

Abstract
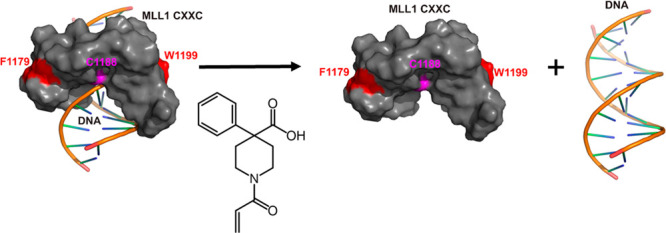
The CXXC domain is a reader of DNA methylation which preferentially binds to unmethylated CpG DNA motifs. Chromosomal translocations involving the MLL1 gene produce in-frame fusion proteins in which the N-terminal portion of the MLL1 protein harboring its CXXC domain is fused to the C-terminal portion of multiple partners. For the MLL-AF9 fusion, mutations which disrupt CXXC domain–DNA binding abrogate the ability to cause leukemia in mice. Based on this, we initiated an effort to develop small-molecule inhibitors of the MLL1 CXXC domain as a novel approach to therapy. We developed a fluorescence polarization-based assay for MLL CXXC domain–DNA binding and screened a library of Cys-reactive molecules. For the most potent hit from this screen, we have synthesized a library of analogs to explore the structure–activity relationship, defined the binding site using chemical shift perturbations in NMR spectra, and explored the selectivity of compounds across the CXXC domain family.
Keywords: MLL, CXXC domain, DNA methylation, leukemia
Epigenetic signaling requires proteins that add epigenetic marks (writers), proteins that erase epigenetic marks (erasers), and proteins that recognize the epigenetic marks (readers). There has been widespread interest in targeting these epigenetic proteins, particularly in the realm of cancer therapy, with numerous agents proceeding to clinical trials. The paradigm for small-molecule inhibition of a reader domain is bromodomain inhibitors, which started with a tool compound developed in the Bradner laboratory1 and has translated into multiple clinical candidates being evaluated.2,3 Efforts to target reader domains have also been extended to plant homeodomain (PHD) fingers, WD40 repeat domains, and Royal family readers (MBT, chromodomain, Tudor domain, PWWP domain).4−6 Thus, there is strong precedent for the targeting of reader domains for drug development.
Readout of the methylation state of DNA is carried out by small protein domains capable of binding to CpG dinucleotides and very specifically discriminating methylated versus unmethylated DNA. Methylated CpG dinucleotides are recognized by the well-characterized methyl binding domains (MBDs). The five members of the MBD-containing family of proteins are MBD1, MBD2, MBD3, MBD4, and MeCP2. To date, the only characterized domain capable of selective binding to unmethylated CpG dinucleotides is the CXXC domain. The CXXC domains from several proteins, including MLL1,7,8 MBD1, and CGBP,9 have been shown to bind DNA and recognize unmethylated CpG dinucleotides. CXXC domains have a highly conserved spacing of eight cysteines that coordinate two zinc ions and fold into a saddle-like structure that centers over an unmethylated CpG residue when bound to DNA.10,11 CXXC domains are present in 12 different mammalian proteins including MLL1, MLL2, DNMT1, CGBP, TET1, TET3, KDM2A, and KDM2B.12
All 12 of the human CXXC domain proteins are involved in epigenetic signaling. They can be broken down into various subgroups based on their epigenetic signaling functions: H3K4 methylation (CFP1, MLL1, MLL2), H3K36 demethylation (KDM2A, KDM2B), DNA methylation (DNMT1), and DNA hydroxymethylation (TET1, TET3, CXXC4 (IDAX)). A recent analysis of the binding specificities of the members of the CXXC family classified them into four groups: CpGpG (CFP1), CpG (MLL1, MLL2, FBXL19, KDM2A, KDM2B, MBD1), CpN (TET1, TET3, CXXC4, CXXC5), and weak binding (DNMT1).13
The Mixed Lineage Leukemia 1 (MLL1) gene (approved name KMT2A) at the 11q23 locus was first identified by its involvement in chromosomal translocations associated with acute leukemia. Patients with MLL1 rearrangements develop either AML, ALL, or mixed-lineage leukemia.14−16 MLL1 fusion leukemia accounts for around 10% of AML and ALL. Chromosomal translocations involving the MLL1 gene produce in-frame fusion proteins in which the N-terminal portion of the MLL1 protein harboring the CXXC domain is fused to the C-terminal portion of one of many protein partners. Regardless of the particular MLL1 fusion, MLL1 leukemia is associated with early relapse, and patients are generally classified as poor-risk.17,18
