

Abstract
Using cycloalkyl and electron-donating groups to decrease the carbonyl electrophilicity, a novel series of 2-(quinoline-4-yloxy)acetamides was synthesized and evaluated as in vitro inhibitors of Mycobacterium tuberculosis (Mtb) growth. Structure–activity relationship studies led to selective and potent antitubercular agents with minimum inhibitory concentrations in the submicromolar range against drug-sensitive and drug-resistant Mtb strains. An evaluation of the activity of the lead compounds against a spontaneous qcrB mutant strain indicated that the structures targeted the cytochrome bc1 complex. In addition, selected molecules inhibited Mtb growth in a macrophage model of tuberculosis infection. Furthermore, the leading compound was chemically stable depending on the context and showed good kinetic solubility, high permeability, and a low rate of in vitro metabolism. Finally, the pharmacokinetic profile of the compound was assessed after oral administration to mice. To the best of our knowledge, for the first time, a 2-(quinoline-4-yloxy)acetamide was obtained with a sufficient exposure, which may enable in vivo effectiveness and its further development as an antituberculosis drug candidate.
Keywords: Mycobacterium tuberculosis, Drug design, Multidrug-resistant strains, Structure−activity relationship, Intracellular activity, Cytochrome bc1 complex
Tuberculosis (TB) is an infectious disease caused by Mycobacterium tuberculosis (Mtb), its main etiological agent in humans. Recognized as a global health emergency since the 1990s, in 2020 it was estimated that about 9.9 million people have fallen ill with TB and 1.5 million have died from this disease worldwide.1 These data have placed TB among the 10 leading causes of death in the world. Complicating this scenario, it has also been reported that a quarter of the world’s population is infected with Mtb and therefore are at risk of developing the active forms of the disease. In addition, in 2019, close to half a million people developed the disease due to drug-resistant strains.1 These strains have shown resistance to drugs such as isoniazid and rifampicin (MDR-TB) or only to rifampicin (RR-TB), components of the first line of TB drug management. Furthermore, pressure on the health system and interference with diagnostic and treatment services has suggested that the coronavirus (COVID-19) pandemic could reverse years of advances made in TB control.1
Interestingly, the 21st century has seen the approval of new therapeutic alternatives for TB treatment. Bedaquiline (2012),2 delamanid (2014),3 and pretomanid (2019)4 are now approved drugs for the treatment of drug-resistant TB. Despite being welcomed after a long period without therapeutic innovations for the treatment of the disease, the clinical use of these drugs has been restricted by potentially dangerous side effects5 and the already reported emergence of resistance to bedaquiline and delamanid.6 These difficulties highlight the need for continuous efforts to obtain new TB treatment alternatives.
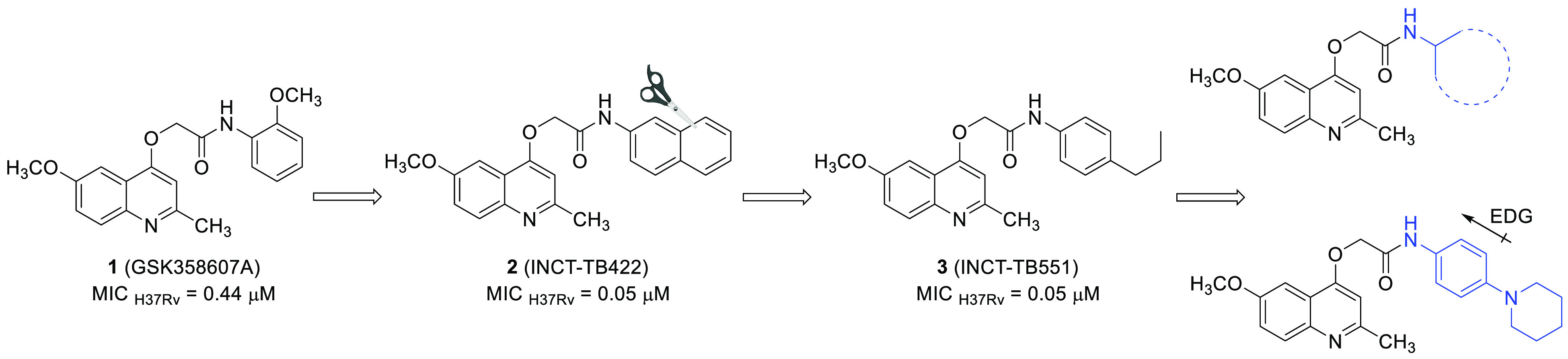
As part of our research program, we are interested in obtaining compounds capable of inhibiting drug-susceptible and drug-resistant Mtb strains. Among the evaluated structures, 2-(quinolin-4-yloxy)acetamides have shown some encouraging results.7,8 According to phenotypic assays,9 this chemical class has provided lead-like compounds endowed with potent and selective antimycobacterial activities. Furthermore, the mechanism of action of these structures has been studied using whole-genome sequencing of selected spontaneous mutants.10 Such data suggest that the QcrB subunit of menaquinol cytochrome c oxidoreductase (bc1 complex) is related to the antimycobacterial activity of these molecules. Despite the potency and selectivity observed in the in vitro experiments, lead-like compounds have exhibited moderate metabolic stability along with acceptable aqueous solubility.7,8 These parameters can compromise the pharmacokinetic exposure and in vivo activity of the molecules. The above instability has been attributed to the amide group, which likely undergoes an enzymatic-mediated hydrolysis reaction.11 In fact, molecular simplification, with the removal of the amide group, or the insertion of α-carbonyl protecting groups has led to compounds with greater metabolic stabilities but reduced activities against the mycobacteria.11,12
To overcome the reduced antimycobacterial activity while maintaining the metabolic stability, our goal was to maintain the amide function and use substituents that would reduce the electrophilicity of the carbonyl group. For this, two strategies were used to increase the electronic availability at the carbonyl group: (i) attaching cycloalkyl groups to the primary amides and (ii) attaching N-heteroalkyl groups to the 4-position of the benzene ring (Figure 1).
