scholar]
- Dee C.; Zinna F.; Kitzmann W. R.; Pescitelli G.; Heinze K.; Di Bari L.; Seitz M. Strong Circularly Polarized Luminescence of an Octahedral Chromium(III) Complex. Chem. Commun. 2019, 55, 13078–13081. 10.1039/C9CC06909G. [DOI] [PubMed] [Google Scholar]
- Wang C.; Otto S.; Dorn M.; Kreidt E.; Lebon J.; Sršan L.; Di Martino-Fumo P.; Gerhards M.; Resch-Genger U.; Seitz M.; Heinze K. Deuterated Molecular Ruby with Record Luminescence Quantum Yield. Angew. Chem., Int. Ed. 2018, 57, 1112–1116. 10.1002/anie.201711350. [DOI] [PubMed] [Google Scholar]
- Sinha N.; Jiménez J.; Pfund B.; Prescimone A.; Piguet C.; Wenger O. S. A Near-Infrared-II Emissive Chromium(III) Complex. Angew. Chem., Int. Ed. 2021, 60, 23722–23728. 10.1002/anie.202106398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto S.; Grabolle M.; Förster C.; Kreitner C.; Resch-Genger U.; Heinze K. [Cr(ddpd)2]3+: A Molecular, Water-Soluble, Highly NIR-Emissive Ruby Analogue. Angew. Chem., Int. Ed. 2015, 54, 11572–11576. 10.1002/anie.201504894. [DOI] [PubMed] [Google Scholar]
- Otto S.; Scholz N.; Behnke T.; Resch-Genger U.; Heinze K. Thermo-Chromium: A Contactless Optical Molecular Thermometer. Chem. - Eur. J. 2017, 23, 12131–12135. 10.1002/chem.201701726. [DOI] [PubMed] [Google Scholar]
- Treiling S.; Wang C.; Förster C.; Reichenauer F.; Kalmbach J.; Boden P.; Harris J. P.; Carrella L. M.; Rentschler E.; Resch-Genger U.; Reber C.; Seitz M.; Gerhards M.; Heinze K. Luminescence and Light-Driven Energy and Electron Transfer from an Exceptionally Long-Lived Excited State of a Non-Innocent Chromium(III) Complex. Angew. Chem., Int. Ed. 2019, 58, 18075–18085. 10.1002/anie.201909325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenauer F.; Wang C.; Förster C.; Boden P.; Ugur N.; Báez-Cruz R.; Kalmbach J.; Carrella L. M.; Rentschler E.; Ramanan C.; Niedner-Schatteburg G.; Gerhards M.; Seitz M.; Resch-Genger U.; Heinze K. Strongly Red-Emissive Molecular Ruby [Cr(bpmp)2]3+ Surpasses [Ru(bpy)3]2+. J. Am. Chem. Soc. 2021, 143, 11843–11855. 10.1021/jacs.1c05971. [DOI] [PubMed] [Google Scholar]
- Jiménez J.; Poncet M.; Míguez-Lago S.; Grass S.; Lacour J.; Besnard C.; Cuerva J. M.; Campaña A. G.; Piguet C. Bright Long-Lived Circularly Polarized Luminescence in Chiral Chromium(III) Complexes. Angew. Chem., Int. Ed. 2021, 60, 10095–10102. 10.1002/anie.202101158. [DOI] [PubMed] [Google Scholar]
- Becker P. M.; Förster C.; Carrella L. M.; Boden P.; Hunger D.; van Slageren J.; Gerhards M.; Rentschler E.; Heinze K. Spin Crossover and Long-Lived Excited States in a Reduced Molecular Ruby. Chem. - Eur. J. 2020, 26, 7199–7204. 10.1002/chem.202001237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson S. M.; Shores M. P.; Ferreira E. M. Photooxidizing Chromium Catalysts for Promoting Radical Cation Cycloadditions. Angew. Chem., Int. Ed. 2015, 54, 6506–6510. 10.1002/anie.201501220. [DOI] [PubMed] [Google Scholar]
- Higgins R. F.; Fatur S. M.; Shepard S. G.; Stevenson S. M.; Boston D. J.; Ferreira E. M.; Damrauer N. H.; Rappé A. K.; Shores M. P. Uncovering the Roles of Oxygen in Cr(III) Photoredox Catalysis. J. Am. Chem. Soc. 2016, 138, 5451–5464. 10.1021/jacs.6b02723. [DOI] [PubMed] [Google Scholar]