We have previously shown that MLL1 and MLL-AF9, an MLL1 fusion protein driver of leukemia, bind to specific clusters of CpG residues in the Hoxa9 locus and regulate expression of two transcripts within this locus, Hoxa9 and miR-196b.19,20 MLL1 functions to protect this specific region from DNA methylation.19 We subsequently solved the structure of the MLL1 CXXC domain–DNA complex using NMR spectroscopy,10 showing how the CXXC domain distinguishes non-methylated from methylated CpG DNA (Figure 1). Point mutations were identified that disrupt the CXXC domain DNA binding ability, in particular the Cys1188Asp mutation, which completely abrogated DNA binding. In the context of MLL-AF9, the Cys1188Asp mutation resulted in increased DNA methylation of the specific Hoxa9 region, increased H3K9 methylation (a silencing histone mark), decreased expression of the Hoxa9-locus transcripts, and abrogated both immortalization potential and ability to cause leukemia in mice.10 Thus, we established that the MLL1 CXXC domain interaction with DNA is a valid target for therapeutic intervention in MLL1 fusion leukemia. In addition, recent studies of MLL1 in the context of other cancers have established a critical role in cancers of the colon, salivary gland, and head and neck.21,22
Figure 1.

(A) Ribbon representation of the structure of the MLL1 CXXC domain–DNA complex (RCSB PDB ID: 2KKF). (B) Kd determination of the binding of GST-MLL1 CXXC domain binding to DNA using the fluorescence polarization assay. (C) Kd values and standard deviations (SD) for GST-MLL1 CXXC domain, the Cys1188Ala mutant of GST-MLL1 CXXC domain (MLL1m), and 10 other CXXC domain family members.
Based on the validated role of MLL1 CXXC domain–DNA binding in MLL-AF9 fusion protein-driven leukemia, we initiated an effort to develop small-molecule inhibitors of the MLL1 CXXC domain as a novel approach to therapy. Furthermore, small-molecule inhibitors of CXXC domains, epigenetic readers of DNA methylation, have not been described, so inhibitors would provide tools to effectively probe the functional role of this domain in different biological settings. There are very few examples of well-characterized small-molecule inhibitors of protein–DNA interactions which target the protein. Reports of inhibitors of the far-upstream element binding protein (FBP),23 a ssDNA binding protein, and of FOXM1 inhibitors24 are among the few examples of which we are aware. Successful targeting of a protein–DNA interaction would add an additional example of successful targeting of this class of interactions.
In order to be able to express and purify the MLL1 CXXC domain, MLL1 residues 1147–1203 were cloned into the pGEX-4T1 vector and expressed in BL21 E. coli cells. The protein was purified first on a glutathione affinity column followed by ion exchange chromatography. In order to be able to assess the specificity of inhibitors we developed, we also cloned and expressed the CXXC domains of 10 additional family members: CGBP: 161–217; MLL2: 958–1014; KDM2A: 562–618; KDM2B: 604–660; TET1: 583–633; TET3: 49–98; CXXC5: 255–305; IDAX: 130–181; DNMT1: 645–700; FBXL19: 31–86. These were expressed and purified using the same methods as for the MLL1 CXXC domain.
To measure DNA binding of the CXXC domains, we employed a fluorescence polarization (FP)-based assay with a fluorescein-labeled dsDNA oligonucleotide (5′-GGGTCGCGGGAG-3′), the sequence of which was based on our previous studies of binding of the MLL1 CXXC domain to DNA.19KD values were measured for all the CXXC domains via addition of increasing concentrations of the CXXC domain to a fixed concentration of the oligonucleotide (see Figure 1 for binding data for the MLL1 CXXC domain and measured values for all the CXXC domains). As mentioned above, there are some differences in binding preference for different CXXC domains, so the oligonucleotide used for the assays may not be optimal for other CXXC domains, but it does bind to all the proteins; we have done competition assays for compounds at a fixed multiple of the determined KD value for each CXXC domain (2KD).