Figure 1.
Scaffold evolution of 2-(quinolin-4-yloxy)acetamides and the novel compounds designed with the aim of reducing the amide carbonyl electrophilicity, which could increase stability in hydrolysis mediated-reactions.
Therefore, seeking to obtain new compounds with activities against drug-susceptible and mostly drug-resistant Mtb strains, a new series of 2-(quinolin-4-yloxy)acetamides was synthesized. First, the structural requirements for the potency of the compounds (structure–activity relationship, SAR) were evaluated using minimum inhibitory concentration (MIC) values. Subsequently, the most active structures against the M. tuberculosis H37Rv strain were tested against a panel of well-characterized multidrug-resistant strains. Furthermore, the mechanism of action was studied using a spontaneous mutant strain with a fully sequenced genome, while the viabilities of HepG2 and Vero cells were used as preliminary indicators of the toxicities and selectivities of the compounds. Additionally, intracellular activity was evaluated in a macrophage model of Mtb infection. Finally, the chemical stability, kinetic solubility, passive permeability, metabolic stability, and in vivo pharmacokinetic profiles of the most active compound are presented.
The designed cycloalkyl amide-containing compounds were obtained in three synthetic steps according to previously described procedures.7,8 First, 4-hydroxyquinoline (2) was synthesized using the classical Conrad–Limpach cyclocondensation reaction between ethyl 3-oxobutanoate and 4-methoxyaniline. Afterward, the acylation reaction of cycloalkyl amines or anilines with bromoacetyl chloride furnished the 2-bromo-N-(cycloalkyl)acetamides 5a–o. Finally, a second-order nucleophilic substitution reaction with concomitant O-alkylation between 4-hydroxyquinoline (4) and 2-bromo-N-(cycloalkyl)acetamides 5a–o led to the desired 2-(quinolin-4-yloxy)acetamides 6a–o. The reaction mixtures were stirred for 18 h at 25 °C in the presence of potassium carbonate (K2CO3) as a base and dimethylformamide (DMF) as the solvent, leading to products 6a–o with 44–80% yields (Scheme 1). Importantly, the chosen cycloalkyl substituents covered a wide range of molecular volumes, including stereogenic centers. Regarding the increase in the electron density of the amide, the piperidinic group and its derivatives were chosen as substituents attached at the 4-positions of the anilines. In addition to electronic effects, piperidine could mimic the steric and hydrophobic requirements of the most potent antitubercular compounds of this chemical class (Figure 1).
Scheme 1.

Conditions and reagents are as follows: i = K2CO3 and DMF at 25 °C for 18 h. Minimum inhibitory concentration (MIC) against the M. tuberculosis H37Rv strain. INH = isoniazid. RIF = rifampin.