- Stevenson S. M.; Higgins R. F.; Shores M. P.; Ferreira E. M. Chromium Photocatalysis: Accessing Structural Complements to Diels-Alder Adducts with Electron-Deficient Dienophiles. Chem. Sci. 2017, 8, 654–660. 10.1039/C6SC03303B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins R. F.; Fatur S. M.; Damrauer N. H.; Ferreira E. M.; Rappé A. K.; Shores M. P. Detection of an Energy-Transfer Pathway in Cr-Photoredox Catalysis. ACS Catal. 2018, 8, 9216–9225. 10.1021/acscatal.8b03013. [DOI] [Google Scholar]
- Büldt L. A.; Wenger O. S. Chromium Complexes for Luminescence, Solar Cells, Photoredox Catalysis, Upconversion, and Phototriggered NO Release. Chem. Sci. 2017, 8, 7359–7367. 10.1039/C7SC03372A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto S.; Nauth A. M.; Ermilov E.; Scholz N.; Friedrich A.; Resch-Genger U.; Lochbrunner S.; Opatz T.; Heinze K. Photo-Chromium: Sensitizer for Visible-Light-Induced Oxidative C–H Bond Functionalization—Electron or Energy Transfer?. ChemPhotoChem 2017, 1, 344–349. 10.1002/cptc.201700077. [DOI] [Google Scholar]
- Sittel S.; Naumann R.; Heinze K. Molecular Rubies in Photoredox Catalysis. Front. Chem. 2022, 10, 887439 10.3389/fchem.2022.887439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe Y.; Sugano S. On the Absorption Spectra of Complex Ions. I. J. Phys. Soc. Jpn. 1954, 9, 753–766. 10.1143/JPSJ.9.753. [DOI] [Google Scholar]
- Kitzmann W. R.; Ramanan C.; Naumann R.; Heinze K. Molecular Ruby: Exploring the Excited State Landscape. Dalton Trans. 2022, 51, 6519. 10.1039/D2DT00569G. [DOI] [PubMed] [Google Scholar]
- Scarborough C. C.; Sproules S.; Weyhermüller T.; DeBeer S.; Wieghardt K. Electronic and Molecular Structures of the Members of the Electron Transfer Series [Cr(tbpy)3]n (n = 3+, 2+, 1+, 0): An X-ray Absorption Spectroscopic and Density Functional Theoretical Study. Inorg. Chem. 2011, 50, 12446–12462. 10.1021/ic201123x. [DOI] [PubMed] [Google Scholar]
- Scarborough C. C.; Lancaster K. M.; Debeer S.; Weyhermüller T.; Sproules S.; Wieghardt K. Experimental Fingerprints for Redox-Active Terpyridine in [Cr(tpy)2](PF6)n (n = 3-0), and the Remarkable Electronic Structure of [Cr(tpy)2]1–. Inorg. Chem. 2012, 51, 3718–3732. 10.1021/ic2027219. [DOI] [PubMed] [Google Scholar]
- Rehm D.; Weller A. Kinetik und Mechanismus der Elektronübertragung bei der Fluoreszenzlöschung in Acetonitril. Ber. Bunsen-Ges. Phys. Chem. 1969, 73, 834–839. 10.1002/bbpc.19690730818. [DOI] [Google Scholar]
- Rehm D.; Weller A. Kinetics of Fluorescence Quenching by Electron and H-Atom Transfer. Isr. J. Chem. 1970, 8, 259–271. 10.1002/ijch.197000029. [DOI] [Google Scholar]
- Dodsworth E. S.; Lever A. B. P. Correlations between Electrochemical Potentials and Optical Charge Transfer Energies in Ruthenium Bipyridine Derivatives. Chem. Phys. Lett. 1986, 124, 152–158. 10.1016/0009-2614(86)85135-1. [DOI] [Google Scholar]