As mentioned above, there are very few examples of successful targeting of protein–DNA binding. This is likely the result of the fact that the surfaces that mediate binding to DNA are typically convex and highly positively charged, making them more difficult surfaces to find drug-like molecules which can bind. Interestingly, many DNA binding domains have Cys residues located on or near their DNA binding interfaces, typically as a mechanism to regulate activity via redox signaling. Because of the unique nucleophilicity of the Cys sulfur atom, these Cys residues present an opportunity to identify compounds which can covalently react with the Cys to mediate inhibition of DNA binding. Similar approaches were used in the development of the covalent kinase inhibitors afatinib (targeting EGFR) and ibrutinib (targeting Bruton’s tyrosine kinase) which were approved by the FDA for non-small-cell lung cancer and chronic lymphocytic leukemia, respectively.25−27 These inhibitors covalently react with a Cys on the target kinase in each case. As described above, we have solved the structure of the MLL1 CXXC domain–DNA complex using NMR.10 The structure shows that all but one of the Cys residues (Cys1188) are chelated by one of the two Zn atoms to stabilize the structure. Cys1188 is exposed and located on the DNA binding interface. As mentioned above, replacement of Cys1188 with an Asp results in a complete loss of binding to DNA, and an MLL1-AF9 fusion protein with the Cys1188Asp mutation was unable to cause leukemia in vivo.
Based on the critical location of Cys1188 on the DNA binding interface, we screened a 1000 compound Cys reactive library from Enamine using the FP assay. This resulted in the identification of six compounds (structures in Figure S1) which showed inhibition of the CXXC domain–DNA interaction but were not active in screens against 3 other unrelated targets, indicating they show specificity of action. The most potent compound identified in this screen, 1, is shown in Figure 2A, with an IC50 of 7 μM in the FP assay (Figure 2B, Table 1). To distinguish whether a compound is acting as a reversible or irreversible inhibitor, it is necessary to analyze the kinetics of the reaction between the compound and the protein.28 We have carried out a kinetics analysis of the inhibition of the CXXC domain with this compound by monitoring the FP versus time. We did not see any time dependence of the reaction (Figure S2), indicating that this compound interacts with the CXXC domain reversibly. In addition, MALDI mass spectrometry analysis does not show a stable adduct being formed (Figure S3), consistent with reversible binding. In order to confirm that the compound does bind to the MLL1 CXXC domain, we recorded 15N–1H and 13C–1H HSQC spectra of the CXXC domain in the presence of 1. While we observe only modest chemical shift changes in the 15N–1H HSQC or in the aliphatic region of the 13C–1H HSQC, we clearly observe significant chemical shift perturbations for resonances belonging to the aromatic ring of Phe1179 (Figure 2C), confirming binding of the protein to the CXXC domain and identifying the site on the protein where binding occurs. This site of binding clearly overlaps with the surface on the protein which mediates DNA binding (Figure 2D), indicating the compound acts by competitively blocking the binding of the DNA. Finally, we mutated Cys1188 to Ala and assayed the compound, and we do not see a reduction in activity (Table 1), indicating it is not interacting with the Cys1188 sulfur. It is possible it is covalently interacting in a reversible manner with some other residue on the protein, but the simplest rationale for our data is that the compound is acting as a standard reversible inhibitor.
Figure 2.

(A) Structure of the initial hit. (B) Results of IC50 determination using the competition FP assay for the initial hit 1. (C) Selected region of the 13C–1H HSQC spectrum of the MLL1 CXXC domain alone (red) and in the presence of compound 1 (blue). Text shows assignments for the peaks in the MLL1 CXXC domain. (D) Surface representation of the MLL1 CXXC domain bound to DNA (stick representation), with the side chain of Phe1179 highlighted in red and that of Cys1188 in magenta.
Table 1. (Top) Measured IC50 Values for Inhibitors with the MLL1 CXXC Domain and (Bottom) Measured IC50 Values for Selected Compounds with CXXC Domains from 10 Additional Members of the CXXC Domain Protein Familya.