2-(Quinolin-4-yloxy)acetamides 6a–o were evaluated for their ability to inhibit the growth of the M. tuberculosis H37Rv strain using isoniazid and rifampin as positive controls.13 In general, cycloalkylamine and alkylamine derivative compounds 6a–l presented MICs ranging from 3.3 to 33.3 μM (Scheme 1). In contrast, the presence of a piperidine ring (6m) and its derivatives (6n–o) yielded the molecules of this series most effective against Mtb. Interestingly, the increase of the volume of the cycloalkyl group appears to be positively correlated with the ability to inhibit the growth of the Mtb. Indeed, 2-(quinolin-4-yloxy)acetamide 6a with a a four-membered carbocycle present at the acetamide group had a MIC of 33.3 μM, while its seven-membered counterpart 6d was able to inhibit Mtb with a MIC value of 3.7 μM. One can conclude that increasing the size of the cycloalkyl chain from four to seven carbons improved the antimycobacterial activity ninefold. The presence of a methyl group at the 4-position of cyclohexyl was well tolerated, as structure 6e exhibited a MIC of 7.3 μM, which differed only slightly from that of nonsubstituted 6c (7.6 μM). By contrast, the methyl group attached at the 2-position of the cyclohexyl ring reduced the activity approximately twofold, as the MIC of molecule 6f was 14.6 μM. The use of an amine containing a methylene spacer was also well-tolerated. Compound 6g had a MIC value of 7.3 μM, which was similar to the cyclohexyl-containing structure 6c. Additionally, the two molecules containing a methyl group at the β-position, 6h and 6i, were ineffective in terms of inhibiting Mtb growth when evaluated at the highest concentrations allowed by the solubility. As described previously,8 the primary amine appears to be imperative for the potent antimycobacterial activity of 2-(quinolin-4-yloxy)acetamides. The presence of a methyl group, regardless of the stereochemistry, can cause steric hindrance at NH, hindering a possible intermolecular interaction. An interesting finding was obtained with the isopinocampheylamine derivatives 6j and 6k. The compound containing the (−)-isopinocampheyl nucleus (6j) was more effective (eightfold) than the derivative containing its mirror image (6k) in terms of inhibiting the M. tuberculosis H37Rv strain. The MIC values obtained for structures 6j and 6k were 3.3 and 26.1 μM, respectively. Finally, the presence of the 2-adamantyl group in the structure of 6l yielded a molecule with a MIC of 6.6 μM. This result showed once again7,8,14 that the planarity and correct positioning of the substituents at the acetamide moiety are detrimental to the potent activity against Mtb.
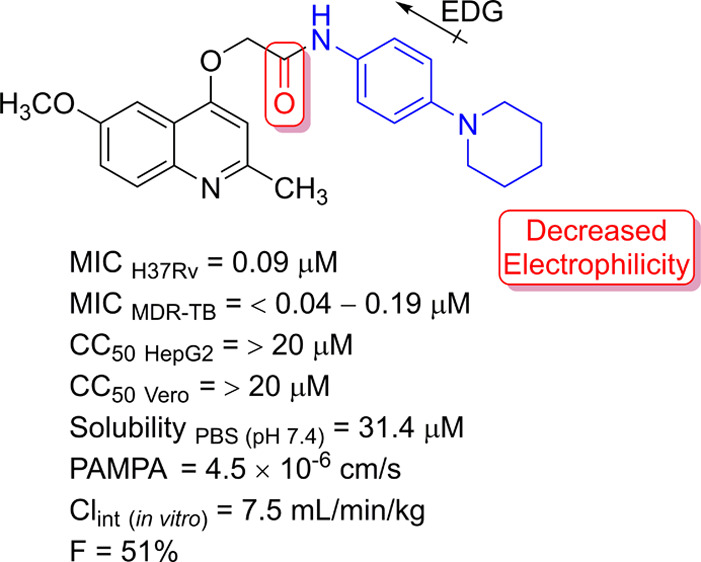
Our next step to try to obtain 2-(quinolin-4-yloxy)acetamides endowed with antimycobacterial potencies similar to lead-like compounds 2 (INCT-TB422)7 and 3 (INCT-TB551)8 (Figure 1) was to synthesize and evaluate piperidine as a substituent at the 4-position of aniline. This five-membered nonaromatic heterocycle has been described as a privileged structure and was ranked as the most frequent nitrogen heterocycle among U.S. Food and Drug Administration-approved drugs.15 The presence of a piperidine moiety produced a structure with a MIC in the submicromolar range. In fact, molecule 6m was able to inhibit the M. tuberculosis H37Rv strain with a MIC value of 0.09 μM. Compared with a 2-(quinolin-4-yloxy)acetamide containing a nonsubstituted benzene ring, which was shown to have a MIC of 0.48 μM,7 compound 6m was at least fivefold more potent against the Mtb. As expected, the increase in polarity that resulted from the substitution of piperidine by morpholine (6n) and thiomorpholine (6o) significantly reduced the antimycobacterial activity. The structures 6n and 6o exhibited MIC values of 3.1 and 0.37 μM, respectively. These findings reinforce the possible existence of a hydrophobic site of interaction in the molecular target surrounding the amide substituents. It is noteworthy that isoniazid and rifampin, the main components of the first-line tuberculosis treatment, showed MIC values of 2.3 and 0.05 μM, respectively, against the M. tuberculosis H37Rv strain when evaluated under the same experimental conditions (Scheme 1).