- Sreenath K.; Thomas T. G.; Gopidas K. R. Cu(II) Mediated Generation and Spectroscopic Study of the Tris(4-anisyl)amine Radical Cation and Dication. Unusually Shielded Chemical Shifts in the Dication. Org. Lett. 2011, 13, 1134–1137. 10.1021/ol103167m. [DOI] [PubMed] [Google Scholar]
- Neumann S.; Kerzig C.; Wenger O. S. Quantitative Insights into Charge-Separated States from One- and Two-Pulse Laser Experiments Relevant for Artificial Photosynthesis. Chem. Sci. 2019, 10, 5624–5633. 10.1039/C9SC01381D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzani V.; Campagna S.. Photochemistry and Photophysics of Coordination Compounds I; Springer: Berlin, Heidelberg, New York, 2007. [Google Scholar]
- Aydogan A.; Bangle R. E.; Cadranel A.; Turlington M. D.; Conroy D. T.; Cauët E.; Singleton M. L.; Meyer G. J.; Sampaio R. N.; Elias B.; Troian-Gautier L. Accessing Photoredox Transformations with an Iron(III) Photosensitizer and Green Light. J. Am. Chem. Soc. 2021, 143, 15661–15673. 10.1021/jacs.1c06081. [DOI] [PubMed] [Google Scholar]
- Schmid L.; Kerzig C.; Prescimone A.; Wenger O. S. Photostable Ruthenium(II) Isocyanoborato Luminophores and Their Use in Energy Transfer and Photoredox Catalysis. JACS Au 2021, 1, 819–832. 10.1021/jacsau.1c00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid L.; Glaser F.; Schaer R.; Wenger O. S. High Triplet Energy Iridium(III) Isocyanoborato Complex for Photochemical Upconversion, Photoredox and Energy Transfer Catalysis. J. Am. Chem. Soc. 2022, 144, 963–976. 10.1021/jacs.1c11667. [DOI] [PubMed] [Google Scholar]
- Ohkubo K.; Mizushima K.; Iwataa R.; Fukuzumi S. Selective Photocatalytic Aerobic Bromination with Hydrogen Bromide via an Electron-Transfer State of 9-Mesityl-10-Methylacridinium Ion. Chem. Sci. 2011, 2, 715–722. 10.1039/c0sc00535e. [DOI] [Google Scholar]
- Romero N. A.; Nicewicz D. A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]
- Karges J.; Kuang S.; Maschietto F.; Blacque O.; Ciofini I.; Chao H.; Gasser G. Rationally Designed Ruthenium Complexes for 1- and 2-Photon Photodynamic Therapy. Nat. Commun. 2020, 11, 3262 10.1038/s41467-020-16993-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourdon L.; Cariou K.; Gasser G. Phototherapeutic Anticancer Strategies with First-Row Transition Metal Complexes: A Critical Review. Chem. Soc. Rev. 2022, 51, 1167–1195. 10.1039/D1CS00609F. [DOI] [PubMed] [Google Scholar]
- Knoll J. D.; Albani B. A.; Turro C. New Ru(II) Complexes for Dual Photoreactivity: Ligand Exchange and 1O2 Generation. Acc. Chem. Res. 2015, 48, 2280–2287. 10.1021/acs.accounts.5b00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teegardin K.; Day J. I.; Chan J.; Weaver J. Advances in Photocatalysis: A Microreview of Visible Light Mediated Ruthenium and Iridium Catalyzed Organic Transformations. Org. Process Res. Dev. 2016, 20, 1156–1163. 10.1021/acs.oprd.6b00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilate B.; Fischer C.; Sparr C. Design and Application of Aminoacridinium Organophotoredox Catalysts. Chem. Commun. 2020, 56, 1767–1775. 10.1039/C9CC08524F. [DOI] [PubMed] [Google Scholar]