| compound
IC50 (μM) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| CXXC domain | 1 | 1a | 1c | 1e | 3 | 3a | 3b | 3c | 4 |
| MLL1 | 7.1 ± 0.4 | 41 ± 4 | 43 ± 4 | 3.3 ± 0.4 | 4.9 ± 0.1 | 15 ± 1 | 22 ± 1 | 7.7 ± 0.7 | 47 ± 3 |
| MLL1 (C1188A) | 3.6 ± 0.4 | 108 ± 6 | 51 ± 3 | 15.4 ± 0.6 | 20 ± 1 | 59 ± 1 | 104 ± 12 | 43 ± 2 | 52 ± 2 |
| MLL2 | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| TET1 | 11.3 ± 1.1 | 166 ± 23 | 81 ± 6 | 6.3 ± 1.4 | 11 ± 2 | 31 ± 6 | 24 ± 3 | 10.7 ± 0.9 | 107 ± 17 |
| TET3 | 5.1 ± 1.1 | 48 ± 1 | 30 ± 3 | 5.8 ± 0.8 | 5.5 ± 0.2 | 15.5 ± 0.4 | 21 ± 2 | 7 ± 1 | 33 ± 4 |
| KDM2A | 3.9 ± 0.5 | 33 ± 4 | 30 ± 2 | 4.2 ± 0.8 | 4.4 ± 0.7 | 16.4 ± 0.6 | 36 ± 0.2 | 7.4 ± 0.9 | 30 ± 3 |
| KDM2B | 6.4 ± 0.8 | 53 ± 3 | 53 ± 3 | 3.6 ± 0.4 | 8.3 ± 0.8 | 32 ± 4 | 30 ± 4 | 9.4 ± 0.7 | 35 ± 5 |
| IDAX | 4.0 ± 0.4 | 33 ± 1 | 35 ± 3 | 1.2 ± 0.3 | 2.4 ± 0.7 | 8.3 ± 1.2 | 8.9 ± 1.6 | 2.2 ± 0.7 | 23 ± 3 |
| FBXL19 | 1.8 ± 0.2 | 31 ± 4 | 17 ± 1 | 2.4 ± 0.5 | 3.1 ± 0.3 | 8.9 ± 1.9 | 10.9 ± 0.8 | 2.6 ± 0.1 | 21.7 ± 0.4 |
| CXXC5 | 4.8 ± 0.3 | 53 ± 5 | 47 ± 3 | 16.2 ± 0.3 | 28 ± 5 | 56 ± 7 | 131 ± 17 | 40 ± 3 | 53 ± 7 |
| CGBP | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| DNMT1 | NA | NA | NA | NA | NA | NA | NA | NA | NA |
NA = no activity. Standard deviations are calculated from three separate measurements.
We tested this compound for its ability to inhibit the growth of an MLL1 fusion containing leukemia cell line (MV4-11) as well as a non-MLL1 fusion leukemia cell line (K562). We do not observe activity for the free acid form of the compound, likely due to poor cellular permeability of this very polar molecule. We then made the ethyl ester derivative 1h, which was active against the MV4-11 cell line but not against the K562 cell line (Figure 3), likely via cellular esterase-mediated hydrolysis to produce the active acid form of the compound. The observed selectivity is consistent with activity occurring through targeting of the MLL1 CXXC domain and suggests there will be specificity of action in targeting MLL1 fusion-containing cells.
Figure 3.