From the results described above, the second round of synthetic procedures kept the 4-(piperidin-1-yl)aniline system as a substituent at the acetamide moiety and varied the substituents at the 2- and 6-positions of the quinoline ring (Scheme 2). Once again, the synthesis of 4-hydroxyquinolines 7a–h was performed using the Conrad–Limpach cyclocondensation reaction, as reported previously.8 The O-alkylation reaction was carried out with bromoacetamide 8 under the same reaction conditions described above, providing the 2-(quinolin-4-yloxy)acetamides 9a–h with 58–88% yields (Scheme 2). Halogens were chosen as substituents to reduce the activation of the quinoline scaffold, which could also help increase the metabolic stabilities of the final products. The synthesized compounds were evaluated for their ability to inhibit the growth of the M. tuberculosis H37Rv strain (Scheme 2). Overall, the presence of piperidine maintained the sub-micromolar antimycobacterial activity potency. However, the presence of halogens at the 6-position and the increase of the alkyl chain length at the 2-position of the quinoline reduced the inhibitory activity toward the Mtb when compared with that of the 6m structure. Chlorine- and bromine-containing molecules 9a and 9b exhibited similar MIC values of 0.75 and 0.68 μM, respectively. These values demonstrated that replacing the methoxy group at the 6-position of the quinoline ring in 6m with halogens reduced the activity against Mtb more than 7.5-fold. An alkyl chain extension at the 2-position of the 2-(quinolin-4-yloxy)acetamides 9c–e led to compounds with MIC values of 0.19–0.37 μM. Interestingly, exchanging the methyl group with a trifluoromethyl group at the 2-position of the heterocyclic ring decreased the potency of the structure almost twofold. In fact, trifluoromethylated molecule 9f exhibited a MIC of 0.17 μM. Finally, when two electron-withdrawing groups were positioned at the 2- and 6-positions of the quinoline, the antimycobacterial activity was greatly reduced. The MIC values of 2-(quinolin-4-yloxy)acetamides 9g and 9h were 86.2 and 78.7 μM, respectively, which represented a decrease of more than 870-fold compared with that of 6m. These findings indicate that the electronic characteristics of the substituents attached to the quinoline ring significantly influence the antitubercular activity. One hypothesis is that the electron-donating groups (methoxy and methyl) can stabilize the protonation of the quinoline nitrogen, which may be detrimental to its efficient interaction with the molecular target. In the presence of electron-withdrawing groups (halogens and trifluoromethyl), quinoline becomes less basic, hindering such interactions. It is noteworthy that the molecular docking of compound 3 (INCT-TB551) (Figure 1) at the QcrB active site positioned the quinoline nitrogen at distances and angles that allowed interactions with the T313 and M342 residues.10 Corroborating our hypothesis, the predicted pKa of the quinoline ring of molecule 6m was 6.5 (strongest basic pKa), whereas the predicted pKa value of its analogues 9g and 9h was 1.3.16
Scheme 2.

Conditions, reagents, and reactants are as follows: i = K2CO3 and DMF at 25 °C for 18 h. Minimum inhibitory concentration (MIC) against the M. tuberculosis H37Rv strain.
On the basis of the obtained results, the compounds 6m and 9c were chosen as representatives of the series of synthesized molecules for subsequent assays. Although the observed MIC value for the structure 9f was promising, its very low aqueous solubility discouraged further evaluation. The molecules 6m and 9c were evaluated against a panel of drug-resistant clinical isolates (Table 1). Importantly, these genomes of these MDR-TB strains have been sequenced, and the genotypic changes related to resistance are known.17 2-(Quinolin-4-yloxy)acetamides 6m and 9c were even more effective against drug-resistant clinical isolates PT2 and PT20 when compared with the MIC values presented against the M. tuberculosis H37Rv strain. In addition, against the PT12 strain, the MIC value of 6m was reduced (twofold), whereas the antimycobacterial activity elicited by 9c was identical compared to that against the drug-sensitive H37Rv strain. Notably, the proximity of results to those of structure 6m did not characterize a change to a resistance phenotype. Altogether, these findings demonstrate that the molecules have promising activities against drug-resistant MDR-TB strains and do not share resistance mechanisms with the main clinically useful drugs, probably acting via distinct biochemical pathways. Furthermore, the involvement of the qcrB gene in the antitubercular activity of 2-(quinolin-4-yloxy)acetamides 6m and 9c was evaluated by determining the MICs of the compounds against a spontaneous 2-(quinolin-4-yloxy)acetamide-resistant M. tuberculosis strain (Table 1). This resistant strain has been shown to have a mutation in the qcrB gene that results in T313A amino acid exchange.10 The MIC values presented by structures 6m and 9c were at least 274- and 125-fold higher than those displayed against the M. tuberculosis H37Rv strain, respectively. These data suggested the participation of the b-subunit of the cytochrome bc1 complex, which is