- Holmberg-Douglas N.; Nicewicz D. A. Photoredox-Catalyzed C–H Functionalization Reactions. Chem. Rev. 2022, 122, 1925–2016. 10.1021/acs.chemrev.1c00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuzumi S.; Kotani H.; Ohkubo K.; Ogo S.; Tkachenko N. V.; Lemmetyinen H. Electron-Transfer State of 9-Mesityl-10-Methylacridinium Ion with a Much Longer Lifetime and Higher Energy Than That of the Natural Photosynthetic Reaction Center. J. Am. Chem. Soc. 2004, 126, 1600–1601. 10.1021/ja038656q. [DOI] [PubMed] [Google Scholar]
- Kerzig C.; Wenger O. S. Reactivity Control of a Photocatalytic System by Changing the Light Intensity. Chem. Sci. 2019, 10, 11023–11029. 10.1039/C9SC04584H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkubo K.; Nanjo T.; Fukuzumi S. Photocatalytic Oxygenation of Olefins with Oxygen. Isolation of 1,2-Dioxetane and the Photocatalytic O-O Bond Cleavage. Catal. Today 2006, 117, 356–361. 10.1016/j.cattod.2006.05.056. [DOI] [Google Scholar]
- Nicholls T. P.; Constable G. E.; Robertson J. C.; Gardiner M. G.; Bissember A. C. Brønsted Acid Cocatalysis in Copper(I)-Photocatalyzed α-Amino C-H Bond Functionalization. ACS Catal. 2016, 6, 451–457. 10.1021/acscatal.5b02014. [DOI] [Google Scholar]
- Wang C.; Reichenauer F.; Kitzmann W. R.; Kerzig C.; Heinze K.; Resch-Genger U. Efficient Triplet-Triplet Annihilation Upconversion Sensitized by a Chromium(III) Complex via an Underexplored Energy Transfer Mechanism. Angew. Chem., Int. Ed. 2022, e202202238 10.1002/anie.202202238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goerner H. Triplet States of Phenylethylenes in Solution. Energies, Lifetimes, and Absorption Spectra of 1,1-Diphenyl-, Triphenyl-, and Tetraphenylethylene Triplets. J. Phys. Chem. 1982, 86, 2028–2035. 10.1021/j100208a023. [DOI] [Google Scholar]
- Zou Y.-Q.; Chen J.-R.; Liu X.-P.; Lu L.-Q.; Davis R. L.; Jørgensen K. A.; Xiao W.-J. Highly Efficient Aerobic Oxidative Hydroxylation of Arylboronic Acids: Photoredox Catalysis Using Visible Light. Angew. Chem., Int. Ed. 2012, 51, 784–788. 10.1002/anie.201107028. [DOI] [PubMed] [Google Scholar]
- Pitre S. P.; McTiernan C. D.; Ismaili H.; Scaiano J. C. Mechanistic Insights and Kinetic Analysis for the Oxidative Hydroxylation of Arylboronic Acids by Visible Light Photoredox Catalysis: A Metal-Free Alternative. J. Am. Chem. Soc. 2013, 135, 13286–13289. 10.1021/ja406311g. [DOI] [PubMed] [Google Scholar]
- Xie H. Y.; Han L. S.; Huang S.; Lei X.; Cheng Y.; Zhao W.; Sun H.; Wen X.; Xu Q.-L. N-Substituted 3(10H)-Acridones as Visible-Light, Water-Soluble Photocatalysts: Aerobic Oxidative Hydroxylation of Arylboronic Acids. J. Org. Chem. 2017, 82, 5236–5241. 10.1021/acs.joc.7b00487. [DOI] [PubMed] [Google Scholar]
- Barbante G. J.; Kebede N.; Hindson C. M.; Doeven E. H.; Zammit E. M.; Hanson G. R.; Hogan C. F.; Francis P. S. Control of Excitation and Quenching in Multi-Colour Electrogenerated Chemiluminescence Systems through Choice of Co-Reactant. Chem. - Eur. J. 2014, 20, 14026–14031. 10.1002/chem.201403767. [DOI] [PubMed] [Google Scholar]
- Pavlishchuk V. V.; Addison A. W. Conversion Constants for Redox Potentials Measured versus Different Reference Electrodes in Acetonitrile Solutions at 25 °C. Inorg. Chim. Acta 2000, 298, 97–102. 10.1016/S0020-1693(99)00407-7. [DOI] [Google Scholar]