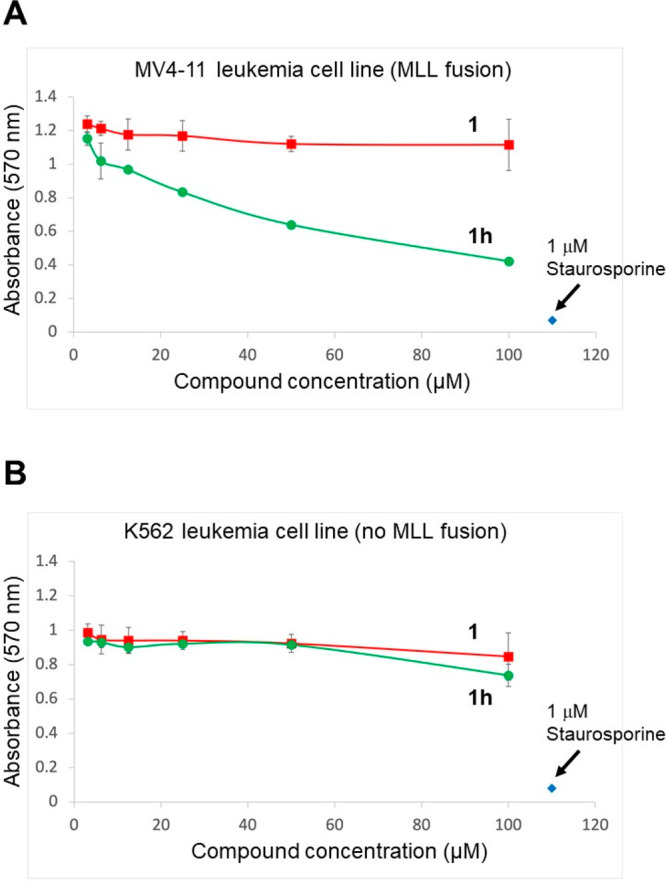
(A) Concentration-dependent effects of compounds 1 and 1h on the growth of the MV4-11 leukemia cell line after 72 h, measured using an MTT assay. Results of three replicates are shown. (B) Concentration-dependent effects of compounds 1 and 1h on the growth of the K562 leukemia cell line after 72 h, measured using an MTT assay. Results of three replicates are shown.
Our initial hit 1 was chosen to explore optimization based on its favorable ligand efficiency (LE) value of 0.42. Table 1 shows the initial library of analogs synthesized to explore structure–activity relationships (SARs) as well as the effects of tetrazole and sulfonamide bioisostere replacements for the carboxylate moiety. Multiple substitutions for the acrylamide moiety were explored (compounds 2a–2j), including propionamide 2a, but all proved to be inactive, indicating substitutions at this site are not tolerated. The compound series 1–1h explored a number of substitutions on the phenyl ring. Introduction of 4-Cl, 4-Me, 4-OMe, and 4-N(CH3)2 substitutions on the phenyl ring decreased activity. Introduction of ethoxy functionality at the 4 position improved activity, whereas introduction at the 3 position decreased activity, likely indicating a steric clash with the 3 substitution. It is notable that the 4-ethoxy substitution improved activity, whereas the 4-methoxy substitution decreased activity, suggesting introduction of more lipophilic substituents at this position may be favored. Modification of the carboxylate to an amide (1g) or an ethyl ester (1h) resulted in complete loss of activity.
As the carboxylate moiety is likely to be the primary determinant for the lack of cellular activity due to an inability to cross the cell membrane, we have also explored bioisostere replacements for the carboxylate, namely tetrazole (compounds 3a–3c) and sulfonamide (compounds 4a–4e). Introduction of the tetrazole improved activity however additional substitutions on the phenyl ring all decreased activity. Unlike the carboxylate series, a 3-ethoxy substituent on the phenyl was more active than the 4-ethoxy substitution, implying a different orientation of the tetrazole compound in the binding site on the CXXC domain. Tetrazole analog 3 was not active on cells, so introduction of this moiety did not alter the putative lack of cellular permeability. All of the sulfonamide derivatives (4a–4e) were significantly weaker in activity than 1.