part of the respiratory electron transport chain, in the antitubercular activity exhibited by these molecules. The selectivity and preliminary toxicity assessment of 2-(quinolin-4-yloxy)acetamides 6m and 9c were performed by evaluating the viability of HepG2 and Vero cell lines after exposure to the compounds (Table 1). After 72 h of incubation, cell line viability was not significantly altered. These data show that the selectivity indexes (CC50/MIC; CC50 was defined as the concentration required to reduce cell viability by 50%) of 6m and 9c are greater than 222 and 105, respectively, considering the MIC values for the M. tuberculosis H37Rv strain. It is noteworthy that cell viability was determined using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)18 and neutral red19 procedures. While the MTT method assesses mitochondrial activity, the neutral red assay infers the lysosomal integrity of cells. Additionally, compounds 6m and 9c were evaluated against Staphylococcus aureus ATCC 25923, Escherichia coli ATCC 25922, and the multidrug-resistant clinical isolates Acinetobacter baumannii and Klebsiella pneumoniae. At a concentration of 20 μM, none of the structures were able to inhibit bacterial growth, suggesting the possible selectivity of these molecules for Mtb inhibition (data not shown). In the next stage, the ability of 2-(quinolin-4-yloxy)acetamides 6m and 9c to inhibit the bacterium when internalized into macrophages was determined using an in vitro model of Mtb infection (Table 1). Compounds 6m and 9c significantly reduced the number of colony-forming units (CFU) when compared to that in the cell group treated with vehicle (near 1 log10 CFU). This result demonstrates that the molecules have structural, electronic, and physicochemical properties that allow them to cross biological barriers and inhibit the growth of the bacterium intracellularly.
Table 1. In Vitro Activities of the Selected 2-(Quinolin-4-yloxy)acetamides 6m and 9c against M. tuberculosis strains H37Rv and MDR-TB and the 2-(Quinolin-4-yloxy)acetamide-Resistant Strain, Evaluation of the Viability of HepG2 and Vero cells, and Intracellular Activity in a Macrophage Model of Mtb Infectiona.
| Entry | MIC H37Rv (μM) | MIC PT2 (μM) | MIC PT12 (μM) | MIC PT20 (μM) | MICbqcrB-T313A (μM) | CC50c HepG2 (μM) | CC50c Vero (μM) | log10 CFU/well (mean ± SD) |
|---|---|---|---|---|---|---|---|---|
| 6m | 0.09 | <0.04 | 0.19 | <0.04 | >24.7 | >20 | >20 | 4.13 ± 0.05d |
| 9c | 0.19 | <0.04 | 0.19 | <0.04 | >23.8 | >20 | >20 | 4.13 ± 0.05d |
| INH | 2.3 | 291.7 | 145.8 | 291.7 | 2.3 | |||
| RIF | 0.05 | >48.6 | >48.6 | >48.6 | ||||
| UNT | 5.10 ± 0.10 |
Data were expressed as the mean ± standard deviation for compounds evaluated at 2.5 μM. INH = isoniazid. RIF = rifampin. UNT = untreated (0.5% DMSO).
2-(Quinolin-4-yloxy)acetamide-resistant spontaneous mutant containing a unique alteration in the qcrB gene (ACC to GCC at nucleotide position 937 or a T313A amino substitution).
The selectivity and toxicity of the compounds were preliminarily studied on HepG2 and Vero cells using MTT and neutral red assays. CC50 was defined as the concentration required to reduce cell viability by 50%.
*P < 0.05 compared to untreated group (Dunnett post-test).
It is important to mention that results describing potency, inhibition of resistant strains, selectivity, and intracellular activity have already been reported for other 2-(quinolin-4-yloxy)acetamides.7−9,11,14 Therefore, to evaluate the impact of the design on the drug-like features of the synthesized compounds, molecule 6m was chosen for in vitro ADME profiling (Table 2). The stability of 6m was not altered after incubation at pH 7.4 for 24 h. By contrast, the use of an acidic (pH 1.2) or basic (pH 9.1) medium reduced the concentration of the molecule by approximately 40%. As expected, the aqueous solubility was significantly higher under acidic conditions. At pH 1.2, the solubility was greater than 2.5 mM. Interestingly, the solubility of 2-(quinolin-4-yloxy)acetamide 6m in the pH 7.4 solution was 31.4 μM. This value is above the cutoff of 25 μM described in early drug discovery programs.20 In addition, this solubility value was determined to be above those of lead-like compounds 2 and 3 (Figure 1). Using a quantification methodology similar to that employed for 6m, structure 2 was not detected in a pH 7.4 solution.7 In addition, molecule 3 has been described to exhibit a solubility of 2.4 μM, which is 13-fold lower than that of 2-(quinolin-4-yloxy)acetamide 6m determined using a similar protocol.8 Furthermore, the passive permeability of compound 6m was assessed using the parallel artificial membrane permeability assay (PAMPA; Table 2). The results showed that the structure 6m presents a high permeability under the experimental conditions used. Finally, the metabolic stability of 2-(quinolin-4-yloxy)acetamide 6m was determined in the presence of rat liver microsomes. This parameter is one of the main attrition rates for this chemical class. Notably, compound 6m displayed a low metabolism rate (Clint < 16 mL/min/kg),21 which resulted in a half-life of 176 min. To the best of our knowledge, this was the first 2-(quinolin-4-yloxy)acetamide to show a low rate of metabolism.