- Achord J. M.; Hussey C. L. Determination of Dissolved Oxygen in Nonaqueous Electrochemical Solvents. Anal. Chem. 1980, 52, 601–602. 10.1021/ac50053a061. [DOI] [Google Scholar]
- Noble A.; MacMillan D. W. C. Photoredox α-Vinylation of α-Amino Acids and N-Aryl Amines. J. Am. Chem. Soc. 2014, 136, 11602–11605. 10.1021/ja506094d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J.; Besnard C.; Cruz C. M.; Piguet C.; Jiménez J.-R. Heteroleptic mer-[Cr(N∩N∩N)(CN)3] Complexes: Synthetic Challenge, Structural Characterization and Photophysical Properties. Dalton Trans. 2022, 51, 4297–4309. 10.1039/D2DT00126H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indelli M. T.; Ballardini R.; Scandola F. Experimental Investigation of Highly Exergonic Outer-Sphere Electron-Transfer Reactions. J. Phys. Chem. 1984, 88, 2547–2551. 10.1021/j150656a023. [DOI] [Google Scholar]
- Woodhouse M. D.; McCusker J. K. Mechanistic Origin of Photoredox Catalysis Involving Iron(II) Polypyridyl Chromophores. J. Am. Chem. Soc. 2020, 142, 16229–16233. 10.1021/jacs.0c08389. [DOI] [PubMed] [Google Scholar]
- Joyce J. P.; Portillo R. I.; Rappé A. K.; Shores M. P. Doublet Ground State in a Vanadium(II) Complex: Redox and Coordinative Noninnocence of Tripodal Ligand Architecture. Inorg. Chem. 2022, 61, 6376–6391. 10.1021/acs.inorgchem.1c03418. [DOI] [PubMed] [Google Scholar]
- Hoffman M. Z. Cage Escape Yields from the Quenching of Excited Tris(bipyridyl)ruthenium(2+) by Methylviologen in Aqueous Solution. J. Phys. Chem. 1991, 95, 2606. 10.1021/j100159a091. [DOI] [Google Scholar]
- Georgopoulos M.; Hoffman M. Z. Cage Escape Yields in the Quenching of *Ru(bpy)32+ by Methylviologen. Presence of Triethanolamine as a Sacrificial Electron Donor. J. Phys. Chem. 1991, 95, 7717–7721. 10.1021/j100173a031. [DOI] [Google Scholar]
- Neshvad G.; Hoffman M. Z. Reductive Quenching of the Luminescent Excited State of Tris(2,2′-bipyrazine)ruthenium(2+) Ion in Aqueous Solution. J. Phys. Chem. 1989, 93, 2445–2452. 10.1021/j100343a044. [DOI] [Google Scholar]
- Senz A.; Gsponer H. E. Yield of Ions Produced in the Luminescence Quenching of Tris (2,2′-bipyridine)ruthenium(II) by Dichlorophenolate Ions. J. Phys. Org. Chem. 1995, 8, 706–712. 10.1002/poc.610081103. [DOI] [Google Scholar]
- Miedlar K.; Das P. K. Tris(2,2′-bipyridine)ruthenium(II)-Sensitized Photooxidation of Phenols. Environmental Effects on Electron Transfer Yields and Kinetics. J. Am. Chem. Soc. 1982, 104, 7462–7469. 10.1021/ja00390a012. [DOI] [Google Scholar]
- Neumann S.; Wenger O. S.; Kerzig C. Controlling Spin-Correlated Radical Pairs with Donor–Acceptor Dyads: A New Concept to Generate Reduced Metal Complexes for More Efficient Photocatalysis. Chem. - Eur. J. 2021, 27, 4115–4123. 10.1002/chem.202004638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troian-Gautier L.; Swords W. B.; Meyer G. J. Iodide Photoredox and Bond Formation Chemistry. Acc. Chem. Res. 2019, 52, 170–179. 10.1021/acs.accounts.8b00373. [DOI] [PubMed] [Google Scholar]