As described above, we have expressed and purified the CXXC domains from 11 proteins and established FP-based assays with all of them. This makes it possible to assess the specificity of the compounds we have synthesized. Table 1 shows a tabulation of the IC50 values with the 11 CXXC domains for all the compounds with IC50 values of 50 μM or better for the MLL1 CXXC domain. The most striking difference observed is the complete lack of activity of these compounds on the CXXC domains of MLL2, CGBP, and DNMT1, indicative of some degree of specificity within the CXXC domain family. After replication, DNA methylation at CpG sites is maintained by DNMT1,29 so the lack of activity against the DNMT1 CXXC domain is likely to be advantageous. Indeed, loss of DNMT1 has been shown to lead to genomic instability and cancer.30 Generally speaking, the compounds shown are active against the other eight CXXC domains, with some limited degree of selectivity for various family members. It is notable that 1e shows approximately 5-fold selectivity for MLL1 versus CXXC5, whereas 1 does not show such selectivity. This suggests that introduction of more lipophilic groups at the 4 position of the phenyl ring may not only improve activity but may also improve specificity to some degree. It is notable that the compounds shown retain good activity against the TET1 CXXC domain. TET1 has been shown to be essential for MLL fusion driven leukemia and has been suggested as a therapeutic target.31 Therefore, a CXXC domain inhibitor with activity against both the MLL1 and TET1 CXXC domains is likely to be more efficacious by virtue of hitting two relevant targets in the disease. Furthermore, such polypharmacology is likely to be much more detrimental to MLL fusion leukemia cells than normal cells, so it is also likely to improve the therapeutic index.
Experimental Procedures
Detailed experimental procedures and characterization data for all compounds are provided in the Supporting Information.
Glossary
Abbreviations
- MLL1
mixed lineage leukemia 1 (KMT2A, lysine methyltransferase 2A)
- MLL1-AF9
fusion protein of MLL1 and AF9 in subset of leukemia patients
- HSQC
heteronuclear single-quantum coherence
- LE
ligand efficiency
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00198.
Figures S1–S3, biochemical and cell culture procedures, procedures for chemical synthesis, and characterization data for all compounds (PDF)
Author Present Address
† Medicines For All Institute, Virginia Commonwealth University, Richmond, VA 23298, USA
Author Present Address
‡ Apellix, 2180 Emerson St., Jacksonville, FL 32207, USA
Author Contributions
A.B. carried out the screen of the Enamine library. H.P.K., C.L., and P.R. synthesized inhibitors. I.P. purified proteins and characterized compound effects using the FP assay. A.K. collected and analyzed NMR data and characterized compound effects with the FP assay. Y.G. characterized effects on cell lines. J.H.B. analyzed data and wrote the manuscript.
This work was supported by a grant from the National Cancer Institute (R21 CA241005).
The authors declare no competing financial interest.
Special Issue
Published as part of the ACS Medicinal Chemistry Letters special issue “Epigenetics 2022”.
Supplementary Material
References
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective inhibition of BET bromodomains. Nature 2010, 468 (7327), 1067–1073. 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alqahtani A.; Choucair K.; Ashraf M.; Hammouda D. M.; Alloghbi A.; Khan T.; Senzer N.; Nemunaitis J. Bromodomain and extra-terminal motif inhibitors: a review of preclinical and clinical advances in cancer therapy. Future Sci. OA 2019, 5 (3), FSO372. 10.4155/fsoa-2018-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran A. G.; Conery A. R.; Sims R. J. III Bromodomains: a new target class for drug development. Nat. Rev. Drug Discovery 2019, 18 (8), 609–628. 10.1038/s41573-019-0030-7. [DOI] [PubMed] [Google Scholar]