Table 2. Chemical Stability, Solubility, Permeability, and Metabolic Stability of 2-(Quinolin-4-yloxy)acetamide 6m.
| chemical
Stabilitya |
solubilityb |
permeability | metabolic
stability |
|||||
|---|---|---|---|---|---|---|---|---|
| entry | pH 1.2c (%) | pH 7.4d (%) | pH 9.1e (%) | pH 1.2c (mM) | pH 7.4d (μM) | PAMPA (10–6 cm/s) | Clintf (mL/min/kg) | t1/2g (min) |
| 6m | 58.3 | 100 | 62.1 | >2.5 | 31.4 | 4.5 | 7.5 | 176 |
Percentage of remaining compound after incubation at 37 °C for 24 h.
Solubility was determined after incubation at 25 °C for 4 h.
Using a 0.1 M HCl solution.
Using PBS.
Using a 0.1 M NH4HCO3 solution.
Intrinsic clearance of rat liver microsomes.
Half-life.
To shed light on the possible in vivo antitubercular activities of the compounds under study, pharmacokinetic experiments were performed in mice. Following intravenous and oral dosing of structure 6m, the parameters were obtained using the naïve polled approach (Table 3). The clearance for both dosing regimens were similar (CLiv 0.088 L/h/kg and CLoral 0.115 L/h/kg), and the same can be said for the volume of distribution (Vdss = V1 + V2; Vdssiv 0.304 L/kg and Vdssoral 0.234 L/kg), confirming the models selected. The terminal half-lives determined in vivo were around 103 (oral) and 136 min (iv), indicating the possible contributions of other mechanisms of drug elimination besides metabolism. Interestingly, the mean maximum plasma concentration (Cmax) obtained after oral administration was 8.3 μg/mL (20.5 μM). This concentration is more than 227-fold higher than the MIC value presented by molecule 6m. In addition, plasma quantification 24 h after oral administration revealed a mean concentration of 1.14 ± 0.82 μg/mL (2.8 μM). Despite the great variability of the data, this value was 31-fold higher than the MIC value obtained for 2-(quinolin-4-yloxy)acetamide 6m. Finally, the oral bioavailability of compound was estimated at 51%. For the first time, these data described a 2-(quinolin-4-yloxy)acetamide with a pharmacokinetic exposure that may enable antimycobacterial activity in a rodent model of tuberculosis infection.
Table 3. Pharmacokinetic Parameters of 2-(Quinolin-4-yloxy)acetamide 6m after Intravenous (25 mg/kg, 61.6 μmol/kg) and Oral (121.6 mg/kg, 300 μmol/kg) Administrationa.
| group | parameters | NPA | RSE (%) |
|---|---|---|---|
| intravenous | CL (L/h/kg) | 0.088 | 11.7 |
| V1 (L/kg) | 0.215 | 22.6 | |
| Q (L/h/kg) | 0.339 | 33 | |
| V2 (L/kg) | 0.0892 | 24.1 | |
| oral | CL/F (L/h/kg) | 0.226 | 18.0 |
| V1/F (L/kg) | 0.460 | 29.0 | |
| Ka (h–1) | 3.01 | 58.0 | |
| F (%) | 51.0 |
NPA = naïve pooled approach. CL = clearance. V1 = volume of central the compartment. Q = intercompartmental clearance. V2 = volume of the peripheral compartment. Ka = absorption rate constant. F = bioavailability.
Herein, the design and synthesis of a new 2-(quinolin-4-yloxy)acetamide series were shown, and we demonstrated the in vitro antimycobacterial activities of the synthesized compounds. The synthetic procedures were accomplished using readily accessible reagents and reactants under known and straightforward methodologies. In addition, the compounds showed selective activities against drug-sensitive and MDR-TB strains. Interestingly, the leading molecules carry out their antitubercular activity by targeting the cytochrome bc1 complex. When evaluated in a macrophage model of Mtb infection, these structures were able to inhibit intracellular M. tuberculosis growth. Furthermore, the design strategy of concomitantly increasing of the amide carbonyl’s electron density to reduce its reactivity in hydrolysis-mediated reactions provided a compound that was chemically stable depending on context medium, showed good kinetic solubility and high permeability, and was metabolically stable in the presence of rat microsomes. Finally, a pharmacokinetic profile of the leading compound showed, for the first time, a 2-(quinolin-4-yloxy)acetamide endowed with good in vivo exposure after oral administration to mice, suggesting that this chemical class may yield new anti-TB drug candidates. A rodent model of TB and new structural modifications, including an evaluation of the possible bioisosterism between Telacebec’s scaffold imidazo[1,2-a]pyridine and the quinoline ring, are currently underway, and these data will be presented in due course.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00254.