- Adams R. E.; Schmehl R. H. Micellar Effects on Photoinduced Electron Transfer in Aqueous Solutions Revisited: Dramatic Enhancement of Cage Escape Yields in Surfactant Ru(II) Diimine Complex/[Ru(NH3)6]2+ Systems. Langmuir 2016, 32, 8598–8607. 10.1021/acs.langmuir.6b02193. [DOI] [PubMed] [Google Scholar]
- McLauchlan K. A.; Steiner U. E. The Spin-Correlated Radical Pair as a Reaction Intermediate. Mol. Phys. 1991, 73, 241–263. 10.1080/00268979100101181. [DOI] [Google Scholar]
- Kavarnos G. J.; Turro N. J. Photosensitization by Reversible Electron Transfer: Theories, Experimental Evidence, and Examples. Chem. Rev. 1986, 86, 401–449. 10.1021/cr00072a005. [DOI] [Google Scholar]
- Xu J.; Jarocha L. E.; Zollitsch T.; Konowalczyk M.; Henbest K. B.; Richert S.; Golesworthy M. J.; Schmidt J.; Déjean V.; Sowood D. J. C.; Bassetto M.; Luo J.; Walton J. R.; Fleming J.; Wei Y.; Pitcher T. L.; Moise G.; Herrmann M.; Yin H.; Wu H.; Bartölke R.; Käsehagen S. J.; Horst S.; Dautaj G.; Murton P. D. F.; Gehrckens A. S.; Chelliah Y.; Takahashi J. S.; Koch K. W.; Weber S.; Solov’yov I. A.; Xie C.; Mackenzie S. R.; Timmel C. R.; Mouritsen H.; Hore P. J. Magnetic Sensitivity of Cryptochrome 4 from a Migratory Songbird. Nature 2021, 594, 535–540. 10.1038/s41586-021-03618-9. [DOI] [PubMed] [Google Scholar]
- Miura T.; Scott A. M.; Wasielewski M. R. Electron Spin Dynamics as a Controlling Factor for Spin-Selective Charge Recombination in Donor-Bridge-Acceptor Molecules. J. Phys. Chem. C 2010, 114, 20370–20379. 10.1021/jp103441n. [DOI] [Google Scholar]
- Rawls M. T.; Kollmannsberger G.; Elliott C. M.; Steiner U. E. Spin Chemical Control of Photoinduced Electron-Transfer Processes in Ruthenium(II)-Trisbipyridine-Based Supramolecular Triads: 2. The Effect of Oxygen, Sulfur, and Selenium as Heteroatom in the Azine Donor. J. Phys. Chem. A 2007, 111, 3485–3496. 10.1021/jp070221s. [DOI] [PubMed] [Google Scholar]
- Mims D.; Herpich J.; Lukzen N. N.; Steiner U. E.; Lambert C. Readout of Spin Quantum Beats in a Charge-Separated Radical Pair by Pump-Push Spectroscopy. Science 2021, 374, 1470–1474. 10.1126/science.abl4254. [DOI] [PubMed] [Google Scholar]
- Serpone N.; Jamieson M. A.; Hoffman M. Z. 4T2→2E Intersystem-Crossing Efficiency in the Cr(bpy)33+ and Cr(phen)33+ Ions in Aqueous Media. J. Chem. Soc., Chem. Commun. 1980, 1006–1007. 10.1039/C39800001006. [DOI] [Google Scholar]
- Bolletta F.; Maestri M.; Balzani V. Efficiency of the Intersystem Crossing from the Lowest Spin-Allowed to the Lowest Spin-Forbidden Excited State of Some Chromium(III) and Ruthenium(II) Complexes. J. Phys. Chem. 1976, 80, 2499–2503. 10.1021/j100563a014. [DOI] [Google Scholar]
- Kirk A. D.; Porter G. B. Luminescence of Chromium(III) Complexes. J. Phys. Chem. 1980, 84, 887–891. 10.1021/j100445a020. [DOI] [Google Scholar]
- Trost B. M. On Inventing Reactions for Atom Economy. Acc. Chem. Res. 2002, 35, 695–705. 10.1021/ar010068z. [DOI] [PubMed] [Google Scholar]
- Sheldon R. A. The E Factor: Fifteen Years On. Green Chem. 2007, 9, 1273–1283. 10.1039/b713736m. [DOI] [Google Scholar]