- Cipriano A.; Sbardella G.; Ciulli A. Targeting epigenetic reader domains by chemical biology. Curr. Opin. Chem. Biol. 2020, 57, 82–94. 10.1016/j.cbpa.2020.05.006. [DOI] [PubMed] [Google Scholar]
- Arrowsmith C. H.; Schapira M. Targeting non-bromodomain chromatin readers. Nat. Struct. Mol. Biol. 2019, 26 (10), 863–869. 10.1038/s41594-019-0290-2. [DOI] [PubMed] [Google Scholar]
- Mio C.; Bulotta S.; Russo D.; Damante G. Reading Cancer: Chromatin Readers as Druggable Targets for Cancer Treatment. Cancers (Basel) 2019, 11 (1), 61. 10.3390/cancers11010061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade P. A. Methyl CpG-binding proteins and transcriptional repression. Bioessays 2001, 23 (12), 1131–1137. 10.1002/bies.10008. [DOI] [PubMed] [Google Scholar]
- Birke M.; Schreiner S.; Garcia-Cuellar M. P.; Mahr K.; Titgemeyer F.; Slany R. K. The MT domain of the proto-oncoprotein MLL binds to CpG-containing DNA and discriminates against methylation. Nucleic Acids Res. 2002, 30 (4), 958–965. 10.1093/nar/30.4.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H.; Voo K. S.; Skalnik D. G. Identification and characterization of the DNA binding domain of CpG-binding protein. J. Biol. Chem. 2001, 276 (48), 44669–44676. 10.1074/jbc.M107179200. [DOI] [PubMed] [Google Scholar]
- Cierpicki T.; Risner L. E.; Grembecka J.; Lukasik S. M.; Popovic R.; Omonkowska M.; Shultis D. D.; Zeleznik-Le N. J.; Bushweller J. H. Structure of the MLL CXXC domain-DNA complex and its functional role in MLL-AF9 leukemia. Nat. Struct. Mol. Biol. 2010, 17 (1), 62–68. 10.1038/nsmb.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen M. D.; Grummitt C. G.; Hilcenko C.; Min S. Y.; Tonkin L. M.; Johnson C. M.; Freund S. M.; Bycroft M.; Warren A. J. Solution structure of the nonmethyl-CpG-binding CXXC domain of the leukaemia-associated MLL histone methyltransferase. EMBO J. 2006, 25, 4503. 10.1038/sj.emboj.7601340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H. K.; Blackledge N. P.; Klose R. J. ZF-CxxC domain-containing proteins, CpG islands and the chromatin connection. Biochem. Soc. Trans. 2013, 41 (3), 727–740. 10.1042/BST20130028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C.; Liu K.; Lei M.; Yang A.; Li Y.; Hughes T. R.; Min J. DNA Sequence Recognition of Human CXXC Domains and Their Structural Determinants. Structure 2018, 26 (1), 85–95. 10.1016/j.str.2017.11.022. [DOI] [PubMed] [Google Scholar]
- Stock W.; Thirman M. J.; Dodge R. K.; Rowley J. D.; Diaz M. O.; Wurster-Hill D.; Sobol R. E.; Davey F. R.; Larson R. A.; Westbrook C. A.; et al. Detection of MLL gene rearrangements in adult acute lymphoblastic leukemia. A Cancer and Leukemia Group B study. Leukemia 1994, 8 (11), 1918–1922. [PubMed] [Google Scholar]
- Rowley J. D. Rearrangements involving chromosome band 11Q23 in acute leukaemia. Seminars Cancer Biol. 1993, 4 (6), 377–385. [PubMed] [Google Scholar]
- Thirman M. J.; Gill H. J.; Burnett R. C.; Mbangkollo D.; McCabe N. R.; Kobayashi H.; Ziemin-van der Poel S.; Kaneko Y.; Morgan R.; Sandberg A. A.; et al. Rearrangement of the MLL gene in acute lymphoblastic and acute myeloid leukemias with 11q23 chromosomal translocations. New Engl. J. Med. 1993, 329 (13), 909–914. 10.1056/NEJM199309233291302. [DOI] [PubMed] [Google Scholar]
- van der Linden M. H.; Creemers S.; Pieters R. Diagnosis and management of neonatal leukaemia. Seminars Fetal Neonatal Med. 2012, 17 (4), 192–195. 10.1016/j.siny.2012.03.003. [DOI] [PubMed] [Google Scholar]
- Khasawneh M. K.; Abdel-Wahab O. Recent discoveries in molecular characterization of acute myeloid leukemia. Curr. Hematol. Malignancy Rep. 2014, 9 (2), 93–99. 10.1007/s11899-014-0200-y. [DOI] [PubMed] [Google Scholar]