Synthetic procedures, analytical data, and bioassay protocols (PDF)
Author Contributions
The manuscript was written through contributions of all authors.
This work was supported by CNPq/FAPERGS/CAPES/BNDES (INCT-TB) (421703-2017-2/17-1265-8/14.2.0914.1). S.D.O. (309933/2020-0), T.DC. and C.V.B. (310344/2016-6), L.A.B. (520182/99-5), and P.M. (305203/2018-5) are research career awardees of the National Council for Scientific and Technological Development of Brazil (CNPq). This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior Brasil (CAPES), finance code 001.
The authors declare no competing financial interest.
Supplementary Material
References
- Global Tuberculosis Report 2021; World Health Organization: Geneva, Switzerland, 2021. https://www.who.int/publications/i/item/9789240037021 (accessed 2022-05).
- Diacon A. H.; Pym A.; Grobusch M.; Patientia R.; Rustomjee R.; Page-Shipp L.; Pistorius C.; Krause R.; Bogoshi M.; Churchyard G.; Venter A.; Allen J.; Palomino J. C.; De Marez T.; van Heeswijk R. P.; Lounis N.; Meyvisch P.; Verbeeck J.; Parys W.; de Beule K.; Andries K.; Mc Neeley D. F. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N. Engl. J. Med. 2009, 360, 2397–2405. 10.1056/NEJMoa0808427. [DOI] [PubMed] [Google Scholar]
- Gler M. T.; Skripconoka V.; Sanchez-Garavito E.; Xiao H.; Cabrera-Rivero J. L.; Vargas-Vasquez D. E.; Gao M.; Awad M.; Park S. K.; Shim T. S.; Suh G. Y.; Danilovits M.; Ogata H.; Kurve A.; Chang J.; Suzuki K.; Tupasi T.; Koh W. J.; Seaworth B.; Geiter L. J.; Wells C. D. Delamanid for multidrug-resistant pulmonary tuberculosis. N. Engl. J. Med. 2012, 366, 2151–2160. 10.1056/NEJMoa1112433. [DOI] [PubMed] [Google Scholar]
- Stover C. K.; Warrener P.; VanDevanter D. R.; Sherman D. R.; Arain T. M.; Langhorne M. H.; Anderson S. W.; Towell J. A.; Yuan Y.; McMurray D. N.; Kreiswirth B. N.; Barry C. E.; Baker W. R. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 2000, 405, 962–966. 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- Avorn J. Approval of a tuberculosis drug based on a paradoxical surrogate measure. J. Am. Med. Assoc. 2013, 309, 1349–1350. 10.1001/jama.2013.623. [DOI] [PubMed] [Google Scholar]
- Bloemberg G. V.; Keller P. M.; Stucki D.; Trauner A.; Borrell S.; Latshang T.; Coscolla M.; Rothe T.; Homke R.; Ritter C.; Feldmann J.; Schulthess B.; Gagneux S.; Bottger E. C. Acquired resistance to bedaquiline and delamanid in therapy for tuberculosis. N. Engl. J. Med. 2015, 373, 1986–1988. 10.1056/NEJMc1505196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pissinate K.; Villela A. D.; Rodrigues-Junior V.; Giacobbo B. C.; Grams E. S.; Abbadi B. L.; Trindade R. V.; Nery L. R.; Bonan C. D.; Back D. F.; Campos M. M.; Basso L. A.; Santos D. S.; Machado P. 2-(Quinolin-4-yloxy)acetamides are active against drug-susceptible and drug-resistant Mycobacterium tuberculosis strains. ACS Med. Chem. Lett. 2016, 7, 235–239. 10.1021/acsmedchemlett.5b00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacobbo B. C.; Pissinate K.; Rodrigues-Junior V.; Villela A. D.; Grams E. S.; Abbadi B. L.; Subtil F. T.; Sperotto N.; Trindade R. V.; Back D. F.; Campos M. M.; Basso L. A.; Machado P.; Santos D. S. New insights into the SAR and drug combination synergy of 2-(quinolin-4-yloxy)acetamides against Mycobacterium tuberculosis. Eur. J. Med. Chem. 2017, 126, 491–501. 10.1016/j.ejmech.2016.11.048. [DOI] [PubMed] [Google Scholar]