- Trost B. M. The Atom Economy - A Search for Synthetic Efficiency. Science 1991, 254, 1471–1477. 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]
- Ford P. C. From Curiosity to Applications. A Personal Perspective on Inorganic Photochemistry. Chem. Sci. 2016, 7, 2964–2986. 10.1039/C6SC00188B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser A.; Reber C. Spectroscopy and Chemical Bonding in Transition Metal Complexes. Struct. Bonding 2017, 172, 291–312. 10.1007/430_2015_195. [DOI] [Google Scholar]
- DeLeo M. A.; Ford P. C. Photoreactions of Coordinated Nitrite Ion. Reversible Nitric Oxide Labilization from the Chromium(III) Complex [trans-Cr(cyclam)(ONO)2]+. Coord. Chem. Rev. 2000, 208, 47–59. 10.1016/S0010-8545(00)00271-X. [DOI] [Google Scholar]
- Kalmbach J.; Wang C.; You Y.; Förster C.; Schubert H.; Heinze K.; Resch-Genger U.; Seitz M. Near-IR to Near-IR Upconversion Luminescence in Molecular Chromium Ytterbium Salts. Angew. Chem., Int. Ed. 2020, 59, 18804–18808. 10.1002/anie.202007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu U.; Otto S.; Heinze K.; Gasser G. Biological Evaluation of the NIR-Emissive Ruby Analogue [Cr(ddpd)2][BF4]3 as a Photodynamic Therapy Photosensitizer. Eur. J. Inorg. Chem. 2019, 2019, 37–41. 10.1002/ejic.201801023. [DOI] [Google Scholar]
- Lenz S.; Bamberger H.; Hallmen P. P.; Thiebes Y.; Otto S.; Heinze K.; Van Slageren J. Chromium(III)-Based Potential Molecular Quantum Bits with Long Coherence Times. Phys. Chem. Chem. Phys. 2019, 21, 6976–6983. 10.1039/C9CP00745H. [DOI] [PubMed] [Google Scholar]
- Abrahamsson M.; Jäger M.; Kumar R. J.; Österman T.; Persson P.; Becker H. C.; Johansson O.; Hammarström L. Bistridentate Ruthenium(II)polypyridyl-Type Complexes with Microsecond 3MLCT State Lifetimes: Sensitizers for Rod-Like Molecular Arrays. J. Am. Chem. Soc. 2008, 130, 15533–15542. 10.1021/ja804890k. [DOI] [PubMed] [Google Scholar]
- Herr P.; Kerzig C.; Larsen C. B.; Häussinger D.; Wenger O. S. Manganese(I) Complexes with Metal-to-Ligand Charge Transfer Luminescence and Photoreactivity. Nat. Chem. 2021, 13, 956–962. 10.1038/s41557-021-00744-9. [DOI] [PubMed] [Google Scholar]
- Förster C.; Heinze K. Photophysics and Photochemistry with Earth-Abundant Metals-Fundamentals and Concepts. Chem. Soc. Rev. 2020, 49, 1057–1070. 10.1039/C9CS00573K. [DOI] [PubMed] [Google Scholar]
- Wegeberg C.; Wenger O. S. Luminescent First-Row Transition Metal Complexes. JACS Au 2021, 1, 1860–1876. 10.1021/jacsau.1c00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zobel J. P.; González L. The Quest to Simulate Excited-State Dynamics of Transition Metal Complexes. JACS Au 2021, 1, 1116–1140. 10.1021/jacsau.1c00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptista F. A.; Krizsan D.; Stitch M.; Sazanovich I. V.; Clark I. P.; Towrie M.; Long C.; Martinez-Fernandez L.; Improta R.; Kane-Maguire N. A. P.; Kelly J. M.; Quinn S. J. Adenine Radical Cation Formation by a Ligand-Centered Excited-State of an Intercalated Chromium Polypyridyl Complex Leads to Enhanced DNA Photooxidation. J. Am. Chem. Soc. 2021, 143, 14766–14779. 10.1021/jacs.1c06658. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.