- Erfurth F. E.; Popovic R.; Grembecka J.; Cierpicki T.; Theisler C.; Xia Z. B.; Stuart T.; Diaz M. O.; Bushweller J. H.; Zeleznik-Le N. J. MLL protects CpG clusters from methylation within the Hoxa9 gene, maintaining transcript expression. Proc. Natl. Acad. Sci. U.S.A. 2008, 105 (21), 7517–7522. 10.1073/pnas.0800090105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic R.; Riesbeck L. E.; Velu C. S.; Chaubey A.; Zhang J.; Achille N. J.; Erfurth F. E.; Eaton K.; Lu J.; Grimes H. L.; Chen J.; Rowley J. D.; Zeleznik-Le N. J. Regulation of mir-196b by MLL and its overexpression by MLL fusions contributes to immortalization. Blood 2009, 113 (14), 3314–3322. 10.1182/blood-2008-04-154310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grinat J.; Heuberger J.; Vidal R. O.; Goveas N.; Kosel F.; Berenguer-Llergo A.; Kranz A.; Wulf-Goldenberg A.; Behrens D.; Melcher B.; Sauer S.; Vieth M.; Batlle E.; Stewart A. F.; Birchmeier W. The epigenetic regulator Mll1 is required for Wnt-driven intestinal tumorigenesis and cancer stemness. Nat. Commun. 2020, 11 (1), 6422. 10.1038/s41467-020-20222-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q.; Fang L.; Heuberger J.; Kranz A.; Schipper J.; Scheckenbach K.; Vidal R. O.; Sunaga-Franze D. Y.; Muller M.; Wulf-Goldenberg A.; Sauer S.; Birchmeier W. The Wnt-Driven Mll1 Epigenome Regulates Salivary Gland and Head and Neck Cancer. Cell Rep. 2019, 26 (2), 415–428. 10.1016/j.celrep.2018.12.059. [DOI] [PubMed] [Google Scholar]
- Huth J. R.; Yu L.; Collins I.; Mack J.; Mendoza R.; Isaac B.; Braddock D. T.; Muchmore S. W.; Comess K. M.; Fesik S. W.; Clore G. M.; Levens D.; Hajduk P. J. NMR-driven discovery of benzoylanthranilic acid inhibitors of far upstream element binding protein binding to the human oncogene c-myc promoter. J. Med. Chem. 2004, 47 (20), 4851–4857. 10.1021/jm0497803. [DOI] [PubMed] [Google Scholar]
- Gormally M. V.; Dexheimer T. S.; Marsico G.; Sanders D. A.; Lowe C.; Matak-Vinkovic D.; Michael S.; Jadhav A.; Rai G.; Maloney D. J.; Simeonov A.; Balasubramanian S. Suppression of the FOXM1 transcriptional programme via novel small molecule inhibition. Nat. Commun. 2014, 5, 5165. 10.1038/ncomms6165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating G. M. Afatinib: a review of its use in the treatment of advanced non-small cell lung cancer. Drugs 2014, 74 (2), 207–221. 10.1007/s40265-013-0170-8. [DOI] [PubMed] [Google Scholar]
- Dungo R. T.; Keating G. M. Afatinib: first global approval. Drugs 2013, 73 (13), 1503–1515. 10.1007/s40265-013-0111-6. [DOI] [PubMed] [Google Scholar]
- Cramer P.; Hallek M.; Eichhorst B. State-of-the-Art Treatment and Novel Agents in Chronic Lymphocytic Leukemia. Oncol. Res. Treatment 2016, 39 (1–2), 25–32. 10.1159/000443903. [DOI] [PubMed] [Google Scholar]
- Strelow J. M. A Perspective on the Kinetics of Covalent and Irreversible Inhibition. SLAS Discovery: Adv. Life Sci. R&D 2017, 22 (1), 3–20. 10.1177/1087057116671509. [DOI] [PubMed] [Google Scholar]
- Goll M. G.; Bestor T. H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- Gaudet F.; Hodgson J. G.; Eden A.; Jackson-Grusby L.; Dausman J.; Gray J. W.; Leonhardt H.; Jaenisch R. Induction of tumors in mice by genomic hypomethylation. Science 2003, 300 (5618), 489–492. 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- Huang H.; Jiang X.; Li Z.; Li Y.; Song C. X.; He C.; Sun M.; Chen P.; Gurbuxani S.; Wang J.; Hong G. M.; Elkahloun A. G.; Arnovitz S.; Wang J.; Szulwach K.; Lin L.; Street C.; Wunderlich M.; Dawlaty M.; Neilly M. B.; Jaenisch R.; Yang F. C.; Mulloy J. C.; Jin P.; Liu P. P.; Rowley J. D.; Xu M.; He C.; Chen J. TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc. Natl. Acad. Sci. U.S.A. 2013, 110 (29), 11994–11999. 10.1073/pnas.1310656110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.