- Ballell L.; Bates R. H.; Young R. J.; Alvarez-Gomez D.; Alvarez-Ruiz E.; Barroso V.; Blanco D.; Crespo B.; Escribano J.; Gonzalez R.; Lozano S.; Huss S.; Santos-Villarejo A.; Martín-Plaza J. J.; Mendoza A.; Rebollo-Lopez M. J.; Remuiñan-Blanco M.; Lavandera J. L.; Pérez-Herran E.; Gamo-Benito F. J.; García-Bustos J. F.; Barros D.; Castro J. P.; Cammack N. Fueling Open-Source Drug Discovery: 177 Small-Molecule Leads against Tuberculosis. ChemMedChem. 2013, 8, 313–321. 10.1002/cmdc.201200428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subtil F. T.; Villela A. D.; Abbadi B. L.; Rodrigues-Junior V. S.; Bizarro C. V.; Timmers L. F. S. M.; de Souza O.N.; Pissinate K.; Machado P.; López-Gavín A.; Tudó G.; González-Martín J.; Basso L. A.; Santos D. S. Activity of 2-(quinolin-4-yloxy)acetamides in Mycobacterium tuberculosis clinical isolates and identification of their molecular target by whole genome sequencing. Int. J. Antimicrob. Agents. 2018, 51, 378–384. 10.1016/j.ijantimicag.2017.08.023. [DOI] [PubMed] [Google Scholar]
- Pitta E.; Rogacki M. K.; Balabon O.; Huss S.; Cunningham F.; Lopez-Roman E. M.; Joossens J.; Augustyns K.; Ballell L.; Bates R. H.; Van der Veken P. Searching for New Leads for Tuberculosis: Design, Synthesis, and Biological Evaluation of Novel 2-Quinolin-4-yloxyacetamides. J. Med. Chem. 2016, 59, 6709–6728. 10.1021/acs.jmedchem.6b00245. [DOI] [PubMed] [Google Scholar]
- Borsoi A. F.; Paz J. D.; Abbadi B. L.; Macchi F. S.; Sperotto N.; Pissinate K.; Rambo R. S.; Ramos A. S.; Machado D.; Viveiros M.; Bizarro C. V.; Basso L. A.; Machado P. Design, synthesis, and evaluation of new 2-(quinoline-4-yloxy)acetamide-based antituberculosis agents. Eur. J. Med. Chem. 2020, 192, 112179. 10.1016/j.ejmech.2020.112179. [DOI] [PubMed] [Google Scholar]
- Palomino J.; Martin A.; Camacho M.; Guerra H.; Swings J.; Portaels F. Resazurin Microtiter Assay Plate: Simple and Inexpensive Method for Detection of Drug Resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2002, 46, 2720–2722. 10.1128/AAC.46.8.2720-2722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phummarin N.; Boshoff H. I.; Tsang P. S.; Dalton J.; Wiles S.; Barry C. E. 3rd.; Copp B. R. SAR and identification of 2-(quinolin-4-yloxy)acetamides as Mycobacterium tuberculosis cytochrome bc1 inhibitors. Med. Chem. Commun. 2016, 7, 2122–2127. 10.1039/C6MD00236F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitaku E.; Smith D. T.; Njardarson J. T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- Chemicalize was used for the pKa prediction in July 2022.; Chemicalize; ChemAxon: Budapest, Hungary. https://chemicalize.com/.
- Perdigão J.; Silva H.; Machado D.; Macedo R.; Maltez F.; Silva C.; Jordao L.; Couto I.; Mallard K.; Coll F.; Hill-Cawthorne G. A.; McNerney R.; Pain A.; Clark T. G.; Viveiros M.; Portugal I. Unraveling Mycobacterium tuberculosis genomic diversity and evolution in Lisbon, Portugal, a highly drug resistant setting. BMC Genomics. 2014, 15, 991. 10.1186/1471-2164-15-991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meerloo J.; Kaspers G. J.; Cloos J. Cell Sensitivity Assays: The MTT Assay. Methods Mol. Biol. 2011, 731, 237–245. 10.1007/978-1-61779-080-5_20. [DOI] [PubMed] [Google Scholar]
- Repetto G.; del Peso A.; Zurita J. L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. 10.1038/nprot.2008.75. [DOI] [PubMed] [Google Scholar]
- Singh V.; Chibale K. Strategies to Combat Multi-Drug Resistance in Tuberculosis. ACS Chem. Res. 2021, 54, 2361–2376. 10.1021/acs.accounts.0c00878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerns E. H.; Di L.. Drug-Like Properties: Concepts, Structure Design and Methods; Academic Press: Cambridge, MA, 2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.